

Abstract
Addiction is believed to result from drug-elicited alterations of synaptic plasticity mechanisms in the dopaminergic reward circuitry. Impaired metabotropic glutamate receptor (mGluR)-dependent long-term depression (LTD) and accumulation of synaptic Ca2+-permeable AMPA receptors (CP-AMPARs) following drug exposure have emerged as important mechanisms underlying drug craving and relapse. Whether this malplasticity occurs in the prefrontal cortex (PFC), a major dopaminergic target in the reward circuitry that regulates executive and affective functions, remains largely unexplored. Here we show that repeated cocaine exposure in vivo causes transient but complete loss of mGluR1-LTD and accumulation of CP-AMPARs in mouse layer 5 PFC pyramidal neurons, which in turn mediates an abnormal form of cocaine-enabled Hebbian synaptic plasticity. Following repetitive cocaine injections, mGluR1- and mTOR (mammalian target of rapamycin)-dependent LTD was abolished and remained impaired for approximately one week. This mGluR1-LTD impairment was prevented by in vivo administration of an mGluR1 positive allosteric modulator (PAM) and rescued by inhibition of dopamine D1 receptors, suggesting that impaired mGluR1 tone and excessive D1 signaling underlie this LTD deficit. Concurrently, CP-AMPARs were generated, indicated by increased sensitivity to the CP-AMPAR inhibitor Naspm and rectification of synaptic AMPAR currents, which were reversed by PAM in cocaine-exposed mice. Finally, these CP-AMPARs mediate an abnormal spike-timing-dependent long-term potentiation enabled by cocaine exposure. Our findings reveal a mechanism by which cocaine impairs LTD and remodels synaptic AMPARs to influence Hebbian plasticity in the PFC. Failure to undergo LTD may prevent the reversal of drug-potentiated brain circuits to their baseline states, perpetuating addictive behaviors.
Keywords: Addiction, prefrontal cortex, synaptic plasticity, long-term depression, long-term potentiation, metabotropic glutamate receptors
Introduction
Drugs of abuse impair dopaminergic reward circuitry by usurping synaptic plasticity mechanisms that underlie normal learning, causing pathological reward and addictive behavior (Dong and Nestler, 2014; Luscher and Malenka, 2011; Wolf, 2016). The prefrontal cortex (PFC) is a major dopamine (DA) terminal field and a key site of the reward circuitry that exerts top-down executive control of motivational and reward processes, and is strongly implicated in drug-craving and relapse (Kalivas, 2009; Koob and Volkow, 2016). Although extensive studies have established that cocaine exposure modifies the efficacy of excitatory synapses in the ventral tegmental area (VTA) and nucleus accumbens (NAc), two other major structures of the reward circuitry (Dong et al., 2017; Francis et al., 2019; Luscher and Malenka, 2011), relatively limited studies have examined how cocaine modifies the synaptic strength and plasticity in the PFC. Prolonged cocaine self-administration has been reported to induce delayed potentiation of glutamatergic synapses onto layer 5 (L5) output neurons in the rat medial PFC (mPFC) that correlates with an addicted state (Kasanetz et al., 2013). In contrast, short-term noncontingent cocaine administration does not produce changes in the strength of these synapses (Huang et al., 2007a; Lu et al., 2010; Lu et al., 2009; Ruan and Yao, 2017). However, short-term drug exposure readily modifies a Hebbian synaptic learning rule by reducing the induction threshold for a spike-timing dependent long-term potentiation (t-LTP) of these synapses in the mouse mPFC (Ruan and Yao, 2017). In addition, cocaine-induced conditioned place preference produces differential changes in synapses onto layer 5/6 neurons in rat prelimbic and infralimbic regions of the PFC (Pena-Bravo et al., 2017). Finally, repeated cocaine exposure also impairs long-term depression (LTD) mediated by group II metabotropic glutamate receptors (mGluR2/3) in deep layer PFC neurons (Huang et al., 2007b; Kasanetz et al., 2013).
A major form of cocaine-induced synaptic plasticity is the emergence of GluA2-lacking AMPA receptors (Bellone and Mameli, 2012; Luscher and Malenka, 2011; Wolf and Tseng, 2012) that display Ca2+ permeability (thus termed CP-AMPARs), larger conductance, inward rectification and sensitivity to the intracellular spermine (Kwak and Weiss, 2006). While largely unexplored in the PFC, this plasticity has been extensively studied in the VTA and NAc. Specifically, a single cocaine injection induces the insertion of CP-AMPARs to synapses onto DA neurons in the mouse VTA via PICK1 (protein interacting with C kinase-1)-dependent endo- and exocytosis (Bellone and Luscher, 2006), which returns to the baseline after one week; this was suggested to be driven by a defensive mGuR1-LTD mechanism that involves mammalian target of rapamycin (mTOR)-dependent local GluA2 synthesis and exchange of GluA2-lacking for GluA2-containing AMPARs (Luscher and Malenka, 2011; Mameli et al., 2007). In NAc medium spiny neurons (MSNs), CP-AMPARs are generated at synapses onto MSNs during prolonged withdrawal (incubation) from extended access cocaine self-administration in a rat model of relapse, and inhibiting these receptors block the incubation of cocaine craving (Conrad et al., 2008; Ma et al., 2014). The accumulation of these NAc CP-AMPARs is due to a decreased mGluR1 level, thus an impaired mGluR1-LTD that hinders a timely removal of surface CP-AMPARs (Loweth et al., 2014). CP-AMPARs are also generated in the mouse NAc in an MSN subtype- and input-specific manner following withdrawal from non-contingent- and contingent- cocaine exposure (Pascoli et al., 2014; Terrier et al., 2016). Little is known about effects of cocaine on CP-AMPARs in PFC synapses, although CP-AMPARs are implicated in cue-induced relapse to heroin-seeking in rats (Van den Oever et al., 2008).
Here, we examined how cocaine exposure modifies mGluR-LTD of glutamatergic synapses onto L5 pyramidal neurons in the mouse mPFC. We found that repetitive daily in vivo cocaine exposure impaired a previously unknown mGluR1-dependent LTD, which in turn leads to generation of CP-AMPARs and facilitation of Hebbian t-LTP in synapses onto these neurons. Our results uncover a novel form of drug-induced neuroadaptation that may play a role in certain prefrontal-related addictive behavior.
Materials and Methods
Animal use.
Male or female C57BL/6J mice (RRID:IMSR_JAX: 000664) were used. Animal experiments were approved by institutional animal care and use committees at the State University of New York, Upstate Medical University.
Electrophysiology.
Male or female mice at postnatal day 21-23 (P21-23) were randomly assigned into cocaine or saline groups. Saline (0.9% NaCl) or cocaine (20 mg/kg, 2 mg/ml in saline) was injected intraperitoneally (i.p.) to pre-weighted mice once per day for up to 7 days in their home cages. This cocaine regimen is known to induce robust locomotor sensitization (Vanderschuren and Kalivas, 2000; Wolf, 1998; Yao et al., 2004) and has been established in our previous study for juvenile mice (Ruan and Yao, 2017). After the last injection, mice were allowed 1 to 7 days of withdrawal before they were euthanized for electrophysiological recording. To activate mGluR1 in vivo, Ro67-7476 (Tocris), a positive allosteric modulator (PAM) of mGluR1, was dissolved in dimethyl sulfoxide (DMSO) at a concentration of 8 mg/ml, and injected (i.p., 4 mg/kg) to mice that had received a cocaine injection 12 hours before. DMSO (50 μl/g) was used as a vehicle control in this experiment.
Medial PFC (mPFC; containing prelimbic and anterior cingulate cortical areas) slices were prepared as described previously (Xu and Yao, 2010)(Ruan et al., 2014; Ruan and Yao, 2017). Briefly, brains were removed into ice-cold artificial cerebrospinal fluid (ACSF) containing (in mM): 126 NaCl, 2.5 KCl, 2.5 CaCl2, 1.2 MgCl2, 25 NaHCO3, 1.2 NaH2PO4, and 11 D-glucose pre-oxygenated with 95% O2 and 5% CO2, and mPFC slices (300 μm) were cut with a Leica S1200 vibratome. Slices were recovered in an incubation chamber (Warner Instruments) at 32°C for 30-40 min, followed by > 1 hour of incubation at room temperature (21-23°C). Slices were transferred to a recording chamber (Warner Instruments) mounted to a BX51WI upright microscope (Olympus). The chamber was perfused with continuously oxygenated ACSF that was maintained at 32°C with a dual-channel temperature controller (Warner Instruments).
