

Abstract
The acute respiratory distress syndrome (ARDS) is a major healthcare problem, accounting for significant mortality and long-term disability. Approximately 25% of patients with ARDS will develop an overexuberant fibrotic response, termed fibroproliferative ARDS (FP-ARDS) that portends a poor prognosis and increased mortality. The cellular pathological processes that drive FP-ARDS remain incompletely understood. We have previously shown that the transmembrane receptor-type tyrosine phosphatase protein tyrosine phosphatase-α (PTPα) promotes pulmonary fibrosis in preclinical murine models through regulation of transforming growth factor-β (TGF-β) signaling. In this study, we examine the role of PTPα in the pathogenesis of FP-ARDS in a preclinical murine model of acid (HCl)-induced acute lung injury. We demonstrate that although mice genetically deficient in PTPα (Ptpra−/−) are susceptible to early HCl-induced lung injury, they exhibit markedly attenuated fibroproliferative responses. In addition, early profibrotic gene expression is reduced in lung tissue after acute lung injury in Ptpra−/− mice, and stimulation of naïve lung fibroblasts with the BAL fluid from these mice results in attenuated fibrotic outcomes compared with wild-type littermate controls. Transcriptomic analyses demonstrate reduced extracellular matrix (ECM) deposition and remodeling in mice genetically deficient in PTPα. Importantly, human lung fibroblasts modified with a CRISPR-targeted deletion of PTPRA exhibit reduced expression of profibrotic genes in response to TGF-β stimulation, demonstrating the importance of PTPα in human lung fibroblasts. Together, these findings demonstrate that PTPα is a key regulator of fibroproliferative processes following acute lung injury and could serve as a therapeutic target for patients at risk for poor long-term outcomes in ARDS.
Keywords: acute lung injury, ARDS, fibroproliferative ARDS, PTPα
INTRODUCTION
The acute respiratory distress syndrome (ARDS) is a major healthcare problem, affecting over 190,000 people in the United States annually and accounting for more than 74,000 deaths, 3.6 million hospital stays, and over $10 billion in healthcare costs each year (1–7). Recent epidemiological data demonstrate mortality rates as high as 40%, despite advances in the supportive care of patients with ARDS (8). The global COVID-19 pandemic has highlighted the importance of this clinical problem, with some studies suggesting at least a twofold excess in the annual incidence of ARDS due to COVID-19 as compared with the prepandemic incidence of ARDS (9). Although some specific therapies have emerged for the treatment of ARDS due to SARS-CoV-2 infection (10–12), in general, treatments for ARDS remain limited to lung-protective ventilatory strategies and conservative fluid management (1, 3–7). Furthermore, impaired health-related quality of life is frequent in survivors of ARDS (13–17), with over 50% of survivors demonstrating abnormal pulmonary physiology (18) and up to 25% with persistent fibrotic changes on computed tomography (CT) scan (16, 17).
The pathogenesis of acute lung injury (ALI) and ARDS is complex. ARDS develops after exposure to an inciting stimulus, which may act directly (e.g., pneumonia or gastric acid aspiration) or indirectly (e.g., sepsis, trauma, shock, and others) to induce lung injury. The early exudative phase of ARDS involves the activation of alveolar macrophages, cytokine and chemokine release, neutrophil recruitment, and activation of the coagulation system. This results in disruption of the alveolar-capillary unit with increased permeability of both the endothelial and epithelial surfaces and subsequent leakage of protein-rich fluid into the alveolar space (19). Furthermore, the release of cytotoxic mediators from immune cells, including matrix metalloproteinases (MMPs) and reactive oxygen and nitrogen species, may result in structural damage to the lung (19–23). For most patients, the exudative phase of ARDS subsides in a timely manner, leading to a transition to the resolution phase of ARDS, which is characterized by removal of the protein-rich edema fluid, alveolar epithelial cell proliferation, and re-establishment of the alveolar-capillary barrier. However, in a subset of patients, ongoing injury and/or dysregulated repair results in persistence of inflammation, myofibroblast hyperplasia, and dysregulated deposition of extracellular matrix (ECM) components, such as collagen and fibronectin (19). An imbalance of profibrotic and antifibrotic mediators results in the development of fibrosis with subsequent physiologic consequences, such as impaired gas exchange and restrictive physiology. This pathologic response is termed fibroproliferative ARDS (FP-ARDS).
Cellular homeostasis in the lungs and other organs is tightly controlled, in part by the process of reversible phosphorylation of proteins on tyrosine residues by protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs). Dysregulation of these enzymes is pivotal to the pathogenesis of various pulmonary disease states (24). Although the role of PTKs has been well studied in both acute lung injury and fibrosis, much less is known about the role of PTPs in these pathophysiologic states. An emerging literature demonstrates the importance of PTPs in ALI/ARDS, in both the initial inflammatory response and the processes that control resolution and repair (24). Deficiency in SH2 domain-containing tyrosine phosphatase-2 (SHP2) results in enhancement of the development of both acute lung injury and pulmonary fibrosis in animal models (25, 26). Similarly, deficiency of serine/threonine protein phosphatase 2A (PP2A) in myeloid cells enhances both acute lung injury and fibrotic outcomes in LPS and bleomycin murine models (27).
We have previously reported that protein tyrosine phosphatase-α (PTPα), a ubiquitously expressed transmembrane receptor-type tyrosine phosphatase encoded by the Ptpra gene, plays important roles in regulating pulmonary fibrosis in preclinical murine models (27). Mice genetically deficient in PTPα (Ptpra−/−) are grossly normal in the naïve state other than subtle neurologic defects (28–31). Importantly, Ptpra−/− mice were protected from bleomycin-induced pulmonary fibrosis without alterations in the initial inflammatory responses. Using cell-type specific knockout mice, we determined that the phenotype was conferred by Ptpra in fibroblasts and that Ptpra−/− fibroblasts exhibited attenuated responses to transforming growth factor-β (TGF-β) in vitro (32). The best known function of PTPα is physiologic regulation of Src family kinases (SFKs) through dephosphorylation of an inhibitory tyrosine residue on Src, resulting in Src activation. Through this regulation of Src, PTPα participates in various cell processes including cell adhesion, spreading, and motility (30, 33–39). Our working model is that PTPα drives fibrogenic responses indirectly through effects on TGF-β signaling pathways.
To date, no pharmacologic treatments have been shown to be effective in reducing fibroproliferative responses in patients with ARDS. The role of PTPα in the development and resolution of FP-ARDS remains unknown. An improved understanding of the cellular regulators that contribute to the development of FP-ARDS may allow for new treatment options targeting these tyrosine phosphorylation-dependent pathways. This goal of this study is to investigate the role of PTPα in fibroproliferative responses to ARDS.
METHODS
Animal Studies
All animal experiments were performed in accordance with National Institutes of Health and the ARRIVE guidelines (40) and were approved locally by the National Jewish Health Institutional Animal Care and Use Committee. Both male and female mice matched for age were used in all experiments. Ptpraf/f mice, generated as previously described (41), were crossed to the “delete” strain, CMV-cre (B6.C-Tg(CMV-cre)1Cgn/J) mice (Jackson Laboratory, Bar Harbor, ME), to generate a global deletion of exon 4 of Ptpra. The Cre transgene was bred out of experimental animals to limit any potential toxicity from Cre expression. These CMV-cre/Ptpraf/f mice are distinct from the Ptpra−/− mice developed by colleagues (Dr. Jan Sap), which are global Ptpra knockouts generated through germline deletion of exon 3 (30). The CMV-cre/Ptpraf/f mice were developed in addition to the Ptpra−/− line to circumvent seasonal variations in breeding in the original line (30). Mice generated from either construct result in a global deletion of Ptpra with absence of PTPα protein (Supplemental Fig. S1; all Supplemental material is available at https://doi.org/10.6084/m9.figshare.16835302). Since the two strains are phenotypically identical and are devoid of any PTPα protein, we will refer to both as Ptpra−/−. Wild-type (WT) littermate control mice are referred to as Ptprawt/wt. Genotyping was performed by nonquantitative PCR on gDNA isolated from tail clips as previously described (41).
Ptpra−/− and wild-type littermate control mice (Ptprawt/wt) were treated with 0.19 N HCl to induce acute lung injury. Briefly, isoflurane (3%–5%) was used to achieve sedation and anesthesia, at which point a plastic gel loading tip bent to the appropriate angle was inserted translaryngeally into the trachea and 0.19 N HCl or saline control was instilled (40 µL volumes for female mice, 50 µL volumes for male mice). Eight to twenty animals were used for each group and animals were euthanized at days 2 or 14 following treatment. After euthanasia, three serial 1-mL bronchoalveolar lavages were performed with PBS containing 0.6 mM EDTA and pooled. Cell counts with differentials and IgM enzyme-linked immunosorbent assay (ELISA: Bethyl Laboratories, Montgomery, TX) were performed on the bronchoalveolar lavage (BAL) fluid. Following lavage, the chest was opened, and lungs were perfused with 10 mL of phosphate-buffered saline (PBS) through the right ventricle. The left lung was inflated to 25 cmH2O with 10% buffered formalin to be used for subsequent histological analyses. Embedding, sectioning, and hematoxylin-eosin (H&E) and Picrosirius Red staining were performed by the National Jewish Health histology core laboratory. The right lung was flash frozen and stored at −80°C for collagen analysis using a hydroxyproline assay as previously described (42–46). Hydroxyproline values are reported as total microgram of collagen per right upper lobe. In some cases, following perfusion, the lungs were used for isolation of primary lung fibroblasts as previously described (23). Briefly, lungs were perfused with ice-cold saline, excised, and placed into prescored 100-mm dishes. The tissue was finely minced, pressed into the surface of the dish, and covered with complete growth media. Cultures were kept at 37°C and media was changed every 2–3 days until a confluent monolayer was achieved.
Generating CRISPR Deletion of PTPRA in Human Lung Fibroblasts
A stable CRISPR-based deletion of the PTPRA gene in hTERT-transformed immortalized human lung fibroblasts (IHLFs) was performed by Applied Biological Materials, Inc. (ABM) (Richmond, BC, Canada). Briefly, three different lentivirus vectors encoding guide RNAs were screened for efficacy in disrupting the human PTPRA locus. One of these, termed sg2, was the most effective and was selected for further work. Sg2 is of the following sequence: caatttcaccaaatggaacg, which targets a sequence in the second coding exon (Exon 9 in the gene), located at nt 974–993 in the cDNA RefSeq (NM_002836.3).
The cells containing the putative deletion were cultured in Prigrow media (ABM, Richmond, BC, Canada) with 10% Fetal Select Bovine Serum (FSBS, Atlas Biologicals, Ft. Collins, CO) on rat-tail collagen-coated tissue culture plates at a density of 600 cells/well, and then subjected to limited dilution. After 24 h and then every 2–3 days, medium was replaced with 50% Prigrow + 10% FSBS and 50% fibroblast-conditioned media. Cells were expanded from wells seeded with 5 cells/well; wells seeded with fewer than 5 cells/well did not proliferate. After adequate expansion, cells were harvested for sequencing and Western blotting to confirm deletion of PTPRA and absence of protein expression. Different groups of cells were screened by PCR with two different primer sets (Table 1).
Table 1.
Primer sets
| Primer Set | Forward | Reverse | Amplicon Size, bp |
|---|---|---|---|
| 1 | attggtctttgcttggtctc | tgccactccatatctaactg | 988 |
| 2 | ggggattggtctttgcttgg | ggatcccacctagcagaaaga | 676 |
Approximately 100 amplicons from both primer sets were screened by direct sequencing. Seven distinct changes were found at the CRISPR targeting site, from single base changes to a 594 bp deletion. All these genetic alterations disrupted the PTPRA coding sequence. Certain amplicons were subcloned into the TA Cloning Vector pCR2.1-TOPO (Invitrogen, Thermo Fisher, Carlsbad, CA) and re-sequenced to isolate the various mutations. Western blotting confirmed the absence of PTPα protein in subcloned cell lines. One of these cell lines was chosen for further characterization and assays as described in the following Methods sections: Tissue Culture, qPCR/Array, and Immunoblotting. Cells with CRISPR-targeted deletion of PTPRA will be referred to as “PTPRA-KD” (knockdown), while control hTERT-transformed IHLFs will be referred to as “PTPRA-sufficient” cells.
