

Abstract
Antibodies are the cardinal effector molecules of the immune system and are being leveraged with enormous success as biotherapeutic drugs. A key part of the adaptive immune response is the production of an epitope-diverse, polyclonal antibody mixture that is capable of neutralizing invading pathogens or disease-causing molecules through binding interference and by mediating humoral and cellular effector functions. Avidity — the accumulated binding strength derived from the affinities of multiple individual non-covalent interactions — is fundamental to virtually all aspects of antibody biology, including antibody–antigen binding, clonal selection and effector functions. The manipulation of antibody avidity has since emerged as an important design principle for enhancing or engineering novel properties in antibody biotherapeutics. In this Review, we describe the multiple levels of avidity interactions that trigger the overall efficacy and control of functional responses in both natural antibody biology and their therapeutic applications. Within this framework, we comprehensively review therapeutic antibody mechanisms of action, with particular emphasis on engineered optimizations and platforms. Overall, we describe how affinity and avidity tuning of engineered antibody formats are enabling a new wave of differentiated antibody drugs with tailored properties and novel functions, promising improved treatment options for a wide variety of diseases.
Subject terms: Antibody therapy, Drug discovery
Antibody function is dependent on avidity — the accumulated strength of multiple affinity interactions between the antibody, antigen, cell surface receptors and other antibodies. In this Review, Oostindie et al. discuss the role of avidity in eliciting antibody functional responses and review the current engineering strategies for manipulating avidity interactions in antibody-based therapies.
Introduction
Antibodies are a critical component of the humoral adaptive immune response for the neutralization and destruction of disease-causing molecules, viruses and cells. Upon initial exposure to a foreign antigen, the immune system mounts a diverse polyclonal B cell response, producing a repertoire of antibodies that recognize multiple overlapping and non-overlapping antigenic epitopes. Although these initial antibodies have low binding affinities, activated antibody-producing B cells undergo a conserved affinity maturation process through somatic hypermutation and clonal selection to generate antibodies with successively greater affinities1–3. Target neutralization and elimination is subsequently triggered through multiple tightly regulated processes, including protein-mediated and cell-mediated effector functions.
The discovery of the hybridoma technology by Köhler and Milstein4, followed by the development of standardizable, high-yield recombinant antibody discovery, production and purification platforms, ushered in a new era of antibody-based drugs. Indeed, antibodies have grown into an established and exceptionally versatile class of therapeutic agents for treating a wide spectrum of human diseases, owing to their high specificity, modular and adaptable architecture, predictable bioavailability and pharmacokinetics and availability of standardized manufacturing platforms. In May 2022, an estimated 1,000 therapeutic antibodies were at various stages of clinical development and 119 antibodies had obtained first marketing approval in Europe and the USA (excluding biosimilars; a list of antibody therapeutics approved or in regulatory review in the EU or US is available through the Antibody Society).
Many factors contribute to the success of antibody-based treatments, but central to the pharmacodynamic mechanism is the interaction of antibodies with their targets, and more specifically the affinity and avidity of binding interactions5,6. Avidity therein is defined as the accumulated binding strength of multiple affinities contributed by individual non-covalent interactions in crosslinking events. For example, antibody-based effector functions are triggered by multivalent target binding on the cell surface7–11 in which avidity-driven surface clustering of IgG molecules serves as a threshold for effector function activation. Indeed, single antibody–antigen binding interactions may be insufficient for successfully eliminating pathogens or diseased cells11–14 and neutralization of soluble proteins based on single binding interactions display a limited potency ceiling that can be surpassed by orders of magnitude using antibody cocktails15–17. Recent innovative antibody formats capable of leveraging antibody avidity, such as bispecific and multispecific antibodies and antibodies with engineered Fc-mediated effector functions, have steered the antibody landscape towards more tailored drug design strategies5,18–21. Notably, the recent decision of the World Health Organization to revise their generic naming scheme for antibodies and create separate categories for Fc-engineered and multispecific antibodies, in addition to changing nomenclature for unmodified antibodies and antibody fragments, highlights the success and maturity of these novel formats as human therapeutic agents22.
In this Review, we describe recent advances in understanding avidity interactions in the context of both natural immunity and therapeutic antibody-based mechanisms of action. Special emphasis is placed on engineering strategies and platforms that tune avidity to boost therapeutic activity, including multispecific and multiparatopic targeting, valency modulation and the optimization of functional interactions with effector molecules. We do not discuss the vast field of obligate bispecific antibodies owing to space constraints and we refer readers to a recent review by Labrijn et al.20 for more information. Within our conceptual framework, we discuss current translational efforts regarding avidity-based antibody concepts in the clinic and provide perspectives that we believe will aid the development of the next wave of differentiated biotherapeutics.
The role of avidity in antibody biology
The strength of a single binding interaction between an antibody Fab fragment and antigen is defined by the term affinity, which we term zero-order avidity for the purpose of this Review. Avidity arising from different affinity interactions mediated by the antibody Fab and Fc domains and modulated by the hinge region (Box 1) can be grouped into tiers depending on the level of multivalency achieved: first-order avidity, referring to bivalent Fab interactions with antigen; second-order avidity, referring to simultaneous Fab–antigen and Fc–Fc or Fab–Fab interactions; and third-order avidity, referring to interactions between immune complexes and immune effector molecules. The term avidity is used below as an overarching term to describe the binding strength that results from these multidimensional affinity interactions23. It is noted that all tiers of avidity interactions may be increased for polyclonal sera and antibody cocktails binding a multitude of non-overlapping epitopes.
Box 1 Human immunoglobulin architecture.
Human immunoglobulins consist of three distinct regions: an antigen-binding fragment (Fab) region that binds antigen, a crystallizable fragment (Fc) region that interacts with immune effector molecules and a hinge region that links the Fab to the Fc and defines conformational flexibility. A monomeric immunoglobulin molecule is Y-shaped and is usually composed of four distinct protein chains, including two identical heavy chains (HCs) and light chains (LCs) (dark and light red, respectively, in the figure). HCs are composed of three or four constant domains (CH), depending on the antibody isotype, and LCs contain a single constant domain (CL). The constant domains together form the Fc region, the hinge region and the base of the Fab region. The Fab region comprises the variable domains that determine antigen binding specificity and affinity. Disulfide bonds shape the overall quaternary structure of the molecule by linking the two HCs in the hinge region and the HC and LC in the Fab region. In humans, variations in the HC constant domain during an immune response generate five different isotypes; IgM, IgD, IgG, IgA and IgE, with IgG and IgA further subdivided into subclasses 1–4 and 1–2, respectively. IgG, IgD and IgE exist as monomers, whereas IgA and IgM may contain an additional polypeptide J-chain that allows the formation of dimers and pentamers, respectively. A small fraction of IgM antibodies exists as hexamers in which the J-chain protein is replaced by an extra monomer subunit. IgA may further contain a secretory component that provides protection against proteolytic degradation. The CH2 and CH3 domains of IgG interact with the neonatal Fc receptor (FcRn), which protects the antibody from catabolism and thereby substantially increases its plasma half-life202,203. Immunoglobulin isotypes and subclasses can further be distinguished by variation in the hinge length, the number and location of disulfide bridges and N-linked or O-linked glycosylation within the HC. The hinge region serves as a connector and also modulates effector function potency; indeed, modification of the hinge length and flexibility has allowed for fine tuning of IgG effector activity180. Together, changes in antibody valency, hinge length and flexibility, as well as glycosylation status can influence avidity interactions and overall functional response kinetics.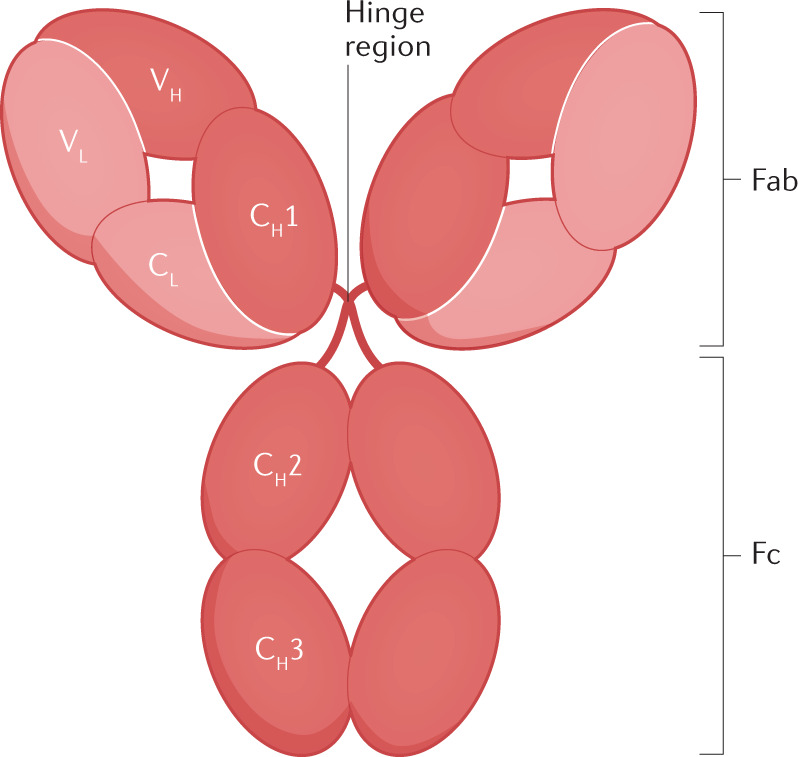
Antibody functional responses
The importance of avidity for antibody–antigen recognition in both natural immunity and antibody therapies has long been recognized6,23,24. The primary mechanism by which antibodies combat infection is through binding and directly neutralizing a pathogenic target antigen by preventing its interaction with host receptors and blocking its function, such as mediating viral entry into a cell. As early as 1937, Burnet et al. recognized that a single antibody binding event was likely to be insufficient to inactivate viruses and argued that neutralization of viruses by antibodies occurs through multivalent antibody binding to a high proportion of viral epitopes12,25. This polyclonal immune complexation, rather than single molecular binding events, is the central trigger for downstream humoral and cellular responses. Further, antibody Fc domains allow for interactions with multiple immune effector molecules present in human plasma or expressed on immune cells and a single binding interaction between an antigen-bound antibody and an effector molecule is generally insufficient to induce an effective Fc-mediated response. All together, avidity binding interactions through both Fab and Fc — with the hinge in a modulatory role — constitute a remarkably sensitive adaptor system that enables the rapid initiation and amplification of different effector mechanisms to combat disease (Box 1).