L5 pyramidal neurons were visually identified with differential interference contrast (DIC) microscopy. Glass electrodes (5-8 MΩ) were pulled with a P-87 micropipette puller (Sutter Instrument). For current clamping, electrodes were filled with (in mM) 130 K-gluconate, 8 NaCl, 10 HEPES, 0.4 EGTA, 2 Mg-ATP, and 0.25 GTP-Tris, pH 7.25. For voltage clamping, electrodes were filled with (in mM) 142 Cs-gluconate, 8 NaCl, 10 HEPES, 0.4 EGTA, 2.5 QX-314, 2 Mg-ATP, and 0.25 GTP-Tris, pH 7.25 with CsOH. The BAPTA intracellular solution contained (in mM) 15 BAPTA, 115 K-gluconate, 8 NaCl, 10 HEPES, 2 Mg-ATP, and 0.25 GTP-Tris, pH 7.25 with KOH. Whole-cell patch-clamping was done using an Axoclamp 2B or Multiclamp 700B amplifier (Molecular Devices). A concentric metal electrode (FHC) was used to evoke EPSCs or EPSPs by stimulating layer 2/3 synapses (0.033 Hz, 200 μs).
In LTD experiments, stable EPSCs were first obtained for 10 min as the baseline, followed by 10 min application of mGluR drugs to the bath, and EPSCs were then recorded for another 60 min. LTD was measured as the percentage of the averaged EPSCs between 50 and 60 min post drug application to the baseline EPSC. For Naspm (1-naphthylacetyl spermine trihydrochloride) sensitivity measurement, stable EPSCs were recorded for 10 min as the baseline, followed by 10-15 min application of the drug delivered to the bath to measure the reduction of EPSC. For the rectification measurement, EPSCs were recorded at −60 mV and +60 mV, respectively. Rectification index was calculated as the EPSC amplitude at +60 mV divided by that at −60 mV. EPSC reversal potential was assumed to be ~0 mV for mPFC L5 neurons in our preparation. Cells were otherwise held at −60 mV during voltage-clamp experiments.
t-LTP experiments were conducted at resting membrane potentials (~ −67 mV(Ruan and Yao, 2017; Xu and Yao, 2010)) under current-clamp. Stable EPSPs were recorded for 10 min as the baseline, followed by a spike-timing-dependent plasticity (STDP) induction protocol, and EPSPs were further recorded for 60 min post induction. The STDP protocol consisted of a train of 60 pairs (0.1 Hz) of single EPSP followed by a single action potential (AP) with the time interval (Δt) set at +30 ms (pre-post). APs were evoked by injecting current to the soma through the patch electrode. Input resistance was monitored throughout the experiment with a 200 pA hyperpolarizing current. LTP was measured as the percentage of the average EPSP between 50 and 60 min to the baseline EPSP.
Signals were filtered at 1 kHz, digitized at 10 kHz, and analyzed with pClamp version 10.2 (Molecular Devices). Data are expressed as mean ± SEM. Statistical comparisons were made with two-tailed t tests or ANOVA with appropriate post hoc tests.
Drugs.
The following drugs were used: cocaine (Sigma-Aldrich), (S)-3,5-Dihydroxyphenylglycine (DHPG, Tocris Bioscience), LY367385 (Tocris Bioscience), 3-((2-Methyl-1,3-thiazol-4-yl)ethynyl)pyridine hydrochloride (MTEP, Tocris Bioscience), 1-Naphthyl acetyl spermine trihydrochloride (Naspm, Sigma-Aldrich), picrotoxin (Sigma-Aldrich), PKI6-22 (Sigma-Aldrich), QX-314 (Sigma-Aldrich), rapamycin (Sigma-Aldrich), Ro67-7476 (Tocris Bioscience), SCH23390 (Tocris Bioscience), SKF81297 (Sigma-Aldrich), tetrodotoxin (Sigma-Aldrich), and U73122 (Tocris Bioscience). Picrotoxin was dissolved directly in the ACSF used for perfusion. LY367385 and MTEP were dissolved in DMSO with a 1:1000 ratio as stock solutions, which were added directly to ASCF to a desired final concentration during experiments. All other drugs were dissolved in distilled water with a 1:1000 ratio as stock solutions. Drugs were delivered to the recording chamber through a perfusion system (Harvard Apparatus).
Results
A DHPG-induced, mGluR1-mediated LTD in the mPFC
We first examined whether group I mGluR-dependent LTD exists in the mPFC as this form of LTD is directly related to CP-AMPARs (Bellone and Mameli, 2012; Luscher and Malenka, 2011; Mameli et al., 2007; Wolf and Tseng, 2012). Using whole-cell patch-clamping, we recorded AMPAR-mediated excitatory postsynaptic currents (EPSCs) from L5 pyramidal neurons in mPFC slices, the major cortical output neurons that regulate top-down executive functions (Miller and Cohen, 2001). Presynaptic stimulation was delivered at layer 2/3 (Ruan and Yao, 2017; Xu and Yao, 2010), and GABAergic transmission was blocked by picrotoxin (100 μM) included in the bath to isolate EPSCs. To induce LTD, we applied the group I mGluR agonist DHPG (75 μM) after establishing a stable baseline EPSC. DHPG induced a robust and persistent depression of EPSCs that lasted at least 60 min (−38.64 ± 6.43%, Fig. 1A,F). Since group I mGluRs contain mGluR1 and mGluR5, we examined which subtype mediated this previously uncharacterized LTD in the mPFC using subtype-specific antagonists. Co-application of the mGluR1 antagonist LY367385 (LY, 100 μM, Fig. 1B,F, −3.00 ± 10.11%), but not the mGluR5 antagonist MTEP (10 μM, Fig. 1C,F, −27.74 ± 2.52%), abolished DHPG-induced LTD. These results indicate that this mPFC DHPG-LTD was mainly mediated by mGluR1, but not mGluR5.
Figure 1. A DHPG-induced, mGluR1-mediated LTD in the mPFC.
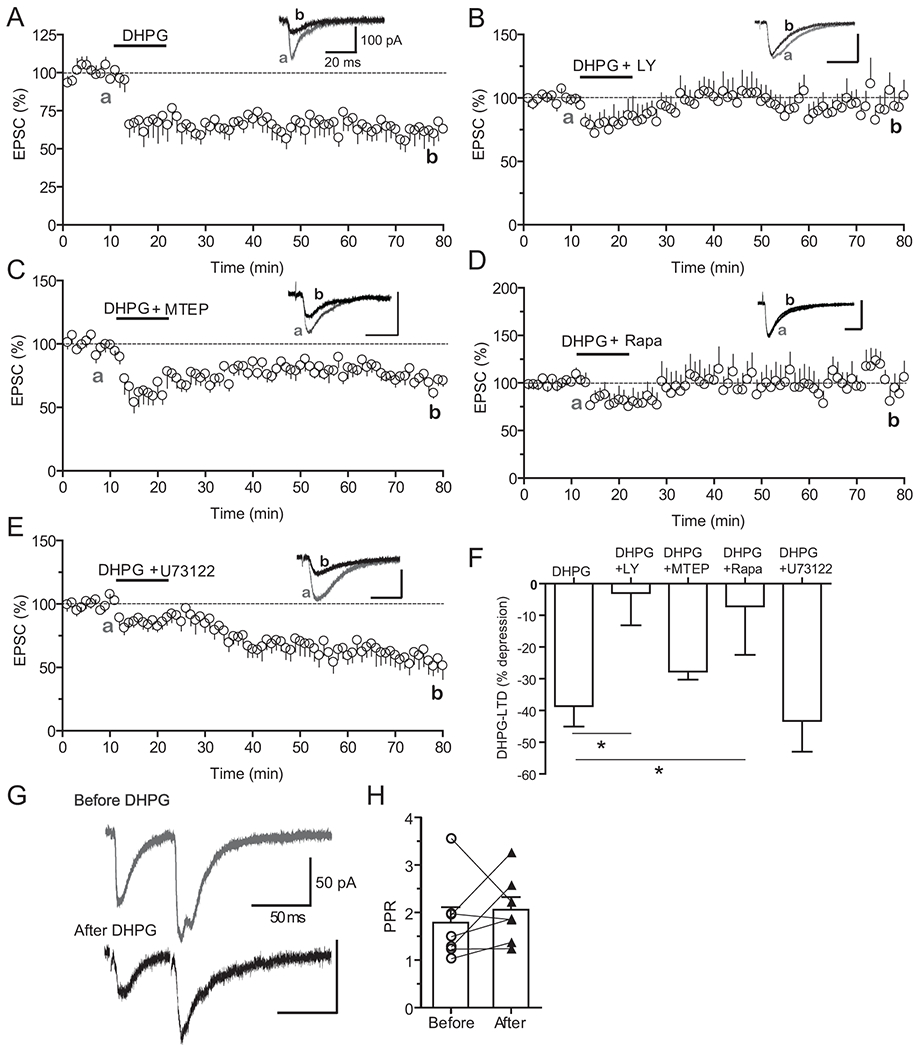
A. Persistent depression of EPSCs in L5 pyramidal neurons following bath-applied DHPG (10 min, 75 μM) in slices prepared from saline-treated mice (n = 8 cells from 7 mice). In this and subsequent figures, sample traces (insets) are average EPSCs recorded from a representative cell during the 5-min period before DHPG (a) and between 55 to 60 min post-DHPG application (b).
