

Abstract
tRNA molecules are post‐transcriptionally modified by tRNA modification enzymes. Although composed of different chemistries, more than 40 types of human tRNA modifications play pivotal roles in protein synthesis by regulating tRNA structure and stability as well as decoding genetic information on mRNA. Many tRNA modifications are conserved among all three kingdoms of life, and aberrations in various human tRNA modification enzymes cause life‐threatening diseases. Here, we describe the class of diseases and disorders caused by aberrations in tRNA modifications as ‘tRNA modopathies’. Aberrations in over 50 tRNA modification enzymes are associated with tRNA modopathies, which most frequently manifest as dysfunctions of the brain and/or kidney, mitochondrial diseases, and cancer. However, the molecular mechanisms that link aberrant tRNA modifications to human diseases are largely unknown. In this review, we provide a comprehensive compilation of human tRNA modification functions, tRNA modification enzyme genes, and tRNA modopathies, and we summarize the elucidated pathogenic mechanisms underlying several tRNA modopathies. We will also discuss important questions that need to be addressed in order to understand the molecular pathogenesis of tRNA modopathies.
Keywords: RNA, RNA modification, translation, tRNA, tRNA modopathy
Human tRNAs are decorated with over 40 chemical modifications (tRNA modifications), which are synthesized by more than 70 identified enzymes. tRNA modifications play pivotal roles in protein translation, and aberration of over 50 modification enzymes causes diseases, collectively termed here as ‘tRNA modopathies’. This review provides comprehensive lists of human tRNA modifications, enzymes, and tRNA modopathies, and discusses important questions that need to be addressed to understand molecular pathogenesis.
Abbreviations
- ADAT1
tRNA‐specific adenosine deaminase 1
- CDK5RAP1
CDK5 regulatory subunit‐associated protein 1
- CDKAL1
CDK5 regulatory subunit‐associated protein 1‐like 1
- CTU1
cytoplasmic tRNA 2‐thiolation protein 1
- DUS2L
dihydrouridine synthase 2‐like
- eIF2α
eukaryotic initiation factor 2 subunit alpha
- ELP1
elongator complex protein 1
- FTSJ1
ftsJ homolog 1
- GTPBP3
GTP‐binding protein 3
- HIF1α
hypoxia‐inducible factor 1‐alpha
- METTL1
methyltransferase‐like 1
- MTO1
mitochondrial translation optimization protein 1 homolog
- NAD
nicotinamide adenine dinucleotide
- NSUN2
NOP1/NOP2/Sun domain family member 2
- PUS1
pseudouridine synthase 1
- QTRT1
queuine tRNA‐ribosyltransferase 1
- SNP
single nucleotide polymorphism
- THG1L
tRNA‐histidine guanylyltransferase 1‐like
- TRIT1
tRNA isopentenytransferase 1
- TRMT1
tRNA methyltransferase 1
- WDR4
WD repeat‐containing protein 4. The conventional symbols of modified nucleosides can be found at RNA Modification Database (https://mods.rna.albany.edu)
Introduction and definition of ‘tRNA modopathy’
The precise and efficient translation of genetic information into proteins is essential for life. tRNA molecules function as adaptor molecules that translate transcribed genetic information in the form of mRNA into 20 amino acids that form proteins [1, 2]. Protein synthesis occurs in the cytoplasm using hundreds of human cytoplasmic tRNA species, which are transcribed from more than 400 tRNA genes encoded in the nuclear chromosomes [3]. Protein synthesis also takes place within mitochondria, where 13 oxidative phosphorylation (OXPHOS) complex proteins are translated using 22 tRNAs transcribed from mitochondrial DNA [4].
tRNA molecules are composed of a highly conserved cloverleaf secondary structure, which consists of an acceptor stem, dihydrouridine (D) loop, D arm, anticodon loop, anticodon arm, variable loop, TΨC (T) loop, and T arm (Fig. 1A,B). tRNA molecules form an L‐shaped tertiary structure via multiple hydrogen bonds between loops and helices. In addition to the characteristic L‐shape, another structural feature of tRNAs is their chemically modified nucleosides (Fig. 1C). tRNA modifications are post‐transcriptionally added to tRNA by specific modifying enzymes. By counting one chemical structure as one modification (e.g., pseudouridines at various positions incorporated by different enzymes are counted as one modification), we counted 43 types of known stable tRNA modifications that exist in humans. These modifications are incorporated by at least 73 human enzymes and partner proteins (including confirmed proteins and widely accepted candidates). Due to their importance in protein synthesis, the dysfunction and aberrant expression of more than 50 tRNA modification enzymes are known to be associated with human diseases. The diseases caused by aberrations in RNA modification were collectively named ‘RNA modopathies’ by the Tsutomu Suzuki group and our group [5]. Although several RNA modopathies occur due to aberrations in mRNA or rRNA modifications, in this review, we will focus on ‘tRNA modopathies’, which are the diseases and disorders caused by aberrations in tRNA modifications.
Fig. 1.
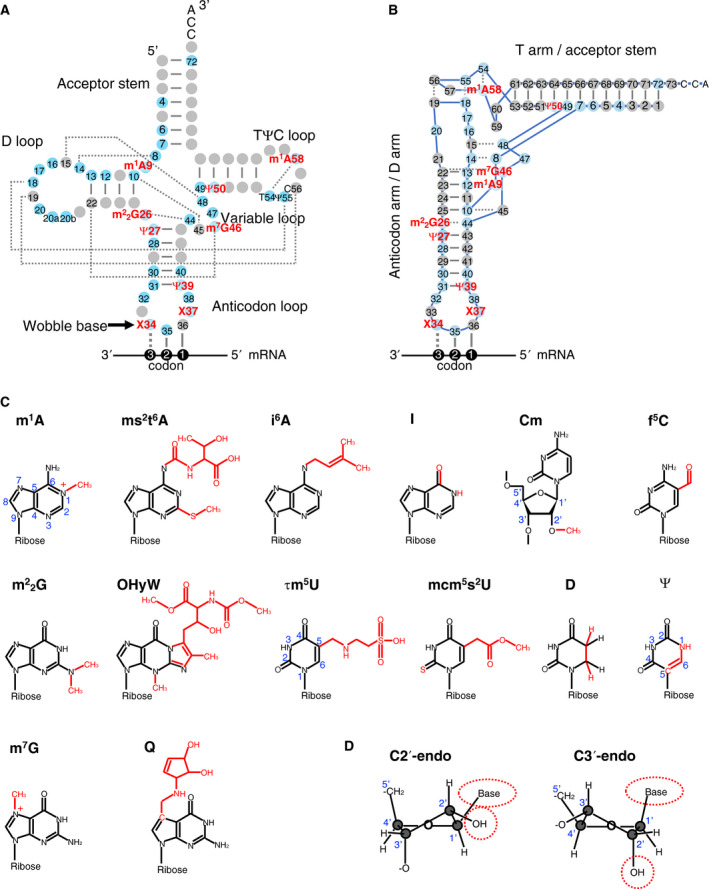
tRNA structure and tRNA modifications. (A) tRNA secondary structure depicted in a cloverleaf form. Nucleoside positions are numbered following conventional guidelines [223]. Red‐lettered tRNA modifications affect tRNA structure in at least some tRNA species. Gray circle, unmodified nucleoside; blue circle, nucleoside known to be modified in at least one tRNA species; straight line between bases, Watson‐Crick base pairs; dotted line between bases, hydrogen bond observed in yeast tRNAPhe tertiary structure [21]. (B) tRNA secondary structure depicted in the L‐shape, based on the yeast tRNAPhe crystal structure [21, 22]. Note that in the actual tertiary structure, a base‐paired stem forms a helix. (C) Chemical structures of various tRNA modifications. (D) Ribose ring C2′‐endo conformation and C3′‐endo conformation. Note that in the C2′‐endo form, the base and the 2′hydroxyl group are in close proximity, and Nm or xm5s2U modifications induce steric repulsion between the base and 2′hydroxyl group to favor the C3′‐endo form. 1‐methyladenosine (m1A), ms2t6A (2‐methylthio‐N6‐threonyl carbamoyladenosine), i6A (N6‐isopentenyladenosine), I (inosine), Cm (2'‐O‐methylcytidine), f5C (5‐formylcytidine), m2 2G (N2,N2‐dimethylguanosine), OHyW (hydroxywybutosine), τm5U (5‐taurinomethyluridine), mcm5s2U (5‐methoxycarbonylmethyl‐2‐thiouridine), D (dihydrouridine), Ψ (pseudouridine), m7G (7‐methylguanosine), Q (queuosine), and X (various modifications).
In this review, we provide a comprehensive compilation of human tRNA modification functions, tRNA modification enzymes, and tRNA modopathies and discuss the important questions that need to be addressed to elucidate the pathogenic molecular mechanisms underlying tRNA modopathies. To understand how tRNA modopathies are caused, it is essential to understand the chemical properties and molecular functions of tRNA modifications. The most important functions of tRNA modifications are tRNA stability regulation and codon recognition. Therefore, we will start by considering these two functions. In this review, we introduce insights that were derived mostly from the study of mammalian cells and animals. Many tRNA modifications, however, are conserved or sometimes functionally converged across the three domains of life. Please refer to other excellent reviews for general information on the codon table [6], anticodon modifications [7], modification‐mediated tRNA stabilization [8], methylation [9], pseudouridine [10], and modification pathways [11].
Function of tRNA modifications: regulation of the physical and biochemical stability of tRNA
tRNA modifications regulate the stability of the tRNA structure in three ways: (a) stabilization of overall tRNA structure, (b) regulation of tRNA local structure, and (c) inhibition of RNase‐mediated tRNA degradation.
Stabilization of overall tRNA structure
The overall tRNA structure can be compromised upon a loss of human tRNA modification, such as 1‐methyladenosine (m1A) at position 9 (m1A9) and N2,N2‐dimethylguanosine (m2 2G) at position 26 (m2 2G26). m1A and m2 2G possess methyl groups in their Watson‐Crick faces (Fig. 1C). Thus, m1A9 and m2 2G26 prevent the Watson‐Crick base pairing of A–U and G–C, respectively. A classic example is human mitochondrial (mt) tRNALys. An unmodified in vitro transcript of tRNALys forms a rod‐like structure by making an aberrant A9‐U64 base pair. The introduction of m1A9, a single methyl group, leads to the disruption of the A9‐U64 pair and enables the formation of a functional tRNA structure [12]. The correct tRNA structure is further stabilized by m2G10, although m2G10 alone cannot correct the tRNA structure [13]. The TRMT10C and HSD17B10 proteins cooperatively incorporate mt tRNA m1A9 [14], and a mutation in either protein can result in mitochondrial dysfunction‐associated disease that sometimes results in infantile death [15, 16]. Many cytoplasmic tRNAs have m1G and not m1A at position 9. As the methyl group of m1G also disrupts the G‐C base pair, m1G9 may also be involved in maintaining the tRNA structure, and experimental studies of this possibility are required.
Another example of a tRNA modification that stabilizes the overall tRNA structure is m2 2G26 of the human cytoplasmic tRNAAsn. Without m2 2G26, an aberrant G26‐C11 base pair forms in tRNAAsn and disrupts the tRNA structure. m2 2G26 prevents the formation of that abnormal base pair and instead forms a hydrogen bond with A44 to stabilize the tRNA structure (Fig. 1A,B) [17]. m2 2G26 is incorporated by tRNA methyltransferase 1 (TRMT1) [18, 19], and mutations in TRMT1 result in microcephaly and intellectual disability [20].
A network of hydrogen bonds collectively contributes to maintaining the L‐shaped tRNA tertiary structure (dotted lines in Fig. 1A,B). In the model case of yeast tRNAPhe, such tertiary interactions include G18‐Ψ55, G19‐C56, and T(m5U)54‐m1A58 interactions. In addition, the tRNA structure is stabilized by base triplets, in which a canonical Watson‐Crick base pair further interacts with a third base using the space in the major groove of the helix. Base triplets form between bases 25–10–45, 9–12–23, and 13–22–46 (Fig. 1B) [21]. Among these interactions, tRNA modifications of m1A58 and m7G46 contribute by increasing the binding energies of T54‐m1A58 and 13–22–m7G46 interactions [22].
Regulation of tRNA local structure
In addition to the stabilization of the overall tRNA structure, tRNA modifications regulate the local tRNA structure in two main ways: (a) strengthening/weakening the rigidity of the RNA helical structure and (b) shaping the ‘U‐turn’ anticodon loop structure.
Four types of tRNA modifications are known to affect RNA helix structural rigidity: (a) 2′‐O‐methylation (Nm, N = any bases), (b) 2‐thiolation of xm5s2U derivatives [5‐methoxycarbonylmethyl‐2‐thiouridine (mcm5s2U), or τm5s2U in humans], (c) pseudouridine, and (d) dihydrouridine. The ribose ring can form two conformations, namely the C2′‐endo conformation and C3′‐endo conformation (Fig. 1D) [23]. When the ribose is in the C2′‐endo form, the base and the 2′‐hydroxyl group are in proximity (Fig. 1D). In the presence of Nm or 2‐thiolation of xm5s2U derivatives, the C3′‐endo form is predominant due to the increased steric repulsion between the even enlarged 2′ hydroxyl moiety and the base [23, 24].
Pseudouridine (Ψ) is a C–C glycosidic isomer of uridine (Fig. 1C). This isomerization exposes the N1 hydrogen, which can bridge with the phosphate backbone via a water molecule. Pseudouridylation improves base stacking in a helical environment, favoring the ribose C3′‐endo conformation [25, 26]. These Ψ‐mediated structural stabilizations are the likely cause of the tRNA thermal stabilization observed in the presence of Ψ27, Ψ39, or Ψ50 [27, 28, 29]. Together, Nm, the 2‐thiolation of xm5s2U derivatives, and Ψ can stabilize the RNA helical structure in the tRNA arm or codon–anticodon minihelix. Many of these modifications are essential for health. For instance, mutations in the genes of the enzymes responsible for 2‐thiolation of xm5s2U derivatives, Gm34, or Ψ39 result in microcephaly and/or intellectual disability and/or nephropathy [30, 31, 32].
In contrast to Nm, 2‐thiolation, and Ψ that stabilize the RNA helical structure, dihydrouridine (D) destabilizes the helical structure. Indeed, D is observed only within loops, namely at D loop positions 16, 17, 20, 20a, and variable loop position 47 (Fig. 2). D is formed by the addition of two hydrogens to the C5=C6 bond (Fig. 1C), which break the planar structure of the uridine base, resulting in a predominance of the ribose C2′‐endo conformation over the C3′‐endo conformation [33].
Fig. 2.
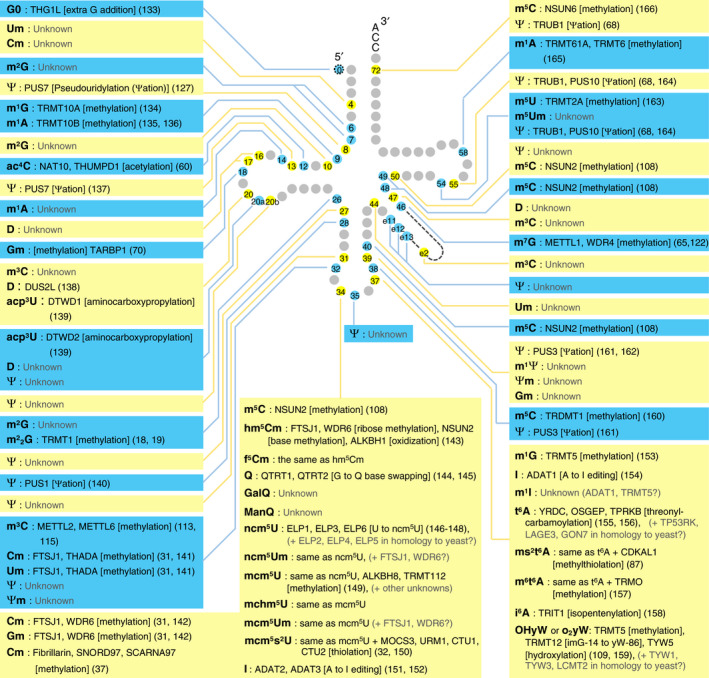
Mammalian cytoplasmic tRNA modifications and modification enzymes. The name of the modification enzyme, the reaction the enzyme is responsible for (in brackets), and the reference (in parentheses) is written next to the species of tRNA modification. Insights are derived mostly from human studies and in part from other mammalian species studies. Note that the strength of evidence varies between different studies, ranging from checking only that the protein is necessary for modification to completely confirming that the protein is both necessary and sufficient for the modification. For the structures of modifications not depicted in Fig. 1, please refer to the RNA Modification Database (https://mods.rna.albany.edu). Abbreviations not described in Fig. 1: G0 (Guanosine added post‐transcriptionally), Um (2′‐O‐methyluridine), m2G (N2‐methylguanosine), m1G (1‐methylguanosine), ac4C (N4‐acetylcytidine), Gm (2′‐O‐methylguanosine), m3C (3‐methylcytidine), acp3U (3‐(3‐amino‐3‐carboxypropyl)uridine), Ψm (2′‐O‐methylpseudouridine), m5C (5‐methylcytidine), hm5Cm (5‐hydroxymethyl‐2′‐O‐methylcytidine), f5Cm (5‐formyl‐2′‐O‐methylcytidine), GalQ (galactosyl‐queuosine), ManQ (mannosyl‐queuosine), ncm5U (5‐carbamoylmethyluridine), mcm5U (5‐methoxycarbonylmethyluridine), mchm5U (5‐(carboxyhydroxymethyl)uridine methyl ester), mcm5Um (5‐methoxycarbonylmethyl‐2′‐O‐methyluridine), m1I (1‐methylinosine), m6t6A (N6‐methyl‐N6‐threonylcarbamoyladenosine), o2yW (peroxywybutosine), m1Ψ (1‐methylpseudouridine), m5U (5‐methyluridine), and m5Um (5,2′‐O‐dimethyluridine).
