

ABSTRACT
Inducing a full antitumor immune response in the tumor microenvironment (TME) is essential for successful cancer immunotherapy. Here, we report that an oncolytic adenovirus carrying mIL-15 (Ad-IL15) can effectively induce antitumor immune response and inhibit tumor growth in a mouse model of cancer. We found that Ad-IL15 facilitated the activation and infiltration of immune cells, including dendritic cells (DCs), T cells and natural killer (NK) cells, in the TME. Unexpectedly, we observed that Ad-IL15 also induced vascular normalization and tertiary lymphoid structure formation in the TME. Moreover, we demonstrated these Ad-IL15-induced changes in the TME were depended on the Ad-IL15-induced activation of the STING-TBK1-IRF3 pathway in DCs. Taken together, our findings suggest that Ad-IL15 is a candidate for cancer immunotherapy that promotes immune cell activation and infiltration, tumor vascular normalization and tertiary lymphoid structure formation in the TME.
KEYWORDS: Oncolytic virus, vascular normalization, tertiary lymphoid structure, STING, DC
Introduction
The incidence and mortality rates of cancer are increasing, becoming a heavy burden worldwide1. Traditional chemotherapy, radiotherapy and targeted therapy have great limitations in clinical application because of their high toxicity and side effects. Immunotherapy can harness the antitumor immune response to kill cancer cells, which has become a focus in the tumor therapy field.2 The tumor microenvironment (TME) is closely related to the development of tumors and comprises tumor cells, immune cells, blood vessels, lymphatic vessels, stromal cells and various cytokines.3 However, tumor cells interact with TME components to form an inhibitory immune microenvironment, which hinders the effect of immunotherapy or results in therapeutic resistance.4
Oncolytic viruses (OVs), as a new immunotherapeutic agent, can specifically infect and kill tumor cells, leading to the direct destruction of tumors.5 In this process, damage-associated molecular patterns (DAMPs) and immunogenic mediators produced by tumor cell lysis induce immunogenic cell death in tumor cells.6 Moreover, the release of tumor-associated antigens (TAAs) can enhance the presentation of tumor antigens by dendritic cells (DCs), which consequently promotes the initiation and activation of T cells and ultimately leads to the formation of an immunostimulatory microenvironment.7 Interestingly, OVs can stimulate the host immune system to produce antiangiogenic factors and antagonize angiogenesis by tumor-related endothelial cells.6,8 At present, many viruses such as adenovirus,9 herpes simplex virus,10 and vaccinia virus,11 have been studied in preclinical, translational and clinical research. However, the results of these clinical trials have also shown that there are limitations to treatment with OVs.6
The TME is closely related to the development of tumors and mainly includes immune cells, blood vessels, lymphatic vessels, stromal cells and various cytokines.12 The formation of abnormal or immature tumor blood vessels leads to interstitial fluid reflux, which is an obstacle that results in an increase in interstitial fluid pressure (IFP), severe hypoxia and lactic acid accumulation and significantly contributes to the formation of an immunosuppressive microenvironment.13 Traditional anti-vascular therapy was expected to induce tumor death or dormancy by blocking tumor angiogenesis. However, clinical trials have shown various restrictions related to simple anti-vascular therapy, such as serious adverse reactions, drug resistance, reductions in blood perfusion, increases in tumor hypoxia, induction of vasculogenic mimicry (VM), and distant metastasis.14 Therefore, pruning redundant vascular structures and stabilizing and remodeling abnormal vessels, to achieve vascular normalization (VN), have become new tumor treatment strategies.15 Interestingly, the normal vascular system can enhance the antitumor immune response,16 and enhanced immune responses also promote VN.17 Additionally, stimulating the TME may promote the formation of tertiary lymphoid structures (TLS), which are spatially ordered structures that produce an in situ adaptive immune response.18 Mature TLSs contain various cells required to produce adaptive antitumor immunity, mainly including T cells, B cells and the follicular dendritic cell (FDC) network.19 However, the formation of TLS requires the presence of a variety of cytokines and chemokines at optimal concentrations.20 Therefore, promoting the formation of TLS in tumor tissue has become a new direction for tumor immunotherapy.
In this study, we describe a new hTERT promoter regulated, E1ACR2-deficient Ad (hTERT-AdΔE1ACR2), and this Ad demonstrated high tumor specificity, exhibited good replication, and effectively elicited antitumor immune responses in a number of tumor models.21,22 We armed the virus with mouse interleukin-15 (mIL-15), a multifunctional cytokine that is similar to IL-2 and can stimulate the proliferation and activation of natural killer (NK) cells, cytotoxic T lymphocytes and memory T cells to promote the generation and maintenance of antitumor immune responses,23 to create hTERT-AdΔE1ACR2-IL15 (hereafter referred to as Ad-IL15). In our evaluation, Ad-IL15, which stably expressed IL-15, profoundly changed the TME by enhancing immune cell activation, and inducing VN improvement and local nonclassical TLS formation; these effects were mediated via the activation of the stimulator of interferon genes (STING) pathway in DCs and resulted in an obvious systemic antitumor effect. In conclusion, our study establishes a novel OV that can limit tumor growth by facilitating an antitumor response, VN and nonclassical TLS formation.
Materials and Methods
Viruses
The recombinant adenovirus used in this study was based on human adenovirus type 5 (Ad5), which has been previously describe.9 Briefly, the fragment containing the target gene (mIL-15) was amplified by PCR and the amplification products were evaluated by agarose electrophoresis. Subsequently, the PXC1 vector containing CMV-EGFP-hTERT-E1AΔCR2 was linearized and the PCR amplification products were ligated into the linearized vector. The ligation products were introduced into competent DH5α cells, which were expanded, plated on LB solid agar medium and cultured at constant temperature for 12–16 h. The positive clone was selected for confirmation by sequencing, and the shuttle plasmid CMV-IL15-EGFP-hTERT-E1AΔCR2 was obtained. Finally, the shuttle plasmid CMV-EGFP-HTERT-E1AΔCR2 or CMV-IL15-EGFP-hTERT-E1AΔCR2 was cotransfected with the assistant packaging plasmid (PBHGE3) (Fig S1A) to obtain oncolytic adenovirus Ad (Fig S1B) or Ad-IL15 (Fig S1C). The viruses were then stored at −80°C until use.
Cell lines
The mouse melanoma cell line B16-F10, and the mouse lung cancer cell line LLC were obtained from the National Collection of Authenticated Cell Cultures (Shanghai, China). The human glioma cell line U87, human lung cancer cell line A549, human breast cancer cell line MDA-MB-231 and mouse colon cancer cell line MC-38 were all kept in our laboratory. A549, U87, MDA-MB-231 and LLC cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco) containing 10% fetal bovine serum and 1% penicillin/streptomycin. B16-F10 and MC-38 cells were cultured in Roswell Park Memorial Institute (RPMI) (Gibco) 1640 medium containing 10% fetal bovine serum and 1% penicillin/streptomycin. The cells were maintained at 37°C in 5% CO2 and confirmed to be free of mycoplasma.
Animal experiments
Pathogen-free female C57BL/6 mice aged 6 to 8 weeks old were purchased from the Animal Center of Slaccas (Shanghai, China). Rag-1−/− mice were obtained from GemPharmatech Co., Ltd. (Jiangsu, China). The mice were maintained in a pathogen-free facility and all the animal experiments were approved by Animal Ethics Committee of The Second Affiliated Hospital Zhejiang University School of Medicine (Hangzhou, China). During the whole experiment, mice were housed in a constant-temperature environment and maintained on a standard diet with free access to drinking water. The length (L) and width (W) of subcutaneous tumors in mice were measured with a digital Vernier caliper, and tumor volume was calculated with the formula L× W2 × 0.5. Tumor volume was continuously monitored.
To evaluate the effects of Ad and Ad-IL15, unilateral B16-F10 and MC-38 tumor models were established in immunocompetent mice. In these models, 2*105 B16-F10 cells or 1*106 MC-38 cells were subcutaneously injected into the right flanks of the mice. Unless otherwise indicated, the mice were randomly divided into a treatment group and a control group when the tumors reached approximately 50 mm3. Approximately 5 × 108 plaque-forming units (PFU) of virus suspension or sterile phosphate-buffered saline (PBS) as a control (40 µL/injection) was injected into each tumor every other day, for a total of three doses. Eleven days after the last treatment, the mice were sacrificed, and tumor tissues were collected for follow-up experiments. To evaluate the effect of Ad and Ad-IL15 on tumor VN and TLS formation, the treatment regimen was performed as described above, and tumor tissues, blood and spleens were collected on the 7th day after the last treatment. To study of functional tumor vessels, 200 µL of 1 mg/mL Alexa Fluor 488-conjugated Lycopersicon esculentum (a.k.a. lectin) (Vector Laboratories) was injected into the caudal vein before the mice were euthanized.
In vitro infection experiments and mIL-15 measured
Human tumor cell lines (1 × 104 cells/well) or mouse tumor cell lines (3 × 103 cells/well) were inoculated in 96-well plates and infected with Ad or Ad-IL15 at various multiplicities of infection (MOIs). After culturing at 37°C for 72 h, cell viability was measured with a cell counting kit-8 (CCK-8) (Meitune, China) according to the manufacturer’s recommended protocols. To evaluate the expression of mIL-15 after tumor cell lines were infected with Ad-IL15, we collected the supernatant of tumor cells infected with Ad-IL15 (MOI = 50) for 24–96 h. Then, a mouse IL-15 ELISA kit (Neobioscience, China) was used to measure the levels of mIL-15 in the supernatants. The values were measured on a multifunctional microplate reader (Bio-Tek). To measure the levels of IL-15 secreted by Ad-IL15-infected tumor tissues, the tumor tissues were collected on the 7th day after the last treatment. The tumor tissues were weighed, cut into small pieces, added to 1 mg/5 mL 1640 medium supplemented 10% FBS for incubated for 24 h. After the supernatant was collected, mIL-15 concentration was detected using an ELSIA kit. Mouse cancer cell lines (1 × 105) were plated in 6-well plates and treated with Ad-IL15 at the indicated MOIs. After 72 h of culture, the cells were collected for protein analysis experiments (see Western blotting).
Bone marrow harvest and DC culture
Mice aged 6–8 weeks old were killed by spinal cord transection, the tibia and femur were separated, the bone marrow cavity was washed repeatedly with RPMI-1640 medium, and the obtained bone marrow cells were purified with erythrocyte lysate as previously described.24 Subsequently, the bone marrow cells were seeded in 6-well plates at a density of 2 × 106 cells/5 mL and cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum + rmGM-CSF (20 ng/mL, Peprotech) + rmIL-4 (20 ng/mL, Peprotech) at 37°C in a 5% CO2 atmosphere. The medium was replaced with fresh medium on day 3, and loosely adherent bone marrow-derived dendritic cells (BMDCs) were collected on day 5. For RNA-seq analysis, Ad or Ad-IL15 (MOI = 50) was added to CD11c+ BMDCs and cultured for 12 h at 37°C. For TBK1 inhibition experiments, BMDCs were pretreated with 150 µg/ml Amlexanox (MedChemExpress) at 37°C for 1 h before Ad or Ad-IL15 (MOI = 100) were added to CD11c+ BMDC.