B. Co-application of LY367385 (LY, 100 μM) in the bath blocked DHPG-LTD. N = 7 cells from 6 mice.
C. Co-application of MTEP (20 μM) failed to block DHPG-LTD. N = 6 cells from 5 mice.
D. Co-application of rapamycin (rapa, 40 nM) blocked DHPG-LTD. N = 6 cells from 4 mice.
E. Co-application of U73122 (10 μM) failed to block DHPG-LTD. N = 7 cells from 4 mice.
F. Summary of DHPG-LTD in different conditions. DHPG + LY vs. DHPG, * p < 0.05, Student’s t-test; DHPG + Rapa vs. DHPG, * p < 0.05, Mann-Whitney test.
G. Representative EPSCs evoked by paired-pulse stimuli, separated by 50 ms inter-pulse interval, before and 40 min after DHPG bath-application.
H. Summary of PPR before and after DHPG (n = 7 cells from 3 mice).
We further investigated the downstream signaling mechanism underlying this mPFC mGluR1-LTD. Bath application of rapamycin (40 nM), a potent inhibitor of mTOR that mediates several forms of mGluR1/5-LTD in a protein synthesis dependent manner (Collingridge et al., 2010; Hou and Klann, 2004; Klann and Dever, 2004; Mameli et al., 2007), fully blocked the DHPG-LTD (Fig. 1D,F, −7.18 ± 15.29%). Interestingly, co-application of U73122 (10 μM), a specific inhibitor of phospholipase C (PLC) that mediates much of the canonical mGluR1/5 signaling and mGluR1/5-LTD in several brain regions (Collingridge et al., 2010), failed to inhibit this DHPG-LTD (Fig. 1E,F, −43.27 ± 9.68%). Thus, this signaling pathway is unlikely to be involved in this mPFC mGluR1-LTD uncovered here. Finally, we examined whether this mGluR1-LTD is expressed pre- or postsynaptically. Using a paired-pulse stimulation protocol that assesses changes in presynaptic release mechanism (Zucker and Regehr, 2002), we found that the paired-pulse ratio (PPR) was similar before and after DHPG application (Fig. 1G,H, before: 1.79 ± 0.32 vs. after 2.06 ± 0.27), indicating unaltered presynaptic transmitter release following DHPG application. Taken together, our results uncovered a previously unreported mGluR1-dependent, mTOR-mediated, and postsynaptically expressed LTD in deep-layer neurons of the mouse mPFC.
Repeated cocaine treatments transiently block mGluR1-LTD
We investigated if in vivo cocaine exposure alters mGluR1-LTD uncovered here in the mPFC. We employed a cocaine-induced locomotor sensitization protocol, a behavior model often used to investigate early neural plasticity mechanisms following drug exposure (Hyman et al., 2006; Nestler, 2001; Wolf, 1998). Mice were injected with cocaine (20 mg/kg, i.p.) or saline for up to 7 days and withdrew from the drug for various days before electrophysiology (Fig. 2A). On slices prepared from mice treated with seven daily cocaine injections followed by one day of withdrawal, bath-applied DHPG failed to induce any sustained level of LTD (Fig. 2B,F, −2.46 ± 7.92%). A partial DHPG-LTD appeared following two days of withdrawal (Fig. 2C,F, −19.46 ± 5.94%), and this partially recovered LTD was blocked by co-application of LY367385 (Fig. 2E, −8.87 ± 10.09%), consistent with the notion that it is mGluR1-dependent. DHPG-LTD recovered to a level (−29.97 ± 6.04%) that was statistically insignificant to that of saline control (−38.64 ± 6.43%) after seven days of withdrawal (Fig. 2D,F), suggesting that this cocaine-induced malplasticity lasted approximately one week (Fig. 2F). Interestingly, this DHPG-LTD appeared intact on slices from mice received one or four, but not six, daily cocaine injections (Fig. 2F), suggesting that the effect of cocaine exposure on DHPG-LTD is cumulative. Together, these results demonstrated that cocaine exposure in vivo resulted in transient and reversible impairment of mGluR1-LTD in the mPFC in a regimen-dependent manner.
Figure 2. Repeated cocaine treatments transiently block mGluR1-LTD.
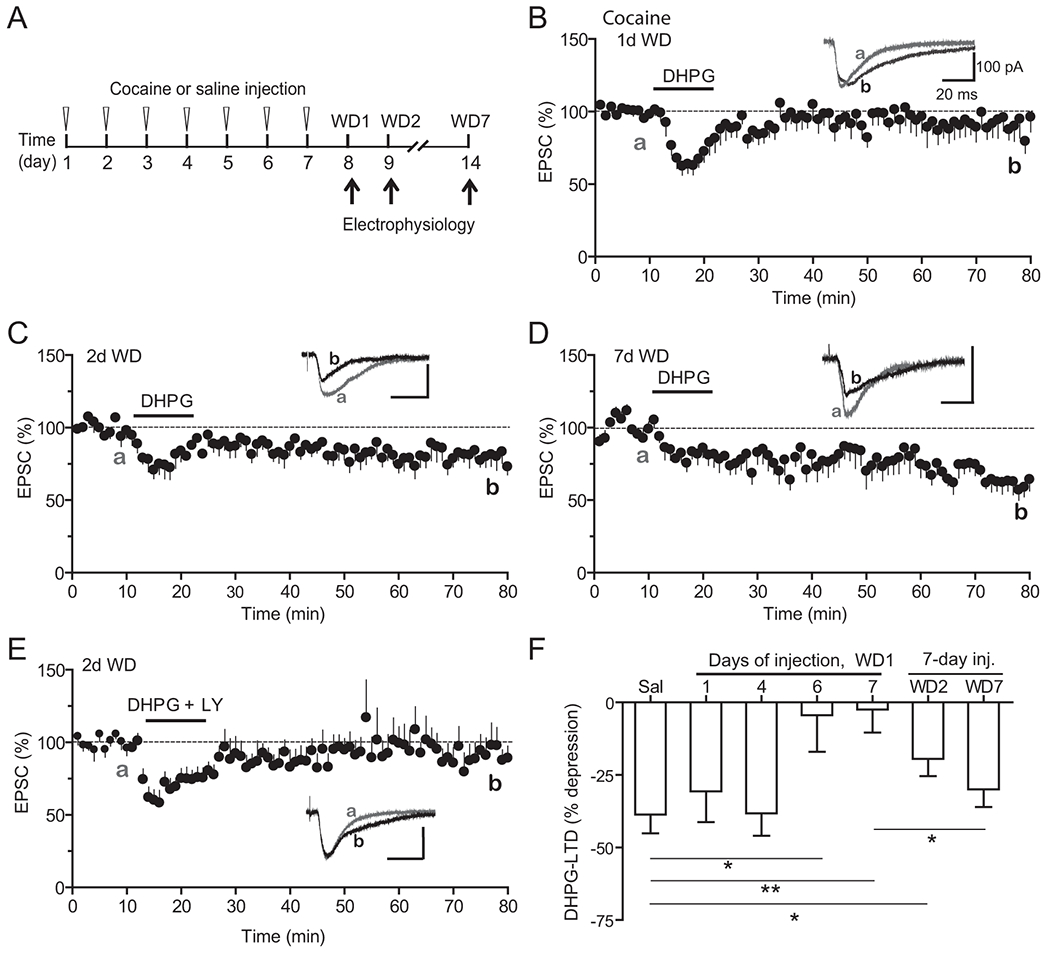
A. Schematic of repeated cocaine or saline injections and withdrawal schedule.
B. Lack of persistent DHPG-induced depression in slices prepared from mice treated with 7 daily cocaine injections followed by 1 day of withdrawal. N = 11 cells from 11 mice.
C. Significantly impaired DHPG-LTD in slices from mice treated with 7 daily cocaine injections followed by 2 days of withdrawal. N = 6 cells from 5 mice.
D. Recovery of DHPG-LTD in slices from mice treated with 7 daily cocaine injections followed by 7 days of withdrawal. N = 7 cells from 7 mice.
E. LY367385 blocks the partially recovered DHPG-LTD in mice withdrew for 2 days after 7-day cocaine injections. N = 7 cells from 5 mice.