Tissue Culture
PTPRA-KD and PTPRA-sufficient fibroblasts, NIH 3T3 (ATCC, Manassas, VA), or normal human lung fibroblasts (NHLF: Lonza, Basel, Switzerland) cells were grown in Prigrow (ABM, Richmond, BC, Canada), DMEM GlutaMAX (GibCo, Thermo Fisher, Carlsbad, CA), or FGM-2 (Lonza, Morristown, NJ) media, respectively, and supplemented with 10% serum and 10 U/mL penicillin-streptomycin (Gibco, Waltham, MA). NIH 3T3 and NHLF cells were plated on tissue culture plastic coated with fibronectin (Sigma-Aldrich, St. Louis, MO) at a concentration of 1:100 in PBS. PTPRA-KD and PTPRA-sufficient cells were grown on nontissue culture plastic coated with rat tail collagen at a concentration of 1:250 in PBS. Media was changed every 3–5 days. Cells were cultured at 37°C in 5% CO2, 95% air.
For TGF-β stimulations of PTPRA-KD and PTPRA-sufficient fibroblasts, cells were growth-arrested by reducing the concentration of serum to 1% charcoal-stripped FBS for 24 h before stimulation with 2 ng/mL recombinant human TGF-β (R&D Systems, Minneapolis, MN or PeproTech, Rocky Hill, NJ).
For stimulations with human BAL fluid, naïve human lung fibroblasts were serum-starved in 0.25% FBS media for 6 or 24 h. Human ARDS or normal control BAL fluid was diluted to 25% in low serum media. BAL fluid was derived from an NIH-sponsored SCCOR project investigating the efficacy of GMCSF in a double-blind randomized controlled trial for ARDS. Recruitment of patients with ARDS and healthy volunteers, and bronchoscopy to obtain BAL fluid was carried out as previously described (47).
qPCR/Array
Cells and/or lung tissue were lysed according to manufacturer’s instructions using RLT Buffer, RNeasy Mini extraction kits, and a QIAcube machine (Qiagen, Valencia, CA). RNA was reverse transcribed into cDNA using qScript XLT cDNA SuperMix (Quantabio, Beverly, MA) according to manufacturer’s instructions. cDNA was analyzed by real-time PCR using individual probes optimized for each of the following genes: Acta2, Col1a1, Serpine1, EDA-fibronectin, and Ctgf (Taqman, Thermo Fisher Scientific, Waltham, MA). qPCR was performed for 40 cycles on a CFX96 instrument (Bio-Rad, Hercules, CA). Relative mRNA expression levels were calculated using the 2−ΔΔCt method (48). As outlined in the figure legends, in some cases the data were natural-log transformed for analysis. In indicated experiments, cDNA was subjected to a Qiagen RT2 Profiler PCR array.
Immunoblotting
Cells were lysed in cold radioimmunoprecipitation assay (RIPA) lysis buffer. Protein concentrations were determined using a bicinchoninic acid (BCA) protein assay (Pierce Protein Biology Products, Rockford, IL). Cell lysates were analyzed by SDS-PAGE and immunoblotted for PTPα (1:1,000) (Millipore, Burlington, MA), β-actin (1:1,000) (Sigma-Aldrich, St. Louis, MO), SMAD3 (1:1,000) (Abcam), phospho-SMAD3 (1:1,000), p38 (1:1,000), or phospho-p38 (1:1,000) (Cell Signaling Technology, Danvers, MA), followed by fluorescently labeled secondary antibodies (1:15,000) (Li-Cor, Lincoln, NE) and imaged using an Odyssey imager (Li-Cor). Densitometry was performed using Image Studio Lite software (Li-Cor).
RNA Sequencing and Analysis
WT and Ptpra−/− murine embryonic fibroblasts (MEFs) were cultured with and without stimulation with TGF-β in quadruplicate. Total RNA was isolated using standard kits from Qiagen (Valencia, CA). The isolated total RNA was processed as previously described for next-generation sequencing (NGS) library construction as developed in the National Jewish Health (NJH) Genomics Facility (41). One replicate from the WT/TGF-β group was observed to have relatively high PCR duplication rates and unusually high expression in mitochondrial genes. It was removed as an outlier, leaving three replicates in the group for downstream analyses. The gene-level RNA counts were statistically modeled as a function of genotype, TGF-β stimulation, and a genotype-stimulation interaction term. Pathway analysis was performed using the Mouse Genome Informatics’ annotations to Gene Ontology (GO) biological process terms (downloaded July 2021) (49). Enrichment statistics of gene set overlaps was done using hypergeometric tests, correcting P values for the total number of gene sets using the Benjamini–Hochberg method. Heatmaps of the RNA-seq data were created using the Seaborn Python library (v. 0.11.2) (50) with Ward hierarchical clustering. Principal component analysis (PCA) was performed using the Scikit-learn library (v. 1.0.2) (51). Both PCA and hierarchical clustering were performed on the log-normalized RNA-seq counts, standardized for each gene across samples.
Statistical Analysis
Data are expressed as means ± SE, or geometric means ± 95% CI, as indicated. Multiple comparisons were performed by one-way analysis of variance (ANOVA) with Tukey’s (post hoc) test for determination of differences between groups and P values <0.05 were considered to be statistically significant. For qPCR experiments, relative gene expression was analyzed using linear mixed models, with genotype (Ptprawt/wt or Ptpra−/−) and condition (acid or saline), and genotype × condition as predictors. The tests of primary interest were acid versus saline within the Ptprawt/wt genotype, and Ptprawt/wt or Ptpra−/− within the acid condition. Random intercept terms were included for plate run and experiment. Right-skewed variables were natural-log transformed before analysis unless otherwise indicated. Comparisons between specific treatment combinations were performed using t tests derived from this model. All data were analyzed from n ≥ 3 independent experiments.
RESULTS
Ptpra−/− Mice Manifest Global Deletion of Ptpra
We generated mice with a global deletion of Ptpra by crossing animals expressing a floxed Ptpra allele to a deleter strain (B6.C-Tg(CMV-cre)1Cgn/J). Deletion of exon 4 of the Ptpra locus was confirmed by PCR genotyping. The CMV-cre/Ptpraf/f mice were back-crossed to wild-type C57BL/6 mice to breed out the CMV-Cre transgene and then bred back to homozygosity for the mutant Ptpra locus. The resulting mice, Ptpra exon4 delete/delete, will be designated as Ptpra−/− in the text and figures that follow. Littermate control mice will be designated as Ptprawt/wt. Genomic DNA (from tail clips) demonstrated recombination and deletion of exon 4 of Ptpra (Fig. 1A). Lung fibroblasts isolated from Ptpra−/− mice demonstrated complete absence of the Ptpra protein product, PTPα (Fig. 1B). WT and Ptpra−/− MEFs obtained from Dr. Jan Sap (Université de Paris) (30) are shown as positive and negative controls.
Figure 1.

CMV-cre/Ptpraf/f (Ptpra−/−) mice do not express protein tyrosine phosphatase-α (PTPα). A: tail clips from CMV-cre/Ptpraf/f (Ptpra−/−) mice were genotyped by endpoint PCR for the Ptpra allele. Genotyping results for two representative mice are shown on the left (1 and 2), with control alleles for the recombinant (RC), wild type (WT) and floxed alleles shown on the right. B: isolated lung fibroblasts from Ptpra−/− mice were lysed for Western blot analysis of expression of PTPα protein. WT and Ptpra−/− murine embryonic fibroblasts (MEFs) are shown as positive and negative controls.
Absence of PTPα Does Not Alter the Severity of Early Inflammatory Responses or Lung Injury after Treatment with HCl
To model acute lung injury/ARDS in humans, Ptpra−/− and littermate control Ptprawt/wt mice were treated with 0.19 N HCl given by intratracheal instillation. This model mimics many features of gastric aspiration injury, a common cause of ARDS in patients (52, 53). Furthermore, the HCl injury model results in the development of fibrotic changes in the lungs by 10–14 days following injury, including increased ECM protein expression, reduced compliance, and histological evidence of fibrosis, making it a useful model for the study of FP-ARDS (54–57).
Forty-eight hours after induction of lung injury with intratracheal HCI, we observed that both Ptprawt/wt mice and Ptpra−/− mice exhibited a robust acute inflammatory response. There was no difference between Ptprawt/wt mice and Ptpra−/− mice in the magnitude of the inflammatory response at this time point, as assessed by total BAL cell counts and the absolute numbers of neutrophils, macrophages, or lymphocytes present in BAL fluid (Fig. 2, A–D). To determine the extent of injury to the alveolo-capillary unit, we measured IgM levels in BAL fluid as an indicator of lung permeability (53). As illustrated in Fig. 2E, no differences were observed in Ptpra−/− mice compared with Ptprawt/wt mice. Finally, we assessed the degree of lung injury histologically and observed no differences in the extent of injury between Ptpra−/− and Ptprawt/wt mice (Fig. 2F). These findings confirm that PTPα does not influence the early inflammatory response after acute lung injury.
Figure 2.
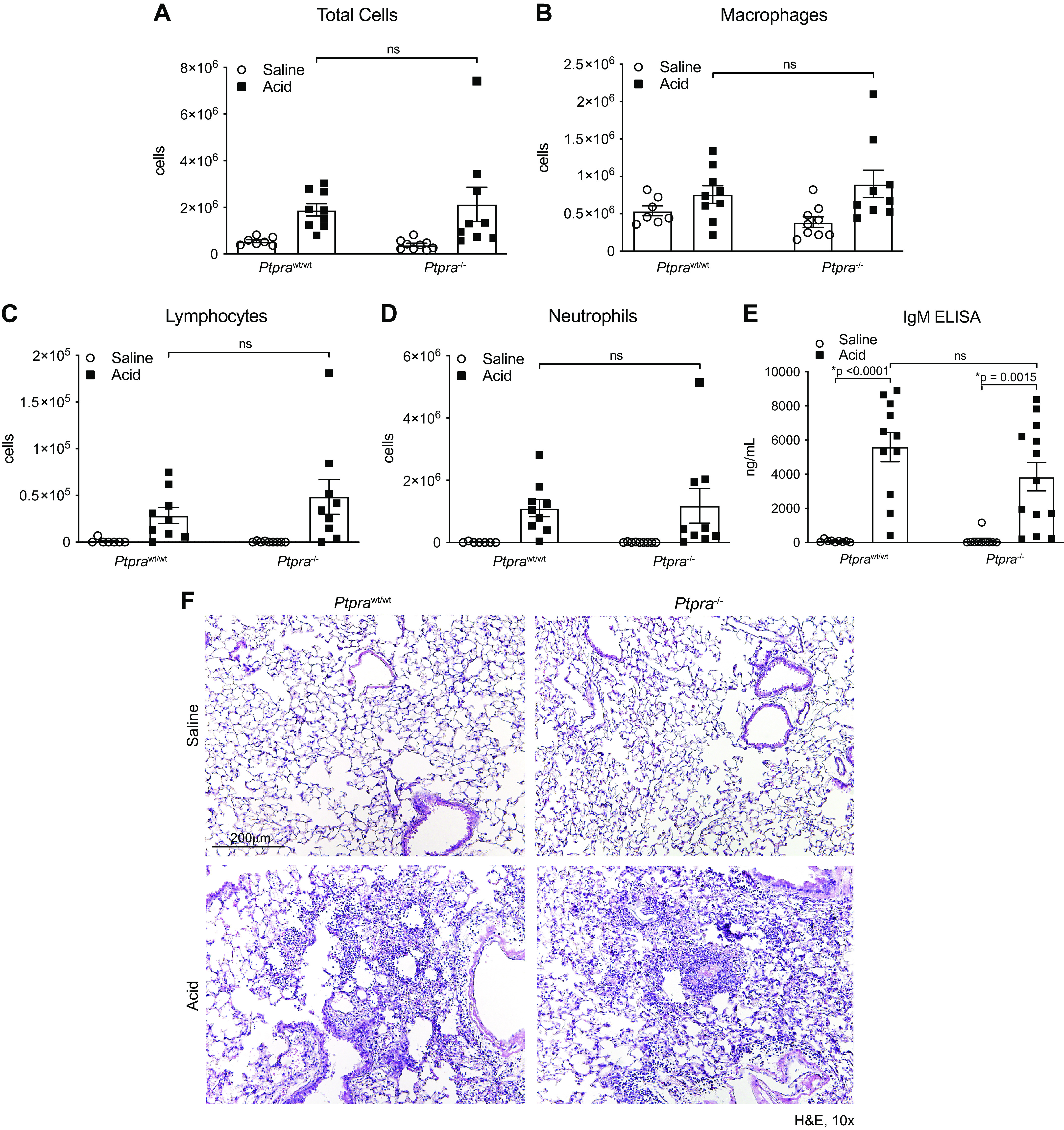
Early lung injury is not altered by the presence or absence of Ptpra. Wild-type (Ptprawt/wt) and Ptpra−/− mice were treated with 0.19 N HCl or saline and harvested at 48 h. Bronchoalveolar lavage (BAL) cell counts were performed for total cells (A), macrophages (B), lymphocytes (C), and neutrophils (D). E: IgM ELISA was performed on BAL fluid collected at the 48-h time point. F: digital images of hematoxylin-eosin (H&E) staining at 48 h after treatment with HCl or saline. Magnification ×10. In A–E, each symbol represents an individual mouse and horizontal lines represent means and SE. Values in each group represent results from pooled independent experiments with a total of 7–13 mice/group. *P < 0.05; one-way ANOVA.