Another important structural role of tRNA modifications is the shaping of a defined 7‐nt anticodon loop structure, called the ‘U‐turn’. Different tRNAs have different anticodon loop sequences. However, every anticodon loop entering the ribosomal A‐site must have a similar conformation to allow for efficient protein synthesis. This is accomplished by making the tRNA anticodon loops of all tRNAs adopt a highly similar U‐turn structure with the help of tRNA modifications, especially at positions 34 and 37, which are heavily modified. Position 34 has complex modifications, such as mcm5s2U, 5‐taurinomethyluridine (τm5U), or Q, and position 37 also often possesses complex modifications, including i6A, t6A, 2‐methylthio‐N6‐threonyl carbamoyladenosine (ms2t6A), and hydroxywybutosine (OHyW; Figs 1, 2, 3). tRNA modifications at positions 34 and 37, such as mcm5U, mcm5s2U, Q, N6‐isopentenyladenosine (i6A), t6A, and m1G, increase the stacking interactions of bases and restrict movement of the anticodon loop [34]. In addition, many modifications in the anticodon loop prevent unwanted intraloop base pairing that would disrupt the U‐turn structure [35]. Another very important function of tRNA modification at positions 34 and 37 is to enable precise and efficient decoding, which will be discussed in a later section.
Fig. 3.
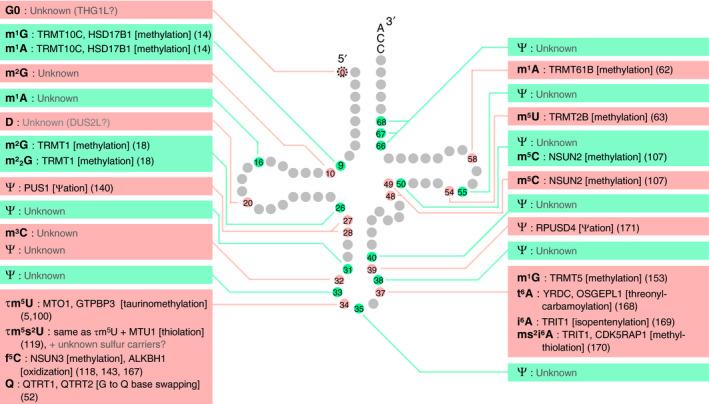
Human mitochondrial (mt) tRNA modifications and modification enzymes. The name of the modification enzyme, the reaction the enzyme is responsible for (in brackets), and the reference (in parentheses) is written next to the species of tRNA modification [52]. Note that the strength of evidence varies between different studies, ranging from checking only that the protein is necessary for modification to fully confirming that the protein is both necessary and sufficient for the modification. For the structures of modifications not depicted in Fig. 1C, please refer to the RNA Modification Database (https://mods.rna.albany.edu). The secondary structures of many mt tRNAs are different from the canonical cloverleaf structure in three ways [13, 46, 224, 225, 226, 227]: (a) mt tRNASer(AGY) lacks the entire D loop, (b) mt tRNASer(UCN) lacks U8 and has a small D loop, a small variable loop, and an extended anticodon stem, and (c) several mt tRNAs do not have canonical D loop/T loop interactions and instead have alternative interactions. Abbreviations not described in Figs. 1 and 2: τm5s2U (5‐taurinomethyl‐2‐thiouridine) and ms2i6A (2‐methylthio‐N6‐isopentenyladenosine).
Inhibition of RNase‐mediated degradation
In response to stress‐inducing stimuli, human cytoplasmic tRNAs are frequently cleaved within the anticodon loop by angiogenin, an endoribonuclease belonging to the RNaseA family [36]. Several anticodon tRNA modifications, namely Cm34 and Q34, are known to prevent angiogenin‐mediated cleavage [37, 38]. Cm34 in tRNAeMet is incorporated by the Fibrillarin/snoRNA machinery and inhibits angiogenin‐mediated cleavage [37]. This protection is provided presumably because 2′‐O‐methylation precludes the deprotonation of the ribose 2′ OH, which is a process needed for RNases such as angiogenin to cut the phosphate backbone. The mechanism of how Q34 prevents angiogenin‐mediated cleavage remains unelucidated. In addition, NOP1/NOP2/Sun domain family member 2 (NSUN2)‐mediated m5C formation inhibits angiogenin‐mediated tRNA cleavage. This protection from angiogenin is likely due to reduced angiogenin‐binding affinity in the presence of m5C [39], although it is not yet clear which of the m5C modifications at positions 34, 48, 49, and 50 inhibits angiogenin. Mutations in the NSUN2 gene cause microcephaly, intellectual disability, and growth retardation [40, 41]; m5C‐mediated tRNA protection from angiogenin is important for health, as cellular stresses in the brain caused by m5C deficiency can be rescued by inhibiting angiogenin [39].
Compared to the endonucleolytic tRNA cleavage mechanism, the exonucleolytic tRNA decay mechanism is poorly characterized in humans. In yeast, when tRNAiMet lacks m1A58, the tRNA is subjected to 3′–5′ decay by the TRAMP complex [42], and tRNAVal AAC lacking both m7G46 and m5C (at positions 34, 40, 48, and/or 49) is subjected to 5′–3′ decay by the rapid tRNA degradation (RTD) pathway [43]. In humans, although such exonucleolytic pathways have not been formally characterized, the existence of similar pathways has been suggested [44]. The molecular characterization of human exonuclease‐mediated decay pathways for hypomodified tRNA is awaited.
Function of tRNA modifications: decoding
The 20 universal amino acids are encoded by 61 codons (43 = 64 codons, minus three stop codons). Most of these codons are organized in codon family boxes, in which synonymous codons code for the same amino acid. In the decoding process, the codon triplet (codon positions 1, 2, and 3) base pairs with the three anticodon bases of the tRNA in positions 36, 35, and 34, respectively (Fig. 1A). Codon positions 1 and 2 base pair with tRNA positions 36 and 35 in normal Watson‐Crick base pairs (A–U, G–C). In contrast, the formation of a nonstandard base pair between the 3rd base of the codon and tRNA position 34 (the so‐called wobble base pair) is permitted [45]. Consequently, one tRNA molecule can often decode several synonymous codons. For example, human mitochondrial tRNA with an unmodified U at position 34 (U34) decodes four synonymous codons in a four‐codon box (e.g., one tRNAGly decodes GGA, GGU, GGG, and GGC codons to incorporate glycine) [46].
The modification at tRNA position 34 ensures restricted or, sometimes, relaxed codon recognition by the tRNA anticodon [11, 47]. An xm5s2U modification at position 34, such as human mcm5s2U (Fig. 1C) or τm5s2U, largely fixes its ribose in the C3′‐endo form and leads to preferential base pairing with A‐ or G‐ending codons and not to U‐ or C‐ending codons [46, 48, 49]. Queuosine (Q, Fig. 1C) and its sugar‐added derivatives (ManQ, GalQ) are present at position 34 of cytoplasmic tRNATyr, tRNAHis, tRNAAsn, and tRNAAsp; these base pair with U‐ or C‐ending codons and not A‐ or G‐ending codons. Q prevents frameshifting and promotes efficient translation of these codons, although the precise mechanism is unknown [50, 51]. Q34 is also present in mitochondrial tRNAs and likely promotes translation of tyrosine in mitochondria [52]. Inosine (I, Fig. 1C) is synthesized by the post‐transcriptional deamination of adenosine (A), and I at position 34 (I34) expands tRNA decoding capacity. I34 facilitates tRNA base pairing not only with U‐ending codons but also with C‐ and A‐ending codons [45]. Similarly, 5‐formylcytidine (f5C, Fig. 1C) and 5‐hydroxymethylcytidine (hm5C) also expand tRNA decoding capacity. Although an unmodified C34 can only decode G‐ending codons, f5C or hm5C can decode both A‐ and G‐ending codons [53].
tRNA position 37, located at the 3′ side of the anticodon, often possesses a bulky modification, such as OHyW or ms2t6A (Figs 1, 2, 3). These position 37 modifications play a critical role in the stabilization of codon–anticodon pairing and maintain the reading frame by increasing base‐stacking interactions and/or preventing unwanted base pairing within the anticodon loop [34, 50, 54, 55, 56]. Aberration of tRNA modifications in anticodon positions 34 and 37 induces various tRNA modopathies, including brain disorders, mitochondrial diseases, diabetes, and cancer (Table 1), which will each be discussed in detail in later sections.
Table 1.
tRNA modopathies. The tRNA modopathy genes are ordered by the nucleoside position of the tRNA modification that the gene product incorporates. Note that the strength of evidence varies between different studies, ranging from simply correlation studies to thorough investigation studies using both human patient cells and mouse disease models. The severity of disorders may vary between patients with mutations in the same gene. In the ‘Modification’ column, an ‘m’ is added to the position number of mitochondrial tRNA modifications. tRNA modifications for which modifying enzymes have not been formally investigated in humans but are commonly predicted by researchers in the field are indicated with a question mark. For cancers in the ‘tRNA modopathy’ column, a note is written in parenthesis to indicate the status of the modification enzyme in the cancer tissue. In the ‘B’, ‘K’, ‘S’, ‘M’, and ‘C’ columns, these letters indicate the occurrence of most frequently occurring tRNA modopathies or symptoms in the forms of brain‐related disorders (B), kidney‐related diseases (K), short stature (S), mitochondrial diseases (M), and cancer (C). Abbreviations: autism spectrum disorder (ASD), intellectual disability (ID), amyotrophic lateral sclerosis (ALS).
| Gene | Modification (position) | Enzymatic activity | tRNA modopathy | B | K | S | M | C | Ref |
|---|---|---|---|---|---|---|---|---|---|
| THG1L | G (0) | Extra G addition | Microcephaly, cerebellar ataxia, ID, nephropathy, short stature | B | K | S | [172, 173, 174] | ||
| PUS7 | Ψ (8, 13) | Pseudouridylation | Microcephaly, ID, ASD, aggressive behavior, short stature | B | S | [137, 175, 176] | |||
| TRMT10A | m1G (9) | Methylation | Microcephaly, ID, diabetes, short stature | B | S | [134] | |||
| TRMT10C | m1G, m1A (9m) | Methylation | Lactic acidosis, hypotonia, polymicrogyria, deafness, early death | B | M | [15] | |||
| HSD17B10 | m1G, m1A (9m) | Partner protein of TRMT10C | Neurodegeneration, cardiomyopathy, early death | B | M | [16] | |||
| NAT10 | ac4C (12) | Acetylation | Colon cancer (mislocalized), liver cancer (high expression) | C | [177, 178] | ||||
| THUMPD1 | ac4C (12) | Partner protein of NAT10 | Breast cancer (mislocalized, high expression) | C | [179] | ||||
| TARBP1 | Gm (18) | Methylation | Liver/skin cancers (high expression) | C | [180, 181] | ||||
| DUS2L | D (20) | Hydrogen addition to U | Lung cancer (high expression) | C | [182] | ||||
| TRMT1 |
m2 2G (26, 26m) m2G (26?, 26m) |
Methylation | Microcephaly, ID | B | [20] | ||||
| PUS1 | Ψ (27, 28, 30, 27m, 28m) | Pseudouridylation | Mitochondrial myopathy, sideroblastic anemia (MLASA) | M | [140, 183] | ||||
| METTL6 | m3C (32) | Methylation | Breast cancer (gene amplification) | C | [184] | ||||
| THADA | Nm (32) | Partner protein of FTSJ1 | Diabetes | [185] | |||||
| FTSJ1 | Nm (32, 34) | Methylation | ID | B | [31, 186] | ||||
| NSUN2 | m5C (34, 48, 49, 50, 48m, 49m, 50m) | Methylation | ID, Dubowitz‐like syndrome, short stature, breast cancer (high expression) | B | S | C | [40, 41, 187] | ||
| ADAT3 | I (34) | A to I editing | ID, strabismus | B | [188] | ||||
| QTRT1 | Q (34, 34m) | G to Q base swapping | Colon cancer (absence) | C | [78, 80] | ||||
| ELP1 | ncm5U (34) | U to ncm5U (as a component of catalytic ELPS complex) | Familial dysautonomia, male infertility, skin cancer (high expression) | C | [77, 148, 189] | ||||
| ELP2 | ncm5U? (34) | U to ncm5U modification | ID, ASD | B | [190, 191] | ||||
| ELP3 | ncm5U (34) | U to ncm5U modification | ALS, skin/breast cancers (high expression) | C | [77, 192, 193] | ||||
| ELP4 | ncm5U? (34) | U to ncm5U modification | ID, ASD | B | [194] | ||||
| ELP5 | ncm5U? (34) | U to ncm5U modification | Diabetes | [195] | |||||
| ALKBH8 | mcm5U (34), mchm5U (34) | cm5U to mcm5U, then to mchm5U modification | ID, bladder cancer (high expression) | B | C | [196, 197] | |||
| CTU1 | mcm5s2U (34) | 2‐thiolation (with CTU2) | Skin/breast cancers (high expression) | C | [77, 192] | ||||
| CTU2 | mcm5s2U (34) | 2‐thiolation (with CTU1) | Microcephaly, ID, nephropathy, ambiguous genitalia, short stature, skin/breast cancers (high expression), early death | B | K | S | C | [32, 77, 192] | |
| MTO1 | τm5U (34m) | Taurinomethylation (with GTPBP3) | Hypertrophic cardiomyopathy, lactic acidosis, ID, short stature, early death | B | S | M | [98, 198] | ||
| GTPBP3 | τm5U (34m) | Taurinomethylation | MELAS, ID, hearing loss, short stature, early death | B | S | M | [99] | ||
| MTU1 | τm5s2U (34m) | Thiolation | Hepatopathy, lactic acidosis, Leigh syndrome, hearing loss, early death | B | M | [199, 200, 201, 202, 203] | |||
| NSUN3 | f5C (34m) | Methylation (followed by oxidization by ALKBH1) | Microcephaly, seizure, lactic acidosis, muscle weakness, short stature, 5‐AZA‐resistant leukemia (high expression) | B | S | M | C | [117, 118, 204] | |
| ALKBH1 | hm5Cm (34), f5Cm (34), f5C (34m) | Oxidization (m5C to f5C, m5Cm to hm5Cm to f5Cm) | Gastric cancer (low expression) | C | [205] | ||||
| ADAT1 | I (37) | A to I editing | Coronary artery disease | [206] | |||||
| TRMT5 | m1G (37, 37m), OHyW? (37) | Methylation | Cardiomyopathy, lactic acidosis, demyelinating neuropathy, renal tubulopathy, cirrhosis, short stature | B | K | S | M | [207] | |
| TRMT12 | OHyW (37) | imG‐14 to yW‐86 | Colon cancer (low expression) | C | [54] | ||||
| TYW3 | OHyW? (37) | yW‐86 to yW‐72? | ALS | [208] | |||||
| LCMT2 | OHyW? (37) | OHyW‐72 to OHyW? | Colon cancer (frameshift) | C | [111] | ||||
| YRDC | t6A (37, 37m) | Threonylcarbamoylation of A | Microcephaly, nephropathy, short stature, liver cancer (high expression), early death | B | K | S | C | [155, 209] | |
| OSGEP | t6A (37) | Threonylcarbamoylation of A | Microcephaly, nephropathy, short stature, early death | B | K | S | [156] | ||
| TP53RK | t6A? (37) | Threonylcarbamoylation? | Microcephaly, nephropathy, short stature, early death | B | K | S | [156] | ||
| TPRKB | t6A (37) | Threonylcarbamoylation of A | Microcephaly, nephropathy, short stature, early death | B | K | S | [156] | ||
| LAGE3 | t6A? (37) | Threonylcarbamoylation? | Microcephaly, nephropathy, short stature, early death | B | K | S | [156] | ||
| GON7 | t6A? (37) | Threonylcarbamoylation? | Microcephaly, nephropathy | B | K | [155] | |||
| CDKAL1 | ms2t6A (37) | Methylthiolation of t6A | Diabetes | [104] | |||||
| TRIT1 | i6A (37, 37m) | Isopentenylation of A | Microcephaly, ID, cardiomyopathy, lung cancer (low expression), short stature | B | S | M | C | [81, 210] | |
| CDK5RAP1 | ms2i6A (37m) | Methylthiolation of i6A | Glioma (high ms2i6A) | C | [211] | ||||
| TRDMT1 | m5C (38) | Methylation | Gastric cancer (SNP association) | C | [212] | ||||
| PUS3 | Ψ (38, 39) | Pseudouridylation | Microcephaly, ID, nephropathy, short stature | B | K | S | [30, 162, 213] | ||
| TRMT44 | Um? (44?) | Methylation | Partial epilepsy with pericentral spikes | B | [214] | ||||
| METTL1 | m7G (46) | Methylation | Multiple sclerosis | [215] | |||||
| WDR4 | m7G (46) | Methylation | Microcephaly, ID, nephropathy, short stature | B | K | S | [216, 217] | ||
| TRMT2A | m5U (54) | Methylation | Breast cancer (high expression) | C | [218] | ||||
| PUS10 | Ψ (54, 55) | Pseudouridylation | Crohn's disease, celiac disease | [219] | |||||
| TRMT6 | m1A (58) | Partner protein of TRMT61A | Colon cancer (frameshift), liver cancer (high expression) | C | [111, 220] | ||||
| TRMT61A | m1A (58) | Methylation | Bladder cancer (high expression) | C | [221] | ||||
| TRMT61B | m1A (58m) | Methylation | Breast cancer (High expression) | C | [222] |
Other functions of tRNA modifications and modifying enzymes
In addition to tRNA stabilization and decoding, some tRNA modifications and tRNA modifying enzymes perform additional functions. Such functions should not be disregarded, as a disease mutation in a tRNA modification enzyme gene may disrupt these additional functions and drive pathogenesis.