In our view, the response kinetics and thresholds governing functional antibody responses can be defined as the net result of distinct activation phases, including scanning of the antibody over the antigen (zero-order avidity), antigen–antibody binding and complex formation (first-order avidity), clustering of antibody–antigen complexes on the cell surface, potentially facilitated by the formation of simultaneous intermolecular Fc–Fc and Fab–Fab contacts (second-order avidity), and Fc-mediated binding of immune effector molecules up to a threshold that triggers an amplification of the functional response (third-order avidity). These stages are followed by the regulation and contraction of the response (Fig. 1). Association and dissociation rate constants and local concentration determine the on-rate and off-rate of antibody binding kinetics and complex formation is favoured when the on-rate is faster than the off-rate. The conditions required to reach an activation threshold may vary between effector systems; for example, different types of effector mechanisms require different antibody concentrations because they are triggered at distinct target occupancy levels26 (Fig. 2). Other factors, including antigen expression and distribution, different immunoglobulin subclasses, and effector elements such as complement factors, effector cell subtypes, Fc receptor subtypes, polymorphisms and expression levels, can also affect the overall avidity of antibody interactions and the consequent amplification of a functional response27,28. Regulation and contraction of the response is induced by target-related outputs such as the elimination of target cells, target densities dropping below the amplification threshold or inhibition by regulatory molecules expressed on the target or effector cell or recruited from plasma. Similarly, systemic regulation can occur when there is a shortage of effector molecules or cells, or by the presence of interfering immunoglobulin subclasses29–31. Conversely, insufficient regulation may cause autoimmunity through excessive or constitutive activation of the antibody functional response.
Fig. 1. Response kinetics governing antibody functional responses.

Avidity arising from combinations of affinity interactions are grouped in distinct tiers that integrate the common biological mechanisms of input, output and feedback. Monovalent antibody binding events, termed zero-order avidity interactions for the purpose of this Review, vary from highly transient to long-lasting, depending on affinity. This antibody scanning mode progresses to first-order avidity binding through bivalent Fab–antigen interactions and second-order avidity binding through concomitant Fab–Fab or Fc–Fc interactions. Third-order avidity is engaged when antibody oligomerization passes a threshold for Fc-mediated binding of soluble or cell-bound immune effector molecules, including configurations allowing interactions with IgG Fc receptors (FcγRs) or the complement component C1. The antibody functional response may be regulated or dampened at any avidity tier by, for example, elimination of target cells, target densities dropping below the amplification threshold or regulatory molecules expressed on either the target cell or the effector cell or recruited from plasma.
Fig. 2. Factors affecting antibody functional response activation.

a | Schematic representation of dose–response relationships for different Fc-mediated effector mechanisms. Functional responses reach the activation threshold at antibody doses that vary per effector mechanism; here illustrated in order of increasing antibody concentrations required for antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP) and complement-dependent cytotoxicity (CDC). b | Increasing antibody doses result in increasing target occupancy and antibody densities on the target cell surface, thereby favouring distinct tiers of avidity binding; saturation may favour monovalent antibody binding. Fc-mediated effector functions are triggered at different target occupancy levels. IgG Fc receptor (FcγR)-induced ADCC involves the release of cytotoxic granules containing granzymes and perforin (an example of a natural killer (NK) cell is shown); ADCP involves the uptake and lysosomal degradation of target cells (an example of a macrophage is shown); CDC involves the triggering of an amplifiable cascade of complement proteins present in blood, terminating in the generation of a lytic membrane-attack complex. In addition to direct killing, the production of cytokines or bioactive complement fragments may contribute to additional attraction and activation of effector cells. The antibody density required for reaching the activation threshold is defined by different parameters including antibody affinity, valency and concentration; structural constraints related to epitope recognized and antibody isotype, antigen expression and distribution; and the type and presence of effector molecules and regulatory molecules.
The humoral adaptive immune response
The role of avidity in humoral immunity is evident early in the initiation of the immune response. Upon entry of foreign antigens into lymphoid tissues, naive B cells present in lymph node follicles are activated through antigen binding to their IgM B cell receptors (BCRs) (Box 2). BCRs expressed on mature B cells are membrane-bound immunoglobulin molecules that are activated following antigen-induced aggregation32. Although several different models have been proposed for BCR activation, considerable evidence suggests that BCRs undergo activation-dependent localization to membrane microdomains, probably facilitating higher-order BCR clustering and signal transduction33,34. Upon activation, some B cells become plasma cells, which switch from primarily expressing membrane-associated IgM to abundantly secreting it by a mechanism of alternative RNA processing35. Secreted IgM antibodies are predominantly pentameric molecules (hexameric molecules comprise <5% of serum IgM)36,37 that, despite binding with characteristically low intrinsic affinity (KD of 10–4 to 10–6 mol l–1), are able to avidly engage immunogens owing to their ten identical antigen binding sites and high conformational flexibility38. IgM antibodies are especially effective in protecting against microbes with highly expressed, closely spaced surface epitopes, which is in part a consequence of efficient avidity-based interactions between surface antigen-bound IgM and the complement system.
In an ongoing immune response, B cell clonal selection and affinity maturation can drive the intrinsic affinity of the Fab domain up to 10–10 mol l–1 (refs.39,40). Additionally, class switching from IgM to IgG, IgA, IgE and their subtypes results in swapping of antibody Fc domains and associated functional properties. Conversely, B cell activation is subject to physiological control mechanisms through Fc-mediated antibody feedback, where secreted antibodies inhibit B cell activation by forming antibody–antigen immune complexes that, in the context of interaction with BCR-bound antigen, co-engage the inhibitory IgG Fc receptor IIb (FcγRIIB). Avidity ligation of both the BCR and FcγRIIB results in inhibitory feedback of BCR complex signalling through phosphatases recruited by the immunoreceptor tyrosine-based inhibitory motif (ITIM) in the cytoplasmic tail of FcγRIIB32,41,42 (Fig. 3a).
Fig. 3. Avidity in antibody biology.

a | B cell receptors (BCRs) constitute antibodies expressed with a transmembrane region on the B cell surface. First-order avidity interactions induce BCR crosslinking and phosphorylation of the BCR-associated transducer molecules that recruit SYK and LYN tyrosine kinases, resulting in protein kinase C (PKC) activation, mitogen-activated protein kinase (MAPK) activation and calcium release. Second-order and third-order avidity binding of antibody-antigen immune complexes recruit FcγRIIB, which comprises an immunoreceptor tyrosine-based inhibitory motif (ITIM motif) that activates phosphatases and reduces BCR signalling. b | Fab-domain-mediated neutralization of pathogen structures, preventing interactions with host cells and blocking pathogen entry into the cell. Binding of a protective antibody against the repetitive P. falciparum circumsporozoite protein (PfCSP) epitope consisting of repeats of the amino acids NPNA is facilitated by second-order avidity Fab–Fab interactions between Fabs from neighbouring IgGs (bound in pairs). Neighbouring Fabs each bind a two NPNA repeat and are shifted 77° along the NPNA spiral, with five IgGs completing a full circle. The Fabs of the IgGs are oriented non-symmetrically in a repeating light chain-bottom/heavy chain-top orientation. facilitating Fab–Fab contacts (left). HIV-1 virions escape second-order avidity binding by sparse surface expression of HIV-1 envelope glycoprotein spikes. The neutralizing antibody 2G12 evolved a domain-swapped structure in which each heavy chain contacts both light chains, thereby creating a large rigid surface that facilitates avid binding of conserved carbohydrate clusters on the HIV-1 envelope spike (right). c | Clustering of immune complexes is initiated by IgG molecules that assemble into ordered hexameric structures through non-covalent Fc–Fc interactions, which facilitate third-order avidity binding and activation of C1 or intracellular signalling through recruitment of signalling molecules such as cIAP1 and TRAF2. IgG hexamer formation proceeds through recruitment of additional IgG molecules through second-order avidity interactions mediated by the Fc domains until ring closure. d | Antibodies that predominantly bind monovalently (such as those with moderate affinity or those engineered for monovalent binding) may elicit stronger effector functions because higher cell surface densities of Fc domains can be achieved. Part a adapted from ref.41, Springer Nature Limited.
Box 2 Human immunoglobulin functional properties.
BCR
Each of the immunoglobulin classes and subclasses may be expressed in monomeric form with an integral membrane domain to form a B cell receptor (BCR) on the surface of a B cell. Expression of the membrane and secreted forms of antibody is regulated by alternative RNA processing. Secreted immunoglobulin is identical in sequence to the BCR except for the absence of the transmembrane region and potential post-translational modifications or quaternary structures.
IgM
In plasma, IgM largely exists as pentamers linked together by disulfide bonds and a polypeptide J-chain, but a small fraction exists as a covalent IgM hexamer in absence of a J-chain. Owing to their structural and functional differences, pentameric and hexameric IgM might be viewed as distinct IgM subclasses68. Studies have shown that both pentameric and hexameric IgM adopt a hexagonal platform conformation supporting high-avidity binding interactions with C1q and efficient complement activation upon antigen binding204,205. Pentameric IgM can be exported at mucosal surfaces by the polymeric immunoglobulin receptor to form secretory IgM in complex with a fragment derived from the polymeric immunoglobulin receptor, the secretory component206.
IgG
IgG is the most abundant immunoglobulin class in human serum and accounts for approximately 10–20% of plasma protein. The four different subclasses, in order of decreasing abundance, are IgG1, IgG2, IgG3 and IgG4. Each subclass contains a unique profile with respect to antigen binding, complement binding and binding to Fc receptors for IgG (FcγRs). Structural determinants in the core hinge region can influence antibody function; for example, increasing hinge length and flexibility enhances binding to antigen for immune complex formation and binding to the complement component C1q94,180,207. The flexibility of the Fab arms affects the relative binding of subclasses to FcγRs and C1q, with IgG3 having the strongest binding, followed by IgG1, IgG4 and IgG2. Furthermore, modification of glycans attached to amino acid N297 affects FcγR binding. Notably, human polyclonal IgG4 antibodies undergo a process named Fab-arm exchange under natural physiological conditions, in which half-molecules (HC–LC pairs) recombine with half-molecules from other IgG4 antibodies. This is a continuous and stochastic process resulting in bispecific, functionally monovalent IgG4 antibodies in blood64.
IgD
IgD is primarily expressed as a transmembrane antigen receptor on naive mature B cells. IgD possesses a long hinge region with high flexibility and is capable of acquiring a T-shaped structure. This structural flexibility potentially contributes to the regulation of B cell responsiveness to different types of antigens owing to preferential binding of IgD to multimeric antigens over monomeric antigens208,209. Secreted IgD weakly interacts with complement or FcδRs. It binds basophils, mast cells, monocytes and dendritic cells in an FcδR-independent manner, contributing to mucosal homeostasis through the production of antimicrobial peptides and inflammatory cytokines.
