

Abstract
Inhibitors targeting the epidermal growth factor receptor (EGFR) are an effective therapy for patients with non-small cell lung cancer harboring drug-sensitive activating mutations in the EGFR kinase domain. Drug resistance due to treatment-acquired mutations has motivated development of successive generations of inhibitors that bind in the ATP-site. Third-generation agent osimertinib is now a first-line treatment for this disease. Recently, allosteric inhibitors have been developed to overcome drug-resistant mutations that confers resistance to osimertinib. Here, we present the structure-guided design and synthesis of a mutant-selective lead compound, which consists of a pyridinyl-imidazole fused benzylisoindolinedione scaffold that simultaneously occupies the orthosteric and allosteric sites. The compound potently inhibits enzymatic activity in L858R/T790M/C797S mutant EGFR (4.9 nM), with significantly lower activity for wild-type EGFR (47 nM). Additionally, this compound achieves modest cetuximab-independent, mutant-selective cellular efficacy on the L858R (1.2 μM) and L858R/T790M (4.4 μM) variants.
Keywords: EGFR-inhibitor, mutant selective, drug design, non-small cell lung cancer, chimeric
Graphical Abstract
Introduction
The epidermal growth factor receptor (EGFR) is one of the most investigated receptor tyrosine kinases and its link to non-small cell lung cancer (NSCLC) is well established.1 However, over 75% of patients succumb to this disease within 5 years after their diagnosis.2 Tumors driven by activating mutations within the EGFR tyrosine kinase domain, e.g. point-mutation L858R or in-frame exon-19 deletions (ex19del), are initially sensitive to first and second generation EGFR tyrosine kinase inhibitors (TKIs) such as gefitinib, erlotinib, and afatinib,3,4 but become resistant in the vast majority of cases due to the acquisition of the secondary ‘gatekeeper’ T790M mutation.5–9 Third-generation inhibitors overcome T790M-mediated resistance. Osimertinib and other third-generation EGFR TKIs are mutant-selective, and rely on formation of a covalent bond with C797 for their potency.10,11 Osimertinib was initially approved for treatment of patients whose tumors harbored the T790M resistance mutation, but is now also approved as a front-line therapy in untreated EGFR mutant NSCLC patients.12 Not surprisingly, patients can become resistant to osimertinib through the acquisition of the C797S mutation that precludes the ability of the drug to form its essential covalent bond with the receptor.13
As an extension of our on-going efforts on inhibitors of p38 MAP kinases, we discovered that trisubstituted imidazole compounds are capable of inhibiting the EGFR kinase.14–16 Medicinal chemistry optimization aided by molecular docking of these scaffolds allowed for the development of a series of inhibitors that can productively inhibit EGFR and exhibited low nanomolar potency against osimertinib-resistant L858R/T790M/C797S triple-mutant EGFR due to their additional strong noncovalent interactions (e.g. LN2057). Structural characterization of a series of trisubstituted imidazole inhibitors in complex with EGFR indicated that the enhanced reversible binding of these inhibitors is due to intramolecular interactions with the kinase residues K745 and D855 in the αC-helix outward inactive conformation.17 Although they are potent against L858R/T790M/C797S EGFR, they have limited therapeutic potential because they also potently inhibit wt EGFR. Mutant-selectivity, i.e. relative sparing of wt EGFR, is required for achieving a therapeutic window with EGFR TKIs in NSCLC.18
As an alternative to the aforementioned ATP-site inhibitors, highly mutant-selective inhibitors with an allosteric mechanism of action have recently been described.19,20 These EGFR allosteric inhibitors (EAIs) bind within an allosteric pocket created by the outward displacement of the αC-helix in the inactive conformation of the kinase. Binding of these allosteric agents is antagonized by formation of active EGFR dimers, and the initial allosteric agent EAI045 required co-administration of the dimer-blocking antibody cetuximab for efficacy. A much more potent allosteric inhibitor, JBJ-04–125-02, is effective in vivo without a requirement for cetuximab.21 Importantly, allosteric inhibitors can bind EGFR simultaneously with certain ATP-site inhibitors, including osimertinib. As such, JBJ-04–125-02 synergizes in vivo with osimertinib in a potentially cooperative mechanism involving simultaneous binding of both agents.
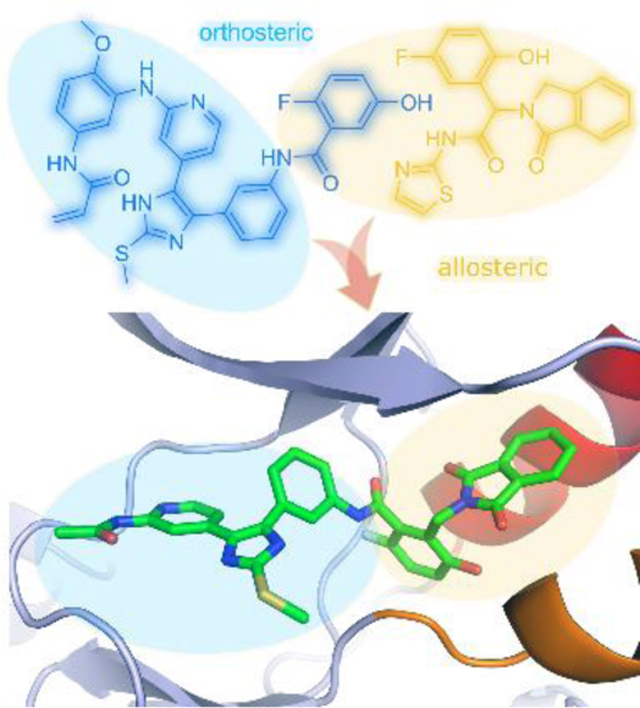
Considering the adjacency of the orthosteric and allosteric sites (Figure 1) and the potential for development of a more effective therapeutic, we sought to design inhibitors that span both binding pockets. With the goal of developing a compound that was both potent against the T790M and C797S resistance mutations and selective for the mutant over wt EGFR, we designed and synthesized a series of compounds that fused portions of the allosteric inhibitor EAI045 with ATP-site inhibitor LN2057 (Figure 2). Here we describe the structure-guided design, medicinal chemistry optimization, and characterization of ATP-allosteric site chimeric inhibitors leading to a mutant-selective agent that potently inhibits L858R/T790M/C797S EGFR (4.9 nM) and significantly lower potency against the wt kinase (47 nM).
Figure 1.

A) Structural superposition of the ATP-site binding LN2057 (PDB code 6V6K) and allosteric inhibitor EAI045 (yellow, PDB code 6P1L) illustrating the overlaying features of the 4-Fluorophenyl residue of LN2057 with the aminothiazole ring of EAI045. B) Chemical structures of LN2057 and EAI045.
Figure 2.

Structural features and the structure-guided design of a novel EGFR inhibitor targeting both ATP and allosteric binding sites.
Results
Guided by structural superpositions that showed close correspondence in the binding poses of the 4-fluorophenyl moiety of LN2057 and the thiazole of EAI045 (Figure 1), we prepared a series of compounds fusing successively larger portions of the allosteric inhibitor with the pyridinyl imidazole scaffold (Figure 2). By linking the two scaffolds via the phenyl group of the ATP-site scaffold, rather than the aminothiazole of EAI045, we avoided introducing a chiral center (Schemes 1–2). The reversible binding derivatives were obtained via amide coupling of the pyridinyl imidazole scaffold with previously prepared carboxylic acids of the benzylisoindoline based (allosteric) motifs and subsequent deprotection reactions (Scheme 3). Compound 2a combines the pyridinyl imidazole scaffold with the 2-fluoro-5-hydroxyphenyl moiety of EAI045. In compound 2b, we installed the 1-oxoisoindolin-2-yl, and in 2c we introduced a 1,3-dioxoisoindolin-2-yl instead to further probe the structure-activity relationship in the allosteric site. To assess the influence of the C797-targeting covalent capacity of these chimeric compounds, we introduced the N-(4-methoxyphenyl) acrylamide warhead via Buchwald-Hartwig amination with the aminopyridine of the orthosteric binding scaffold, followed by amide coupling with a 1,3-dioxoisoindolin-2-yl holding allosteric motif, by means of an adequate protection group strategy, to produce compound 1 (Scheme 4), a dual-site inhibitor expected to form a covalent bond with C797, as done in previous trisubstituted imidazole EGFR inhibitors.14–16
Scheme 1.

Preparation of the orthosteric binding pyridinyl imidazole scaffold via previously described conditions14,15,17 and adaptions of Sämann et al.22 and Feldman et al.23 a
aReagents and conditions: (i) NaH, SEM-Cl, THF, 0 °C, 91 %; (ii) n-BuLi, DMDS, THF, −75 °C, 98 %; (iii) NBS, CHCl3, −10 °C, 73 % (iv) n-BuLi, THF, −75 °C, 95 %; (v) 3-Nitrobenzoic acid, K3PO4, P(t-Bu)3 Pd G3, 1,4-dioxane/H2O, 50 °C, 88 %; (vi) NBS, ACN, −30 °C, 94 %; (vii) N-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)acetamide K3PO4, P(t-Bu)3 Pd G3, 1,4-dioxane/H2O, 50 °C, 90 %; (viii) Zn, NH4HCO2, MeOH, rt, 85 %; (ix) a) Boc2O, t-BuOH, 60 °C, b) 3 N NaOH, MeOH, 60 °C, 70 %.
Scheme 2.

Preparation of allosteric binding motifs a
aReagents and conditions: (i) H2SO4, 60 °C, 42 %; (ii) Sn, HCl/AcOH, 50 °C, 85 %; (iii) Ac2O, 4-DMAP, rf, 75 %; (iv) TBDMS-Cl, Et3N, ACN, rt, 98 %/46 %.
Scheme 3.

Preparation of reversible binding inhibitors 2a, 2b and 2c a
aReagents and conditions: (i) 16, TBTU, Et3N, DMF, rt; 17/18 (COCl)2, DMF cat., Et3N, THF, rt, 60 %/87 %; (ii) NaHCO3 (aq), MeOH and TFA, DCM, rt or TFA, DCM, rt and TBAF, THF, rt, 48–57 %.
Scheme 4.

Preparation of irreversible binding inhibitor 1 a
aReagents and conditions: (i) a) N-(3-Bromo-4-methoxyphenyl) acrylamide, Cs2CO3, BrettPhos Pd G3, 1,4-dioxane/t-BuOH, rf, b) TFA/DCM 5 %, 41 %; (ii) 17, (COCl)2, DMF cat., Et3N, THF, rt, 70 %; (iii) TFA, DCM, H2SO4, 1,4-dioxane, rt, 80 %.
X-ray co-crystal structures of 2a, 2c, and 24 with EGFR(T790M/V948R) confirmed that these compounds bound as expected (Figure 3, Figure S1). Overall, the binding modes of these three compounds are largely comparable, and we focus our discussion on the 1.8 Å resolution structure of 2c (Figure 3). The aminopyridine moiety anchors the inhibitor to the ATP site via H-bonds with the kinase hinge (at residue M793) and the imidazole is engaged in the expected H-bond with K745 at N3, which is critical for the strong reversible binding of the imidazole scaffold.17 The phenylamide linkage extending toward the allosteric pocket is positioned directly against the T790M gatekeeper mutation with the amide nitrogen forming hydrogen bonds with the side chains of T854 and D855. The interactions of 2c in the allosteric pocket are analogous to those of EAI045; the 2-fluoro-5-hydroxyphenyl and 1,3-dioxoisoindolin-2-yl groups closely superimpose with the corresponding regions of EAI045 (Figure 3C).19,21 Despite extensive efforts, our attempts to obtain an X-ray crystal structure of 1 in complex with EGFR were unsuccessful. Computer-aided docking of 1 into the EGFR(T790M/V948R) kinase domain results in a pose that is practically identical to 2c with the methoxyphenyl acrylamide positioned for covalent bond formation with C797 (Figure S7). These inhibitor binding modes are consistent with our present and previously reported crystal structures of a variety of trisubstituted imidazole variants where all C797-targeting covalent imidazole inhibitors superimpose with the ATP-allosteric site chimeric inhibitors 2c and 24 (Figure S8).14–17 Therefore, the structural correspondence of a variety of covalent inhibitors as well as experimental confirmation of 1 forming a covalent bond with C797 with LC-MS (Figure S3) indicate that the binding mode of 1 in complex with the EGFR kinase domain is analogous to 2c. Furthermore, we do not observe covalent labeling of the L858R/T790M/C797S mutant kinase domain by 1, confirming that it is a reversible inhibitor of this mutant (Figure S4). These findings also imply that the methoxyphenyl acrylamide group contributes to reversible binding of 1 with EGFR(L858R/T790M/C797S).