Western blotting
Collected cells were lysed with 1× RIPA lysis buffer containing phosphatase and protease inhibitors. The cell lysates were centrifuged at a high speed and 4°C for 30 min, and the protein concentrations of the supernatants were measured with a BCA Protein Assay Kit (Beyotime, China). Then, 30 μg of boiled protein was separated on a 10% SDS-PAGE gel. Then, the protein was transferred to a PVDF membrane, which was blocked for 1 h with TBS supplemented with 5% fresh nonfat milk at room temperature (RT). After being incubated with a primary antibody (listed in online supplemental table S1) overnight at 4°C, the membrane was incubated with the corresponding horseradish peroxidase (HRP)-conjugated secondary antibodies at RT for 1 h. Enhanced chemiluminescence (ECL) was used to detect target proteins on the membrane.
Immunofluorescence staining of mouse samples
Tumor tissues were collected, fixed in 4% (wt/vol) paraformaldehyde (PFA) overnight and embedded in paraffin blocks. Then, 5 μm-thick paraffin sections were prepared, dewaxed in xylene, rehydrated with ethanol, and washed with distilled water. Then, antigen repair was performed in a microwave oven with EDTA antigen repair buffer (pH 8.0). After 3% BSA was added for blocking at RT for 30 min, primary antibody (listed in online supplemental table S1) staining was performed at 4°C overnight. After the sections were washed with PBS (pH 7.4), a corresponding secondary antibody was added and incubated in the dark at RT for 1 h. Next, DAPI was added and incubated for 10 min at RT to stain nuclei. Finally, epifluorescence multispectral images of whole sections were acquired on a NIKON ECLIPSE C1 system (Nikon company), and scanned with a Panoramic SCAN II system (3DHISTECH Ltd.) with high resolution. For image acquisition, 5–6 images were randomly collected for each tumor sample for quantitative analysis at 40× magnification. The number of nucleated cells was counted with ImageJ software (V1.53). Then the numbers of CD133+ or Jarid1b+ cells were manually counted, and finally the proportions of CD133+ or Jarid1b+ cells were calculated. The quantitative expression of PDGFRβ, VCAM-1 and Lectin in tumor blood vessels was calculated with the ROI Manager function of ImageJ software. Quantitative analysis of T/B cell numbers and DC-T interactions was performed by manual counting. All immunofluorescence images were derived from representative images of three or more tumor samples.
Multicolor flow cytometry
Subcutaneous tumor tissues were collected from mice, chopped and digested by incubation in RPMI-1640 medium supplemented 1 mg/mL collagenase I (Gibco) and 1 mg/ml collagenase IV (Gibco) at 37°C for 1–2 h. Then, the cell suspension was passed through a 70-μm cell filter and resuspended in cell staining buffer (BioLegend) after washing with PBS. Finally, the single cell suspension was incubated with different fluorochrome-conjugated antibodies for 15 min at RT. Single cell suspensions derived from mouse spleen and peripheral blood were prepared as described25 and stained according to the scheme described above. For intracellular cytokine detection, single cell suspensions of cells from spleens or tumor tissues were distributed into 12-well plates and stimulated with a 1× PMA/ionomycin mixture and 1× BFA/monensin mixture at 37°C for 4–5 h. Then, surface antibody staining was performed as previously described. Subsequently, the cells were collected, permeabilized and fixed by incubation with Fixation/Permeabilization Solution (BD Biosciences) at 4°C for 20 min. The cells were then washed with Perm/Wash Buffer (BD Biosciences) and stained with intracellular factor antibodies at 4°C for 30 min. To determine the effects of OVs on normal tissue, a single-cell suspension of C57 mouse lung tissue was collected and infected with Ad/Ad-IL15 (MOI = 50) for 72 h. The proportion of dead cells was determined after 7-AAD staining for 10 min. Surface and intracellular factor staining was performed using antibodies recognizing the following markers: mouse CD45 (30-F11), mouse CD3 (17A2), mouse CD4 (GK1.5), mouse CD8 (53–6.7), mouse CD69 (H1.2F3), mouse CD103 (2E7), mouse CD11c (N418), mouse CD86 (GL-1), mouse CD54 (YN1/1.7.4), mouse PD-1 (29 F.1A12), mouse MHCII (M5/114.15.2), mouse CCR7 (4B12), mouse CD19 (6D5), mouse Granzyme B (QA16A02), mouse Perforin (S16009A), mouse TNF-α (MP6-XT22), mouse IFN-γ (XMG1.2) (all from Biolegend) and mouse NK1.1 (PK136, Thermo Fisher Scientific). Data were acquired on a FACSCanto II system or FACSFortessa system (BD Biosciences) and analyzed using FlowJo X.
Quantitative real-time PCR (qPCR)
Total RNA was extracted from CD11c+ DCs or enzymatically dissociated tumors using an RNA-Quick Purification Kit (Esunbio, China). Then, 1 μg of total RNA was converted into cDNA with PrimeScript RT Master Mix (Takara). The cDNA products were used as a template for qPCR amplification using TB Green Premix Ex Taq (Takara) on 7500 Fast Real-Time PCR System (Applied Biosystems). According to the manufacturer’s protocol, the cycle conditions were 95°C for 30 seconds followed by 40 cycles of 95°C for 5 seconds and 60°C for 34 seconds. GAPDH was used as a housekeeping gene for the standardization of target gene expression. The fold changes in target gene expression were determined using the 2–ΔΔct method. The primer sequences are shown in online supplemental table S2.
RNA-seq and analysis
Total RNA was extracted from frozen treated cells by the TRIzol-chloroform extraction method. Subsequently, the isolated total RNA was qualified and quantified using a Nano Drop and Agilent 2100 bioanalyzer (Thermo Fisher Scientific, MA, USA). mRNA was purified by Oligo(dT)-attached magnetic beads and fragmented at RT. First-strand cDNA was generated by random hexamer-reverse transcription, and second-strand cDNA was synthesized. Then, the generated cDNA was amplified and purified with Ampure XP Beads, and the final library was obtained by splint oligonucleotide sequence circulation. In vitro, sscir cDNA was rolled and amplified under the action of phi29. When it was amplified to 100–1000 copies, it formed a DNA nanoball (DNB) and was run on a Bgiseq500 sequencing platform. Bgiseq500 contains patterned nanoarrays, which are a kind of binding site array chip formed and processed on the surface of a silicon wafer. The spacing of modification sites on the chip is uniform, and only one DNB is fixed at each site, to ensure that the optical signals of each DNB will not interfere with those of the others and to improve the accuracy of subsequent sequencing. Subsequently, a single-ended 50-base reading was generated on the Bgiseq500 platform (BGI, Shenzhen, China).
The sequencing data were filtered with SOAPnuke (v1.5.2) by (1) removing reads containing sequencing adaptors; (2) removing reads whose low-quality base ratio (base quality less than or equal to 5) was more than 20%; (3) removing reads whose unknown base (‘N’ base) ratio was more than 5%, afterward clean reads were obtained and stored in FASTQ format. The clean reads were mapped to the reference genome using HISAT2 (v2.0.4). Bowtie2 (v2.2.5) was applied to align the clean reads to the reference coding gene set, then expression level of gene was calculated by RSEM (v1.2.12). The heatmap was drawn by pheatmap (v1.0.8) according to the gene expression in different samples. Essentially, differential expression analysis was performed using the DESeq2(v1.4.5) with Q value ≤ 0.05. The significant levels of terms and pathways were corrected by Q value with a rigorous threshold (Q value ≤ 0.05) by Bonferroni.
Bioinformatic analysis
The “Gene-DE” module in TIMER2.026,27 was used to analyze IL-15 mRNA differential expression in date from The Cancer Genome Atlas (TCGA) database, and the Wilcoxon test was applied to assess statistical significance. In the R environment, Kaplan-Meier curves were generated using the “survival” and “survivminer” packages, and Cox analysis was performed. P < .05 was considered to be significant, and significant results were visualized. The “Immune-Gene” module of TIMER2.0 was used to evaluate the relationship between the mRNA expression level of IL-15 and the degree of immune cell infiltration into tumors, and the “TIMER” algorithm was used to quantify it.27,28 The p value and partial correlation value (cor) were obtained by Spearman’s rank correlation test with purity adjustment.
Statistical analysis
Except for the bioinformatics analysis, all the statistical analyses were carried out with GraphPad Prism version 8.3 (GraphPad Software). The corresponding statistical test method is described in each figure. Results are expressed as the mean ± SEM. P values <.05 were considered significant.
Results
A modified oncolytic adenovirus successfully proliferates and produces IL15 in tumor cells
IL-15 is a pleiotropic cell stimulating factor that plays important roles in innate and adaptive immunity. By analyzing the mRNA expression of different cancer and normal tissues in the TCGA database, we found that the expression of IL-15 differed between the tumor group and normal group for many cancer types (Figure 1a). Moreover, the expression level of IL-15 was positively correlated with a better survival rate in patients with skin cutaneous melanoma (SKCM) (Figure 1b) or rectum adenocarcinoma (READ) (Figure 1c). Subsequently, we analyzed the correlation between the expression level of IL-15 and the degree of tumor infiltration by immune cells. We found that in patients with SKCM, high expression of IL-15 was associated with the infiltration of CD8+T cells, CD4+T cells and DCs (Figure 1d). In READ patients, high expression of IL-15 was associated with the infiltration of CD8+T cells and DCs (Figure 1e). These results suggest that the expression of IL-15 may be associated, at least in part, with more tumor-infiltrating lymphocytes (TILs). However, the use of recombinant IL-15 in the treatment of solid tumors has disadvantages, such as its short half-life and the potential toxicity associated with high-dose administration.29 Therefore, arming an OV with IL-15 to establish stable IL-15 expression has become a feasible strategy.
Figure 1.
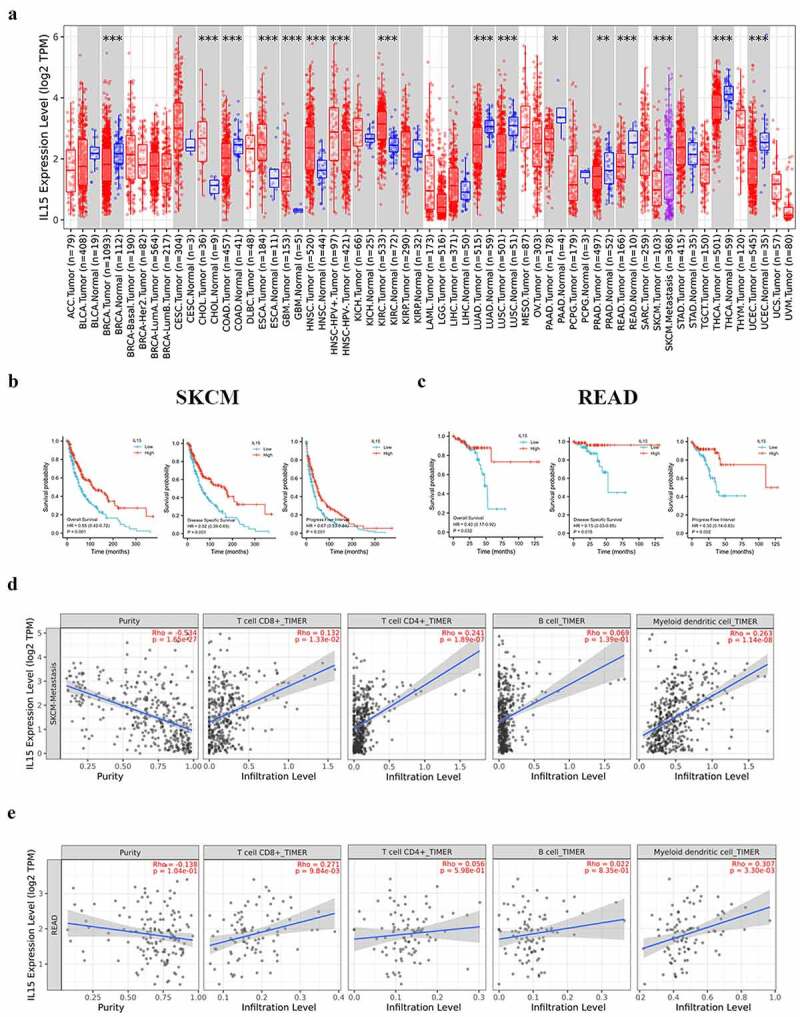
Expression of IL-15 and its relationship with tumor prognosis and immune cell infiltration. (a) The expression status of the IL-15 gene in different cancers was analyzed using TIMER2.0. Tumor abbreviations are shown in Table S3. Red represents tumor tissue, blue represents corresponding normal tissue, and purple represents melanoma metastasis. (b - c) Correlation between the expression level of IL-15 and the survival rate of patients with SKCM (b) or READ (c). (d - e) The correlation between the expression level of IL-15 and immune cell infiltration in SKCM (d) or READ (e) was analyzed by TIMER2.0.
