

ABSTRACT
A self-hardening three-dimensional (3D)-porous composite bone graft consisting of 65 wt% hydroxyapatite (HA) and 35 wt% aragonite was fabricated using a 3D-Bioplotter®. New tetracalcium phosphate and dicalcium phosphate anhydrous/aragonite/gelatine paste formulae were developed to overcome the phase separation of the liquid and solid components. The mechanical properties, porosity, height and width stability of the end products were optimised through a systematic analysis of the fabrication processing parameters including printing pressure, printing speed and distance between strands. The resulting 3D-printed bone graft was confirmed to be a mixture of HA and aragonite by X-ray diffraction, Fourier transform infrared spectroscopy and energy dispersive X-ray spectroscopy. The compression strength of HA/aragonite was between 0.56 and 2.49 MPa. Cytotoxicity was assessed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in vitro. The osteogenicity of HA/aragonite was evaluated in vitro by alkaline phosphatase assay using human umbilical cord matrix mesenchymal stem cells, and in vivo by juxtapositional implantation between the tibia and the anterior tibialis muscle in rats. The results showed that the scaffold was not toxic and supported osteogenic differentiation in vitro. HA/aragonite stimulated new bone formation that bridged host bone and intramuscular implants in vivo. We conclude that HA/aragonite is a biodegradable and conductive bone formation biomaterial that stimulates bone regeneration. Since this material is formed near 37°C, it will have great potential for incorporating bioactive molecules to suit personalised application; however, further study of its biodegradation and osteogenic capacity is warranted. The study was approved by the Animal Ethical Committee at Tongji Medical School, Huazhong University of Science and Technology (IACUC No. 738) on October 1, 2017.
Key Words: biofabrication, cytotoxicity, hydroxyapatite/aragonite, osteogenesis
Introduction
Trauma, cancer, aging and genetic diseases can result in bone lesions and bone defects. An important step in the repair of such defects is to provide suitable scaffolds to retain mechanical and functional integrity for bone regeneration. Hydroxyapatite (HA) is the most studied biomaterial used as a bone graft substitute, with its clinical application dating back to the 1970s.1-3
Porous HA has now replaced the dense form due to its better integration into bone. After HA is implanted into bone defects, new bone grows into the pores.4, 5 However, HA has a low biodegradation rate.2, 3, 6 Another widely-studied inorganic material is calcium carbonate, which is low cost, safe, accessible, biocompatible, bioresorptive and osteoconductive.7, 8 Calcium carbonate has three crystalline polymorphs, namely calcite, aragonite and vaterite.9, 10 The differences in the morphological forms of calcium carbonate may be related to their synthetic conditions. Calcite is the stable form and exists as a trigonal crystalline form in nature. Vaterite is the least stable and exists as a hexagonal crystalline form. In contact with water, vaterite slowly dissolves and recrystallises to a stable form.11
Aragonite occurs as the orthorhombic form and has been the exclusive focus of research attention due to its biocompatible properties.12 Aragonite is biocompatible and can be integrated into and replaced by bone.13, 14 However, due to its fast biodegradation rate, aragonite alone is not suitable as a bone graft.
Our previous study demonstrated that a partially-converted coralline HA/calcium carbonate can be completely biodegraded with great osteogenic capacity.15 Since coral is an endangered species and a limited resource, a composite bone graft created by addition of aragonite into porous HA may enhance the biodegradation of HA whilst retaining ideal osteogenic capacity. However, it is a technical challenge to fabricate porous HA/aragonite using conventional methods.
There are a variety of techniques for fabricating porous bone scaffolds. Traditional technologies include chemical foaming16 and foam-gel technology,17, 18 solvent casting and particle leaching technology,19 freeze drying20 as well as thermally-induced phase separation.21 Pore magnitude, shape, and interconnectivity, however, are not completely controllable using these techniques. Furthermore, an ideal bone graft should be biocompatible and biodegradable. It should also have suitable mechanical properties. The interconnected porous structure of bone grafts allows vascularization, cell spreading and effective transport of nutrients, oxygen, and waste, as well as growth factors. This process favours continuous ingrowth of bone tissue from the surface into the inner part of the scaffold.22 However, scaffolds with porosity designed for particular defects are hard to fabricate using most of these techniques.23-26
Additive manufacturing, also termed three-dimensional (3D)-printing, can be one solution to the design and fabrication of such bone scaffolds.27-29 Various additive manufacturing processing techniques allow the building of complex form scaffolds directly from a computer-aided design model in stereolithography file format.30 At present, a variety of additive manufacturing techniques involving stereolithography, fused deposition modelling, and selective laser sintering have been developed for tissue engineering applications.22, 29, 31 However, the products fabricated using these techniques may suffer from the effects of the high temperatures involved, which prevent the incorporation of bioactive molecules.28
The use of low temperature 3D-printing of calcium phosphate cements has been demonstrated in the literature.32, 33 The binders used for calcium phosphate powders are acidic, which pose issues for biocompatibility.27, 28 In recent work, collagen or hydrogel has been used to develop 3D-printed scaffolds, either alone or combined with other materials.34, 35 However, crosslinking is required to improve the mechanical strength and accuracy of 3D-printed scaffolds.35, 36 In recent years, a new form of HA called HA cement has been developed for treating cranial defects.32 Fast-setting calcium phosphate cements such as tetracalcium phosphate (TTCP) and dicalcium phosphate anhydrous (DCPA) use a sodium phosphate solution as the liquid phase and the setting time is around 5 minutes.32, 37
Natural bone forms at body temperature (37°C) and is a composite consisting of HA, calcium carbonate and other minerals deposited onto an organic extracellular matrix. The crystalline structure and composition differ from those of artificial calcium phosphate ceramics such as HA, β-tricalcium phosphate (β-TCP) and their hybrid scaffolds (HA/TCP), which take longer than natural bone to degrade after implantation.6 We hypothesise that to fabricate HA under near-physiological conditions (37°C) in combination with calcium carbonate may not only improve the mechanical strength of the scaffolds without crosslinking, but will also enhance the osteogenic and biodegradation properties.
In this study, we report a novel HA/aragonite bone graft substitute fabricated by low temperature 3D bio-printing on a 3D bio-plotter®. The effect of printing parameters on the porosity and compression strength was analysed. The cytotoxicity and osteogenic capacity of the HA/aragonite was assessed in vitro using human umbilical cord matrix mesenchymal stem cells. Soft tissue responses to HA/aragonite were evaluated in vivo after implantation between the tibia and the tibialis anterior muscle in a rat model.
Methods
Preparation of HA/aragonite
TTCP/DCPA paste
TTCP (Shanghai Rebone Biomaterials Co., Ltd., Shanghai, China), DCPA (Shanghai Rebone Biomaterials Co., Ltd.) and aragonite powders (Sigma-Aldrich, St. Louis, MO, USA) were ground together for 20 minutes using a pestle and mortar. The molar ratio of TTCP/DCPA was 1:1, according to the equation
| (1) |
In brief, after being ground, 10 g of the combined powders were mixed with 5 g carrier liquid consisting of a gelatine solution in water. The paste was loaded into a 3D-Bioplotter® (Envisiontec GmbH, Gladbeck, Germany) through cartridges and printed within 30 minutes.
Design and optimization of parameters for bioplotting
In this study computer-aided design software (Visual Machines, Envisiontec GmbH) was used to design the grid-like structure of HA/aragonite, which was formed into 10 mm × 10 mm × 5 mm blocks (Figure 1). The paste was printed by a 3D-Bioplotter® (Envision Developer, Envisiontec GmbH), using a 0.4 mm nozzle, with layer height of 0.4 mm.
Figure 1. (A) Bioplotter®. (B) The design of hydroxyapatite/aragonite. (C) The end product of fabrication. Scale bar: 10 mm.

