

ABSTRACT
The improved survival rate of ovarian cancer (OC) is related to the action of infiltrating cytotoxic T lymphocytes (CTLs). Recently, oncolytic adenoviruses (OAds) have emerged as a key player in treating solid tumors; however, the immunosuppressive tumor microenvironment (TME) and the body-mediated antiviral immune response limit their therapeutic effect. In this study, we tested the hypothesis that bispecific T-cell engagers (BiTEs) could activate and redirect CTLs to increase the anti-tumor effect of OAds. We modified the parental OAd to express a MUC16-targeting BiTE antibody (OAd-MUC16-BiTE), which retained its oncolytic properties and replication ability in vitro. This BiTE secreted from infected tumor cells into the microenvironment binds to MUC16 on target cells and cross‐links them to CD3 on T cells, leading to activation, proliferation, and toxicity of T cells against MUC16+ tumor cells. In cell coculture assays, OAd-MUC16-BiTE–mediated oncolysis enhanced T-cell–mediated tumor cell killing and bystander effect. In ex vivo tumor cultures freshly derived from OC patients, OAd-MUC16-BiTE overcame the suppressed immune TME, achieving stronger toxicity than the parental virus. Moreover, in the cell-derived xenograft and patient-derived xenograft model, OAd-MUC16-BiTE showed stronger antitumor activity and increased the number of CTLs, compared with the parental virus. Further, we demonstrated that the OAd-MUC16-BiTE-mediated anti-tumor activity is related to the reversal of the TME and improved MHC I antigen presentation. Overall, our results show how arming OAds with BiTE can overcome limitations in oncolytic virotherapy, yielding a potent therapy that is ready for clinical assessment.
KEYWORDS: Oncolytic virus, adenovirus, bispecific T-cell engagers, cytotoxic T lymphocyte, tumor microenvironment, immunotherapy, ovarian cancer, PDX
Introduction
Ovarian cancer (OC) is the most fatal tumor of the female reproductive system.1 Recent advancements in cancer screening, diagnosis, and treatment have improved the survival rates of many cancer types; however, those of patients with OC remain poor.2 Therefore, there is an urgent need to develop novel methods for OC treatment. Accumulating evidence suggests that the presence of cytotoxic T lymphocytes (CTLs) is associated with better prognosis and improved survival for patients with OC,3–5 implying that they could potentially benefit from immunotherapy.
Oncolytic adenoviruses (OAds) have been successfully engineered to selectively enter and lyse tumor cells. OAds can induce anti-tumor immune responses in the host by releasing tumor-associated antigens, pathogen-associated molecular pattern molecules, damage-associated molecular pattern molecules, cytokines, and chemokines during infection.6–8 Various OAds have been investigated in preclinical and clinical studies and have shown tolerable safety and promising efficacy;9,10however, the therapeutic effect of OAd monotherapy is limited by the body-mediated antiviral response, and immunosuppressive tumor microenvironment (TME).11,12
Bispecific T-cell engager (BiTE) is an antibody construct comprising two single-chain antibody fragments (scFvs), one of which is for tumor-associated antigen (TAA) on target cells and the other for CD3 on T cells.13 Blinatumomab, a BiTE-targeting CD3 and CD19, gained FDA approval for the treatment of relapsed/refractory precursor B cell acute lymphoid leukemia.14 BiTE is independent of human leukocyte antigen presentation and can activate T cells to bind and subsequently kill adjacent target cells.15 In addition, BiTE-mediated T cell activation can overcome the immunosuppressive TME, leading to the activation and proliferation of exhausted tumor-specific T cells.16,17 However, BiTE has a limited effect on solid tumors owing to toxicity caused by systemic administration and penetration to the TME.18,19 If BiTEs can be continuously expressed locally in the tumor, it can stimulate tumor-infiltrating lymphocytes (TILs) without systemic toxicity. Interestingly, the use of oncolytic viruses as vectors of BiTE can address this issue.17,20,21
MUC16 is a highly glycosylated mucin overexpressed in most OCs. It consists of a large cleavage and release domain termed CA-125, composed of multiple repeat sequences and a retention domain (MUC-CD).22 CA-125 is a serum marker for the diagnosis of OC. MUC-CD is retained on the cell surface and is expressed at low levels in normal tissues, making it an attractive target.23
This study, constructed an oncolytic adenovirus, OAd-MUC16-BiTE, that secretes BiTE targeting MUC16 (exactly MUC-CD) when replicating in cancer cells. We hypothesized that after OAd-MUC16-BiTE infection induces a tumor immune response – various signals, such as chemokines, released by infected cells in response to the virus attract T cells, and then BiTEs direct T cells to MUC16+ tumor cells, leading the attracted activated T cells to increase attraction. More and more T cells are attracted, which addresses the limitations of OAd treatment. Conversely, BiTE can be continuously expressed locally in the tumor, and be optimized to reduce systemic exposure.
Materials and methods
Cell lines and culture
The human OC cell lines SKOV3, OVCAR3, and CAOV3 were purchased from the American Type Culture Collection (ATCC), whereas A2780, HEY, and HEK293T cell lines were provided by the Chinese Academy of Sciences (Shanghai, China). HEK293A cells were kindly provided by Fubio (Shanghai, China). SKOV3 and A2780 cells were cultured in RPMI 1640 (Gibco, USA) containing 10% (v/v) fetal bovine serum (FBS). OVCAR3 cells were cultured in RPMI 1640 containing 20% FBS. HEY, HEK293T, and HEK293A cells were cultured in Dulbecco’s modified eagle medium (DMEM; Gibco) containing 10% FBS. CAOV3 cells were cultured in DMEM with 15% FBS. All cell lines were maintained at 37°C with 5% CO2 in a humidified incubator. Cell lines were authenticated by the ATCC human STR profiling cell authentication service and routinely checked for Mycoplasma.
Stable transfection
The DNA sequence encoding MUC16 (exactly MUC-CD) was synthesized by Genechem (Shanghai, China) and ligated into the plasmid GV260 (Genechem) expressing firefly luciferase. The lentivirus was obtained using the HEK293T cell line packaged with pMD2.G (Addgene) and psPAX2 (Addgene) vectors. After adding the lentivirus with the multiplicity of infection (MOI) value ranging from 20 to 40 to the cells to be infected for 24 h, they were selected with a medium containing 2 μg/mL puromycin (Sigma-Aldrich) for 1 week, and the cell lines HEY-LUC and HEY-MUC16-ffLuc (abbreviated as HEY-MUC16) and SKOV3-LUC, and SKOV3-MUC16-ffLuc (abbreviated as SKOV3-MUC16) overexpressing ffLuc and MUC16-ffLuc, respectively, were obtained.
Preparation of T cell and single-cell tumor digests
Blood samples were obtained from healthy volunteers. Peripheral blood mononuclear cells (PBMCs) were isolated from Ficoll (Solarbio) by density-gradient centrifugation. CD3+ cells were extracted by depleting non-CD3 cells using the MojoSort™ Human CD3 T Cell Isolation Kit (BioLegend). T cells were cultured with CD3/CD28-activating Dynabeads (Thermo Fisher Scientific) at a bead-to-cell ratio of 3:1 for stimulation. T cells were cultured in RPMI 1640 supplemented with 10% FBS, 20 mM HEPES buffer (Gibco), 1% penicillin, and streptomycin (Gibco). T cells used in all experiments were purified via magnetic bead cell sorting unless otherwise stated.
Fresh OC tumor tissues were isolated in the operating room, placed in the culture medium, and immediately sent to a super clean bench. OC tumors were diced into 1–5 mm3 pieces and placed in a 50 mL falcon tube containing RPMI 1640 supplemented with 1% pen/strep, 100 mg/L collagenase type I (Diamond), 100 mg/L collagenase type IV (Diamond), and 10 mg/L DNase I (BBI) for 4–8 h enzymatic digestion with rocking at 37°C. After digestion, the cell suspension was filtered with a 100 μm filter and treated with 1× RBC Lysis Buffer (BioLegend) to remove undigested fragments and red cells. Fresh cells were resuspended in DMEM containing 20% FBS supplemented with 1% pen/strep. The complete clinical characteristic of these patients is reported in Supplemental Table S1.
Generation of OAd expressing BiTE
The parental virus OAd without the transgene was purchased from Fubio. Based on the type 5 adenovirus, the specific promoter hTERT is used to start the E1A/B region, which controls the replication of the virus24 and inserts the RGD sequence into fiber to increase the affinity of the virus.25
A MUC16-targeted BiTE was produced by joining the DNA encoding two scFvs recognizing human MUC16 and CD3ε with a sequence encoding a flexible (glycine)4-serine (G4S) linker, in which anti-MUC16 scFv (4H11) sequence is from patent application US9790283, and the anti-CD3 scFv sequence is from patent application WO2004106381. An N-terminal human IgK signal peptide for mammalian secretion and a C-terminal 6× His tag for detection were added.
Modified OAd was produced by the direct insertion of the BiTE cassette under the control of the CMV promoter into the parental OAd cloning plasmid. Plasmid DNA was linearized by restriction digestion with AscI (New England Biolabs) and transfected into HEK293A cells to produce OAd-MUC16-BiTE. Once an extensive cytopathic effect was observed, the virus was harvested from HEK293A cells using three freeze–thaw cycles. Single clones were selected by serial dilution and amplified by serial infection, followed using an Adeno-X™ Virus Purification Kit (Takara) to produce concentrated and purified virus stocks. These stocks were titrated using the TCID50 assay (PFU/ml).26
Replication activity of recombinant virus
HEY cells were infected with OAd or OAd-MUC16-BiTE (MOI = 100). After 6 h, the virus-free medium was replaced. At 0, 1, 2, 3, and 4 days post-infection, 200 μL of the culture supernatant was drawn, and the DNA was extracted using a TIANamp Virus DNA/RNA Kit (TIANGEN, China). The DNA concentration was adjusted to 100 ng/µL.