Here, we designed a new OV, Ad-IL15, and its structure is shown in Figure 2a. Next, to investigate whether Ad-IL15 can infect tumor cells and stably secrete mIL-15, we infected three human tumor cell lines (A549, U87, and MDA-MB-231) and three mouse tumor cell lines (LLC, MC38, and B16-F10) with Ad-IL15 at an MOI of 50. As shown in Figure 2b, the IL-15 levels in the supernatant of Ad-IL15-treated cells were detected with ELISA kits, which proved that mIL-15 was efficiently produced by and released from Ad-IL15-infected human and mouse tumor cells. The concentration of IL-15 reached higher values at 96 h after infection of human tumor cells with Ad-IL15, while the peak of IL-15 secretion appeared to occur at 72 h after Ad-IL15 infection in the three mouse tumor cells lines. Subsequently, to evaluate the oncolytic ability of Ad-IL15, three human tumor cell lines (A549, U87, and MDA-MB-231) and three mouse tumor cell lines (LLC, MC38, and B16-F10) were infected with Ad or Ad-IL15 at different MOIs for 72 h. Both Ad and Ad-IL15 induced time-dependent and dose (MOI)-dependent killing activity against the human and mouse tumor cells (Figure 2c). However, the human tumor cell lines were more easily infected than the mouse tumor cell lines, which probably occurred because human cells are natural hosts of adenovirus.30 After the indicated cell line was infected with an increasing amount of Ad-IL15, the upregulated expression of E1A and fibers (early and late regulatory proteins in the virus cycle, respectively) was found (Figure 2d). In addition, to evaluate the safety of OV, we infected mouse lung tissue cells with Ad or Ad-IL15 at an MOI = 50 for 72 h. We found that Ad and Ad-IL15 did not kill normal tissue cells (Fig S2A). These results suggest that Ad-IL15 can exert an oncolytic effect on both mouse tumor cells and human tumor cells in vitro, can stably release IL-15, and is very safe.
Figure 2.
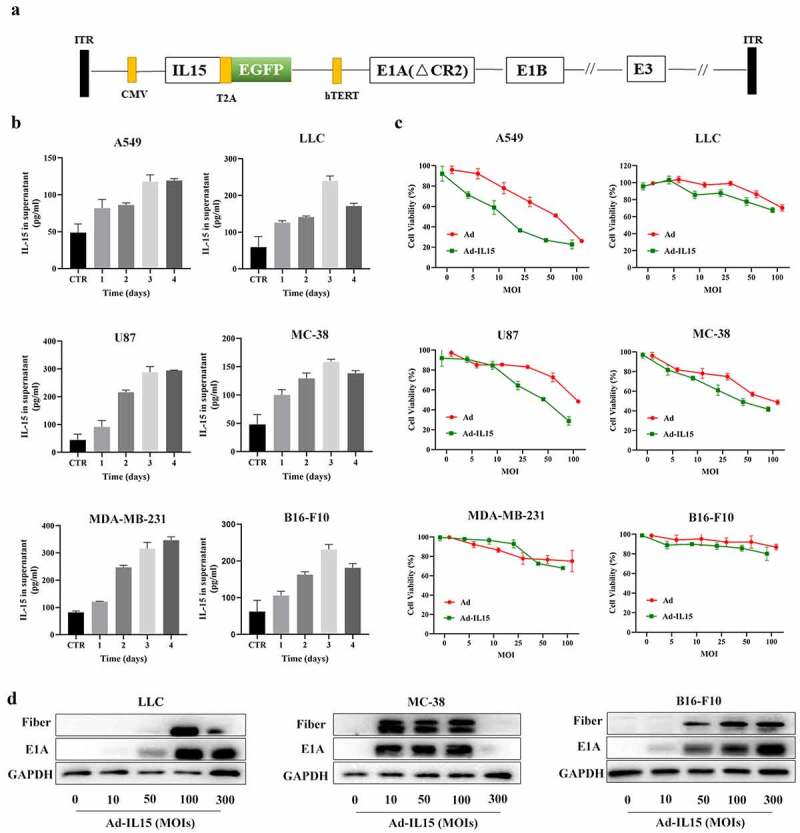
A modified oncolytic adenovirus successfully proliferates and produces IL15in tumor cells. (a) A schematic diagram of recombinant Ad-IL15. (b) The expression of IL-15. ELISA was used to measure IL-15 levels in the supernatant of cell cultures. Different human and mouse tumor cell lines were treated with Ad-IL15 for 24, 48, 72 or 96 h at the indicated dose (MOI = 50). The supernatant of untreated cell cultures was used as a control (CTR). (c) Cell proliferation analyses of Ad/Ad-IL15-infected human and mouse tumor cell lines. A CCK-8 assay was performed to evaluate the viability of different human and mouse cell lines treated with Ad/Ad-IL15 at different MOIs for 3 days. (d) Assessment of viral protein expression in three mouse cell lines infected with Ad-IL15 by western blotting.
Ad-IL15 plays an antitumor role by increasing TILs in vivo
To characterize the effects of the oncolytic adenovirus, we evaluated its use in two different immunocompetent mouse models. B16-F10 and MC-38 tumor bearing C57BL/6 mice were intratumorally injected with PBS, Ad or Ad-IL15 every other day for a total of 3 times. Compared with that of PBS treated mice, the tumor volume of Ad/Ad-IL15-treated mice was significantly smaller, although Ad-IL15 treatment significantly reduced tumor volume compared with Ad treatment in only the B16-F10 model, not in the MC-38 model (Figure 3a). Additionally, these results also indicated that there was no correlation between the in vivo efficacy and the in vitro cytotoxicity of these murine cell lines, suggesting that direct oncolytic activity was not the only factor that contributed to the antitumor effects observed in mouse models; for example the immune response in the TME might be involved.31 To evaluate virus-induced immune responses, treated tumors were harvested on day 14 and subjected to multicolor flow cytometry for TIL analysis. As expected, our FACS analysis revealed a significant increase in the frequency of CD45high lymphocytes after the administration of Ad or Ad-IL15 in both models (Figure 3b,). In the B16-F10 model, compared with PBS- or Ad-treated mice, Ad-IL15-treated mice had a significantly higher proportion of lymphocytes including CD45+ cells, CD8+ T cells, CD4+T cells, NK cells and natural killer T (NKT) cells in the TME (Figure 3b). In addition, we examined whether Ad-IL15 can express mIL-15 in tumor tissues. Consistent with the in vitro results, we observed the release of mIL-15 in tumor tissues after Ad-IL15 infection (Fig S3A). In the MC-38 model, similar trends were observed for infiltratingCD45+, CD4+, and CD8+ T cells as well as NKT cells (Figure 3c). However, we did not observe an increase in infiltrating NK cells. Notably, the frequency of tissue-resident memory (TRM) cells in Ad-IL15-treatedmice was also elevated compared to PBS-treated mice and had an increased tendency in comparision with Ad-treated mice in both models (Figure 3b,). The infiltration of TRM cells into tumors correlates with an enhanced response to immunotherapy and is often associated with a favorable clinical outcome in patients with cancer.32 Interestingly, intratumoral administration of Ad-IL15 increased the rates of tumor-infiltration by CD8+ T cells in an activated state, as evidenced by their increased CD69 expression. However, it also increased the expression of inhibitory checkpoint molecules, such as PD-1 in CD8+ T cells (Figure 3d,), indicating that there may be a negative feedback loop mediated by PD-1 to limit the expansion of effector T cells during the immune response. Therefore, is the difference in therapeutic efficacy between Ad and Ad-IL15 due to host immunity? We monitored changes in the subcutaneous B16-F10 tumor volume in C57 and Rag-1−/− mice (lack T/B cell, Fig S3B) treated with Ad or Ad-IL15. The results showed that the efficacy of Ad and Ad-IL15 in Rag-1−/− mice was significantly inhibited compared with that in C57 mice (Fig S3C). This suggests that Ad-IL15 can elicit stronger host immune system activation. Overall, these results suggested that the Ad-IL15–mediated inhibition of tumor growth might stem from the ability of this OV to increase TILs.
Figure 3.
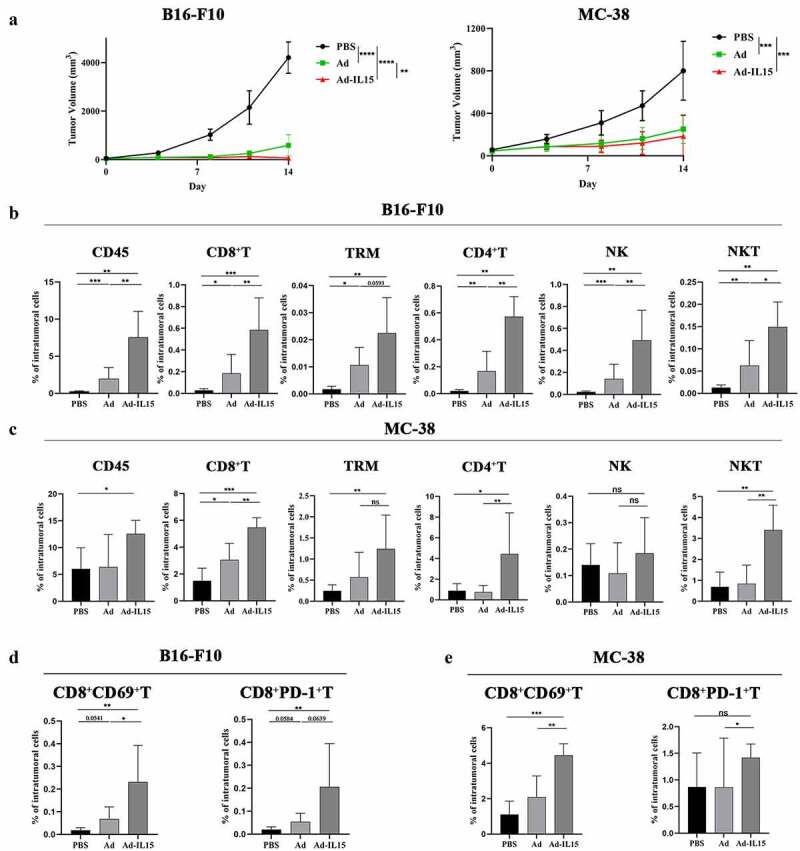
Ad-IL15 plays an antitumor role by increasing TILs. Mice were subcutaneously inoculated with B16-F10 or MC-38 cells. When tumors reached approximately 50 mm3, they were directly injected with PBS, Ad (5 × 108 PFU) or Ad-IL15 (5 × 108 PFU) every other day for a total of three injections. (a)The tumor growth in mice treated with PBS, Ad or Ad-IL15 was shown. n = 7–8. **P < .01, ***P < .001 and ****P < .0001 by an unpaired t test. (b - e) Eleven days after the last treatment, tumors were collected and analyzed by flow cytometry to calculate the percentages of TILs in the tumors. (b to c) Intratumoral CD45+ cells, CD8+ T cells, TRM cells, CD4+ T cells, NK cells, and NKT cells in B16-F10 (b) and MC-38 (c) tumor models. (d - e) Intratumoral CD8+CD69+T cells and CD8+PD-1+ T cells in B16-F10 (d) and MC-38 (e) tumor models. n = 5–8. * P < .05, **P < .01 and ***P < .001 by the Mann-Whitney U test. ns, not significant.