Absence of PTPα Attenuates Fibroproliferative Responses after Lung Injury
We next evaluated whether genetic deletion of Ptpra altered fibrogenic responses in the lung after lung injury. We examined the lungs of Ptprawt/wt mice and Ptpra−/− mice 14 days following acid-induced lung injury. Figure 3A shows H&E and Picrosirius Red staining of lung tissue. Areas of dense fibrosis were evident in Ptprawt/wt mice 14 days after intratracheal HCl instillation, whereas Ptpra−/− mice showed preserved lung architecture. Biochemical analysis of lung collagen content using the hydroxyproline assay demonstrated a significant increase in collagen content following HCl treatment only in wild-type mice (Fig. 3B). No difference in lung compliance was observed between Ptprawt/wt mice and Ptpra−/− mice at 14 days (Supplemental Fig. S2). Together, these data demonstrate that while early inflammatory responses and degree of acute lung injury are comparable between Ptprawt/wt mice and Ptpra−/− mice, the latter are highly protected from the development of experimental fibroproliferative acute lung injury.
Figure 3.
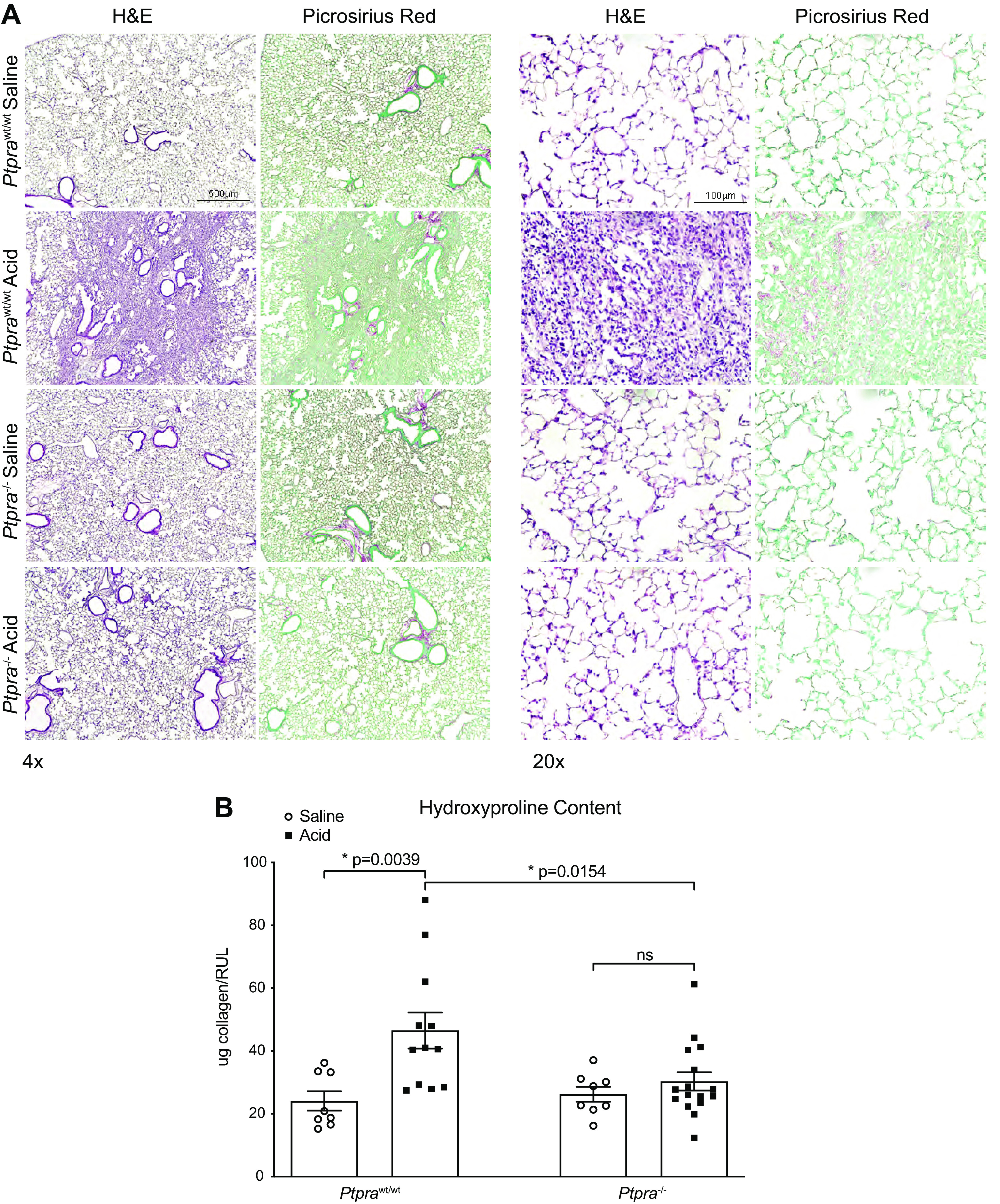
Ptpra−/− mice are protected from the development of fibrosis. Wild-type and Ptpra−/− mice were treated with 0.19 N HCl or saline and harvested at 14 days. A: digital images of hematoxylin-eosin (H&E) and Picrosirius Red staining for collagen deposition (red) were obtained. Magnification ×4 (left) and ×20 (right). B: effect of deletion of Ptpra as reflected by changes in lung hydroxyproline content. Each symbol represents an individual mouse and horizontal lines represent means and SE. Values in each group represent results from pooled independent experiments with a total of 8–16 mice/group. *P < 0.05; one-way ANOVA.
Absence of PTPα Reduces Profibrotic Gene Expression Early in the Development of FP-ARDS
In patients with FP-ARDS, inflammatory cytokines and profibrotic mediators are detectable in BAL fluid and lung tissue early in the course of disease (19, 58–60). We evaluated the homogenized lung tissue from Ptprawt/wt mice and Ptpra−/− mice 48 h after treatment with HCl or saline for the expression of various profibrotic genes. Figure 4A demonstrates increased expression of TGF-β-dependent profibrotic genes Eda-fibronectin, Ctgf, and Acta2 in the lungs of Ptprawt/wt mice treated with HCl compared with saline control. In contrast, in Ptpra−/− mice, HCl treatment did not induce a significant increase in profibrotic gene expression.
Figure 4.
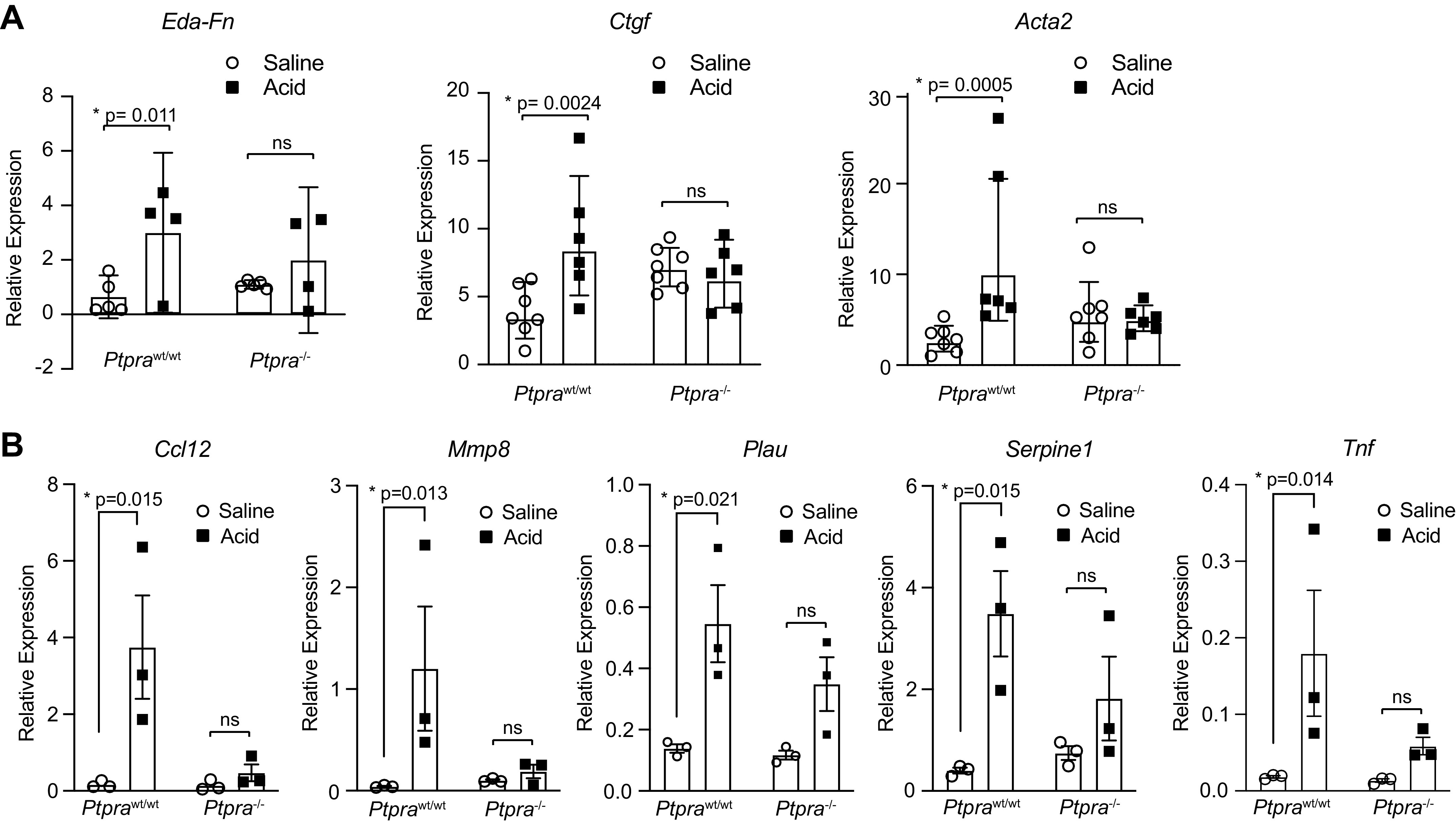
Profibrotic gene expression is reduced in the absence of Ptpra. Ptprawt/wt and Ptpra−/− mice were treated with 0.19 N HCl or saline and harvested at 48 h. A: mRNA expression of fibrotic markers Eda-fibronectin, Ctgf, and Acta2 was assessed in homogenized lung tissue using qPCR. Right-skewed variables were natural-log transformed prior to analysis for significance. Each symbol represents an individual mouse and horizontal lines represent geometric mean and 95% confidence interval (CI). Values in each group represent results from pooled independent experiments with a total of 4–7 mice/group. *P < 0.05; t test between specific treatment combinations. B: a fibrosis-focused qPCR array was performed on homogenized lung tissue. Data were normalized to the housekeeping gene GusB. qPCR array plates were performed in triplicate and data are represented as means ± SE. *P < 0.05.