First, a tRNA modification can serve as the recognition determinant for another tRNA modification enzyme. For example, Cm32 modification of cytoplasmic tRNAPhe promotes the formation of OHyW [31]. In yeast, Cm32 and m1G37 are required for Gm34 formation, and Gm34 is required for yW37 formation [57]. The same recognition mechanisms might also work in the human OHyW37 formation. In addition, the Q34 modification of cytoplasmic tRNAAsp promotes the efficient modification of m5C38 [51].
Second, a tRNA modification enzyme can function not only as a tRNA modification enzyme but also as a modification enzyme for different RNA species. For example, NAT10, a cytoplasmic tRNA acetyltransferase, also acetylates 18S rRNA and various mRNAs [58, 59, 60]. Mitochondrial (mt) tRNA methylases TRMT2B and TRMT61B also methylate mt 12S rRNA and mt 16S rRNA, respectively [61, 62, 63]. In addition, methyltransferase‐like 1 (METTL1), a cytoplasmic tRNA m7G46 methylase, also methylates the precursor of let‐7 microRNA [64, 65].
Third, a tRNA modification enzyme can sometimes perform two functions, and one function can be completely different from tRNA modification. A prominent example is TRMT10C, a mt tRNA m1A9/m1G9 methylase that also functions as an essential component of mt RNaseP, an endoribonuclease complex that cleaves the 5′ end of a tRNA from the precursor RNA [14, 66]. Additionally, TRUB1, a cytoplasmic tRNA pseudouridylase for Ψ54, Ψ55, and Ψ72, binds to the let‐7 microRNA precursor but does not modify it. Instead, TRUB1 promotes cleavage of the let‐7 microRNA precursor by enhancing the interaction between the let‐7 microRNA precursor and an endoribonuclease complex [67, 68]. In yeast, Trm2 functions not only as a tRNA m5U54 methylase but also as a tRNA chaperone [69]. Whether the human homologs of Trm2, namely, TRMT2A and TRMT2B, have similar tRNA chaperone activity is still unknown.
Fourth, a tRNA modification can affect immune responses. Transfection of human total tRNA deficient in Gm18 induces innate immune responses by stimulating Toll‐like receptors TLR7/8, whereas total tRNA of wild‐type cells does not stimulate immune responses [70]. In addition, whereas bacterial tRNA usually induces interferon‐α secretion from human peripheral blood mononuclear cells, bacterial tRNATyr is not immunostimulatory because bacterial tRNATyr has a Gm18 modification that functions as an antagonist of TLR7 [71, 72].
Fifth, tRNA modifications affect precursor tRNA splicing in some eukaryotes. In several precursor tRNAs, an intron in the anticodon stem‐loop is post‐transcriptionally removed by tRNA splicing. In Trypanosoma brucei, unusual RNA modifications, namely G–A editing and A–U editing, within the intron of pre‐tRNATyr GUA promote intron removal from pre‐tRNATyr GUA [73]. Conversely, tRNA splicing can affect tRNA modification. The intron in human pre‐tRNALeu CAA needs to be removed for NSUN2‐mediated m5C34 formation [74]. Thus, aberration of various tRNA‐related pathways should not be overlooked as a potential cause of tRNA modopathies.
tRNA modification enzyme genes
To understand how tRNA modopathies are caused by aberrations in tRNA modification, identification of modification enzymes is essential, as tRNA modopathies are often caused by mutations in tRNA modification enzyme genes. To the best of our knowledge, 43 different types of tRNA modifications are incorporated into human tRNA molecules by at least 73 enzymes and their partner proteins. Mammalian cytoplasmic tRNA modifications and their modification enzymes are shown in Fig. 2, and human mitochondrial tRNA modifications and their responsible enzymes are shown in Fig. 3.
tRNA modopathies
Mutations or expression changes in 54 tRNA modification enzymes and their partner proteins are known as the direct, or strong, candidate causes of various tRNA modopathies. In addition to a previous study that compiled human tRNA modifications and modopathies [75], we added many of the latest insights and provided more detailed information to the list of tRNA modopathies (Table 1). This number represents 72% of the 75 modification proteins (73 confirmed proteins or strong candidates, plus two weak candidates) and demonstrates the biological importance of tRNA modifications. The organ that is most frequently affected by tRNA modification deficiencies is the brain. Of the 54 tRNA modopathy‐associated proteins, dysfunction of 28 proteins can cause or are associated with brain disorders (Table 1). Relatively severe brain disorders, such as microcephaly, are usually associated with intellectual disability and often associated with kidney disorders and/or short stature (Table 1). Relatively moderate brain disorders, such as intellectual disability or autism spectrum disorder, often occur without other apparent symptoms.
Our compilation shows that aberrations of 24 tRNA modification enzymes cause or are associated with cancer (Table 1). Cancer is often associated with a high rate of tRNA modification or high expression of tRNA modification enzymes. For example, mcm5s2U34 is necessary for the efficient translation of the AAA, GAA, and CAA codons, and high mcm5s2U34 is required for melanoma cells to survive [76]. The hypoxia‐inducible factor 1‐alpha (HIF1α) protein, which is enriched with these codons, requires the mcm5s2U34 modification enzymes ELP3, cytoplasmic tRNA 2‐thiolation proteins 1, and 2 (CTU1 and CTU2) to be efficiently translated and to exert HIF1α‐dependent metabolic reprogramming in melanoma [77]. In contrast, in several cases, a lower modification rate, including modifications such as i6A, OHyW, or Q (Table 1), is associated with or sometimes directly promotes cancer formation [54, 78, 79, 80, 81]. The mechanism by which OHyW hypomodification causes colon cancer is described in a later section [54]. Compared to other tRNA modopathies in which mutations are usually inherited from parent(s), cancer‐causing aberrations usually occur after birth, making it more difficult to distinguish a cancer‐causing aberration from a mere cancer‐associated aberration. To formally show that the aberration of a tRNA modification enzyme gene (upregulation, downregulation, or mutation) causes cancer, it is necessary to show at least two things: (a) The aberration of the gene is associated with poor survival in cancer patients, and (b) the aberration of the gene in a cell line increases virulence (e.g., cell proliferation, metastasis, or drug resistance).
Mitochondrial aberrations cause dysfunction in high‐energy demand organs such as the brain and heart, and these diseases are collectively called ‘mitochondrial diseases’. Dysfunction of at least 9 mitochondrial tRNA modification enzymes causes mitochondrial diseases, comprising a major group of tRNA modopathies (Table 1) [82]. In addition to mutations in mitochondrial tRNA modification enzymes, numerous mt tRNA mutations result in mt tRNA modification deficiencies and mitochondrial diseases (MITOMAP, https://www.mitomap.org/MITOMAP).
Codon‐specific translational aberration in tRNA modopathies
In contrast to the diseases caused by mutations in general translation factors such as eukaryotic initiation factor 2 subunit alpha (eIF2α) and eukaryotic elongation factor 1 alpha‐2, which decrease the overall translation rate, one feature of tRNA modopathies is their codon‐specific pathogenic mechanisms. For example, the pathogenic mitochondrial (mt) DNA A3243G mutation (mt tRNALeu(UUR) mutation) specifically causes the hypomodification of the τm5U34 modification of mt tRNALeu(UUR), which specifically reduces the translation of the UUG codon, resulting in mitochondrial disease [83]. The lack of the mt tRNALeu(UUR) τm5U34 modification, and not the tRNA mutation itself, is responsible for translational deficiency [84]. Of the 13 proteins translated in mitochondria, the translation of ND6 mRNA is specifically and markedly reduced in A3243G mutant cells [85], likely due to the enrichment of the UUG codon in ND6 mRNAs [84].
Anticodon modifications at positions 34 and 37 directly regulate decoding, and mutations in the modification enzymes affect translation in a codon‐specific manner and cause various diseases, such as diabetes, neurodegenerative diseases, and mitochondrial diseases (Table 1). In yeast and nematodes, the loss of cytoplasmic tRNA mcm5s2U34 slowed translation specifically at the AAA, CAA, or GAA codons, inducing protein aggregation. The codon translation rates and protein homeostasis were restored in yeast by overexpressing mcm5s2U‐less tRNA, showing that the optimal codon translation rate is critical for maintaining proteome integrity [86]. In mammals, as described in a previous section, melanoma cells require cytoplasmic tRNA mcm5s2U34 for the efficient translation of the AAA, GAA, and CAA codons to enable efficient translation of NAA codon‐rich HIF1α and HIF1α‐dependent metabolic reprogramming [77].
As another example of codon‐specific translational aberration in tRNA modopathy, a loss of the tRNALys UUU‐specific ms2t6A37 modification decreased the translation of lysine codons, causing unfolded‐protein responses and inducing the onset of type 2 diabetes [87].
In yeast, several tRNA modifications are required for cell survival under stressed conditions [88]. For example, in response to H2O2 exposure, an increased m5C34 level of tRNALeu CAA, which decodes the UUG codon, is observed; among the 38 UUG‐enriched mRNAs in yeast, the m5C‐dependent translation of ribosomal protein Rpl22a was especially required for the cells to survive under stress [89]. In another case, certain DNA damage response genes in yeast are enriched with codons that are decoded by tRNAs containing mcm5U34 or mcm5s2U34; Trm9, an enzyme required for these modifications, is essential for yeast cells to survive through DNA damage [90]. If similar tRNA modification‐dependent stress‐response mechanisms are identified in mammals, it would expand our understanding of the role of tRNA modifications in health and disease.
Of the many tRNA modopathies, pathogenic mechanisms have been thoroughly elucidated in only a few. The next sections will describe the pathogenic mechanisms of four relatively well‐understood tRNA modopathies.
Μitochondrial (mt) diseases caused by a deficiency in mt tRNA taurine modification at position 34
The first identified tRNA modopathies were mitochondrial (mt) diseases caused by deficiencies in taurine modifications. In healthy individuals, two taurine‐containing modifications are present at position 34 in five mt tRNAs: τm5U (Fig. 1C) in tRNALeu(UUR) and tRNATrp and 5‐taurinomethyl‐2‐thiouridine (τm5s2U) in tRNAGlu, tRNALys, and tRNAGln [91]. These taurine modifications promote accurate mitochondrial translation of A‐ and G‐ending codons and prevent misreading of C‐ or U‐ending codons [46].
Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke‐like episodes (MELAS) and myoclonus epilepsy associated with ragged red fibers (MERRF) are severe mitochondrial diseases with various symptoms, including muscle weakness and epilepsy [92, 93]. A majority of MELAS patients carry an A3243G mutation in the mt tRNALeu(UUR ) gene on mtDNA [83], and many MERRF patients carry an A8344G mutation in the mt tRNALys gene on mtDNA [94]. mt tRNALeu(UUR) with that MELAS mutation and mt tRNALys with that MERRF mutation lack taurine modifications and show deficiencies in recognizing their cognate codons [95, 96, 97]. Moreover, the lack of a τm5U modification, and not the tRNA mutation itself, is responsible for disruption of translation [84]. MELAS‐ or MERRF‐associated pathogenic tRNA mutations are presumed to prevent tRNA recognition by taurine modification enzymes, but formal studies have not been conducted.
In addition to mutations in mt tRNA genes, various mutations in taurine modification enzyme genes, namely the MTO1 and GTPBP3 genes, are observed in mitochondrial disease patients [98, 99]. The mitochondrial translation optimization protein 1 homolog (MTO1)‐GTP‐binding protein 3 (GTPBP3) complex uses 5,10‐methylenetetrahydrofolate and taurine as metabolic substrates for τm5U formation [5]. Patients with MTO1 or GTPBP3 mutations show diverse symptoms starting in infancy or early childhood, including optic neuropathy and cognitive disability, with cardiomyopathy being the most frequent symptom. To understand the role of the taurine modification in vivo and to recapitulate the pathogenesis of mitochondrial disease, animal models have been generated [100] (Fig. 4A).
Fig. 4.