Figure 3.

X-ray crystallographic definition of the binding mode of an ATP-Allosteric chimeric inhibitor. A) Binding mode of 2c in complex with EGFR(T790M/V948R). The V948R mutation allows for the crystallization of the EGFR kinase domain in the inactive conformation. B) Positive Fo-Fc electron density map contoured at 3 sigma generated in PHENIX through molecular replacement from 1.8 Å resolution data collected on EGFR crystals soaked with 2c and coordinates of an apo EGFR(T790M/V948R) kinase domain. C) Superposition of crystal structures of 2c (green) with EAI045 (yellow, PDB code 6P1L).
We assessed inhibitory activity of these compounds against wt EGFR and L858R, L858R/T790M and L858R/T790M/C797S mutants using an HTRF-based biochemical activity assay [Table 1]. 2a exhibited ~5–32 nanomolar inhibition across all enzyme variants, indicating that incorporation of the 2-fluoro-5-hydroxyphenyl group alone did not confer mutant-selectivity. The potency trends for 2a are consistent with lower potency for L858R/T790M, which may be due to steric clashing with the M790 with the inhibitor phenyl ring (Figure S1A), and slightly improved potency for the L858R/T790M/C797S variant that could gain extra binding due to more favorable interactions to the S797 through a water-mediated H-bond. The addition of the oxoisoindolin-2-yl moiety in compound 2b resulted in a decrease in the IC50 value, while exhibiting some potency against the L858R mutant. Introduction of an additional carbonyl group in 2c restored inhibitor potency. This desired improvement in enzymatic potency resulting from the oxoisoindoline-to-dioxoisoindoline replacement is challenging to rationalize, but could be due to enhanced structural rigidity of the compound in the active conformation. Puzzlingly, wt EGFR kinase activity was not fully inhibited by 2c at concentrations approaching 1000 nM (Figure S2). Furthermore, the inclusion of the acrylamide warhead in 1 resulted in low nanomolar potency for all three EGFR mutants. The potency of 1 for wt EGFR (IC50 = 47 nM) indicated a moderate degree of mutant-selectivity. It was not clear what factors allow for 1 to exhibit mutant-selectivity, in comparison to the analogous reversible compound 2c, although we speculate that it may be due to enhanced interactions from the methoxyphenyl acrylamide. To assess the kinase selectivity of 1, a kinome screen was performed, which included 335 wt kinases (Wildtype Profiler, ProQinase GmbH, Freiburg, Germany). 1 exhibited a high degree of selectivity across the kinome with a selectivity score of 0.006 at an inhibitor concentration of 1 μM [Table S2]. Further kinases, which were inhibited to a similar degree were MKK6 SDTD and ERBB4. Kinases MAP4K5, MINK1, TGFBR2, ACK1, ERBB2 and MST2 showed a residual activity of ≤ 70 %. While the overall percent inhibition values indicated partial inhibition in the radiometric 33PanQuinase® assay, we expect that optimized variants of 1 with improved enzyme and cellular potencies will be more suitable for robust kinome selectivity analysis, which is the subject of current research efforts.
Table 1.
Biochemical activities against wt and mutant EGFR.
| EGFR IC50 [nM]a | ||||
|---|---|---|---|---|
|
| ||||
| Compound | wt | LRd | LR/TMd | LR/TM/CSd |
|
| ||||
| 1 | 47 ± 8 | 2.0 ± 0.5 | 1.5 ± 0.3 | 4.9 ± 1.0 |
| 2a | 6.2 ± 1 | 5.5 ± 0.3 | 32 ± 3 | 8.0 ± 1 |
| 2b | > 1000 | 280 ± 22 | > 1000 | > 1000 |
| 2c | 5.8 ± 1.0c | 1.2 ± 0.4 | 51 ± 3 | 32 ± 6 |
| LN2057 | 6.5 ± 1.0 | 0.26 ± 0.03 | 0.27 ± 0.05 | 130 ± 40 |
| EAI045 b | > 1000 | 8.8 ± 0.9 | 2.0 ± 0.5 | 13 ± 0.8 |
| Osimertinib | 17 ± 2 | 1.5 ± 0.4 | 0.35 ± 0.04 | > 1000 |
IC50 values were measured from a single experiment in triplicate. ATP concentration was 100 μM. Errors are reported as ± standard error.
Data from De Clercq and Heppner ACS Med Chem Lett.20
Value is EC50 due to incomplete tyrosine kinase activity inhibition at [2c] = 1000 nM.
L858R (LR), T790M (TM), C797S (CS).
We next evaluated the anti-proliferative activity of these compounds in Ba/F3 cell lines stably transfected with wt EGFR, L858R, L858R/T790M and L858R/T790M/C797S EGFR mutants [Table 2]. Our previous studies on allosteric EGFR inhibitors have indicated that Ba/F3 cells, and related cell line models, are potentially resistant to EAIs due to EGFR dimerization, but can be re-sensitized by co-administration of the anti-EGFR antibody, cetuximab (see EAI045 in Table 2).20,21 Therefore, we compared the cellular activity of our inhibitors in the presence and absence of cetuximab. Overall, the reversible chimeric inhibitors (2b and 2c) did not impact cell proliferation at concentrations below 10 μM irrespective of cetuximab treatment. 2a and 1 showed anti-proliferative effects on wt EGFR Ba/F3 only in combination with cetuximab, which is most likely due to these inhibitors exhibiting anti-proliferative activity due to drug synergy with the EGF blocking cetuximab antibody. However, compound 1 did show anti-proliferative effects in L858R and L858R/T790M mutant cell lines with micromolar-level IC50 values both with and without cetuximab treatment. Interestingly, the IC50 value of 1 against L858R Ba/F3 cells (1100 nM) was comparable to the IC50 value of EAI045 in combination with cetuximab (Table 2). We hypothesized that the lack of cetuximab sensitivity of 1 likely stem from its irreversible binding mode, which may allow it to overcome formation of the asymmetric dimer interaction of the kinase domain by forcing the outward rotated inactive conformation of the αC-helix. In general, the biochemical potencies of compounds 1, 2a, 2b, and 2c do not straightforwardly correlate to their modest Ba/F3 anti-proliferative activity. To better understand the divergent potencies between HTRF kinase activity and Ba/F3 anti-proliferative assays, a monolayer Caco-2 experiment was performed (Eurofins Panlabs Inc., St. Charles, MO, USA) to evaluate the permeability properties of 1 and 2c (Table S3). The chimeric inhibitors 1 and 2c were found to have limited cellular permeability as consistent with their modest cellular activity.
Table 2.
Anti-proliferative activities on proliferation of Ba/F3 cell lines of wt EGFR and selected mutants. Measured without and (with) dimerization preventing antibody cetuximab.
| EGFR IC50 [nM]a | ||||
|---|---|---|---|---|
|
| ||||
| Compound | wt | LR | LR/TM | LR/TM/CS |
|
| ||||
| 1 | >1 × 104
(3700 ± 400) |
1200 ± 70 (1100 ± 100) |
4400 ± 500 (3600 ± 300) |
>1 × 104
(>1 × 104) |
| 2a | >1 × 104
(800 ± 100) |
>1 × 104
(>1 × 104) |
>1 × 104
(>1 × 104) |
>1 × 104
(>1 × 104) |
| 2b | >1 × 104
(>1 × 104) |
>1 × 104
(>1 × 104) |
>1 × 104
(>1 × 104) |
>1 × 104
(>1 × 104) |
| 2c | >1 × 104
(>1 × 104) |
>1 × 104
(>1 × 104) |
>1 × 104
(>1 × 104) |
>1 × 104
(>1 × 104) |
| LN2057 | 20 ± 6 (2.3 ± 0.6) |
< 1d
(< 1)d |
22 ± 1 (3 ± 0.5) |
1600 ± 200 (780 ± 200) |
| EAI045 b | >1 × 104
(>1 × 104) |
>1 × 104
(840 ± 700) |
>1 × 104
(470 ± 200) |
> 1 × 104 (250 ± 200) |
| Osimertinib | 110 ± 40 (16 ± 4) |
3.3 ± 0.6 (3 ± 0) |
8 ± 0 (2 ± 0) |
1200 ± 130 (800 ± 200) |
IC50 values are averages of at least three independent experiments with each experiment performed in triplicate. Errors are reported as ± standard deviation.
Data from De Clercq and Heppner ACS Med Chem Lett.20
L858R (LR), T790M (TM), C797S (CS).
values below the resolution limit of the assay.
To assess target engagement with wt EGFR in a cell-based assay, we employed nanoluciferase bioluminescence resonance energy transfer (NanoBRET) live cell target engagement assays in transiently transfected HEK293T cells. This assay measures inhibitor-induced displacement of a sapitinib-BODIPY tracer molecule from the NanoLuc-tagged receptor (Figure S9). The NanoBRET results confirm wt EGFR target engagement by 1, 2a and control compounds EAI045, osimertinib, and LN2057 in living cells (Table 3). These findings are largely consistent with the anti-proliferative effects observed in Ba/F3 cells, as well as Caco-2 permeability experiments, with the exception of 2a that shows engagement in the NanoBRET assay but is not effective against Ba/F3 cells.
Table 3.
Target engagement of chimeric and control inhibitors on wt EGFR from NanoBRET assays.
| EGFR IC50 [ nM] | |
|---|---|
|
| |
| Compound | wt |
|
| |
| 1 | 6700 ± 4900 |
| 2a | 330 ± 130 |
| 2b | > 40000 |
| 2c | > 40000 |
| LN2057 | 330 ± 190 |
| EAI045 | 15000 ± 11000 |
| Osimertinib | 1700 ± 1400 |
Discussion
Overcoming acquired drug resistance in EGFR-mutant NSCLC remains challenging despite development of third-generation agents such as osimertinib and more recently described mutant-selective allosteric inhibitors. The potency of trisubstituted imidazole inhibitors against EGFR(L858R/T790M/C797S), together with recent structural studies of both these ATP-competitive compounds and phenylglycine-based allosteric inhibitors, suggested preparation of chimeric orthosteric/allosteric molecules as a route to enhanced mutant-selectivity (Figure 1). In this study, we have reported the structure-guided design and characterization of mutant-selective EGFR inhibitor 1, which occupies both the ATP (orthosteric) and allosteric sites and forms a covalent bond with C797.
Structurally, our novel lead compound 1 shares some resemblance to other ATP-site inhibitors that comprise functional groups that extend into the allosteric site. Certain approved first and second-generation EGFR/ErbB2 inhibitors (e.g. lapatinib and neratinib, respectively) also span the ATP-site and the hydrophobic pocket occupied by allosteric inhibitors, and bind the αC-helix out conformation of the kinase (Figure S5). However, these compounds are not clinically effective against the L858R/T790M or L858R/T790M/C797S mutations. We demonstrated that targeting the allosteric back pocket as well as the ATP binding site, as exemplified in this study, enables the design of mutant selective EGFR inhibitors.
The discovery of EGFR allosteric inhibitors19 and the observation that ATP-site osimertinib synergizes with JBJ-04–125-02 in vivo by potentially co-binding the EGFR kinase domain21 has motivated efforts to explore inhibitor scaffolds that more completely span both ATP and allosteric sites. Accordingly, recent examples of chimeric compounds have been reported for ErbB-family based on known ATP-site scaffolds.24,25 An initial example was an ATP-allosteric hybrid compound consisting of a combination of a reversible binding first-generation EGFR TKI amino quinazoline scaffold hybridized with EAI045-like functional groups.24 While high potency for the drug-resistant L858R/T790M/C797S mutation was shown in anti-proliferative cellular assays, the lead compound from this study lacked mutant selectivity.24 More recently, a structure-based approach was used to develop covalent inhibitors of the ErbB2-insYVMA variant that feature functional groups similar to EGFR allosteric inhibitors.25 Structurally resembling the chimera inhibitors in this work, a subset of these compounds where assayed against EGFR in HTRF and cellular assays (A431 and H1975) including binding mode characterization with X-ray crystallography of EGFR(T790M/V948R). Ultimately, these inhibitors were less effective against EGFR variants and did not exhibit selectivity over wt, potentially due to non-ideal binding modes of the back pocket functional groups as compared to our well-optimized chimeric inhibitors (Figure S6).25 Although a robust head-to-head analysis of these compounds and the ones reported here is not available, we expect that ideal anchoring of functional groups of the allosteric inhibitors is necessary for eliciting mutant selectivity in covalent chimeric compounds.