F. Summary of DHPG-LTD in saline and cocaine treated mice in different injection and withdrawal conditions. Data for saline are replotted from Figure 1 for direct comparisons. * < 0.05; ** p < 0.01, unpaired Student’s t-tests.
Systemic administration of mGluR1 PAM restores mGluR1-LTD
Previous studies have shown that in vivo activation of mGluR1 by systemic administration of positive allosteric modulators (PAMs) can reverse cocaine-induced synaptic malplasticity (Bellone and Luscher, 2006; Loweth et al., 2014). We therefore examined whether the cocaine-induced loss of mPFC mGluR1-LTD could be prevented by a mGluR1 PAM, Ro67-7476. We administered Ro67-7476 (i.p., 4 mg/kg in DMSO) or DMSO (as vehicle control) 12 hours after the cocaine injection each day on days 2 through 7 (6 injections total) during the cocaine treatment regimen (Fig. 3A). This repeated PAM administration regimen was designed to chronically activate mGluR1 to reduce CP-AMPAR accumulation. PAM treatments fully restored mGluR1-LTD (Fig. 3B,D, −36.07 ± 6.52%) whereas injections of DMSO did not (−0.64 ± 19.63%) (Fig. 3 C,D). These results suggest that activation of mGluR1 function by Ro67-7476 during exposure to cocaine can mitigate the negative effects of cocaine on mGluR1-LTD.
Figure 3. Systemic administration of mGluR1 PAM restores DHPG-LTD.
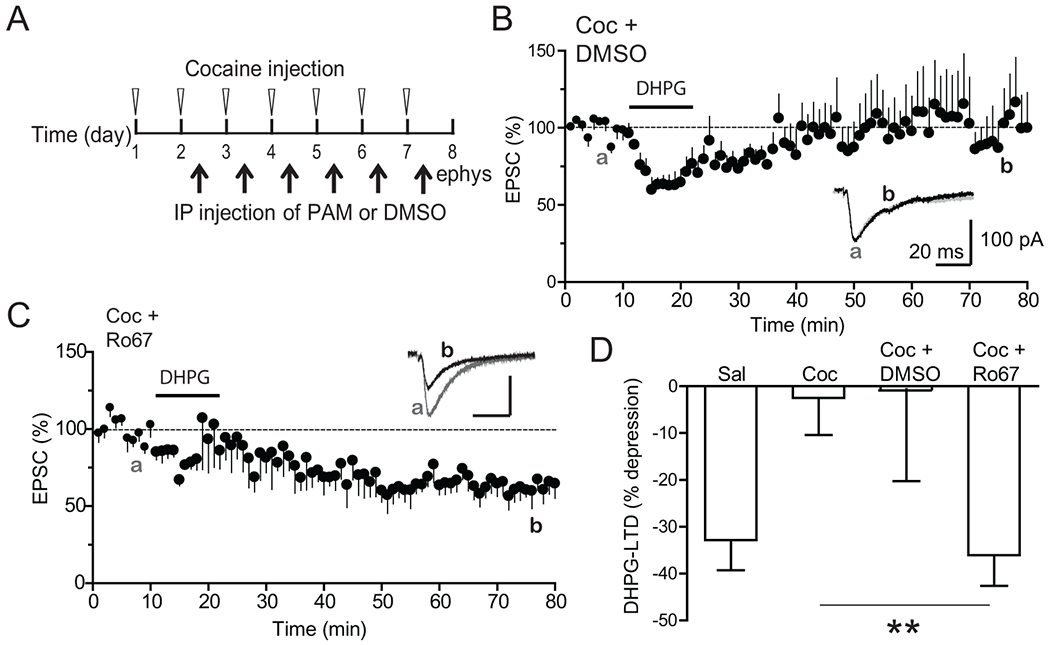
A. PAM injection schedule. Six daily in vivo administration of DMSO or PAM (4 mg/kg, IP) was given 12 hours after cocaine injection on indicated days during cocaine injection regimen.
B. Lack of effect of DMSO injections on DHPG-LTD in cocaine-treated mice. N = 8 cells from 4 mice.
C. Rescue of DHPG-LTD in PAM-injected, cocaine-treated mice. N = 6 cells from 3 mice.
D. Summary of DHPG-LTD in difference conditions. Data for saline are replotted from Figure 1 for direct comparisons. ** p < 0.01, unpaired Student’s t-test.
Excessive D1 DA signaling underlies cocaine-induced impairment of mGluR1-LTD
We next investigated the underlying mechanisms of impaired mGluR1-LTD in cocaine-exposed mice. We and others have shown that repeated cocaine exposure results in sensitization of D1 signaling in mPFC pyramidal neurons, which promotes mPFC LTP (Ruan and Yao, 2017). DA D1 signaling also was reported to mask an mGluR-dependent LTD in striatal MSNs (Shen et al., 2008). We thus tested the possibility that the overactive D1 signaling led to the loss of mPFC mGluR1-LTD in cocaine-treated mice. Bath application of the D1 antagonist SCH23390 (SCH, 10 μM) fully restored the DHPG-induced mGluR1-LTD in slices from repeated cocaine-treated mice (Fig. 4A, −36.76 ± 7.75%). Importantly, a single administration of SCH23390 in vivo (1mg/kg in 0.01% saline, subcutaneously or s.c.) 30 min before scarifying the mice also restored mGluR1-LTD on slices (Fig. 4B, −46.76 ± 12.11%). Furthermore, including the membrane-impermeable PKA inhibitor, PKI (20 μM), in the recording patch electrode rescued the DHPG-induced mGluR1-LTD from cocaine mice (Fig. 4C, −65.30 ± 8.83%). These results indicate that the impaired mGluR1-LTD in cocaine mice is mediated by an excessive DA D1/PKA signaling in pyramidal neurons. However, acute activation of D1 signaling by SKF81297 (2 μM) on slices prepared from saline-treated control mice did not block DHPG-induced mGluR1-LTD (Fig. 4D, −42.18 ± 7.48%). This data suggests that additional mechanisms downstream of D1/PKA signaling might be recruited by chronic cocaine exposure and contribute to the cocaine-induced mGluR1-LTD impairment.
Figure 4. Excessive D1-PKA signaling underlies cocaine-induced impairment of mGluR1-LTD.
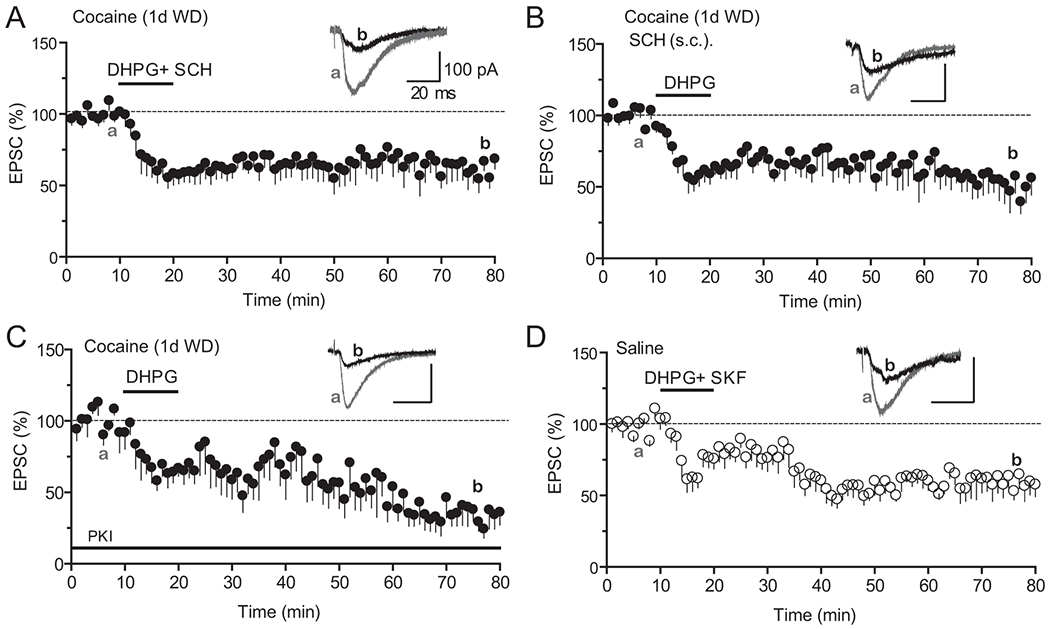
A. Co-application of SCH23390 (SCH, 10 μM) in the bath restored DHPG-LTD in slices prepared from cocaine mice. N = 8 cells from 8 mice.