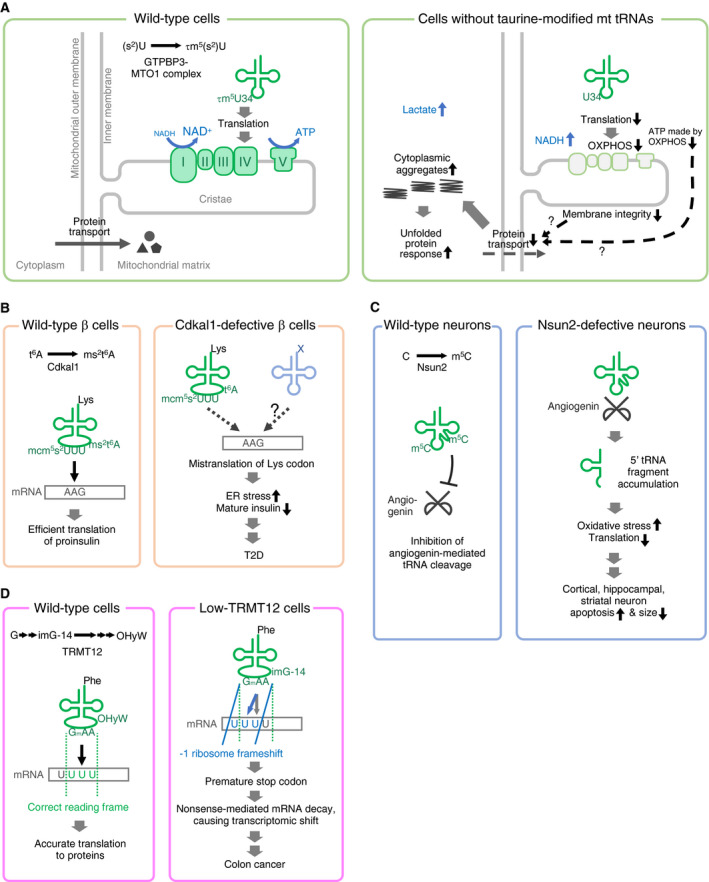
Pathogenic molecular mechanisms of tRNA modopathies. (A) Μitochondrial (mt) diseases caused by deficiencies of mt tRNA taurine modifications at position 34. The GTPBP3–MTO1 complex incorporates τm5U34 modification into five mt tRNAs. Without the taurine modification, the translation rate of OXPHOS complex proteins declines, causing a metabolic shift as well as a proteostasis shift, especially affecting energy‐demanding organs such as the brain and muscle. (B) Type 2 diabetes caused by a deficiency of CDKAL1‐mediated thiomethylation of cytoplasmic tRNALys UUU at position 37. Cdkal1 incorporates the ms2 modification to t6A37 of tRNALys UUU and promotes translation of lysine from the AAA and AAG codons. Cdkal1 is especially important in pancreatic β cells, in which lysine‐containing proinsulin is rapidly and massively translated upon glucose stimulus. (C) Neurodevelopmental disorder caused by a deficiency of NSUN2‐mediated m5C modifications. NSUN2 incorporates m5C into several sites within tRNAs and inhibits angiogenin‐mediated tRNA cleavage. NSUN2 deficiency induces the accumulation of 5′ tRFs, which evokes reduced translation rates and activated stress responses and is the cause of brain disorders, including microcephaly and intellectual disability. (D) Colon cancer caused by epigenetic loss of TRMT12‐mediated OHyW modification of tRNAPhe at position 37. Epigenetic silencing of TYW2 is a cause of colon cancer via the loss of the OHyW37 modification, inducing a −1 ribosome frameshift to downregulate various mRNAs, conferring enhanced migration properties and epithelial‐to‐mesenchymal features to the cells.
Whole‐body Mto1 knockout in mice was embryonic lethal at an early developmental stage (approximately E9.0). Mto1 KO embryonic stem cells showed a > 80% reduction in mitochondrial protein synthesis, poor assembly and activity of mitochondrial respiratory complexes, increased lactate levels, and increased NADH/NAD+ ratios. Interestingly, the total ATP level in KO cells was only slightly decreased compared with that in WT cells due to increased cytoplasmic ATP production by glycolysis, as evidenced by increased lactate levels and NADH/NAD+ ratios. Heart‐specific Mto1 knockout mice developed normally during the embryonic stage but could not survive more than 24 h after birth and showed elevated expression of the heart failure marker genes Anp and Bnp.
Mto1 knockout cells underwent not only metabolic changes but also protein homeostasis changes [100] (Fig. 4A). In healthy cells, more than 1000 nucleus‐encoded proteins are translated in the cytoplasm by cytoplasmic ribosomes and then efficiently transported into mitochondria from the cytoplasm [101]. In Mto1 knockout cells, the transport of mitochondria‐targeted proteins across the inner membrane was defective. The defective mitochondrial transport of proteins from the cytoplasm may be due to decreased mitochondrial inner membrane integrity and/or decreased mitochondrial ATP generation that may decrease ATP‐driven transmembrane protein transport by mitochondrial Hsp70 proteins; these possibilities, however, need to be investigated further. As a consequence of decreased mitochondrial transport from the cytoplasm, mitochondria‐targeted proteins formed cytoplasmic protein aggregates and induced a cytotoxic unfolded‐protein response.
Intriguingly, tauroursodeoxycholic acid (TUDCA), a chemical chaperone that improves protein folding and prevents protein aggregation, suppresses protein aggregation and moderately improves respiratory activity in both cell cultures and tissue‐specific Mto1 knockout mice [100]. The safety of TUDCA has been proven in humans, and the effect of TUDCA has been tested in clinical trials for diseases such as diabetes and amyloidosis [102, 103]. As a symptomatic therapy, future clinical assessments need to be performed in mitochondrial disease patients to investigate whether TUDCA can mitigate the symptoms of mitochondrial diseases.
Type 2 diabetes caused by a deficiency in CDKAL1‐mediated thiomethylation of cytoplasmic tRNALys UUU at position 37
Cdkal1 is a mammalian methylthiotransferase that synthesizes 2‐methylthio‐N6‐threonylacarbamoyladenosine (ms2t6A, Fig. 1C) at position 37 of cytoplasmic tRNALys UUU [87]. The ms2 modification of t6A37 stabilizes the interaction between tRNALys UUU and its cognate codon AAG as well as AAA and increases the translation rate of these codons (Fig. 4B) [87]. Whole‐genome association studies identified a number of genes associated with type 2 diabetes (T2D). Among these risk genes, CDKAL1 is one of the most common genes across different ethnicities [104]. Among the various tRNA modifications, T2D caused by CDKAL1 single nucleotide polymorphisms (SNPs) may affect the largest human population, as suggested by genome‐wide association studies. Various SNPs in the CDKAL1 gene influence the risk of T2D, and CDKAL1 SNPs are associated with decreased insulin secretion but not peripheral insulin sensitivity [105].
To understand the pathophysiology and pathogenesis of T2D, pancreatic β‐cell‐specific Cdkal1 KO mice were studied [87] (Fig. 4B). In β‐cell‐specific Cdkal1 KO mice, a deficiency in ms2t6A caused the mistranslation of proinsulin Lys codons, one of which is present at the proinsulin processing site, resulting in improper proinsulin processing. The mice showed pancreatic islet hypertrophy, decreased insulin secretion, and impaired blood glucose control. Mistranslation was associated with the endoplasmic reticulum (ER) stress response, and the mice were hypersensitive to high‐fat diet‐induced ER stress. Consistent with this model, human proinsulin conversion was decreased in homozygous carriers of CDKAL1 risk SNPs [106].
Neurodevelopmental disorder caused by a deficiency in Nsun2‐mediated m5C modification
Autosomal recessive mutations in the human NSUN2 gene were found to cause intellectual disability, microcephaly, behavioral deficits, speech delay, unusual facies, and growth retardation [40, 41]. NSUN2 is a 5‐methylcytidine (m5C) modification enzyme for m5C at cytoplasmic tRNA positions 34, 48, 49, 50, and at mitochondrial tRNA positions 48, 49, and 50 [107, 108]. Nsun2 knockout mouse models and human cells obtained from Dubowitz‐like syndrome individuals were studied as disease models in order to understand the pathophysiology and pathogenesis [39] (Fig. 4C). In human cells and mouse tissues without functional NSUN2, an accumulation of 5′ tRNA fragments (tRFs) was observed. In the Nsun2 KO mice, angiogenin‐mediated tRNA cleavage resulted in 5′ tRF accumulation. The accumulation of 5′ tRFs reduced protein synthesis rates and activated stress responses and was accompanied by increased apoptosis of cortical, hippocampal, and striatal neurons. Importantly, the increased sensitivity of Nsun2‐deficient brains to oxidative stress could be rescued by inhibiting angiogenin. To the best of our knowledge, this is the first case of detailed (although not complete) elucidation of the molecular pathogenesis of a brain tRNA modopathy. Further studies are needed to understand why the phenotypes of a whole‐body Nsun2 KO manifest mainly in the brain and not in other tissues.
Colon cancer caused by epigenetic loss of TRMT12 (TYW2)‐mediated OHyW modification of tRNAPhe at position 37
Human tRNAPhe contains a tRNAPhe‐specific, bulky tRNA modification at position 37, called OHyW (Fig. 1C), or an oxidized derivative, peroxywybutosine (o2yW). Similar to the well‐characterized yeast wybutosine synthesis pathway, human wybutosine derivatives are presumed to be synthesized by six enzymes, namely TRMT5, TYW1, TRMT12 (TYW2), TYW3, LCMT2 (TYW4), and TYW5 [109, 110]. A comprehensive analysis of the Cancer Genome Atlas revealed that the TRMT12 promoter CpG island was methylated in many primary colorectal carcinoma cases, and TRMT12 epigenetic inactivation was correlated with poor overall survival in patients with early‐stage colorectal cancer [54]. In human cell lines, the TRMT12 knockout induced the hypomodification of OHyW and increased −1 ribosome frameshifts at certain Phe codons. Those ribosome frameshifts created premature termination codons, resulting in transcript degradation via nonsense‐mediated mRNA decay (Fig. 4D). Increased nonsense‐mediated mRNA decay caused imbalances in the transcriptome, including in the mRNA levels of cell mobility‐related genes, conferring migration properties and epithelial‐to‐mesenchymal features to TRMT12‐deficient cells [54]. Interestingly, a frameshift mutation in the LCMT2 (TYW4) gene, which encodes another enzyme presumed to be required to synthesize OHyW, is also found in colon cancers [111]. Thus, loss of OHyW derivatives might generally be involved in the formation of a subset of colon cancers.
Important questions to be addressed in order to understand the molecular pathogenesis of tRNA modopathies
Other than the four tRNA modopathies described above, the pathogenic mechanisms of most tRNA modopathies are poorly understood, especially in diseases caused by aberrant cytoplasmic tRNA modifications. In the next sections, we will raise and discuss four questions and problems that need to be addressed to elucidate the pathogenic mechanisms of various tRNA modopathies.
Mapping all human tRNA modifications
Due to accumulating knowledge regarding tRNA modification enzymes, we know which specific tRNA modification enzyme modifies specific positions of representative tRNA species. However, we do not have a complete understanding of which tRNA species are modified by each enzyme because we do not have a complete map of all the tRNA modifications of all tRNAs. Without knowing all the tRNA species that each enzyme modifies, it is difficult to understand the consequences of a specific modification enzyme dysfunction. A milestone study in this field is the complete identification of all mitochondrial tRNA modifications in all 22 human mitochondrial tRNAs [52]. This work serves as an important foundation for understanding the molecular pathogenesis of mitochondrial tRNA modopathies. In contrast, the tRNA modifications of hundreds of cytoplasmic tRNA species transcribed in the nucleus are yet to be completely identified. Although the identification of all the modifications of all human cytoplasmic tRNA species requires tremendous work, such insight would greatly contribute to our understanding of the molecular pathogenesis of cytoplasmic tRNA modopathies.
Elucidation of the unidentified tRNA modification enzymes
Approximately 40 tRNA modification sites are modified by unknown enzymes (Figs 2 and 3). A fraction of these tRNA modifications are associated with strong candidate modification enzymes. This is because many of these modifications are located in the same positions as in yeast tRNAs, and the corresponding yeast tRNA modification enzymes have already been identified. In comparison with those in yeast, many additional tRNA modification enzymes exist in humans, many of which are likely generated by gene duplications from yeast homologs. Diversified human enzymes usually target different cytoplasmic tRNA species or different cellular compartments. For example, whereas only a single m3C32 methylase, Trm140, exists in Saccharomyces cerevisiae, Saccharomyces pombe has two homologs, and humans have three homologs, namely METTL2, METTL6, and METTL8 [112, 113]. The three human homologs are functionally differentiated. METTL2 synthesizes m3C32 of cytoplasmic tRNAThr and tRNAArg [114]. METTL6 synthesizes m3C32 of cytoplasmic tRNASer [115]. METTL8 localizes in mitochondria [116] and awaits investigation of whether it is responsible for m3C32 of mitochondrial tRNAThr and tRNASer [52]. m3C32 is not present in tRNAArg of S. cerevisiae but is present in human tRNAArg. Gene duplication and divergence expanded the substrate tRNA species. Even if a human tRNA modification is not conserved from yeast, it is still important to identify the responsible enzyme. Indeed, mt tRNA modifications such as τm5U34 and f5C34 are not present in yeast, but mutations in the responsible human enzymes cause severe mitochondrial diseases [98, 99, 117, 118]. The cytoplasmic tRNA ms2t6A37 is also not present in yeast, but the dysfunction of the responsible human enzyme causes type 2 diabetes and affects a large human population [87, 104].
tRNA modifications, such as GalQ34, ManQ34, and Nm39, are not associated with clear candidate enzymes. In addition, which pseudouridylases are responsible for Ψ at various positions remains unidentified.
Even for the tRNA modifications that are mediated by identified enzymes, it is possible that these enzymes may need additional partner proteins or upstream proteins to function. For instance, although the 2‐thiolation of mitochondrial τm5s2U34 is catalyzed by MTU1 [119], how sulfur is carried to MTU1 is unknown. Analogous to the S. cerevisiae or Escherichia coli 2‐thiolation pathways [120, 121], it is likely that specific mitochondrial proteins relay sulfur from cysteine desulfurase to MTU1. Elps complex proteins (ELP1–6) and ALKBH8 are essential for forming cytoplasmic tRNA mcm5U34 modification, but additional enzyme(s) are expected to form an intermediate cm5U and remain unidentified [75].
Identifying how each tRNA modification affects mRNA translation and other steps of gene expression
To understand how a tRNA modopathy is caused, it is necessary to understand how translation is affected by the loss of tRNA modification. Although specific enzymes can modify various tRNAs, tRNA modifications often critically affect the translation of only a fraction of modified tRNAs. If tRNA modification deficiency affects anticodon:codon interactions or critically affects tRNA in other ways, tRNA modification deficiency would decrease the ribosome transition rate at the corresponding codon. Thus, techniques such as ribosome profiling would be useful for elucidating how the translation of a specific codon is affected following the loss of a tRNA modification enzyme. For instance, ribosome profiling revealed the codons at which ribosomes slow down upon loss of Wdr4, a protein required for m7G46 modification [122]. Ribosome profiling can also identify which tRNAs are less frequently bound by ribosomes following the loss of specific species of tRNA modifications [123].
When the loss of a tRNA modification enzyme results in the decreased translation of an mRNA codon, one possibility is that the corresponding tRNA may degrade more easily without the modification. To investigate the effect of a tRNA modification on the tRNA steady‐state level in an unbiased manner, quantification of the tRNA transcriptome is useful. As tRNAs are difficult to reverse‐transcribe due to the presence of base pair‐inhibitory modifications such as m1A58, m1G37, m3C32, m2 2G26, and m1G9, it is helpful to use techniques such as demethylation via AlkB demethylase [124].
Recently, tRFs have become recognized to affect various steps of gene expression [125]. The generation of tRFs is affected by tRNA modifications such as Ψ, m1G, Q, and m5C [38, 39, 126, 127]. Thus, we should not forget the possibility that some tRNA modopathies might be caused not only by dysfunctional tRNA but also by increased tRFs.
What causes the tissue specificity of tRNA modopathies?
Aberrations in various tRNA modification enzymes affect our body, often in tissue‐specific or tissue‐preferential ways (Table 1). The effects of mutations in important mitochondrial tRNA modification enzymes appear mostly in high‐energy demand organs such as the heart and brain. This makes sense given that mitochondrial tRNA modification contributes to the translation of mitochondrial respiratory complex proteins used for ATP production.
Mutations in cytoplasmic tRNA modification enzymes most frequently affect the brain (Table 1); the mutations that affect brain development also often affect kidney development and overall body development (Table 1). We currently do not know why the brain is the most strongly affected organ. One clue may be that mutations in various other general translation regulatory proteins such as eIF2α cause neurological diseases, such as microcephaly, while having little effect to other tissues [128, 129], and this is a highly similar phenotype seen in many tRNA modopathies. Perhaps neurons are extremely sensitive to relatively small changes in translational competency because these polarized cells require rapid and local protein synthesis for synaptic plasticity [128, 129]. Neurons have long axons, and protein synthesis occurs not only in the cell body but also near synapses, which can be located at the end of long axons far from the cell body [130]. tRNA stability and translational efficiency may be especially important for translating proteins at such synaptic terminals, where tRNAs and ribosomes may not be transported from the cell body in abundance. Such possibilities merit investigation in order to understand the brain‐biased phenotypes of various tRNA modopathies.
Although mutations in many cytoplasmic tRNA modification enzymes affect the brain, there are many exceptions. For example, CDKAL1 SNPs are mainly correlated with type 2 diabetes, and CDK5 regulatory subunit‐associated protein 1‐like 1 (CDKAL1) dysfunction is not known to affect the brain, except for the role it plays in hormone biosynthesis in pituitary adenomas [131]. Why CDKAL1 mainly affects insulin biogenesis in pancreatic β‐cells and not in other tissues is not fully understood. Dysfunction of β‐cells in the context of CDKAL1 dysfunction, however, may at least in part be attributed to the heavy demand for the translation of proinsulin in β‐cells. In a bacterial lysine translation reporter model, upon knockout of the bacterial CDKAL1‐homolog, an increased translation rate led to decreased lysine translation fidelity [87]. As the translation of proinsulin comprises nearly 50% of total protein production upon glucose stimulation, and lysine is located at an important site within the proinsulin protein, it may be logical that β‐cells are more affected by CDKAL1 deficiency than other tissues.