While 1 is potent and selective in biochemical assays, this selectivity was achieved at a high cost with respect to molecular weight and number of hydrogen bond donors and acceptors, which potentially lead to low cellular permeability (Table S3). These medicinal chemistry liabilities limit its cellular potency, and likely explain its lack of cellular efficacy in the context of L858R/T790M/C797S, with which it cannot bind irreversibly. Despite extensive interactions across both the ATP- and allosteric sites, our chimeric compounds do not confer higher potency in biochemical assays than that achieved with prior ATP-site or allosteric-site inhibitors. The reasons for this are unclear, but it could stem from many factors including non-ideal binding geometries of the orthosteric and allosteric portions of the inhibitors and/or constrained access of these large compounds to bind the dual site.
Conclusions
In summary, we have rationally designed and synthesized a mutant-selective EGFR inhibitor that simultaneously binds to the EGFR kinase ATP- and allosteric sites. The addition of allosteric inhibitor elements to an ATP-site scaffold contributes to the mutant-selectivity of these compounds, and the incorporation of an C797-targeting acrylamide warhead yields anti-proliferative effects in Ba/F3 cell line models. Unlike allosteric site inhibitor EAI045, our lead compound is capable of inducing anti-proliferative activity in the L858R and L858R/T790M Ba/F3 cell models independent of co-administration of cetuximab. Future directions will involve structure-guided medicinal chemistry optimization of this lead compound to improve its cellular activity.
Experimental Section
General Information
All starting materials, reagents and (anhydrous) solvents were commercially available and were used as received without any further purification or drying procedures unless otherwise noted. All NMR spectra were obtained with Bruker Avance 200 and Bruker Avance 400 or with Bruker Avance 600 MHz spectrometer (NMR Department, Institute of Organic Chemistry, Eberhard-Karls-Universität Tübingen). Solvents for NMR are noted in the experimental procedures for each compound. Residual solvent peaks were used to calibrate the chemical shifts. Chemical shifts (δ) are reported in parts per million. Mass spectra were obtained by Advion TLC-MS (ESI) and from the MASS Spectrometry Department (ESI-HRMS), Institute of Organic Chemistry, Eberhard-Karls-Universität Tübingen. The purity of tested compounds was determined via HPLC analysis on an Agilent 1100 Series LC with Phenomenex Luna C8 column (150 × 4.6 mm, 5 μm) and detection was performed with a UV diode array detector (DAD, at 254 nm and 230 nm wavelength) and was > 95 %. Elution was carried out with the following gradient: 0.01 M KH2PO4, pH 2.30 (solvent A), MeOH (solvent B), 40 % B to 85 % B in 8 min, 85 % B for 5 min, 85 % to 40 % B in 1 min, 40 % B for 2 min, stop time 16 min; 5 μl injection volume, flow 1.5 ml/min, 25 °C oven temperature. Thin-layer-chromatography (TLC) analyses were performed on fluorescent silica gel 60 F254 plates (Merck) and visualized via UV illumination at 254 and 366 nm. Column chromatography was performed on Davisil LC60A 20−45 μm silica from Grace Davison as stationary phase and Geduran Si60 63−200 μm silica from Merck for the precolumn using an Interchim PuriFlash XS 420 automated flash chromatography system.
Synthesis
1-((2-(Trimethylsilyl)ethoxy)methyl)-1H-imidazole (3)
10.02 g (147.2 mmol) of imidazole was dissolved in 250 ml of THF under nitrogen atmosphere and cooled down to −5 °C. 6.77 g (169.3 mmol) of a 60 % dispersion in oil of sodium hydride was added portion wise to the solution maintaining temperature under 0 °C. 27.20 ml (154.5 mmol) SEM-Cl was added dropwise to the stirred reaction mixture maintaining the temperature under 10 °C. After full addition, the mixture was warmed up to room temperature and stirred there for 1 h until complete conversion. 200 ml of brine was added and the organic layer was separated. The aqueous phase was extracted with EtOAc twice and the combined organic layers were dried over Na2SO4. After evaporation of the solvent the residue was distilled to obtain a colorless liquid (bp: 85 °C, p = 8×10−3 mbar) in 91 % yield (26.47 g, 133.9 mmol). 1H NMR (200 MHz, CDCl3) δ 7.5 (s, 1H), 7.06 – 7.00 (m, 1H), 7.00 – 6.95 (m, 1H), 5.21 (s, 2H), 3.46 – 3.35 (m, 2H), 0.89 – 0.78 (m, 2H), −0.09 (s, 9H). 13C NMR (50 MHz, CDCl3) δ 137.4, 129.9, 118.9, 75.9, 66.4, 17.7, −1.4.
2-(Methylthio)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazole (4)
19.73 g (99.5 mmol) 3 was dissolved in 500 ml THF under argon atmosphere and cooled down to −75 °C. 39.8 ml (99.5 mmol) of a 2.5 M n-BuLi in n-hexane solution was added via dropping funnel over 20 min maintaining the temperature at −75 °C. The solution was quenched with 8.84 ml (99.5 mmol) of dimethyl disulfide and was then slowly warmed up to room temperature. 200 ml of brine was added and the organic layer was separated. The aqueous phase was extracted with EtOAc three times. The combined organic layers were dried over Na2SO4 and the solvent was removed in vacuo. The product was obtained as a yellow oil (98 %, 24.15 g, 98.5 mmol) and was used without further purification in the next step. 1H NMR (200 MHz, CDCl3) δ 7.07 (d, J = 1.0 Hz, 1H), 7.04 (d, J = 0.9 Hz, 1H), 5.25 (s, 2H), 3.54 – 3.45 (m, 2H), 2.60 (s, 3H), 0.95 – 0.85 (m, 2H), −0.02 (s, 9H). 13C NMR (50 MHz, CDCl3) δ 143.9, 129.3, 121.2, 75.1, 66.6, 17.9, 16.5, −1.3.
4,5-Dibromo-2-(methylthio)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazole (5)
10.30 g (42.1 mmol) 4 was dissolved in 85 ml CHCl3 and cooled down to −5 °C. 15.0 g (84.3 mmol) of N-bromosuccinimide was added portion wise maintaining the temperature under +5 °C. After complete conversion, the precipitating succinimide was filtered off and the filtrate was quenched with saturated sodium sulfite solution. After vigorous stirring for 10 min the organic layer was separated, and the aqueous layer was extracted with DCM twice. Combined organic layers were dried over Na2SO4 and solvent was removed in vacuo. The residue was purified via flash chromatography (SiO2; isocratic: n-hexane/EtOAc 95:5). The product was obtained as yellow oil in 73 % yield (12.40 g, 30.8 mmol). 1H NMR (200 MHz, CDCl3) δ 5.28 (s, 2H), 3.62 – 3.51 (m, 2H), 2.62 (s, 3H), 0.97 – 0.88 (m, 2H), −0.00 (s, 9H). 13C NMR (50 MHz, CDCl3) δ 145.5, 117.5, 104.2, 74.6, 67.1, 17.9, 16.1, −1.3.
4-Bromo-2-(methylthio)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazole (6)
12.40 g (30.8 mmol) 5 was dissolved in 155 ml THF under argon atmosphere and cooled down to −75 °C. 12.33 ml (30.8 mmol) of a 2.5 M n-BuLi in n-hexane solution was added via dropping funnel maintaining the temperature at −75 °C. After complete addition, the reaction mixture was quenched with 50 ml MeOH and warmed up to room temperature. Organic layer was separated and the aqueous layer was extracted twice with EtOAc. Combined organic layers were dried over Na2SO4 and solvent was removed in vacuo. The product was obtained as yellow oil in 95 % yield (9.54 g, 29.5 mmol) and was used without further purification in the next step. 1H NMR (200 MHz, CDCl3) δ 6.99 (s, 1H), 5.18 (s, 2H), 3.55 – 3.41 (m, 2H), 2.59 (s, 3H), 0.95 – 0.84 (m, 2H), −0.03 (s, 9H). 13C NMR (50 MHz, CDCl3) δ 144.1, 120.1, 115.4, 75.1, 66.7, 17.8, 16.3, −1.4.