To optimise the bioplotting process, a multi-level experimental trial was performed to analyse the effect of printing pressure, printing speed and distance between printed strands, and in total nine tests were performed. To compare the differently-printed 3D bio-printed bone graft samples, the resulting mechanical properties, specifically the compressive strength and the porosity, were assessed.
Characterization of HA/aragonite
X-ray powder diffraction of HA/aragonite and CaCO3 was performed using an X’pert3 powder X-ray diffractometer (Malvern PANalytical B.V., Almelo, Netherlands) with a Pixel detector. The dataset was collected using Cu Kα radiation in the 2θ range of 9°-36° at a scan speed of 3°/min.
Fourier transform infrared (FTIR) spectroscopy of HA/aragonite was carried out using a VERTEX 70 (Bruker, Kontich, Belgium). Chemical changes to the micro-structure were scrutinised by analysing the absorbances and positions of the infrared bands.
The surface morphology of HA/aragonite was observed by scanning electron microscopy (Sirion 200 and Quanta 200 FEI; FEI Company, Hillsboro, OR, USA) following platinum sputter coating for 300 seconds to avoid charging.
A universal material testing machine (Wuhan Guoliang Instrument Co. Ltd., Wuhan, China) was used to carry out the compression loading properties with a test standard GB/T4740-1999.38 The porosity of HA/aragonite was tested by Archimedes’ method.
The theoretical porosity of the HA/aragonite (φ) can be calculated from equation (2)
| (2) |
Where m1 is the dry weight of the HA/aragonite, m2 is the weight of the HA/aragonite soaked in water, v is the volume of the HA/aragonite, and ρ is the density of water.
Thermal analysis of the HA/aragonite was performed using a thermogravimetric analyser (Pyris 1; PerkinElmer, Waltham, MA, USA) in the range of 0-1000°C at a heating rate of 10 K/min.
In vitro assessments
The design of the in vitro tests is shown in Figure 2.
Figure 2. Experimental design of in vitro and in vivo tests. The blocks of HA/aragonite were sterilised by autoclaving at 121°C, 15 psi (1 psi = 6894.76 Pa) for 30 minutes. Further in vitro and in vivo tests were designed to compare between HA/aragonite and clinically-applied gelatine sponge. HA: hydroxyapatite; U & Pb: uranyl acetate and lead citrate staining.

Cytotoxicity
Test samples of HA/aragonite and gelatine sponge were diced into 2 mm × 2 mm × 2 mm size cubes and soaked in Dulbecco’s modified Eagle medium/F12 (DMEM/F12 medium; Life Technology; Thermo Fisher Scientific, Waltham, MA, USA) overnight.
Human umbilical cord matrix mesenchymal stem cells were isolated from umbilical cord of healthy pregnancies during normal deliveries at the end of gestation with informed consent (South Wales REC No. 11/WA/0040). Umbilical cords were washed with DMEM/F12, diced into 2 mm2, explanted into T25 culture flasks containing 0.5 mL fetal bovine serum (FBS) for 24 hours then transferred to complete culture medium which was DMEM/F12 supplemented with 10% FBS, and 1% penicillin/streptomycin (Life Technology, Thermo Fisher Scientific) in a 5% CO2 atmosphere at 37°C. After 2 weeks, adherent cells were trypsinised for subpassage. Cells at passage three were seeded onto each of the test samples at 5 × 104 cells/well in a 96-well plate, while negative control cultures containing the same number of cells alone were cultured as a monolayer in wells of a 96-well plate without scaffolds. At 1, 3 and 7 days, the medium was replaced with 100 μL medium containing 10 μL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich). After incubating for 4 hours, the supernatant was carefully removed and 100 μL dimethyl sulfoxide was added to each well to dissolve the formazan crystals. The plates were wrapped in foil and incubated for 1 hour at room temperature. The supernatant was then transferred to a new 96-well plate, and the absorbance of each well was measured using a microplate reader at 570 nm wavelength (series No. 415-1387, BMG Labtech, Ortenberg, Germany).
Alkaline phosphatase assay
Alkaline phosphatase (ALP) activity of human umbilical cord matrix mesenchymal stem cells on HA/aragonite and control samples was quantified according to the manufacturer’s protocol (ab83369, Abcam, Cambridge, UK). In brief, cells at 5 × 104/well were seeded onto HA/aragonite, gelatine sponge or into blank wells and cultured for 3 days in DMEM/F12 supplemented with 10% FBS and 1% penicillin/streptomycin. The DMEM/F12 was then replaced by osteogenic medium containing dexamethasone (0.01 μM), β-glycerophosphate (0.1 M), ascorbic acid (0.5 mM), 10% FBS and 1% penicillin/streptomycin.
For ALP assay, HA/aragonite samples with attached cells were washed twice in cold phosphate-buffered saline, then cells were lysed in ALP assay buffer on ice for 2 hours with shaking, then processed according to the manufacturer’s protocol. ALP was quantified using a microplate reader (FLUOstar Omega; BMG LabTech) at an optical density of 405 nm. ALP activity (μmol/min/min or U/mL) was then calculated from the following equation
| (3) |
Where B is the amount of p-nitrophenol in the sample well, calculated from the standard curve; ΔT is the reaction time; V is the original sample volume added to each sample well and D is the sample dilution factor.
Degradation test
The degradation test was carried out following a standard testing method (ISO 10993-14).39 The HA/aragonite was ground into particles and the particle size was controlled to 355-415 μm by sieving. The particles were soaked in Tris-HCl buffer (pH 7.4) for 3, 7 or 14 days with a mixing speed of 120 r/min at 37°C. After 3, 7 or 14 days, the particles were filtered (0.2 μm) and washed twice with deionised water. The filtered particles were then dried in an oven for 24 hours to constant weight.
In vivo implantation
Six Sprague-Dawley male rats with a weight in the range of 200-250 g bought from Laboratory Animal Center, Huazhong University of Science and Technology (licence No. SCXK2016-0057) were employed in this study. The HA/aragonite scaffolds and control gelatine sponges were implanted juxtapositionally between the tibia and the anterior tibialis muscle (Figure 2) of six adult rats (three per group) to observe any soft/hard tissue reaction to the HA/aragonite in comparison with gelatine sponge controls.
The procedure was approved by the Animal Ethical Committee at Tongji Medical School, Huazhong University of Science and Technology on October 1, 2017 (IACUC No.738).
Six weeks after operation, the rats were euthanised and the implanted tissues were harvested and fixed immediately in 4% glutaraldehyde/0.1 M phosphate-buffered saline overnight, washed in deionised water and stored in 70% ethanol for MicroCT (Scanco VivaCT40; Scanco Medical, Bassersdorf, Switzerland), then processed for histology and transmission electron microscopy.
All samples were dehydrated and embedded into LR White resin (London Resin Company, Reading, UK) at 50°C for a minimum of 48 hours. Undecalcified 10 µm sections were obtained using a Leica RM2155 motorised microtome and stained with 1% toluidine blue in 50 mM Tris buffer pH 7.3. Stained sections were examined using an Olympus BX51 research light microscope (Olympus, Tokyo, Japan) and digital photomicrographs were captured using a Zeiss Axiocam and Axiovision software (Carl Zeiss Vision GmbH, Hallbergmoos, Germany).
The areas of interest identified by light microscopy were selected, post fixed in 1% osmium tetroxide and re-embedded in LR White resin, then 100 nm ultrathin sections were obtained using a glass knife on an Ultracut E ultramicrotome (Leica Microsystems Ltd, Wetzlar, Germany) stained with uranyl acetate and lead citrate, and examined in a Philips CM12 transmission electron microscope (FEI U.K. Ltd., Milton, UK) at 80 kV. Images were captured with a Megaview III camera and AnalySIS software (Soft Imaging System GmbH, Münster, Germany).