Intratumoral administration of Ad-IL15 promotes VN
Many studies have confirmed that OVs are powerful antiangiogenic agents that can significantly improve the hypoxic state of the TME.14 However, whether OVs can achieve tumor VN remains to be further studied. To determine the effect of Ad-IL15 on tumor vessels, we collected tumor tissues from the B16-F10 model on day 7 after treatment. Compared with the PBS group, the intratumoral Ad or Ad-IL15 treatment group exhibited significantly decreased expression of HIF-1α, which suggested that OV significantly diminished the hypoxia state in the TME (Figure 4a). CD133 and Jarid1b are considered markers of hypoxia-responsive cancer stem cells,33,34 so we measured the expression of CD133 and Jarid1b in the TME. Immunofluorescence staining showed that the levels of CD133 (Figure 4b) and Jarid1b (Figure 4c) were significantly decreased in the intratumoral Ad-IL15 treatment group compared with the PBS and Ad treatment groups. Moreover, compared with PBS or Ad treatment, Ad-IL15 treatment promoted the expression of antiangiogenic (CXCL10 and TNFsf15) and vascular maturation (CXCL9) factors (Figure 4a). This evidence supported our hypothesis that Ad-IL15 treatment can diminish the hypoxic state in the TME and promote tumor VN. To further evaluate the effect of Ad-IL15 on indices of VN, immunofluorescence analysis was performed and showed that the percentage of platelet-derived growth factor receptor β (PDGFRβ) + pericyte-covered blood vessels was significantly higher in Ad-IL15-treated mice, which indicated that more mature blood vessels were present in the tumor tissues in these mice (Figure 4d). In addition, in the Ad-IL15-treatment group, there was a significant increase in the number of lectin+ functional vessels (Figure 4e) and vascular cell adhesion protein-1 (VCAM-1) + blood vessels (figure 4f), which indicated improvements in vascular perfusion and maturation. VCAM-1+ vascular endothelial cells (VECs) can respond to exposure to foreign inflammatory factors and can promote leukocyte adhesion to VECs and leukocyte entry into the parenchyma via diapedesis.35 Lymphatic vessels are another important component of the TME and are responsible for the return of interstitial fluid and the delivery of antigen-presenting cells (APCs) to initiate adaptive immunity. Therefore, we evaluated the lymphatic vessel density of the TME in the three groups and found that the density of Lyve+ lymphatic endothelial cells was significantly higher in the Ad-IL15 treatment group than in the control group (Figure 4g). In conclusion, our data suggest that intratumoral Ad-IL15 therapy can increase tumor VN and tumor lymphatic vessel density.
Figure 4.
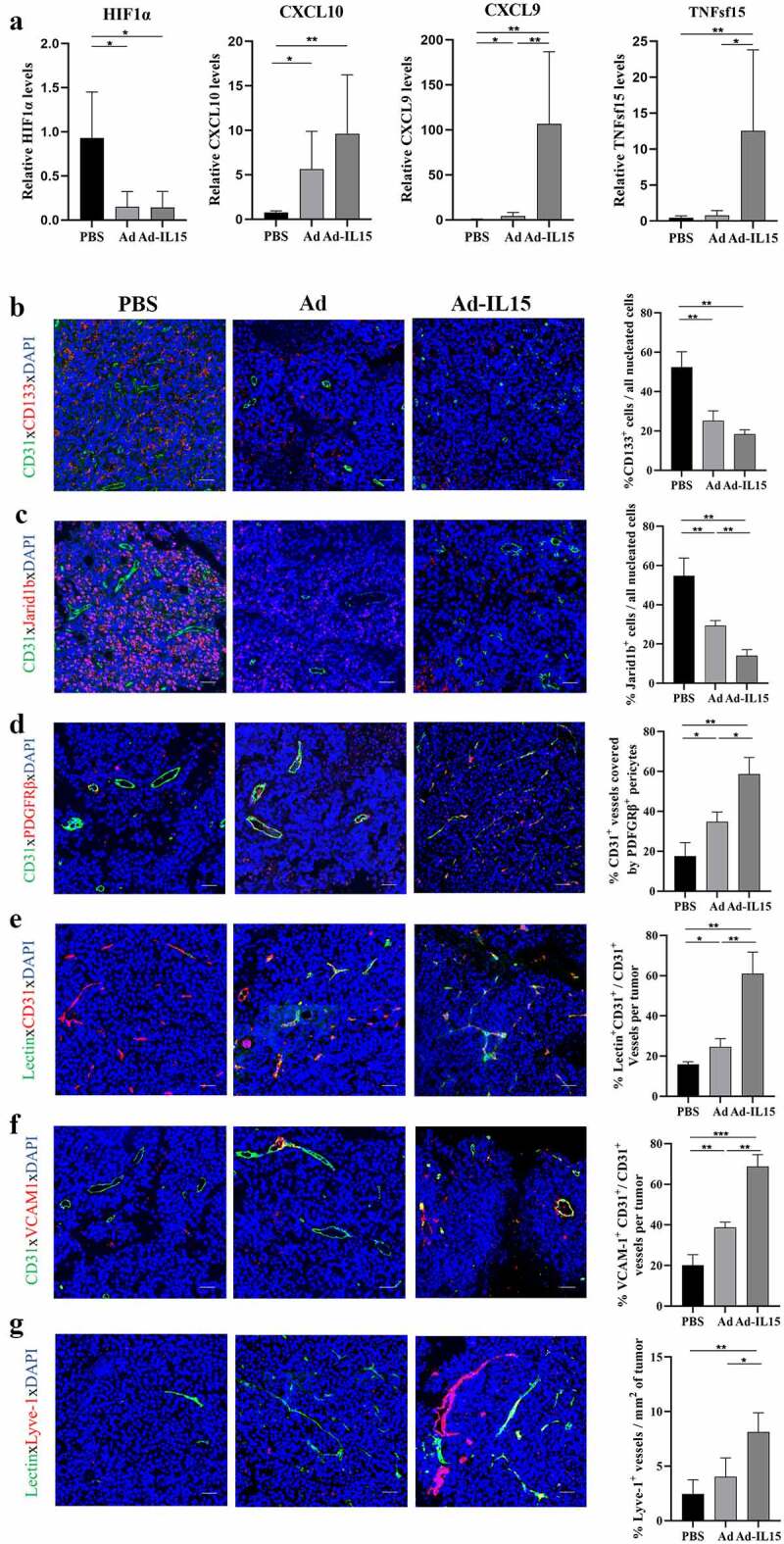
Intratumoral administration of Ad-IL15 promotes VN. Mice bearing subcutaneous B16-F10 tumors (approximately 50 mm3) were intratumorally injected every other day for a total of three times with PBS, Ad or Ad-IL15 (5 × 108 PFU). (a) Seven days after the last treatment, tumor tissues were collected, and RNA was isolated. RT-PCR was used to analyze gene expression. The mRNA levels of antiangiogenic factors (CXCL10 and TNFsf15), vascular maturation factors (CXCL9) and HIF1α were measured. n = 5–6. *P ≤ .05 and **P ≤ .01 by the Mann-Whitney U test. (b-c) Representative immunofluorescence staining and image quantification of the hypoxia-reactive tumor stem cell markers CD133 (b) and Jarid1b (c). (d) Representative immunofluorescence staining and image quantification of PDGFRβ+ pericyte coverage on tumor VECs. (e) Representative immunofluorescence staining and image quantification of lectin-perfused functional vessels. (f) Representative immunofluorescence staining and image quantification of VCAM-1 expression on tumor VECs. (g) Representative immunofluorescence staining and image quantification of Lyve-1+ lymphatic endothelial cells. Statistical significance was assessed by an Unpaired Student’s t test. *P ≤ .05, **P ≤ .01. Scale bars: 50 μm.
Intratumoral injection of Ad-IL15 induces nonclassical TLS formation
To date, TLSs have been found in a variety of human tumor tissues,36 and studies have confirmed that TLSs are related to a favorable prognosis in patients.37 While evaluating the effect of intratumoral Ad-IL15 therapy on TLS formation, we observed that Ad-IL15 treatment increased LTβR agonists (LTα, LTβ and TNFsf14) (Figure 5a) and lymphoid tissue steady-state cytokines (CCL19, CCL21 and CXCL13) (Figure 5b) in the TME. These data suggest that the tumor locally produces “soil” that initiates and promotes TLS formation. It has been reported that PANd+ high endothelial venules (HEV) are crucial structures in TLS formation.38 Therefore, we explored the expression of CD45 and PNAd. The results showed that compared with control treatment, intratumoral treatment with either Ad or Ad-IL15 could promote the infiltration of CD45+ lymphocytes, and the lymphocytes were spatially arranged in clusters (Figure 5c). Interestingly, compared with the control and Ad groups, intratumoral treatment with Ad-IL15 promoted the formation of PNAd+ HEVs, and CD45+ lymphocytes were spatially more closely associated with PNAd+ HEVs to form a TLS-like structure (Figure 5c). These results suggest that Ad-IL15 treatment may induce the formation of classical TLSs. Subsequently, we used CD3, B220 and PNAd to characterize classical TLSs. We found that Ad-IL15 induced the formation of structures similar to classical TLSs (Figure 5d) and increased CD3+T cells infiltration (Fig S4A); however, the B cell infiltration in this group was lower than expected and scattered, although the numbers of infiltrating B cells were increased compared with those in the Ad and PBS groups (Figure 5d). This suggests that this TLS may be in the early stage and that its function is not perfect. Therefore, we continued to explore whether Ad-IL15 can induce the formation of nonclassical TLSs including CD11c+ DCs, CD3+ T cells and CD31+ blood vessels in the TME, and add an isotype control comparison (Fig S4B). Interestingly, we found nonclassical TLSs in the TME after Ad-IL15 treatment (Figure 5e), and there was spatial contact between CD11c+ DCs and CD3+ T cells in the TME, which provided the spatial possibility for antigen presentation by DCs to T cells and subsequent T cell activation. Subsequently, we further verified the existence of nonclassical TLSs using CD11c, CD3 and PNAd. As expected, we observed the presence of nonclassical TLSs containing CD11c+ DCs, CD3+ T cells, and PNAd+ HEVs in the TME after Ad-IL15 treatment (Fig S4C). Altogether, our data suggest that Ad-IL15 treatment may mainly stimulate the formation of nonclassical TLSs to exert antitumor effects.