2-(Methylthio)-4-(3-nitrophenyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazole (7)
3.00 g (9.3 mmol) 6, 2.01 g (12.1 mmol) 3-nitrophenylboronic acid and 5.91 g (27.8 mmol) of K3PO4 were dissolved in 90 ml 1,4-dioxane and 20 ml demineralized water. The solution was degassed with three cycles of evacuation and backfilling with argon. 130 mg (2.5 mol%) P(t-Bu)3 Pd G3 were added to the solution and another three cycles of evacuation and argon backfilling were carried out. The reaction mixture was warmed up to 50 °C and stirred overnight. After cooling to room temperature, the mixture was diluted with DCM, washed once with brine, dried over Na2SO4, filtered and evaporated to dryness. The crude product was purified via flash chromatography (SiO2; n-hexane/EtOAc 60:40) obtaining a yellow oil in 88 % yield (2.99 g, 8.2 mmol). 1H NMR (400 MHz, CDCl3) δ 8.58 (t, J = 1.9 Hz, 1H), 8.12 – 8.08 (m, 1H), 8.05 (ddd, J = 8.2, 2.3, 1.0 Hz, 1H), 7.50 (t, J = 8.0 Hz, 1H), 7.45 (s, 1H), 5.29 (s, 2H), 3.60 – 3.52 (m, 2H), 2.69 (s, 3H), 0.96 – 0.90 (m, 2H), −0.01 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 148.9, 145.2, 140.2, 135.8, 130.6, 129.5, 121.5, 119.6, 117.8, 75.3, 66.8, 17.9, 16.5, −1.3. TLC-MS (ESI+): calcd. m/z 365.12 for C16H23N3O3SSi. Found 366.2 [M+H]+
5-Bromo-2-(methylthio)-4-(3-nitrophenyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazole (8)
2.93 g (8.0 mmol) 7 was dissolved in 80 ml ACN under argon atmosphere. The solution was cooled to −30 °C. 1.50 g (8.4 mmol) N-bromosuccinimide dissolved in 40 ml ACN was added dropwise under vigorous stirring maintaining −30 °C. The reaction mixture was stirred for 1 h at −30 °C and was then slowly warmed up to room temperature. The reaction was quenched by the addition of an aqueous, saturated Na2SO3 solution. The product was partitioned between water and DCM. The organic layer was washed with brine, dried over Na2SO4, filtered and evaporated to dryness. The crude product was purified via flash chromatography (SiO2; n-hexane/EtOAc 60:40) to obtain a yellow oil in 94 % yield (3.35 g, 7.5 mmol). 1H NMR (200 MHz, CDCl3) δ 8.89 (t, J = 1.8 Hz, 1H), 8.35 (ddd, J = 7.9, 1.6, 1.1 Hz, 1H), 8.14 (ddd, J = 8.2, 2.3, 1.0 Hz, 1H), 7.56 (t, J = 8.0 Hz, 1H), 5.37 (s, 2H), 3.70 – 3.58 (m, 2H), 2.70 (s, 3H), 1.01 – 0.91 (m, 2H), 0.01 (s, 9H). 13C NMR (50 MHz, CDCl3) δ 148.6, 146.3, 137.1, 134.8, 132.3, 129.4, 122.0, 121.5, 101.6, 74.0, 67.0, 18.0, 16.0, −1.3. TLC-MS (ESI+): calcd. m/z 443.03 for C16H22BrN3O3SSi. Found 498.4/500.3 [M+MeOH+Na]+
N-(4-(2-(Methylthio)-4-(3-nitrophenyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-5-yl)pyridin-2-yl)acetamide (9)
2.60 g (5.9 mmol) 8, 2.30 g (8.8 mmol) N-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)acetamide and 3.73 g (17.0 mmol) of K3PO4 were suspended in 58 ml 1,4-dioxane and 12 ml demineralized water. The solution was degassed with three cycles of evacuation and backfilling with argon. 83 mg (2.5 mol%) P(t-Bu)3 Pd G3 were added to the solution and another three cycles of evacuation and argon backfilling were carried out. The reaction mixture was warmed up to 50 °C and stirred overnight. After cooling to room temperature, the mixture was diluted with EtOAc, washed once with brine, dried over Na2SO4, filtered and evaporated to dryness. The crude product was purified via flash chromatography (SiO2; n-hexane/EtOAc/MeOH 35:60:5) obtaining a yellow oil in 90 % yield (2.65 g, 5.3 mmol) with residues of pinacol. 1H NMR (400 MHz, CDCl3) δ 9.06 (s, 1H), 8.39 – 8.35 (m, 1H), 8.34 – 8.25 (m, 2H), 8.03 – 7.97 (m, 1H), 7.78 – 7.72 (m, 1H), 7.36 (t, J = 8.0 Hz, 1H), 7.06 (dd, J = 5.2, 1.2 Hz, 1H), 5.18 (s, 2H), 3.55 – 3.46 (m, 2H), 2.73 (s, 3H), 2.17 (s, 3H), 0.93 – 0.86 (m, 2H), −0.05 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 169.0, 152.6, 148.5, 148.5, 146.8, 140.4, 137.8, 135.6, 133.0, 129.2, 129.0, 122.1, 121.8, 121.1, 115.3, 73.2, 66.7, 24.6, 17.9, 16.2, −1.4. TLC-MS (ESI+): calcd. m/z 499.17 for C23H29N5O4SSi. Found 522.2 [M+Na]+
N-(4-(4-(3-Aminophenyl)-2-(methylthio)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-5-yl)pyridin-2-yl)acetamide (10)
2.10 g (4.2 mmol) 9 was dissolved in MeOH and 1.37 g (21.0 mmol) zinc powder was added. 1.32 g (21.0 mmol) ammonium formate were added portion wise over 15 min to the suspension. After TLC indicated complete conversion, the crude mixture was filtered over celite and washed with MeOH. The filtrate was evaporated to dryness. The thereby obtained yellowish oily residue was dissolved in MeOH and precipitated in iced water. Filtration and washing with iced water led to a white solid with 85 % yield (1.68 g, 3.5 mmol) after drying. 1H NMR (200 MHz, DMSO-d6) δ 10.61 (s, 1H), 8.47 – 8.25 (m, 1H), 8.11 (s, 1H), 7.09 – 6.97 (m, 1H), 6.91 – 6.76 (m, 2H), 6.47 – 6.31 (m, 2H), 5.09 (s, 2H), 5.01 (s, 2H), 3.49 – 3.37 (m, 2H), 2.65 (s, 3H), 2.08 (s, 3H), 0.78 (t, J = 7.7 Hz, 2H), −0.08 (s, 9H). 13C NMR (50 MHz, DMSO-d6) δ 169.3, 152.6, 148.5, 148.3, 144.4, 140.0, 139.3, 134.1, 128.5, 127.4, 120.8, 114.7, 114.5, 112.8, 112.6, 72.5, 65.5, 23.9, 17.2, 15.6, −1.5. TLC-MS (ESI+): calcd. m/z 469.20 for C23H31N5O2SSi. Found 470.1 [M+H]+
tert-Butyl(3-(5-(2-aminopyridin-4-yl)-2-(methylthio)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-4-yl)phenyl)carbamate (11)
1.2 g (2.55 mmol) 10 was suspended in t-BuOH and 585 mg of di-tert-butyl dicarbonate was added in one portion. The reaction mixture was stirred overnight at 60 °C. After complete conversion [identification of intermediate via TLC-MS (ESI+): calcd. m/z 569.25 for C28H39N5O4SSi. Found 592.3 [M+Na]+] t-BuOH was evaporated and the residue was dissolved in 25 ml of MeOH and 5 ml of a 3 N NaOH solution was added. The mixture was stirred at 60 °C for 5 h, celite was added and the solvents were removed in vacuo. After purification via flash chromatography (SiO2; n-hexane/EtOAc 40:60) a white solid in 70 % yield (950 mg, 1.79 mmol) was obtained. 1H NMR (400 MHz, CDCl3) δ 8.10 – 8.01 (m, 1H), 7.47 – 7.35 (m, 2H), 7.18 – 7.12 (m, 1H), 7.12 – 7.07 (m, 1H), 6.72 – 6.66 (m, 1H), 6.59 (s, 1H), 6.53 (s, 1H), 5.14 (s, 2H), 4.66 (s, 2H), 3.55 (t, J = 7.9 Hz, 2H), 2.71 (s, 3H), 1.49 (s, 9H), 0.92 (t, J = 7.9 Hz, 2H), −0.00 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 158.8, 152.9, 148.2, 145.6, 140.3, 139.6, 138.5, 134.6, 129.1, 128.5, 122.4, 117.8, 117.7, 115.7, 110.2, 80.6, 73.1, 66.7, 28.5, 18.1, 16.4, −1.3. TLC-MS (ESI+): calcd. m/z 527.24 for C26H37N5O3SSi. Found 550.3 [M+Na]+
N-(3-((4-(4-(3-Aminophenyl)-2-(methylthio)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-5-yl)pyridin-2-yl)amino)-4-methoxyphenyl)acrylamide (12)
600 mg (1.13 mmol) 11, 363 mg (1.42 mmol) N-(3-bromo-4-methoxyphenyl) acrylamide and 1.85 g Cs2CO3 (5.68 mmol) in 7.5 ml of a mixture of 1,4-dioxane/t-BuOH (4 + 1) was degassed three times by evacuating and backfilling with argon under stirring. Under an atmosphere of argon, 51 mg BrettPhos Pd G3 (5 mol %, 0.05 mmol) was added. The solution was stirred under reflux for 4 h. After cooling down to room temperature, celite was added to the mixture and solvents were removed in vacuo. Purification via flash chromatography (SiO2; n-hexane/EtOAc 45:55) yielded 500 mg (62 %, 0.70 mmol) of a white solid [identification of intermediate via TLC-MS (ESI+): calcd. m/z 702.30 for C36H46N6O5SSi. Found 703.3 [M+H]+], which was then dissolved in a mixture of TFA/DCM 5 % v/v and stirred overnight at room temperature. After quenching the reaction with saturated aqueous NaHCO3 solution and extraction of the aqueous layer with EtOAc, the combined organic layers were dried over Na2SO4, filtered and the solvents removed in vacuo. The crude product was purified via flash chromatography (SiO2; n-hexane/EtOAc 15:85) to give the pure product as a white solid in 41 % yield (285 mg, 0.46 mmol) over two steps. 1H NMR (600 MHz, DMSO-d6) δ 9.97 (s, 1H), 8.39 (d, J = 2.4 Hz, 1H), 8.21 (s, 1H), 8.19 (d, J = 5.2 Hz, 1H), 7.43 (dd, J = 8.8, 2.4 Hz, 1H), 7.02 (s, 1H), 6.95 (d, J = 8.9 Hz, 1H), 6.90 – 6.88 (m, 1H), 6.87 – 6.84 (m, 1H), 6.69 (dd, J = 5.2, 1.2 Hz, 1H), 6.48 – 6.43 (m, 2H), 6.40 (dd, J = 7.9, 1.4 Hz, 1H), 6.22 (dd, J = 17.0, 1.9 Hz, 1H), 5.70 (dd, J = 10.2, 1.9 Hz, 1H), 5.13 (s, 2H), 5.00 (s, 2H), 3.79 (s, 3H), 3.39 – 3.35 (m, 2H), 2.65 (s, 3H), 0.78 – 0.74 (m, 2H), −0.09 (s, 9H). 13C NMR (151 MHz, DMSO-d6) δ 162.7, 156.3, 148.6, 147.6, 145.4, 144.0, 139.2, 138.8, 134.3, 132.2, 131.9, 129.7, 128.5, 127.7, 126.1, 115.9, 114.6, 113.0, 112.7, 112.6, 112.4, 111.8, 110.8, 72.5, 65.6, 55.8, 17.2, 15.6, −1.5. TLC-MS (ESI+): calcd. m/z 602.25 for C31H38N6O3SSi. Found 625.5 [M+Na]+
2-(Hydroxymethyl)isoindoline-1,3-dione (13)
[Adapted from dos Santos et al.26]: 6.36 g (43 mmol) of phthalimide were suspended in demineralized water and 4.45 ml of formalin solution (35 % v/v, 52 mmol) was added. The flask was sealed and heated for 4 h at 95 °C. The reaction mixture was cooled down to room temperature and ice was added to the suspension. The precipitate was filtered and washed with cold water, obtaining 96 % yield (7.28 g, 41.3 mmol) of white crystals. 1H NMR (200 MHz, DMSO-d6) δ 8.03 – 7.74 (m, 4H), 6.39 (t, J = 7.0 Hz, 1H), 4.96 (d, J = 7.0 Hz, 2H). 13C NMR (50 MHz, DMSO-d6) δ 154.3, 121.7, 118.4, 110.2, 47.1.
2-((1,3-Dioxoisoindolin-2-yl)methyl)-6-fluoro-3-hydroxybenzoic acid (14)
3.33 g (21.3 mmol) 2-fluoro-5-hydroxybenzoic acid was dissolved in 50 ml of fresh concentrated sulfuric acid and warmed up to 65 °C. 3.78 g (21.3 mmol) 13 was added portion wise over 30 min to the reaction mixture. The mixture was cooled to room temperature and addition of 300 ml of iced water precipitated a pinkish solid, which was filtered and dried. Recrystallization in 45 ml AcOH yielded 42 % (2.85 g, 8.96 mmol) of a white solid. 1H NMR (400 MHz, DMSO-d6) δ 13.38 (br s, 1H), 9.86 (s, 1H), 7.82 (s, 4H), 7.02 (t, J = 9.1 Hz, 1H), 6.84 (dd, J = 9.0, 4.8 Hz, 1H), 4.82 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 167.1, 165.8, 151.8 (d, J = 1.8 Hz), 151.7 (d, J = 237.3 Hz), 134.2, 131.6, 123.5 (d, J = 19.5 Hz), 122.9, 120.3 (d, J = 2.8 Hz), 116.8 (d, J = 8.2 Hz), 115.0 (d, J = 23.2 Hz), 34.3. TLC-MS (ESI+): calcd. m/z 315.05 for C16H10FNO5. Found 315.9 [M+H]+
6-Fluoro-3-hydroxy-2-((1-oxoisoindolin-2-yl)methyl)benzoic acid (15)
1.00 g (3.2 mmol) 14 was suspended in 15 ml acetic acid and 15 ml concentrated hydrochloric acid and warmed up to 50 °C. 2.09 g (17.6 mmol) of tin powder was added portion wise over 3 h under stirring at 50 °C until complete conversion via TLC was indicated. The reaction mixture was cooled down to room temperature and the aqueous layer was extracted with EtOAc. The combined organic layers were washed three times with 0.1 N hydrochloric acid and dried over Na2SO4. The solvent was removed in vacuo and the residue was purified via flash chromatography (SiO2; n-hexane /EtOAc/MeOH 50:47.5:2.5 + 1 % acetic acid) to obtain a yellowish solid in 85 % yield (0.82 g, 2.7 mmol). 1H NMR (200 MHz, DMSO-d6) δ 10.02 (br s, 1H), 7.71 – 7.64 (m, 1H), 7.56 – 7.40 (m, 3H), 7.11 (t, J = 9.0 Hz, 1H), 6.93 (dd, J = 8.9, 4.8 Hz, 1H), 4.73 (s, 2H), 4.22 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 167.5, 166.2, 152.6 (d, J = 1.2 Hz), 151.7 (d, J = 237.7 Hz), 142.0, 132.1, 131.5, 128.0, 124.9 (d, J = 19.1 Hz), 123.6, 122.9, 120.6 (d, J = 2.4 Hz), 117.4 (d, J = 8.1 Hz), 116.2 (d, J = 22.9 Hz), 49.4, 38.2. TLC-MS (ESI-): calcd. m/z 301.08 for C16H12FNO4. Found 300.1 [M-H]-
5-Acetoxy-2-fluorobenzoic acid (16)
550 mg (3.52 mmol) 2-fluoro-5-hydroxybenzoic acid and 5 mg (0.04 mmol) 4-dimethylaminopyridine were suspended in 7 ml acetic anhydride and the mixture was refluxed overnight. After cooling down to room temperature a saturated aqueous NH4Cl solution was added to the reaction mixture and the aqueous layer was extracted with EtOAc. Combined organic layers were dried over Na2SO4 and the solvents were removed in vacuo. Purification via flash chromatography (SiO2; DCM/MeOH 90:10 + 1% formic acid) yielded 75 % (530 mg, 2.67 mmol) of a white solid. 1H NMR (200 MHz, CDCl3) δ 8.70 (br s, 1H), 7.75 (dd, J = 5.9, 2.9 Hz, 1H), 7.39 – 7.28 (m, 1H), 7.25 – 7.13 (m, 1H), 2.32 (s, 3H). 13C NMR (50 MHz, CDCl3) δ 169.3, 168.3 (d, J = 3.7 Hz), 160.1 (d, J = 260.8 Hz), 146.3 (d, J = 3.3 Hz), 129.0 (d, J = 9.3 Hz), 125.6 (d, J = 0.7 Hz), 118.5 (d, J = 3.6 Hz), 118.1 (d, J = 10.1 Hz), 21.1.