Statistical analysis
All data are presented as mean ± standard error (SE) and statistical analysis was performed by analysis of variance using GraphPad Prism 6 (GraphPad Software Inc., La Jolla, CA, USA). The differences between the two groups at each time point were analysed by two-way analysis of variance with the criterion α = 0.5.
Results
Optimization of 3D bio printing processing parameters
The effects of bioprinting parameters of pressure, printing speed and distance between strands on the compression strength and porosity of the end product are shown in Table 1. The results of analysis of the data are shown in Table 2, and the original calculation and data leading to the results in Table 2 can be referred to in the Additional Table 1 (10.7KB, xlsx) .
Table 1. The effect of bioplotting parameters on hydroxyapatite/aragonite end products.
| Sample No. | Factor | Compression (MPa) | Porosity (%) | ||
|---|---|---|---|---|---|
| (A) Pressure (MPa) | (B) Printing speed (mm/s) | (C) Distance between strands (mm) | |||
| 1 | 0.6 | 25 | 1 | 2.49 ± 0.14 | 37.0 ± 1.4 |
| 2 | 0.6 | 27 | 1.1 | 2.43 ± 0.23 | 37.5 ± 1.5 |
| 3 | 0.6 | 30 | 1.2 | 1.11 ± 0.07 | 42.8 ± 1.8 |
| 4 | 0.7 | 25 | 1.1 | 1.13 ± 0.11 | 41.1 ± 1.1 |
| 5 | 0.7 | 27 | 1.2 | 1.34 ± 0.09 | 30.2 ± 1.5 |
| 6 | 0.7 | 30 | 1 | 1.58 ± 0.13 | 40.7 ± 1.3 |
| 7 | 0.8 | 25 | 1.2 | 1.03 ± 0.08 | 41.8 ± 1.1 |
| 8 | 0.8 | 27 | 1 | 0.56 ± 0.05 | 40.2 ± 1.2 |
| 9 | 0.8 | 30 | 1.1 | 0.75 ± 0.07 | 35.5 ± 1.6 |
Note: Data are expressed as the mean ± SE.
Table 2. Range analysis of compression strength and porosity of hydroxyapatite/aragonite.
| Factors | Pressure (MPa) | Printing speed (mm/s) | Distance between strands (mm) |
|---|---|---|---|
| Compression strength | |||
| k1 | 2.01 | 1.55 | 1.54 |
| k2 | 1.35 | 1.44 | 1.44 |
| k3 | 0.78 | 1.15 | 1.16 |
| R | 1.23 | 0.4 | 0.38 |
| Porosity | |||
| k1 | 39.1 | 40.0 | 39.3 |
| k2 | 27.3 | 36.0 | 38.0 |
| k3 | 39.2 | 39.7 | 38.3 |
| R | 11.9 | 3.0 | 1.0 |
Table 2 shows the range analysis of variance tests. Mean k1, k2 and k3 are the mean values of the sum of compression strength at the same level. For the same factor, range R is the average value difference between the maximum value and the minimum value of k at different levels.
In Table 2, k1, k2 and k3 are the mean values of the sum of compression strength at the same level, which can be calculated from Equation (4)
| (4) |
Where S1 is the average compression strength of pressure (factor A), S2 is the average compression strength of printing speed (factor B) and S3 is the average compression strength of distance between strands (factor C). For the same factor, range R is the average value difference between the maximum value and the minimum value of K at the different levels. According to the range analysis of compression strength, without any interaction between the three factors, the results of RA > RB > RC shown in Table 2 indicate that the printing pressure has the greatest effect on compression strength, printing speed takes second place, and distance between strands has the least effect. For printing pressure (factor A), kA1 > kA2 > kA3, which indicates that as the printing pressure increases, the compression strengthdecreases. For printing speed (factor B), kB1 > kB2 > kB3, indicating that as the printing speed increases, the compression strength decreases. For printing speed (factor B), kB1 > kB2 > kB3, indicating that as the printing speed increases, the compression strength decreases. For distance between strands (factor C), kC1 > kC2 > kC3, which means that as distance between strands increases, the compression strength decreases. According to these results, the best combination of the three factors should be A1B1C1, namely a printing pressure of 0.6 MPa, printing speed of 25 mm/s and distance between strands of 1 mm.
According to the results of the range analysis for porosity, without any interaction between the three factors, the result of R1 > R2 > R3, shown in Table 2, indicates that the printing pressure has the greatest effect, followed by printing speed in second place, while distance between strands has the least effect. For printing pressure (factor A), kA3 > kA1 > kA2, which means that as printing pressure increases, porosity decreases but porosity is the lowest when printing pressure reaches an intermediate value. For printing speed (factor B), kB1 > kB3 > kB2 which indicates that as the printing speed increases, the porosity decreases, but porosity is the lowest when printing speed reaches an intermediate value. For distance between strands (factor C), kC1 > kC3 > kC2, indicating that when distance between strands increases, porosity decreases, but porosity is the lowest when distance between strands reaches an intermediate value. According to these results, the best combination of the three factors should be A1B3C3, namely a printing pressure of 0.6 MPa, printing speed of 30 mm/s anddistance between strands of 1.2 mm.
In general, the compression strength decreases as porosity increases. At a fixed printing pressure, the higher the printing speed the higher the porosity, which implies a lower compression strength. It can therefore be seen that the porosity exerts a lesser effect on compression strength compared with the printing pressure from 2. Following the above tests, for the in vivo and in vitro characterisation, samples were fabricated adopting processing parameters from test sample 1 in view of the resultant higher compressive strength.
Material characterization of HA/aragonite
The surface structure, cross-section and X-ray spectroscopy analysis of HA/aragonite, visualised by scanning electron microscopy, are shown in Figure 3. The pore size was around 300 μm with some visible micro-pores on the surface and within the scaffolds.
Figure 3. (A) Scanning electron microscopic image of the cross section of hydroxyapatite/aragonite and measurement of pore size. (B) Higher magnification of the scanning electron microscopic image shown in A. (C) Energy dispersive X-ray spectroscopy analysis of the cross section of hydroxyapatite/aragonite. Scale bars: 100 μm in A, 10 μm in B.

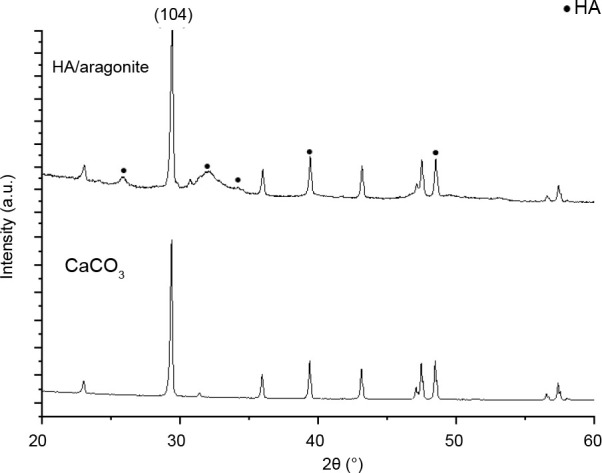
The results of X-ray diffraction analysis of HA/aragonite and calcium carbonate are shown in Figure 4. In Figure 4, the reflection peaks visible at a 2θ value of 104 in the calcium carbonate spectrum indicate that the calcium carbonate in the HA/aragonite belongs to aragonite.12, 13 In the X-ray diffraction patterns of the HA/aragonite, the diffraction peaks of calcium carbonate in the HA/aragonite are still visible. More importantly, the diffraction peaks of HA appear at 2θ values of 29.1°, 32.9°, 34.1°, 39.6° and 49.5° which correspond to (002), (211), (300), (130) and (213) planes, respectively. These diffraction peaks are in good agreement with the diffraction standard data of pure HA (JCPDS PDF#09-0432).40
Figure 4. X-ray diffraction analysis of hydroxyapatite/aragonite and calcium carbonate (CaCO3). The (104) in 2θ indicates that the CaCO3 in the HA/aragonite belongs to aragonite. HA: hydroxyapatite.