To explore in more detail the alterations in profibrotic gene expression between Ptpra−/− and Ptprawt/wt mice, we used a fibrosis-focused qPCR array (RT2 Profiler PCR Array Mouse Fibrosis, Qiagen) to evaluate the expression of 84 genes involved in fibrogenesis. Several genes emerged as having a pattern of increased expression 2 days after HCl treatment in Ptprawt/wt mice, with no change in expression in Ptpra−/− mice (Fig. 4B). These genes are members of cytokines and chemokines families (Ccl12, Tnf), and are molecules that are known to participate in the formation and remodeling of the extracellular matrix (Mmp8, Serpine1, and Plau).
Absence of Ptpra Attenuates Extracellular Matrix Organization and Profibrotic Responses to TGF-β in MEFs
Given the profibrotic gene expression array findings earlier, which revealed several differentially expressed genes involved in ECM remodeling, we performed bulk RNA sequencing on WT and Ptpra−/− MEFs treated with 2 ng/mL of TGF-β or buffer control solution to further interrogate the group of genes included in the Gene Ontology (GO) term Extracellular Matrix Organization. TGF-β was chosen as the stimulus as it is known to drive ECM deposition that contributes to tissue fibrosis (61–68). The transcriptional profiles of the four groups were very distinct in principal component analysis of the top 500 genes based on coefficient of variation (Fig. 5A). The first principal component (PC1) clearly separated WT from Ptpra mutants, whereas the second principal component (PC2) separated TGF-β stimulation from buffer solution. Indeed, the top 25 loadings of PC2 were enriched for ECM organization and related GO terms (Supplemental Table S1).
Figure 5.
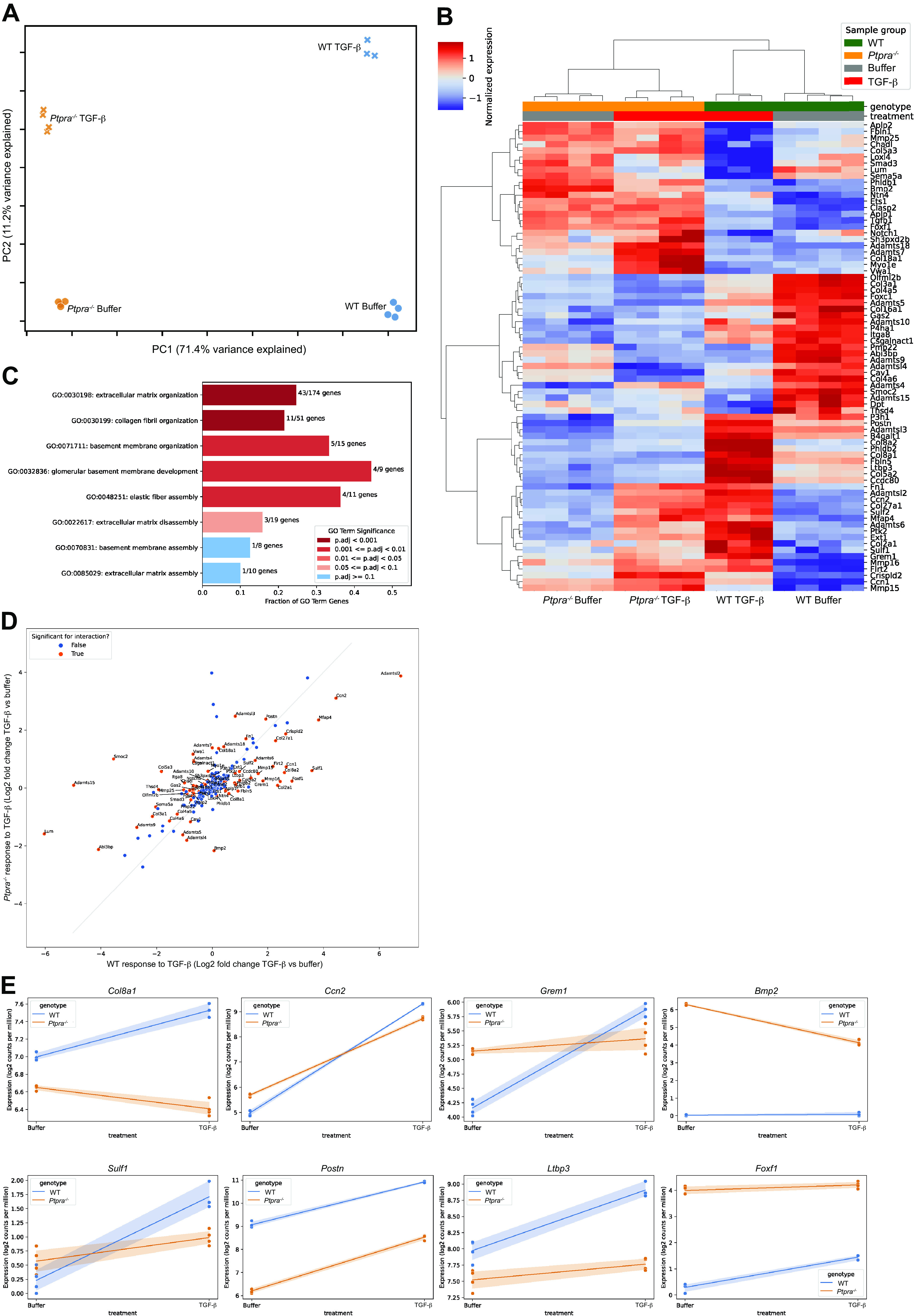
Extracellular matrix responses to transforming growth factor-β (TGF-β) stimulation are altered in the absence of Ptpra. Wild type WT and Ptpra−/− murine embryonic fibroblasts (MEFs) were stimulated with 2 ng/mL of TGF-β for 24 h and harvested for bulk RNA sequencing. The “ECM Organization” GO term pathway was interrogated. A: principal component analysis (PCA) shows clear clustering of transcriptomic data. A single sample (WT, TGF-β, 24 h stimulation) was excluded due to poor quality RNA. B: heatmap of differentially expressed genes (DEGs) within the “ECM Organization” GO term pathway that were significant for the interaction comparison between WT and Ptpra−/− after stimulation with TGF-β at a false discovery rate (FDR) of 0.05. C: bar plot of the “parent” ECM Organization GO Term (GO:0030198) and its “children” ordered by P value significance on the y-axis, with x-axis showing the fraction of overlap between the GO term’s annotated genes and the significant genes in the analysis. D: scatterplot of genes within the ECM Organization GO term pathway. The x-axis shows log2-fold change for TGF-β over buffer in the WT fibroblasts, and the y-axis shows log2-fold change for TGF-β over buffer in the Ptpra−/− cells. Genes that reached significance are shown in orange. E: a subset of genes within the ECM Organization GO term that demonstrate differential responses to TGF-β based on genotype (WT vs. Ptpra−/−) are shown.
A total of 2,060 genes were found to respond differently to TGF-β stimulation in Ptpra−/− MEFs compared with WT MEFs [significant for the interaction term of the statistical model using a false discovery rate (FDR) of 0.05]. Enrichment analysis demonstrated a statistically significant level of overlap between these differentially responding genes and the 174 genes annotated to the ECM Organization GO term (adjusted P value <0.001), with nearly a quarter of the ECM term’s genes also showing differential regulation to TGF-β stimulation. A heatmap of the differentially expressed genes (DEGs) in this pathway is shown in Fig. 5B. Figure 5C further examines the “children” of the parent ECM Organization GO term and demonstrates the significance level (based on P value), and the fraction of overlap between the significant genes in the analysis and annotated genes in the GO term. Qualitatively, DEGs within this pathway tended to lie on the axis demonstrating upregulation in the WT cells after TGF-β stimulation, with little to no change in the Ptpra−/− fibroblasts, as demonstrated in the scatterplot (Fig. 5D). Representative plots for DEGs that are relevant to the pathogenesis of pulmonary fibrosis, and that are significant for the interaction at 24 h after TGF-β stimulation, are shown in Fig. 5E. Of note, Bmp2 and Foxf1, which are known to be antifibrotic, show significantly higher baseline expression in the Ptpra−/− MEFs.
Genetic Deletion of PTPRA in Human Lung Fibroblasts Attenuates Profibrotic Responses to TGF-β
Although the antifibrotic effect of deletion of Ptpra in mice is compelling, to date, no small molecule inhibitors of this enzyme exist, making the in vitro study of its effects in human systems challenging. To compare the responses of human fibroblasts to our prior findings in murine fibroblasts, we generated human lung fibroblasts with a stable CRISPR-targeted deletion of the PTPRA gene (PTPRA-KD). These cells do not have any detectable expression of the PTPα protein product (Fig. 6A). PTPRA-KD fibroblasts, along with control PTPRA-sufficient fibroblasts, were stimulated with 2 ng/mL of TGF-β for 24 h, and fibrogenic gene expression was assessed using qPCR analysis. Figure 6B demonstrates significant attenuation in the TGF-β-induced mRNA expression of SERPINE1, CTGF, and CYR61/CCN1. These findings further validate the results of the RNA-seq analysis (Fig. 5), in which these genes show a similar pattern of expression based on presence or absence of Ptpra.
Figure 6.