3-((tert-Butyldimethylsilyl)oxy)-2-((1,3-dioxoisoindolin-2-yl)methyl)-6-fluorobenzoic acid (17)
2.25 g (7.13 mmol) 14 was suspended in 20 ml of ACN and cooled down to 0 °C. After slow addition of 2.5 ml (18.0 mmol) triethylamine, 1.29 g (8.56 mmol) TBDMS-Cl was added in one portion. The mixture was warmed up to room temperature and stirred overnight to complete conversion. 10 ml of a 2 N HCl in 1,4-dioxane and 50 ml EtOAc were added. 10 ml of an aqueous 0.01 N hydrochloric acid solution was added under dissolution of the precipitate. Layers were separated, the organic layer was dried with Na2SO4 and solvents were removed in vacuo. After precipitation in n-pentane a white solid in 98 % yield (3.03 g, 6.98 mmol) was obtained. 1H NMR (400 MHz, DMSO-d6) δ 13.41 (s, 1H), 7.83 (s, 4H), 7.11 (t, J = 9.0 Hz, 1H), 6.95 (dd, J = 9.0, 4.7 Hz, 1H), 4.84 (s, 2H), 0.95 (s, 9H), 0.28 (s, 6H). 13C NMR (101 MHz, DMSO-d6) δ 167.2, 165.6, 152.6 (d, J = 239.5 Hz), 49.7 (d, J = 2.2 Hz), 134.3, 131.6, 124.5 (d, J = 3.3 Hz), 123.6 (d, J = 20.4 Hz), 123.0, 120.1 (d, J = 8.3 Hz), 115.2 (d, J = 23.1 Hz), 34.3, 25.7, 18.0, −4.4. TLC-MS (ESI+): calcd. m/z 429.14 for C22H24FNO5Si. Found 430.2 [M+H]+
3-((tert-Butyldimethylsilyl)oxy)-6-fluoro-2-((1-oxoisoindolin-2-yl)methyl)benzoic acid (18)
780 mg (2.58 mmol) 15 was suspended in 12 ml ACN and cooled down to 0 °C. After slow addition of 1.08 ml (7.76 mmol) triethylamine, 507 mg (3.36 mmol) TBDMS-Cl was added portion wise and the reaction mixture was warmed up to room temperature. Stirring over 4 h led to complete conversion, whereupon water was added to the mixture and 0.1 N hydrochloric acid was used to adjust to pH 4. After extraction of the aqueous layer multiple times with EtOAc, the organic layers were dried with Na2SO4 and the solvents were removed in vacuo. Purification via flash chromatography (SiO2; EtOAc/MeOH 85:15) yielded 46 % (500 mg, 1.19 mmol) of a yellowish solid. 1H NMR (200 MHz, DMSO-d6) δ 7.70 – 7.60 (m, 1H), 7.57 – 7.35 (m, 3H), 7.00 (t, J = 8.7 Hz, 1H), 6.72 (dd, J = 8.8, 4.6 Hz, 1H), 4.66 (s, 2H), 4.14 (s, 2H), 0.82 (s, 9H), 0.17 (s, 6H). TLC-MS (ESI+): calcd. m/z 415.16 for C22H26FNO4Si. Found 438.6 [M+Na]+
N-(3-(5-(2-((5-acrylamido-2-methoxyphenyl)amino)pyridin-4-yl)-2-(methylthio)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-4-yl)phenyl)-3-((tert-butyldimethylsilyl)oxy)-2-((1,3-dioxoisoindolin-2-yl)methyl)-6-fluorobenzamide (19)
138 mg (0.32 mmol) 17 was dissolved in 2 ml THF and 5 drops DMF were added. 32 μl (0.37 mmol) oxalyl chloride was added drop wise under gas formation and the mixture was stirred for 1 h at room temperature, whereupon the excess of oxalyl chloride was removed in vacuo. 130 mg (0.21 mmol) 12 was dissolved in 2 ml THF, 90 μl (0.64 mmol) triethylamine was added and the mixture was cooled down to 0 °C. The beforehand prepared acid chloride dissolved in 2 ml THF was slowly added, whereupon the mixture was warmed up to room temperature and stirred for 1 h. Brine was added and the aqueous layer was extracted with EtOAc. The combined organic layers were dried over Na2SO4 and the solvents were removed in vacuo. Purification via flash chromatography (SiO2; n-hexane/EtOAc 10:90) yielded 70 % (154 mg, 0.15 mmol) of a white solid. 1H NMR (400 MHz, CDCl3) δ 9.55 (s, 1H), 8.07 (s, 1H), 7.80 – 7.75 (m, J = 2.6 Hz, 2H), 7.73 – 7.68 (m, 3H), 7.67 – 7.63 (m, 1H), 7.57 – 7.53 (m, 1H), 7.53 – 7.49 (m, 1H), 7.40 (s, 1H), 7.38 – 7.32 (m, 1H), 7.04 (s, 1H), 6.97 – 6.69 (m, 6H), 6.46 – 6.39 (m, 2H), 5.81 – 5.70 (m, 1H), 5.10 (s, 2H), 4.69 (s, 2H), 3.78 (s, 3H), 3.52 (t, J = 8.0 Hz, 2H), 2.69 (s, 3H), 0.90 – 0.85 (m, 2H), 0.81 (s, 9H), 0.20 (s, 6H), −0.04 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 168.3, 164.0, 163.6, 155.9, 154.5, 152.1, 150.8, 150.8, 146.8, 146.3, 139.9, 139.6, 136.8, 134.8, 134.1, 132.2, 131.8, 131.5, 129.6, 128.8, 127.7, 127.5, 126.9, 125.1, 123.4, 123.4, 122.8, 122.7, 120.7, 120.3, 120.2, 119.7, 116.4, 116.1, 115.6, 111.6, 111.2, 109.2, 73.1, 66.7, 56.0, 36.8, 26.1, 18.9, 18.0, 16.5, −1.3, −3.3. TLC-MS (ESI+): calcd. m/z 1013.38 for C53H60FN7O7SSi2. Found 1037.0 [M+Na]+
N-(3-(5-(2-((5-acrylamido-2-methoxyphenyl)amino)pyridin-4-yl)-2-(methylthio)-1H-imidazol-4-yl)phenyl)-2-((1,3-dioxoisoindolin-2-yl)methyl)-6-fluoro-3-hydroxybenzamide (1)
50 mg (0.05 mmol) 19 was dissolved in 1.5 ml DCM and 0.5 ml TFA was added. The mixture was stirred at room temperature for 20 h [identification of intermediate via TLC-MS (ESI+): calcd. m/z 883.30 for C47H46FN7O6SSi. Found 906.3 [M+Na]+]. 4 ml 1,4-dioxane and 1 ml of an aqueous 50 % v/v sulfuric acid were added until the complete conversion was indicated via HPLC. A saturated aqueous NaHCO3 solution was added adjusting to pH 8. The emerging precipitate was filtered, washed with demineralized water and n-pentane and dried via high vacuum pump yielding 89 % (34 mg, 0.045 mmol) of an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ 12.92 (br s, 1H), 10.62 (s, 1H), 10.10 (s, 1H), 9.91 (s, 1H), 8.19 – 7.73 (m, 3H), 7.68 – 7.60 (m, 5H), 7.54 – 7.49 (m, 1H), 7.49 – 7.45 (m, 1H), 7.41 – 7.23 (m, 2H), 7.15 – 7.11 (m, 1H), 7.06 (t, J = 8.8 Hz, 2H), 6.87 (dd, J = 8.9, 4.8 Hz, 1H), 6.82 (s, 1H), 6.43 (dd, J = 17.0, 10.1 Hz, 1H), 6.23 (dd, J = 17.0, 1.8 Hz, 1H), 5.72 (dd, J = 10.3, 1.5 Hz, 1H), 4.82 (s, 2H), 3.76 (s, 3H), 2.65 (s, 3H). HRMS (ESI): exact mass calcd for C41H32FN7O6S [M+H]+: 770.21916. Found: 770.21941.
N-(3-(5-(2-acetamidopyridin-4-yl)-2-(methylthio)-1H-imidazol-4-yl)phenyl)-2-fluoro-5-hydroxybenzamide (2a)
60 mg (0.12 mmol) 10, 33 mg (0.16 mmol) 16 and 62 mg (0.19 mmol) TBTU were dissolved in 2 ml DMF and 53 μl (0.38 mmol) triethylamine was added. The reaction mixture was stirred overnight at 40 °C [identification of intermediate via TLC-MS (ESI+): calcd. m/z 649.22 for C32H36FN5O5SSi. Found 672.2 [M+Na]+]. MeOH and a saturated aqueous NaHCO3 solution were added and stirring proceeded for further 1.5 h. MeOH was evaporated and the aqueous layer was extracted with EtOAc. The combined organic layers were dried over Na2SO4 and solvents were removed in vacuo. The residue was purified via flash chromatography (SiO2; DCM/MeOH 90:10) [identification of intermediate via TLC-MS (ESI+): calcd. m/z 607.21 for C30H34FN5O4SSi. Found 630.3 [M+Na]+] and then directly dissolved in a 25 % TFA/DCM mixture. After stirring overnight at room temperature, a saturated aqueous NaHCO3 solution was added and the aqueous layer was extracted with EtOAc. Combined organic layers were dried over Na2SO4 and solvents were removed in vacuo. The residue was purified via flash chromatography (SiO2; DCM/MeOH 90:10) yielding 57 % (35 mg, 0.07 mmol) of a white solid. As mixture of tautomers: 1H NMR (400 MHz, DMSO-d6) δ 12.93 – 12.62 (m, 1H), 10.48 – 10.38 (m, 1H), 10.31 (s, 1H), 9.77 (s, 1H), 8.38 – 8.08 (m, 2H), 7.92 – 7.80 (m, 1H), 7.79 – 7.64 (m, 1H), 7.44 – 7.23 (m, 1H), 7.18 – 7.09 (m, 2H), 7.08 – 7.00 (m, 1H), 6.98 – 6.86 (m, 2H), 2.62 (s, 3H), 2.04 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 169.3, 169.1, 163.0, 162.8, 153.5, 152.5, 152.2 (d, J = 239.2 Hz), 148.2, 148.2, 147.6, 143.8, 142.4, 139.5, 139.3, 139.0, 134.9, 134.4, 131.1, 130.7, 129.4, 128.7, 128.7, 126.1, 126.1, 125.4, 125.2, 125.1, 124.1, 119.7, 119.2, 118.7 (d, J = 6.3 Hz), 117.3, 117.0 (d, J = 24.0 Hz), 116.6, 115.3 (d, J = 2.2 Hz), 111.0, 110.6, 23.9, 15.2. HRMS (ESI): exact mass calcd for C24H20FN5O3S [M+H]+: 478.13437. Found: 478.13482.