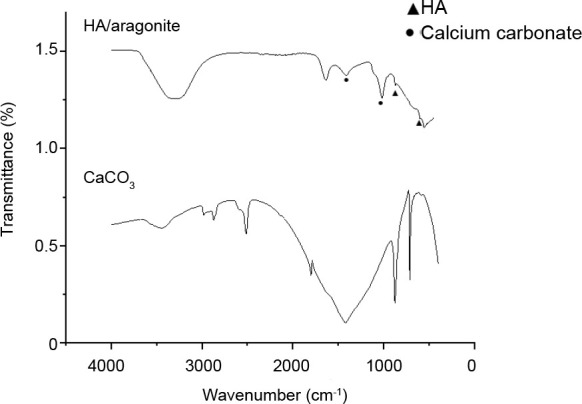
The FTIR spectra of HA/aragonite and calcium carbonate are shown in Figure 5. The FTIR spectra of the reference samples show absorptions at v1—963 cm-1, v3—1036 and 1095 cm-1, v4— 568 and 600 cm-1 which are due to PO43- ions, while OH groups are visible at 630 cm-1.15, 41, 42 The FTIR spectra of the reference samples show absorptions at a ν3 peak of 1453.7 cm-1, a ν2 peak of 853.8 cm-1, a ν1 peak of 1083.8 cm-1 and ν4 peaks of 699.2 and 712.2 cm-1 corresponding to CO32-. The ν4 absorption peak of CO32- is a single peak for calcite calcium carbonate, but a double peak for aragonite calcium carbonate.40
Figure 5. Comparation of the Fourier-transform infrared spectra of HA/aragonite. and calcium carbonate. Triangle indicates that the peak corresponding to PO43-; dot indicates that the peak corresponding to CO32-.
The results of thermogravimetric analysis of HA/aragonite are shown in Figure 6. The thermal decomposition process consists of three stages. In the first stage, as the temperature increases from 0°C to 200°C, gelatine in the HA/aragonite begins to decompose, resulting in weight loss. The decomposition products, CO2 and H2O, are emitted as gases. As the temperature continues to increase to 600°C, the trend of weight loss decreases until the weight reaches around 92%. At the second stage, as the temperature increases from 600°C to 800°C, CaCO3 in the HA/aragonite begins to dramatically decompose, resulting in massive weight loss. The decomposition product, CO2, is emitted as a gas. In the third stage, when the temperature increases to 1000°C, there is little weight loss and the residual weight tends to be stable because the residual components are CaO and HA with higher melting temperatures, which are stable at 1000°C, so no further weight loss will occur even as the temperature increases.43 The weight percentage of CaCO3 can be calculated from the following equation
| (5) |
Figure 6. Thermogravimetric analysis results of hydroxyapatite/aragonite. Stage 1, gelatine in the HA/aragonite begins to decompose; stage 2, aragonite begins to dramatically decompose; stage 3, residual components of CaO and HA are stable, there are no mass weight loss.
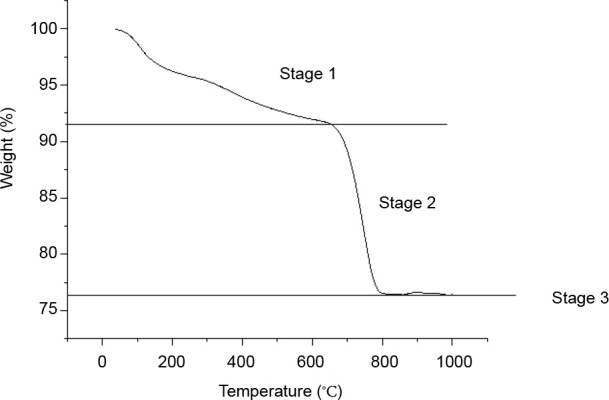
Thus, the weight percentage of CaCO3 is around 35%.
MTT and ALP assays
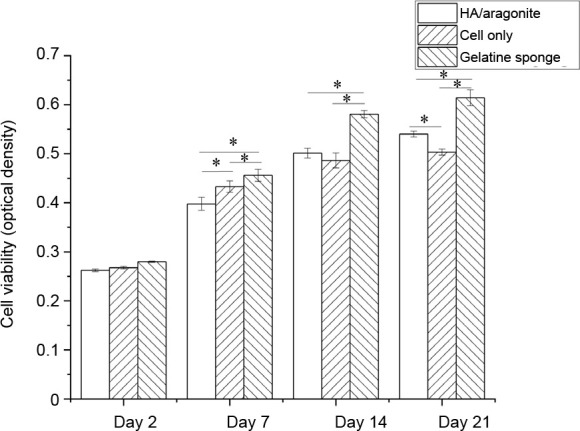
MTT assay demonstrated that there were no significant differences in cell viability between cells grown on HA/aragonite and cells alone at days 7 or 14 (Figure 7), indicating that HA/aragonite is nontoxic to human umbilical cord matrix mesenchymal cells. However, the result of MTT assay were significantly higher in the gelatine sponge group than the HA/aragonite group at days 7, 14 and 21.
Figure 7. Effect of HA/aragonite on the viability of human umbilical cord matrix mesenchymal stem cells detected by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Data are expressed as the mean ± SE. *P < 0.05 (two-way analysis of variance). HA: hydroxyapatite.
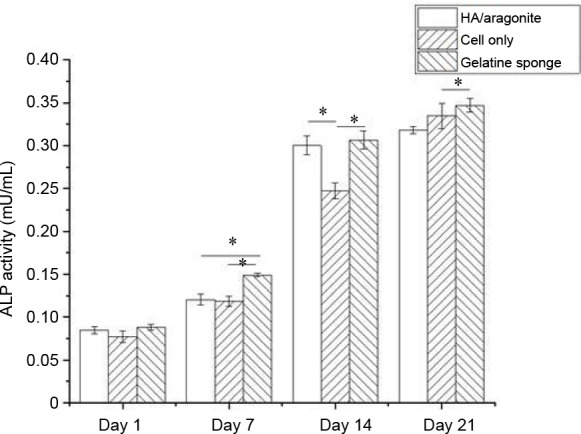
The osteogenic capacity of the HA/aragonite was confirmed by ALP assay. The ALP activity of cells seeded on HA/aragonite, on gelatine sponge and of cells alone is shown in Figure 8. After day 1, the ALP activity increased significantly on gelatine sponge, HA/aragonite and in cells alone between day 1 and day 14 (P < 0.05). However, ALP activity in the gelatine sponge group was higher than that of cells on HA/aragonite at days 7 and 21.
Figure 8. Effect of HA/aragonite on the ALP activity of human umbilical cord matrix mesenchymal stem cells. Data are expressed as the mean ± SE. *P < 0.05 (two-way analysis of variance. ALP: alkaline phosphatase; HA: hydroxyapatite.
In vitro degradation test of HA/aragonite
The degradation of the HA/aragonite was confirmed by degradation testing. The percentage degradation of the HA/aragonite was determined by weight loss which can be defined by equation
| (6) |
The weight loss of HA/aragonite at day 3 was around 3.5%, increasing to 25.58% after day 14. The percentage weight loss of HA/aragonite increased significantly over time, while the weight loss of HA alone showed no significant difference with increasing time (Figure 9).