Total viral genomes were quantified using absolute quantitative qPCR against the Ad5 E4 gene with specific primers (forward, 5′-GGAGTGCGCCGAGACAAC and reverse, 5′-ACTACGTCCGGCGTTCCAT). A standard curve was made by measuring the threshold cycle (Ct) value of plasmid pHelper (GeneChem) (Supplemental Figure S1.).
Expression of BiTE in recombinant virus
HEY cells were infected with OAd or OAd-MUC16-BiTE (MOI = 10) or without the virus as a blank control. After 48 h, cells were collected and lysed in RIPA lysis buffer (Beyotime, China) with 1% PMSF and 1% NaF by incubating on ice for 30 min. The supernatant was obtained by centrifugation, and protein concentration was determined using a BCA Assay Kit (Thermo Fisher Scientific). Protein samples (30 μg per well) were separated by SDS-PAGE (5.5% stacking gel and 11% separation gel), transferred onto PVDF membranes (Millipore) by BIO-RAD Trans-blot (15 V, 90 min), and blocked in 5% non-fat milk solution at 25°C for 2 h. Next, the membranes were incubated overnight at 4°C in diluted 6× His (BioLegend, 1:1000 dilution) primary antibodies and rinsed with TBST before incubation with the appropriate horseradish peroxidase-linked secondary antibodies for 1.5 h at 25°C. Band signals were detected using an enhanced chemiluminescence detection kit (PerkinElmer) with Image Quant LAS 4000 (GE Healthcare Life Sciences). β-Actin was used as an endogenous control.
Flow cytometry analysis
Flow cytometry was performed on a CytoFLEX flow cytometer (Beckman Coulter) and FACSCalibur™ flow cytometer (BD Biosciences), and data were processed using the FlowJo software version 10.6.2 (TreeStar Inc., USA). A representative example of flow cytometry data analysis is shown in Supplemental Figure S2. Incubation with mouse anti-MUC-CD recombinant antibody (clone 4H11; Creative Biolabs) and goat anti-mouse IgG (Abcam) coupled with Alexa Fluor 488 was performed to detect the expression of MUC16. For the analysis of T cell populations, the following antibody clones conjugated with different fluorophores were used: CD3, CD4, CD8, CD69, CD25, CD107a, LAG3, TIGIT, PD-1, TIM-3 and Ki67 (all from BioLegend). Matched isotype control antibodies were also used where appropriate.
To evaluate cytokines in vitro, appropriate CBA Flex Sets were used, and FCAP Array version 3.0 software (Becton Dickinson, Franklin Lakes, NJ, USA) was used for data analysis. In one case, OC tumor single cells from fresh OC tumor tissues (1 × 106) were infected with OAd or OAd-MUC16-BiTE (MOI = 100), and the supernatant was collected 72 h after infection. Cytokines were assessed using the human IL-10 and TGF-β CBA Flex Sets (BD Biosciences).
The antibodies are listed in Supplemental Table S2.
Production of supernatants
HEY cells (1 × 107) were infected with OAd or OAd-MUC16-BiTE (MOI = 20), and the supernatant was collected 72 h after infection. Supernatants were concentrated (approximately 20×) with Amicon Ultra-15 filter units with a molecular weight cutoff of 30 kDa (Merck Millipore). MUC16-BiTE protein concentration was determined by His Tag ELISA Detection Kit (Genscript, China). Supernatants from uninfected cells were used as the negative controls.
In vitro co-culture experiments
A total of 2 × 104 SKOV3/SKOV3-MUC16 or HEY/HEY-MUC16 cells/well (SKOV3-LUC or HEY-LUC cells were used when appropriate) and 1 × 105 unstimulated T cells (E: T = 5:1) were seeded in 96-well plates in 100 μL medium. The co-cultures were mixed with 100 μL of the supernatant and incubated for the indicated times to assess T-cell activation by the supernatants from virus-infected cells. For T-cell activation assays, co-cultures were incubated for 24 h as described above, and cells were stained with antibodies specific for CD3, CD8, CD4, CD69 and CD25. For proliferation assays, T cells were labeled with 1 μL 5 mM CFSE (BioLegend) and co-cultured as abovementioned for 3 days. The cells were then stained with CD3 antibodies. Ki67 also was stained when co-cultured as abovementioned for 3 days for further validation of T cells proliferation. Intracellular Ki67 was stained after the cells were fixed-permeabilized with Foxp3/Transcription Factor Staining Buffer Set (eBioscience). For cytokine production assays, supernatants were obtained 24 h after co-culture, and cytokines, including the human IL-2, TNF-α, IFN-γ, granzyme B, GM-CSF, IL-10 and TGF-β were assessed using CBA Flex Sets (BD Biosciences). For cytotoxicity to target cells, co-cultures were incubated for 24 h as described above, and cell viability was assessed using the Bright-Lumi™ II Firefly Luciferase Assay Kit (Beyotime). In addition, 4 h before the end, CD107a antibody was added to the co-cultures, and protein transport inhibitors (BD Biosciences) were added 1 h later, followed by incubation with CD3, CD4, and CD8 antibodies. T cells stimulated by Dynabeads CD3/CD28 were used as the positive control, whereas untreated T cells were used as the negative control.
Virus- and cell-mediated cytotoxicity assays
OC cell lines (5 × 103 cells/well) or tumor single-cells from fresh OC tumor tissues (5 × 104 cells/well) were plated in 96-well plates and infected with OAd or OAd-MUC16-BiTE. At the indicated time points post-infection, 10 μL CCK-8 was added to each well. The cells were then incubated for 2 h at 37°C. Cell viability was determined by measuring the absorbance at 450 nm using a microplate reader (Thermo Fisher Scientific).
For the oncolysis-mediated T-cell assays, SKOV3-LUC/SKOV3-MUC16 or HEY-LUC/HEY-MUC16 cells were infected with OAd or OAd-MUC16-BiTE (MOI = 10). After 24 h, unstimulated T cells were added (E:T = 5:1). The co-cultures were incubated for 24 h, and cell viability was assessed using the Bright-Lumi™ II Firefly Luciferase Assay Kit.
For bystander killing assays, HEK293A cells in suspension were infected with OAd or OAd-MUC16-BiTE (MOI = 20) for 4 h. The excess virus was then washed with PBS. Approximately 2 × 104 virus-infected HEK293A cells per well and 2 × 104 SKOV3-MUC16 or HEY-MUC16 per well were co-cultured with 1 × 105 unstimulated T cells (E:T = 5:1) in 96-well plates for 24 h and 48 h. Finally, the Bright-Lumi™ II Firefly Luciferase Assay Kit was used to determine the survival of tumor cells. Co-cultures without T cells were used as controls for virus-mediated cytotoxicity. The concentration of MUC16-BiTE secreted by HEK293A was determined by His Tag ELISA Detection Kit (Genscript).
Results were normalized to 100% viability.
In vivo treatments
Four- to six-week-old female NCG (NOD-Prkdcem26Cd52IL2rgem26Cd22/Gpt) mice were purchased from the Nanjing Biomedical Research Institute of the Nanjing University (Nanjing, China). All the NCG mice were housed in specific-pathogen-free environments. 1 × 107 HEY-MUC16 cells were injected subcutaneously into the lower dorsal flank or axilla of NCG mice to establish the cell-derived xenograft (CDX) model. The establishment of the patient-derived xenograft (PDX) model was as previously described.27 Passage 2 (P2) were harvested, washed with PBS solution, homogenized, suspended in an isometric PBS solution, and mixed with Matrigel Matrix (Corning). The homogenate was injected subcutaneously into the lower dorsal flank or axilla of NCG mice. After 1 week, mice were randomized into six (CDX) or five (PDX) groups (n = 5 each) and were treated with a multi-center intratumoral injection of 5 × 109 PFU of the indicated viruses or PBS on days 0 and 3. One day after each round of virus injection, PBS or 1 × 107 preactivated T cells (activated by CD3/CD28-activating Dynabeads for 48 hours) were administered to mice by intravenous injection. In both models, tumor mass and volume were measured every 2–3 days. Tumor volume was calculated as V (mm3) = W2 × L/2, where W and L are the width and the length of the tumor, respectively. Finally, the tumors were collected, photographed, weighed, and sectioned.
For the PDX model, proteins in the tumor tissue were extracted, and cytokines were evaluated by Luminex liquid suspension chip detection (Wayen Biotechnologies, China). Differentially expressed proteins (DEPs) were evaluated using quantitative proteomics based on isobaric tags for relative and absolute quantitation (iTRAQ) (BGI Tech).
The measurement of toxicity study
Female C57/BL6 mice at 6 to 8 weeks of age were used in this study. The toxicity was examined after systemic injection of 1 × 109 PFU of the indicated viruses or PBS. At 3 days posttreatment, mice were sacrificed and organs (heart, liver, spleen, lung, kidney, and stomach) were harvested. The organs were sectioned, and representative sections were stained with hematoxylin and eosin (H&E) to check for toxicity.
Immunohistochemistry (IHC)
The fresh tumor tissues were fixed by formalin for at least 24 h and cut into 4-μm-thick sections. Xylene and ethanol were used to deparaffinize and rehydrate. After that, antigenic retrieval was proceeded by microwave heating. Endogenous peroxidase and nonspecific binding were blocked with 3% hydrogen peroxide and goat serum respectively. The primary antibodies are anti-MUC-CD (1:1000 diluted), HEXON (Sigma Aldrich, 1:100 dilution), 6× His (Abcam, 1:500 dilution), CD8 (Proteintech, 1:10,000 dilution), PD-1(CST,1:100 dilution) and LAG-3(CST,1:100 dilution), which were incubated in humid chamber overnight at 4°C. The next day, the slides were incubated with the corresponding secondary antibody. Expression was detected by I–View 3,3′-diaminobenzidine (DAB) staining detection. The number of CD8+ cells and tissue area (mm2) were determined using an image analysis system (Halo v3.0.311.314).