B. Administration of SCH23390 in vivo restored DHPG-LTD in cocaine-treated mice. SCH23390 (1mg/kg, IP) was injected 30 min before mice were sacrificed for recording. N = 7 cells from 5 mice.
C. Inclusion of PKI 6-22 (PKI, 20 μM) in the patch pipette fully restored DHPG-LTD in cocaine mice. N = 6 cells from 3 mice.
D. Co-application of SKF81297 (2 μM) in the bath failed to block DHPG-LTD in saline mice. N = 7 cells from 4 mice. All recordings were performed on slices derived from mice treated with 7-day cocaine or saline injections followed by 1 day of withdrawal.
Accumulation of CP-AMPARs by repeated cocaine exposure
Synaptic insertion of CP-AMPARs has emerged as a hallmark mechanism in cocaine-induced synaptic plasticity in the VTA and NAc, but has not been explored in the mPFC. We thus examined whether repeated cocaine-treatments alter CP-AMPARs at mPFC synapses. CP-AMPARs are characterized by the inward rectification and sensitivity to blockade by the polyamine, such as Naspm. We found that the rectification index of AMPAR-EPSCs was significantly reduced in slices prepared from mice withdrew 1 day after repeated cocaine treatment (0.60 ± 0.05), compared to saline-treated control (0.85 ± 0.08, Fig. 5A). In addition, AMPAR-EPSCs showed a significantly increased sensitivity to Naspm (200 μM) blockade in cocaine-treated mice (0.42 ± 0.04 vs. 0.18 ± 0.06, Fig. 5B). These data suggest that CP-AMPARs are inserted into mPFC synapses immediately following chronical cocaine treatment.
Figure 5. Cocaine exposure accumulates CP-AMPARs at mPFC synapses in an mGluR1-dependent manner and these CP-AMPARs mediate cocaine-enabled t-LTP.
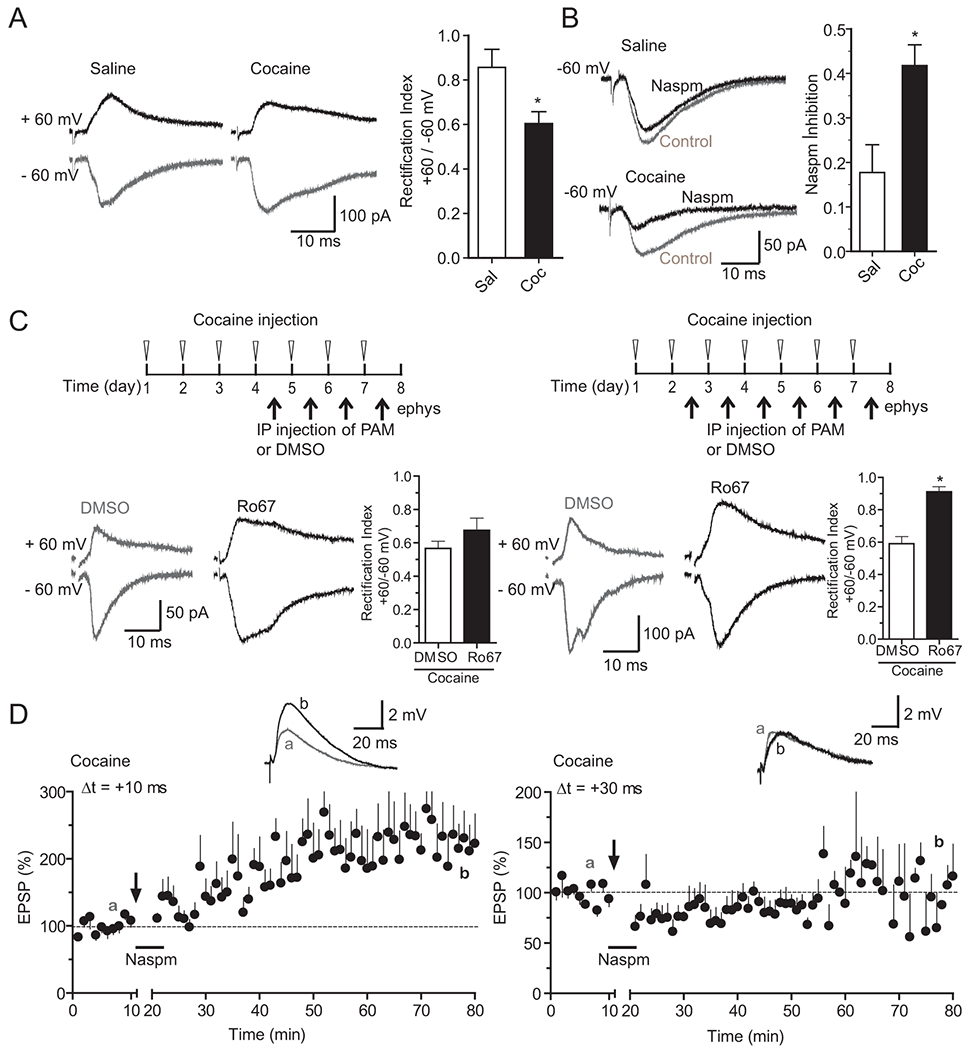
A, B. Repeated cocaine exposure generated CP-AMPARs at mPFC synapses. A, Reduced rectification of EPSCs in repeated cocaine-treated animals. Left, representative EPSCs at +60 mV and −60 mV from saline and cocaine treated mice. Right, summary of rectification index defined as the ratio of EPSC+60/EPSC−60. N = 9 cells from 3 mice. B, Increased Naspm sensitivity of EPSCs in cocaine-treated mice. Left, representative EPSCs before and after bath-application of Naspm (200 μM) from saline and cocaine treated mice. Right, summary of Naspm inhibition of EPSCs defined as the ratio of EPSCafter/EPSCbefore. N = 8 cells from 5 saline mice and n= 7 cells from 5 cocaine mice. * p < 0.05, unpaired Student’s t-tests.
C. Cocaine-generated CP-AMPARs were removed by in vivo Ro67-7476 administration. Significant increase of rectification index following 6, but not 4 daily Ro67-7476 injections in cocaine-exposed mice. Treatment regimen are shown on the top. Representative EPSCs from DMSO or Ro67-7476-treated cocaine mice for each regimen are shown to the left. Summaries of rectification index are shown to the right. N = 6-8 cells from 2 DMSO- or Ro67-7476-injected cocaine mice in each group. *p < 0.05, unpaired Student’s t-tests.
D. CP-AMPARs are required for the extended timing t-LTP in repeated cocaine-treated mice. Left, Application of Naspm (200 μM) during EPSP-AP pairings did not block t-LTP at +10 ms, a normal timing interval. N = 5, from 4 mice. Right, Naspm application blocked t-LTP at +30 ms, an extended timing interval. N = 6, from 5 mice. Arrows indicate the start of t-LTP induction. Naspm was applied to the extracellular bath during EPSP–AP pairings.
Ro67-7476 administration in vivo removes cocaine-induced CP-AMPARs
Previous studies have shown that systemic administration of Ro67-7476 removes abnormally generated CP-AMPARs in other brain regions following cocaine exposure and/or withdrawal (Bellone and Luscher, 2006; Loweth et al., 2014). Thus, we next examined whether the cocaine-induced insertion of CP-AMPARs in mPFC synapses could be reversed by Ro67-7476. Similar to PAM rescue of mGluR1-LTD, six daily injections of Ro67-7476 during the cocaine injection regimen significantly increased AMPA-EPSC rectification index (0.91 ± 0.03 vs. 0.59 ± 0.04; Fig. 5C), and in fact restored the index to the saline control level (0.85 ± 0.08). In contrast, four daily Ro67-7476 injections did not result in a significant change in AMPA-EPSC rectification index compared to the vehicle control (0.68 ± 0.07 vs. 0.57 ± 0.04, Fig. 5C). Thus, chronic PAM administration can antagonize cocaine-induced impairments in both mGluR1-LTD and CP-AMPARs, likely in a regimen-dependent manner. These results support the ideas that mGluR1-LTD mediates removal of CP-AMPARs from mPFC synapses, that cocaine exposure impairs mPFC mGluR1 function and mGluR1-LTD, and that in vivo activation of mGluR1 by Ro67-7476 treatments rescue both processes.