IgA
IgA has two subclasses — IgA1 and IgA2 — that contain a heavily glycosylated T-shaped hinge and a more rigid Y-shaped hinge, respectively. IgA in serum exists as a monomer (±90%) and a J-chain-linked dimer (±10%)210. The latter is the precursor for secretory IgA, which is exported across mucosal surfaces in copious amounts by the polymeric immunoglobulin receptor to form IgA in complex with the secretory component. IgA immune complexes efficiently interact with FcαRI on myeloid cells, including neutrophils and macrophages211.
IgE
IgE is the least abundant and fastest-clearing isotype in human plasma (its half-life is less than a day, compared to about 3 weeks for IgG). IgE binds to the high-affinity FcεRI on mast cells and the low-affinity CD23 on haematopoietic cell types212. IgE glycosylation is essential for IgE–FcεRI interactions and this requirement is primarily attributed to one of seven N-linked glycan sites (N394)213. IgE can remain bound to FcεRI for weeks to months, thereby contributing to long tissue persistence214.
Fc-mediated effector functions
Classical antibody Fc-mediated effector mechanisms such as complement-dependent cytotoxicity (CDC), antibody-dependent cellular cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP) are key mechanisms for the elimination of pathogens or diseased cells such as infected or cancerous cells. Although the processes of antigen binding by the Fab domain and effector function activation by the Fc domain were long thought to act independently, multiple studies have challenged this view and an optimal configuration of immune complexes probably exists for different types of effector functions. In addition to different Fc-density requirements for activation (Fig. 2), there is accumulating evidence for allosteric or intramolecular cooperativity28,44; for example, the optimal immune complex stoichiometry required for complement activation may be facilitated by IgG Fc–Fc interactions and modulated by their Fab domains45. Fab and Fc function may even be considered interdependent, given that efficient complement activation requires clustering of cell-bound IgGs through concerted antigen–Fab and Fc–Fc interactions, thereby forming optimal docking sites for recruitment of C1q46. Furthermore, the binding of IgG–antigen complexes to FcγRIIIA was recently shown to be stabilized through contacts with the IgG light-chain constant region, as well as by Fab and Fc conformational changes44. First-order and second-order avidity interactions are therefore a prerequisite for third-order avidity interactions and resulting functional responses.
Neutralization
Fab-domain-mediated neutralization is the most direct mechanism by which antigens and pathogens can be inactivated. Indeed, binding affinity is a principal driver of potency for some highly successful therapeutic antibodies that neutralize cytokines, growth factors or complement factors. Binding of the Fab domain to specific pathogenic structures prevents interactions with host cells, thereby blocking toxin activity or viral entry into the cell. Although efficient neutralization fundamentally depends on strong affinity, recent studies provide compelling evidence for additional avidity mechanisms for optimizing pathogen recognition. For example, the repetitive nature of certain Plasmodium falciparum circumsporozoite protein (PfCSP) epitopes drives the selection of homotypic Fab–Fab interactions between PfCSP repeat region-specific antibodies47,48 (Fig. 3b). These second-order avidity interactions presumably increase B cell receptor clustering and selective clonal expansion of PfCSP-reactive B cells.
One of the most striking illustrations of the selective pressure of repeating microbial epitopes involves the heavy chain variable (VH)-domain swapping observed in anti-HIV-1 human antibody 2G12, which enables multivalent interactions with conserved carbohydrate clusters on the HIV-1 envelope glycoprotein subunit gp120 (ref.49). A single large and rigid binding domain is formed by the two heavy chains cross-contacted by both light chains in which the novel VH–VH interface creates an additional antigen binding site for increased contacts with glycan structures (Fig. 3b, right). Notably, further studies demonstrated the existence of a supramolecular antibody structure consisting of 2G12 dimers with two large (Fab)2 binding units and two Fc domains providing >50-fold greater neutralization potency over monomeric 2G12 (ref.50). We note that although the germline precursor to this antibody adopts a conventional structure, five somatic mutations were sufficient for domain exchange, suggesting that this unique structural rearrangement could be exploited for generating therapeutic antibodies against (2G12-like) carbohydrate epitopes on other viruses51,52.
The crucial importance of avidity in antibody-mediated neutralization of toxins and viruses is further exemplified by HIV-1 itself, which escapes neutralizing antibodies by impeding bivalent high-avidity antibody binding, in addition to other mechanisms such as its rapid mutation rate and shielding of conserved epitopes on the trimeric spike protein. As a result, antibodies against HIV-1 envelope glycoproteins are thought to predominantly bind the viral surface monovalently53–55. Several studies suggest that evasion of bivalent antibody binding occurs owing to the low spike protein density on the virion surface and an unfavourable distribution of epitopes on the viral spike protein trimer, thereby limiting interspike and intraspike crosslinking, respectively56 (Fig. 3b, right). By contrast, antibodies targeting viruses such as influenza and respiratory syncytial virus can bind bivalently and thereby take advantage of second-order avidity effects57,58.
Often, Fc-domain-mediated third-order avidity interactions are required to augment toxin or pathogen inactivation through recruitment of complement or FcγRs on immune cells (Fig. 2). In the case of HIV-1, in vivo protection by broadly neutralizing antibodies (bNAbs) was shown to be augmented for antibodies with Fc domains capable of engaging activating FcγRs59,60. Similarly, in an elegant Fc engineering approach, Gunn and colleagues recently used a library of Fc variants with identical Fab domains to demonstrate the critical importance of FcγR-mediated effector functions and complement activation in antibody-mediated protection against Ebola virus infection61.
The potentially deleterious effects of effector functions, such as antibody-dependent enhancement of viral infection, represent an important design consideration. Interactions between virus-bound antibodies and FcγRs or complement receptors may enhance viral entry and replication in phagocytic cells, as has been demonstrated for flaviviruses such as dengue virus62. A cocktail of antibodies targeting distinct epitopes on dengue virus envelope glycoprotein E and engineered to reduce FcγR binding (L234A–L235A mutations) induced efficient dengue virus neutralization without precipitating infection63. This indicates that potentiating first-order and second-order avidity interactions in a polyclonal antibody mixture while minimizing third-order avidity may be crucial for obtaining optimal protection in certain cases.
Autoimmunity
The avidity of autoantibodies against self-antigens can increase their pathogenicity, as demonstrated by the induction of symptoms of myasthenia gravis in monkeys by bivalent IgG1 antibodies against the acetylcholine receptor when infused alone, but not when combined with IgG4 antibodies. This can be explained by the finding that IgG4 antibodies in plasma cannot engage in first-order Fab-mediated avidity interactions as they become functionally monovalent in vivo by the dynamic process of Fab-arm exchange between IgG4 antibodies of unrelated specificity64. Notably, interspecies exchange between human and macaque IgG4 antibodies occurs with higher efficiency than intraspecies exchange because of the increased stability of the human/macaque IgG4 heterodimer65,66, which may have contributed to the observed effects. Paradoxically, avidity binding may also ameliorate autoimmune disease, as was demonstrated in the case of myasthenia gravis caused by autoantibodies against muscle-specific kinase (MuSK). Signalling through MuSK has an essential role in maintaining neuromuscular junctions and a recent study demonstrated that monospecific, functionally bivalent anti-MuSK antibodies act as partial agonists and are therefore less pathogenic than bispecific, functionally monovalent IgG4 anti-MuSK antibodies, which abolished MuSK signalling. These findings suggest that a lack of first-order avidity of IgG4 molecules could be a pathogenic mechanism in other IgG4-mediated autoimmune diseases67.
Complement activation
The complement system is a potent innate immune defence mechanism composed of an amplifiable enzymatic cascade that kills pathogens and attracts immune effector cells. Complement is activated through three distinct pathways: the classical pathway, the lectin pathway and the alternative pathway, which depend on the binding of C1q, mannan-binding lectin or spontaneously activated complement to pathogenic surfaces, respectively. All pathways ultimately converge in the generation of C3 and C5 convertases that cooperate in the production of opsonins, anaphylatoxins, chemoattractants and the formation of the membrane attack complex, which breaches the target cell membrane to kill the cell68.
The classical pathway is triggered upon binding of C1q to the Fc region of cell-bound IgG or IgM. C1q consists of six collagen-like triple-helical stalks connected to globular headpieces that resemble the shape of a bunch of tulips69. The binding affinity of monomeric IgG Fc for C1q is weak (KD ≈ 10–4 M), resulting in highly transient interactions. Consequently, functional C1q binding and activation requires second-order and third-order avidity interactions, leading to an increase in KD by four orders of magnitude70. IgM antibodies naturally exist in a multimeric form, which allows them to interact with C1q to activate complement following a conformational change36,37,71. By contrast, IgGs require assembly into ordered antigen-bound hexamers to form an optimal structure for binding and activating C1 (ref.7). Mass spectrometry, cryo-electron tomography and mutational studies have given compelling evidence for the requirement of higher-order IgG oligomers for optimal complement activation, demonstrating that IgG monomers, dimers and trimers do not contribute much to CDC and that engagement of least four C1q binding sites is required for C1 activation14,45,46. The assembly of IgG hexamers on antigenic surfaces results from an intricate process in which incoming IgGs are predominantly recruited from solution through second-order avidity events in which Fc–Fc interactions facilitate antigen binding (Fig. 3c). At low antigen densities or high IgG concentrations, monovalently bound IgGs may contribute to oligomerization mediated through lateral diffusion46,72. Interestingly, functionally monovalent antibodies, such as bispecific antibodies comprising only a single Fab arm that is capable of antigen binding on a target cell, were shown to induce CDC more potently than the corresponding bivalent (parent) antibodies, potentially owing to optimal geometric positioning and a superior ability to form hexamers and engage C1 (refs.7,14,45,46,72). The high amplification potential of the IgG hexamer-complement system is counterbalanced by strong regulation through numerous soluble factors — for example, C1 inhibitor, C4Bp, and complement factors I and H — and cell-surface-expressed proteins such as CD46, CD55 and CD59, which block the complement cascade from progressing at multiple checkpoints73.
As discussed earlier, an ideal immune complex stoichiometry probably exists for efficient activation and amplification of Fc-mediated effector functions such as the complement cascade. Aside from optimal immunoglobulin oligomerization, a number of additional factors can influence triggering of complement activity. For example, there are several structural prerequisites that affect IgG Fc–Fc interactions and optimal positioning of IgG hexamers on the cell surface, including antigen-dependent constraints such as their size and distribution, and epitope geometry and orientation. Antigen-dependent and epitope-dependent constraints are most clearly exemplified by the enigmatic existence of two functional types of antibodies against the established therapeutic target CD20 in B cell malignancies and inflammatory diseases. Type I CD20 antibodies induce CD20 translocation into lipid rafts and induce cell killing through complement activation and ADCC, whereas type II CD20 antibodies do not cluster CD20 and instead kill through apoptosis and ADCC74,75. Recent studies employing structural and thermodynamic analyses have shed light on the mechanistic basis of this class distinction, showing that CD20 forms a dimer that provides two binding sites for type I CD20 antibodies such as rituximab and ofatumumab, facilitating avidity interactions and complement activation. These interactions include Fab–Fab and Fc–Fc second-order avidity interactions enabled by the two binding sites on the CD20 dimer. By contrast, each antigen binding site of the type II CD20 antibody obinutuzumab can bind only a single CD20 dimer, therefore precluding second-order avidity interactions that facilitate complement activation9,76. These insights suggest that besides Fc–Fc interactions, Fab–Fab homotypic interactions can also be exploited to avidity-engineer therapeutic antibody function.