Statistical analysis
Quantitative data were tested for normality and variance. All statistical analyses were performed using GraphPad Prism version 9.1.0. Non-parametric tests were used for data with skewed distribution. Two-tailed unpaired or paired t-tests were used to compare normally distributed data between two groups. Welch’s correction was used when appropriate. Welch’s ANOVA tests were used for multi-group comparison, and Dunnett T3 tests were used for post-hoc analysis. P < 0.05 was considered statistically significant (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001).
Results
Generation and in vitro characterization of MUC16-targeting BiTE-armed oncolytic adenovirus
Both the parental OAd and the recombinant OAd-MUC16-BiTE contain the hTERT gene promoter, which allows tumor-specific regulation of the gene expression of E1A and E1B required for viral replication. The arginine-glycine-aspartic acid (RGD) sequence was inserted into the fiber gene to promote virus infection and eGFP for visualization. MUC16-BiTE can simultaneously target human CD3 and MUC16, which is encoded within the E1 region under the transcriptional control of a CMV promoter, using a shuttle vector inserted into the virus backbone by Gibson assembly (Figure 1(a)). Western blotting results confirmed the expression of the encoded MUC16-BiTE after infection of tumor cells (Figure 1(b)).
Figure 1.
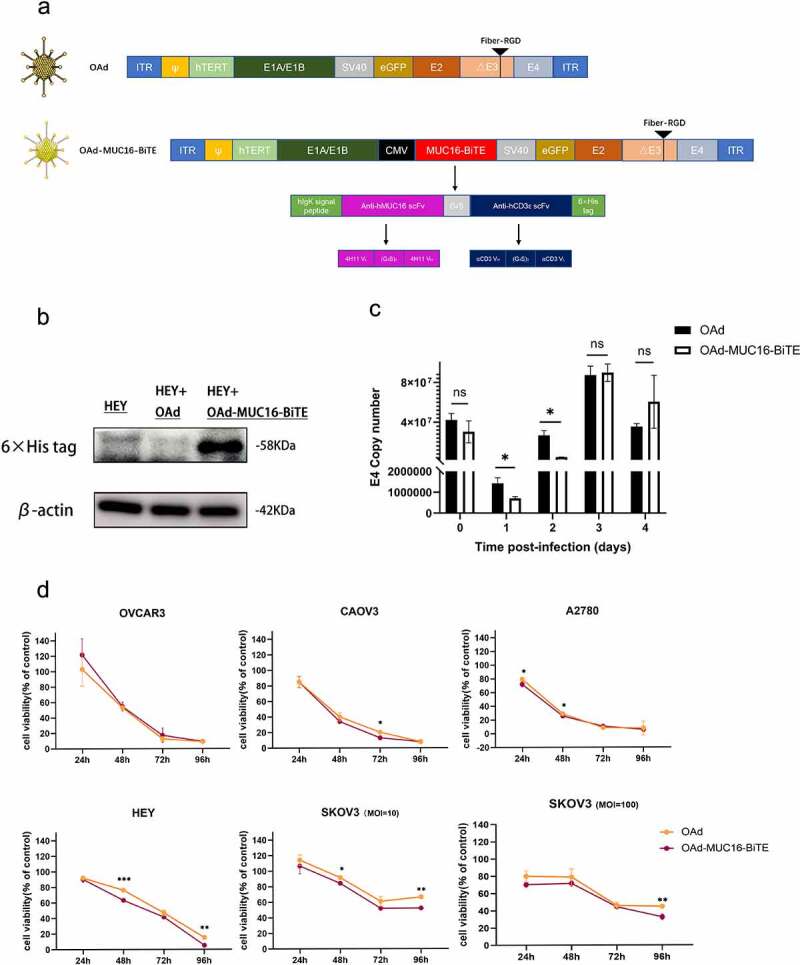
Generation and in vitro characterization of OAd-MUC16-BiTE. (a) Graphical illustration of the features of OAd-MUC16-BiTE and its parental OAd. (b) HEY cells were infected with the specified virus (MOI = 10). Cells were collected 48 h after infection, and western blotting was used to analyze 6× His. HEY cells without the virus as a blank control. β-actin was used as the endogenous control. (c) HEY cells were infected with the indicated virus (MOI = 100). The conditioned medium was harvested at 0 d, 1 d, 2 d, 3 d, and 4 d after infection. DNA extraction and qPCR was performed to detect the copy number of the E4 gene as a measure of viral replication. (d) Cell viability was assessed using CCK8 assays 24 h, 48 h, 72 h, and 96 h after infection. Data are shown as the percentage of cells alive after infection with OAd or OAd-MUC16-BiTE at the indicated MOIs relative to the non-infected cells (control values were set to 100% viability). Data represents the mean ± SD in triplicates (c, d). OAd, oncolytic adenovirus; BiTE, bispecific T-cell engager; ITR, inverted terminal repeats; ψ, virus packaging signal; CMV, cytomegalovirus promoter; SA, Simian virus; Ig, immunoglobulin; H, heavy chain; L, light chain; h, human; MOI, multiplicity of infection; ns, not significant; qPCR, quantitative PCR.
We investigated viral replication ability and oncolytic property in the absence of human T cells. Infection of HEY cells with parental OAd and the recombinant OAd-MUC16-BiTE virus yielded similar amounts of viral genomes as measured by qPCR, which peaked on the third day after infection (Figure 1(c) and Supplemental Figure S3). The cytotoxicity of the recombinant virus was also comparable with that of parental OAd in most cases (Figure 1(d)). Therefore, the BiTE transgene slightly affected viral replication kinetics and oncolytic activity. Notably, OAd showed strong oncolytic activity in all tested OC cell lines, except for SKOV3 cells that showed partial resistance and were killed more slowly;17 increasing the dose of viruses did not influence this resistance (Figure 1(d)).
MUC16-BiTE mediated T cell activation and target cell lysis is antigen-specific
We evaluated MUC16 expression (exactly MUC-CD) in each OC cell line by RT-PCR (Supplemental Figure S4). To assess the tumor selectivity of MUC16-BiTE, cell lines SKOV3-MUC16 and HEY-MUC16 stably overexpressing MUC16 (exactly MUC-CD) were established and verified using flow cytometry (Figure 2(a)).
Figure 2.
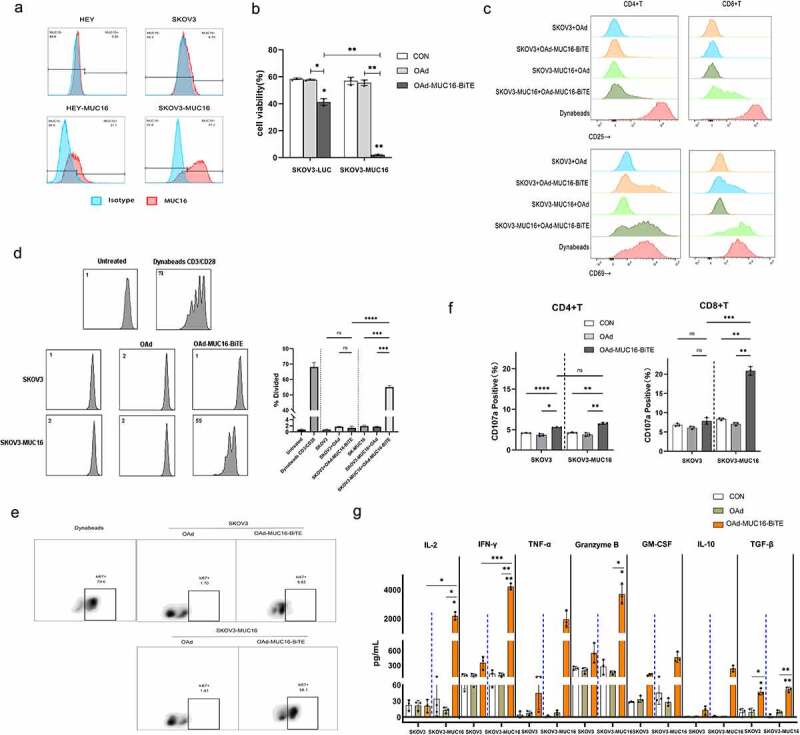
MUC16-BiTE expressed by OAd-MUC16-BiTE–infected cells enhances PBMC-derived T-cell functions. (a)Flow cytometry results showing the expression of MUC16 (MUC-CD) on SKOV3/SKOV3-MUC16 and HEY/HEY-MUC16 cells. (b) Results of luciferase activity-based cytotoxicity tests. (c) Flow cytometry results showing the surface expression of CD69 and CD25 on CD4+ and CD8 + T cells. (d) Proliferative response of CFSE-stained T cells co-cultured with SKOV3 or SKOV3-MUC16 cells and incubated with or without a specified supernatant. Fluorescence was measured by FACS analysis after incubation for 72 h. Untreated T ceslls were used as the negative control, whereas Dynabeads CD3/CD28-stimulated T cells were used as the positive control. (e) Representative flow cytometric plot showing the Ki67 expression of CD3 + T cells. (f) CD107a‐specific antibodies were added to the culture media during co-culture, and degranulation was assessed by flow cytometry. (g) Levels of human IL-2, TNF-α, IFN-γ, granzyme B, GM-CSF, IL-10 and TGF-β as assessed by flow cytometry. (b-c) and (f-g) In the presence of different supernatants, PBMC-derived unstimulated T cells were co-cultured with SKOV3/SKOV3-LUC or SKOV3-MUC16 (E:T = 5:1) for 24 h, whereas (e) for 72 h. (c, e) T cells stimulated by Dynabeads CD3/CD28 were used as the positive control. (b, f-g) co-cultures without supernatants were used as empty controls. (b-g) Each condition was measured in biological triplicates, and data represent the mean ± SD. CON, control; FACS, fluorescence-activated cell sorting; IL-2, interleukin 2; IL-10, interleukin 10; TNF-α, tumor necrosis factor alpha; IFN-γ, interferon gamma; GM-CSF, granulocyte macrophage-colony stimulating factor; TGF-β, transforming growth factor β.