Figure 5.
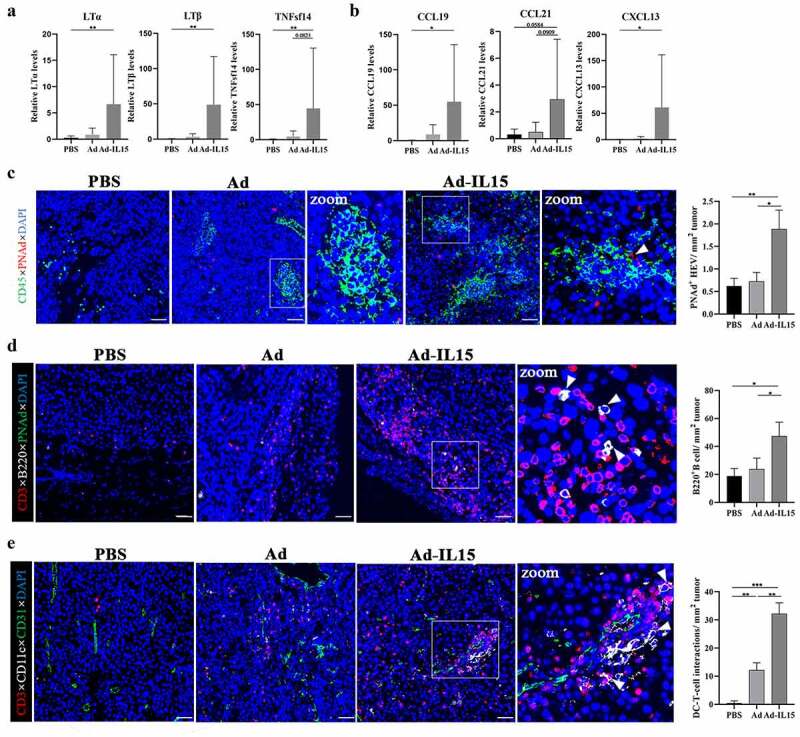
Intratumoral injection of Ad-IL15 induces nonclassical TLS formation. Mice bearing subcutaneous B16-F10 tumors (about 50 mm3) were intratumorally injected every other day for a total of three times with PBS, Ad or Ad-IL15 (5 × 108 PFU). (a-b) Seven days after the last treatment, tumor tissues were collected, and RNA was isolated. RT-PCR was used to analyze gene expression. The mRNA levels of TLS-inducing LTβR agonists (LTα, LTβ and TNFsf14) (a) and TLS-inducing homeostatic chemokines (CCL19, CCL21 and CXCL13) (b) were measured. n = 5–6. *P ≤ .05 and **P ≤ .01 by the Mann-Whitney U test. (c) Representative immunofluorescence staining images showing CD45 (green), PNAd (red) and DAPI (nuclei, blue) staining in PBS, Ad and Ad-IL15 treated tumors. PANd+ HEV area was quantitatively analyzed. The arrow in C indicates PANd+ HEV. *P ≤ .05 and **P ≤ .01 by Unpaired Student’s t test. Scale bars: 50 μm. (d) Representative immunofluorescence staining images showing CD3 (red), B220 (white), PNAd (green) and DAPI (nuclei, blue) staining in PBS, Ad and Ad-IL15-treated tumors. B220+ B cell numbers were quantitatively analyzed. Arrows in D indicate B220+ cells. *P ≤ .05 by Unpaired Student’s t test. Scale bars: 50 μm. (e) Representative immunofluorescence staining images showing nonclassical TLSs composed of CD11c+ DC, CD3+ T cell and CD31+ blood vessel staining in PBS, Ad and Ad-IL15-treated tumors. Quantification of the physical contact between DC-T cells. Arrows in E indicate the physical contact between DC-T cells. *P ≤ .05, **P ≤ .01 and ***P < .001 by Unpaired Student’s t test. Scale bars: 50 μm.
Ad-IL15 induces VN and TLS formation by activating the STING pathway in CD11c+ DCs
DCs not only function as activators of CD8+ T cells but also maintain the phenotype and function of HEVs in adult mouse lymph nodes (LNs)39 to support the formation of TLSs. Therefore, we hypothesized that Ad-IL15 promotes VN improvement and TLS formation by activating DCs. We performed RNA-seq analysis of mouse CD11c+ BMDCs treated with Ad-IL15 or PBS for 12 h. A list of 3808 differentially expressed genes (1705 downregulated and 2103 upregulated, |log2FC|>1 and p < .05) is shown in Fig S5A. Gene Ontology (GO) enrichment analysis of the differentially expressed genes showed that Ad-IL15 induced vascular development, lymphatic development and IL-15-mediated signaling pathway changes in DCs (Figure 6a). Subsequently, we further evaluated the specific differential gene changes induced by Ad-IL15. Consistent with our hypothesis, Ad-IL15 treatment significantly downregulated the expression of angiogenic factors (VEGFA, VEGFB and Ang-2) and significantly upregulated the expression of vascular homeostasis factors (CXCL9, CXCL10, TNFSF15 and VCAM-1) in DCs (Figure 6b). Moreover, a biomarker panel related to the promotion of local TLS formation in tumors was also markedly increased, as previously described40 (Figure 6b). In the Ad-IL15 stimulation group, CD11c+DCs expressed more maturation factors and costimulatory molecules (Figure 6c). Moreover, the expression of CD54hi+, CD86hi+, MHCIIhi+ and CCR7hi+ of DCs was confirmed by flow cytometry (Figure 6d). Considering that Ad-IL15 is a DNA virus, the host can sense double-stranded DNA (dsDNA) through cGAs to activate the SITNG signaling pathway and subsequently induce the downstream secretion of IFN and other cytokines, which can activate antitumor immune responses.41 Recently, a number of studies have confirmed that treatment with low-doses of STING agonists can promote tumor VN and TLS formation.24,42 Therefore, we explored whether Ad-IL15 mediates VN and the formation of TLSs by activating the STING pathway in DCs. STING oligomerizes after binding of cGAMP, leading to the recruitment and activation of the kinase TBK1.43 We tested our hypothesis with Amlexanox, which is a small-molecule drug that can bind to the TBK1 protein in the STING-TBK1-IRF3 signaling pathway and inhibit its phosphorylation, resulting in inhibited STING signaling.44 We observed that the expression levels of LTα, CXCL10, IFN-β and IL-6 were significantly increased, and this effect could be blocked by Amlexanox. (Figure 6e). The expression of STING, p-TBK1/TBK1 and p-IRF3/IRF3 was also increased in DCs treated with Ad-IL15 compared with those treated with PBS, and inhibition of TBK1 reduced the expression of these proteins (figure 6f). In summary, our data suggest that Ad-IL15-activated DCs may serve as sponsors for VN and TLS formation within the TME.
Figure 6.
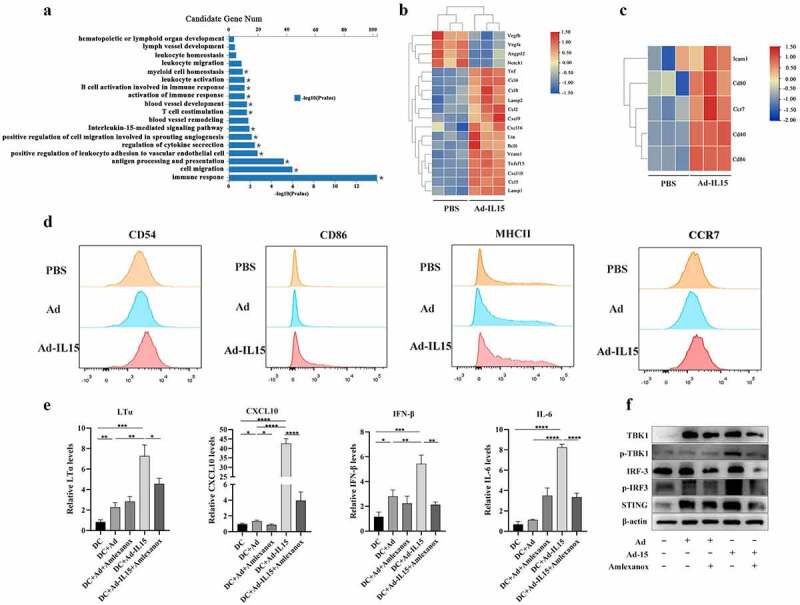
Ad-IL15 induces VN improvement and TLS formation by activating the STING pathway in CD11c+ DCs. BMDCs were isolated from C57 mice and cultured with rmGM-CSF (20 ng/mL) + rmIL-4 (20 ng/mL). The BMDCs were collected and purified on day 5, and then stimulated in vitro . (a-c) BMDCs were collected and stimulated with PBS, Ad or Ad-IL15 for 12 h. RNA was isolated and analyzed by RNA-seq. GO analysis of differentially expressed genes (a). VN-induced and TLS-induced gene expression (b). DC maturation related gene expression (c). (d) BMDCs were stimulated with PBS Ad or Ad-IL15 for 12 h, and the state of DC maturation was analyzed by flow cytometry . (e-f) BMDCs were stimulated with PBS, Ad, Ad + Amlexanox, Ad-IL15 or Ad-IL15 + Amlexanox for 12 h . Gene expression of LTα, CXCL10, IFN-β and IL-6 (e). Protein expression of components of the STING-TBK1-IRF3 pathway (f). Statistical significance was assessed by Unpaired Student’s t test. *P ≤ .05, **P ≤ .01, ***p < .001, ****p < .0001.
Intratumoral administration of Ad-IL15 leads to DC maturation and T-cell activation
We observed that CD11c+ DCs activated by Ad-IL15 in vitro showed VN-inducing and TLS-inducing properties (Figure 6b). Therefore, we next evaluated the effect of Ad-IL15 on DCs in vivo. We used the unilateral B16-F10 subcutaneous tumor model and collected the spleen, blood and tumor tissue of mice on day 7 after the last intratumoral injection. We observed that Ad-IL15 treatment increased the numbers of DCs and CD86+ DCs in the TME (Figure 7a). We found that the Ad group had the highest number of DCs in the peripheral blood, while the Ad-IL15 group had the highest number of CD86+ DCs in peripheral blood (Figure 7b). In addition, there was no significant difference in the number of DCs in mouse spleen tissue, but the Ad group had the highest number of CD86+ DCs in the spleen (Figure 7c), indicating that local treatment with Ad or Ad-IL15 may induce a systemic immune response, promote the proliferation and activation of DCs in the spleen, and recruit mature DCs to local tumor tissues through the peripheral blood. Consistent with these results, RT-PCR analysis of tumor tissues further confirmed the upregulated expression of genes involved in DC-specific costimulatory signaling (CD40) and DC maturation (CD86) (Figure 7d). Subsequently, we evaluated functional changes in CD8+ T cells using flow cytometry and found that Ad-IL15 treatment significantly increased the number of activated-CD8+T cells, which were characterized by high expression of IFN-γ, TNF-α, Granzyme B and Perforin compared with Ad or PBS treatment (Figure 7e). Collectively, these findings suggest that local Ad-IL15 treatment can induce systemic DC maturation and ultimately restore the effector function of CD8+ T cells in the TME, resulting in strong antitumor immunity.