To understand the tissue specificity of tRNA modopathies, a global intertissue comparison of protein synthesis and tRNA status is essential. Some tissues may have a higher demand for the translation of specific mRNA codons, and some tissues may have a limited supply of tRNAs that translate those amino acids. The absence of a tRNA modification may greatly alter tRNA stability in different tissues, due to, for example, different expression levels of angiogenin (which can cleave the hypomodified tRNA anticodon loop) and its inhibitor RNH1 [39, 132]. Therefore, to understand the tissue specificity of tRNA modifications, it would be useful to generate animal disease models and perform intertissue comparisons of the transcriptome (via RNA‐seq) as well as protein synthesis (e.g., via ribosome profiling) between wild‐type and disease model animals.
Concluding remarks
In recent decades, the identities and functions of many human tRNA modifications and the enzymes that cause these modifications have been elucidated. Moreover, tRNA modopathies resulting from aberrations in more than 50 tRNA modification enzyme genes have been discovered. Presently, the molecular pathogenesis of most tRNA modopathies remains unelucidated. In the next decade, the identification of all the tRNA modifications and modifying enzymes, as well as the intertissue comparison of protein synthesis in animal models, would elucidate these pathogenic mechanisms and provide evidence to support the development of treatments for these diseases.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
TC and KT wrote the review.
Acknowledgements
We thank the present and past members of the Tomizawa laboratory, in particular Fan‐Yan Wei, for fruitful discussions. This work was supported by Grants‐in‐Aid for Scientific Research from the Japanese Society for the Promotion of Science (JSPS) and the Ministry of Education, Culture, Sports, Science, and Technology of Japan to KT and TC (18H02865 and 17KT0054 to KT, and 20H03187 to TC).
The conventional symbols of modified nucleosides can be found at RNA Modification Database (https://mods.rna.albany.edu).
References
- 1. Crick F (1958) On protein synthesis. Symp Soc Exp Biol 12, 138–163. [PubMed] [Google Scholar]
- 2. Hoagland MB, Stephenson ML, Scott JF, Hecht LI & Zamecnik PC (1958) A soluble ribonucleic acid intermediate in protein synthesis. J Biol Chem 231, 241–257. [PubMed] [Google Scholar]
- 3. Chan PP & Lowe TM (2016) GtRNAdb 2.0: an expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res 44, D184–D189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F et al. (1981) Sequence and organization of the human mitochondrial genome. Nature 290, 457–465. [DOI] [PubMed] [Google Scholar]
- 5. Asano K, Suzuki T, Saito A, Wei FY, Ikeuchi Y, Numata T, Tanaka R, Yamane Y, Yamamoto T, Goto T et al. (2018) Metabolic and chemical regulation of tRNA modification associated with taurine deficiency and human disease. Nucleic Acids Res 46, 1565–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Grosjean H & Westhof E (2016) An integrated, structure‐ and energy‐based view of the genetic code. Nucleic Acids Res 44, 8020–8040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Agris PF, Eruysal ER, Narendran A, Vare VYP, Vangaveti S & Ranganathan SV (2018) Celebrating wobble decoding: half a century and still much is new. RNA Biol 15, 537–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Motorin Y & Helm M (2010) tRNA stabilization by modified nucleotides. Biochemistry 49, 4934–4944. [DOI] [PubMed] [Google Scholar]
- 9. Hori H (2014) Methylated nucleosides in tRNA and tRNA methyltransferases. Front Genet 5, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Spenkuch F, Motorin Y & Helm M (2014) Pseudouridine: still mysterious, but never a fake (uridine)!. RNA Biol 11, 1540–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. El Yacoubi B, Bailly M & de Crecy‐Lagard V (2012) Biosynthesis and function of posttranscriptional modifications of transfer RNAs. Annu Rev Genet 46, 69–95. [DOI] [PubMed] [Google Scholar]
- 12. Helm M, Giege R & Florentz C (1999) A Watson‐Crick pair‐disrupting methyl group (m1A9) is sufficient for cloverleaf folding of human mitochondrial tRNA Lys. Biochemistry 38, 13338–13346. [DOI] [PubMed] [Google Scholar]
- 13. Kobitski AY, Hengesbach M, Seidu‐Larry S, Dammertz K, Chow CS, van Aerschot A, Nienhaus GU & Helm M (2011) Single‐molecule FRET reveals a cooperative effect of two methyl group modifications in the folding of human mitochondrial tRNA(Lys). Chem Biol 18, 928–936. [DOI] [PubMed] [Google Scholar]
- 14. Vilardo E, Nachbagauer C, Buzet A, Taschner A, Holzmann J & Rossmanith W (2012) A subcomplex of human mitochondrial RNase P is a bifunctional methyltransferase‐extensive moonlighting in mitochondrial tRNA biogenesis. Nucleic Acids Res 40, 11583–11593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Metodiev MD, Thompson K, Alston CL, Morris AAM, He L, Assouline Z, Rio M, Bahi‐Buisson N, Pyle A, Griffin H et al. (2016) Recessive mutations in TRMT10C cause defects in mitochondrial RNA processing and multiple respiratory chain deficiencies. Am J Hum Genet 98, 993–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Oerum S, Roovers M, Leichsenring M, Acquaviva‐Bourdain C, Beermann F, Gemperle‐Britschgi C, Fouilhoux A, Korwitz‐Reichelt A, Bailey HJ, Droogmans L et al. (2017) Novel patient missense mutations in the HSD17B10 gene affect dehydrogenase and mitochondrial tRNA modification functions of the encoded protein. Biochim Biophys Acta Mol Basis Dis 1863, 3294–3302. [DOI] [PubMed] [Google Scholar]
- 17. Steinberg S & Cedergren R (1995) A correlation between N2‐dimethylguanosine presence and alternate tRNA conformers. RNA 1, 886–891. [PMC free article] [PubMed] [Google Scholar]
- 18. Dewe JM, Fuller BL, Lentini JM, Kellner SM & Fu D (2017) TRMT1‐catalyzed tRNA modifications are required for redox homeostasis to ensure proper cellular proliferation and oxidative stress survival. Mol Cell Biol 37, e00214–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu J & Straby KB (2000) The human tRNA(m(2)(2)G(26))dimethyltransferase: functional expression and characterization of a cloned hTRM1 gene. Nucleic Acids Res 28, 3445–3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Blaesius K, Abbasi AA, Tahir TH, Tietze A, Picker‐Minh S, Ali G, Farooq S, Hu H, Latif Z, Khan MN et al. (2018) Mutations in the tRNA methyltransferase 1 gene TRMT1 cause congenital microcephaly, isolated inferior vermian hypoplasia and cystic leukomalacia in addition to intellectual disability. Am J Med Genet A 176, 2517–2521. [DOI] [PubMed] [Google Scholar]
- 21. Kim SH, Suddath FL, Quigley GJ, McPherson A, Sussman JL, Wang AH, Seeman NC & Rich A (1974) Three‐dimensional tertiary structure of yeast phenylalanine transfer RNA. Science 185, 437–440. [DOI] [PubMed] [Google Scholar]
- 22. Oliva R, Cavallo L & Tramontano A (2006) Accurate energies of hydrogen bonded nucleic acid base pairs and triplets in tRNA tertiary interactions. Nucleic Acids Res 34, 865–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yokoyama S, Inagaki F & Miyazawa T (1981) Advanced nuclear magnetic resonance lanthanide probe analyses of short‐range conformational interrelations controlling ribonucleic acid structures. Biochemistry 20, 2981–2988. [DOI] [PubMed] [Google Scholar]
- 24. Kawai G, Yamamoto Y, Kamimura T, Masegi T, Sekine M, Hata T, Iimori T, Watanabe T, Miayazawa T & Yokoyama S (1992) Conformational rigidity of specific pyrimidine residues in tRNA arises from posttranscriptional modifications that enhances steric interaction between the base and the 2’‐hydroxyl group. Biochemistry 31, 1040–1046. [DOI] [PubMed] [Google Scholar]
- 25. Arnez J & Steitz T (1994) Crystal structure of unmodified tRNA Gln complexed with glutaminyl‐tRNA synthetase and ATP suggests a possible role for pseudo‐uridines in stabilization of RNA structure. Biochemistry 33, 7560–7567. [DOI] [PubMed] [Google Scholar]
- 26. Davis D (1995) Stabilization of RNA stacking by pseudouridine. Nucleic Acids Res 23, 5050–5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jones CI, Spencer AC, Hsu JL, Spremulli LL, Martinis SA, DeRider M & Agris PF (2006) A counterintuitive Mg2+‐dependent and modification‐assisted functional folding of mitochondrial tRNAs. J Mol Biol 362, 771–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sipa K, Sochacka E, Kazmierczak‐Baranska J, Maszewska M, Janicka M, Nowak G & Nawrot B (2007) Effect of base modifications on structure, thermodynamic stability, and gene silencing activity of short interfering RNA. RNA 13, 1301–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yarian CS, Basti MM, Cain RJ, Ansari G, Guenther RH, Sochacka E, Czerwinska G, Malkiewicz A & Agris PF (1999) Structural and functional roles of the N1‐ and N3‐protons of pseudouridine at tRNA’s position 39. Nucleic Acids Res 27, 3543–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. de Paiva ARB, Lynch DS, Melo US, Lucato LT, Freua F, de Assis BDR, Barcelos I, Listik C, de CastroDosSantos D, Macedo‐Souza LI et al. (2019) PUS3 mutations are associated with intellectual disability, leukoencephalopathy, and nephropathy. Neurol Genet 5, e306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guy MP, Shaw M, Weiner CL, Hobson L, Stark Z, Rose K, Kalscheuer VM, Gecz J & Phizicky EM (2015) Defects in tRNA anticodon loop 2’‐O‐methylation are implicated in nonsyndromic X‐linked intellectual disability due to mutations in FTSJ1. Hum Mutat 36, 1176–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shaheen R, Mark P, Prevost CT, Alkindi A, Alhag A, Estwani F, Al‐Sheddi T, Alobeid E, Alenazi M, Ewida N et al. (2019) Biallelic variants in CTU2 cause DREAM‐PL syndrome and impair thiolation of tRNA wobble U34. Hum Mutat 40, 2108–2120. [DOI] [PubMed] [Google Scholar]
- 33. Dalluge JJ, Hashizume T, Sopchik AE & McCloskey JA (1996) Conformational flexibility in tRNA: the role of dihydrouridine. Nucleic Acids Res 24, 1073–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Agris PF (2008) Bringing order to translation: the contributions of transfer RNA anticodon‐domain modifications. EMBO Rep 9, 629–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lorenz C, Lunse CE & Morl M (2017) tRNA modifications: impact on structure and thermal adaptation. Biomolecules 7, 1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yamasaki S, Ivanov P, Hu GF & Anderson P (2009) Angiogenin cleaves tRNA and promotes stress‐induced translational repression. J Cell Biol 185, 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vitali P & Kiss T (2019) Cooperative 2'‐O‐methylation of the wobble cytidine of human elongator tRNA(Met)(CAT) by a nucleolar and a Cajal body‐specific box C/D RNP. Genes Dev 33, 741–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang X, Matuszek Z, Huang Y, Parisien M, Dai Q, Clark W, Schwartz MH & Pan T (2018) Queuosine modification protects cognate tRNAs against ribonuclease cleavage. RNA 24, 1305–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Blanco S, Dietmann S, Flores JV, Hussain S, Kutter C, Humphreys P, Lukk M, Lombard P, Treps L, Popis M et al. (2014) Aberrant methylation of tRNAs links cellular stress to neuro‐developmental disorders. EMBO J 33, 2020–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Abbasi‐Moheb L, Mertel S, Gonsior M, Nouri‐Vahid L, Kahrizi K, Cirak S, Wieczorek D, Motazacker MM, Esmaeeli‐Nieh S, Cremer K et al. (2012) Mutations in NSUN2 cause autosomal‐recessive intellectual disability. Am J Hum Genet 90, 847–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Martinez FJ, Lee JH, Lee JE, Blanco S, Nickerson E, Gabriel S, Frye M, Al‐Gazali L & Gleeson JG (2012) Whole exome sequencing identifies a splicing mutation in NSUN2 as a cause of a Dubowitz‐like syndrome. J Med Genet 49, 380–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kadaba S, Krueger A, Trice T, Krecic AM, Hinnebusch AG & Anderson J (2004) Nuclear surveillance and degradation of hypomodified initiator tRNAMet in S. cerevisiae . Genes Dev 18, 1227–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Alexandrov A, Chernyakov I, Gu W, Hiley SL, Hughes TR, Grayhack EJ & Phizicky EM (2006) Rapid tRNA decay can result from lack of nonessential modifications. Mol Cell 21, 87–96. [DOI] [PubMed] [Google Scholar]
- 44. Wilusz JE, Whipple JM, Phizicky EM & Sharp AS (2011) tRNAs Marked with CCACCA are targeted for degradation. Science 334, 817–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Crick F (1966) Codon‐anticodon pairing. J Mol Biol 19, 548–555. [DOI] [PubMed] [Google Scholar]
- 46. Suzuki T, Nagao A & Suzuki T (2011) Human mitochondrial tRNAs: biogenesis, function, structural aspects, and diseases. Annu Rev Genet 45, 299–329. [DOI] [PubMed] [Google Scholar]
- 47. Grosjean H, de Crecy‐Lagard V & Marck C (2010) Deciphering synonymous codons in the three domains of life: co‐evolution with specific tRNA modification enzymes. FEBS Lett 584, 252–264. [DOI] [PubMed] [Google Scholar]
- 48. Johansson MJ, Esberg A, Huang B, Bjork GR & Bystrom AS (2008) Eukaryotic wobble uridine modifications promote a functionally redundant decoding system. Mol Cell Biol 28, 3301–3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yokoyama S, Watanabe T, Murao K, Ishikura H, Yamaizumi Z, Nishimura S & Miyazawa T (1985) Molecular mechanism of codon recognition by tRNA species with modified uridine in the first position of the anticodon. Proc Natl Acad Sci USA 82, 4905–4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Carlson BA, Kwon SY, Chamorro M, Oroszlan S, Hatfield DL & Lee BJ (1999) Transfer RNA modification status influences retroviral ribosomal fameshifting. Virology 255, 2–8. [DOI] [PubMed] [Google Scholar]