The supernatant of the OAd-MUC16-BiTE-infected HEY cells in which MUC16-BiTE concentration was 60.43 ng/ml (Supplemental Figure S5) was added to cultures of SKOV3/SKOV3-MUC16 or HEY/HEY-MUC16 cells, with human PBMC-derived unstimulated T cells, to evaluate the function of MUC16-BiTE secreted by the recombinant virus. T cells co-cultured with SKOV3-MUC16 or HEY-MUC16 cells exhibited potent target cell lysis (Figure 2(b) and Supplemental Figure S6a) and expressed activation-related surface markers CD69 (Figure 2(c) and Supplemental Figure S6b) and CD25 (Figure 2(c) and Supplemental Figure S6c) upon incubation with supernatant of OAd-MUC16-BiTE–infected cells. Another important indicator of T-cell activation is their proliferative capacity. PBMC-derived T cells underwent multiple rounds of proliferation only upon co-culture with SKOV3-MUC16 (Figure 2(d,e)) or HEY-MUC16 (Supplemental Figure S6d and S6e) cells and OAd-MUC16-BiTE supernatant. T-cell activation by BiTEs leads to degranulation-mediated cytotoxicity. After co-culture with SKOV3-MUC16 or HEY-MUC16 cells and OAd-MUC16-BiTE supernatant, the expression of surface CD107a was upregulated (Figure 2(f) and Supplemental Figure S6f), and the production of cytokines, including IL-2, IFN-γ, TNF-α, granzyme B, GM-CSF, IL-10, and TGF-β increased (Figure 2(g) and Supplemental Figure S6g). These results indicate that the cytotoxicity of MUC16-BiTE is antigen-specific.
We also observed that both CD4 + and CD8 + cells showed higher levels of CD69 (Figure 2(c) and Supplemental Figure S6b) and CD25 (Figure 2(c), and Supplemental Figure S6c) expression. Although, the expression of the degranulation indicator CD107a was higher in CD8+ cells (Figure 2(f) and Supplemental Figure S6f). These results show that both CD4+ and CD8+ cells contribute to BiTE-mediated cytotoxicity.
OAd-MUC16-BiTE–mediated oncolysis enhanced T-cell–mediated tumor cell killing and bystander effect
To study OAd-MUC16-BiTE in a setting more closely resembling the oncolytic process, we infected the co-cultures of PBMC-derived T cells with SKOV3/SKOV3-MUC16 or HEY/HEY-MUC16 cell lines at an MOI of 10. We found that upon co-culture with PBMC-derived T cells, both viruses induced rapid tumor cell killing; however, OAd-MUC16-BiTE showed a stronger cytotoxicity against SKOV3-MUC16 or HEY-MUC16 cells (Figure 3(a)).
Figure 3.
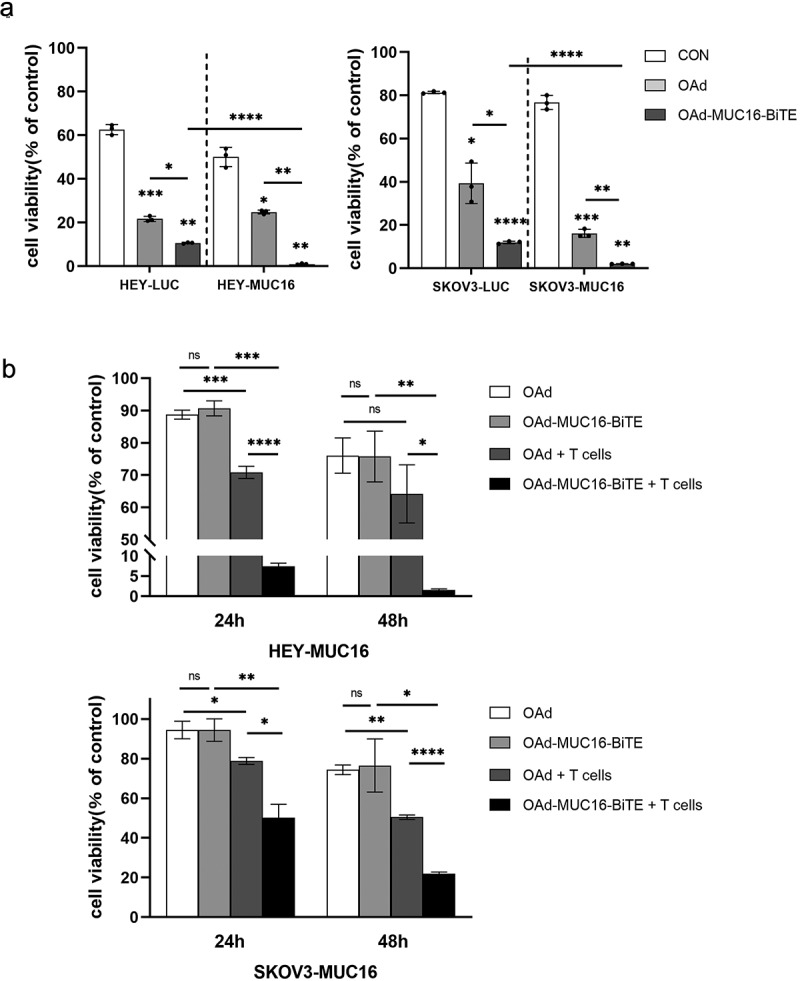
OAd-MUC16-BiTE–mediated oncolysis enhances T-cell–mediated tumor cell killing and bystander effect. (a) SKOV3-LUC/SKOV3-MUC16 or HEY-LUC/HEY-MUC16 cells were infected with OAd or OAd-MUC16-BiTE; uninfected cells were used as the control. PBMC-derived T cells (5:1) was added 24 h after infection and incubated for 24 h.(b) HEK293A cells infected with OAd or OAd-MUC16-BiTE were co-cultured with HEY-MUC16 or SKOV3-MUC16 cells and PBMC-derived T cells (5:1) for 24 h and 48 h. A co-culture without T cells served as the negative control. (a-b) Luciferase activity was measured using a microplate reader. Data represent the mean ± SD in triplicates. CON, control.
Another important feature of a secreted BiTE is its bystander effect. HEY-MUC16 or SKOV3-MUC16 cells were co-cultured with OAd- or OAd-MUC16-BiTE–infected HEK293A cells in the presence or absence of PBMC-derived T cells for 24 h and 48 h. In this setting, HEK293A cells act as BiTEs producers, the concentration of MUC16-BiTEs produced after 24 hours was 1.14 ± 0.32 ng/ml, and after 48 hours was 3.03 ± 1.02 ng/ml, measured by ELISA (Supplemental Figure S5). OAd-MUC16-BiTE–infected HEK293A cells significantly decreased in the number of live HEY-MUC16/SKOV3-MUC16 cells compared with OAd-infected cells when co-cultured with T cells (Figure 3(b)). This increased cytotoxicity was dependent on the presence of T cells, as OAd and OAd-MUC16-BiTE induced similar levels of cell death in the absence of T cells.
Oncolytic adenovirus reduced the viability of OC patient ex vivo tumor cultures
We collected ex vivo tumor cultures from five patients with OC, who expressed high levels of MUC-CD (Figure 4(a)). PD-1 expression was found to average 46.66% of T cells in the tumors (up to 80.7%), TIM-3 29.38% (up to 71%), LAG-3 23.33% (up to 47%), and TIGIT 49.66% (up to 69.9%) (Figure 4(b)), indicating that the immune TME was highly suppressed. Then, we investigated whether virus-mediated killing is affected by the TME. OAd and OAd-MUC16-BiTE showed similar anti-tumor killing effects in most cases (Figure 4(c)). In particular, OAd and OAd-MUC16-BiTE were the most effective against tumors in patients 1 and 3, but they were lightly effective for patient 2, and the survival rate was reduced by 40% (OAd) at 96 h (Figure 4(c)). IL-10 and TGF-β levels decreased upon co-incubation with oncolytic adenoviruses (Figure 4(d)). These results indicate that oncolytic adenoviruses can overcome the immunosuppressive TME and exert cytotoxicity.
Figure 4.