Combinations of IgG antibodies simultaneously targeting CD20 and CD37 antigens can induce synergistic CDC of tumour B cells by forming mixed hetero-hexameric complexes on the cell surface, even where the monoclonal parent antibodies are unable to activate complement77. This synergy between antibodies targeting two different antigens is probably caused by a favourable distribution of target epitopes on the cell surface that facilitates avidity binding. Conversely, structural constraints such as hinge length and glycan heterogeneity influence IgG oligomerization and complement activation78–81. The composition of polyclonal mixtures of IgG subclasses or including other antibody classes such as IgA and IgE may therefore affect complement activation through competition for antigen binding and differences in flexibility, conformation and valency (Box 2).
FcR-mediated cellular effector functions
FcγRs are bound by the different human IgG isotypes with varying affinities (Box 3). Only FcγRI can bind monomeric IgG with high (nanomolar range) affinity and as a consequence may hamper avidity interactions within immune complexes in vivo, given that the receptor is probably nearly saturated by the high IgG content in serum. Cytokine stimulation during inflammation may induce FcγRI clustering in membrane microdomains, thereby facilitating avidity binding of immune complexes over monomeric IgG82. All other FcγRs exhibit low, micromolar affinities for monomeric IgG, and as a consequence require third-order avidity interactions for IgG binding under physiological conditions83, thereby preventing inappropriate effector cell activation in the absence of a pathogenic trigger. Multimerization of cell-bound IgG strengthens FcγR binding interactions over a threshold that triggers receptor signalling through phosphorylation of immunoreceptor tyrosine activating motif (ITAM) domains, which in turn leads to elimination of target cells through ADCC or ADCP. Notably, FcγRIIIA — which is responsible for mediating ADCC by natural killer (NK) cells — binds afucosylated IgG, with 40-fold greater affinity than fucosylated IgG84. The relevance of this for immune regulation was demonstrated by the observation that afucosylated IgG is selectively produced against membrane antigens on enveloped viruses — triggered by an as-yet-unknown mechanism — and the degree of afucosylation correlates with disease severity in COVID-19 and dengue virus haemorrhagic fever85,86. Similar observations have been made in antibody responses against red blood cells and platelets in pregnancy-associated alloimmunization and related pathologies87. This suggests that the tuning of third-order avidity interactions through modulating affinity for Fc receptors is used as a natural mechanism to protect against infection and that disease risk is increased if regulation is insufficient.
Non-IgG antibody subclasses bind to corresponding FcRs, including FcαRI (IgA), FcεRI and FcεRII/CD23 (IgE), FcµR (IgM), IgδR (IgD) and Fcα/µR (IgA/IgM). IgA has a key role in mucosal tissues by regulating the tolerance to and protection from exposure to antigens, food, commensal microorganisms and enteric pathogens, whereas IgE has a role in the host defence against parasites such as helminths and is known for its role in allergic reactions through the high-affinity binding to FcεRI of mast cells (Box 2). IgA immune complexes bind FcαRI expressed on myeloid cells (including neutrophils, eosinophils, monocytes and macrophages) through third-order avidity interactions. Indeed, IgA antibodies have been reported to recruit neutrophils through release of leukotriene B4 and induce ADCC of tumour cells via the FcαRI88–92. Neutrophil-mediated ADCC may be mediated through a process called trogocytosis, which involves a stepwise destruction of the plasma membrane and uptake of the resulting plasma membrane fragments93.
A multitude of factors influence the strength of avidity and degree of antibody–FcR crosslinking, including antibody and FcR binding affinity, location of the antigenic epitope, and cell surface rigidity10. Several studies have characterized FcγR polymorphic variants that display varying binding affinities for different IgG subclasses, which may impact FcγR crosslinking and activation83,94. Mazor et al. showed that antibodies of intermediate affinity targeting CD4, epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 2 (HER2) elicited stronger ADCC and ADCP (as assessed by higher maximum kill) than high-affinity antibody variants43. The observed difference was attributed to increased cell-surface opsonization owing to a greater propensity for monovalent binding through faster off-rates compared to high-affinity antibodies, leading to a higher local density of Fc domains (Fig. 3d). Indeed, dynamic, monovalent binding allows greater antibody mobility and flexibility95, which may favour the formation of third-order avidity interactions with FcγR on cells beyond a threshold for functional activation. This is supported by previously mentioned studies demonstrating that monovalent binding may facilitate third-order avidity interactions with C1, thereby enhancing complement activation, whereas high-affinity binding can lead to stagnation7,72,96. Further, at low IgG concentrations, phagocytosis of emulsion droplets was shown to be more efficient compared to solid particles97; the authors speculated that lateral diffusion of IgGs attached to the emulsion droplet surface was prevented in solid particles displaying higher cell surface rigidity. These examples illustrate the interplay between affinity and avidity interactions in tuning antibody effector functions and their response to immune complexes.
Box 3 IgG Fc receptors.
The Fc domain of antibodies interacts with a family of FcRs expressed on various immune cells to mediate effector functions such as antibody-dependent cellular cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP). IgG binds to FcγRs (Box 2), which are broadly classified as either activating or inhibitory, depending on whether their intracellular signalling domain contains an immunoreceptor tyrosine activating motif (ITAM) or immunoreceptor tyrosine inhibitory motif (ITIM), respectively. Activating FcγRs in humans include FcγRI (CD64), FcγRIIA (CD32a) and FcγRIIIA (CD16a) and the less well characterized FcγRIIC (CD16c). By contrast, FcγRIIB (CD32b) represents the sole inhibitory FcγR, which regulates the function of activating FcγRs, controls antibody production by B cells and serves as the main scavenging receptor in the liver. Finally, FcγRIIIB (CD16b) uniquely contains no intracellular signalling domain and is instead anchored to the membrane by glycosylphosphatidylinositol. FcγRs are differentially expressed on lymphoid-derived and myeloid-derived effector cells, although the receptor distribution is unique to each cell type215. FcγRIIIA is expressed on monocytes, macrophages and natural killer (NK) cells, and is the main receptor for mediating ADCC, promoting release of cytotoxic granules and pro-apoptotic signalling molecules after engaging IgG-opsonized target cells. Phagocytes such as macrophages and dendritic cells express a combination of different FcγRs, including FcγRI, FcγRIIA, FcγRIIIA and FcγRIIB, that mediate ADCP for innate destruction of IgG-opsonized targets cells and also promote cross-presentation to T cells to elicit immunological memory216–218. Studies have shown that the balance of signals between activating FcγRIIA and inhibitory FcγRIIB are important determinants of the ability of antibody–antigen immune complexes to activate monocytes and dendritic cells186,216,219,220. Although the link between FcγRs and cellular immunity has historically been viewed as indirect through antigen presentation by cells, it was recently demonstrated that FcγRIIA is capable of directly promoting activation of CD4+ T cells by IgG immune complexes221.
Avidity engineering of antibody therapeutics
Antibodies have become widely established as therapies for numerous diseases, including cancer, infectious diseases, inflammatory diseases and autoimmunity13. The design of therapeutics against hard-to-hit targets that provide meaningful improvements for patients compared to contemporary drug regimens is shifting antibody drug development from canonical IgG antibodies towards formats with novel functionalities. The search for approaches to enhance antibody function is not surprising considering the limitations imposed by monospecificity; indeed, the use of monoclonal antibodies as a single agent for therapy seems contradictory in light of the polyclonal, avidity-tuned nature of antibodies generated during the natural immune response.
The avidity engineering toolbox
A broad spectrum of novel antibody engineering strategies and formats that enhance or tune avidity are emerging. These strategies include those that optimize physiological immune pathways, such as effector function enhancement, and those that enable non-native mechanisms of action, for example effector cell redirection or receptor agonism.
Below, we describe an ‘avidity engineering toolbox’ and discuss the impact of different strategies on the different phases of antibody functional response activation as defined above and in Fig. 1. Avidity engineering strategies include targeting multiple epitopes; targeting multiple cell surface receptors in cis; tuning antibody valency; and tuning binding strength for complement or FcγRs. Antibody therapies employing these engineering strategies have advanced into clinical development and as of May 2022, the commercial clinical pipeline includes 35 programmes leveraging avidity to increase antibody function (Table 1). In the subsections below, we highlight some of these key engineering strategies exploiting avidity to boost antibody functional responses.
Table 1.