N-(3-(5-(2-acetamidopyridin-4-yl)-2-(methylthio)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-4-yl)phenyl)-3-((tert-butyldimethylsilyl)oxy)-6-fluoro-2-((1-oxoisoindolin-2-yl)methyl)benzamide (20)
86 mg (0.18 mmol) 18 was dissolved in 3 ml THF and 5 drops DMF were added. 19 μl (0.21 mmol) oxalyl chloride was added drop wise under gas formation and the mixture was stirred for 1 h at room temperature, whereupon the excess of oxalyl chloride was removed in vacuo. 75 mg (0.16 mmol) 10 was dissolved in 3 ml THF, 66 μl (0.48 mmol) triethylamine was added and the mixture was cooled down to 0 °C. The beforehand prepared acid chloride dissolved in 2 ml THF was slowly added, whereupon the mixture was warmed up to room temperature and stirred for 1 h. Brine was added and the aqueous layer was extracted with EtOAc. The combined organic layers were dried over Na2SO4 and solvents were removed in vacuo. Purification via flash chromatography (SiO2; n-hexane/EtOAc 10:90) yielded 75 % (105 mg, 0.12 mmol) of a white solid. 1H NMR (200 MHz, CDCl3) δ 9.00 (s, 1H), 8.55 (s, 1H), 8.33 – 8.20 (m, 2H), 7.84 (s, 2H), 7.60 – 7.52 (m, 1H), 7.45 – 7.27 (m, 5H), 7.12 – 7.06 (m, 1H), 6.98 (t, J = 8.7 Hz, 1H), 6.88 (dd, J = 8.7, 3.5 Hz, 1H), 5.22 (s, 2H), 4.92 (s, 2H), 4.42 (s, 2H), 3.59 – 3.48 (m, 2H), 2.75 (s, 3H), 2.15 (s, 3H), 0.99 (s, 9H), 0.96 – 0.89 (m, 2H), 0.22 (s, 6H), −0.00 (s, 9H). 13C NMR (50 MHz, CDCl3) δ 169.2, 167.5, 166.5 (d, J = 2.6 Hz), 153.5 (d, J = 252.8 Hz), 152.0, 148.1, 146.8 (d, J = 3.1 Hz), 146.1, 140.8, 140.0, 138.4, 136.5, 135.3, 134.4, 133.8, 133.7, 130.7, 129.1 (d, J = 6.7 Hz), 128.0, 127.7, 123.8, 122.8 (d, J = 7.4 Hz), 121.7, 121.0, 120.7, 119.3, 119.2, 116.3 (d, J = 21.4 Hz), 115.9, 73.1, 66.5, 49.3, 43.9, 25.7 24.6, 18.2, 17.9, 16.4, −1.3, −4.2. TLC-MS (ESI+): calcd. m/z 866.35 for C45H55FN6O5SSi2. Found 890.0 [M+Na]+
N-(3-(5-(2-acetamidopyridin-4-yl)-2-(methylthio)-1H-imidazol-4-yl)phenyl)-6-fluoro-3-hydroxy-2-((1-oxoisoindolin-2-yl)methyl)benzamide (2b)
90 mg (0.10 mmol) 20 was dissolved in a 33 % TFA/DCM mixture and stirred overnight at room temperature. A saturated aqueous NaHCO3 solution was added and the aqueous layer was extracted with DCM. The combined organic layers were dried over Na2SO4 and solvents were removed in vacuo [identification of intermediate via TLC-MS (ESI+): calcd. m/z 736.27 for C39H41FN6O4SSi Found 759.6 [M+Na]+]. The residue was dissolved in 3 ml THF and cooled down to 0 °C. 205 μl (0.20 mmol) of a 1 M TBAF in THF solution were added and the mixture was stirred overnight at room temperature. Brine was added and the organic layer was extracted with EtOAc. The combined organic layers were dried over Na2SO4 and solvents were removed in vacuo. The crude product afforded two purification steps via flash chromatography (SiO2; n-hexane/EtOAc/MeOH 0:95:5; DCM/MeOH 90:10) to yield 48 % (31 mg, 0.05 mmol) of an off-white solid. As mixture of tautomers: 1H NMR (400 MHz, DMSO-d6) δ 12.87 – 12.60 (m, 1H), 10.62 – 10.27 (m, 2H), 9.96 (s, 1H), 8.40 – 8.07 (m, 2H), 8.00 – 7.86 (m, 1H), 7.82 – 7.68 (m, 1H), 7.63 – 7.54 (m, 1H), 7.51 – 7.25 (m, 4H), 7.20 – 7.13 (m, 1H), 7.12 – 7.01 (m, 2H), 6.98 – 6.88 (m, 1H), 4.84 (s, 2H), 4.27 (s, 2H), 2.63 (s, 3H), 2.12 – 1.97 (m, 3H). 13C NMR (101 MHz, DMSO-d6) δ 169.1, 168.9, 167.2, 167.1, 164.9 (d, J = 1.8 Hz), 152.4, 151.3 (d, J = 259.3 Hz), 148.6 (d, J = 2.1 Hz), 148.1, 147.5, 143.7, 143.3, 142.2, 139.6, 139.5, 139.4, 139.2, 136.4, 136.3, 135.2, 134.8, 134.3, 131.0, 130.6, 130.4, 130.3, 129.3, 129.2, 128.5, 128.1, 128.0, 127.7, 127.3, 127.2, 126.0, 123.8, 123.1, 119.7, 119.6, 119.6, 119.0 (d, J = 7.0 Hz), 117.2, 116.5, 115.8 (d, J = 21.2 Hz), 110.9, 110.5, 47.6, 43.0, 23.8, 15.1, 15.0. HRMS (ESI): exact mass calcd for C33H27FN6O4S [M+H]+: 623.18713. Found: 623.18759.
N-(3-(5-(2-acetamidopyridin-4-yl)-2-(methylthio)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-4-yl)phenyl)-3-((tert-butyldimethylsilyl)oxy)-2-((1,3-dioxoisoindolin-2-yl)methyl)-6-fluorobenzamide (21)
131 mg (0.30 mmol) 17 was dissolved in 3 ml DCM and 3 drops DMF were added. 31 μl (0.42 mmol) oxalyl chloride was added drop wise under gas formation and the mixture was stirred for 1.5 h at room temperature, whereupon the excess of oxalyl chloride was removed in vacuo. 144 mg (0.30 mmol) 10 was dissolved in 3 ml THF, 128 μl (0.92 mmol) triethylamine was added and the mixture was cooled down to 0 °C. The beforehand prepared acid chloride dissolved in 2 ml DCM was slowly added, whereupon the mixture was warmed up to room temperature and stirred for 0.5 h. Solvents were removed in vacuo and purification via flash chromatography (SiO2; DCM/MeOH 97:3) yielded 87 % (235 mg, 0.27 mmol) of a white solid. 1H NMR (400 MHz, CDCl3) δ 9.56 (s, 1H), 8.71 (s, 1H), 8.40 (s, 1H), 7.92 – 7.82 (m, 1H), 7.79 – 7.73 (m, 2H), 7.66 – 7.61 (m, 1H), 7.54 – 7.49 (m, 2H), 7.47 – 7.41 (m, 1H), 7.24 (s, 1H), 7.06 – 7.00 (m, 1H), 6.99 – 6.95 (m, 1H), 6.73 – 6.59 (m, 2H), 5.12 (s, 2H), 4.94 (s, 2H), 3.58 – 3.50 (m, 2H), 2.73 (s, 3H), 2.20 (s, 3H), 0.95 – 0.88 (m, 11H), 0.23 (s, 6H), −0.03 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 169.4, 168.7, 162.4, 154.6, 151.1 (d, J = 234.9 Hz), 152.0, 147.7, 145.9, 141.3, 139.3, 139.2, 138.3, 134.2, 133.9, 131.9, 129.3, 127.8, 123.9 (d, J = 2.4 Hz), 123.6, 123.0, 120.8, 120.1 (d, J = 8.1 Hz), 118.1, 116.5, 115.4, 115.2 (d, J = 23.5 Hz), 73.0, 66.5, 36.4, 26.0, 24.7, 18.6, 18.0, 16.5, −1.3, −3.7. TLC-MS (ESI+): calcd. m/z 880.33 for C45H53FN6O6SSi2. Found 903.0 [M+Na]+
N-(3-(5-(2-acetamidopyridin-4-yl)-2-(methylthio)-1H-imidazol-4-yl)phenyl)-2-((1,3-dioxoisoindolin-2-yl)methyl)-6-fluoro-3-hydroxybenzamide (2c)
100 mg (0.11 mmol) 21 was dissolved in a 33 % TFA/DCM mixture and stirred overnight at room temperature. A saturated aqueous NaHCO3 solution was added and the aqueous layer was extracted with DCM. The combined organic layers were dried over Na2SO4 and solvents were removed in vacuo [identification of intermediate via TLC-MS (ESI+): calcd. m/z 750.25 for C39H39FN6O5SSi Found 773.9 [M+Na]+]. The residue was dissolved in 3 ml THF and cooled down to 0 °C. 340 μl (0.34 mmol) of a 1 M TBAF in THF solution were added and the mixture was stirred overnight at room temperature. Brine was added and the organic layer was extracted with EtOAc. The combined organic layers were dried over Na2SO4 and the solvents removed in vacuo. The crude product afforded two purification steps via flash chromatography (SiO2; DCM/MeOH 90:10; n-hexane/EtOAc/MeOH 35:55:10) to yield 49 % (36 mg, 0.05 mmol) of an off-white solid. As mixture of tautomers: 1H NMR (400 MHz, DMSO-d6) δ 12.80 – 12.64 (m, 1H), 10.58 – 10.30 (m, 2H), 9.94 – 9.81 (m, 1H), 8.43 – 8.08 (m, 2H), 7.74 – 7.52 (m, 5H), 7.51 – 7.40 (m, 1H), 7.28 – 6.97 (m, 4H), 6.90 – 6.81 (m, 1H), 4.82 (s, 2H), 2.62 (s, 3H), 2.16 – 1.95 (m, 3H). 13C NMR (101 MHz, DMSO-d6) δ 169.2, 168.9, 167.2, 162.1, 162.0, 152.6, 152.5, 151.8, 151.7, 151.7 (d, J = 235.4 Hz), 148.0, 147.4, 143.7, 143.2, 142.1, 139.6, 139.4, 139.1, 138.8, 134.3, 134.0, 131.6, 131.0, 130.4, 128.9, 128.2, 126.1, 125.9, 125.9, 123.7, 122.8, 122.8, 122.8, 120.5 (d, J = 2.6 Hz), 119.2 (d, J = 8.7 Hz), 118.8, 117.2, 116.4, 116.4, 115.0 (d, J = 23.2 Hz), 110.8, 110.6, 34.4, 23.9, 15.2. HRMS (ESI): exact mass calcd for C33H25FN6O5S [M+H]+: 637.16639. Found: 637.16676.
HTRF Assay
Biochemical assays for WT EGFR (5 nM) and each mutant (L858R, 0.1 nM; L858R/T790M and L858R/T790M/C797S 0.02 nM) were carried out using a homogeneous time-resolved fluorescence (HTRF) KinEASE-TK (Cisbio) assay as described previously.20 Assays were optimized for each [ATP] = 100 μM final concentration. Inhibitor compounds in DMSO were dispensed directly in 384-well plates with the D300 digital dispenser (Hewlett Packard) followed immediately by the addition of aqueous buffered solutions using the Multidrop Combi Reagent Dispenser (Thermo Fischer). Compound IC50 values were determined by 11-point inhibition curves (from 1.0 to 0.00130 μM) in triplicate. The data was graphically displayed using GraphPad Prism version 8.0 (GraphPad software). The curves were fitted using a non-linear regression model with a sigmoidal dose response.
EGFR protein purification.
Constructs spanning residues 696–1022 of the human EGFR (including WT and T790M/V948R mutant sequences) were prepared in a GST-fusion format using the pTriEX system (Novagen) for expression in Sf9 insect cells as described previously.6,27 Briefly, EGFR kinase proteins were purified by Ni-NTA and glutathione-affinity chromatography in 50 mM Tris-HCl (pH = 8.0), 500 mM NaCl, 5% glycerol, 5 mM TCEP followed by size-exclusion chromatography after cleavage with Tomato etch virus (TEV) to remove the GST fusion partner following established procedures.
Structure determination.