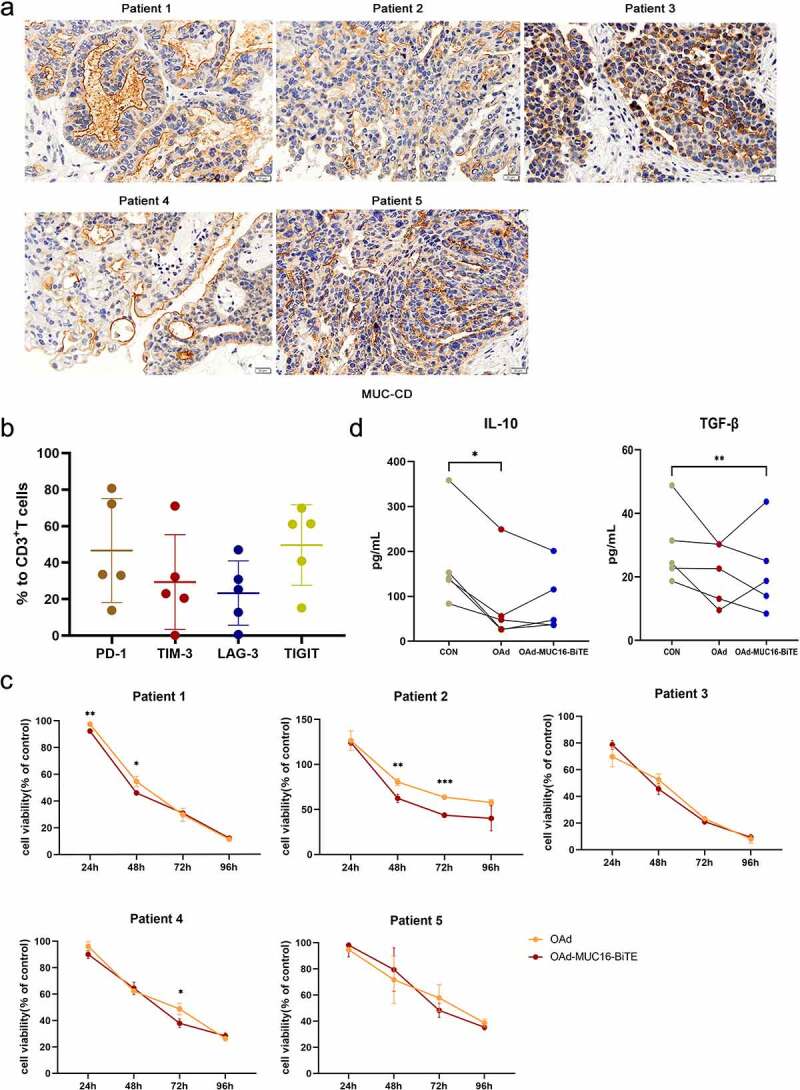
Lytic capability of oncolytic adenovirus in ex vivo tumor cultures from patients with resected OC. (a) Representative IHC images showing MUC-CD expression of OC patients. Scale bar: 50 μm. (b) PD-1, TIM-3, LAG-3, and TIGIT expression in T cells were evaluated by flow cytometry. Data are shown as percentages of CD3+ cells. Data are mean ± SD, n = 5. (c) Ex vivo tumor cultures were incubated with oncolytic adenovirus for 96 h (MOI = 100). Viability was assessed on the indicated days, non-infected as negative control. Data represent the mean ± SD in triplicates. (d) Ex vivo tumor cultures were incubated with or without oncolytic adenovirus for 72 h (MOI = 100). IL-10 and TGF-β levels of the supernatant were analyzed (n = 5). OC, Ovarian cancer; PD-1, programmed death 1; LAG3, lymphocyte activation gene-3; Tim-3, T cell immunoglobulin and mucin domain-3; TIGIT, T cell immunoglobulin and ITIM domain.
OAd-MUC16-BiTE enhanced in vivo tumor control on a CDX and PDX OC model
First, 1 × 107 HEY-MUC16 cells were injected into NCG mice to construct a CDX model. Since patient’s OC tissue provided a better simulation environment for the progression of OC than cell line, the PDX model was further established. NCG mice were injected intratumorally with PBS, parental OAd, or OAd-MUC16-BiTE. One day after virus administration, mice were intravenously injected with 1 × 107 pre-stimulated T cells derived from PBMCs or PBS, with two cycles of treatment (Figure 5(a)). The PBS group showed the fastest tumor growth, whereas the OAd-MUC16-BiTE group exhibited an antitumor efficacy similar to that of the parental virus in the absence of T cells. Notably, T cells administration significantly enhanced the antitumor efficacy of OAd-MUC16-BiTE in terms of tumor volume or bodyweight variation, either in the CDX model (Figure 5(b-d)) or in the PDX model (Figure 5(e-g)). IHC analysis revealed the expression of the HEXON protein in all virus treatment groups (Figure 5(h) and Supplemental Figure S7a), and the expression of MUC16-BiTE was detected in tumors injected with OAd-MUC16-BiTE (Figure 5(i) and Supplemental Figure S7b), indicating that T cell-mediated cytotoxicity did not affect virus persistence in the tumor. In addition, after 3 days of various treatments, HE staining was performed on the main organs, respectively (Figure 5(j)). No obvious lesions and organic injuries were detected in major organs (heart, liver, spleen, lung, kidney, stomach), indicating that OAds had no obvious side effects on mice.
Figure 5.
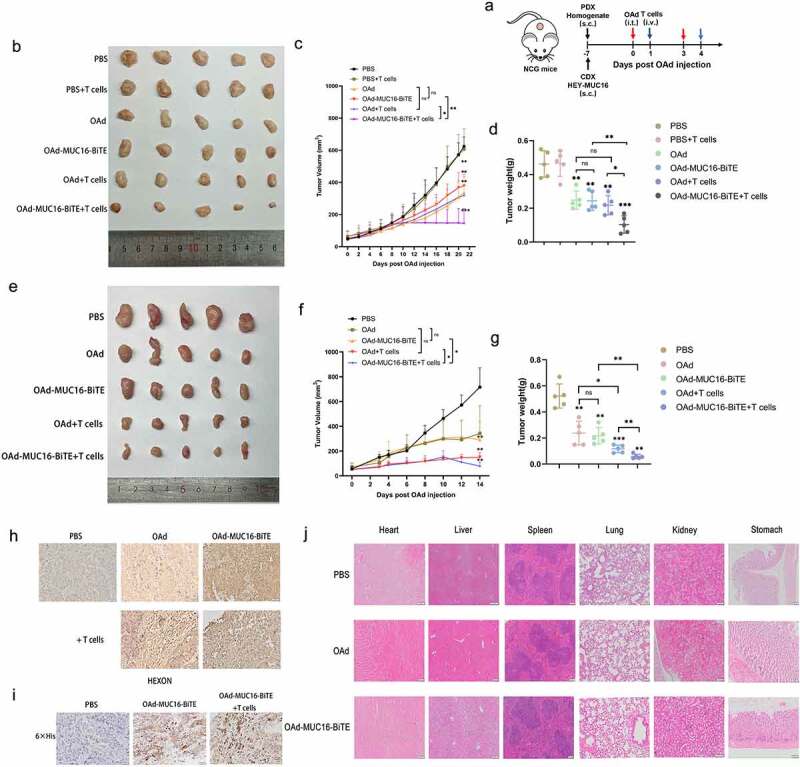
OAd-MUC16-BiTE enhances antitumor efficacy in vivo. (a) Schematic experimental design. After treatment, tumor tissues were collected at the indicated time points. (b, e) Photographs of tumors after treatment, (b) the CDX model, (d) the PDX model. (c, f) Tumor volume and (d, g) mass were measured (Data represent the mean ± SD, n = 5). (c, d) the CDX model, (f, g) the PDX model. (h, i) IHC-stained tumor sections of the PDX model. Representative images of the virus HEXON (h) and 6× His tag (i). Scale bar: 50 μm.(j) HE staining of the major organs (heart, liver, spleen, lung, kidney and stomach) of C57/BL6 mice after different treatments. The scale bars are 100 μm. s.c. subcutaneous; i.v., intravenous; i.t., intratumoral; CDX, cell-derived xenograft; PDX, patient-derived xenograft; IHC, immunohistochemistry; HE, hematoxylin and eosin.
OAd-MUC16-BiTE facilitated CTL infiltration and reversed the immunosuppressive TME
T cell infiltration has a significant influence on the natural course of many cancers. First, flow cytometry was used to analyze the CD4+ and CD8+ ratio of TILs, and high infiltration CD8+ and little CD4+ lymphocytes were found (Supplemental Figure S8). Compared to OAd, we found more CD8 + T cells in the tissues treated with OAd-MUC16-BiTE by IHC analysis, either in the PDX model (Figure 6(a)) or the CDX model (Supplemental Figure S7c). Similarly, OAd-MUC16-BiTE mediated an increase in cells immuno-positive for the immune checkpoints PD-1 and LAG-3 (Figure 6(b)).
Figure 6.