CP-AMPARs contribute to an extended timing t-LTP in cocaine mice
CP-AMPARs can conduct Ca2+ and have been shown to contribute to LTP in the hippocampus (Asrar et al., 2009; Park et al., 2016) and mediate an abnormal cocaine-induced anti-Hebbian LTP in the VTA (Mameli et al., 2011). We have previously reported that repeated cocaine exposure facilitates mPFC t-LTP induction by extending the timing window under a STDP induction protocol (Ruan and Yao, 2017). Specifically, t-LTP is induced by 60 EPSP-AP pairs at Δt = +30 ms in slices prepared from repeated cocaine-, but not saline-treated mice. In contrast, t-LTP at Δt = +10 ms is induced in both saline and cocaine mice (Ruan and Yao, 2017). We thus asked whether CP-AMPARs contributed to the cocaine-specific +30 ms t-LTP. We found that in the presence of Naspm applied during EPSP-AP pairing, t-LTP could be induced by EPSP-AP pairing at +10 ms (227.2 ± 47.7%), but not + 30 ms (85.3 ± 10.2%, Fig. 5D) in slices prepared from cocaine-treated mice. These data indicate that CP-AMPARs inserted by cocaine are not required for the normal timing +10-ms t-LTP, but are necessary for the extended timing +30-ms t-LTP enabled specifically by cocaine exposure in the mPFC.
Discussion
In this study, we report a previously uncharacterized postsynaptic LTD in deep-layer mPFC neurons that depends on activation of mGluR1 and downstream mTOR signaling. Repeated cocaine exposure transiently abolishes this mGluR1-LTD in mGluR1- and DA D1R signaling-dependent manner. Concomitantly, the cocaine-induced mGluR1-LTD impairment is associated with emergence of synaptic CP-AMPARs, a deficit that is rescued by in vivo mGluR1 PAM administration. Finally, these abnormally inserted CP-AMPARs contribute to a cocaine-specific Hebbian synaptic plasticity. Together, these results uncover a mechanism by which repeated cocaine exposure impairs synaptic depression to alter synaptic AMPAR composition and plasticity which may play an important role in cocaine addictive behavior.
Several forms of mGluR-dependent LTD have been documented in rodent PFC. Activation of group II mGluR2/3 receptors by their agonists, e.g. DCG IV and LY379268, has been shown to induce robust LTD in both mouse and rat PFC (Huang et al., 2007b; Joffe et al., 2019; Kasanetz et al., 2013; Walker et al., 2015). In addition, mGluR5 also contributes to a mGluR3-dependent LTD (Joffe et al., 2019). Although mGluR1-LTD induced by DHPG has been well studied in several mouse brain regions, including the hippocampus, primary cortices, VTA, and the bed nucleus of the stria terminalis (Bhakar et al., 2012; Collingridge et al., 2010; Luscher and Huber, 2010), it has not been reported for rodent mPFC L5 neurons. It was previously shown that DHPG when applied alone failed to induce persistent LTD but co-activation of either DA D1 (Otani et al., 1999) or the 5-HT2A/2C receptors (Zhong et al., 2008) with DHPG induced LTD. The reasons for the discrepancy between these and our results are not immediately clear but likely were due to differences in species (rat vs. mouse), sex, and/or experimental conditions (e.g. methodology details and associated monoamine activation states). The robust and persistent postsynaptic mGluR1-LTD induced by DHPG in our preparation did not involve the canonic PLC signaling downstream of mGluR1, but depends on mTOR, suggesting involvement of protein synthesis. Such mGluR1- and mTOR-dependent LTD in other brain regions (e.g. VTA) has been shown to result from the rapid translation and synaptic insertion of CP-AMPARs (Mameli et al., 2007). Similar mechanism may mediate the mGluR1-LTD observed reported (see below). Taken together, our study uncovered a previously uncharacterized LTD in deep-layer neurons in the mouse mPFC.
Our results indicate that the impairment of mPFC mGluR1-LTD by in vivo cocaine exposure lasts for approximately one week and requires multiple cocaine injections. DHPG-induced postsynaptic mGluR5-dependent LTD is also disrupted transiently by repeated cocaine administration in the mouse bed nucleus of the stria terminalis (Grueter et al., 2008). A similar cocaine exposure regimen has been reported to induce a delayed impairment of mGluR5-LTD in the NAc (Huang et al., 2011). In comparison, mGluR2/3-LTD in the rat PFC is impaired following both non-contingent (Huang et al., 2007b) and contingent cocaine administration (Kasanetz et al., 2013). These studies indicate that cocaine exposure inhibits multiple forms of mGluR-LTD, and the underlying mechanisms and the nature of impairments may differ. Whether and how cocaine exposure alters NMDAR-dependent Hebbian LTD remains to be examined, but a previous study has shown that PFC t-LTD remains intact following prenatal cocaine exposure (Lu et al., 2009).
Several potential mechanisms may underlie the impairment of mPFC mGluR1-LTD following chronic cocaine exposure. First, membrane-bond mGluR1 could be decreased following cocaine exposure. A reduced mGluR1 tone (30 - 40%) has been shown for NAc mGluR1 following prolonged withdrawal from cocaine self-administration and can be rescued by repeated administrations of PAM (Loweth et al., 2014). Consistent with this idea, our data also shows that repeated PAM injections rescues mGluR1-LTD in the mPFC. However, a partial decrease in mGluR1 tone may not fully account for the complete loss of mGluR-LTD consistently observed by us and others (Grueter et al., 2008; Huang et al., 2007b; Huang et al., 2011; Kasanetz et al., 2013). Our observation that the acute phase of DHPG-LTD appears intact suggests largely preserved surface mGluR1 activity. Thus, certain signaling components downstream of mGluR1 that control GluA2 synthesis and trafficking, i.e. the maintenance phase of mGluR-LTD, may be altered by chronic cocaine exposure. For example, cocaine is known to dysregulate the levels of mTOR (Sutton and Caron, 2015), homer (Loweth et al., 2014) and PICK1 (Bellone and Luscher, 2006) in the NAc, all of which can regulate mGluR1/5-LTD (Collingridge et al., 2010). Whether cocaine exposure may induce adaptations in these proteins and contribute to impaired mPFC mGluR1-LTD remains to be investigated.
Regardless of the potential mGluR-related mechanisms mentioned above, our results suggest an involvement of dopaminergic signaling. Specifically, our observation that inhibiting D1/PKA signaling both in vitro and in vivo completely rescues mGluR1-LTD suggests that excessive D1-cAMP-PKA signaling resulted from chronic cocaine exposure (Dong et al., 2005; Ford et al., 2009; Ruan and Yao, 2017) may exert an inhibitory influence on the normal mGluR1-LTD. It is worth noting that this repeated cocaine exposure-induced D1-PKA signaling elevation can occur independent of endogenous dopamine levels in slices. A previous study in the NAc has shown that mGluR-LTD is absent unless the D1 signaling is inhibited, suggesting a masking effect of LTD by D1 receptors in these neurons (Shen et al., 2008). In certain cells and under certain conditions, activation of D1 signaling can promote or induce some forms of synaptic potentiation to compete against on-going LTD (Zhang et al., 2009). Finally, it has been shown that suppressing D1 signaling in PFC neurons rescues the lost mGluR2-LTD in cocaine-sensitized rats through an adenosine A3 receptor/PKC-dependent mechanism (Huang et al., 2007b). Thus, the excessively activated D1/PKA signaling in mPFC neurons in our preparation may suppress mGluR-LTD through masking, by promoting a competing LTP or by interfering with some signaling components that drive mGluR1-LTD. Delineating the precise mechanisms underlying the loss of mPFC mGluR1-LTD following cocaine exposure may prove challenging, but the D1-dependent rescue of this plasticity we observed is robust and novel and provides insights in how some cocaine-induced malplasticity can be reversed by curtailing DA D1 signaling.
The appearance of CP-AMPARs following drug exposure, particular cocaine, has emerged as common synaptic plasticity in reward circuits (Luscher, 2016; Wolf, 2016), although the underlying mechanistic details may differ. In VTA DA neurons, single or repeated cocaine injections rapidly induce the insertion of synaptic CP-AMPARs, which depends on PICK1-mediated GluA2 trafficking and is reversed by in vivo activation of mGluR1 signaling that drives LTD (Bellone and Luscher, 2006; Mameli et al., 2009). CP-AMPARs are also generated following prolonged withdrawal from cocaine self-administration in rat NAc MSNs (Conrad et al., 2008; Loweth et al., 2014), resulting from reduced surface mGluR1 level that precedes and enables the gradual accumulation of CP-AMPARs at late incubation phase presumably due to attenuated mGluR1-LTD (Loweth et al., 2014). The emergence of CP-AMPARs in mPFC pyramidal neurons shown in our study is rapid (following one day of withdrawal), consistent with what is seen in VTA neurons (Mameli et al., 2009), and is rescued by PAM, consistent with both VTA and NAc neurons (Bellone and Luscher, 2006; Loweth et al., 2014; Mameli et al., 2009), which supports the involvement of an mGluR1-LTD mechanism. Interestingly, CP-AMPARs are also implicated in heroin addiction in rats, as re-exposure to heroin-associated cues results in internalization of GluA2 associated with reduced AMPA/NMDA ratio and increased rectification (Van den Oever et al., 2008). Whether mGluR-LTD is involved in this plasticity is unknown. Regardless of mechanistic differences, CP-AMPARs are generated by multiple drugs of abuse in different brain regions and targeting these receptors or underlying mechanisms has been employed to reverse several drug-elicited addictive behaviors (Luscher, 2016; Wolf, 2016).