Figure 9. (A, B) Weight loss percentage of HA/aragonite (A) and HA (B). Data are expressed as the mean ± SE. *P < 0.05 (two-way analysis of variance). HA: hydroxyapatite.

Juxtapositional implantation between tibia and tibialis anterior muscle in a rat model
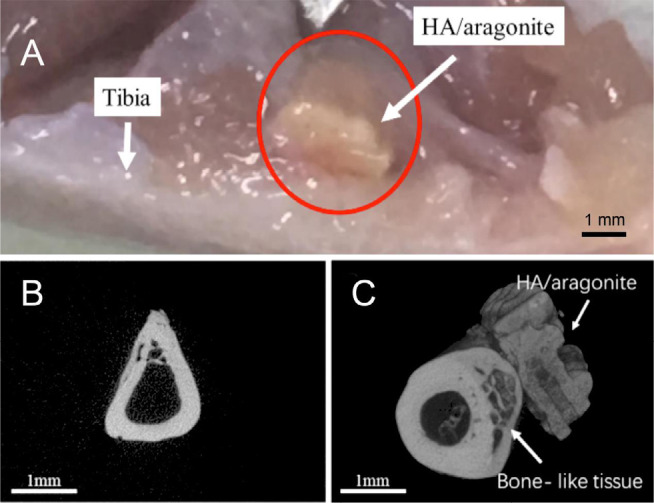
The position of the HA/aragonite implanted between the tibia and the tibialis anterior muscle in a rat is indicated in Figure 10A. The HA/aragonite was firmly integrated into the tibia after 6 weeks. Figure 10B shows a microCT image of a gelatine sponge implanted in a rat model, in which only the tibia is visible, with no mineralisation of the gelatine. Figure 10C shows HA/aragonite implanted in a rat model. The image shows that the HA/aragonite has the same density as bone, and HA/aragonite is firmly integrated with the indicated bone-like tissue.
Figure 10. Implantation of HA/aragonite in a rat model. (A) A specimen of the HA/aragonite (red circle) implanted between the tibia and the tibialis anterior muscle in a rat model. After 6 weeks the sample has become well integrated. (B) MicroCT of gelatine sponge implanted in a rat model. Only the tibia is visible, with no mineralised tissue formed in the sponge. (C) HA/aragonite implanted in a rat model. Interestingly, formation of bone-like tissue (arrow) can be seen between the tibia and the HA/aragonite. Scale bars: 1 mm. HA: hydroxyapatite.
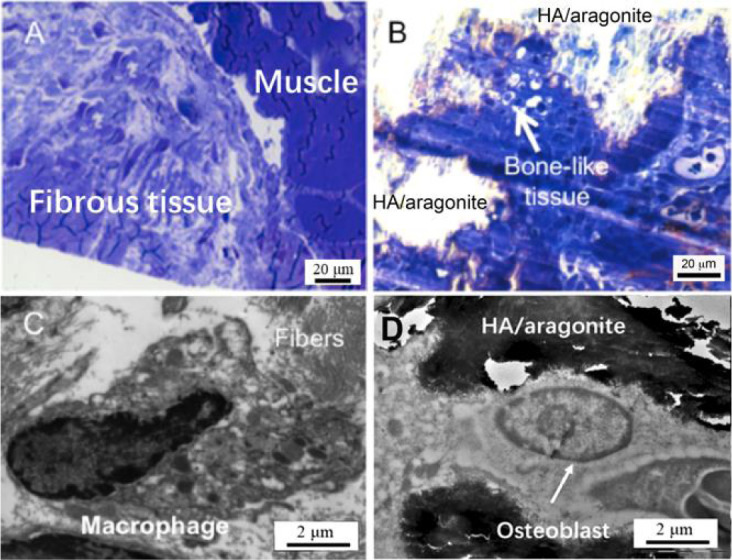
As shown in Figure 11A, light microscopy revealed that at 6 weeks after gelatine sponge implantation, fibrous tissue was formed between the tibia and the tibialis anterior muscle. The HA/aragonite samples implanted at the same time were covered mainly by connective tissue containing blood vessels, fibroblasts and some macrophages; in particular, there were patches of bone-like tissue formation and minimal inflammatory response, as shown in Figure 11B. The TEM results confirmed the findings of light microscopy. In the gelatine sponge implantation group (Figure 11C) macrophagic responses were observed with fibroblast infiltration for tissue regeneration; whereas within the patch of bone-like tissue in the HA/aragonite group (Figure 11D), typical osteoblast-like cells with calcified lacunae and canaliculi-like structure were observed, which confirmed that the marked area is likely to be new bone formation or callus formation.
Figure 11. (A) Light microscopy (1 mm section stained with Toluidine blue) revealed that implantation of a gelatine sponge resulted in formation of fibrous tissue between the tibia and the tibialis anterior muscle. (B) At 6 weeks after HA/aragonite implantation, the materials were covered by fibroblasts and macrophages; interestingly, there was a small patch of bone-like tissue formation. Transmission electron microscopy (100 nm section with uranyl acetate and lead citrate staining) observation confirmed the findings of light microscopy. In the control gelatine sponge implantation group (C), macrophagic responses were observed with fibroblast infiltration for tissue regeneration; whereas with implantation of the HA/aragonite (D), typical osteoblast-like cells with canaliculi-like structures within calcified lacunae were observed in the patch of bone-like tissue. Scale bars: 20 μm in A, B, 2 μm in C, D. HA: hydroxyapatite.
Discussion
The results of this study demonstrated the ability to fabricate a composite HA/aragonite bone graft substitute that supports osteogenesis in vitro and in vivo.
In this study, the purpose of adding aragonite into HA was to enhance the biodegradation of the bone graft. It is well known that HA is a bone graft material which is slow to degrade and its degradation period can be more than 100 months44 which is far beyond the bone remodelling cycle of between 6 and 24 months. In order to increase biodegradation, a more biodegradable ceramic, β-TCP is added into bone graft materials.44, 45 Even though HA/β-TCP is significantly more degradable than HA alone, the degradation period of HA/β-TCP is also rather long, reported to be more than 36 months.44, 45 It is therefore necessary to introduce more biodegradable bone graft materials, such as composites containing both HA and more soluble calcium carbonate, but apart from coralline HA, it is difficult to form HA composites containing calcium carbonate that also have the desired mechanical properties.
HA ceramics are normally sintered at between 1200-1300°C, whereas calcium carbonate undergoes thermal decomposition above 840°C:
| (7) |
Therefore it is not possible to synthesise HA/aragonite through sintering. It has been reported that when TTCP and DCPA are mixed in double-distilled water, the dissolved ions precipitate on the surface of the material and can self-set into various forms of HA.31 By adding calcium carbonate into this mixture, it may be possible to produce bulk self-setting HA/aragonite without pores.
Additive manufacturing provides the ideal solution to producing a controllable porous structure. However, it is well known that the filter-pressing phenomenon leads to the separation of solid and liquid phases when the mixture of powders and aqueous solution are injected through a syringe.46, 47
The addition of gelatine solution formed a viscous and cohesive paste and created a strong combination between solid and liquid phases, thereby improving the injectability of the paste. The setting time of TTCP/DCPA is 6-10 minutes, but the addition of gelatine increases the setting time to around 30 minutes which makes the pastes ‘printable’ and allows the fabrication of desired features. The precipitated particles were crystalline in texture which is similar to the observations reported in previous publications.48 Strong evidence for the presence of HA was provided by the peak which appeared at around 961 cm-1. The ν4 absorption peak of CO32- was seen in the HA/aragonite, which indicated that calcium carbonate was present in the HA/aragonite after all the processing. The peaks had all shifted from the reference peaks but by a reasonable value. The absorption peak of HPO42- at 895 cm-1 caused by the decreasing TTCP/DCPA ratio did not appear in the HA/aragonite, indicating that there was no Ca9(HPO4)(PO4)5(OH) present in the HA/aragonite.49
The compression strength of cancellous bone is 1-12 MPa, and the porosity is 50-90%.50 The compression strength of the HA/aragonite we fabricated was 2.49 MPa and the highest porosity of HA/aragonite is 42.8%, indicating that the strength and porosity of HA/aragonite are comparable to those of cancellous bone. The pore size of HA/aragonite was around 280 μm, which is suitable for tissue ingrowth.51 As the porosity of the HA/aragonite increases, its compression strength decreases. Recent studies have reported the same issue,52 and consequently further research is needed to improve the mechanical strength.