Avidity-based therapeutic antibody concepts in the clinic and engineering platforms
| Agent (company) | Target | Format | Indication (selected) | Development status (selected trials) |
|---|---|---|---|---|
| Targeting multiple epitopes | ||||
| Sym015 (Symphogen) | MET | Mixture of two IgG1 antibodies binding non-overlapping epitopes on the Semaphorin domain of MET | Solid tumours (NSCLC) | Phase IIa completed (NCT02648724) |
| REGN5093 (Regeneron) | MET | Biparatopic, bispecific IgG4 antibody targeting two non-overlapping epitopes on MET (Veloci-Bi bispecific technology) | Solid tumours (NSCLC) | Phase I/II (NCT04077099) |
| REGN5093-M114 (Regeneron) | MET | Biparatopic, bispecific IgG4 antibody targeting two non-overlapping epitopes on MET (Veloci-Bi bispecific technology) conjugated to a maytansinoid | Solid tumours (NSCLC) | Phase I/II (NCT04077099) |
| Sym004 (Symphogen) | EGFR | Mixture of two IgG1 antibodies (futuximab, modotuximab) binding non-overlapping epitopes on EGFR | Solid tumours (mCRC) | Phase III completed (NCT02083653) |
| KN026 (Alphamab) | HER2 | Biparatopic, bispecific IgG1 antibody targeting two non-overlapping epitopes on HER2 (CRIB technology) | Solid tumours (breast and gastric cancer) | Phase I (NCT03619681, NCT03847168) |
| MBS301 (Beijing Mabworks Biotech) | HER2 | Afucosylated, biparatopic, bispecific IgG1 antibody targeting two non-overlapping epitopes on HER2 (knobs-into-holes bispecific technology) | Solid tumours (HER2+) | Phase I (NCT03842085) |
| Zanidatamab, ZW25 (Zymeworks) | HER2 | Biparatopic, bispecific IgG1 antibody targeting two non-overlapping epitopes on HER2 (Azymetric bispecific technology) | Solid tumours (HER2+) | Phase II (NCT04466891, NCT03929666, NCT04224272) |
| ZW49 (Zymeworks) | HER2 | Biparatopic, bispecific IgG1 antibody targeting two non-overlapping epitopes on HER2 (Azymetric bispecific technology) conjugated to a maytansinoid (Zymelink antibody-drug condensate technology) | Solid tumours (HER2+) | Phase I (NCT03821233) |
| REGN-COV, casirivimab/imdevimab (Regeneron) | SARS-CoV-2 spike | Mixture of two human IgG1 antibodies, binding non-overlapping epitopes on the SARS-CoV-2 viral spike | COVID-19 | EUA, Phase III completed (NCT04381936) |
| Evusheld, tixagevimab/cilgavimab, (AstraZeneca, VanderBilt) | SARS-CoV-2 spike | Mixture of two human IgG1 antibodies with mutations that extend half-life (M252Y,S254T,T256E) and reduce Fc receptor and C1q interactions (L234F, L235E, P331S), binding non-overlapping epitopes on the SARS-CoV-2 viral spike | COVID-19 | EUA, Phase III (NCT04625725) |
| BMS-986414/BMS-986413 (Bristol Myers Squibb/Rockefeller University) | SARS-CoV-2 spike | Mixture of two human IgG1 antibodies with mutations that extend half-life (M428L/N434S), binding non-overlapping epitopes on the SARS-CoV-2 viral spike | COVID-19 | Phase II/III (NCT04518410) |
| ADM03820 (Ology Bioservices) | SARS-CoV-2 spike | Mixture of two human IgG1 antibodies binding non-overlapping epitopes on the SARS-CoV-2 viral spike | COVID-19 | Phase I (NCT04592549) |
| SAR441236 (Sanofi) | HIV-1 envelope | Triparatopic IgG1 bNAb from the VRC01-LS, PGDM1400 and 10E8v4 therapeutic antibodies, targeting the CD4bs, gp41 MPER and V1/V2 glycan-directed binding sites on HIV-1 (CODV-Ig technology) | HIV-1 | Phase I (NCT03705169) |
| Inmazeb, atoltivimab/maftivimab/odesivimab (Regeneron) | Zaire ebolavirus (ZEBOV) glycoprotein | A mixture of three afucosylated IgG1 antibodies that each bind to different, non-overlapping epitopes on the ZEBOV glycoprotein | ZEBOV | Approved (NCT03576690) |
| Targeting multiple cell surface receptors | ||||
| LAVA-051 (Lava Therapeutics) | CD1d, Vδ2 TCR | A trispecific γδ T cell engager comprised of a single-domain antibody (VHH) that binds both CD1d and the iNKT TCR, and a VHH targeting the Vγ9Vδ2 TCR, which triggers iNKT and Vγ9Vδ2 T cell activation | Haematologic malignancies (CLL, MM, AML) | Phase I /II (NCT04887259) |
| Sym013 (Symphogen) | EGFR, HER2, HER3 | A mixture of six humanized monoclonal IgG1 antibodies (three pairs) that bind to non-overlapping epitopes on EGFR, HER2 and HER3 | Advanced epithelial malignancies | Phase I/II terminated (NCT02906670) |
| Tuning antibody valency | ||||
| IGM-8444 (IgM Biosciences) | DR5 | Pentameric IgM antibody with ten binding sites specific for DR5 | Solid tumours | Phase I (NCT04553692) |
| INBRX-109 (Inhibrx) | DR5 | Four single-domain antibodies (tetravalent) fused to an Fc domain (sdAb technology; binding units derived from heavy-chain-only antibodies) | Solid tumours | Phase I (NCT03715933, NCT04950075) |
| INBRX-106 (Inhibrx) | OX40 | Six single-domain antibodies (hexavalent) fused to an Fc domain (sdAb technology; binding units derived from heavy-chain-only antibodies) | Solid tumours | Phase I (NCT04198766) |
| ABBV-621, APG350, Eftozanermin alpha (Abbvie/Apogenix) | TRAIL | Hexavalent TNFRSF agonist comprising a fusion protein composed of three receptor binding domains in a single chain arrangement, linked to a silenced human IgG1 Fc-domain (HERA-ligand technology) | Multiple myeloma | Phase Ib (NCT04570631) |
| PF-06755347, GL-2045 (Pfizer/Gliknik) | C1q | Recombinant human IgG1-based Fc multimer; fusion of the complete IgG1 hinge-CH2-CH3 coding region to the IgG2 hinge region (Stradomer technology) | Autoimmune diseases | Phase I (NCT03275740) |
| Tuning antibody oligomerization | ||||
| GEN3014, HexaBody-CD38 (Genmab) | CD38 | IgG1 antibody containing an E430G hexamerization-enhancing Fc mutation (HexaBody technology) | Haematologic malignancies (MM) | Phase I/II (NCT04824794) |
| Targeting multiple epitopes and tuning antibody oligomerization | ||||
| GEN3009, DuoHexaBody-CD37 (Genmab/AbbVie) | CD37 | Biparatopic, bispecific IgG1 antibody targeting two non-overlapping epitopes on CD37 containing an E430G hexamerization-enhancing Fc mutation (DuoHexaBody technology) | Haematologic malignancies (B cell NHL, CLL) | Phase I /II (NCT04358458) |
| GEN1029, HexaBody DR5/DR5 (Genmab) | DR5 | Mixture of two IgG1 antibodies targeting non-overlapping epitopes on DR5, both containing an E430G hexamerization-enhancing Fc mutation (HexaBody technology) | Solid tumours | Phase I (terminated) (NCT03576131) |
| Tuning binding to Fc effector receptors | ||||
| GS-1811, JXT-1811 (Gilead Sciences/Jounce Therapeutics) | CCR8 | IgG1 antibody afucosylated for enhanced ADCC | Solid tumours | Phase I (NCT05007782) |
| BMS-986340 (Bristol Myers Squibb) | CCR8 | IgG1 antibody afucosylated for enhanced ADCC | Solid tumours | Phase I (NCT04895709) |
| SEA-CD70 (Seattle Genetics) | CD70 | IgG1 containing afucosylated for enhanced ADCC (SEA technology) | Myeloid malignancies (MDS, AML) | Phase I (NCT04227847) |
| Elipovimab, GS-9722 (Gilead Sciences) | HIV-1 envelope | IgG1 bNAb (derived from PGT121) containing S239D/I332E and M428L/N434S Fc mutations for improved PK, enhanced ADCC and ADCP (high cytotoxicity XmAb and Xtend Fc domain technology) | HIV-1 | Phase Ib (GS-US-420-3902a) |
| Monjuvi, tafasitamab (MorphoSys/Incyte) | CD19 | IgG1 antibody containing S239D/I332E Fc mutations for enhanced ADCC and ADCP (high-cytotoxicity XmAb Fc domain technology) | Haematologic malignancies (DLBCL) | Approved |
| Margenza, margetuximab (Macrogenics) | HER2 | IgG1 antibody containing L235V/F243L/R292P/Y300L/P396L Fc mutations for enhanced ADCC | Solid tumours (HER2+ breast cancer) | Approved |
| Poteligeo, mogamulizumab (Kyowa Hakko Kirin) | CCR4 | IgG1 antibody afucosylated for enhanced ADCC (Potelligent Technology) | Adult T cell leukaemia or lymphoma | Approved |
| Gazyva, obinutuzumab (Roche) | CD20 | IgG1 antibody afucosylated for enhanced ADCC (GlycoMab Technology) | Haematologic malignancies (CLL, FL) | Approved |
| Fasenra, benralizumab (Astra Zeneca/Kyoa Hakko Kirin) | IL-5Rα | IgG1 antibody afucosylated for enhanced ADCC (Potelligent Technology) | Asthma | Approved |
| Targeting multiple cell surface receptors and tuning binding to Fc effector receptors | ||||
| Rybrevant, amivantamab (Janssen/JNJ) | EGFR, MET | IgG1 bispecific antibody afucosylated for enhanced ADCC (DuoBody technology) | Solid tumours (metastatic NSCLC with EGFR exon 20 insertion mutations) | Approved |
| MCLA-129 (Merus/Betta pharmaceuticals) | EGFR, MET | IgG1 bispecific antibody afucosylated for enhanced ADCC (Biclonics and GlymaxX technology) | Solid tumours (NSCLC) | Phase I/II (NCT04868877) |
Data available as of 1 May 2022. Engineering data were obtained from public documents (scientific literature, abstracts, posters and patent publications). This overview table excludes clinical programmes investigating bispecific antibodies, for which we refer to Labrijn et al.20. ADCC, antibody-dependent cellular cytotoxicity; ADCP, antibody-dependent cellular phagocytosis; AML, acute myeloid leukaemia; bNAb, broadly neutralizing antibody; CCR, CC-chemokine receptor; CLL, chronic lymphocytic leukaemia; CODV-Ig, crossover dual variable Ig-like protein; COVID-19, coronavirus disease 2019; CRIB, charge repulsion-induced bispecificity; DLBCL, diffuse large B cell lymphoma; DR5, death receptor 5; EGFR, epidermal growth factor receptor; EUA, emergency use authorization; Fc, crystallizable fragment; FL, follicular lymphoma; HER2, human epidermal growth factor receptor 2; iNKT cell, type I natural killer T cell; mCRC, metastatic colorectal cancer; MDS, myelodysplastic syndrome; MET, tyrosine-protein kinase MET; MM, multiple myeloma; MPER, membrane-proximal external region; NHL, non-Hodgkin lymphoma; NSCLC, non-small-cell lung cancer; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; sdAb, single-domain antibody; TCR, T cell receptor; TNFRSF, tumour necrosis factor receptor superfamily; TRAIL, tumour necrosis factor-related apoptosis-inducing ligand; VHH, variable domain of a heavy chain-only antibody. aAdinsight entry, trial not listed in ClinicalTrials.gov.
Multispecific targeting
In recent years, a growing number of antibody format technologies have been introduced that enable increased antibody binding by targeting more than one epitope or target. Although these antibodies were initially developed to improve selectivity by targeting multiple disease-related antigens or signalling pathways, multi-targeting concepts may simultaneously enhance functional activity by promoting avidity interactions in early phases of the antibody functional response. Multi-targeting concepts include combinations of two or more antibody molecules with different defined specificities — for example, antibody mixtures or designer polyclonals — and antibody architectures and formats combining two or more specificities in a single antibody molecule such as bispecific or multispecific antibodies.