Conventionally, Hebbian LTP is triggered by Ca2+ influx through the NMDARs (Bi and Poo, 2001; Malenka and Bear, 2004). In the normal brain, most AMPARs do not conduct Ca2+ as levels of endogenous CP-AMPARs are low, and are rarely directly involved in LTP induction (Liu and Zukin, 2007). However, CP-AMPARs can contribute to LTP induction in certain cell types. For example, CP-AMPARs are required for tetanic stimulation-induced LTP in amygdala interneurons (Mahanty and Sah, 1998) and for a PKA-dependent component of LTP induced by spaced theta burst stimulation in hippocampal CA1 synapses (Park et al., 2016). CP-AMPARs become elevated under certain pathophysiological conditions, and can potently influence LTP inductions via Ca2+ entry through these receptors (Kullmann and Lamsa, 2008). A prominent example is provided by Luscher and colleagues (Mameli et al., 2011) who showed that following cocaine exposure, CP-AMPARs are generated in the VTA which in turn mediate a hyperpolarization-induced “anti-Hebbian” LTP not present in normal conditions, thus inverting rules for synaptic plasticity. Here, we provide evidence that cocaine-generated CP-AMPARs in PFC neurons are required for a cocaine-induced extended-timing t-LTP (+30 ms), but not normal-timing t-LTP (+10 ms), thus facilitating a classical Hebbian plasticity. Taken together, through the generation of Ca2+-conducting CP-AMPARs and other Ca2+ sources, e.g. LTCC (Ruan and Yao, 2017), cocaine can effectively alter Hebbian synaptic learning rules that underlie certain addictive behaviors.
Cocaine-induced loss of mGluR1-LTD in our study may have several consequences on synaptic and neural circuit function and plasticity. First, it could impair normal reward and motivational process that depends on intact mGluR1-LTD. Secondly, although transient, it can lead to rapid accumulation of the larger conductance CP-AMPARs, which can directly drive addictive behavior, such as craving (Conrad et al., 2008; Lee et al., 2013; Loweth et al., 2014). Thirdly, it could indirectly facilitate and contribute through CP-AMPARs to lasting potentiation of synapses and circuits (this study). Finally, it could hinder reversal or depotentiation of synaptic potentiation following drug exposure, making the potentiation more persistent (Mameli et al., 2009). Drug-induced synaptic potentiation is a major neuroplasticity in the reward circuitry that perpetuates addictive behavior (Luscher, 2016; Wolf, 2016). Through these potential mechanisms, drug-induced mGluR1-LTD may drive synapse and circuit malplasticity in addiction.
Highlights.
A mGluR1- and mTOR-dependent LTD is present in the mouse medial prefrontal cortex.
Repeated cocaine exposure in vivo temporally but completely abolishes prefrontal mGluR1-LTD.
Impaired mGluR1 function and excessive D1 DA signaling likely underlie cocaine impairment of mGluR1-LTD.
Ca2+-permeable AMPA receptors are generated by cocaine exposure, likely resulting from mGluR1-LTD impairment, and contribute to a cocaine-induced extended spike timing LTP.
Acknowledgements:
We thank members of the laboratory for comments and discussions.
Funding and Disclosure
This work was supported by the National Institutes of Health Grant DA032283, MH106489, and NS093097 to W.-D.Y. The authors declare no competing financial interests.
References
- Asrar S, Zhou Z, Ren W, and Jia Z (2009). Ca(2+) permeable AMPA receptor induced long-term potentiation requires PI3/MAP kinases but not Ca/CaM-dependent kinase II. PLoS One 4, e4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellone C, and Luscher C (2006). Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent long-term depression. Nature neuroscience 9, 636–641. [DOI] [PubMed] [Google Scholar]
- Bellone C, and Mameli M (2012). mGluR-Dependent Synaptic Plasticity in Drug-Seeking. Front Pharmacol 3, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhakar AL, Dolen G, and Bear MF (2012). The pathophysiology of fragile X (and what it teaches us about synapses). Annual review of neuroscience 35, 417–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi G, and Poo M (2001). Synaptic modification by correlated activity: Hebb’s postulate revisited. Annual review of neuroscience 24, 139–166. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Peineau S, Howland JG, and Wang YT (2010). Long-term depression in the CNS. Nature reviews Neuroscience 11, 459–473. [DOI] [PubMed] [Google Scholar]
- Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y, Marinelli M, and Wolf ME (2008). Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature 454, 118–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Nasif FJ, Tsui JJ, Ju WY, Cooper DC, Hu XT, Malenka RC, and White FJ (2005). Cocaine-induced plasticity of intrinsic membrane properties in prefrontal cortex pyramidal neurons: adaptations in potassium currents. The Journal of neuroscience : the official journal of the Society for Neuroscience 25, 936–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, and Nestler EJ (2014). The neural rejuvenation hypothesis of cocaine addiction. Trends in pharmacological sciences 35, 374–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Taylor JR, Wolf ME, and Shaham Y (2017). Circuit and Synaptic Plasticity Mechanisms of Drug Relapse. The Journal of neuroscience : the official journal of the Society for Neuroscience 37, 10867–10876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford KA, Wolf ME, and Hu XT (2009). Plasticity of L-type Ca2+ channels after cocaine withdrawal. Synapse 63, 690–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis TC, Gantz SC, Moussawi K, and Bonci A (2019). Synaptic and intrinsic plasticity in the ventral tegmental area after chronic cocaine. Current opinion in neurobiology 54, 66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grueter BA, McElligott ZA, Robison AJ, Mathews GC, and Winder DG (2008). In vivo metabotropic glutamate receptor 5 (mGluR5) antagonism prevents cocaine-induced disruption of postsynaptically maintained mGluR5-dependent long-term depression. The Journal of neuroscience : the official journal of the Society for Neuroscience 28, 9261–9270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou L, and Klann E (2004). Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. The Journal of neuroscience : the official journal of the Society for Neuroscience 24, 6352–6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Lin HJ, and Hsu KS (2007a). Repeated cocaine administration promotes long-term potentiation induction in rat medial prefrontal cortex. Cereb Cortex 17, 1877–1888. [DOI] [PubMed] [Google Scholar]
- Huang CC, Yang PC, Lin HJ, and Hsu KS (2007b). Repeated cocaine administration impairs group II metabotropic glutamate receptor-mediated long-term depression in rat medial prefrontal cortex. The Journal of neuroscience : the official journal of the Society for Neuroscience 27, 2958–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Yeh CM, Wu MY, Chang AY, Chan JY, Chan SH, and Hsu KS (2011). Cocaine withdrawal impairs metabotropic glutamate receptor-dependent long-term depression in the nucleus accumbens. The Journal of neuroscience : the official journal of the Society for Neuroscience 31, 4194–4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, and Nestler EJ (2006). Neural mechanisms of addiction: the role of reward-related learning and memory. Annual review of neuroscience 29, 565–598. [DOI] [PubMed] [Google Scholar]
- Joffe ME, Santiago CI, Stansley BJ, Maksymetz J, Gogliotti RG, Engers JL, Nicoletti F, Lindsley CW, and Conn PJ (2019). Mechanisms underlying prelimbic prefrontal cortex mGlu3/mGlu5-dependent plasticity and reversal learning deficits following acute stress. Neuropharmacology 144, 19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW (2009). The glutamate homeostasis hypothesis of addiction. Nature reviews Neuroscience 10, 561–572. [DOI] [PubMed] [Google Scholar]
- Kasanetz F, Lafourcade M, Deroche-Gamonet V, Revest JM, Berson N, Balado E, Fiancette JF, Renault P, Piazza PV, and Manzoni OJ (2013). Prefrontal synaptic markers of cocaine addiction-like behavior in rats. Mol Psychiatry 18, 729–737. [DOI] [PubMed] [Google Scholar]
- Klann E, and Dever TE (2004). Biochemical mechanisms for translational regulation in synaptic plasticity. Nature reviews Neuroscience 5, 931–942. [DOI] [PubMed] [Google Scholar]
- Koob GF, and Volkow ND (2016). Neurobiology of addiction: a neurocircuitry analysis. Lancet Psychiatry 3, 760–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann DM, and Lamsa K (2008). Roles of distinct glutamate receptors in induction of anti-Hebbian long-term potentiation. J Physiol 586, 1481–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak S, and Weiss JH (2006). Calcium-permeable AMPA channels in neurodegenerative disease and ischemia. Current opinion in neurobiology 16, 281–287. [DOI] [PubMed] [Google Scholar]