- 51. Tuorto F, Legrand C, Cirzi C, Federico G, Liebers R, Muller M, Ehrenhofer‐Murray AE, Dittmar G, Grone HJ & Lyko F (2018) Queuosine‐modified tRNAs confer nutritional control of protein translation. EMBO J 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Suzuki T, Yashiro Y, Kikuchi I, Ishigami Y, Saito H, Matsuzawa I, Okada S, Mito M, Iwasaki S, Ma D et al. (2020) Complete chemical structures of human mitochondrial tRNAs. Nat Commun 11, 4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Takemoto C, Spremulli LL, Benkowski LA, Ueda T, Yokogawa T & Watanabe K (2009) Unconventional decoding of the AUA codon as methionine by mitochondrial tRNAMet with the anticodon f5CAU as revealed with a mitochondrial in vitro translation system. Nucleic Acids Res 37, 1616–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rossello‐Tortella M, Llinas‐Arias P, Sakaguchi Y, Miyauchi K, Davalos V, Setien F, Calleja‐Cervantes ME, Pineyro D, Martinez‐Gomez J, Guil S et al. (2020) Epigenetic loss of the transfer RNA‐modifying enzyme TYW2 induces ribosome frameshifts in colon cancer. Proc Natl Acad Sci USA 117, 20785–20793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Urbonavicius J, Qian Q, Durand JM, Hagervall TG & Bjork GR (2001) Improvement of reading frame maintenance is a common function for several tRNA modifications. EMBO J 20, 4863–4873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wilson RK & Roe BA (1989) Presence of the hypermodified nucleotide N6‐(delta 2‐isopentenyl)‐2‐methylthioadenosine prevents codon misreading by Escherichia coli phenylalanyl‐transfer RNA. Proc Natl Acad Sci USA 86, 409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hirata A, Okada K, Yoshii K, Shiraishi H, Saijo S, Yonezawa K, Shimizu N & Hori H (2019) Structure of tRNA methyltransferase complex of Trm7 and Trm734 reveals a novel binding interface for tRNA recognition. Nucleic Acids Res 47, 10942–10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Arango D, Sturgill D, Alhusaini N, Dillman AA, Sweet TJ, Hanson G, Hosogane M, Sinclair WR, Nanan KK, Mandler MD et al. (2018) Acetylation of cytidine in mRNA promotes translation efficiency. Cell 175, 1872–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ito S, Horikawa S, Suzuki T, Kawauchi H, Tanaka Y, Suzuki T & Suzuki T (2014) Human NAT10 is an ATP‐dependent RNA acetyltransferase responsible for N4‐acetylcytidine formation in 18 S ribosomal RNA (rRNA). J Biol Chem 289, 35724–35730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sharma S, Langhendries JL, Watzinger P, Kotter P, Entian KD & Lafontaine DL (2015) Yeast Kre33 and human NAT10 are conserved 18S rRNA cytosine acetyltransferases that modify tRNAs assisted by the adaptor Tan1/THUMPD1. Nucleic Acids Res 43, 2242–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bar‐Yaacov D, Frumkin I, Yashiro Y, Chujo T, Ishigami Y, Chemla Y, Blumberg A, Schlesinger O, Bieri P, Greber B et al. (2016) Mitochondrial 16S rRNA is methylated by tRNA methyltransferase TRMT61B in all vertebrates. PLoS Biol 14, e1002557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chujo T & Suzuki T (2012) TRMT61B is a methyltransferase responsible for 1‐methyladenosine at position 58 of human mitochondrial tRNAs. RNA 18, 2269–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Powell CA & Minczuk M (2020) TRMT2B is responsible for both tRNA and rRNA m(5)U‐methylation in human mitochondria. RNA Biol 17, 451–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pandolfini L, Barbieri I, Bannister AJ, Hendrick A, Andrews B, Webster N, Murat P, Mach P, Brandi R, Robson SC et al. (2019) METTL1 promotes let‐7 microRNA processing via m7G methylation. Mol Cell 74, 1278–1290.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhang LS, Liu C, Ma H, Dai Q, Sun HL, Luo G, Zhang Z, Zhang L, Hu L, Dong X et al. (2019) Transcriptome‐wide mapping of internal N(7)‐methylguanosine methylome in mammalian mRNA. Mol Cell 74, 1304–1316.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Holzmann J, Frank P, Loffler E, Bennett KL, Gerner C & Rossmanith W (2008) RNase P without RNA: identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell 135, 462–474. [DOI] [PubMed] [Google Scholar]
- 67. Kurimoto R, Chiba T, Ito Y, Matsushima T, Yano Y, Miyata K, Yashiro Y, Suzuki T, Tomita K & Asahara H (2020) The tRNA pseudouridine synthase TruB1 regulates the maturation of let‐7 miRNA. EMBO J 39, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Schwartz S, Bernstein DA, Mumbach MR, Jovanovic M, Herbst RH, Leon‐Ricardo BX, Engreitz JM, Guttman M, Satija R, Lander ES et al. (2014) Transcriptome‐wide mapping reveals widespread dynamic‐regulated pseudouridylation of ncRNA and mRNA. Cell 159, 148–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Johansson MJ & Bystrom AS (2002) Dual function of the tRNA(m5U54) methyltransferase in tRNA maturation. RNA 8, 324–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Freund I, Buhl DK, Boutin S, Kotter A, Pichot F, Machand V, Vierbuchen T, Heine H, Motorin Y, Helm M et al. (2019) 2′‐O‐methylation within prokaryotic and eukaryotic tRNA inhibits innate immune activation by endosomal Toll‐like receptors but does not affect recognition of whole organisms. RNA 25, 869–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gehrig S, Eberle ME, Botschen F, Rimbach K, Eberle F, Eigenbrod T, Kaiser S, Holmes WM, Erdmann VA, Sprinzl M et al. (2012) Identification of modifications in microbial, native tRNA that suppress immunostimulatory activity. J Exp Med 209, 225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jockel S, Nees G, Sommer R, Zhao Y, Cherkasov D, Hori H, Ehm G, Schnare M, Nain M, Kaufmann A et al. (2012) The 2'‐O‐methylation status of a single guanosine controls transfer RNA‐mediated Toll‐like receptor 7 activation or inhibition. J Exp Med 209, 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Rubio MA, Paris Z, Gaston KW, Fleming IM, Sample P, Trotta CR & Alfonzo JD (2013) Unusual noncanonical intron editing is important for tRNA splicing in Trypanosoma brucei . Mol Cell 52, 184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Brzezicha B, Schmidt M, Makalowska I, Jarmolowski A, Pienkowska J & Szweykowska‐Kulinska Z (2006) Identification of human tRNA:m5C methyltransferase catalysing intron‐dependent m5C formation in the first position of the anticodon of the pre‐tRNA Leu (CAA). Nucleic Acids Res 34, 6034–6043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. de Crecy‐Lagard V, Boccaletto P, Mangleburg CG, Sharma P, Lowe TM, Leidel SA & Bujnicki JM (2019) Matching tRNA modifications in humans to their known and predicted enzymes. Nucleic Acids Res 47, 2143–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zinshteyn B & Gilbert WV (2013) Loss of a conserved tRNA anticodon modification perturbs cellular signaling. PLoS Genet 9, e1003675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Rapino F, Delaunay S, Rambow F, Zhou Z, Tharun L, De Tullio P, Sin O, Shostak K, Schmitz S, Piepers J et al. (2018) Codon‐specific translation reprogramming promotes resistance to targeted therapy. Nature 558, 605–609. [DOI] [PubMed] [Google Scholar]
- 78. Gunduz U, Elliott MS, Seubert PH, Houghton JA, Houghton PJ, Trewyn RW & Katze JR (1992) Absence of tRNA‐guanine transglycosylase in a human colon adenocarcinoma cell line. Biochim Biophys Acta 1139, 229–238. [DOI] [PubMed] [Google Scholar]
- 79. Kuchino Y, Borek E, Grunberger D, Mushinski JF & Nishimura S (1982) Changes of post‐transcriptional modification of wye base in tumor‐specific tRNA Phe. Nucleic Acids Res 10, 6421–6432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Pathak C, Jaiswal YK & Vinayak M (2005) Hypomodification of transfer RNA in cancer with respect to queuosine. RNA Biol 2, 143–148. [DOI] [PubMed] [Google Scholar]
- 81. Spinola M, Galvan A, Pignatiello C, Conti B, Pastorino U, Nicander B, Paroni R & Dragani TA (2005) Identification and functional characterization of the candidate tumor suppressor gene TRIT1 in human lung cancer. Oncogene 24, 5502–5509. [DOI] [PubMed] [Google Scholar]
- 82. Tomizawa K & Wei FY (2020) Posttranscriptional modifications in mitochondrial tRNA and its implication in mitochondrial translation and disease. J Biochem 168, 435–444. [DOI] [PubMed] [Google Scholar]
- 83. Goto Y, Nonaka I & Horai S (1990) A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 348, 651–653. [DOI] [PubMed] [Google Scholar]
- 84. Kirino Y, Yasukawa T, Ohta S, Akira S, Ishihara K, Watanabe K & Suzuki T (2004) Codon‐specific translational defect caused by a wobble modification deficiency in mutant tRNA from a human mitochondrial disease. Proc Natl Acad Sci USA 101, 15070–15075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hayashi JI, Ohta S, Takai D, Miyabayashi S, Sakuta R, Goto Y & Nonaka I (1993) Accumulation of mtDNA with a mutation at position 3271 in tRNA(Leu)(UUR) gene introduced from a MELAS patient to HeLa cells lacking mtDNA results in progressive inhibition of mitochondrial respiratory function. Biochem Biophys Res Commun 197, 1049–1055. [DOI] [PubMed] [Google Scholar]
- 86. Nedialkova DD & Leidel SA (2015) Optimization of codon translation rates via tRNA modifications maintains proteome integrity. Cell 161, 1606–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wei FY, Suzuki T, Watanabe S, Kimura S, Kaitsuka T, Fujimura A, Matsui H, Atta M, Michiue H, Fontecave M et al. (2011) Deficit of tRNA(Lys) modification by Cdkal1 causes the development of type 2 diabetes in mice. J Clin Invest 121, 3598–3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Gu C, Begley TJ & Dedon PC (2014) tRNA modifications regulate translation during cellular stress. FEBS Lett 588, 4287–4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Chan CT, Pang YL, Deng W, Babu IR, Dyavaiah M, Begley TJ & Dedon PC (2012) Reprogramming of tRNA modifications controls the oxidative stress response by codon‐biased translation of proteins. Nat Commun 3, 937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Begley U, Dyavaiah M, Patil A, Rooney JP, DiRenzo D, Young CM, Conklin DS, Zitomer RS & Begley TJ (2007) Trm9‐catalyzed tRNA modifications link translation to the DNA damage response. Mol Cell 28, 860–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Suzuki T, Suzuki T, Wada T, Saigo K & Watanabe K (2002) Taurine as a constituent of mitochondrial tRNAs: new insights into the functions of taurine and human mitochondrial diseases. EMBO J 21, 6581–6589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Fukuhara N, Tokiguchi S, Shirakawa K & Tsubaki T (1980) Myoclonus epilepsy associated with ragged‐red fibres (mitochondrial abnormalities): disease entity or a syndrome? J Neurol Sci 47, 117–133. [DOI] [PubMed] [Google Scholar]
- 93. Pavlakis SG, Phillips PC, DiMauro S, De Vivo D & Rowland LP (1984) Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes; a distinctive clinical syndrome. Ann Neurol 16, 481–488. [DOI] [PubMed] [Google Scholar]
- 94. Yoneda M, Horai YT, Ozawa T, Miyatake T & Tsuji S (1994) A common mitochondrial DNA mutation in the t‐RNA(Lys) of patients with myoclonus epilepsy associated with ragged‐red fibers. Biochem Int 21, 789–796. [PubMed] [Google Scholar]
- 95. Kirino Y, Goto Y, Campos Y, Arenas J & Suzuki T (2005) Specific correlation between the wobble modification deficiency in mutant tRNAs and the clinical features of a human mitochondrial disease. Proc Natl Acad Sci USA 102, 7127–7132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Yasukawa T, Suzuki T, Ishii N, Ueda T, Ohta S & Watanabe K (2000) Defect in modification at the anticodon wobble nucleotide of mitochondrial tRNA(Lys) with the MERRF encephalomyopathy pathogenic mutation. FEBS Lett 467, 175–178. [DOI] [PubMed] [Google Scholar]
- 97. Yasukawa T, Suzuki T, Suzuki T, Ueda T, Ohta S & Watanabe K (2000) Modification defect at anticodon wobble nucleotide of mitochondrial tRNAs(Leu)(UUR) with pathogenic mutations of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke‐like episodes. J Biol Chem 11, 4251–4257. [DOI] [PubMed] [Google Scholar]
- 98. Ghezzi D, Baruffini E, Haack TB, Invernizzi F, Melchionda L, Dallabona C, Strom TM, Parini R, Burlina AB, Meitinger T et al. (2012) Mutations of the mitochondrial‐tRNA modifier MTO1 cause hypertrophic cardiomyopathy and lactic acidosis. Am J Hum Genet 90, 1079–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kopajtich R, Nicholls TJ, Rorbach J, Metodiev MD, Freisinger P, Mandel H, Vanlander A, Ghezzi D, Carrozzo R, Taylor RW et al. (2014) Mutations in GTPBP3 cause a mitochondrial translation defect associated with hypertrophic cardiomyopathy, lactic acidosis, and encephalopathy. Am J Hum Genet 95, 708–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Fakruddin M, Wei FY, Suzuki T, Asano K, Kaieda T, Omori A, Izumi R, Fujimura A, Kaitsuka T, Miyata K et al. (2018) Defective mitochondrial tRNA taurine modification activates global proteostress and leads to mitochondrial disease. Cell Rep 22, 482–496. [DOI] [PubMed] [Google Scholar]
- 101. Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK et al. (2008) A mitochondrial protein compendium elucidates complex I disease biology. Cell 134, 112–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Hawkins PN, Ando Y, Dispenzeri A, Gonzalez‐Duarte A, Adams D & Suhr OB (2015) Evolving landscape in the management of transthyretin amyloidosis. Ann Med 47, 625–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Kars M, Yang L, Gregor MF, Mohammed BS, Pietka TA, Finck BN, Patterson BW, Horton JD, Mittendorfer B, Hotamisligil GS et al. (2010) Tauroursodeoxycholic acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women. Diabetes 59, 1899–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Steinthorsdottir V, Thorleifsson G, Reynisdottir I, Benediktsson R, Jonsdottir T, Walters GB, Styrkarsdottir U, Gretarsdottir S, Emilsson V, Ghosh S et al. (2007) A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat Genet 39, 770–775. [DOI] [PubMed] [Google Scholar]
- 105. Wei FY & Tomizawa K (2012) Development of type 2 diabetes caused by a deficiency of a tRNA(lys) modification. Islets 4, 71–73. [DOI] [PubMed] [Google Scholar]
- 106. Ruchat SM, Elks CE, Loos RJ, Vohl MC, Weisnagel SJ, Rankinen T, Bouchard C & Perusse L (2009) Association between insulin secretion, insulin sensitivity and type 2 diabetes susceptibility variants identified in genome‐wide association studies. Acta Diabetol 46, 217–226. [DOI] [PubMed] [Google Scholar]
- 107. Shinoda S, Kitagawa S, Nakagawa S, Wei FY, Tomizawa K, Araki K, Araki M, Suzuki T & Suzuki T (2019) Mammalian NSUN2 introduces 5‐methylcytidines into mitochondrial tRNAs. Nucleic Acids Res 47, 8734–8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Tuorto F, Liebers R, Musch T, Schaefer M, Hofmann S, Kellner S, Frye M, Helm M, Stoecklin G & Lyko F (2012) RNA cytosine methylation by Dnmt2 and NSun2 promotes tRNA stability and protein synthesis. Nat Struct Mol Biol 19, 900–905. [DOI] [PubMed] [Google Scholar]
- 109. Noma A, Ishitani R, Kato M, Nagao A, Nureki O & Suzuki T (2010) Expanding role of the jumonji C domain as an RNA hydroxylase. J Biol Chem 285, 34503–34507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Noma A, Kirino Y, Ikeuchi Y & Suzuki T (2006) Biosynthesis of wybutosine, a hyper‐modified nucleoside in eukaryotic phenylalanine tRNA. EMBO J 25, 2142–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Yeon SY, Jo YS, Choi EJ, Kim MS, Yoo NJ & Lee SH (2018) Frameshift mutations in repeat sequences of ANK3, HACD4, TCP10L, TP53BP1, MFN1, LCMT2, RNMT, TRMT6, METTL8 and METTL16 genes in colon cancers. Pathol Oncol Res 24, 617–622. [DOI] [PubMed] [Google Scholar]