Targeting multiple epitopes
Multi-targeting antibody technologies directed towards non-overlapping epitopes on the same target come closest to exploiting the potentiation of first-order and second-order avidity interactions as they occur in natural polyclonal antibody responses (Fig. 4a). These technologies can increase target occupancy and local Fc-domain density to enhance antibody clustering and overall functional activity, and some may even lead to novel functions. For example, in separate studies targeting the oncogenic drivers EGFR and MET, combinations of two antibodies against non-overlapping epitopes on the same cell surface receptor showed enhanced antitumour activity over individual antibodies in preclinical and clinical studies98–100. Both EGFR and MET antibody combinations promoted receptor internalization and degradation, which for EGFR could be attributed to enhanced receptor crosslinking and clustering within detergent-insoluble, plasma-membrane-associated tubules98,101. Increased receptor internalization can also be achieved using multispecific antibody formats. Biparatopic antibodies individually targeting HER2 and MET are currently being developed to induce increased degradation of target receptors or alternatively, as biparatopic antibody–drug conjugates, which rely on efficient internalization of the conjugated drug for optimal cytotoxicity (Table 1). Biparatopic antibodies are also being developed to enhance neutralization, as described in a recent study in which a biparatopic antibody provided superior inhibition of the ectonucleotidase activity of CD73 compared to mixtures of the parent antibodies by freezing the ectoenzyme in an inactive conformation102.
Fig. 4. Antibody avidity engineering strategies.

a | Enhancing first-order and second-order avidity binding by dual-epitope targeting or biparatopic antibodies facilitates antibody clustering and increased functional responses (left). Targeting multiple antigens by designer polyclonals may increase antibody and antigen clustering through Fc–Fc interactions. Examples of hetero-hexamer formation between two antibodies are shown to generate assemblies for dual target-mediated C1 binding and complement activation (right). b | Multivalent/multiligand molecules such as the hexavalent tumour necrosis factor superfamily (TNFSF)–Fc (HERA) technology and multivalent antibody architectures induce tumour necrosis factor receptor superfamily (TNFRSF) member clustering and agonism (left). IgG molecules engineered for an increased ability for on-target hexamerization may trigger complement activation or act as signalling agonists by inducing cell surface receptor clustering. A dimer of an IgG engineered for enhanced self-association is shown with exemplary amino acid residues that facilitate Fc–Fc interactions (shown in red) (right). c | The activation threshold for effector functions may be decreased by affinity tuning. Exemplary combinations of amino acid residues that can be mutated to enhance C1q binding affinity are shown in red and purple. Complement activation remains conditional on first-order and second-order avidity binding (left). FcγR binding may be tuned by protein engineering and glycoengineering (right). Exemplary combinations of amino acid residues that can be mutated to enhance FcγR binding affinity are shown in red (as in tafasitamab) (Table 1) and purple (as in margetuximab) (Table 1), respectively. IgG molecules lacking a fucose residue in one or both heavy chains in the Fc domain results in increased FcγRIIIA binding and ADCC (for approved glycoengineered antibodies, see Table 1). Part b adapted with permission from ref.159, MDPI.
Classical Fc-mediated effector functions may be enhanced through multi-epitope targeting. Antibody combinations targeting multiple epitopes of either MET or EGFR were reported to more potently trigger complement activation than monoclonal antibodies targeting a single epitope of either protein98,103,104. Potentiation of CDC through multi-epitope targeting has been reported for a number of other cell surface targets including CD37, human leukocyte antigen (HLA), the Neisseria meningitides factor H-binding protein, folate receptor alpha (FRα)105–108, and against multiple myeloma and Burkitt lymphoma cell lines using engineered biparatopic heavy-chain-only antibody constructs targeting CD38 (ref.109). We speculate that avidity engineering of multispecific antibodies may effectively overcome complement defence mechanisms through increased second-order avidity interactions, exploiting the natural capability of antibodies to hexamerize and induce CDC.
Targeting non-overlapping epitopes using multi-specific antibody technologies has been explored to reduce virus resistance and lower the risk of viral escape. Trispecific antibody formats combining Fab-derived binding domains of broadly neutralizing antibodies (bNAbs) directed against different functional HIV-1 epitopes on the envelope spike exhibited greater neutralization potency and activity against a larger set of diverse HIV-1 isolates compared to combinations of the parental bNAbs both in vitro and in vivo110,111.
Formats that enhance antibody binding by targeting multiple, non-overlapping epitopes on a single antigen represent the largest group of clinical programmes leveraging avidity tuning (Table 1). Such formats range from mixtures of two or more non-competing antibodies to bispecific or multispecific antibody architectures that combine different epitope binding specificities into a single molecule. For example, multiple clinical programs are currently investigating therapeutics that target multiple epitopes on EGFR family members overexpressed in many solid tumour indications100,112,113. Clinical data show that patients who initially respond to EGFR-specific antibodies eventually become resistant. A proof-of-concept study using the anti-EGFR antibody cocktail Sym004 consisting of the antibodies futuximab and modotuximab (Symphogen) showed clinical activity in a patient with metastatic colorectal cancer with acquired EGFR mutation-mediated resistance to cetuximab114. Unselected patients treated with Sym004 did not show an increase in overall survival in a phase II study, although a retrospective analysis indicated a survival benefit for a patient subgroup negative for RAS/BRAF/EGFR extracellular domain mutations (NCT02083653)99.
Multi-epitope-targeting antibody cocktails are being explored for prophylaxis or treatment of infection with the highly variant RNA virus severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). A combination of casirivimab and imdevimab (Regeneron, NCT04381936) targeting non-overlapping epitopes on the SARS-CoV-2 S glycoprotein was shown to prevent in vitro selection of escape mutants of a pseudovirus expressing the SARS-CoV-2 S glycoprotein, whereas neutralization escape quickly developed in the presence of the individual antibodies or combinations of antibodies against overlapping epitopes115–118. The cocktail was found to be inactive against the omicron BA.1 variant of SARS-COV-2 although it did neutralize the emerging omicron BA.2 subvariant, albeit with a reduction in potency of two orders of magnitude119,120. A recent study using in vitro and in vivo models of SARS-CoV-2 infection demonstrated synergism between two potently neutralizing non-competing antibodies121. These antibodies were optimized for extended half-life and reduced FcγR and C1q binding and are being developed as tixagevimab and cilgavimab (AstraZeneca, NCT04625725). It should be noted that Fc-mutated antibodies with a reduced potential for third-order avidity interactions would be unable to leverage the contribution of Fc-mediated effector functions to in vivo therapy122–124, although these mutations would decrease the potential risk for Fc-mediated enhancement of disease125.
Multi-epitope targeting is being explored in a phase I clinical trial of a triparatopic antibody that contains binding domains from three bNAbs (VRC01LS, PGDM1400 and 10E8v4 (Sanofi)) against the HIV-1 envelope spike (NCT03705169)111. Further, a cocktail of the three non-competing IgG1 antibodies atoltivimab, maftivimab and odesivimab (REGN-EB3, Regeneron) was recently approved for treatment of Zaire Ebola virus (ZEBOV) (NCT03576690)126. The antibodies were selected for their ability to bind the ZEBOV glycoprotein simultaneously and were produced as afucosylated IgGs owing to the role of Fc-mediated effector functions in protecting against Ebola virus infection127–129.
Targeting multiple cell surface receptors in cis
Antibody cocktails targeting two or more cell surface receptors represent an archetypical example of avidity engineering by employing the learnings of natural polyclonal antibody responses. Targeting multiple cell surface receptors may increase both antibody occupancy (Fig. 3d) and antigen clustering through crosslinking and Fc–Fc interactions, resulting in enhanced third-order avidity interactions. We recently described a mixture of two antibodies targeting CD20 and CD37 antigens on tumour B cells that co-engaged in hetero-hexameric complexes, thereby potentiating complement activation61 (Fig. 4a, right). Jacobsen and colleagues reported a mixture of six antibodies which, in synergistic pairs, target each of the HER family members EGFR, HER2 and HER3 (ref.130). This mixture, known as Pan-HER (Sym013, Symphogen) was shown to downregulate the expression of all three targets and overcome acquired resistance in HER family-expressing tumours caused by compensatory receptor upregulation; the cocktail entered clinical development, but was terminated after phase I (NCT02906670).
Other approaches to targeting multiple cell surface receptors include combining multiple binding specificities into a single molecule in the form of bispecific or multispecific antibodies that bridge different receptors on the same cell (in cis) or on different cell types (in trans). One of the strengths of bispecific or multispecific antibodies is their capacity to activate novel ‘designed’ functions that cannot be achieved by antibody mixtures (for bispecific antibodies, these are referred to as ‘obligate’ mechanisms of action)20. Further, their therapeutic index can be increased through the localized tethering of receptors, which can focus therapeutic activity to preferred cells or tissues and limit on-target and off-tumour toxicity131. This concept makes use of a particular aspect of avidity binding that has been referred to as cross-arm binding, in which the first binding event restricts the second binding arm to a small volume, thereby increasing its effective concentration and probability of binding132,133. Notably, the increase in avidity achievable by cross-arm binding is highly dependent on the epitope, target expression levels, antibody format and valency.
The most widely studied ‘obligate’ bispecific antibodies employ in trans bridging to redirect the cytotoxic activity of T cells or other effector cell types to eliminate tumour cells. We refer the reader to recent reviews for a deeper discussion of this important therapeutic antibody class20,134.
An interesting example of how in cis bridging can unlock obligate mechanisms of action is represented by amivantamab (Table 1), a recently FDA-approved and EMA-approved bispecific antibody targeting EGFR and MET (Janssen). Crosslinking of MET using bivalent antibodies causes undesired tumour cell activation and amivantamab circumvents this by combining a single, non-crosslinking MET-binding arm with an EGFR-binding arm in a bispecific configuration that effectively blocks both MET and EGFR signalling135,136. The potential to engage in third-order avidity interactions was increased by engineering the bispecific antibody for low fucosylation. This example is noteworthy in terms of avidity tuning as it illustrates the importance of understanding both antibody and target biology in the design of effective antibody-based therapeutics.
Avidity binding to a protein complex can be achieved by a single binding domain. Lameris et al. recently described a single-domain antibody, VHH1D12, against the antigen-presenting glycoprotein CD1d, a glycolipid-presenting molecule closely related to the HLA class I proteins. VHH1D12 can simultaneously interact with CD1d and the type I natural killer T cell receptor (TCR), which triggers antitumour activity by type I natural killer T (NKT) cells owing to CD1d–TCR crosslinking137. The ability of a single antibody binding site to interact with multiple proteins or structures has previously been described for antibodies against HIV-1, for which polyreactivity increased the observed affinity for the virion138. Specifically, bNAbs against the HIV envelope glycoprotein membrane-proximal region (MPER) were shown to interact with both the gp41 subunit and the viral lipid membrane139.