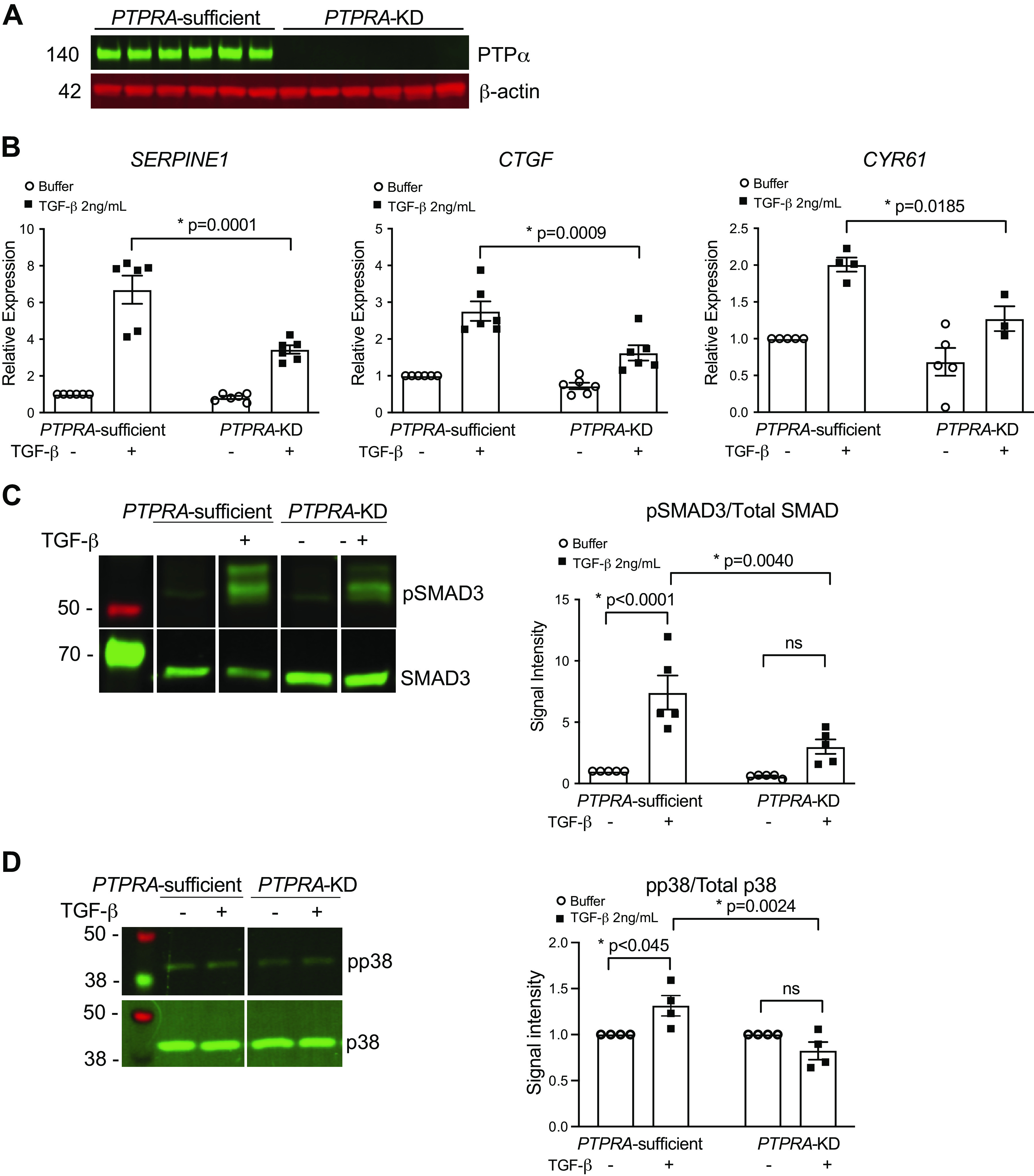
CRISPR-targeted knockdown of PTPRA results in attenuated responses of human fibroblasts to transforming growth factor-β (TGF-β) stimulation. A: CRISPR-targeted knockdown of PTPRA in hTERT-transformed immortalized human lung fibroblasts (IHLFs) results in efficient deletion of the protein tyrosine phosphatase-α (PTPα) protein by Western blotting. B: PTPRA-sufficient and PTPRA-KD fibroblasts were treated with 2 ng/mL of TGF-β, or buffer for 24 h and harvested to analyze the expression of SERPINE1, CTGF, and CYR61 by qPCR. C: PTPRA-sufficient and PTPRA-KD fibroblasts were treated with 2 ng/mL of TGF-β for 30 min and harvested, after which phosphorylation status of SMAD3 was assessed by Western blotting. Densitometry shows expression of phospho-SMAD3 over total SMAD3. D: PTPRA-sufficient and PTPRA-KD fibroblasts were treated with 2 ng/mL of TGF-β for 15 min and harvested, after which phosphorylation status of p38 was assessed by Western blotting. Densitometry shows expression of phospho-p38 over total p38. Data represent mean values ± SE from at least three independent experiments. *P < 0.05; one-way ANOVA.
Next, PTPRA-KD and PTPRA-sufficient fibroblasts treated with buffer or exogenous TGF-β for 15 or 30 min were harvested for protein analysis. In the PTPRA-KD cells, we observed significantly reduced phosphorylation of SMAD3 (Fig. 6C), a key step in the canonical TGF-β signaling cascade. Similarly, we observed reduced signaling through noncanonical TGF-β pathways, demonstrated by attenuation of p38 phosphorylation in PTPRA-KD cells (Fig. 6D). These findings provide strong evidence that genetic deletion of PTPRA attenuates TGF-β signaling in human lung fibroblasts.
Human ARDS- and Murine Acute Lung Injury-BAL Fluid Induces Expression of TGF-β-Dependent Profibrotic Genes in Lung Fibroblasts, Which Is Attenuated in the Absence of PTPα
We next sought to determine the importance of PTPα in fibrogenic responses of fibroblasts to BAL fluid obtained from mechanically ventilated patients with ARDS at 2–3 days after onset of respiratory failure (47). BAL fluid from normal human subjects was used as a control. Etiology of ARDS for each patient is reported in Supplemental Table S2. qPCR analysis was performed to assess expression of TGF-β-dependent profibrotic genes in normal human lung fibroblasts (NHLFs). We observed a wide range of alterations in gene expression in response to the ARDS BAL fluid. A trend was seen toward increased expression of all genes tested after treatment with BAL fluid from patient with ARDS, with statistically significant differences in expression of COL1A1 and SERPINE1 compared with control patient BAL fluid (Fig. 7, A and B). Expression of nonsignificant genes is shown in Supplemental Fig. S3.
Figure 7.

Bronchoalveolar lavage (BAL) fluid from patients with acute respiratory distress syndrome (ARDS) and murine lung injury-BAL fluid induces expression of transforming growth factor-β (TGF-β)-dependent profibrotic genes in lung fibroblasts, which is attenuated in the absence of protein tyrosine phosphatase-α (PTPα). BAL fluid collected from patients with ARDS or normal human controls was used to stimulate cultured normal human lung fibroblasts (NHLFs). After 24 h, cells were harvested for qPCR expression of COL1A1 (A) and SERPINE1 (B). BAL fluid from Ptprawt/wt and Ptpra−/− mice treated for 48 h with HCl or saline was used to stimulate cultured NIH 3T3 fibroblasts. After 6 h, cells were harvested for qPCR expression of Ctgf (C) and Acta2 (D). Right-skewed variables were natural-log transformed prior to analysis for significance. Each symbol represents an individual patient (n = 14 ARDS patients, 4 controls) or experimental animal (n = 4–8 mice/group), and horizontal lines represent geometric mean and 95% confidence interval (CI). *P ≤ 0.05; t test between specific treatment combinations.
We performed similar experiments using the BAL fluid from HCl-treated Ptprawt/wt and Ptpra−/− mice (48-h time point after HCl instillation) as a stimulus for cultured murine NIH 3T3 fibroblasts. Like BAL fluid from patients with ARDS, we found that BAL fluid from wild-type mice subjected to HCl instillation induced significant alterations in profibrotic gene expression and myofibroblast differentiation in NIH 3T3 cells, including increased expression of TGF-β-dependent profibrotic genes (Ctgf, Acta2). By contrast, the BAL fluid from HCl-treated Ptpra−/− mice produced significantly attenuated responses (Fig. 7, C and D, and Supplemental Fig. S3). This suggests that the absence of PTPα attenuates the fibrogenic properties of the distal airway and alveolar environment in the setting of acute lung injury.
DISCUSSION
In this manuscript, we observed that genetic deletion of Ptpra is protective against the development of lung fibrosis in a model of fibroproliferative-ARDS and have characterized some of the cellular fibrogenic pathways regulated by Ptpra that can drive fibroproliferation in this preclinical model of fibroproliferative ALI/ARDS. We demonstrated that although the early inflammatory responses and the magnitude of lung injury are comparable in Ptprawt/wt and Ptpra−/− mice, the pathological fibrotic response that transpires after the acute lung injury phase is markedly attenuated in Ptpra−/− mice. The preclinical model of acid-induced ALI was found to be inflammatory in nature, and therefore highly relevant to the pathogenesis of ARDS and its fibroproliferative sequelae. The molecular basis for the differences in fibroproliferation was notably manifest early during lung injury, with mRNA expression of key profibrotic genes attenuated in Ptpra−/− mice within 48 h following the onset of lung injury. We also demonstrated that the BAL fluid of mice genetically deficient in Ptpra induced a significantly reduced fibrogenic response in cultured murine lung fibroblasts compared with BAL fluid from wild-type mice subjected to acid-induced ALI.
The common underpinnings of these responses to lung injury lie in alterations to TGF-β signaling and changes in expression of genes that affect ECM deposition and remodeling. Fibrogenesis is highly dependent on signaling pathways downstream of growth factors such as TGF-β, a fundamental driver of fibrosis in the lung and other organs (69–77). ECM deposition and organization is one of the many processes that is regulated by TGF-β, in part through canonical and noncanonical signaling events involving phosphorylation of SMAD proteins and mitogen-activated protein (MAP) kinases, respectively (61–66). The development of FP-ARDS is also likely due in part to excess TGF-β expression in injured lungs (67, 68). We have previously assessed mechanisms by which PTPα promotes the fibrotic process, including promoting TGF-β signaling pathways through activation of Src kinase (27) and via expression of MMP3 in fibroblasts (38). Here, we demonstrate that absence of PTPα protein reduces canonical and noncanonical TGF-β signaling in human fibroblasts by attenuating SMAD and p38 MAPK phosphorylation, respectively.
We have demonstrated that although PTPα does not play a major role in control of the acute inflammatory response to acid instillation, it does function as a key checkpoint in the process of fibrogenesis, specifically through its actions in fibroblasts. Fibroblasts are extremely sensitive to changes in the ECM and the effects of mechanical strain (78). PTPα plays important roles in cell adhesion, spreading, and motility. Specifically, it is involved in mechanotransduction and the regulation of integrin-mediated events. In fibroblasts, PTPα colocalizes with αv integrins during cell spreading, is required for αvβ3 integrin-mediated cell-matrix connections, and contributes to force-dependent signal transduction pathways through activation of SFKs (36). It is known that activation of TGF-β in vitro can occur via binding of αv-containing integrins, including αvβ3, to the RGD sequence on the latency-associated peptide (LAP) of TGF-β1. Interestingly, αvβ3-null mice are not resistant to bleomycin-induced fibrosis (79). This integrin is ubiquitously expressed, as is PTPα. It is possible that in the absence of Ptpra, the reduced integrin-mediated cell-matrix connections that results from lack of colocalization and binding to αv integrins causes impaired mechanotransduction, attenuated downstream signaling, and reduced activation of TGF-β, all of which results in reduced myofibroblast differentiation and attenuated fibrogenesis.
Further evidence that PTPα controls ECM deposition and remodeling is provided through transcriptomic analyses using qPCR arrays and bulk RNA sequencing. Many ECM-related genes were differentially expressed based on genotype following HCl treatment or TGF-β stimulation. These include Grem1, Ltbp3, Ccn2/Ctgf, and Sulf1, which are TGF-β responsive and have been shown to play roles in the development of pulmonary fibrosis (80–83). Expression of Periostin (Postn), a matricellular protein that binds to ECM components, participates in signal transduction, and is increased in areas of fibrosis in the lungs of patients with idiopathic pulmonary fibrosis (IPF), is also attenuated in the absence of PTPα in fibroblasts (84). Serpine1 is similarly upregulated in the tissue of Ptprawt/wt mice and in normal human and murine fibroblasts, but not in those that lack PTPα. In pathologic conditions, excessive inhibition of urokinase-type plasminogen activator and tissue-type plasminogen activator (uPA and tPA) by Serpine1 results in accumulation of collagen and other ECM proteins, ultimately leading to scar formation (85–89). We have previously shown reduced Serpine1 expression in MEFs that lack PTPα (27). Furthermore, Serpine1 plays roles in regulation of MMPs, which may also contribute to fibrosis. As noted, urokinase-type plasminogen activator (encoded by the Plau gene) under normal circumstances plays roles in tissue homeostasis by controlling cellular proteolytic degradation of ECM proteins (88). Upregulation of Plau in our qPCR arrays may reflect preservation of some normal repair processes following lung injury, which are overwhelmed by dysregulated pathologic fibrotic signals. Notably in our analysis, some genes known to have antifibrotic properties, such as Bmp2 and Foxf1 (90, 91), demonstrated higher baseline levels in the absence of Ptpra. These baseline alterations may indicate a more global attenuation of profibrotic pathways in fibroblasts genetically deficient in Ptpra.
Given our compelling findings in a murine model of FP-ARDS and in vitro responses to TGF-β, we interrogated analogous responses in human fibroblasts. Since there are no small molecular inhibitors for PTPα, this is the first opportunity to evaluate the importance of PTPα in human lung fibroblasts through stable CRISPR-Cas9 targeted silencing of the PTPRA gene. Although transient silencing of PTPRA has been performed in human synovial fibroblasts, we are the first to create a stable deletion of PTPRA in immortalized human lung fibroblasts. Prior studies investigating the role of PTPα in the pathogenesis of rheumatoid arthritis used transient knockdown of PTPRA in fibroblast-like synoviocytes (FLS). Importantly, these studies showed that deletion of PTPRA resulted in impaired spreading, migration, and invasiveness of human FLS in response to various stimuli (92). In our observations of human fibroblasts with CRISPR-targeted deletion of PTPRA, we similarly demonstrated attenuation of responses to stimulation with TGF-β. Notably, the reduced profibrotic gene expression in these fibroblasts mirrors the findings we previously demonstrated in Ptpra−/− MEFs (27).