- 112. Arimbasseri AG, Iben J, Wei FY, Rijal K, Tomizawa K, Hafner M & Maraia RJ (2016) Evolving specificity of tRNA 3‐methyl‐cytidine‐32 (m3C32) modification: a subset of tRNAsSer requires N6‐isopentenylation of A37. RNA 22, 1400–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Noma A, Yi S, Katoh T, Takai Y, Suzuki T & Suzuki T (2011) Actin‐binding protein ABP140 is a methyltransferase for 3‐methylcytidine at position 32 of tRNAs in Saccharomyces cerevisiae . RNA 17, 1111–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Xu L, Liu X, Sheng N, Oo KS, Liang J, Chionh YH, Xu J, Ye F, Gao YG, Dedon PC et al. (2017) Three distinct 3‐methylcytidine (m(3)C) methyltransferases modify tRNA and mRNA in mice and humans. J Biol Chem 292, 14695–14703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Ignatova VV, Kaiser S, Ho JSY, Bing X, Stolz P, Tan YX, Lee CL, Gay FP, Lastres PR, Gerlini F et al. (2020) METTL6 is a tRNA m3C methyltransferase that regulates pluripotency and tumor cell growth. Sci Adv 6, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Rath S, Sharma R, Gupta R, Ast T, Chan C, Durham TJ, Goodman RP, Grabarek Z, Haas ME, Hung WHW et al. (2020) MitoCarta3.0: an updated mitochondrial proteome now with sub‐organelle localization and pathway annotations. Nucleic Acids Res 49(D1), D1541–D1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Paramasivam A, Meena AK, Venkatapathi C, Pitceathly RDS & Thangaraj K (2020) Novel biallelic NSUN3 variants cause early‐onset mitochondrial encephalomyopathy and seizures. J Mol Neurosci 70, 1962–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Van Haute L, Dietmann S, Kremer L, Hussain S, Pearce SF, Powell CA, Rorbach J, Lantaff R, Blanco S, Sauer S et al. (2016) Deficient methylation and formylation of mt‐tRNA(Met) wobble cytosine in a patient carrying mutations in NSUN3. Nat Commun 7, 12039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Umeda N, Suzuki T, Yukawa M, Ohya Y, Shindo H, Watanabe K & Suzuki T (2005) Mitochondria‐specific RNA‐modifying enzymes responsible for the biosynthesis of the wobble base in mitochondrial tRNAs. Implications for the molecular pathogenesis of human mitochondrial diseases. J Biol Chem 280, 1613–1624. [DOI] [PubMed] [Google Scholar]
- 120. Ikeuchi Y, Shigi N, Kato J, Nishimura A & Suzuki T (2006) Mechanistic insights into sulfur relay by multiple sulfur mediators involved in thiouridine biosynthesis at tRNA wobble positions. Mol Cell 21, 97–108. [DOI] [PubMed] [Google Scholar]
- 121. Noma A, Sakaguchi Y & Suzuki T (2009) Mechanistic characterization of the sulfur‐relay system for eukaryotic 2‐thiouridine biogenesis at tRNA wobble positions. Nucleic Acids Res 37, 1335–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Lin S, Liu Q, Lelyveld VS, Choe J, Szostak JW & Gregory RI (2018) Mettl1/Wdr4‐mediated m(7)G tRNA methylome is required for normal mRNA translation and embryonic stem cell self‐renewal and differentiation. Mol Cell 71, 244–255.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Chen CW & Tanaka M (2018) Genome‐wide translation profiling by ribosome‐bound tRNA capture. Cell Rep 23, 608–621. [DOI] [PubMed] [Google Scholar]
- 124. Clark WC, Evans ME, Dominissini D, Zheng G & Pan T (2016) tRNA base methylation identification and quantification via high‐throughput sequencing. RNA 22, 1771–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Kumar P, Kuscu C & Dutta A (2016) Biogenesis and function of transfer RNA‐related fragments (tRFs). Trends Biochem Sci 41, 679–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Cosentino C, Toivonen S, Diaz Villamil E, Atta M, Ravanat JL, Demine S, Schiavo AA, Pachera N, Deglasse JP, Jonas JC et al. (2018) Pancreatic beta‐cell tRNA hypomethylation and fragmentation link TRMT10A deficiency with diabetes. Nucleic Acids Res 46, 10302–10318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Guzzi N, Ciesla M, Ngoc PCT, Lang S, Arora S, Dimitriou M, Pimkova K, Sommarin MNE, Munita R, Lubas M et al. (2018) Pseudouridylation of tRNA‐derived fragments steers translational control in stem cells. Cell 173, 1204–1216.e26. [DOI] [PubMed] [Google Scholar]
- 128. Kapur M & Ackerman SL (2018) mRNA translation gone awry: translation fidelity and neurological disease. Trends Genet 34, 218–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Kapur M, Monaghan CE & Ackerman SL (2017) Regulation of mRNA translation in neurons‐A matter of life and death. Neuron 96, 616–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Hafner AS, Donlin‐Asp PG, Leitch B, Herzog E & Schuman EM (2019) Local protein synthesis is a ubiquitous feature of neuronal pre‐ and postsynaptic compartments. Science 364, eaau3644. [DOI] [PubMed] [Google Scholar]
- 131. Takesue Y, Wei FY, Fukuda H, Tanoue Y, Yamamoto T, Chujo T, Shinojima N, Yano S, Morioka M, Mukasa A et al. (2019) Regulation of growth hormone biosynthesis by Cdk5 regulatory subunit associated protein1‐like1 (CDKAL1) in pituitary adenomas. Endocr J 66, 807–816. [DOI] [PubMed] [Google Scholar]
- 132. Gamazon ER, Segre AV, van de Bunt M, Wen X, Xi HS, Hormozdiari F, Ongen H, Konkashbaev A, Derks EM, Aguet F et al. (2018) Using an atlas of gene regulation across 44 human tissues to inform complex disease‐ and trait‐associated variation. Nat Genet 50, 956–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Hyde SJ, Eckenroth BE, Smith BA, Eberley WA, Heintz NH, Jackman JE & Doublie S (2010) tRNA(His) guanylyltransferase (THG1), a unique 3'‐5' nucleotidyl transferase, shares unexpected structural homology with canonical 5'‐3' DNA polymerases. Proc Natl Acad Sci USA 107, 20305–21310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Igoillo‐Esteve M, Genin A, Lambert N, Desir J, Pirson I, Abdulkarim B, Simonis N, Drielsma A, Marselli L, Marchetti P et al. (2013) tRNA methyltransferase homolog gene TRMT10A mutation in young onset diabetes and primary microcephaly in humans. PLoS Genet 9, e1003888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Howell NW, Jora M, Jepson BF, Limbach PA & Jackman JE (2019) Distinct substrate specificities of the human tRNA methyltransferases TRMT10A and TRMT10B. RNA 25, 1366–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Vilardo E, Amman F, Toth U, Kotter A, Helm M & Rossmanith W (2020) Functional characterization of the human tRNA methyltransferases TRMT10A and TRMT10B. Nucleic Acids Res 48, 6157–6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. de Brouwer APM, Abou Jamra R, Kortel N, Soyris C, Polla DL, Safra M, Zisso A, Powell CA, Rebelo‐Guiomar P, Dinges N et al. (2018) Variants in PUS7 cause intellectual disability with speech delay, microcephaly, short stature, and aggressive behavior. Am J Hum Genet 103, 1045–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Bou‐Nader C, Pecqueur L, Bregeon D, Kamah A, Guerineau V, Golinelli‐Pimpaneau B, Guimaraes BG, Fontecave M & Hamdane D (2015) An extended dsRBD is required for post‐transcriptional modification in human tRNAs. Nucleic Acids Res 43, 9446–9456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Takakura M, Ishiguro K, Akichika S, Miyauchi K & Suzuki T (2019) Biogenesis and functions of aminocarboxypropyluridine in tRNA. Nat Commun 10, 5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Patton JR, Bykhovskaya Y, Mengesha E, Bertolotto C & Fischel‐Ghodsian N (2005) Mitochondrial myopathy and sideroblastic anemia (MLASA): missense mutation in the pseudouridine synthase 1 (PUS1) gene is associated with the loss of tRNA pseudouridylation. J Biol Chem 280, 19823–19828. [DOI] [PubMed] [Google Scholar]
- 141. Guy MP & Phizicky EM (2015) Conservation of an intricate circuit for crucial modifications of the tRNAPhe anticodon loop in eukaryotes. RNA 21, 61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Li J, Wang YN, Xu BS, Liu YP, Zhou M, Long T, Li H, Dong H, Nie Y, Chen PR et al. (2020) Intellectual disability‐associated gene ftsj1 is responsible for 2'‐O‐methylation of specific tRNAs. EMBO Rep 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Kawarada L, Suzuki T, Ohira T, Hirata S, Miyauchi K & Suzuki T (2017) ALKBH1 is an RNA dioxygenase responsible for cytoplasmic and mitochondrial tRNA modifications. Nucleic Acids Res 45, 7401–7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Boland C, Hayes P, Santa‐Maria I, Nishimura S & Kelly VP (2009) Queuosine formation in eukaryotic tRNA occurs via a mitochondria‐localized heteromeric transglycosylase. J Biol Chem 284, 18218–18227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Chen YC, Kelly VP, Stachura SV & Garcia GA (2010) Characterization of the human tRNA‐guanine transglycosylase: confirmation of the heterodimeric subunit structure. RNA 16, 958–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Bento‐Abreu A, Jager G, Swinnen B, Rue L, Hendrickx S, Jones A, Staats KA, Taes I, Eykens C, Nonneman A et al. (2018) Elongator subunit 3 (ELP3) modifies ALS through tRNA modification. Hum Mol Genet 27, 1276–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Kojic M, Gaik M, Kiska B, Salerno‐Kochan A, Hunt S, Tedoldi A, Mureev S, Jones A, Whittle B, Genovesi LA et al. (2018) Elongator mutation in mice induces neurodegeneration and ataxia‐like behavior. Nat Commun 9, 3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Lin FJ, Shen L, Jang CW, Falnes PO & Zhang Y (2013) Ikbkap/Elp1 deficiency causes male infertility by disrupting meiotic progression. PLoS Genet 9, e1003516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Songe‐Moller L, van den Born E, Leihne V, Vagbo CB, Kristoffersen T, Krokan HE, Kirpekar F, Falnes PO & Klungland A (2010) Mammalian ALKBH8 possesses tRNA methyltransferase activity required for the biogenesis of multiple wobble uridine modifications implicated in translational decoding. Mol Cell Biol 30, 1814–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Schlieker CD, Van der Veen AG, Damon JR, Spooner E & Ploegh HL (2008) A functional proteomics approach links the ubiquitin‐related modifier Urm1 to a tRNA modification pathway. Proc Natl Acad Sci USA 105, 18255–18260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Ramos J, Han L, Li Y, Hagelskamp F, Kellner SM, Alkuraya FS, Phizicky EM & Fu D (2019) Formation of tRNA wobble inosine in humans is disrupted by a millennia‐old mutation causing intellectual disability. Mol Cell Biol 39, e00203–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Torres AG, Pineyro D, Rodriguez‐Escriba M, Camacho N, Reina O, Saint‐Leger A, Filonava L, Batlle E & Ribas de Pouplana L (2015) Inosine modifications in human tRNAs are incorporated at the precursor tRNA level. Nucleic Acids Res 43, 5145–5157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Brule H, Elliott M, Redlak M, Zehner ZE & Holmes WM (2004) Isolation and characterization of the human tRNA‐(N1G37) methyltransferase (TRM5) and comparison to the Escherichia coli TrmD protein. Biochemistry 43, 9243–9255. [DOI] [PubMed] [Google Scholar]
- 154. Maas S, Gerber AP & Rich A (1999) Identification and characterization of a human tRNA‐specific adenosine deaminase related to the ADAR family of pre‐mRNA editing enzymes. Proc Natl Acad Sci USA 96, 8895–8900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Arrondel C, Missoury S, Snoek R, Patat J, Menara G, Collinet B, Liger D, Durand D, Gribouval O, Boyer O et al. (2019) Defects in t(6)A tRNA modification due to GON7 and YRDC mutations lead to Galloway‐Mowat syndrome. Nat Commun 10, 3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Braun DA, Rao J, Mollet G, Schapiro D, Daugeron MC, Tan W, Gribouval O, Boyer O, Revy P, Jobst‐Schwan T et al. (2017) Mutations in KEOPS‐complex genes cause nephrotic syndrome with primary microcephaly. Nat Genet 49, 1529–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Kimura S, Miyauchi K, Ikeuchi Y, Thiaville PC, Crecy‐Lagard V & Suzuki T (2014) Discovery of the beta‐barrel‐type RNA methyltransferase responsible for N6‐methylation of N6‐threonylcarbamoyladenosine in tRNAs. Nucleic Acids Res 42, 9350–9365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Lamichhane TN, Blewett NH & Maraia RJ (2011) Plasticity and diversity of tRNA anticodon determinants of substrate recognition by eukaryotic A37 isopentenyltransferases. RNA 17, 1846–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Rodriguez V, Vasudevan S, Noma A, Carlson BA, Green JE, Suzuki T & Chandrasekharappa SC (2012) Structure‐function analysis of human TYW2 enzyme required for the biosynthesis of a highly modified Wybutosine (yW) base in phenylalanine‐tRNA. PLoS One 7, e39297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Goll MG, Kirpekar F, Maggert KA, Yoder JA, Hsieh AC, Zhang X, Golic KG, Jacobsen SE & Bestor TH (2006) Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 311, 395–398. [DOI] [PubMed] [Google Scholar]
- 161. Chen J & Patton JR (2000) Pseudouridine synthase 3 from mouse modifies the anticodon loop of tRNA. Biochemistry 39, 12723–12730. [DOI] [PubMed] [Google Scholar]
- 162. Shaheen R, Han L, Faqeih E, Ewida N, Alobeid E, Phizicky EM & Alkuraya FS (2016) A homozygous truncating mutation in PUS3 expands the role of tRNA modification in normal cognition. Hum Genet 135, 707–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Carter JM, Emmett W, Mozos IR, Kotter A, Helm M, Ule J & Hussain S (2019) FICC‐Seq: a method for enzyme‐specified profiling of methyl‐5‐uridine in cellular RNA. Nucleic Acids Res 47, e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Deogharia M, Mukhopadhyay S, Joardar A & Gupta R (2019) The human ortholog of archaeal Pus10 produces pseudouridine 54 in select tRNAs where its recognition sequence contains a modified residue. RNA 25, 336–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Ozanick S, Krecic A, Andersland J & Anderson JT (2005) The bipartite structure of the tRNA m1A58 methyltransferase from S. cerevisiae is conserved in humans. RNA 11, 1281–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Haag S, Warda AS, Kretschmer J, Gunnigmann MA, Hobartner C & Bohnsack MT (2015) NSUN6 is a human RNA methyltransferase that catalyzes formation of m5C72 in specific tRNAs. RNA 21, 1532–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Nakano S, Suzuki T, Kawarada L, Iwata H, Asano K & Suzuki T (2016) NSUN3 methylase initiates 5‐formylcytidine biogenesis in human mitochondrial tRNA(Met). Nat Chem Biol 12, 546–551. [DOI] [PubMed] [Google Scholar]
- 168. Lin H, Miyauchi K, Harada T, Okita R, Takeshita E, Komaki H, Fujioka K, Yagasaki H, Goto YI, Yanaka K et al. (2018) CO2‐sensitive tRNA modification associated with human mitochondrial disease. Nat Commun 9, 1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Yarham JW, Lamichhane TN, Pyle A, Mattijssen S, Baruffini E, Bruni F, Donnini C, Vassilev A, He L, Blakely EL et al. (2014) Defective i6A37 modification of mitochondrial and cytosolic tRNAs results from pathogenic mutations in TRIT1 and its substrate tRNA. PLoS Genet 10, e1004424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Wei FY, Zhou B, Suzuki T, Miyata K, Ujihara Y, Horiguchi H, Takahashi N, Xie P, Michiue H, Fujimura A et al. (2015) Cdk5rap1‐mediated 2‐methylthio modification of mitochondrial tRNAs governs protein translation and contributes to myopathy in mice and humans. Cell Metab 21, 428–442. [DOI] [PubMed] [Google Scholar]
- 171. Zaganelli S, Rebelo‐Guiomar P, Maundrell K, Rozanska A, Pierredon S, Powell CA, Jourdain AA, Hulo N, Lightowlers RN, Chrzanowska‐Lightowlers ZM et al. (2017) The Pseudouridine synthase RPUSD4 is an essential component of mitochondrial RNA granules. J Biol Chem 292, 4519–4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Edvardson S, Elbaz‐Alon Y, Jalas C, Matlock A, Patel K, Labbe K, Shaag A, Jackman JE & Elpeleg O (2016) A mutation in the THG1L gene in a family with cerebellar ataxia and developmental delay. Neurogenetics 17, 219–225. [DOI] [PubMed] [Google Scholar]