Combined in cis and in trans bridging of receptors can also be achieved by engineering additional antibody fragments onto bispecific antibodies, thereby creating tri-specific or multi-specific antibodies that enable simultaneous targeting of multiple cell types and same-cell receptors140,141. For example, the trispecific antibody LAVA-051 (Lava therapeutics) — based on VHH1D12 linked to an additional single-domain antibody targeting the Vγ9Vδ2 TCR of γδ T cells — recruits both type I NKT cells and Vγ9Vδ2 T cells for tumour cell killing. LAVA-051 is currently being investigated as a next-generation T cell engager for the treatment of haematological cancers (NCT04887259) (Table 1). In an additional example, Shivange and colleagues recently reported on the generation of a single-agent, bispecific-anchored cytotoxicity-activator (BaCa), an antibody that co-engages death receptor 5 (DR5, also known as TRAIL-R2) and FRα, which are expressed together on ovarian cancer cells. The BaCa antibody was reported to bind FRα and crosslink DR5 both in cis and in trans, by which FRα served as a tumour-specific anchor and primary clustering point for DR5 agonist signalling. Consistent with the potential for an avidity gain resulting from cross-arm binding, in cis killing required lower antibody concentrations than killing in trans142.
Tuning avidity through antibody valency
Over the past few years, engineering strategies for enhancing antibody clustering to improve antibody function have primarily aimed at increasing antibody valency by replicating antibody binding or Fc domains in fragments and IgG-like or IgM-like structures. Indeed, several studies have reported on the design of antibody formats containing multiple Fc domains to enhance FcγR crosslinking and ADCC143–146. In a recent study, Miller and colleagues adapted the tetramerization domain of the tumour suppressor p53 and fused it to different antigen binding domains to create octavalent monospecific and bispecific antibody variants with increased functional activity, known as quads147. Further, CDC can be enhanced for all four human IgG isotypes through covalent hexamerization, which is achieved by genetically fusing the 18-amino-acid µ-tailpiece of IgM to their carboxyl termini, thus generating multivalent structures that efficiently bind and activate complement148.
Some receptors, including those with multimeric cognate ligands or those that interact at cell–cell synapses, require higher-order oligomerization of adjacent complexes for activation of downstream signalling. The most widely studied receptors of this class are members of the tumour necrosis factor receptor superfamily (TNFRSF), which are of growing interest to drug developers owing to their function in regulating cell survival and inflammatory signalling. Regular binding of immunomodulatory antibodies targeting these receptors generally induces insufficient TNFRSF clustering and many require additional third-order avidity binding through FcγRs, in particular FcγRIIB, on immune effector cells to engage in agonist activity149–151. Pioneering work by Ashkenazi, Presta and colleagues demonstrated that avidity engineering by increasing valency using tandem Fab repeats can enhance the forward signalling activity of antibodies targeting the TNFRSF DR5 (ref.152). Previous efforts to develop TNFRSF-targeting antibodies have yielded limited clinical success, possibly owing to insufficient FcγR-mediated crosslinking and in some cases adverse events or toxicity153,154. Interest in targeting TNFRSF has revived building upon the use of high-valency formats that allow for superior crosslinking and signalling independent of third-order avidity interactions with Fc receptors. At present, four valency-increased TNFR-targeting molecules have entered early-stage clinical trials, including IGM-8444 (IGM Biosciences), a pentameric IgM-based antibody construct targeting DR5 (NCT04553692); INBRX-109 (Inhibrx), a tetravalent format containing multiple ligand binding domains fused to an Fc domain targeting DR5 (NCT03715933, NCT04950075); and hexavalent formats INBRX-106 (Inhibrx, NCT04198766) and ABBV-621 (AbbVie, NCT03082209), targeting OX40 and TRAIL, respectively.
Further approaches to enhance TNFRSF activation independently of FcγR crosslinking include the use of Fc fusions with six TNFRSF-binding domains, fusion to trimeric C-propeptide of α1(I) collagen, IgM, and tetravalent biparatopic targeting concepts155–158 (Fig. 4b, left). The hexavalent receptor agonist (HERA) technology makes use of single-chain trimeric tumour necrosis factor superfamily (TNFSF) ligand domains fused to a silenced IgG1-derived Fc domain to enhance hyperclustering of CD40, glucocorticoid-induced TNFRSF-related protein (GITR) and CD27 and boost antigen-specific T cell responses159–161. Likewise, combined multivalent and multi-epitopic engagement of DR5 and OX40 using various tetravalent (2 + 2) biparatopic antibody formats has been shown to induce agonistic signalling without the need for additional crosslinking through FcγR158.
Although tuning avidity through increasing valency represents a promising approach towards enhanced TNFRSF agonism, for some targets activity must be balanced with safety. For example, high-dose agonistic CD40 antibody treatment is generally associated with adverse clinical events, such as cytokine release syndrome and hepatotoxicity162,163. Studies using multiple CD40-targeting antibodies and ligands have demonstrated markedly different biological responses between molecules with distinct binding orientations and valency164. The epitope of CD40 agonistic antibodies is of crucial importance as simultaneous binding of antibodies and the natural trivalent ligand may result in hyperclustering and uncontrolled overstimulation165. Furthermore, extensive FcγR crosslinking may increase Fc-domain-driven side effects such as unwanted depletion of CD40-expressing immune cell populations or overstimulation of the immune response by FcγR-expressing cells. Indeed, the latter was recapitulated in mouse models in which CD40 antibody-mediated hepatic toxicity was abrogated by blocking macrophage activation with antibodies targeting colony-stimulating factor 1 receptor (CSF1R)162. Another example of unexpected toxicity was observed in a phase I study with TAS 266, a tetravalent agonistic nanobody against DR5 in which hepatotoxicity was linked to the presence of pre-existing anti-drug antibodies potentially mediating hypercrosslinking166. All together, these studies demonstrate the importance of careful design and rigorous preclinical investigation for avidity-engineered biotherapeutic agonists.
There has been a steady increase of studies detailing the engineering and clinical translation of naturally multivalent immunoglobulin isotypes such as IgM and IgA, despite being more challenging to develop than IgGs owing to their more complex multichain architectures, more extensive and heterogeneous glycosylation patterns and shorter in vivo half-lives38,167,168. In a unique ocular application, Agard and colleagues used the multivalent nature of IgM to effectively agonize the tyrosine-protein kinase receptor TIE2; this application capitalized on the large size of IgM to reduce vitreal clearance, which was shown to be inversely proportional to the molecule’s hydrodynamic radius169.
Tuning avidity through oligomerization
The ability of IgGs to self-assemble into ordered oligomers on antigenic surfaces through Fc–Fc interactions can be exploited in therapeutic applications and the design and development of IgG antibodies with standard architecture is more straightforward than that of pre-assembled antibody multimers. De Jong and colleagues reported a novel HexaBody technology platform that uses specific single-point mutations in the Fc domain to enhance target-dependent self-assembly of IgG molecules into hexameric complexes on cell surfaces170 (Fig. 4b, right). In a mutational screening approach, they identified two conserved glutamic acid residues in the Fc region of IgG — E345 and E430 — for which the substitution of any other amino acid enhanced IgG hexamer formation and complement activation for a wide range of cell surface targets. Sopp et al. demonstrated that self-assembly dependent on target binding can also be facilitated by extending IgG1 antibodies with the µ-tailpiece mentioned above, but in which the carboxyl-terminal cysteine is mutated to a serine to prevent covalent hexamerization171. By systematically depleting individual complement components, Taylor et al. showed that antibodies containing hexamer-enhancing mutations require a reduced presence of membrane attack complex-forming complement components to promote CDC compared to the non-mutated parent antibodies172. As discussed previously, on-target self-assembly of hexamerization-enhanced antibodies on antigenic surfaces generates an increased density of IgG hexamers72, which therefore probably results in a lower complement activation threshold than regular IgGs.
Enhancement of antibody clustering by promoting self-assembly may also have particular applications in amplifying ‘designed’ antibody functions such as agonistic receptor signalling. The introduction of hexamerization-enhancing mutations in antibodies targeting DR5 has proved to be an effective agonist strategy for inducing tumour cell death; recently, a novel antibody mixture of two non-competing, DR5-targeting antibodies containing an E430G hexamer-enhancing mutation was shown to improve DR5 signalling and tumour cell death independently of FcγR crosslinking in a wide range of tumour types173. Intermolecular Fc–Fc interactions between the two IgG molecules were essential for DR5 agonistic activity and binding of complement component C1q reportedly contributed to the potency observed in vitro and in vivo, potentially resulting from stabilizing IgG hexamers through third-order avidity binding. Combining multi-epitope targeting with enhanced hexamerization is being explored in clinical trials, for example a biparatopic molecule GEN3009 (Genmab/AbbVie) targeting CD37 in haematologic malignancies (NCT04358458)106. A phase I study using a hexamerization-enhanced antibody mixture GEN1029 (Genmab) targeting DR5 on solid tumours was recently terminated (NCT03576131)173.
Besides targeting TRAIL receptors on tumour cells, efforts have been directed towards targeting TNFRSF on immune cells — such as OX40 receptor, CD27, CD40, CD137 and GITR — using agonist antibodies to stimulate T cell proliferation and cytotoxic activity. Tuning the avidity of such antibodies has shown promise in improving their functional effects. For OX40, hexamerization-enhancing Fc mutations in antibodies were reported to increase their agonistic activity174,175.
Tuning binding towards complement and FcγRs
Fc domains are being engineered to enhance their binding affinity for immune effector molecules with the aim to decrease an activation threshold by increasing target occupancy at lower antibody densities or antibody immune complex concentrations, or by extending interaction times (owing to a decreased off-rate). The Fc regions of IgG subclasses differ in their affinity for C1q and their potency to activate complement (Box 2), and reshuffling of segments from different IgG subclasses has therefore proved to be a successful engineering strategy for increasing binding176–179. Several groups have enhanced C1q binding affinity and CDC by mutating amino acid positions in the Fc region, including amino acids at positions S267, H268, S324, K326, E333 (refs.180–184) (Fig. 4c, left). The affinity increase for C1q must be carefully tailored to retain a proper activation threshold in order to avoid unattended complement activation in the absence of avidity binding to antigen. In a comparison of affinity and avidity engineering, Tammen et al. demonstrated that complement-mediated tumour cell lysis was enhanced more effectively with mutations that enhanced on-target IgG hexamerization compared to mutations that enhanced monomeric IgG affinity for C1q, particularly against cells with low EGFR expression levels96.
Extensive mutational analyses have identified amino acid positions in the IgG Fc region that differentially affect binding to distinct FcγRs and that can be modified to enhance ADCC or ADCP activity185–187. S239D/I332E amino acid substitutions in the Fc domain of antibodies targeting EGFR, CD52 and CD20 were shown to enhance FcγRIIIA/FcγRIIB affinity and increase ADCC and ADCP185 (Fig. 4c, right). Glycoengineering the Fc region for reduced fucose content can also enhance effector function by improving binding to FcγRIIIA. Next to improving classical FcγR-mediated effector functions, enhancement of FcγR binding interactions may be used to increase the agonistic activity of immunomodulatory antibodies. For example, enhancing FcγR affinity using S267E/L328F or E233D/G237D/H268D/P271G/A330R Fc mutations was reported to increase the antitumour activity of DR5 and OX40 antibodies149,175,188–190. Further, selective enhancement of antibody binding to FcγRIIB has been leveraged to inhibit BCR-mediated B cell activation and suppress humoral immunity188,191, as well as enhance the clearance of immune complexes192.