We tested the effects of BAL fluid from healthy controls and patients with ARDS on the induction of fibrotic responses in normal lung fibroblasts. We found significantly increased expression of COL1A1 and SERPINE1 in response to incubation of human lung fibroblasts with human ARDS BAL fluid as compared with BAL fluid from healthy controls, and a trend toward increased expression of other TGF-β-dependent profibrotic genes, including ACTA2 and CTGF. We have previously demonstrated that Ptpra−/− MEFs have reduced expression of Acta2, Ctgf, and Col1a1 in response to TGF-β as compared with wild-type MEFs (24), along with other TGF-β-dependent genes. Furthermore, we demonstrated that homogenized lung tissue from HCl-treated Ptpra−/− mice also resulted in reduced expression of a similar profile of genes, suggesting that mechanisms of fibrosis that rely on PTPα activity in mice have correlates in human ARDS. The fact that not every gene tested was significantly different between patients with ARDS and controls may be related to heterogeneity in ARDS severity, differences in mechanism of injury, or the time point at which BAL fluid was collected (2–3 days after onset of ARDS), which may not correlate exactly to the timeframe of acid-induced lung injury in our murine model.
There are several limitations to our current studies. Although we can identify that many of the profibrotic gene expression signatures that can be induced in lung fibroblasts by murine alveolar lavage fluid are similar to those that can be induced by human ARDS alveolar fluid, we did not examine lung tissue in these patients to determine whether expression within the lung tissue was altered. Furthermore, whether PTPα expression or activation is altered in patients with ARDS is unknown. We have previously shown that in patients with idiopathic pulmonary fibrosis, PTPα protein expression is not different from controls and have hypothesized that changes in enzyme activity is likely more important than change in total protein abundance. Future studies with lung tissue from deceased patients with ARDS will permit analysis of both tissue and alveolar fluid.
The recognition in recent years of ARDS as a heterogeneous syndrome has highlighted the increasingly important task of identifying, defining, and understanding the various subphenotypes of ARDS. The heterogeneity within these groups is in part responsible for the failures of multiple clinical trials for therapeutics in ARDS, with previous studies failing to account for the physiologic differences in etiology and outcomes among the disease subgroups (93, 94). Understanding the mechanisms that result in some patients developing fibrotic outcomes following lung injury is key to planning clinical trials and to developing new therapeutic outcomes that target this maladaptive response. This is especially relevant in the current era of COVID-19, as we begin to understand that post-ARDS fibroproliferation is an important consequence in a subset of these patients and as the pandemic continues to result in severe disease worldwide (95, 96).
Our recognition that the absence of PTPα selectively reduces fibroproliferative responses following acute lung injury in a murine model is important, particularly in that the early inflammatory responses are preserved. Specific targeting of PTPα would thus allow for potentially beneficial immune responses to occur, particularly in the case of ARDS that is induced by pneumonia or sepsis, yet would attenuate the detrimental fibroproliferative sequelae. Currently, there are no small molecule inhibitors of PTPα, but antibody-mediated inhibitors are a potential therapeutic approach to inhibit this key control node in pathological fibrogenic responses. In addition, the downstream target of PTPα, Src, can be specifically targeted by small molecule inhibitors, which may inhibit fibroproliferative responses.
DATA AVAILABILITY
The data that support the findings of this study will be made available upon reasonable request from the corresponding author.
SUPPLEMENTAL DATA
Supplemental Figs. S1–S3 and Supplemental Tables S1 and S2: https://doi.org/10.6084/m9.figshare.16835302.
GRANTS
This work was supported by funding from the National Institutes of Health R01HL132950 and R01HL157424 (to G. P. Downey), R24AA019661 (to E. L. Burnham), The Parker B. Francis Foundation (to Y. Aschner), NIH/NCATS Colorado CTSA KL2 TR002534 (to Y. Aschner), and NIH K08HL135279-01A1 (to Y. Aschner).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Y.A., E.L.B., and G.P.D. conceived and designed research; Y.A., K.A.C., K.M.B., D.G.F., H.M.R., M.R.N., C.L.M., K.C.A., P.R.R., and K.W.K. performed experiments; Y.A., K.A.C., K.M.B., D.G.F., C.L.M., M.S., K.C.A., C.M.M., and P.R.R., analyzed data; Y.A., K.A.C., C.M.M., and G.P.D. interpreted results of experiments; Y.A. and K.A.C. prepared figures; Y.A. and G.P.D. drafted manuscript; Y.A., M.S., K.C.A., C.M.M., and G.P.D. edited and revised manuscript; Y.A., K.A.C., K.M.B., D.G.F., H.M.R., M.R.N., C.L.M., M.S., K.C.A., C.M.M., P.R.R., K.W.K., E.L.B., and G.P.D. approved final version of manuscript.
REFERENCES
- 1.Acute Respiratory Distress Syndrome N, Brower RG, Matthay MA, Morris A, Schoenfeld D, Thompson BT, Wheeler A. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med 342: 1301–1308, 2000. doi: 10.1056/NEJM200005043421801. [DOI] [PubMed] [Google Scholar]
- 2.Matthay MA, Zemans RL. The acute respiratory distress syndrome: pathogenesis and treatment. Annu Rev Pathol 6: 147–163, 2011. doi: 10.1146/annurev-pathol-011110-130158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.National Heart L, Blood Institute Acute Respiratory Distress Syndrome Clinical Trials N, Wiedemann HP, Wheeler AP, Bernard GR, Thompson BT, Hayden D, deBoisblanc B, Connors AF Jr, Hite RD, Harabin AL. Comparison of two fluid-management strategies in acute lung injury. N Engl J Med 354: 2564–2575, 2006. doi: 10.1056/NEJMoa062200. [DOI] [PubMed] [Google Scholar]
- 4.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med 353: 1685–1693, 2005. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 5.Rubenfeld GD, Herridge MS. Epidemiology and outcomes of acute lung injury. Chest 131: 554–562, 2007. doi: 10.1378/chest.06-1976. [DOI] [PubMed] [Google Scholar]
- 6.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med 342: 1334–1349, 2000. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 7.Force ADT, Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, Camporota L, Slutsky AS; ARDS Definition Task Force. Acute respiratory distress syndrome: the Berlin definition. JAMA 307: 2526–2533, 2012. doi: 10.1001/jama.2012.5669. [DOI] [PubMed] [Google Scholar]
- 8.Bellani G, Laffey JG, Pham T, Fan E, Brochard L, Esteban A, Gattinoni L, van Haren F, Larsson A, McAuley DF, Ranieri M, Rubenfeld G, Thompson BT, Wrigge H, Slutsky AS, Pesenti A; LUNG SAFE Investigators, ESICM Trials Group. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA 315: 788–800, 2016. doi: 10.1001/jama.2016.0291. [DOI] [PubMed] [Google Scholar]
- 9.Matthay MA, Leligdowicz A, Liu KD. Biological mechanisms of COVID-19 acute respiratory distress syndrome. Am J Respir Crit Care Med 202: 1489–1491, 2020. doi: 10.1164/rccm.202009-3629ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Group RC, Horby P, Lim WS, Emberson JR, Mafham M, Bell JL, Linsell L, Staplin N, Brightling C, Ustianowski A, Elmahi E, Prudon B, Green C, Felton T, Chadwick D, Rege K, Fegan C, Chappell LC, Faust SN, Jaki T, Jeffery K, Montgomery A, Rowan K, Juszczak E, Baillie JK, Haynes R, Landray MJ. Dexamethasone in hospitalized patients with Covid-19. N Engl J Med 384: 693–704, 2021. doi: 10.1056/NEJMoa2021436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beigel JH, Tomashek KM, Dodd LE, Mehta AK, Zingman BS, Kalil AC, et al. Remdesivir for the treatment of Covid-19—final report. N Engl J Med 383: 1813–1826, 2020. doi: 10.1056/NEJMoa2007764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Salama C, Han J, Yau L, Reiss WG, Kramer B, Neidhart JD, Criner GJ, Kaplan-Lewis E, Baden R, Pandit L, Cameron ML, Garcia-Diaz J, Chavez V, Mekebeb-Reuter M, Lima de Menezes F, Shah R, Gonzalez-Lara MF, Assman B, Freedman J, Mohan SV. Tocilizumab in patients hospitalized with Covid-19 pneumonia. N Engl J Med 384: 20–30, 2021. doi: 10.1056/NEJMoa2030340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herridge MS, Cheung AM, Tansey CM, Matte-Martyn A, Diaz-Granados N, Al-Saidi F, Cooper AB, Guest CB, Mazer CD, Mehta S, Stewart TE, Barr A, Cook D, Slutsky AS; Canadian Critical Care Trials Group. One-year outcomes in survivors of the acute respiratory distress syndrome. N Engl J Med 348: 683–693, 2003. doi: 10.1056/NEJMoa022450. [DOI] [PubMed] [Google Scholar]
- 14.Herridge MS, Tansey CM, Matte A, Tomlinson G, Diaz-Granados N, Cooper A, Guest CB, Mazer CD, Mehta S, Stewart TE, Kudlow P, Cook D, Slutsky AS, Cheung AM; Canadian Critical Care Trials Group. Functional disability 5 years after acute respiratory distress syndrome. N Engl J Med 364: 1293–1304, 2011. doi: 10.1056/NEJMoa1011802. [DOI] [PubMed] [Google Scholar]
- 15.Heyland DK, Groll D, Caeser M. Survivors of acute respiratory distress syndrome: relationship between pulmonary dysfunction and long-term health-related quality of life. Crit Care Med 33: 1549–1556, 2005. doi: 10.1097/01.ccm.0000168609.98847.50. [DOI] [PubMed] [Google Scholar]
- 16.Burnham EL, Hyzy RC, Paine R 3rd, Coley C 2nd, Kelly AM, Quint LE, Lynch D, Janssen WJ, Moss M, Standiford TJ. Chest CT features are associated with poorer quality of life in acute lung injury survivors. Crit Care Med 41: 445–456, 2013. doi: 10.1097/CCM.0b013e31826a5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilcox ME, Patsios D, Murphy G, Kudlow P, Paul N, Tansey CM, Chu L, Matte A, Tomlinson G, Herridge MS. Radiologic outcomes at 5 years after severe ARDS. Chest 143: 920–926, 2013. doi: 10.1378/chest.12-0685. [DOI] [PubMed] [Google Scholar]