EGFR(T790M/V948R) pre-incubated with 1 mM AMP-PNP and 10 mM MgCl2 on ice was prepared by hanging-drop vapor diffusion over a reservoir solution containing 0.1 M Bis-Tris (pH = 5.5), 25% PEG-3350, and 5 mM TCEP (buffer A). Drops containing crystals in buffer A were exchanged with solutions of buffer A containing ~1.0 mM 2a, 24 and 2c three times for an hour and then left overnight. Crystals were flash frozen after rapid immersion in a cryoprotectant solution with buffer A containing 25% ethylene glycol. X-ray diffraction data was collected at 100K the Advanced Light Source a part of the Northeastern Collaborative Access Team (NE-CAT) on Beamline 24-ID-C or 24-ID-E. Diffraction data was processed and merged in Xia2 using aimless and XDS. The structure was determined by molecular replacement with the program PHASER using the inactive EGFR kinase from our previous inhibitor work excluding the LN2084 ligand (PDB 6V5N).17 Repeated rounds of manual refitting and crystallographic refinement were performed using COOT and Phenix. The inhibitor was modeled into the closely fitting positive Fo − Fc electron density and then included in following refinement cycles. In one case, the inhibitor binding side within chain A of 6WXN was not modeled as the density is a mixture of LN3844 and ANP-PNP. Statistics for diffraction data processing and structure refinement are shown in [Table S1].
LC-MS/MS sample preparation, instrument methods and data processing.
Proteins were denatured in 8 M urea and incubated in 10 mM TCEP for 1 hr prior to cysteine carbamidomethylation with 12 mM iodoacetamide in the dark at 25°C. TCEP concentration was brought back to 10 mM for an additional 15 min before samples were diluted to 2 M urea and subjected to proteolytic digestion with trypsin (1:50 ratio of trypsin:protein) overnight at 32°C. Digestions were brought to 1% formic acid (FA) and peptides were dried by vacuum centrifugation and desalted over C18 resin. Peptides were resuspended in 2.5% acetonitrile (MeCN)/0.1% FA and analyzed by liquid chromatography tandem mass spectrometry (LC-MS/MS) on an Ultimate 3000 RSLCnano system coupled to an Orbitrap Eclipse mass spectrometer (Thermo Scientific, Waltham, MA, USA). Peptides were separated with a 75-min gradient of 3–48% MeCN in 1% FA over an ES803A column (Thermo Scientific, Waltham, MA, USA) and electrosprayed (spray voltage = 2.15 kV, ion transfer tube = 300°C) into the mass spectrometer with an EasySpray ion source (Thermo Scientific, Waltham, MA, USA). Precursor ion scans (300–2,000 m/z) were obtained in the orbitrap at 120,000 resolution in profile (RF lens % = 30, Max IT = 100 ms, 1 microscan). Precursor scans were followed by fragment ion scans (HCD fragmentation at 30% NCE) over 3 sec in the orbitrap at 30,000 resolution in centroid (intensity threshold = 5.0E4) with dynamic exclusion enabled (exclusion duration = 15 sec, mass tolerance = ± 10 ppm). Raw data were searched against a forward and reverse human proteome (Uniprot 2011) with the EGFR(L858R/T790M) sequence (residues 696–1022) appended to it via the SEQUEST algorithm, with the following modifications allowed: carbamidomethylation of Cys (+ 57.0215 Da), phosphorylation of Ser/Thr/Tyr (+ 79.9663 Da), oxidation of Met (+ 15.9949 Da), compound 1 labeling of Cys (+ 769.2119 Da), carbamylation of Lys (+ 43.0058 Da), ubiquitylation of Lys (+ 114.0429 Da). Resulting peptide hits were filtered to a FDR < 1% with XCorr scores (1.8 for z = 1, 2 for z = 2, 2.2 for z = 3, 2.4 for z = 4, 2.6 for z = 5) and mass accuracy (± 5 ppm).
Intact LC-MS sample preparation and instrument methods
Purified EGFR(L858R/T790M) or EGFR(L858R/T790M/V948R) kinase domain was incubated 1:1 with 1 for 1 hr at room temperature. Proteins were denatured in 8 M urea+1% formic acid and desalted over C4 resin prior to LCMS analysis. Denatured proteins (1 μg per condition) were analyzed by LC-MS via a U3000 RSLC coupled to an Orbitrap Eclipse mass spectrometer. Proteins were eluted off a ES811A 15-cm C4 column with a 5–50% gradient of MeCN in 1% FA. MS scans (range = 600–2000 m/z) were obtained in the orbitrap at 120,000 resolution with 4 microscans. The Xtract function in Freestyle software (Thermo Scientific, Waltham, MA, USA) was used to deconvolute charge states averaged across the elution window.
Cell lines
The parental Ba/F3 cells was a generous gift from the laboratory of Dr. David Weinstock (in 2014) and was used to generate the wildtype EGFR, L858R, L858R/T790M and L858R/T790M/C797S EGFR mutant Ba/F3 cells. These cells were previously characterized as described.19,28 All Ba/F3 cells were cultured in RPMI1640 media with 10% fetal bovine serum and 1% penicillin and streptomycin. Wildtype EGFR Ba/F3 cells were additionally supplemented with 10 ng/ml of EGF purchased from Life Technologies (Cat#PHG0311L). All cell lines were tested negative for Mycoplasma using Mycoplasma Plus PCR Primer Set (Agilent) and were passaged and/or used for no longer than 4 weeks for all experiments. AZD9291 (osimertinib) was purchased from Selleckchem (Houston, TX, Cat#S7297) and Cell Titer Glo reagents were purchased from Promega (Cat# G7570). Cetuximab was purchased from Eli Lilly and Company (Cat#66733–948-23).
Cell viability assays
Ba/F3 cells were plated and treated with increasing concentrations of inhibitors in triplicate for 72 hours. Cellular growth or the inhibition of growth was assessed by Cell Titer Glo Assays per manufacturer’s protocol. For experiments performed using wildtype EGFR Ba/F3 cells and/or in the presence of cetuximab, 10 ng/ml EGF and/or 1 μg/ml of cetuximab were added respectively at the time of drug treatment. All experiments were repeated at least 3 times and values were reported as an average of n=3 with standard deviation.
NanoBRET assay
The assay was performed as described previously.29,30 In brief: Full-length wild-type EGFR was obtained as plasmids cloned in frame with a C-terminal NanoLuc-fusion (Promega, CS1810C131). Plasmids were transfected into HEK293T cells using FuGENE HD (Promega, E2312) and proteins were allowed to express for 20h. Serially diluted inhibitor and NanoBRET EGFR Tracer at 700 nM concentration (Suppl. Fig. S9 - Tracer KD, app was determined to be >500 nM) were pipetted into white 384-well plates (Greiner 784075) using an Echo 550 acoustic dispenser (Labcyte). The corresponding protein-transfected cells were added and reseeded at a density of 2.5 × 105 cells/mL after trypsinization and resuspending in Opti-MEM without phenol red (Life Technologies). The system was allowed to equilibrate for 2 hours at 37°C/5% CO2 prior to BRET measurements. To measure BRET, Nano-Glo Substrate and Extracellular NanoLuc® Inhibitor (Promega, N2540) were added as per the manufacturer’s protocol, and filtered luminescence was measured on a PHERAstar plate reader (BMG Labtech) equipped with a luminescence filter pair (450 nm BP filter (donor) and 610 nm LP filter (acceptor)). Competitive displacement data were then graphed using GraphPad Prism 9 software using a normalized 3-parameter curve fit with the following equation: Y=100/(1+10^(X-LogIC50)).
Compound Docking
Computer-aided compound docking was performed with Glide, Schrödinger, LLC, New York, NY, 2021 release 2021–3 and Maestro 12.8.117. The receptor grid was generated for EGFR(T790M/V948R) kinase domain from Chain A of PDB ID 6WXN and ligands prepared with LigPrep. The best binding poses were ranked on the basis of lowest docking and glide score values.31–33
Safety Statement
No unexpected or unusually high safety hazards were encountered.
Supplementary Material
Acknowledgment
We thank Kristine Schmidt for the language proof-reading.
Funding Sources
This work was supported by the National Institutes of Health grants R01CA201049, R01CA116020, and R35CA242461 (to M.J.E), R35CA220497 (to P.A.J.), and postdoctoral fellowship F32CA247198 (to T.S.B). S.L. and iFIT are funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy - EXC 2180 - 390900677. TüCAD2 is funded by the Federal Ministry of Education and Research (BMBF) and the Baden-Württemberg Ministry of Science as part of the Excellence Strategy of the German Federal and State Governments. This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). The Eiger 16M detector on 24-ID-E is funded by a NIH-ORIP HEI grant (S10OD021527). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. SK, L.M.B, NB, and B-T. B are grateful for support by the SGC, a registered charity (no: 1097737) that receives funds from AbbVie, Bayer AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genentech, Genome Canada through Ontario Genomics Institute [OGI-196], EU/EFPIA/OICR/McGill/KTH/Diamond, Innovative Medicines Initiative 2 Joint Undertaking [EUbOPEN grant 875510], Janssen, Merck KGaA, Merck & Co, Pfizer, São Paulo Research Foundation-FAPESP, Takeda and Wellcome. S.K. is also grateful for funding by the German translational cancer network (DKTK) the Frankfurt Cancer Centre (FCI).
Abbreviations
- DMDS
Dimethyl disulfide
- EAI
EGFR allosteric inhibitors
- EGFR
Epidermal growth factor receptor
- H-Bond
Hydrogen bond
- NBS
N-Bromosuccinimide
- NSCLC
Non-small cell lung cancer
- SEM
2-(Trimethylsilyl)ethoxymethyl
- TBAF
Tetrabutylammonium fluoride
- TBDMS
tert-Butyldimethylsilyl
- TBTU
2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethylaminiumtetra fluoro borate
- TFA
Trifluoroacetic acid
- TKI
Tyrosine kinase inhibitor
Footnotes
Supporting Information
Associated Content
The supporting information is available free of charge on the ACS Publications Website.
X-ray crystallography statistics; supporting structural images; kinome selectivity screening; Biochemical potencies of 2c; MS-experiments; NanoBRET-experiments; synthetical preparation of 24 and the NanoBRET EGFR tracer; HPLC traces.
SMILES molecular formula strings.