Figure 7.
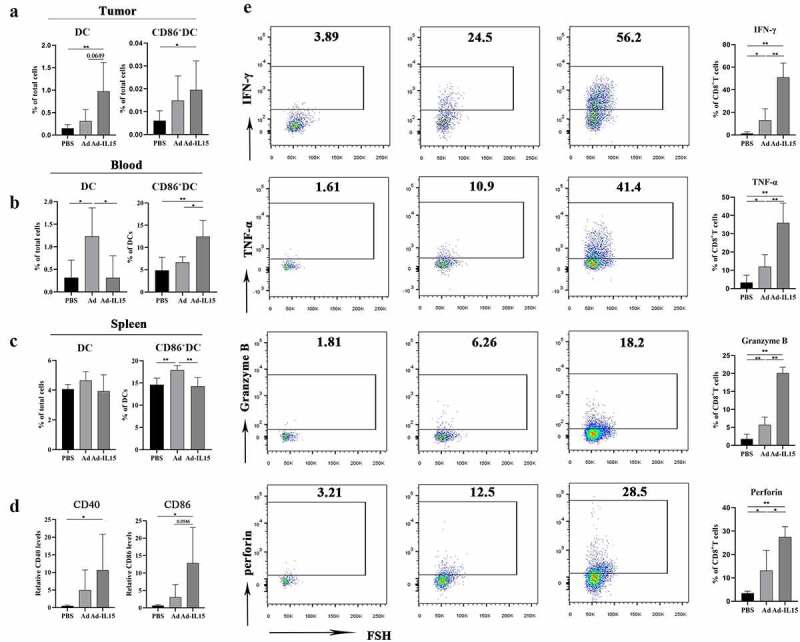
Intratumoral administration of Ad-IL15 leads to systemic DC maturation and T-cell activation. Mice bearing subcutaneous B16-F10 tumors were intratumorally injected every other day, for a total of three times, with PBS, Ad or Ad-IL15 (5 × 108 PFU). (a - c) Seven days after the last treatment, tumors, blood and spleen were collected and analyzed by flow cytometry. Frequencies of DCs and activated DCs (CD86+DCs) in tumors (a), blood (b) and spleen(c) from treated mice. (d) Seven days after the last treatment, tumor tissues were collected and RNA isolated. Quantitative RT-PCR was used to analyze gene expression. The mRNA levels of genes associated with DC-specific costimulatory signaling (CD40) and DC maturation (CD86) were measured. (e) Seven days after the last treatment tumors were collected and analyzed by flow cytometry . Expression of IFN-γ, TNF-α, Granzyme B and Perforin in tumor-infiltrating CD8+ T cells. n =5-6. Statistical significance was assessed by Unpaired Student’s t test. *P ≤ .05, **P ≤ .01, ***P ≤ .001. ns, not significant.
Amlexanox inhibits Ad-IL15-induced immune activation, tumor VN and nonclassical TLS formation
We found that the immune-activating effect induced by Ad-IL15 might be mediated by the STING pathway of activating DCs. Therefore, we next explored whether Amlexanox can inhibit the immune activation induced by Ad-IL15 in vivo. We found that Amlexanox combined with Ad/Ad-IL15 significantly reduced the tumor growth inhibition induced by Ad/Ad-IL15 (Figure 8a). Next, we analyzed the changes in TILs after treatment by flow cytometry. We found that the increased infiltration of CD8+ T cells (Figure 8b), CD4+ T cells (Figure 8c) and NK cells (Figure 8d) induced by Ad-IL15 treatment was significantly inhibited by Amlexanox. Moreover, the number of infiltrating DCs (Figure 8e) and the proportion of DCs expressing CD86 (figure 8f) and CD54 (Figure 8g) were also significantly reduced. Subsequently, we used VN markers (PDGFRβ and VCAM-1) to explore whether Amlexanox can inhibit Ad-IL15-mediated tumor VN. As expected, Amlexanox significantly reduced PDGFRβ+ pericyte coverage (Figure 8h) and VCAM-1 expression (Figure 8i) on the vascular surface after Ad-IL15 treatment. In addition, we also observed that intratumoral Amlexanox treatment hindered the formation of nonclassical TLSs, that DCs and T cells were more spatially dispersed, and that DC-T connections were reduced (Fig S6A). These results further suggest that the activation of the STING pathway plays an important role in Ad-IL15 induced immune activation, tumor VN and nonclassical TLS formation.
Figure 8.
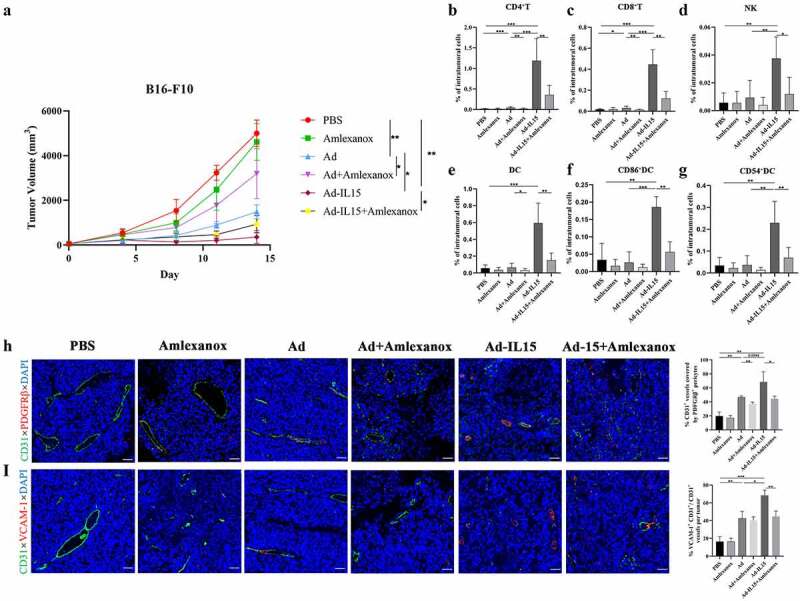
Amlexanox inhibits Ad-IL15-induced immune activation, tumor VN and nonclassical TLS formation. Mice bearing subcutaneous B16-F10 tumors (approximately 50 mm3) were intratumorally injected every other day for a total of three times with PBS, Amlexanox, Ad, Ad + Amlexanox, Ad-IL15, or Ad-IL15 + Amlexanox. (a)Tumor growth in mice treated with PBS, Amlexanox, Ad, Ad + Amlexanox, Ad-IL15, or Ad-IL15 + Amlexanox is shown. n=6–9. * P < .05, **P < .01 by Mann-Whitney U test. (b - g) Eleven days after the last treatment, tumors were collected and the percentage of immune cells was analyzed by flow cytometry. (b - d) Changes in the proportion of CD8+T cells, CD4+T cells and NK cells in the TME. (e - g) Changes in the proportion of DC, CD86+ DC and CD54+ DC in the TME. n = 6-9. * P < .05, **P < .01 and ***P ≤ .001 by Mann-Whitney U test. (h) Representative immunofluorescence staining and quantification of PDGFRβ+ pericyte coverage on tumor VECs. (i) Representative immunofluorescence staining and image quantification of VCAM-1 expression on tumor VECs. Statistical significance was assessed by Unpaired Student’s t test. *P ≤ .05, **P ≤ .01, ****P ≤ .0001. Scale bars: 50 μm.
Discussion
Vascular-targeted therapy is an important part of immunotherapy. The current clinical data show that antiangiogenic therapy alone cannot produce long-term survival benefits.45 At present, the field is more inclined toward the strategy of tumor VN, which can allow effective transport of drugs and oxygen into the TME.46 Here, we constructed and identified a novel oncolytic adenovirus, Ad-IL15, which could directly lyse tumor cells in vitro and enhance host antitumor immunity by promoting the infiltration of TILs. In addition, local Ad-IL15 treatment could also induce tumor VN and the formation of local nonclassical TLSs, which might depend on the activation of the STING pathway in CD11c+ DCs.24,47
As an emerging immunotherapy, a variety of OVs such as Imlygic, H101 and Rigvir,48 have been approved by the Food and Drug Administration (FDA) of various countries. Oncolytic adenoviruses are the most widely studied OV at present, and their transformation mainly focuses on the E1 and E3 regions. E1A is an essential gene for adenovirus replication and can promote S-phase entry in infected cells.49 E1ACR2-deleted mutants can replicate only in Rb/p16 pathway-deficient tumor cells, which has been confirmed in most cancers; they cannot replicate in normal cells.50Telomeres are proteins that stabilize cell chromosomes and are regulated by telomerase. Telomerase is usually highly expressed in tumor cells, but silent in normal cells.51 Therefore, using the human telomerase reverse transcriptase (hTERT) promoter as the promoter of an OV can make the OV replicate specifically in tumor cells.52 Deletion of the CR2 region of E1A and replacement with the hTERT promoter are two common ways to enhance the specific replication of an oncolytic adenovirus in cancer cells. Interestingly, Zhang et al53 constructed an oncolytic adenovirus with the above two strategies. They found that the antitumor effect of the bispecific oncolytic adenovirus was better than that of a monospecific oncolytic adenovirus. In addition, E3 encodes a protein that inhibits the antiviral immune response and participates in the interaction between the virus and host, thus reducing the probability of immune recognition of infected cells.54 The reconstruction of the E3 region is controversial. E3 deletion can allow accommodation of additional foreign genes, but it also leads to a decline in viral gene expression, weakening replication, increasing clearance and reducing antitumor effects.49 Therefore, based on the above strategy, we constructed an oncolytic adenovirus with hTERT initiation, E1ACR2 deletion and E3 retention.
We observed that the therapeutic effect of Ad-IL15 was different in the two mouse subcutaneous tumor models. In TME after Ad-IL15 treatment, the infiltrating T cells and NK cells were also inconsistent. This may be due to tumor heterogeneity. B16-F10 cells are tumor cells with low immunogenicity, while MC-38 cells are tumor cells with high immunogenicity.55 The main role of oncolytic adenovirus is to promote the transformation of “cold tumors” into “hot tumors”. Therefore, the chemotactic effects of Ad-IL15-treated B16-F10 and MC-38 on NK cells may be different. Interestingly, Ad-IL15 inhibited the growth of subcutaneous tumors compared with PBS treatment in the B16-F10 model of Rag-1−/− mice, which may be mediated by Ad-IL15-induced NK cell infiltration. Recently, an oncolytic vaccinia virus armed with IL15-Rα has demonstrated effective tumor growth inhibition in the intraperitoneal model of MC-38 colon cancer.56 The virus induced increased infiltration of immune cells, but depletion experiments showed that its antitumor effect was mainly dependent on CD8+T cells rather than NK cells. This may be due to the fact that the virus can inhibit NK cell functions in the TME through direct infection or the virus encodes multiple genes whose products are to inhibit NK cells via inhibition of the nuclear factor ĸB (NF- ĸB) pathway. This suggests that NK cells may play different roles in different types of viruses and tumor models.