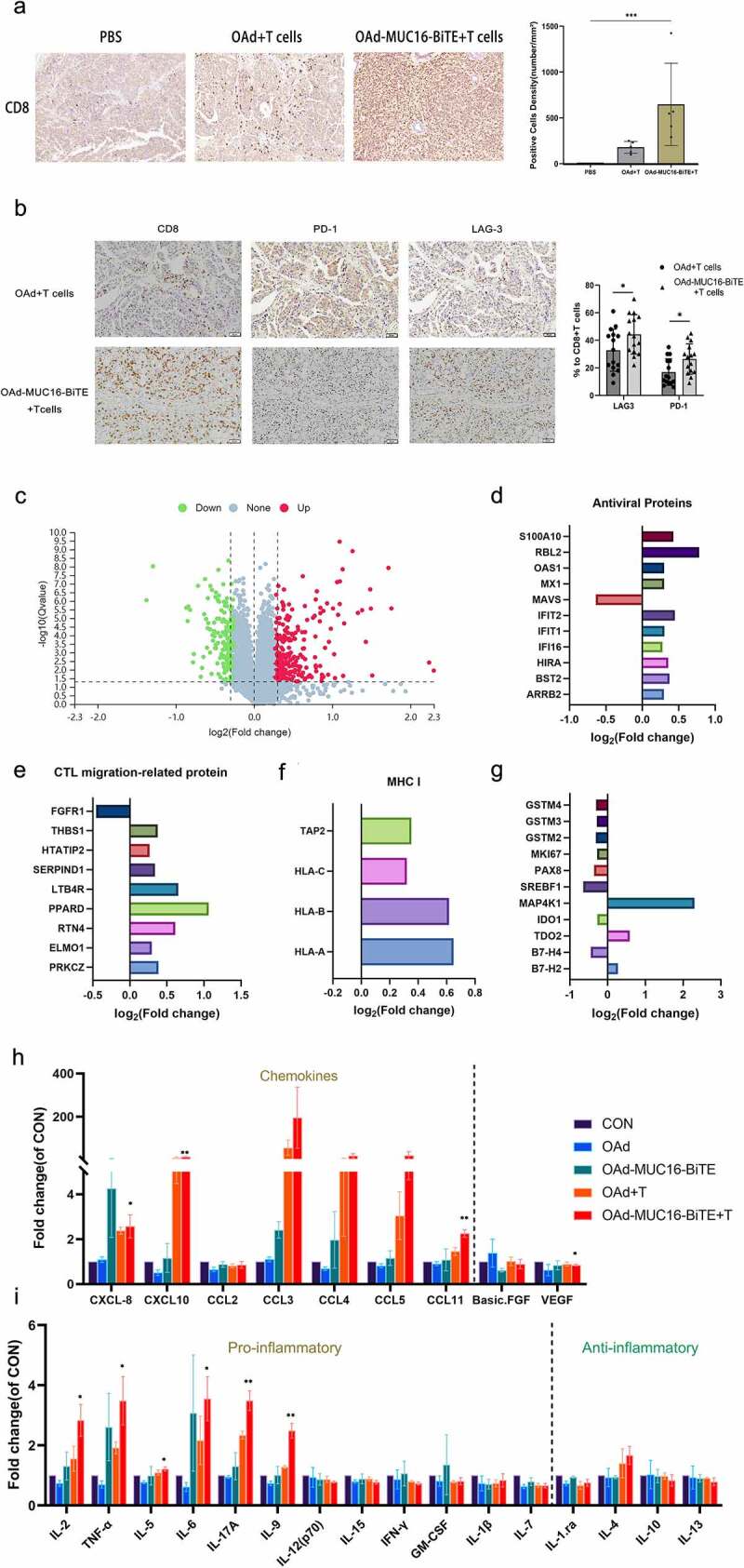
OAd-MUC16-BiTE increases T cell infiltration and reverses TME. (a) Representative IHC images showing CD8 lymphocytes in PDX tumors. Scale bar: 50 μm. CD8+ cell density (cells/mm2) of each tissue slice (n = 5) is shown right. Data are shown as the mean ± SD. (b) IHC of immune markers, CD8, PD-1, and LAG-3 in PDX tumors. Scale bar, 50 μm. Percentages of PD-1+ and LAG-3+ cells among CD8 + T cells are shown right. Number of positive cells from randomly chosen 5 fields/tumor section/mouse were counted, and the mean ± SD of all fields across the mice (N = 3/group) are presented. (c) Volcano plot of DEPs between the two groups (OAd-MUC16-BiTE + T cells vs. PBS). Each point represents a detected protein. The red dots indicate the upregulated proteins; green dots, downregulated proteins; and gray dots, non-significant DEPs. The thresholds for significant differential expression were set at q < 0.05 and fold-change > 1.2. DEPs related to antiviral activity (d), T cell migration (e), and MHC I antigen processing and presentation(f). (g) DEPs related to T cell function and platinum resistance. (h) Chemokines (left) and angiogenic factors (right). (i) Pro-inflammatory factors (left) and anti-inflammatory factors (right). (h, i) Data are shown as the mean ± SD in triplicates. DEP, differentially expressed protein; MHC, major histocompatibility complex; CTL, cytotoxic T lymphocytes; CON, control; CXCL, chemokine CXC motif ligand; CCL, chemokine C-C motif ligand; VEGF, vascular endothelial growth factor; FGF, fibroblast growth factor.
Next, we evaluated the impact of OAd-MUC16-BiTE on the TME using iTRAQ quantitative proteomics and Luminex liquid suspension chip detection. A total of 433 DEPs were screened, of which 261 were upregulated and 172 were downregulated (Figure 6(c)). After OAd-MUC16-BiTE treatment, significant antiviral protein expression was detected, although MAVS protein expression was downregulated (Figure 6(d)). It is worth noting that the expression of proteins related to the promoting T cell migration into tumor tissues (Figure 6(e)) and the processing and presentation of MHC class I antigens (Figure 6(f)) were both upregulated. Consistently, we detected an increase in chemokine levels in tumor tissues treated with OAd-MUC16-BiTE and T cells (Figure 6(h)). Decreased VEGF and basic FGF levels (Figure 6(h)) and increased chemokine levels promote T cell migration into tumor tissues. There is an increase in the concentration of pro-inflammatory factors IL-2, TNF-α, IL-5, IL-6, IL-17A, and IL-9, and a decrease in that of anti-inflammatory factors IL-1.Ra, IL-10, and IL-13, although not significant (Figure 6(i)). In addition, changes in proteins related to T cells function and platinum resistance were also observed (Figure 6(g)).
In summary, our results demonstrate that the OAd-MUC16-BiTE-mediated anti-tumor activity is related to the reversal of the TME and improved MHC I antigen presentation.
Discussion
New treatments are urgently needed to improve the survival rate of patients with OC. Strategies involving the infection of tumor cells using oncolytic viruses, direct lysis, and stimulation of the host immune system to produce anti-tumor responses are gaining increasing attention.28 However, there are still some limitations in the clinical application of OAd, and new strategies are needed to improve its tumor-killing effect. BiTEs can relocate T cells to tumor cells by bidirectionally connecting T cells and tumor cells. We generated an OAd with BiTE targeting MUC16 to achieve a synergistic effect and overcome the limitations of single immunotherapy.
The newly assembled OAd-MUC16-BiTE did not reduce the replication activity and oncolytic ability of the OAd both in vitro and vivo. A previous study21 speculated that competition between BiTE and the viral genes for transcription and translation would decrease oncolytic activity. It is believed that this discrepancy in results may be due to the virus design; thus, the optimal genome sequence and assembly protocol should be considered. Consistent with previous reports,17,21,29 this study also demonstrated that after secretion from infected cells, MUC16-BiTE could successfully activate CD4 and CD8 T cells to kill tumor cells, and BiTE-mediated cytotoxicity is antigen-specific.MUC16-BiTE triggered release of several cytokines, including high levels of IL‐2,IFN-γ, and TNF-α (associated with a Th1 response), a smaller increase of IL-10 (linked to a Th2 response) and TGF-β (a key cytokine of regulatory T cells differentiation30). It was reported that activation of T cells can potentially contribute directly to the induction of IL-10 or TGF-β by T cell subsets, which is a delicate balance.31 Anyhow, the production of Th2 cytokines and possible expansion of regulatory T cells caused by MUC16-BiTE should be seriously considered when clinical translations are performed.
We further evaluated the combined therapeutic effects of OAd and MUC16-BiTE in a CDX and PDX model of OC. Consistent with many studies on oncolytic viruses with BiTE,21,29,32,33 we confirmed that OAd-MUC16-BiTE could provide the most effective tumor regression. In contrast to previous studies, we also used a PDX model of OC. Compared with CDX, the PDX model better retains the heterogeneity and genetic characteristics of OC and has a higher degree of clinical similarity,34 and can better represent the killing effect of OAd-MUC16-BiTE on OC. An important role for co-stimulation during BiTE-engagement has been demonstrated in achieving improved antitumor efficacy.35 The incomplete tumor rejection could be related to the insufficient activation of T cells, although preactivated T cells were injected. Also, the suppressive TME is a critical barrier for the efficacy of cancer immunotherapy. Myeloid-derived suppressor cells(MDSCs) in TME directly contact CD8 + T cells, secrete suppressive cytokines such as IL-10, or consume amino acids around CD8 + T cells to inhibit the proliferation and function of effector T cells, thereby weakening anti-tumor immune response.36 Further carefully conducted trials are needed to account for the effect of MDSCs on the therapeutic efficacy of OAD-MUC16-BiTE by building a more appropriate model.
The most attractive feature of oncolytic virus is that they convert “cold” tumors to “hot” tumors.37–39 We found that OAd-MUC16-BiTE promotes the secretion of pro-inflammatory factors by T cells and decreases anti-inflammatory factors. The increase in chemokine levels40 and the normalization of blood vessels41 promote the transport of T cells to the tumor. In line with this, we found that after OAd-MUC16-BiTE treatment, tumors expressed higher levels of chemokines and lower angiogenesis factor, which promotes more CTL infiltration. The combination of OAd and MUC16-BiTE also has a potential synergistic effect on infiltrating CTL, which may provide a higher number of effector cells for BiTE-mediated cytotoxicity. The survival rate of many tumor patients, such as those with OC, is related to the action of TILs,3–5 which makes the clinical application of OAd-MUC16-BiTE attractive. In addition, this allows OAd-MUC16-BiTE and anti-angiogenic drugs to be used in combination to increase CTL infiltration and achieve a more significant anti-tumor response. OAd-MUC16-BiTE treatment induced slight changes in immune checkpoint levels, likely limiting the antitumor activity. Ribas et al reported the strong enhanced immune recognition of cancer when combined Talimogene laherparepvec oncolytic virus with an anti-PD1 antibody.38 Study results support our rationale for using different immune checkpoint inhibitors in combination with our BiTE-expressing virus.
OAd stimulates the body to induce anti-tumor immunity, which ultimately depends on MHC I antigens expression on the surface of tumor cells. Studies have shown that the lack of MHC expression is an important tumor immune evasion strategy.42 BiTE can act independently of the MHC I antigens of tumor cells;43,44 thus, even if tumor cells lose MHC expression, OAd-MUC16-BiTE can still be used to kill cancer cells. It is also worth noting that our data suggest that MHC I expression enhances tumor antigen presentation in the OAd-MUC16-BiTE treatment group.
In addition, based on the in vitro cultures of OC tumor cells, the heterogeneity of advanced cancer and the multi-faceted cellular interactions were retained, and we found that OAds can overcome the immunosuppressive TME to exert toxic effects. Particular attention should be paid to the reduction of suppressive cytokines (IL-10, TGF-β); increasing levels of these cytokines have been associated with suppressing antitumor TIL activity in OC.45 It is particularly encouraging that the T cells in the biopsy of all tested patients tested positive for PD1 expression, may be anergic, and were easily activated by BiTE to mediate cytotoxicity. In principle, OAd-MUC16-BiTE should exert a better toxic effect in in vitro tumor cultures of OC; however, we only observed this phenomenon in one patient.