Accession Codes
X-ray coordinates and structures factors have been deposited in the Protein Data Bank. PDB accession codes: EGFR(T790M/V948R): 6WA2 (2a), 6WAK (24), 6WXN (2c). Authors will release the atomic coordinates upon article publication
References
- (1).Russo A; Franchina T; Ricciardi GRR; Picone A; Ferraro G; Zanghì M; Toscano G; Giordano A; Adamo V A Decade of EGFR Inhibition in EGFR-Mutated Non Small Cell Lung Cancer (NSCLC): Old Successes and Future Perspectives. Oncotarget 2015, 6 (29), 26814–26825. DOI: 10.18632/oncotarget.4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Howlader N, Noone AM, Krapcho M, Miller D, Brest A, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, Feuer EJ, Cronin KA. SEER Cancer Statistics Review, 1975–2017, National Cancer Institute. Bethesda, MD. https://seer.cancer.gov/csr/1975_2017/. [Google Scholar]
- (3).Lynch TJ; Bell DW; Sordella R; Gurubhagavatula S; Okimoto RA; Brannigan BW; Harris PL; Haserlat SM; Supko JG; Haluska FG; Louis DN; Christiani DC; Settleman J; Haber DA Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non-Small-Cell Lung Cancer to Gefitinib. The New England journal of medicine 2004, 350 (21), 2129–2139. DOI: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- (4).Paez JG; Jänne PA; Lee JC; Tracy S; Greulich H; Gabriel S; Herman P; Kaye FJ; Lindeman N; Boggon TJ; Naoki K; Sasaki H; Fujii Y; Eck MJ; Sellers WR; Johnson BE; Meyerson M EGFR Mutations in Lung Cancer: Correlation with Clinical Response to Gefitinib Therapy. Science (New York, N.Y.) 2004, 304 (5676), 1497–1500. DOI: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- (5).Gazdar AF Activating and Resistance Mutations of EGFR in Non-Small-Cell Lung Cancer:Role in Clinical Response to EGFR Tyrosine Kinase Inhibitors. Oncogene 2009, 28 Suppl 1, S24–31. DOI: 10.1038/onc.2009.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Yun C-H; Mengwasser KE; Toms AV; Woo MS; Greulich H; Wong K-K; Meyerson M; Eck MJ The T790M Mutation in EGFR Kinase Causes Drug Resistance by Increasing the Affinity for ATP. Proceedings of the National Academy of Sciences of the United States of America 2008, 105 (6), 2070–2075. DOI: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kobayashi S; Boggon TJ; Dayaram T; Jänne PA; Kocher O; Meyerson M; Johnson BE; Eck MJ; Tenen DG; Halmos B EGFR Mutation and Resistance of Non-Small-Cell Lung Cancer to Gefitinib. The New England journal of medicine 2005, 352 (8), 786–792. DOI: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- (8).Pao W; Miller VA; Politi KA; Riely GJ; Somwar R; Zakowski MF; Kris MG; Varmus H Acquired Resistance of Lung Adenocarcinomas to Gefitinib or Erlotinib is Associated with a Second Mutation in the EGFR Kinase Domain. PLoS medicine 2005, 2 (3), e73. DOI: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Yu HA; Arcila ME; Rekhtman N; Sima CS; Zakowski MF; Pao W; Kris MG; Miller VA; Ladanyi M; Riely GJ Analysis of Tumor Specimens at the Time of Acquired Resistance to EGFR-TKI Therapy in 155 Patients with EGFR-Mutant Lung Cancers. Clinical cancer research: an official journal of the American Association for Cancer Research 2013, 19 (8), 2240–2247. DOI: 10.1158/1078-0432.CCR-12-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Kwak EL; Sordella R; Bell DW; Godin-Heymann N; Okimoto RA; Brannigan BW; Harris PL; Driscoll DR; Fidias P; Lynch TJ; Rabindran SK; McGinnis JP; Wissner A; Sharma SV; Isselbacher KJ; Settleman J; Haber DA Irreversible Inhibitors of the EGF Receptor May Circumvent Acquired Resistance to Gefitinib. Proceedings of the National Academy of Sciences of the United States of America 2005, 102 (21), 7665–7670. DOI: 10.1073/pnas.0502860102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Cross DAE; Ashton SE; Ghiorghiu S; Eberlein C; Nebhan CA; Spitzler PJ; Orme JP; Finlay MRV; Ward RA; Mellor MJ; Hughes G; Rahi A; Jacobs VN; Red Brewer M; Ichihara E; Sun J; Jin H; Ballard P; Al-Kadhimi K; Rowlinson R; Klinowska T; Richmond GHP; Cantarini M; Kim D-W; Ranson MR; Pao W AZD9291, an Irreversible EGFR TKI, Overcomes T790M-Mediated Resistance to EGFR Inhibitors in Lung Cancer. Cancer discovery 2014, 4 (9), 1046–1061. DOI: 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Soria J-C; Ohe Y; Vansteenkiste J; Reungwetwattana T; Chewaskulyong B; Lee KH; Dechaphunkul A; Imamura F; Nogami N; Kurata T; Okamoto I; Zhou C; Cho BC; Cheng Y; Cho EK; Voon PJ; Planchard D; Su W-C; Gray JE; Lee S-M; Hodge R; Marotti M; Rukazenkov Y; Ramalingam SS Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. The New England journal of medicine 2018, 378 (2), 113–125. DOI: 10.1056/NEJMoa1713137. [DOI] [PubMed] [Google Scholar]
- (13).Thress KS; Paweletz CP; Felip E; Cho BC; Stetson D; Dougherty B; Lai Z; Markovets A; Vivancos A; Kuang Y; Ercan D; Matthews SE; Cantarini M; Barrett JC; Jänne PA; Oxnard GR Acquired EGFR C797S Mutation Mediates Resistance to AZD9291 in Non-Small Cell Lung Cancer Harboring EGFR T790M. Nature medicine 2015, 21 (6), 560–562. DOI: 10.1038/nm.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Günther M; Juchum M; Kelter G; Fiebig H; Laufer S Lung Cancer: EGFR Inhibitors with Low Nanomolar Activity against a Therapy-Resistant L858R/T790M/C797S Mutant. Angewandte Chemie (International ed. in English) 2016, 55 (36), 10890–10894. DOI: 10.1002/anie.201603736. [DOI] [PubMed] [Google Scholar]
- (15).Günther M; Lategahn J; Juchum M; Döring E; Keul M; Engel J; Tumbrink HL; Rauh D; Laufer S Trisubstituted Pyridinylimidazoles as Potent Inhibitors of the Clinically Resistant L858R/T790M/C797S EGFR Mutant: Targeting of Both Hydrophobic Regions and the Phosphate Binding Site. J. Med. Chem 2017, 60 (13), 5613–5637. DOI: 10.1021/acs.jmedchem.7b00316. [DOI] [PubMed] [Google Scholar]
- (16).Juchum M; Günther M; Döring E; Sievers-Engler A; Lämmerhofer M; Laufer S Trisubstituted Imidazoles with a Rigidized Hinge Binding Motif Act as Single Digit nM Inhibitors of Clinically Relevant EGFR L858R/T790M and L858R/T790M/C797S Mutants: An Example of Target Hopping. Journal of medicinal chemistry 2017, 60 (11), 4636–4656. DOI: 10.1021/acs.jmedchem.7b00178. [DOI] [PubMed] [Google Scholar]
- (17).Heppner DE; Günther M; Wittlinger F; Laufer SA; Eck MJ Structural Basis for EGFR Mutant Inhibition by Trisubstituted Imidazole Inhibitors. J. Med. Chem 2020, 63 (8), 4293–4305. DOI: 10.1021/acs.jmedchem.0c00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Chen L; Fu W; Zheng L; Liu Z; Liang G Recent Progress of Small-Molecule Epidermal Growth Factor Receptor (EGFR) Inhibitors against C797S Resistance in Non-Small-Cell Lung Cancer. Journal of medicinal chemistry 2018, 61 (10), 4290–4300. DOI: 10.1021/acs.jmedchem.7b01310. [DOI] [PubMed] [Google Scholar]
- (19).Jia Y; Yun C-H; Park E; Ercan D; Manuia M; Juarez J; Xu C; Rhee K; Chen T; Zhang H; Palakurthi S; Jang J; Lelais G; DiDonato M; Bursulaya B; Michellys P-Y; Epple R; Marsilje TH; McNeill M; Lu W; Harris J; Bender S; Wong K-K; Jänne PA; Eck MJ Overcoming EGFR(T790M) and EGFR(C797S) Resistance with Mutant-Selective Allosteric Inhibitors. Nature 2016, 534 (7605), 129–132. DOI: 10.1038/nature17960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Clercq DJH de Heppner DE; To C; Jang J; Park E; Yun C-H; Mushajiang M; Shin BH; Gero TW; Scott DA; Jänne PA; Eck MJ; Gray NS Discovery and Optimization of Dibenzodiazepinones as Allosteric Mutant-Selective EGFR Inhibitors. ACS medicinal chemistry letters 2019, 10 (11), 1549–1553. DOI: 10.1021/acsmedchemlett.9b00381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).To C; Jang J; Chen T; Park E; Mushajiang M; De Clercq Dries J H; Xu M; Wang S; Cameron MD; Heppner DE; Shin BH; Gero TW; Yang A; Dahlberg SE; Wong K-K; Eck MJ; Gray NS; Jänne PA Single and Dual Targeting of Mutant EGFR with an Allosteric Inhibitor. Cancer discovery 2019, 9 (7), 926–943. DOI: 10.1158/2159-8290.CD-18-0903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Sämann C; Coya E; Knochel P Full Functionalization of the Imidazole Scaffold by Selective Metalation and Sulfoxide/Magnesium Exchange. Angewandte Chemie (International ed. in English) 2014, 53 (5), 1430–1434. DOI: 10.1002/anie.201309217. [DOI] [PubMed] [Google Scholar]
- (23).Feldman KS; Nuriye AY; Li J Extending Pummerer Reaction Chemistry: Studies in the Palau’amine Synthesis Area. The Journal of organic chemistry 2011, 76 (12), 5042–5060. DOI: 10.1021/jo200740b. [DOI] [PubMed] [Google Scholar]
- (24).Li Q; Zhang T; Li S; Tong L; Li J; Su Z; Feng F; Sun D; Tong Y; Wang X; Zhao Z; Zhu L; Ding J; Li H; Xie H; Xu Y Discovery of Potent and Noncovalent Reversible EGFR Kinase Inhibitors of EGFRL858R/T790M/C797S. ACS medicinal chemistry letters 2019, 10 (6), 869–873. DOI: 10.1021/acsmedchemlett.8b00564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Lategahn J; Hardick J; Grabe T; Niggenaber J; Jeyakumar K; Keul M; Tumbrink HL; Becker C; Hodson L; Kirschner T; Klövekorn P; Ketzer J; Baumann M; Terheyden S; Unger A; Weisner J; Müller MP; van Otterlo WAL; Bauer S; Rauh D Targeting Her2-insYVMA with Covalent Inhibitors-A Focused Compound Screening and Structure-Based Design Approach. Journal of medicinal chemistry 2020, 63 (20), 11725–11755. DOI: 10.1021/acs.jmedchem.0c00870. [DOI] [PubMed] [Google Scholar]
- (26).dos Santos JL; Lanaro C; Lima LM; Gambero S; Franco-Penteado CF; Alexandre-Moreira MS; Wade M; Yerigenahally S; Kutlar A; Meiler SE; Costa FF; Chung M Design, Synthesis, and Pharmacological Evaluation of Novel Hybrid Compounds to Treat Sickle Cell Disease Symptoms. J. Med. Chem 2011, 54 (16), 5811–5819. DOI: 10.1021/jm200531f. [DOI] [PubMed] [Google Scholar]
- (27).Yun C-H; Boggon TJ; Li Y; Woo MS; Greulich H; Meyerson M; Eck MJ Structures of Lung Cancer-Derived EGFR Mutants and Inhibitor Complexes: Mechanism of Activation and Insights into Differential Inhibitor Sensitivity. Cancer cell 2007, 11 (3), 217–227. DOI: 10.1016/j.ccr.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Zhou W; Ercan D; Chen L; Yun C-H; Li D; Capelletti M; Cortot AB; Chirieac L; Iacob RE; Padera R; Engen JR; Wong K-K; Eck MJ; Gray NS; Jänne PA Novel Mutant-Selective EGFR Kinase Inhibitors Against EGFR T790 M. Nature 2009, 462 (7276), 1070–1074. DOI: 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Robers MB; Dart ML; Woodroofe CC; Zimprich CA; Kirkland TA; Machleidt T; Kupcho KR; Levin S; Hartnett JR; Zimmerman K; Niles AL; Ohana RF; Daniels DL; Slater M; Wood MG; Cong M; Cheng Y-Q; Wood KV Target Engagement and Drug Residence Time Can be Observed in Living Cells with BRET. Nature communications 2015, 6, 10091. DOI: 10.1038/ncomms10091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Vasta JD; Corona CR; Wilkinson J; Zimprich CA; Hartnett JR; Ingold MR; Zimmerman K; Machleidt T; Kirkland TA; Huwiler KG; Ohana RF; Slater M; Otto P; Cong M; Wells CI; Berger B-T; Hanke T; Glas C; Ding K; Drewry DH; Huber KVM; Willson TM; Knapp S; Müller S; Meisenheimer PL; Fan F; Wood KV; Robers MB Quantitative, Wide-Spectrum Kinase Profiling in Live Cells for Assessing the Effect of Cellular ATP on Target Engagement. Cell chemical biology 2018, 25 (2), 206–214.e11. DOI: 10.1016/j.chembiol.2017.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Friesner RA; Banks JL; Murphy RB; Halgren TA; Klicic JJ; Mainz DT; Repasky MP; Knoll EH; Shelley M; Perry JK; Shaw DE; Francis P; Shenkin PS Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem 2004, 47 (7), 1739–1749. DOI: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- (32).Halgren TA; Murphy RB; Friesner RA; Beard HS; Frye LL; Pollard WT; Banks JL Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem 2004, 47 (7), 1750–1759. DOI: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- (33).Friesner RA; Murphy RB; Repasky MP; Frye LL; Greenwood JR; Halgren TA; Sanschagrin PC; Mainz DT Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem 2006, 49 (21), 6177–6196. DOI: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.