- Lee BR, Ma YY, Huang YH, Wang X, Otaka M, Ishikawa M, Neumann PA, Graziane NM, Brown TE, Suska A, et al. (2013). Maturation of silent synapses in amygdala-accumbens projection contributes to incubation of cocaine craving. Nature neuroscience 16, 1644–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SJ, and Zukin RS (2007). Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci 30, 126–134. [DOI] [PubMed] [Google Scholar]
- Loweth JA, Scheyer AF, Milovanovic M, LaCrosse AL, Flores-Barrera E, Werner CT, Li X, Ford KA, Le T, Olive MF, et al. (2014). Synaptic depression via mGluR1 positive allosteric modulation suppresses cue-induced cocaine craving. Nature neuroscience 17, 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Cheng PL, Lim BK, Khoshnevisrad N, and Poo MM (2010). Elevated BDNF after cocaine withdrawal facilitates LTP in medial prefrontal cortex by suppressing GABA inhibition. Neuron 67, 821–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Lim B, and Poo MM (2009). Cocaine exposure in utero alters synaptic plasticity in the medial prefrontal cortex of postnatal rats. The Journal of neuroscience : the official journal of the Society for Neuroscience 29, 12664–12674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher C (2016). The Emergence of a Circuit Model for Addiction. Annual review of neuroscience 39, 257–276. [DOI] [PubMed] [Google Scholar]
- Luscher C, and Huber KM (2010). Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron 65, 445–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher C, and Malenka RC (2011). Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron 69, 650–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma YY, Lee BR, Wang X, Guo C, Liu L, Cui R, Lan Y, Balcita-Pedicino JJ, Wolf ME, Sesack SR, et al. (2014). Bidirectional modulation of incubation of cocaine craving by silent synapse-based remodeling of prefrontal cortex to accumbens projections. Neuron 83, 1453–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahanty NK, and Sah P (1998). Calcium-permeable AMPA receptors mediate long-term potentiation in interneurons in the amygdala. Nature 394, 683–687. [DOI] [PubMed] [Google Scholar]
- Malenka RC, and Bear MF (2004). LTP and LTD: an embarrassment of riches. Neuron 44, 5–21. [DOI] [PubMed] [Google Scholar]
- Mameli M, Balland B, Lujan R, and Luscher C (2007). Rapid synthesis and synaptic insertion of GluR2 for mGluR-LTD in the ventral tegmental area. Science 317, 530–533. [DOI] [PubMed] [Google Scholar]
- Mameli M, Bellone C, Brown MT, and Luscher C (2011). Cocaine inverts rules for synaptic plasticity of glutamate transmission in the ventral tegmental area. Nature neuroscience 14, 414–416. [DOI] [PubMed] [Google Scholar]
- Mameli M, Halbout B, Creton C, Engblom D, Parkitna JR, Spanagel R, and Luscher C (2009). Cocaine-evoked synaptic plasticity: persistence in the VTA triggers adaptations in the NAc. Nature neuroscience 12, 1036–1041. [DOI] [PubMed] [Google Scholar]
- Miller EK, and Cohen JD (2001). An integrative theory of prefrontal cortex function. Annual review of neuroscience 24, 167–202. [DOI] [PubMed] [Google Scholar]
- Nestler EJ (2001). Molecular basis of long-term plasticity underlying addiction. Nature reviews Neuroscience 2, 119–128. [DOI] [PubMed] [Google Scholar]
- Otani S, Auclair N, Desce JM, Roisin MP, and Crepel F (1999). Dopamine receptors and groups I and II mGluRs cooperate for long-term depression induction in rat prefrontal cortex through converging postsynaptic activation of MAP kinases. The Journal of neuroscience : the official journal of the Society for Neuroscience 19, 9788–9802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park P, Sanderson TM, Amici M, Choi SL, Bortolotto ZA, Zhuo M, Kaang BK, and Collingridge GL (2016). Calcium-Permeable AMPA Receptors Mediate the Induction of the Protein Kinase A-Dependent Component of Long-Term Potentiation in the Hippocampus. The Journal of neuroscience : the official journal of the Society for Neuroscience 36, 622–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascoli V, Terrier J, Espallergues J, Valjent E, O’Connor EC, and Luscher C (2014). Contrasting forms of cocaine-evoked plasticity control components of relapse. Nature 509, 459–464. [DOI] [PubMed] [Google Scholar]
- Pena-Bravo JI, Reichel CM, and Lavin A (2017). Abstinence from Cocaine-Induced Conditioned Place Preference Produces Discrete Changes in Glutamatergic Synapses onto Deep Layer 5/6 Neurons from Prelimbic and Infralimbic Cortices. eNeuro 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan H, Saur T, and Yao WD (2014). Dopamine-enabled anti-Hebbian timing-dependent plasticity in prefrontal circuitry. Frontiers in neural circuits 8, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan H, and Yao WD (2017). Cocaine Promotes Coincidence Detection and Lowers Induction Threshold during Hebbian Associative Synaptic Potentiation in Prefrontal Cortex. The Journal of neuroscience : the official journal of the Society for Neuroscience 37, 986–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Flajolet M, Greengard P, and Surmeier DJ (2008). Dichotomous dopaminergic control of striatal synaptic plasticity. Science 321, 848–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton LP, and Caron MG (2015). Essential role of D1R in the regulation of mTOR complex1 signaling induced by cocaine. Neuropharmacology 99, 610–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrier J, Luscher C, and Pascoli V (2016). Cell-Type Specific Insertion of GluA2-Lacking AMPARs with Cocaine Exposure Leading to Sensitization, Cue-Induced Seeking, and Incubation of Craving. Neuropsychopharmacology 41, 1779–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Oever MC, Goriounova NA, Li KW, Van der Schors RC, Binnekade R, Schoffelmeer AN, Mansvelder HD, Smit AB, Spijker S, and De Vries TJ (2008). Prefrontal cortex AMPA receptor plasticity is crucial for cue-induced relapse to heroin-seeking. Nature neuroscience 11, 1053–1058. [DOI] [PubMed] [Google Scholar]
- Vanderschuren LJ, and Kalivas PW (2000). Alterations in dopaminergic and glutamatergic transmission in the induction and expression of behavioral sensitization: a critical review of preclinical studies. Psychopharmacology (Berl) 151, 99–120. [DOI] [PubMed] [Google Scholar]
- Walker AG, Wenthur CJ, Xiang Z, Rook JM, Emmitte KA, Niswender CM, Lindsley CW, and Conn PJ (2015). Metabotropic glutamate receptor 3 activation is required for long-term depression in medial prefrontal cortex and fear extinction. Proceedings of the National Academy of Sciences of the United States of America 112, 1196–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf ME (1998). The role of excitatory amino acids in behavioral sensitization to psychomotor stimulants. Progress in neurobiology 54, 679–720. [DOI] [PubMed] [Google Scholar]
- Wolf ME (2016). Synaptic mechanisms underlying persistent cocaine craving. Nature reviews Neuroscience 17, 351–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf ME, and Tseng KY (2012). Calcium-permeable AMPA receptors in the VTA and nucleus accumbens after cocaine exposure: when, how, and why? Frontiers in molecular neuroscience 5, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu TX, and Yao WD (2010). D1 and D2 dopamine receptors in separate circuits cooperate to drive associative long-term potentiation in the prefrontal cortex. Proceedings of the National Academy of Sciences of the United States of America 107, 16366–16371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao WD, Gainetdinov RR, Arbuckle MI, Sotnikova TD, Cyr M, Beaulieu JM, Torres GE, Grant SG, and Caron MG (2004). Identification of PSD-95 as a regulator of dopamine-mediated synaptic and behavioral plasticity. Neuron 41, 625–638. [DOI] [PubMed] [Google Scholar]
- Zhang JC, Lau PM, and Bi GQ (2009). Gain in sensitivity and loss in temporal contrast of STDP by dopaminergic modulation at hippocampal synapses. Proceedings of the National Academy of Sciences of the United States of America 106, 13028–13033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong P, Liu W, Gu Z, and Yan Z (2008). Serotonin facilitates long-term depression induction in prefrontal cortex via p38 MAPK/Rab5-mediated enhancement of AMPA receptor internalization. J Physiol 586, 4465–4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, and Regehr WG (2002). Short-term synaptic plasticity. Annu Rev Physiol 64, 355–405. [DOI] [PubMed] [Google Scholar]