Systemic administration of BiTE usually causes systemic effects, some of which can be fatal if treated improperly.46 Through the in vivo CDX and PDX model of OC, we successfully confirmed that MUC16-BiTE is highly expressed locally in the tumor. The local administration of recombinant OAd-MUC16-BiTE can limit the expression of BiTE in the tumor, minimize systemic exposure, and reduce the toxicity of the “outside of the tumor,” thereby overcoming the relatively short circulation dynamics and limited tumor penetration after intravenous injection.47
Generally, the host immune system simultaneously induces anti-viral immunity to eliminate the virus, which counteracts the oncolytic effect of the treatment.48–50 We detected the expression of antiviral proteins IFI16, IFIT1, IFIT2, MX1, OAS1, and S100A10. However, at the end of our study, HEXON expression was detected in all virus-treated tumors, regardless of the presence of T cells. This indicates that MUC16-BiTE-mediated cancer cell death would not affect the persistence of the virus in tumors. We concede that the use of immunodeficient mice has limitations because they cannot simulate the human innate antiviral response and immune memory. However, our experimental results suggest that OAd-MUC16-BiTE activates infiltrating T lymphocytes, redirects them, and lyses MUC16+ cells, thus balancing antiviral and antitumor immunities.
We also observed increased expression of T cell costimulatory signal ICOSLG (B7-H2), immune checkpoint TDO2, T cell exhaustion-related protein MAP4K1,51 and a decrease in that of immune checkpoint IDO1 and B7-H4 and T cell proliferation-related protein SREBF1,52 suggesting that OAd-MUC16-BiTE mediates a more complex tumor immune response. Surprisingly, we found that OAd-MUC16-BiTE treatment reduced the levels of platinum resistance-related proteins GSTM2, GSTM3, and GSTM4. It is speculated that OAd-MUC16-BiTE may be combined with chemotherapy to reduce platinum resistance in patients with OC. We plan to evaluate the combination therapy of OAd-MUC16-BiTE and platinum in our future study.
Overall, the synergistic and cumulative effect achieved by the combination of OAd and MUC16-BiTE outweighs its limitations, representing a novel method for the treatment of OC. Furthermore, it can be used in conjunction with various anti-tumor therapies, such as immune checkpoint inhibitors, chemotherapy and VEGF inhibitors; however, studies assessing clinical evaluation are required.
Supplementary Material
Acknowledgments
We sincerely thank all participants in the study.
Funding Statement
This work was supported by the National Natural Science Foundation of China (82172695, 82072871 & 81874107) and Natural Science Foundation of Shandong Province (ZR2021MH269). This work was also supported by the Taishan Scholars Program of Shandong Province.
Abbreviations
- OC
Ovarian cancer
- CTL
Cytotoxic T lymphocyte
- OAd
Oncolytic adenovirus
- BiTE
Bispecific T-cell engager
- TME
Tumor microenvironment
- scFv
Single-chain antibody fragment
- TIL
Tumor-infiltrating lymphocyte
- CMV
Cytomegalovirus promoter
- RGD
Arginine-glycine-aspartic acid
- PDX
Patient-derived xenograft
- MOI
Multiplicity of infection
- iTRAQ
Isobaric tags for relative and absolute quantitation
- DEP
Differentially expressed protein
- RT-qPCR
Real-time quantitative PCR
- CDX
Cell-derived xenograft
- IHC
Immunohistochemistry
- H&E
Hematoxylin and eosin
- MDSC
Myeloid-derived suppressor cell
Disclosure statement
No potential conflict of interest was reported by the author(s).
Ethics approval and consent to participate
Patients provided informed consent. Ethical approval was obtained from the Ethics Committee of the Qilu Hospital of Shandong University (KYLL-2018(KS)-229). All animal experiments were approved by the Animal Care and Use Committee of the School of Medicine of Shandong University (SDULCLL2019-2-08).
Availability of data and materials
The data used and/or analysis during the current study are available from the corresponding author on reasonable request.
Authors’ contributions
This study was conceived, designed, and interpreted by CZY and BHK. QMW, XYM, and HW undertook the data acquisition, analysis, and interpretation. RRL, SY and YWL were responsible for the comprehensive technical support. QMW wrote original draft. KS and QZ provided the fresh tumor samples during their surgeries. CZ and JYC contributed to the inspection of data and final manuscript. BHK and CZY supervised and provided funding support. All authors have read and approved the final manuscript.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/2162402X.2022.2096362
References
- 1.Siegel RL, Miller KD, Fuchs HE, Jemal A.. Cancer Statistics, 2021. CA Cancer J Clin. 2021;71(1):7–14. doi: 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 2.Lheureux S, Braunstein M, Oza AM. Epithelial ovarian cancer: evolution of management in the era of precision medicine. CA Cancer J Clin. 2019;69(4):280–304. doi: 10.3322/caac.21559. [DOI] [PubMed] [Google Scholar]
- 3.Leffers N, Gooden MJM, de Jong RA, Hoogeboom B-N, ten Hoor KA, Hollema H, Boezen HM, van der Zee AGJ, Daemen T, Nijman HW. Prognostic significance of tumor-infiltrating T-lymphocytes in primary and metastatic lesions of advanced stage ovarian cancer. Cancer Immunol Immunother. 2009;58(3):449–459. doi: 10.1007/s00262-008-0583-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, Makrigiannakis A, Gray H, Schlienger K, Liebman MN, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348(3):203–213. doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- 5.Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, Jungbluth AA, Frosina D, Gnjatic S, Ambrosone C, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A. 2005;102(51):18538–18543. doi: 10.1073/pnas.0509182102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bommareddy PK, Shettigar M, Kaufman HL. Integrating oncolytic viruses in combination cancer immunotherapy. Nat Rev Immunol. 2018;18(8):498–513. doi: 10.1038/s41577-018-0014-6. [DOI] [PubMed] [Google Scholar]
- 7.Shi T, Song X, Wang Y, Liu F, Wei J. Combining oncolytic viruses with cancer immunotherapy: establishing a new generation of cancer treatment. Front Immunol. 2020;11:683. doi: 10.3389/fimmu.2020.00683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pol JG, Workenhe ST, Konda P, Gujar S, Kroemer G. Cytokines in oncolytic virotherapy. Cytokine Growth Factor Rev. 2020;56:4–27. doi: 10.1016/j.cytogfr.2020.10.007. [DOI] [PubMed] [Google Scholar]
- 9.Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. 2015;14(9):642–662. doi: 10.1038/nrd4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heidbuechel JPW, Engeland CE. Oncolytic viruses encoding bispecific T cell engagers: a blueprint for emerging immunovirotherapies. J Hematol Oncol. 2021;14:63. doi: 10.1186/s13045-021-01075-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farrera-Sal M, Fillat C, Alemany R. Effect of transgene location, transcriptional control elements and transgene features in armed oncolytic adenoviruses. Cancers (Basel). 2020;12(4):1034. doi: 10.3390/cancers12041034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–360. doi: 10.1146/annurev.immunol.22.012703.104803. [DOI] [PubMed] [Google Scholar]
- 13.Velasquez MP, Torres D, Iwahori K, Kakarla S, Arber C, Rodriguez-Cruz T, Szoor A, Bonifant CL, Gerken C, Cooper LJN, et al. T cells expressing CD19-specific engager molecules for the immunotherapy of CD19-positive malignancies. Sci Rep. 2016;6:27130. doi: 10.1038/srep27130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Topp MS, Gökbuget N, Stein AS, Zugmaier G, O’Brien S, Bargou RC, Dombret H, Fielding AK, Heffner L, Larson RA, et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol. 2015;16(1):57–66. doi: 10.1016/S1470-2045(14)71170-2. [DOI] [PubMed] [Google Scholar]
- 15.Brischwein K, Schlereth B, Guller B, Steiger C, Wolf A, Lutterbuese R, Offner S, Locher M, Urbig T, Raum T, et al. MT110: a novel bispecific single-chain antibody construct with high efficacy in eradicating established tumors. Mol Immunol. 2006;43(8):1129–1143. doi: 10.1016/j.molimm.2005.07.034. [DOI] [PubMed] [Google Scholar]
- 16.Dao T, Pankov D, Scott A, Korontsvit T, Zakhaleva V, Xu Y, Xiang J, Yan S, de Morais Guerreiro MD, Veomett N, et al. Therapeutic bispecific T-cell engager antibody targeting the intracellular oncoprotein WT1. Nat Biotechnol. 2015;33(10):1079–1086. doi: 10.1038/nbt.3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Freedman JD, Hagel J, Scott EM, Psallidas I, Gupta A, Spiers L, Miller P, Kanellakis N, Ashfield R, Fisher KD, et al. Oncolytic adenovirus expressing bispecific antibody targets T‐cell cytotoxicity in cancer biopsies. EMBO Mol Med. 2017;9(8):1067–1087. doi: 10.15252/emmm.201707567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu M, Wu B, Brandl C, Johnson J, Wolf A, Chow A, Doshi S. Blinatumomab, a bispecific T-cell engager (BiTE(®)) for CD-19 targeted cancer immunotherapy: clinical pharmacology and its implications. Clin Pharmacokinet. 2016;55(10):1271–1288. doi: 10.1007/s40262-016-0405-4. [DOI] [PubMed] [Google Scholar]
- 19.Goebeler M-E, Bargou RC. T cell-engaging therapies - BiTEs and beyond. Nat Rev Clin Oncol. 2020;17(7):418–434. doi: 10.1038/s41571-020-0347-5. [DOI] [PubMed] [Google Scholar]