In this study, significant weight loss was noted between successive time points, indicating that HA/aragonite is a degradable material, which confirmed the previous finding of the in vitro biodegradation of calcium carbonate.53
As a potential bone graft substitute, it is important that this new material is able to enhance osteogenesis and promote bone formation. It is well documented that HA and calcium carbonate, used either alone or combined, have demonstrated osteogenicity and bone conductivity.1, 4, 7, 8, 15 In the present study, HA/aragonite supported ALP activity of mesenchymal stem cells in vitro, which is a sign of enhanced osteogenicity.
HA/aragonite was compared with clinically-used gelatine sponges which consist of gelatine alone. Interestingly, gelatine sponges showed greater cell numbers and ALP activities in vitro compared with HA/aragonite. This may be due to the fact that gelatine sponges are much more porous than HA/aragonite, thus allowing more cell penetration and growth over the 3-week period of cell culture. However, after implantation, no bone formation was observed in the gelatine sponge group in vivo.
Conductive bone formation can only be observed in vivo. In order to observe the tissue response to HA/aragonite, this bone graft material was implanted between the tibialis anterior muscle and the tibia. No bone defects were created, as the initial plan was to exclude any potential adverse effect of this material. Unexpectedly, at the time the implants were harvested, the HA/aragonite was firmly integrated into the tibia. New bone formation between the HA/aragonite and the tibia was confirmed by microCT, with bony material bridging the implant to the tibia bone. The presence of bone-like tissue on the surface of the implants was also confirmed by both light and transmission electron microscopy.
Undifferentiated cells from muscles and blood vessel walls have osteogenic potential.54 The formation of bone on the surface of calcium phosphate scaffolds in intramuscular sites has been reported by several studies.55-57 The precipitation and dissolution of the material’s surface plays an important role in the intramuscular osteogenesis process.57, 58 In this study, the HA/aragonite formed a biodegradable bone graft. When HA/aragonite is implanted in the body, the process of interaction between the implant and the surrounding body fluid or tissue can be activated. The dissolution of aragonite should increase the concentration of Ca2+ on the surface of the material resulting in the formation of apatite-like phosphate by absorbing proteins and growth factors.57 The fast degradation rate of aragonite may be responsible for the early signs of osteogenesis, while other animal studies only showed signs of osteogenesis after 45 days.56, 59 In the current study, we not only observed small patches of bone-like tissue formation on the HA/aragonite when implanted in muscle, but also a large amount of visible bone formation between the HA/aragonite and the tibia.
Our results show that the HA/aragonite did not induce any adverse tissue responses after implantation between the tibia and the tibialis anterior muscle, but demonstrated conductive bone formation between the implant and the tibia. Further study is needed to understand the mechanism involved.
Within this study, there are a number of limitations. The first purpose of this study was to achieve the biofabrication of a novel HA/aragonite bone graft substitute using 3D bioprinting techniques. Even though the material fabrication process was successful, the ratio of raw materials in the formulation may not be optimal in terms of the mechanical strength, biodegradation and osteogenicity of the product. Secondly, the parameters used are designed for use in a 3D-Bioplotter® and may not be directly transferrable to other types of additive manufacturing devices. Thirdly, the favourable results of HA/aragonite in vitro and in vivo are still preliminary. More comprehensive and systematic studies are needed to explore the full potential of HA/aragonite on osteogenesis and biodegradation.
In summary, the 3D-bioprinted bone graft substitute HA/aragonite prepared in this study is a porous, biodegradable and nontoxic HA/calcium carbonate composite material with osteogenic capability. By controlling the printing parameters, product size and porosity were fabricated within the range desired for bone grafts. HA/aragonite can be printed with high resolution to achieve versatile geometries that are critical for fluid exchange and cellular ingrowth during bone healing. The mechanical strength and porosity of the HA/aragonite are comparable to those of cancellous bone, which allows implantation into a non-loading bone defect. In vitro and in vivo experiments were performed successfully showing suitability for bone formation. Since this material is formed around 37°C, there is great potential for the incorporation of bioactive particles to suit personalised application. Future studies will focus on further optimisation of the formulae to strengthen the mechanical properties with extensive evaluation of the effects on osteogenicity and biodegradation.
Funding Statement
This study was supported by the Wuhan International Collaboration Project of China (No. 2017030209020252) and Wuhan Science and Technology Project of China (No. 2018010401011281).
Footnotes
Acknowledgement: We would like to thank for Dr. Christopher Von Ruhland, the Facility Lead (Electron and Light Microscopy), Central Biotechnology Services, Cardiff University for the sample preparation for light and electron microscopy; Dr. Hui Liu and Mr. Dahu Qi at Tongji Hospital, Huazhong University of Science and Technology for assistance with microCT and in vitro biodegradation analysis.
Conflicts of interest statement: The authors declare that they have no competing financial interests or personal relationships that could have appeared to influence the work reported in this study.
Additional file: Additional Table 1 (10.7KB, xlsx) . Original calculation and data leading to the results in Table 2.