- 173. Murphy M, Docherty NG, Griffin B, Howlin J, McArdle E, McMahon R, Schmid H, Kretzler M, Droguett A, Mezzano S et al. (2008) IHG‐1 amplifies TGF‐beta1 signaling and is increased in renal fibrosis. J Am Soc Nephrol 19, 1672–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Shaheen R, Maddirevula S, Ewida N, Alsahli S, Abdel‐Salam GMH, Zaki MS, Tala SA, Alhashem A, Softah A, Al‐Owain M et al. (2019) Genomic and phenotypic delineation of congenital microcephaly. Genet Med 21, 545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Darvish H, Azcona LJ, Alehabib E, Jamali F, Tafakhori A, Ranji‐Burachaloo S, Jen JC & Paisan‐Ruiz C (2019) A novel PUS7 mutation causes intellectual disability with autistic and aggressive behaviors. Neurol Genet 5, e356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Shaheen R, Tasak M, Maddirevula S, Abdel‐Salam GMH, Sayed ISM, Alazami AM, Al‐Sheddi T, Alobeid E, Phizicky EM & Alkuraya FS (2019) PUS7 mutations impair pseudouridylation in humans and cause intellectual disability and microcephaly. Hum Genet 138, 231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Zhang H, Hou W, Wang HL, Liu HJ, Jia XY, Zheng XZ, Zou YX, Li X, Hou L, McNutt MA et al. (2014) GSK‐3beta‐regulated N‐acetyltransferase 10 is involved in colorectal cancer invasion. Clin Cancer Res 20, 4717–4729. [DOI] [PubMed] [Google Scholar]
- 178. Zhang X, Liu J, Yan S, Huang K, Bai Y & Zheng S (2015) High expression of N‐acetyltransferase 10: a novel independent prognostic marker of worse outcome in patients with hepatocellular carcinoma. Int J Clin Exp Pathol 8, 14765–14771. [PMC free article] [PubMed] [Google Scholar]
- 179. Zhang X, Jiang G, Sun M, Zhou H, Miao Y, Liang M, Wang E & Zhang Y (2017) Cytosolic THUMPD1 promotes breast cancer cells invasion and metastasis via the AKT‐GSK3‐Snail pathway. Oncotarget 8, 13357–13366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Sand M, Skrygan M, Georgas D, Arenz C, Gambichler T, Sand D, Altmeyer P & Bechara FG (2012) Expression levels of the microRNA maturing microprocessor complex component DGCR8 and the RNA‐induced silencing complex (RISC) components argonaute‐1, argonaute‐2, PACT, TARBP1, and TARBP2 in epithelial skin cancer. Mol Carcinog 51, 916–922. [DOI] [PubMed] [Google Scholar]
- 181. Ye J, Wang J, Tan L, Yang S, Xu L, Wu X, Deng H & Tan H (2015) Expression of protein TARBP1 in human hepatocellular carcinoma and its prognostic significance. Int J Clin Exp Pathol 8, 9089–9096. [PMC free article] [PubMed] [Google Scholar]
- 182. Kato T, Daigo Y, Hayama S, Ishikawa N, Yamabuki T, Ito T, Miyamoto M, Kondo S & Nakamura Y (2005) A novel human tRNA‐dihydrouridine synthase involved in pulmonary carcinogenesis. Cancer Res 65, 5638–5646. [DOI] [PubMed] [Google Scholar]
- 183. Bykhovskaya Y, Casas K, Mengesha E, Inbal A & Fischel‐Ghodsian N (2004) Missense mutation in pseudouridine synthase 1 (PUS1) causes mitochondrial myopathy and sideroblastic anemia (MLASA). Am J Hum Genet 74, 1303–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Gatza ML, Silva GO, Parker JS, Fan C & Perou CM (2014) An integrated genomics approach identifies drivers of proliferation in luminal‐subtype human breast cancer. Nat Genet 46, 1051–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Vangipurapu J, Stancakova A, Pihlajamaki J, Kuulasmaa TM, Kuulasmaa T, Paananen J, Kuusisto J, Ferrannini E & Laakso M (2011) Association of indices of liver and adipocyte insulin resistance with 19 confirmed susceptibility loci for type 2 diabetes in 6,733 non‐diabetic Finnish men. Diabetologia 54, 563–571. [DOI] [PubMed] [Google Scholar]
- 186. Freude K, Hoffmann K, Jensen LR, Delatycki MB, des Portes V, Moser B, Hamel B, van Bokhoven H, Moraine C, Fryns JP et al. (2004) Mutations in the FTSJ1 gene coding for a novel S‐adenosylmethionine‐binding protein cause nonsyndromic X‐linked mental retardation. Am J Hum Genet 75, 305–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Frye M, Dragoni I, Chin SF, Spiteri I, Kurowski A, Provenzano E, Green A, Ellis IO, Grimmer D, Teschendorff A et al. (2010) Genomic gain of 5p15 leads to over‐expression of Misu (NSUN2) in breast cancer. Cancer Lett 289, 71–80. [DOI] [PubMed] [Google Scholar]
- 188. Alazami AM, Hijazi H, Al‐Dosari MS, Shaheen R, Hashem A, Aldahmesh MA, Mohamed JY, Kentab A, Salih MA, Awaji A et al. (2013) Mutation in ADAT3, encoding adenosine deaminase acting on transfer RNA, causes intellectual disability and strabismus. J Med Genet 50, 425–430. [DOI] [PubMed] [Google Scholar]
- 189. Slaugenhaupt SA, Blumenfeld A, Gill SP, Leyne M, Mull J, Cuajungco MP, Liebert CB, Chadwick B, Idelson M, Reznik L et al. (2001) Tissue‐specific expression of a splicing mutation in the IKBKAP gene causes familial dysautonomia. Am J Hum Genet 68, 598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Cohen JS, Srivastava S, Farwell KD, Lu HM, Zeng W, Lu H, Chao EC & Fatemi A (2015) ELP2 is a novel gene implicated in neurodevelopmental disabilities. Am J Med Genet A 167, 1391–1395. [DOI] [PubMed] [Google Scholar]
- 191. Rossi M, El‐Khechen D, Black MH, Farwell Hagman KD, Tang S & Powis Z (2017) Outcomes of diagnostic exome sequencing in patients with diagnosed or suspected autism spectrum disorders. Pediatr Neurol 70, 34–43.e2. [DOI] [PubMed] [Google Scholar]
- 192. Delaunay S, Rapino F, Tharun L, Zhou Z, Heukamp L, Termathe M, Shostak K, Klevernic I, Florin A, Desmecht H et al. (2016) Elp3 links tRNA modification to IRES‐dependent translation of LEF1 to sustain metastasis in breast cancer. J Exp Med 213, 2503–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Simpson CL, Lemmens R, Miskiewicz K, Broom WJ, Hansen VK, van Vught PW, Landers JE, Sapp P, Van Den Bosch L, Knight J et al. (2009) Variants of the elongator protein 3 (ELP3) gene are associated with motor neuron degeneration. Hum Mol Genet 18, 472–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Addis L, Ahn JW, Dobson R, Dixit A, Ogilvie CM, Pinto D, Vaags AK, Coon H, Chaste P, Wilson S et al. (2015) Microdeletions of ELP4 are associated with language impairment, autism spectrum disorder, and mental retardation. Hum Mutat 36, 842–850. [DOI] [PubMed] [Google Scholar]
- 195. Fadason T, Ekblad C, Ingram JR, Schierding WS & O'Sullivan JM (2017) Physical interactions and expression quantitative traits loci identify regulatory connections for obesity and type 2 diabetes associated SNPs. Front Genet 8, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Monies D, Vagbo CB, Al‐Owain M, Alhomaidi S & Alkuraya FS (2019) Recessive truncating mutations in ALKBH8 cause intellectual disability and severe impairment of wobble uridine modification. Am J Hum Genet 104, 1202–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Shimada K, Nakamura M, Anai S, De Velasco M, Tanaka M, Tsujikawa K, Ouji Y & Konishi N (2009) A novel human AlkB homologue, ALKBH8, contributes to human bladder cancer progression. Cancer Res 69, 3157–3164. [DOI] [PubMed] [Google Scholar]
- 198. O'Byrne JJ, Tarailo‐Graovac M, Ghani A, Champion M, Deshpande C, Dursun A, Ozgul RK, Freisinger P, Garber I, Haack TB et al. (2018) The genotypic and phenotypic spectrum of MTO1 deficiency. Mol Genet Metab 123, 28–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Gaignard P, Gonzales E, Ackermann O, Labrune P, Correia I, Therond P, Jacquemin E & Slama A (2013) Mitochondrial Infantile liver disease due to TRMU gene mutations: three new cases. JIMD Rep 11, 117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Guan MX, Yan Q, Li X, Bykhovskaya Y, Gallo‐Teran J, Hajek P, Umeda N, Zhao H, Garrido G, Mengesha E et al. (2006) Mutation in TRMU related to transfer RNA modification modulates the phenotypic expression of the deafness‐associated mitochondrial 12S ribosomal RNA mutations. Am J Hum Genet 79, 291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Lake NJ, Compton AG, Rahman S & Thorburn DR (2016) Leigh syndrome: One disorder, more than 75 monogenic causes. Ann Neurol 79, 190–203. [DOI] [PubMed] [Google Scholar]
- 202. Wu Y, Wei FY, Kawarada L, Suzuki T, Araki K, Komohara Y, Fujimura A, Kaitsuka T, Takeya M, Oike Y et al. (2016) Mtu1‐mediated thiouridine formation of mitochondrial tRNAs Is required for mitochondrial translation and is involved in reversible infantile liver injury. PLoS Genet 12, e1006355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Zeharia A, Shaag A, Pappo O, Mager‐Heckel AM, Saada A, Beinat M, Karicheva O, Mandel H, Ofek N, Segel R et al. (2009) Acute infantile liver failure due to mutations in the TRMU gene. Am J Hum Genet 85, 401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Cheng JX, Chen L, Li Y, Cloe A, Yue M, Wei J, Watanabe KA, Shammo JM, Anastasi J, Shen QJ et al. (2018) RNA cytosine methylation and methyltransferases mediate chromatin organization and 5‐azacytidine response and resistance in leukaemia. Nat Commun 9, 1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Li Y, Zheng D, Wang F, Xu Y, Yu H & Zhang H (2019) Expression of demethylase genes, FTO and ALKBH1, is associated with prognosis of gastric cancer. Dig Dis Sci 64, 1503–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Yamada Y, Yasukochi Y, Kato K, Oguri M, Horibe H, Fujimaki T, Takeuchi I & Sakuma J (2018) Identification of 26 novel loci that confer susceptibility to early‐onset coronary artery disease in a Japanese population. Biomed Rep 9, 383–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Powell CA, Kopajtich R, D'Souza AR, Rorbach J, Kremer LS, Husain RA, Dallabona C, Donnini C, Alston CL, Griffin H et al. (2015) TRMT5 mutations cause a defect in post‐transcriptional modification of mitochondrial tRNA associated with multiple respiratory‐chain deficiencies. Am J Hum Genet 97, 319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Wei L, Tian Y, Chen Y, Wei Q, Chen F, Cao B, Wu Y, Zhao B, Chen X, Xie C et al. (2019) Identification of TYW3/CRYZ and FGD4 as susceptibility genes for amyotrophic lateral sclerosis. Neurol Genet 5, e375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Huang S, Zhu P, Sun B, Guo J, Zhou H, Shu Y & Li Q (2019) Modulation of YrdC promotes hepatocellular carcinoma progression via MEK/ERK signaling pathway. Biomed Pharmacother 108859, 1–9. [DOI] [PubMed] [Google Scholar]
- 210. Kernohan KD, Dyment DA, Pupavac M, Cramer Z, McBride A, Bernard G, Straub I, Tetreault M, Hartley T, Huang L et al. (2017) Matchmaking facilitates the diagnosis of an autosomal‐recessive mitochondrial disease caused by biallelic mutation of the tRNA isopentenyltransferase (TRIT1) gene. Hum Mutat 38, 511–516. [DOI] [PubMed] [Google Scholar]
- 211. Yamamoto T, Fujimura A, Wei FY, Shinojima N, Kuroda JI, Mukasa A & Tomizawa K (2019) 2‐Methylthio conversion of N6‐isopentenyladenosine in mitochondrial tRNAs by CDK5RAP1 promotes the maintenance of glioma‐initiating cells. iScience 21, 42–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Yang XX, He XQ, Li FX, Wu YS, Gao Y & Li M (2012) Risk‐association of DNA methyltransferases polymorphisms with gastric cancer in the Southern Chinese population. Int J Mol Sci 13, 8364–8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Abdelrahman HA, Al‐Shamsi AM, Ali BR & Al‐Gazali L (2018) A null variant in PUS3 confirms its involvement in intellectual disability and further delineates the associated neurodevelopmental disease. Clin Genet 94, 586–587. [DOI] [PubMed] [Google Scholar]
- 214. Leschziner GD, Coffey AJ, Andrew T, Gregorio SP, Dias‐Neto E, Calafato M, Bentley DR, Kinton L, Sander JW & Johnson MR (2011) Q8IYL2 is a candidate gene for the familial epilepsy syndrome of Partial Epilepsy with Pericentral Spikes (PEPS). Epilepsy Res 96, 109–115. [DOI] [PubMed] [Google Scholar]
- 215. Hadjigeorgiou GM, Kountra PM, Koutsis G, Tsimourtou V, Siokas V, Dardioti M, Rikos D, Marogianni C, Aloizou AM, Karadima G et al. (2019) Replication study of GWAS risk loci in Greek multiple sclerosis patients. Neurol Sci 40, 253–260. [DOI] [PubMed] [Google Scholar]
- 216. Braun DA, Shril S, Sinha A, Schneider R, Tan W, Ashraf S, Hermle T, Jobst‐Schwan T, Widmeier E, Majmundar AJ et al. (2018) Mutations in WDR4 as a new cause of Galloway‐Mowat syndrome. Am J Med Genet A 176, 2460–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Shaheen R, Abdel‐Salam GM, Guy MP, Alomar R, Abdel‐Hamid MS, Afifi HH, Ismail SI, Emam BA, Phizicky EM & Alkuraya FS (2015) Mutation in WDR4 impairs tRNA m(7)G46 methylation and causes a distinct form of microcephalic primordial dwarfism. Genome Biol 16, 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Hicks DG, Janarthanan BR, Vardarajan R, Kulkarni SA, Khoury T, Dim D, Budd GT, Yoder BJ, Tubbs R, Schreeder MT et al. (2010) The expression of TRMT2A, a novel cell cycle regulated protein, identifies a subset of breast cancer patients with HER2 over‐expression that are at an increased risk of recurrence. BMC Cancer 10, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Festen EA, Goyette P, Green T, Boucher G, Beauchamp C, Trynka G, Dubois PC, Lagace C, Stokkers PC, Hommes DW et al. (2011) A meta‐analysis of genome‐wide association scans identifies IL18RAP, PTPN2, TAGAP, and PUS10 as shared risk loci for Crohn's disease and celiac disease. PLoS Genet 7, e1001283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Wang Y, Huang Q, Deng T, Li BH & Ren XQ (2019) Clinical significance of TRMT6 in hepatocellular carcinoma: a bioinformatics‐based study. Med Sci Monit 25, 3894–3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Shi L, Yang X, Tang D, Liu G, Yuan P, Yang Y, Chang L, Zhang L & Song D (2015) Expression and significance of m1A transmethylase, hTrm6p/hTrm61p and its related gene hTrm6/hTrm61 in bladder urothelial carcinoma. Am J Cancer Res 5, 2169–2179. [PMC free article] [PubMed] [Google Scholar]
- 222. Couch FJ, Kuchenbaecker KB, Michailidou K, Mendoza‐Fandino GA, Nord S, Lilyquist J, Olswold C, Hallberg E, Agata S, Ahsan H et al. (2016) Identification of four novel susceptibility loci for oestrogen receptor negative breast cancer. Nat Commun 7, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Steinberg S, Misch A & Sprinzl M (1993) Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res 21, 3011–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Hanada T, Suzuki T, Yokogawa T, Takemoto‐Hori C, Sprinzl M & Watanabe K (2001) Translation ability of mitochondrial tRNAsSer with unusual secondary structures in an in vitro translation system of bovine mitochondria. Genes Cells 6, 1019–1030. [DOI] [PubMed] [Google Scholar]
- 225. Messmer M, Putz J, Suzuki T, Suzuki T, Sauter C, Sissler M & Catherine F (2009) Tertiary network in mammalian mitochondrial tRNAAsp revealed by solution probing and phylogeny. Nucleic Acids Res 37, 6881–6895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Putz J, Dupuis B, Sissler M & Florentz C (2007) Mamit‐tRNA, a database of mammalian mitochondrial tRNA primary and secondary structures. RNA 13, 1184–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Yokogawa T, Watanabe Y, Kumazawa Y, Ueda T, Hirao I, Miura K & Watanabe K (1991) A novel cloverleaf structure found in mammalian mitochondrial tRNA(Ser) (UCN). Nucleic Acids Res 19, 6101–6105. [DOI] [PMC free article] [PubMed] [Google Scholar]