- 18.Masclans JR, Roca O, Munoz X, Pallisa E, Torres F, Rello J, Morell F. Quality of life, pulmonary function, and tomographic scan abnormalities after ARDS. Chest 139: 1340–1346, 2011. doi: 10.1378/chest.10-2438. [DOI] [PubMed] [Google Scholar]
- 19.Burnham EL, Janssen WJ, Riches DW, Moss M, Downey GP. The fibroproliferative response in acute respiratory distress syndrome: mechanisms and clinical significance. Eur Respir J 43: 276–285, 2014. doi: 10.1183/09031936.00196412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dos Santos CC. Advances in mechanisms of repair and remodelling in acute lung injury. Intensive Care Med 34: 619–630, 2008. doi: 10.1007/s00134-007-0963-x. [DOI] [PubMed] [Google Scholar]
- 21.Davey A, McAuley DF, O'Kane CM. Matrix metalloproteinases in acute lung injury: mediators of injury and drivers of repair. Eur Respir J 38: 959–970, 2011. doi: 10.1183/09031936.00032111. [DOI] [PubMed] [Google Scholar]
- 22.Lee WL, Downey GP. Leukocyte elastase: physiological functions and role in acute lung injury. Am J Respir Crit Care Med 164: 896–904, 2001. doi: 10.1164/ajrccm.164.5.2103040. [DOI] [PubMed] [Google Scholar]
- 23.Gonzalez-Lopez A, Astudillo A, Garcia-Prieto E, Fernandez-Garcia MS, Lopez-Vazquez A, Batalla-Solis E, Taboada F, Fueyo A, Albaiceta GM. Inflammation and matrix remodeling during repair of ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol 301: L500–L509, 2011. doi: 10.1152/ajplung.00010.2011. [DOI] [PubMed] [Google Scholar]
- 24.Aschner Y, Downey GP. The importance of tyrosine phosphorylation control of cellular signaling pathways in respiratory disease: pY and pY Not. Am J Respir Cell Mol Biol 59: 535–547, 2018. doi: 10.1165/rcmb.2018-0049TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chichger H, Braza J, Duong H, Harrington EO. SH2 domain-containing protein tyrosine phosphatase 2 and focal adhesion kinase protein interactions regulate pulmonary endothelium barrier function. Am J Respir Cell Mol Biol 52: 695–707, 2015. doi: 10.1165/rcmb.2013-0489OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tzouvelekis A, Yu G, Lino Cardenas CL, Herazo-Maya JD, Wang R, Woolard T, Zhang Y, Sakamoto K, Lee H, Yi JS, DeIuliis G, Xylourgidis N, Ahangari F, Lee PJ, Aidinis V, Herzog EL, Homer R, Bennett AM, Kaminski N. SH2 domain-containing phosphatase-2 is a novel antifibrotic regulator in pulmonary fibrosis. Am J Respir Crit Care Med 195: 500–514, 2017. doi: 10.1164/rccm.201602-0329OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aschner Y, Khalifah AP, Briones N, Yamashita C, Dolgonos L, Young SK, Campbell MN, Riches DW, Redente EF, Janssen WJ, Henson PM, Sap J, Vacaresse N, Kapus A, McCulloch CA, Zemans RL, Downey GP. Protein tyrosine phosphatase α mediates profibrotic signaling in lung fibroblasts through TGF-β responsiveness. Am J Pathol 184: 1489–1502, 2014. doi: 10.1016/j.ajpath.2014.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cohen-Sharir Y, Kuperman Y, Apelblat D, den Hertog J, Spiegel I, Knobler H, Elson A. Protein tyrosine phosphatase α inhibits hypothalamic leptin receptor signaling and regulates body weight in vivo. FASEB J 33: 5101–5111, 2019. doi: 10.1096/fj.201800860RR. [DOI] [PubMed] [Google Scholar]
- 29.Skelton MR, Ponniah S, Wang DZ, Doetschman T, Vorhees CV, Pallen CJ. Protein tyrosine phosphatase α (PTP α) knockout mice show deficits in Morris water maze learning, decreased locomotor activity, and decreases in anxiety. Brain Res 984: 1–10, 2003. doi: 10.1016/S0006-8993(03)02839-7. [DOI] [PubMed] [Google Scholar]
- 30.Su J, Muranjan M, Sap J. Receptor protein tyrosine phosphatase α activates Src-family kinases and controls integrin-mediated responses in fibroblasts. Curr Biol 9: 505–511, 1999. doi: 10.1016/s0960-9822(99)80234-6. [DOI] [PubMed] [Google Scholar]
- 31.Takahashi N, Nielsen KS, Aleksic B, Petersen S, Ikeda M, Kushima I, Vacaresse N, Ujike H, Iwata N, Dubreuil V, Mirza N, Sakurai T, Ozaki N, Buxbaum JD, Sap J. Loss of function studies in mice and genetic association link receptor protein tyrosine phosphatase α to schizophrenia. Biol Psychiatry 70: 626–635, 2011. doi: 10.1016/j.biopsych.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aschner Y, Nelson M, Brenner M, Roybal H, Beke K, Meador C, Foster D, Reynolds PR, Anderson K, Redente EF, Matsuda J, Riches DWH, Groshong SD, Pozzi A, Sap J, Wang Q, Rajshankar D, McCulloch CAG, Zemans RL, Downey GP. Protein tyrosine phosphatase-α amplifies TGF-β-dependent pro-fibrotic signaling in lung fibroblasts. Am J Physiol Lung Cell Mol Physiol 319: L294–L311, 2020. doi: 10.1152/ajplung.00235.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.den Hertog J, Pals CE, Peppelenbosch MP, Tertoolen LG, de Laat SW, Kruijer W. Receptor protein tyrosine phosphatase α activates pp60c-src and is involved in neuronal differentiation. EMBO J 12: 3789–3798, 1993. doi: 10.1002/j.1460-2075.1993.tb06057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herrera Abreu MT, Penton PC, Kwok V, Vachon E, Shalloway D, Vidali L, Lee W, McCulloch CA, Downey GP. Tyrosine phosphatase PTPα regulates focal adhesion remodeling through Rac1 activation. Am J Physiol Cell Physiol 294: C931–C944, 2008. doi: 10.1152/ajpcell.00359.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ponniah S, Wang DZ, Lim KL, Pallen CJ. Targeted disruption of the tyrosine phosphatase PTPα leads to constitutive downregulation of the kinases Src and Fyn. Curr Biol 9: 535–538, 1999. doi: 10.1016/s0960-9822(99)80238-3. [DOI] [PubMed] [Google Scholar]
- 36.von Wichert G, Jiang G, Kostic A, De Vos K, Sap J, Sheetz MP. RPTP-α acts as a transducer of mechanical force on αv/β3-integrin-cytoskeleton linkages. J Cell Biol 161: 143–153, 2003. doi: 10.1083/jcb.200211061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Q, Rajshankar D, Branch DR, Siminovitch KA, Herrera Abreu MT, Downey GP, McCulloch CA. Protein-tyrosine phosphatase-α and Src functionally link focal adhesions to the endoplasmic reticulum to mediate interleukin-1-induced Ca2+ signaling. J Biol Chem 284: 20763–20772, 2009. [Erratum in J Biol Chem 284: 27020, 2009]. doi: 10.1074/jbc.M808828200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Q, Rajshankar D, Laschinger C, Talior-Volodarsky I, Wang Y, Downey GP, McCulloch CA. Importance of protein-tyrosine phosphatase-α catalytic domains for interactions with SHP-2 and interleukin-1-induced matrix metalloproteinase-3 expression. J Biol Chem 285: 22308–22317, 2010. doi: 10.1074/jbc.M110.102426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zheng XM, Wang Y, Pallen CJ. Cell transformation and activation of pp60c-src by overexpression of a protein tyrosine phosphatase. Nature 359: 336–339, 1992. doi: 10.1038/359336a0. [DOI] [PubMed] [Google Scholar]
- 40.Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol 8: e1000412, 2010. doi: 10.1371/journal.pbio.1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aschner Y, Nelson M, Brenner M, Roybal H, Beke K, Meador C, Foster D, Correll KA, Reynolds PR, Anderson K, Redente EF, Matsuda J, Riches DWH, Groshong SD, Pozzi A, Sap J, Wang Q, Rajshankar D, McCulloch CAG, Zemans RL, Downey GP. Protein tyrosine phosphatase-α amplifies transforming growth factor-β-dependent profibrotic signaling in lung fibroblasts. Am J Physiol Lung Cell Mol Physiol 319: L294–L311, 2020. doi: 10.1152/ajplung.00235.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCubbrey AL, Barthel L, Mohning MP, Redente EF, Mould KJ, Thomas SM, Leach SM, Danhorn T, Gibbings SL, Jakubzick CV, Henson PM, Janssen WJ. Deletion of c-FLIP from CD11b(hi) macrophages prevents development of bleomycin-induced lung fibrosis. Am J Respir Cell Mol Biol 58: 66–78, 2018. doi: 10.1165/rcmb.2017-0154OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Redente EF, Keith RC, Janssen W, Henson PM, Ortiz LA, Downey GP, Bratton DL, Riches DW. Tumor necrosis factor-α accelerates the resolution of established pulmonary fibrosis in mice by targeting profibrotic lung macrophages. Am J Respir Cell Mol Biol 50: 825–837, 2014. doi: 10.1165/rcmb.2013-0386OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Junquiera LC, Junqueira LC, Brentani RR. A simple and sensitive method for the quantitative estimation of collagen. Anal Biochem 94: 96–99, 1979. doi: 10.1016/0003-2697(79)90795-4. [DOI] [PubMed] [Google Scholar]
- 45.Yamashita CM, Dolgonos L, Zemans RL, Young SK, Robertson J, Briones N, Suzuki T, Campbell MN, Gauldie J, Radisky DC, Riches DW, Yu G, Kaminski N, McCulloch CA, Downey GP. Matrix metalloproteinase 3 is a mediator of pulmonary fibrosis. Am J Pathol 179: 1733–1745, 2011. doi: 10.1016/j.ajpath.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frankel SK, Moats-Staats BM, Cool CD, Wynes MW, Stiles AD, Riches DW. Human insulin-like growth factor-IA expression in transgenic mice promotes adenomatous hyperplasia but not pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 288: L805–L812, 2005. doi: 10.1152/ajplung.00420.2004. [DOI] [PubMed] [Google Scholar]
- 47.Paine R 3rd, Standiford TJ, Dechert RE, Moss M, Martin GS, Rosenberg AL, Thannickal VJ, Burnham EL, Brown MB, Hyzy RC. A randomized trial of recombinant human granulocyte-macrophage colony stimulating factor for patients with acute lung injury. Crit Care Med 40: 90–97, 2012. doi: 10.1097/CCM.0b013e31822d7bf0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3: 1101–1108, 2008. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 49.Bult CJ, Blake JA, Smith CL, Kadin JA, Richardson JE; Mouse Genome Database Group. Mouse Genome Database (MGD) 2019. Nucleic Acids Res 47: D801–D806, 2019. doi: 10.1093/nar/gky1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Waskom ML. seaborn: statistical data visualization. J Open Source Softw 6: 3021, 2021. doi: 10.21105/joss.03021. [DOI] [Google Scholar]
- 51.Pedregosa F, Gaël V, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Müller A, Nothman J, Louppe G, Prettenhofer P, Weiss R, Dubourg V, Vanderplas J, Passos A, Cournapeau D, Brucher M, Perrot M, Duchesnay É. Scikit-learn: machine learning in Python. arXiv 1201.0490, 2012.