References
- 1.Holmes R. E., Bucholz R. W., Mooney V. Porous hydroxyapatite as a bone-graft substitute in metaphyseal defects. A histometric study. J Bone Joint Surg Am. 1986;68:904–911. [PubMed] [Google Scholar]
- 2.Giannoudis P. V., Dinopoulos H., Tsiridis E. Bone substitutes: an update. Injury. 2005;36(Suppl 3):S20–27. doi: 10.1016/j.injury.2005.07.029. [DOI] [PubMed] [Google Scholar]
- 3.Kao S. T., Scott D. D. A review of bone substitutes. Oral Maxillofac Surg Clin North Am. 2007;19:513–521. doi: 10.1016/j.coms.2007.06.002. vi. [DOI] [PubMed] [Google Scholar]
- 4.Okuda T., Ioku K., Yonezawa I., Minagi H., Gonda Y., Kawachi G., Kamitakahara M., Shibata Y., Murayama H., Kurosawa H., Ikeda T. The slow resorption with replacement by bone of a hydrothermally synthesized pure calcium-deficient hydroxyapatite. Biomaterials. 2008;29:2719–2728. doi: 10.1016/j.biomaterials.2008.03.028. [DOI] [PubMed] [Google Scholar]
- 5.Tonino A. J., van der Wal B. C., Heyligers I. C., Grimm B. Bone remodeling and hydroxyapatite resorption in coated primary hip prostheses. Clin Orthop Relat Res. 2009;467:478–484. doi: 10.1007/s11999-008-0559-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bouler J. M., Pilet P., Gauthier O., Verron E. Biphasic calcium phosphate ceramics for bone reconstruction: A review of biological response. Acta Biomater. 2017;53:1–12. doi: 10.1016/j.actbio.2017.01.076. [DOI] [PubMed] [Google Scholar]
- 7.Wei W., Ma G. H., Hu G., Yu D., McLeish T., Su Z. G., Shen Z. Y. Preparation of hierarchical hollow CaCO3 particles and the application as anticancer drug carrier. J Am Chem Soc. 2008;130:15808–15810. doi: 10.1021/ja8039585. [DOI] [PubMed] [Google Scholar]
- 8.Biradar S., Ravichandran P., Gopikrishnan R., Goornavar V., Hall J. C., Ramesh V., Baluchamy S., Jeffers R. B., Ramesh G. T. Calcium carbonate nanoparticles: synthesis, characterization and biocompatibility. J Nanosci Nanotechnol. 2011;11:6868–6874. doi: 10.1166/jnn.2011.4251. [DOI] [PubMed] [Google Scholar]
- 9.Addadi L., Raz S., Weiner S. Taking advantage of disorder: amorphous calcium carbonate and its roles in biomineralization. Adv Mater. 2003;15:959–970. [Google Scholar]
- 10.Addadi L., Weiner S. A pavement of pearl. Nature. 1997;389:912–913. [Google Scholar]
- 11.Svenskaya Y., Parakhonskiy B., Haase A., Atkin V., Lukyanets E., Gorin D., Antolini R. Anticancer drug delivery system based on calcium carbonate particles loaded with a photosensitizer. Biophys Chem. 2013;182:11–15. doi: 10.1016/j.bpc.2013.07.006. [DOI] [PubMed] [Google Scholar]
- 12.Küther J., Seshadri R., Knoll W., Tremel W. Templated growth of calcite, vaterite and aragonite crystals onself-assembled monolayers of substituted alkylthiols on gold. J Mater Chem. 1998;8:641–650. [Google Scholar]
- 13.Wang C., Zhao J., Zhao X., Bala H., Wang Z. Synthesis of nanosized calcium carbonate (aragonite) via a polyacrylamide inducing process. Powder Technol. 2006;163:134–138. [Google Scholar]
- 14.Islam K. N., Bakar M. Z. B. A., Noordin M. M., Hussein M. Z. B., Rahman N. S. B. A., Ali M. E. Characterisation of calcium carbonate and its polymorphs from cockle shells (Anadara granosa) Powder Technol. 2011;213:188–191. [Google Scholar]
- 15.Fu K., Xu Q., Czernuszka J., Triffitt J. T., Xia Z. Characterization of a biodegradable coralline hydroxyapatite/calcium carbonate composite and its clinical implementation. Biomed Mater. 2013;8:065007. doi: 10.1088/1748-6041/8/6/065007. [DOI] [PubMed] [Google Scholar]
- 16.Kucharska M., Butruk B., Walenko K., Brynk T., Ciach T. Fabrication of in-situ foamed chitosan/β-TCP scaffolds for bone tissue engineering application. Mater Lett. 2012;85:124–127. [Google Scholar]
- 17.Nommeots-Nomm A., Labbaf S., Devlin A., Todd N., Geng H., Solanki A. K., Tang H. M., Perdika P., Pinna A., Ejeian F., Tsigkou O., Lee P. D., Esfahani M. H. N., Mitchell C. A., Jones J. R. Highly degradable porous melt-derived bioactive glass foam scaffolds for bone regeneration. Acta Biomater. 2017;57:449–461. doi: 10.1016/j.actbio.2017.04.030. [DOI] [PubMed] [Google Scholar]
- 18.Yoshikawa H., Tamai N., Murase T., Myoui A. Interconnected porous hydroxyapatite ceramics for bone tissue engineering. J R Soc Interface. 2009;6(Suppl 3):S341–348. doi: 10.1098/rsif.2008.0425.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cao H., Kuboyama N. A biodegradable porous composite scaffold of PGA/beta-TCP for bone tissue engineering. Bone. 2010;46:386–395. doi: 10.1016/j.bone.2009.09.031. [DOI] [PubMed] [Google Scholar]
- 20.Brougham C. M., Levingstone T. J., Shen N., Cooney G. M., Jockenhoevel S., Flanagan T. C., O’Brien F. J. Freeze-drying as a novel biofabrication method for achieving a controlled microarchitecture within large, complex natural biomaterial scaffolds. Adv Healthc Mater. 2017;6:1700598. doi: 10.1002/adhm.201700598. [DOI] [PubMed] [Google Scholar]
- 21.Mi H. Y., Jing X., McNulty J., Salick M. R., Peng X. F., Turng L. S. Approaches to fabricating multiple-layered vascular scaffolds using hybrid electrospinning and thermally induced phase separation methods. Ind Eng Chem Res. 2016;55:882–892. [Google Scholar]
- 22.Bose S., Vahabzadeh S., Bandyopadhyay A. Bone tissue engineering using 3D printing. Mater Today. 2013;16:496–504. [Google Scholar]
- 23.Bracaglia L. G., Smith B. T., Watson E., Arumugasaamy N., Mikos A. G., Fisher J. P. 3D printing for the design and fabrication of polymer-based gradient scaffolds. Acta Biomater. 2017;56:3–13. doi: 10.1016/j.actbio.2017.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loh Q. L., Choong C. Three-dimensional scaffolds for tissue engineering applications: role of porosity and pore size. Tissue Eng Part B Rev. 2013;19:485–502. doi: 10.1089/ten.teb.2012.0437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ngo T. D., Kashani A., Imbalzano G., Nguyen K. T. Q., Hui D. Additive manufacturing (3D printing): A review of materials, methods, applications and challenges. Compos B Eng. 2018;143:172–196. [Google Scholar]
- 26.Sobral J. M., Caridade S. G., Sousa R. A., Mano J. F., Reis R. L. Three-dimensional plotted scaffolds with controlled pore size gradients: Effect of scaffold geometry on mechanical performance and cell seeding efficiency. Acta Biomater. 2011;7:1009–1018. doi: 10.1016/j.actbio.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 27.Trombetta R., Inzana J. A., Schwarz E. M., Kates S. L., Awad H. A. 3D printing of calcium phosphate ceramics for bone tissue engineering and drug delivery. Ann Biomed Eng. 2017;45:23–44. doi: 10.1007/s10439-016-1678-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Inzana J. A., Olvera D., Fuller S. M., Kelly J. P., Graeve O. A., Schwarz E. M., Kates S. L., Awad H. A. 3D printing of composite calcium phosphate and collagen scaffolds for bone regeneration. Biomaterials. 2014;35:4026–4034. doi: 10.1016/j.biomaterials.2014.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brunello G., Sivolella S., Meneghello R., Ferroni L., Gardin C., Piattelli A., Zavan B., Bressan E. Powder-based 3D printing for bone tissue engineering. Biotechnol Adv. 2016;34:740–753. doi: 10.1016/j.biotechadv.2016.03.009. [DOI] [PubMed] [Google Scholar]
- 30.Bose S., Saha S. K. Synthesis of hydroxyapatite nanopowders via sucrose-templated sol-gel method. J Am Ceram Soc. 2003;86:1055–1057. [Google Scholar]