- 20.Scott EM, Duffy MR, Freedman JD, Fisher KD, Seymour LW. Solid tumor immunotherapy with T cell engager-armed oncolytic viruses. Macromol Biosci. 2018;18(1):1700187. doi: 10.1002/mabi.201700187. [DOI] [PubMed] [Google Scholar]
- 21.Fajardo CA, Guedan S, Rojas LA, Moreno R, Arias-Badia M, de Sostoa J, June CH, Alemany R. Oncolytic adenoviral delivery of an EGFR-Targeting T-cell engager improves antitumor efficacy. Cancer Res. 2017;77(8):2052–2063. doi: 10.1158/0008-5472.CAN-16-1708. [DOI] [PubMed] [Google Scholar]
- 22.O’Brien TJ, Beard JB, Underwood LJ, Dennis RA, Santin AD, York L. The CA 125 gene: an extracellular superstructure dominated by repeat sequences. Tumour Biol. 2001;22(6):348–366. doi: 10.1159/000050638. [DOI] [PubMed] [Google Scholar]
- 23.Chekmasova AA, Rao TD, Nikhamin Y, Park KJ, Levine DA, Spriggs DR, Brentjens RJ. Successful eradication of established peritoneal ovarian tumors in SCID-Beige mice following adoptive transfer of T cells genetically targeted to the MUC16 antigen. Clin Cancer Res. 2010;16(14):3594–3606. doi: 10.1158/1078-0432.CCR-10-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kawashima T, Kagawa S, Kobayashi N, Shirakiya Y, Umeoka T, Teraishi F, Taki M, Kyo S, Tanaka N, Fujiwara T. Telomerase-specific replication-selective virotherapy for human cancer. Clin Cancer Res. 2004;10(1):285–292. doi: 10.1158/1078-0432.CCR-1075-3. [DOI] [PubMed] [Google Scholar]
- 25.Taki M, Kagawa S, Nishizaki M, Mizuguchi H, Hayakawa T, Kyo S, Nagai K, Urata Y, Tanaka N, Fujiwara T. Enhanced oncolysis by a tropism-modified telomerase-specific replication-selective adenoviral agent OBP-405 (‘Telomelysin-RGD’). Oncogene. 2005;24(19):3130–3140. doi: 10.1038/sj.onc.1208460. [DOI] [PubMed] [Google Scholar]
- 26.Darling AJ, Boose JA, Spaltro J. Virus assay methods: accuracy and validation. Biologicals. 1998;26(2):105–110. doi: 10.1006/biol.1998.0134. [DOI] [PubMed] [Google Scholar]
- 27.Sun C, Cao W, Qiu C, Li C, Dongol S, Zhang Z, Dong R, Song K, Yang X, Zhang Q, et al. MiR-509-3 augments the synthetic lethality of PARPi by regulating HR repair in PDX model of HGSOC. J Hematol Oncol. 2020;13:9. doi: 10.1186/s13045-020-0844-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Woller N, Gürlevik E, Fleischmann-Mundt B, Schumacher A, Knocke S, Kloos AM, Saborowski M, Geffers R, Manns MP, Wirth TC, et al. Viral infection of tumors overcomes resistance to PD-1-immunotherapy by broadening neoantigenome-directed T-cell responses. Mol Ther. 2015;23(10):1630–1640. doi: 10.1038/mt.2015.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Sostoa J, Fajardo CA, Moreno R, Ramos MD, Farrera-Sal M, Alemany R. Targeting the tumor stroma with an oncolytic adenovirus secreting a fibroblast activation protein-targeted bispecific T-cell engager. J Immunother Cancer. 2019;7:19. doi: 10.1186/s40425-019-0505-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peng Y, Laouar Y, Li MO, Green EA, Flavell RA. TGF-β regulates in vivo expansion of Foxp3-expressing CD4+CD25+ regulatory T cells responsible for protection against diabetes. Proc Natl Acad Sci U S A. 2004;101(13):4572–4577. doi: 10.1073/pnas.0400810101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sapski S, Beha N, Kontermann R, Müller D. Tumor-targeted costimulation with antibody-fusion proteins improves bispecific antibody-mediated immune response in presence of immunosuppressive factors. Oncoimmunology. 2017;6(12):e1361594. doi: 10.1080/2162402X.2017.1361594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu F, Wang X, Guo ZS, Bartlett DL, Gottschalk SM, Song X-T. T-cell engager-armed oncolytic vaccinia virus significantly enhances antitumor therapy. Mol Ther. 2014;22(1):102–111. doi: 10.1038/mt.2013.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barlabé P, de Sostoa J, Fajardo CA, Alemany R, Moreno R. Enhanced antitumor efficacy of an oncolytic adenovirus armed with an EGFR-targeted BiTE using menstrual blood-derived mesenchymal stem cells as carriers. Cancer Gene Ther. 2020;27(5):383–388. doi: 10.1038/s41417-019-0110-1. [DOI] [PubMed] [Google Scholar]
- 34.Kopetz S, Lemos R, Powis G. The promise of patient-derived xenografts: the best laid plans of mice and men. Clin Cancer Res. 2012;18(19):5160–5162. doi: 10.1158/1078-0432.CCR-12-2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arnone CM, Polito VA, Mastronuzzi A, Carai A, Diomedi FC, Antonucci L, Petrilli LL, Vinci M, Ferrari F, Salviato E, et al. Oncolytic adenovirus and gene therapy with EphA2-BiTE for the treatment of pediatric high-grade gliomas. J Immunother Cancer. 2021;9(5):e001930. doi: 10.1136/jitc-2020-001930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol. 2018;19(2):108–119. doi: 10.1038/s41590-017-0022-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garcia-Carbonero R, Salazar R, Duran I, Osman-Garcia I, Paz-Ares L, Bozada JM, Boni V, Blanc C, Seymour L, Beadle J, et al. Phase 1 study of intravenous administration of the chimeric adenovirus enadenotucirev in patients undergoing primary tumor resection. J Immunother Cancer. 2017;5:71. doi: 10.1186/s40425-017-0277-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, Olszanski AJ, Malvehy J, Cebon J, Fernandez E, et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves Anti-PD-1 immunotherapy. Cell. 2017;170(6):1109–1119.e10. doi: 10.1016/j.cell.2017.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tähtinen S, Grönberg-Vähä-Koskela S, Lumen D, Merisalo-Soikkeli M, Siurala M, Airaksinen AJ, Vähä-Koskela M, Hemminki A. Adenovirus improves the efficacy of adoptive T-cell therapy by recruiting immune cells to and promoting their activity at the tumor. Cancer Immunol Res. 2015;3(8):915–925. doi: 10.1158/2326-6066.CIR-14-0220-T. [DOI] [PubMed] [Google Scholar]
- 40.Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. 2017;17(9):559–572. doi: 10.1038/nri.2017.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao Y, Ting KK, Coleman P, Qi Y, Chen J, Vadas M, Gamble J. The tumour vasculature as a target to modulate leucocyte trafficking. Cancers (Basel). 2021;13(7):1724. doi: 10.3390/cancers13071724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garrido F, Aptsiauri N, Doorduijn EM, Garcia Lora AM, van Hall T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr Opin Immunol. 2016;39:44–51. doi: 10.1016/j.coi.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Offner S, Hofmeister R, Romaniuk A, Kufer P, Baeuerle PA. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol Immunol. 2006;43(6):763–771. doi: 10.1016/j.molimm.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 44.Schlereth B, Fichtner I, Lorenczewski G, Kleindienst P, Brischwein K, da Silva A, Kufer P, Lutterbuese R, Junghahn I, Kasimir-Bauer S, et al. Eradication of tumors from a human colon cancer cell line and from ovarian cancer metastases in immunodeficient mice by a Single-Chain Ep-CAM-/CD3-Bispecific antibody construct. Cancer Res. 2005;65(7):2882–2889. doi: 10.1158/0008-5472.CAN-04-2637. [DOI] [PubMed] [Google Scholar]
- 45.McCloskey CW, Rodriguez GM, Galpin KJC, Vanderhyden BC. Ovarian cancer immunotherapy: preclinical models and emerging therapeutics. Cancers (Basel). 2018;10(8):244. doi: 10.3390/cancers10080244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Teachey DT, Rheingold SR, Maude SL, Zugmaier G, Barrett DM, Seif AE, Nichols KE, Suppa EK, Kalos M, Berg RA, et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood. 2013;121(26):5154–5157. doi: 10.1182/blood-2013-02-485623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klinger M, Brandl C, Zugmaier G, Hijazi Y, Bargou RC, Topp MS, Gökbuget N, Neumann S, Goebeler M, Viardot A, et al. Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19/CD3-bispecific BiTE antibody blinatumomab. Blood. 2012;119(26):6226–6233. doi: 10.1182/blood-2012-01-400515. [DOI] [PubMed] [Google Scholar]
- 48.Schirmbeck R, Reimann J, Kochanek S, Kreppel F. The immunogenicity of adenovirus vectors limits the multispecificity of CD8 T-cell responses to vector-encoded transgenic antigens. Mol Ther. 2008;16(9):1609–1616. doi: 10.1038/mt.2008.141. [DOI] [PubMed] [Google Scholar]
- 49.Frahm N, DeCamp AC, Friedrich DP, Carter DK, Defawe OD, Kublin JG, Casimiro DR, Duerr A, Robertson MN, Buchbinder SP, et al. Human adenovirus-specific T cells modulate HIV-specific T cell responses to an Ad5-vectored HIV-1 vaccine. J Clin Invest. 2012;122(1):359–367. doi: 10.1172/JCI60202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li X, Wang P, Li H, Du X, Liu M, Huang Q, Wang Y, Wang S. The efficacy of oncolytic adenovirus is mediated by T-cell responses against virus and tumor in Syrian hamster model. Clin Cancer Res. 2017;23(1):239–249. doi: 10.1158/1078-0432.CCR-16-0477. [DOI] [PubMed] [Google Scholar]
- 51.Si J, Shi X, Sun S, Zou B, Li Y, An D, Lin X, Gao Y, Long F, Pang B, et al. Hematopoietic progenitor kinase1 (HPK1) mediates T cell dysfunction and is a druggable target for T Cell-Based immunotherapies. Cancer Cell. 2020;38(4):551–566.e11. doi: 10.1016/j.ccell.2020.08.001. [DOI] [PubMed] [Google Scholar]
- 52.Kidani Y, Elsaesser H, Hock MB, Vergnes L, Williams KJ, Argus JP, Marbois BN, Komisopoulou E, Wilson EB, Osborne TF, et al. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat Immunol. 2013;14(5):489–499. doi: 10.1038/ni.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data used and/or analysis during the current study are available from the corresponding author on reasonable request.