- 52.B BM, Lawson WE, Oury TD, Sisson TH, Raghavendran K, Hogaboam CM. Animal models of fibrotic lung disease. Am J Respir Cell Mol Biol 49: 167–179, 2013. doi: 10.1165/rcmb.2013-0094TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Matute-Bello G, Frevert CW, Martin TR. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol 295: L379–L399, 2008. doi: 10.1152/ajplung.00010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Amigoni M, Bellani G, Scanziani M, Masson S, Bertoli E, Radaelli E, Patroniti N, Di Lelio A, Pesenti A, Latini R. Lung injury and recovery in a murine model of unilateral acid aspiration: functional, biochemical, and morphologic characterization. Anesthesiology 108: 1037–1046, 2008. doi: 10.1097/ALN.0b013e318173f64f. [DOI] [PubMed] [Google Scholar]
- 55.Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, Yang P, Sarao R, Wada T, Leong-Poi H, Crackower MA, Fukamizu A, Hui CC, Hein L, Uhlig S, Slutsky AS, Jiang C, Penninger JM. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 436: 112–116, 2005. doi: 10.1038/nature03712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marinova M, Solopov P, Dimitropoulou C, Colunga Biancatelli RML, Catravas JD. Acute exposure of mice to hydrochloric acid leads to the development of chronic lung injury and pulmonary fibrosis. Inhal Toxicol 31: 147–160, 2019. doi: 10.1080/08958378.2019.1624895. [DOI] [PubMed] [Google Scholar]
- 57.Patel BV, Wilson MR, Takata M. Resolution of acute lung injury and inflammation: a translational mouse model. Eur Respir J 39: 1162–1170, 2012. doi: 10.1183/09031936.00093911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marshall RP, Bellingan G, Webb S, Puddicombe A, Goldsack N, McAnulty RJ, Laurent GJ. Fibroproliferation occurs early in the acute respiratory distress syndrome and impacts on outcome. Am J Respir Crit Care Med 162: 1783–1788, 2000. doi: 10.1164/ajrccm.162.5.2001061. [DOI] [PubMed] [Google Scholar]
- 59.Meduri GU, Kohler G, Headley S, Tolley E, Stentz F, Postlethwaite A. Inflammatory cytokines in the BAL of patients with ARDS. Persistent elevation over time predicts poor outcome. Chest 108: 1303–1314, 1995. doi: 10.1378/chest.108.5.1303. [DOI] [PubMed] [Google Scholar]
- 60.Krein PM, Sabatini PJ, Tinmouth W, Green FH, Winston BW. Localization of insulin-like growth factor-I in lung tissues of patients with fibroproliferative acute respiratory distress syndrome. Am J Respir Crit Care Med 167: 83–90, 2003. doi: 10.1164/rccm.2201012. [DOI] [PubMed] [Google Scholar]
- 61.Verrecchia F, Mauviel A. Transforming growth factor-β signaling through the Smad pathway: role in extracellular matrix gene expression and regulation. J Invest Dermatol 118: 211–215, 2002. doi: 10.1046/j.1523-1747.2002.01641.x. [DOI] [PubMed] [Google Scholar]
- 62.Massague J, Wotton D. Transcriptional control by the TGF-β/Smad signaling system. EMBO J 19: 1745–1754, 2000. doi: 10.1093/emboj/19.8.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Massague J, Chen YG. Controlling TGF-β signaling. Genes Dev 14: 627–644, 2000. doi: 10.1101/gad.14.6.627. [DOI] [PubMed] [Google Scholar]
- 64.Verrecchia F, Chu ML, Mauviel A. Identification of novel TGF-β/Smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach. J Biol Chem 276: 17058–17062, 2001. doi: 10.1074/jbc.M100754200. [DOI] [PubMed] [Google Scholar]
- 65.Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGF β-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J 17: 3091–3100, 1998. doi: 10.1093/emboj/17.11.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lai CF, Feng X, Nishimura R, Teitelbaum SL, Avioli LV, Ross FP, Cheng SL. Transforming growth factor-β up-regulates the β 5 integrin subunit expression via Sp1 and Smad signaling. J Biol Chem 275: 36400–36406, 2000. doi: 10.1074/jbc.M002131200. [DOI] [PubMed] [Google Scholar]
- 67.Synenki L, Chandel NS, Budinger GR, Donnelly HK, Topin J, Eisenbart J, Jovanovic B, Jain M. Bronchoalveolar lavage fluid from patients with acute lung injury/acute respiratory distress syndrome induces myofibroblast differentiation. Crit Care Med 35: 842–848, 2007. doi: 10.1097/01.CCM.0000257254.87984.69. [DOI] [PubMed] [Google Scholar]
- 68.Budinger GR, Chandel NS, Donnelly HK, Eisenbart J, Oberoi M, Jain M. Active transforming growth factor-β1 activates the procollagen I promoter in patients with acute lung injury. Intensive Care Med 31: 121–128, 2005. doi: 10.1007/s00134-004-2503-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bergeron A, Soler P, Kambouchner M, Loiseau P, Milleron B, Valeyre D, Hance AJ, Tazi A. Cytokine profiles in idiopathic pulmonary fibrosis suggest an important role for TGF-β and IL-10. Eur Respir J 22: 69–76, 2003. doi: 10.1183/09031936.03.00014703. [DOI] [PubMed] [Google Scholar]
- 70.Border WA, Noble NA. Transforming growth factor β in tissue fibrosis. N Engl J Med 331: 1286–1292, 1994. doi: 10.1056/NEJM199411103311907. [DOI] [PubMed] [Google Scholar]
- 71.Khalil N, Bereznay O, Sporn M, Greenberg AH. Macrophage production of transforming growth factor β and fibroblast collagen synthesis in chronic pulmonary inflammation. J Exp Med 170: 727–737, 1989. doi: 10.1084/jem.170.3.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Khalil N, O'Connor RN, Unruh HW, Warren PW, Flanders KC, Kemp A, Bereznay OH, Greenberg AH. Increased production and immunohistochemical localization of transforming growth factor-β in idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol 5: 155–162, 1991. doi: 10.1165/ajrcmb/5.2.155. [DOI] [PubMed] [Google Scholar]
- 73.Khalil N, Parekh TV, O'Connor R, Antman N, Kepron W, Yehaulaeshet T, Xu YD, Gold LI. Regulation of the effects of TGF-β 1 by activation of latent TGF-β 1 and differential expression of TGF-β receptors (T β R-I and T β R-II) in idiopathic pulmonary fibrosis. Thorax 56: 907–915, 2001. doi: 10.1136/thorax.56.12.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li M, Krishnaveni MS, Li C, Zhou B, Xing Y, Banfalvi A, Li A, Lombardi V, Akbari O, Borok Z, Minoo P. Epithelium-specific deletion of TGF-β receptor type II protects mice from bleomycin-induced pulmonary fibrosis. J Clin Invest 121: 277–287, 2011. doi: 10.1172/JCI42090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sime PJ, Xing Z, Graham FL, Csaky KG, Gauldie J. Adenovector-mediated gene transfer of active transforming growth factor-β1 induces prolonged severe fibrosis in rat lung. J Clin Invest 100: 768–776, 1997. doi: 10.1172/JCI119590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Westergren-Thorsson G, Hernnas J, Sarnstrand B, Oldberg A, Heinegard D, Malmstrom A. Altered expression of small proteoglycans, collagen, and transforming growth factor-β 1 in developing bleomycin-induced pulmonary fibrosis in rats. J Clin Invest 92: 632–637, 1993. doi: 10.1172/JCI116631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wynn TA. Integrating mechanisms of pulmonary fibrosis. J Exp Med 208: 1339–1350, 2011. doi: 10.1084/jem.20110551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Duscher D, Maan ZN, Wong VW, Rennert RC, Januszyk M, Rodrigues M, Hu M, Whitmore AJ, Whittam AJ, Longaker MT, Gurtner GC. Mechanotransduction and fibrosis. J Biomech 47: 1997–2005, 2014. doi: 10.1016/j.jbiomech.2014.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tatler AL, Jenkins G. TGF-β activation and lung fibrosis. Proc Am Thorac Soc 9: 130–136, 2012. doi: 10.1513/pats.201201-003AW. [DOI] [PubMed] [Google Scholar]
- 80.Yue X, Li X, Nguyen HT, Chin DR, Sullivan DE, Lasky JA. Transforming growth factor-β1 induces heparan sulfate 6-O-endosulfatase 1 expression in vitro and in vivo. J Biol Chem 283: 20397–20407, 2008. doi: 10.1074/jbc.M802850200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Myllarniemi M, Lindholm P, Ryynanen MJ, Kliment CR, Salmenkivi K, Keski-Oja J, Kinnula VL, Oury TD, Koli K. Gremlin-mediated decrease in bone morphogenetic protein signaling promotes pulmonary fibrosis. Am J Respir Crit Care Med 177: 321–329, 2008. doi: 10.1164/rccm.200706-945OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hinz B. The extracellular matrix and transforming growth factor-β1: tale of a strained relationship. Matrix Biol 47: 54–65, 2015. doi: 10.1016/j.matbio.2015.05.006. [DOI] [PubMed] [Google Scholar]
- 83.Ponticos M, Holmes AM, Shi-Wen X, Leoni P, Khan K, Rajkumar VS, Hoyles RK, Bou-Gharios G, Black CM, Denton CP, Abraham DJ, Leask A, Lindahl GE. Pivotal role of connective tissue growth factor in lung fibrosis: MAPK-dependent transcriptional activation of type I collagen. Arthritis Rheum 60: 2142–2155, 2009. doi: 10.1002/art.24620. [DOI] [PubMed] [Google Scholar]
- 84.Ashley SL, Wilke CA, Kim KK, Moore BB. Periostin regulates fibrocyte function to promote myofibroblast differentiation and lung fibrosis. Mucosal Immunol 10: 341–351, 2017. doi: 10.1038/mi.2016.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Eitzman DT, Krauss JC, Shen T, Cui J. Ginsburg. Lack of plasminogen activator inhibitor-1 effect in a transgenic mouse model of metastatic melanoma. Blood 87: 4718–4722, 1996. [PubMed] [Google Scholar]
- 86.Hattori N, Degen JL, Sisson TH, Liu H, Moore BB, Pandrangi RG, Simon RH, Drew AF. Bleomycin-induced pulmonary fibrosis in fibrinogen-null mice. J Clin Invest 106: 1341–1350, 2000. doi: 10.1172/JCI10531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Eitzman DT, McCoy RD, Zheng X, Fay WP, Shen T, Ginsburg D, Simon RH. Bleomycin-induced pulmonary fibrosis in transgenic mice that either lack or overexpress the murine plasminogen activator inhibitor-1 gene. J Clin Invest 97: 232–237, 1996. doi: 10.1172/JCI118396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sisson TH, Hattori N, Xu Y, Simon RH. Treatment of bleomycin-induced pulmonary fibrosis by transfer of urokinase-type plasminogen activator genes. Hum Gene Ther 10: 2315–2323, 1999. doi: 10.1089/10430349950016960. [DOI] [PubMed] [Google Scholar]
- 89.Bauman KA, Wettlaufer SH, Okunishi K, Vannella KM, Stoolman JS, Huang SK, Courey AJ, White ES, Hogaboam CM, Simon RH, Toews GB, Sisson TH, Moore BB, Peters-Golden M. The antifibrotic effects of plasminogen activation occur via prostaglandin E2 synthesis in humans and mice. J Clin Invest 120: 1950–1960, 2010. doi: 10.1172/JCI38369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Black M, Milewski D, Le T, Ren X, Xu Y, Kalinichenko VV, Kalin TV. FOXF1 inhibits pulmonary fibrosis by preventing CDH2-CDH11 cadherin switch in myofibroblasts. Cell Rep 23: 442–458, 2018. doi: 10.1016/j.celrep.2018.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.De Langhe E, Cailotto F, De Vooght V, Aznar-Lopez C, Vanoirbeek JA, Luyten FP, Lories RJ. Enhanced endogenous bone morphogenetic protein signaling protects against bleomycin induced pulmonary fibrosis. Respir Res 16: 38, 2015. doi: 10.1186/s12931-015-0202-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Stanford SM, Svensson MN, Sacchetti C, Pilo CA, Wu DJ, Kiosses WB, Hellvard A, Bergum B, Muench GR, Elly C, Liu YC, den Hertog J, Elson A, Sap J, Mydel P, Boyle DL, Corr M, Firestein GS, Bottini N. Receptor protein tyrosine phosphatase α-mediated enhancement of rheumatoid synovial fibroblast signaling and promotion of arthritis in mice. Arthritis Rheumatol 68: 359–369, 2016. doi: 10.1002/art.39442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wilson JG, Calfee CS. ARDS subphenotypes: understanding a heterogeneous syndrome. Crit Care 24: 102, 2020. doi: 10.1186/s13054-020-2778-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Reilly JP, Calfee CS, Christie JD. Acute respiratory distress syndrome phenotypes. Semin Respir Crit Care Med 40: 19–30, 2019. doi: 10.1055/s-0039-1684049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Vasarmidi E, Tsitoura E, Spandidos DA, Tzanakis N, Antoniou KM. Pulmonary fibrosis in the aftermath of the COVID-19 era (Review). Exp Ther Med 20: 2557–2560, 2020. doi: 10.3892/etm.2020.8980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Spagnolo P, Balestro E, Aliberti S, Cocconcelli E, Biondini D, Casa GD, Sverzellati N, Maher TM. Pulmonary fibrosis secondary to COVID-19: a call to arms? Lancet Respir Med 8: 750–752, 2020. doi: 10.1016/S2213-2600(20)30222-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figs. S1–S3 and Supplemental Tables S1 and S2: https://doi.org/10.6084/m9.figshare.16835302.
Data Availability Statement
The data that support the findings of this study will be made available upon reasonable request from the corresponding author.