The strategy of promoting tumor VN has a long history. Exogenous factor intervention can improve tumor vascular function, which is a marker of VN.57 In our model, tumor vascular perfusion, hypoxia and pericyte coverage were significantly improved, indicating VN. Moreover, the Ad-IL15-induced tumor VN may be caused by the activation of STING pathway of DCs. These data suggest a mechanism by which Ad-IL15 promotes the stability of tumor blood vessels and lymphatic vessels by changing the TME, to restore tumor blood perfusion and enhance the development of antitumor immunity.24,42 Moreover, the stabilization of tumor blood vessels and lymphatic vessels can reduce tumor IFP and promote the infiltration of antitumor factors and immune cells into tumor tissues, indicating that VN and antitumor immunity can promote each other. Interestingly, studies have confirmed that the growth rate of tumors is not affected by the blood flow velocity, vascular volume or use of oxygen or glucose,15 indicating that VN does not accelerate tumor growth.
TLSs, as the starting site for in situ immunity, often have different formation mechanisms under pathological conditions.47 B-cell follicles and germinal centers (GCs) are key components of TLSs.20,38 CXCL13 secreted by immune cells can chemoattract B cells and initiate TLS formation. However, we did not observe significant B-cell infiltration or GC formation, despite increased CXCL13 gene expression. This shows that the formation of TLSs requires the joint action of many factors. However, studies have shown that nonclassical TLS formation is also associated with a good prognosis.58 Therefore, the combination of OVs and factors promoting the infiltration of B cells and T follicular helper (Tfh) cells may be a feasible strategy to induce the formation of mature TLSs. Additionally, in a mouse influenza virus model,59 CD11b+ DCs-mediate the maintenance of tertiary lymphoid organs (LTOs) by continuously secreting LTβ, as DCs can retain T cells and B cells in dense clusters to maintain GCs, which is also consistent with our data. STING is a cytoplasmic DNA-sensing protein, that plays important roles in apoptosis induction and immune surveillance. STING agonists have shown strong vascular targeting and the ability to induce nonclassical TLS formation in lung cancer,42 melanoma24 and colon cancer60 models. As a dsDNA virus, Ad-IL15 is a powerful activator of the STING pathway and has natural advantages. These features are also related to the reduced DNA repair ability and high mutational load of melanoma cells, which make it easier to transform the TME into a “hot tumor” and form an environment that induces TLS formation.61 In addition, the existence of TLSs is closely related to immune checkpoint blockade (ICB).38 Remark et al.62 found that TLSs were present in the tumors of patients undergoing neoadjuvant therapy (PD-1 inhibitor), which was associated with longer disease-free survival and overall survival. Moreover, B cells in TLSs may contribute to the effective T-cell response after ICB treatment.36 In tumors without TLSs, T cells have a dysfunctional molecular phenotype.63 Mature TLSs may be a marker of the effectiveness of immune checkpoint inhibitors in solid tumors, and TLS formation is related to patient responses to ICB and high survival rates.64 Moreover, this correlation was not related to the PD-L1 expression status and CD8+ T-cell density.65
Taken together, our preclinical study results show that in addition to its effect in activating immune responses, an oncolytic adenovirus (Ad-IL15) can promote VN and nonclassical TLS formation in tumors. Furthermore, Ad-IL15 can transform the TME from “cold” to “hot” and induce VN and TLS formation via STING pathway-mediated activation of tumor-infiltrating DCs. Our findings provide insight into the mechanism underlying OV treatment and suggest that targeting the TME is a promising strategy for further studying antitumor OVs.
Supplementary Material
Acknowledgments
We thank Jian Huang lab for technical assistance and helpful supports. We thank all the members of the Pin Wu and Feng Xu lab for discussions and comments on the manuscript.
Funding Statement
This work was supported by the Fundamental Research Funds for the Central Universities [2019QNA7025]; National Natural Science Foundation of China [81572800, 82073141, 81970004]; the Natural Science Foundation of Zhejiang Province [LR22H160006 and LY15H160041].
Disclosure statement
The authors declare no competing interests.
Author contributions
PW and FX designed the study. TH and ZXH performed murine experiments.TH, ZXH, MJL, ZWX and YYC collected the samples and experimental data. TH and QY designed the figures. TH and PW analyzed the data, interpreted the data, and wrote the manuscript. WOY, CXK, WYZ, HZ provided comments for the writing of the manuscript. FX and PW supervised the project, all authors reviewed and edited the manuscript.
Data and materials availability
All data needed to evaluate the conclusions in the paper are presented in the paper and/or the Supplementary Materials. Additional data related to this paper may requested from authors.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/2162402X.2022.2093054
References
- 1.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F.. Global cancer statistics 2020: GLOBOCAN Estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–17. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Malogolovkin A, Gasanov N, Egorov A, Weener M, Ivanov R, Karabelsky A. Combinatorial approaches for cancer treatment using oncolytic viruses: projecting the perspectives through clinical trials outcomes. Viruses. 2021;13(7):1271. doi: 10.3390/v13071271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24(5):541–550. doi: 10.1038/s41591-018-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21(3):309–322. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 5.Ravirala D, Mistretta B, Gunaratne PH, Pei G, Zhao Z, Zhang X. Co-delivery of novel bispecific and trispecific engagers by an amplicon vector augments the therapeutic effect of an HSV-based oncolytic virotherapy. J Immunother Cancer. 2021;9(7): 10.1136/jitc-2021-002454. doi: 10.1136/jitc-2021-002454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang B, Cheng P. Improving antitumor efficacy via combinatorial regimens of oncolytic virotherapy. Mol Cancer. 2020;19(1):158. doi: 10.1186/s12943-020-01275-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Achard C, Surendran A, Wedge ME, Ungerechts G, Bell J, Ilkow CS. Lighting a fire in the tumor microenvironment using oncolytic immunotherapy. EBioMedicine. 2018;31:17–24. doi: 10.1016/j.ebiom.2018.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berkey SE, Thorne SH, Bartlett DL. Oncolytic virotherapy and the tumor microenvironment. Adv Exp Med Biol. 2017;1036:157–172. doi: 10.1007/978-3-319-67577-0_11. [DOI] [PubMed] [Google Scholar]
- 9.Oberg D, Yanover E, Adam V, Sweeney K, Costas C, Lemoine NR, Halldén G. Improved potency and selectivity of an oncolytic E1ACR2 and E1B19K deleted adenoviral mutant in prostate and pancreatic cancers. Clin Cancer Res. 2010;16(2):541–553. doi: 10.1158/1078-0432.Ccr-09-1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koch MS, Lawler SE, Chiocca EA. HSV-1 oncolytic viruses from bench to bedside: an overview of current clinical trials. Cancers. 2020;12(12):3514. doi: 10.3390/cancers12123514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Torres-Domínguez LE, McFadden G. Poxvirus oncolytic virotherapy. Expert Opin Biol Ther. 2019;19(6):561–573. doi: 10.1080/14712598.2019.1600669. [DOI] [PubMed] [Google Scholar]
- 12.Belli C, Trapani D, Viale G, D’Amico P, Duso BA, Della Vigna P, Orsi F, Curigliano G. Targeting the microenvironment in solid tumors. Cancer Treat Rev. 2018;65:22–32. doi: 10.1016/j.ctrv.2018.02.004. [DOI] [PubMed] [Google Scholar]
- 13.Chauhan VP, Chen IX, Tong R, Ng MR, Martin JD, Naxerova K, Wu MW, Huang P, Boucher Y, Kohane DS, et al. Reprogramming the microenvironment with tumor-selective angiotensin blockers enhances cancer immunotherapy. Proc Natl Acad Sci U S A. 2019;116(22):10674–10680. doi: 10.1073/pnas.1819889116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zheng R, Li F, Li F, Gong A. Targeting tumor vascularization: promising strategies for vascular normalization. J Cancer Res Clin Oncol. 2021;147(9):2489–2505. doi: 10.1007/s00432-021-03701-8. [DOI] [PubMed] [Google Scholar]
- 15.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science (New York, N Y). 2005;307(5706):58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 16.Mpekris F, Voutouri C, Baish JW, Duda DG, Munn LL, Stylianopoulos T, Jain RK. Combining microenvironment normalization strategies to improve cancer immunotherapy. Proc Natl Acad Sci U S A. 2020;117(7):3728–3737. doi: 10.1073/pnas.1919764117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun T, Yang Y, Luo X, Cheng Y, Zhang M, Wang K, Ge C. Inhibition of tumor angiogenesis by interferon-γ by suppression of tumor-associated macrophage differentiation. Oncol Res. 2014;21(5):227–235. doi: 10.3727/096504014x13890370410285. [DOI] [PubMed] [Google Scholar]
- 18.N J, J T, Sl N, Gt B. Tertiary lymphoid structures and B lymphocytes in cancer prognosis and response to immunotherapies. Oncoimmunology. 2021;10(1):1900508. doi: 10.1080/2162402x.2021.1900508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Filderman JN, Appleman M, Chelvanambi M, Taylor JL, Storkus WJ. STINGing the tumor microenvironment to promote therapeutic tertiary lymphoid structure development. Front Immunol. 2021;12:690105. doi: 10.3389/fimmu.2021.690105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johansson-Percival A, Ganss R. Therapeutic induction of tertiary lymphoid structures in cancer through stromal remodeling. Front Immunol. 2021;12:674375. doi: 10.3389/fimmu.2021.674375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cherubini G, Kallin C, Mozetic A, Hammaren-Busch K, Müller H, Lemoine NR, Halldén G. The oncolytic adenovirus AdΔΔ enhances selective cancer cell killing in combination with DNA-damaging drugs in pancreatic cancer models. Gene Ther. 2011;18(12):1157–1165. doi: 10.1038/gt.2011.141. [DOI] [PubMed] [Google Scholar]
- 22.Halldén G. Journal of B.U.ON.: official journal of the Balkan Union of Oncology. Optimisation of replication-selective Oncolytic Adenoviral Mutants in Combination with Chemotherapeutics. 2009;14 Suppl 1:S61–67. [PubMed] [Google Scholar]