- 31.Lee J. W., Kim J. Y., Cho D. W. Solid free-form fabrication technology and its application to bone tissue engineering. Int J Stem Cells. 2010;3:85–95. doi: 10.15283/ijsc.2010.3.2.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu H. H., Wang P., Wang L., Bao C., Chen Q., Weir M. D., Chow L. C., Zhao L., Zhou X., Reynolds M. A. Calcium phosphate cements for bone engineering and their biological properties. Bone Res. 2017;5:17056. doi: 10.1038/boneres.2017.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ozdemir F., Evans I., Bretcanu O. Calcium phosphate cements for medical applications. In: Kaur G., editor. Clinical Applications of Biomaterials: State-of-the-Art Progress, Trends, and Novel Approaches. Springer International Publishing; Cham: 2017. pp. 91–121. [Google Scholar]
- 34.Pimentel C. R., Ko S. K., Caviglia C., Wolff A., Emnéus J., Keller S. S., Dufva M. Three-dimensional fabrication of thick and densely populated soft constructs with complex and actively perfused channel network. Acta Biomater. 2018;65:174–184. doi: 10.1016/j.actbio.2017.10.047. [DOI] [PubMed] [Google Scholar]
- 35.Stanton M. M., Samitier J., Sánchez S. Bioprinting of 3D hydrogels. Lab Chip. 2015;15:3111–3115. doi: 10.1039/c5lc90069g. [DOI] [PubMed] [Google Scholar]
- 36.Gopinathan J., Noh I. Recent trends in bioinks for 3D printing. Biomater Res. 2018;22:11. doi: 10.1186/s40824-018-0122-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Şahin E., Kalyon D. M. The rheological behavior of a fast-setting calcium phosphate bone cement and its dependence on deformation conditions. J Mech Behav Biomed Mater. 2017;72:252–260. doi: 10.1016/j.jmbbm.2017.05.017. [DOI] [PubMed] [Google Scholar]
- 38.General Administration of Quality Supervision Inspection and Quarantine of the People’s Republic of China Standardization Administration of the People’s Republic of China. Standard test method for compressive resistance of ceramic materials. GB/T 4740-1984 [Google Scholar]
- 39.Biological evaluation of medical devices — Part 14: Identification and quantification of degradation products from ceramics. ISO 10993-14:2001. [Google Scholar]
- 40.Ma T., Xia Z., Liao L. Effect of reaction systems and surfactant additives on the morphology evolution of hydroxyapatite nanorods obtained via a hydrothermal route. Appl Surf Sci. 2011;257:4384–4388. [Google Scholar]
- 41.Merry J. C., Gibson I. R., Best S. M., Bonfield W. Synthesis and characterization of carbonate hydroxyapatite. J Mater Sci Mater Med. 1998;9:779–783. doi: 10.1023/a:1008975507498. [DOI] [PubMed] [Google Scholar]
- 42.Wilson R. M., Elliott J. C., Dowker S. E., Rodriguez-Lorenzo L. M. Rietveld refinements and spectroscopic studies of the structure of Ca-deficient apatite. Biomaterials. 2005;26:1317–1327. doi: 10.1016/j.biomaterials.2004.04.038. [DOI] [PubMed] [Google Scholar]
- 43.Aminzare M., Eskandari A., Baroonian M. H., Berenov A., Razavi Hesabi Z., Taheri M., Sadrnezhaad S. K. Hydroxyapatite nanocomposites: synthesis, sintering and mechanical properties. Ceram Int. 2013;39:2197–2206. [Google Scholar]
- 44.Ogose A., Hotta T., Kawashima H., Kondo N., Gu W., Kamura T., Endo N. Comparison of hydroxyapatite and beta tricalcium phosphate as bone substitutes after excision of bone tumors. J Biomed Mater Res B Appl Biomater. 2005;72:94–101. doi: 10.1002/jbm.b.30136. [DOI] [PubMed] [Google Scholar]
- 45.Oh K. J., Ko Y. B., Jaiswal S., Whang I. C. Comparison of osteoconductivity and absorbability of beta-tricalcium phosphate and hydroxyapatite in clinical scenario of opening wedge high tibial osteotomy. J Mater Sci Mater Med. 2016;27:179. doi: 10.1007/s10856-016-5795-1. [DOI] [PubMed] [Google Scholar]
- 46.Bohner M., Baroud G. Injectability of calcium phosphate pastes. Biomaterials. 2005;26:1553–1563. doi: 10.1016/j.biomaterials.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 47.Gbureck U., Barralet J. E., Spatz K., Grover L. M., Thull R. Ionic modification of calcium phosphate cement viscosity. Part I: hypodermic injection and strength improvement of apatite cement. Biomaterials. 2004;25:2187–2195. doi: 10.1016/j.biomaterials.2003.08.066. [DOI] [PubMed] [Google Scholar]
- 48.Venkatesan J., Rekha P. D., Anil S., Bhatnagar I., Sudha P. N., Dechsakulwatana C., Kim S. K., Shim M. S. Hydroxyapatite from cuttlefish bone: isolation, characterizations, and applications. Biotechnol Bioprocess Eng. 2018;23:383–393. [Google Scholar]
- 49.Ishikawa K., Takagi S., Chow L. C., Suzuki K. Reaction of calcium phosphate cements with different amounts of tetracalcium phosphate and dicalcium phosphate anhydrous. J Biomed Mater Res. 1999;46:504–510. doi: 10.1002/(sici)1097-4636(19990915)46:4<504::aid-jbm8>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 50.Manassero M., Decambron A., Guillemin N., Petite H., Bizios R., Viateau V. Coral scaffolds in bone tissue engineering and bone regeneration. In: Goffredo S., Dubinsky Z., editors. The Cnidaria, Past, Present and Future: The world of Medusa and her sisters. Springer International Publishing; Cham: 2016. pp. 691–714. [Google Scholar]
- 51.Sopyan I., Mel M., Ramesh S., Khalid K. A. Porous hydroxyapatite for artificial bone applications. Science and Technology of Advanced Materials. 2007;8:116–123. [Google Scholar]
- 52.Sudarmadji N., Tan J. Y., Leong K. F., Chua C. K., Loh Y. T. Investigation of the mechanical properties and porosity relationships in selective laser-sintered polyhedral for functionally graded scaffolds. Acta Biomater. 2011;7:530–537. doi: 10.1016/j.actbio.2010.09.024. [DOI] [PubMed] [Google Scholar]
- 53.He F., Zhang J., Yang F., Zhu J., Tian X., Chen X. In vitro degradation and cell response of calcium carbonate composite ceramic in comparison with other synthetic bone substitute materials. Mater Sci Eng C Mater Biol Appl. 2015;50:257–265. doi: 10.1016/j.msec.2015.02.019. [DOI] [PubMed] [Google Scholar]
- 54.Gerstenfeld L. C., Cullinane D. M., Barnes G. L., Graves D. T., Einhorn T. A. Fracture healing as a post-natal developmental process: molecular, spatial, and temporal aspects of its regulation. J Cell Biochem. 2003;88:873–884. doi: 10.1002/jcb.10435. [DOI] [PubMed] [Google Scholar]
- 55.Le Nihouannen D., Daculsi G., Saffarzadeh A., Gauthier O., Delplace S., Pilet P., Layrolle P. Ectopic bone formation by microporous calcium phosphate ceramic particles in sheep muscles. Bone. 2005;36:1086–1093. doi: 10.1016/j.bone.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 56.Yang Z. J., Yuan H., Zou P., Tong W., Qu S., Zhang X. D. Osteogenic responses to extraskeletally implanted synthetic porous calcium phosphate ceramics: an early stage histomorphological study in dogs. J Mater Sci Mater Med. 1997;8:697–701. doi: 10.1023/a:1018540024082. [DOI] [PubMed] [Google Scholar]
- 57.Lu J., Yu H., Chen C. Biological properties of calcium phosphate biomaterials for bone repair: a review. RSC Adv. 2018;8:2015–2033. doi: 10.1039/c7ra11278e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Habibovic P., Sees T. M., van den Doel M. A., van Blitterswijk C. A., de Groot K. Osteoinduction by biomaterials--physicochemical and structural influences. J Biomed Mater Res A. 2006;77:747–762. doi: 10.1002/jbm.a.30712. [DOI] [PubMed] [Google Scholar]
- 59.Gedrange T., Mai R., Weingaertner J., Hietschold V., Bourauel C., Pradel W., Lauer G., Proff P. Finite element representation of bone substitute remodelling in the jaw bone. Biomed Tech (Berl) 2008;53:220–223. doi: 10.1515/BMT.2008.039. [DOI] [PubMed] [Google Scholar]