- 23.Waldmann TA, Miljkovic MD, Conlon KC. Interleukin-15 (dys)regulation of lymphoid homeostasis: implications for therapy of autoimmunity and cancer. J Exp Med. 2020;217(1):e20191062. doi: 10.1084/jem.20191062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chelvanambi M, Fecek RJ, Taylor JL, Storkus WJ. STING agonist-based treatment promotes vascular normalization and tertiary lymphoid structure formation in the therapeutic melanoma microenvironment. J Immunother Cancer. 2021;9(2):e001906. doi: 10.1136/jitc-2020-001906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schuijs MJ, Png S, Richard AC, Tsyben A, Hamm G, Stockis J, Garcia C, Pinaud S, Nicholls A, Ros XR, et al. ILC2-driven innate immune checkpoint mechanism antagonizes NK cell antimetastatic function in the lung. Nat Immunol. 2020;21(9):998–1009. doi: 10.1038/s41590-020-0745-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li T, Fu J, Zeng Z, Cohen D, Li J, Chen Q, Li B, Liu XS. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020;48(W1):W509–W514. doi: 10.1093/nar/gkaa407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li T, Fan J, Wang B, Traugh N, Chen Q, Liu JS, Li B, Liu XS. TIMER: a web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 2017;77(21):e108–e110. doi: 10.1158/0008-5472.CAN-17-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li B, Severson E, Pignon JC, Zhao H, Li T, Novak J, Jiang P, Shen H, Aster JC, Rodig S, et al. Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome Biol. 2016;17(1):174. doi: 10.1186/s13059-016-1028-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peter M, Kühnel F. Oncolytic Adenovirus in Cancer Immunotherapy. Cancers. 2020;12(11):3354. doi: 10.3390/cancers12113354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Q, Zhang J, Tian Y, Zhu G, Liu S, Liu F. Efficacy of a novel double-controlled oncolytic adenovirus driven by the Ki67 core promoter and armed with IL-15 against glioblastoma cells. Cell Biosci. 2020;10(1):124. doi: 10.1186/s13578-020-00485-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakao S, Arai Y, Tasaki M, Yamashita M, Murakami R, Kawase T, Amino N, Nakatake M, Kurosaki H, Mori M, et al. Intratumoral expression of IL-7 and IL-12 using an oncolytic virus increases systemic sensitivity to immune checkpoint blockade. Sci Transl Med. 2020;12(526):eaax7992. doi: 10.1126/scitranslmed.aax7992 [DOI] [PubMed] [Google Scholar]
- 32.Okła K, Farber DL, Zou W. Tissue-resident memory T cells in tumor immunity and immunotherapy. J Exp Med. 2021;218(4):e20201605. doi: 10.1084/jem.20201605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chi Sabins N, Taylor JL, Fabian KP, Appleman LJ, Maranchie JK, Stolz DB, Storkus WJ. DLK1: a novel target for immunotherapeutic remodeling of the tumor blood vasculature. Mol Ther. 2013;21(10):1958–1968. doi: 10.1038/mt.2013.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roesch A, Fukunaga-Kalabis M, Schmidt EC, Zabierowski SE, Brafford PA, Vultur A, Basu D, Gimotty P, Vogt T, Herlyn M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell. 2010;141(4):583–594. doi: 10.1016/j.cell.2010.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takakura N. Vascular reconstitution in the tumor for more effective tumor immunotherapy. Cancer Sci. 2021;112(4):1348–1356. doi: 10.1111/cas.14854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, Yizhak K, Sade-Feldman M, Blando J, Han G, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. 2020;577(7791):549–555. doi: 10.1038/s41586-019-1922-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dieu-Nosjean M-C, Antoine M, Danel C, Heudes D, Wislez M, Poulot V, Rabbe N, Laurans L, Tartour E, de Chaisemartin L, et al. Long-term survival for patients with non–small-cell lung cancer with intratumoral lymphoid structures. J Clin Oncol. 2008;26(27):4410–4417. doi: 10.1200/JCO.2007.15.0284. [DOI] [PubMed] [Google Scholar]
- 38.Sautes-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer. 2019;19(6):307–325. doi: 10.1038/s41568-019-0144-6. [DOI] [PubMed] [Google Scholar]
- 39.Girard JP, Moussion C, Förster R. HEVs, lymphatics and homeostatic immune cell trafficking in lymph nodes. Nat Rev Immunol. 2012;12(11):762–773. doi: 10.1038/nri3298. [DOI] [PubMed] [Google Scholar]
- 40.Zhu G, Falahat R, Wang K, Mailloux A, Artzi N, Mule JJ. Tumor-associated tertiary lymphoid structures: gene-expression profiling and their bioengineering. Front Immunol. 2017;8:767. doi: 10.3389/fimmu.2017.00767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang M, Chen P, Wang L, Li W, Chen B, Liu Y, Wang H, Zhao S, Ye L, He Y, et al. cGAS-STING, an important pathway in cancer immunotherapy. J Hematol Oncol. 2020;13(1):81. doi: 10.1186/s13045-020-00916-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang H, Lee WS, Kong SJ, Kim CG, Kim JH, Chang SK, Kim S, Kim G, Chon HJ, Kim C. STING activation reprograms tumor vasculatures and synergizes with VEGFR2 blockade. J Clin Invest. 2019;129(10):4350–4364. doi: 10.1172/jci125413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao B, Du F, Xu P, Shu C, Sankaran B, Bell SL, Liu M, Lei Y, Gao X, Fu X, et al. A conserved PLPLRT/SD motif of STING mediates the recruitment and activation of TBK1. Nature. 2019;569(7758):718–722. doi: 10.1038/s41586-019-1228-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ozasa K, Temizoz B, Kusakabe T, Kobari S, Momota M, Coban C, Ito S, Kobiyama K, Kuroda E, Ishii KJ. Cyclic GMP-AMP triggers asthma in an IL-33-dependent manner that is blocked by amlexanox, a TBK1 inhibitor. Front Immunol. 2019;10:2212. doi: 10.3389/fimmu.2019.02212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mayer RJ. Two steps forward in the treatment of colorectal cancer. N Engl J Med. 2004;350(23):2406–2408. doi: 10.1056/NEJMe048098. [DOI] [PubMed] [Google Scholar]
- 46.Jain RK. Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat Med. 2001;7(9):987–989. doi: 10.1038/nm0901-987. [DOI] [PubMed] [Google Scholar]
- 47.van Hooren L, Vaccaro A, Ramachandran M, Vazaios K, Libard S, van de Walle T, Georganaki M, Huang H, Pietilä I, Lau J, et al. Agonistic CD40 therapy induces tertiary lymphoid structures but impairs responses to checkpoint blockade in glioma. Nat Commun. 2021;12(1):4127. doi: 10.1038/s41467-021-24347-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alberts P, Tilgase A, Rasa A, Bandere K, Venskus D. The advent of oncolytic virotherapy in oncology: the Rigvir® story. Eur J Pharmacol. 2018;837:117–126. doi: 10.1016/j.ejphar.2018.08.042. [DOI] [PubMed] [Google Scholar]
- 49.Cunliffe TG, Bates EA, Parker AL. Hitting the target but missing the point: recent progress towards adenovirus-based precision virotherapies. Cancers. 2020;12(11):3327. doi: 10.3390/cancers12113327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heise C, Hermiston T, Johnson L, Brooks G, Sampson-Johannes A, Williams A, Hawkins L, Kirn D. An adenovirus E1A mutant that demonstrates potent and selective systemic anti-tumoral efficacy. Nat Med. 2000;6(10):1134–1139. doi: 10.1038/80474. [DOI] [PubMed] [Google Scholar]
- 51.Bajaj S, Kumar MS, Peters GJ, Mayur YC. Targeting telomerase for its advent in cancer therapeutics. Med Res Rev. 2020;40(5):1871–1919. doi: 10.1002/med.21674. [DOI] [PubMed] [Google Scholar]
- 52.Chen D, Huang LY, Zhou HY, Zhang YH. Combining IL-10 and oncolytic adenovirus demonstrates enhanced antitumor efficacy through CD8(+) T cells. Front Immunol. 2021;12:615089. doi: 10.3389/fimmu.2021.615089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang W, Cai R, Luo J, Wang Y, Cui Q, Wei X, Zhang H, Qian C. The oncolytic adenovirus targeting to TERT and RB pathway induced specific and potent anti-tumor efficacy in vitro and in vivo for hepatocellular carcinoma. Cancer Biol Ther. 2007;6(11):1726–1732. doi: 10.4161/cbt.6.11.4831. [DOI] [PubMed] [Google Scholar]
- 54.Wang Y, Hallden G, Hill R, Anand A, Liu TC, Francis J, Brooks G, Lemoine N, Kirn D. E3 gene manipulations affect oncolytic adenovirus activity in immunocompetent tumor models. Nat Biotechnol. 2003;21(11):1328–1335. doi: 10.1038/nbt887. [DOI] [PubMed] [Google Scholar]
- 55.Ringel AE, Drijvers JM, Baker GJ, Catozzi A, García-Cañaveras JC, Gassaway BM, Miller BC, Juneja VR, Nguyen TH, Joshi S, et al. Obesity shapes metabolism in the tumor microenvironment to suppress anti-tumor immunity. Cell. 2020;183(7):1848–1866.e26. doi: 10.1016/j.cell.2020.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kowalsky SJ, Liu Z, Feist M, Berkey SE, Ma C, Ravindranathan R, Dai E, Roy EJ, Guo ZS, Bartlett DL. Superagonist IL-15-armed oncolytic virus elicits potent antitumor immunity and therapy that are enhanced with PD-1 blockade. Mol Ther. 2018;26(10):2476–2486. doi: 10.1016/j.ymthe.2018.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang N, Yin R, Zhou P, Liu X, Fan P, Qian L, Dong L, Zhang C, Zheng X, Deng S, et al. DLL1 orchestrates CD8(+) T cells to induce long-term vascular normalization and tumor regression. Proc Natl Acad Sci U S A. 2021;118. doi: 10.1073/pnas.2020057118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stowman AM, Hickman AW, Mauldin IS, Mahmutovic A, Gru AA, Slingluff CL Jr. Lymphoid aggregates in desmoplastic melanoma have features of tertiary lymphoid structures. Melanoma Res. 2018;28(3):237–245. doi: 10.1097/cmr.0000000000000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.GeurtsvanKessel CH, Willart MA, Bergen IM, van Rijt LS, Muskens F, Elewaut D, Osterhaus AD, Hendriks R, Rimmelzwaan GF, Lambrecht BN. Dendritic cells are crucial for maintenance of tertiary lymphoid structures in the lung of influenza virus–infected mice. J Exp Med. 2009;206(11):2339–2349. doi: 10.1084/jem.20090410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee SJ, Yang H, Kim WR, Lee YS, Lee WS, Kong SJ, Lee HJ, Kim JH, Cheon J, Kang B, et al. STING activation normalizes the intraperitoneal vascular-immune microenvironment and suppresses peritoneal carcinomatosis of colon cancer. J Immunother Cancer. 2021;9(6): 10.1136/jitc-2020-002195. doi: 10.1136/jitc-2020-002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Salem D, Chelvanambi M, Storkus WJ, Fecek RJ. Cutaneous melanoma: mutational status and potential links to tertiary lymphoid structure formation. Front Immunol. 2021;12:629519. doi: 10.3389/fimmu.2021.629519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Remark R, Lupo A, Alifano M, Biton J, Ouakrim H, Stefani A, Cremer I, Goc J, Régnard J-F, Dieu-Nosjean M-C, et al. Immune contexture and histological response after neoadjuvant chemotherapy predict clinical outcome of lung cancer patients. Oncoimmunology. 2016;5(12):e1255394–e1255394. doi: 10.1080/2162402X.2016.1255394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, Johansson I, Phung B, Harbst K, Vallon-Christersson J, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 2020;577(7791):561–565. doi: 10.1038/s41586-019-1914-8. [DOI] [PubMed] [Google Scholar]
- 64.Petitprez F, de Reyniès A, Keung EZ, Chen T-W-W, Sun C-M, Calderaro J, Jeng Y-M, Hsiao L-P, Lacroix L, Bougoüin A, et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature. 2020;577(7791):556–560. doi: 10.1038/s41586-019-1906-8. [DOI] [PubMed] [Google Scholar]
- 65.Vanhersecke L, Brunet M, Guegan JP, Rey C, Bougouin A, Cousin S, Moulec SL, Besse B, Loriot Y, Larroquette M, et al. Mature tertiary lymphoid structures predict immune checkpoint inhibitor efficacy in solid tumors independently of PD-L1 expression. Nat Cancer. 2021;2(8):794–802. doi: 10.1038/s43018-021-00232-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.