At present, seven active clinical programs apply Fc engineering to amplify FcγR-mediated effector functions (Table 1). The combination of two Fc-domain single-point mutations (S239D/I332E) enhances ADCC and ADCP of antibodies targeting the envelope spike on HIV-1 and CD19 on B tumour cells. Tafasitamab, an antibody targeting the latter, was recently approved for treatment of relapsed/refractory diffuse large B cell lymphoma (DLBCL) in combination with lenalidomide. Margetuximab, an anti-HER2 antibody Fc engineered with the mutations L235V/F243L/R292P/Y300L/P396L, was recently approved for HER2-positive breast cancer193,194 (Fig. 4c, right). Glycoengineering for effector function enhancement has met with clinical success in five approved afucosylated antibody products, including mogamulizumab (cutaneous T cell lymphoma)195,196, obinutuzumab (chronic lymphocytic leukaemia and follicular lymphoma)197, benralizumab (asthma)198,199, the cocktail of atoltivimab, maftivimab and odesivimab (Zaire Ebola virus infection)127 and the bispecific antibody amivantamab (lung cancer)135,200 (Fig. 4c, right).
Recently, Ravetch and colleagues have demonstrated that antibodies Fc-engineered for selective enhancement to FcγRIIA186 can increase dendritic cell maturation and induce protective CD8+ T cell antiviral response201.
Conclusions and future perspectives
Antibody function relies on complex interactions between antibodies, their targets and their association with effector molecules and cells of the immune system. There is considerable evidence demonstrating the crucial role of avidity in natural antibody biology, from antibody affinity maturation early in the immune response to effector function activation after target engagement. Specific interactions between antibody and antigen, which we have termed zero-order avidity binding, can vary from highly transient (fast off-rate) to long lasting (slow off-rate). Next, avidity interactions critically contribute to antibody biology by increasing the strength of binding until a threshold is reached that triggers functional activity. We distinguish first-order avidity (bivalent Fab–antigen binding), second-order avidity (simultaneous Fab–antigen and Fab–Fab or Fc–Fc interactions) and third-order avidity (interactions between the immune complex and effector molecule). All levels of avidity binding can be potentiated by the polyclonality of antibody responses targeting multiple epitopes. Although the threshold at which antibody avidity interactions translate into an effective functional response varies between effector mechanisms, avidity binding can be viewed as a checkpoint before response amplification that in large part determines the efficacy of the functional response. Understanding the key determinants that shape antibody functional response kinetics, including the balance of multiple levels of antibody avidity interactions and response regulators, is therefore crucial in mastering antibody mechanisms of action and designing more effective antibody-based therapeutics.
The single-agent use of monoclonal antibodies for therapy is in many ways contrary to antibody function as established in natural biology. In traditional drug development, single-agent safety and activity is usually required for a drug to be registered and drug combinations are typically only sought following first registration, often using empirical approaches. However, in the body, a combinatorial, polyclonal response is the starting point of the immune response to an antigen. An appreciation of this paradox has steadily grown, and antibody-based therapeutics have evolved from canonical monoclonal antibodies towards more complex antibody architectures and formats in an effort to enhance functional activity and cocktails are being rationally designed and assessed in combination in clinical trials.
Novel formats are emerging that leverage antibody avidity interactions to boost classical effector functions and novel or ‘designed’ mechanisms. These formats feature avidity improvements, including multiplicities of binding specificities, increased valency, enhanced Fab–Fab or Fc–Fc interactions or enhanced interactions between Fc and the effector molecule. Using this concept, distinct avidity engineering approaches may be combined to achieve incremental avidity effects. For example, multi-epitope or multivalent formats can be combined with self-assembling technologies, which may be further enhanced by effector molecules. We believe that multi-agent approaches will be a major player in the future of antibody therapy, either in the form of bispecific/multispecific antibodies with obligate functions or designer polyclonal antibodies. There is a strong argument for greater efforts towards the rational design of antibody cocktails in response to the growing list of therapeutic antibodies entering or undergoing clinical development. Notably, forward-looking antibody discovery approaches that perform unbiased screening of antibody libraries in their final format will be critical to identifying optimal multispecific antibodies or antibody combinations. Similarly, thoughtful antibody design, together with advances in engineering capabilities and improvements in preclinical characterization, will enable avidity-optimized molecular formats that better achieve tuned potency to balance maximal functional activity with minimizing the risks for adverse events. Collectively, recent novel insights into antibody effector biology together with current antibody design efforts that extend our capabilities beyond the classic monoclonal format are paving the way for novel transformative biotherapeutics that can positively affect patient lives.
Acknowledgements
The authors thank L. Trouw, H. van der Vliet, R. Roovers, A. Labrijn and J. Reichert for helpful comments on the manuscript. The authors are grateful to C. Cottrell, A. Ward and I. Wilson (Scripps Research) for providing cryo-electron microscopy (cryo-EM) structures of IgG-311 in complex with the PfCSP NANP repeat and T. Wilpshaar, X. Xue and R. de Jong (Genmab) for generating annotated IgG monomer, hexamer and C1 structures.
Glossary
- Hybridoma technology
A method for the generation of (monoclonal) antibodies with a single-antigen specificity by fusing a short-lived antibody-producing B cell from an immunized animal with an immortal myeloma cell.
- Antibody cocktails
A mixture of two, three or more antibodies combined into a single formulation.
- Multiparatopic
An antibody containing antigen-binding domains recognizing two, three or more epitopes on the same target protein.
- Immune complexation
The formation of multivalent antibody–antigen complexes.
- Antibody-dependent enhancement
A phenomenon in which non-neutralizing antibodies (or neutralizing antibodies at a suboptimal concentration) bind to pathogens without blocking or clearing infection and instead exacerbate disease by leading to increased infection or enhanced immune activation.
- Afucosylated
Describes an IgG antibody that lacks one or both fucose residues in the N-linked branched glycan moiety of the Fc domain, leading to strongly enhanced affinity for FcγRIIIA.
- Polymorphic
Indicates that the protein occurs as two or more variants or alleles in the population with functional polymorphisms of Fc receptors referring to isoforms with different immunoglobulin-binding properties.
Author contributions
The authors contributed equally to this article.
Peer review
Peer review information
Nature Reviews Drug Discovery thanks the anonymous reviewers for their contribution to the peer review of this work.
Competing interests
J.S. and S.C.O. are employees of Genmab (a biotechnology company developing therapeutic antibodies including bispecific and avidity-engineered molecules), own Genmab warrants and/or stock and are inventors on patent applications relating to bispecific and avidity-engineered molecules assigned to Genmab. J.S. is a board member of the Antibody Society. G.A.L. is an employee of Genentech, a member of the Roche Group that develops and commercializes therapeutics including antibody-based drugs. P.W.H.I.P. is an employee of Lava Therapeutics and Leiden University Medical Center and a former employee of Genmab, owns options and/or stock of Lava Therapeutics and is an inventor on applications relating to bispecific and avidity-engineered molecules assigned to Lava Therapeutics and Genmab. He is a board member of The Antibody Society and provides consulting services on behalf of Sparring Bioconsult and served as an expert witness in a case of Roche versus Takeda. He is also an operational partner at Gilde Healthcare.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Antibody therapeutics approved or in regulatory review in the EU or US: https://www.antibodysociety.org/resources/approved-antibodies/
References
- 1.Eisen HN. Affinity enhancement of antibodies: how low-affinity antibodies produced early in immune responses are followed by high-affinity antibodies later and in memory B-cell responses. Cancer Immunol. Res. 2014;2:381. doi: 10.1158/2326-6066.CIR-14-0029. [DOI] [PubMed] [Google Scholar]
- 2.Rajewsky K. Clonal selection and learning in the antibody system. Nature. 1996;381:751–758. doi: 10.1038/381751a0. [DOI] [PubMed] [Google Scholar]
- 3.Tonegawa S. Somatic generation of antibody diversity. Nature. 1983;302:575–581. doi: 10.1038/302575a0. [DOI] [PubMed] [Google Scholar]
- 4.Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495–497. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- 5.Chiu ML, Goulet DR, Teplyakov A, Gilliland GL. Antibody structure and function: the basis for engineering therapeutics. Antibodies. 2019;8:55. doi: 10.3390/antib8040055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rudnick SI, Adams GP. Affinity and avidity in antibody-based tumor targeting. Cancer Biother. Radiopharm. 2009;24:155–161. doi: 10.1089/cbr.2009.0627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diebolder CA, et al. Complement is activated by IgG hexamers assembled at the cell surface. Science. 2014;343:1260–1263. doi: 10.1126/science.1248943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hiramoto E, et al. The IgM pentamer is an asymmetric pentagon with an open groove that binds the AIM protein. Sci. Adv. 2018;4:eaau1199. doi: 10.1126/sciadv.aau1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rougé L, et al. Structure of CD20 in complex with the therapeutic monoclonal antibody rituximab. Science. 2020;367:1224–1230. doi: 10.1126/science.aaz9356. [DOI] [PubMed] [Google Scholar]
- 10.Patel KR, Roberts JT, Barb AW. Multiple variables at the leukocyte cell surface impact Fcγ receptor-dependent mechanisms. Front. Immunol. 2019;10:223. doi: 10.3389/fimmu.2019.00223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sondermann P, Szymkowski DE. Harnessing Fc receptor biology in the design of therapeutic antibodies. Curr. Opin. Immunol. 2016;40:78–87. doi: 10.1016/j.coi.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 12.Parren PW, Burton DR. The antiviral activity of antibodies in vitro and in vivo. Adv. Immunol. 2001;77:195–262. doi: 10.1016/S0065-2776(01)77018-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carter PJ, Lazar GA. Next generation antibody drugs: pursuit of the ‘high-hanging fruit’. Nat. Rev. Drug Discov. 2018;17:197–223. doi: 10.1038/nrd.2017.227. [DOI] [PubMed] [Google Scholar]
- 14.Ugurlar D, et al. Structures of C1-IgG1 provide insights into how danger pattern recognition activates complement. Science. 2018;359:794–797. doi: 10.1126/science.aao4988. [DOI] [PubMed] [Google Scholar]
- 15.Nowakowski A, et al. Potent neutralization of botulinum neurotoxin by recombinant oligoclonal antibody. Proc. Natl Acad. Sci. USA. 2002;99:11346–11350. doi: 10.1073/pnas.172229899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang XZ, Coljee VW, Maynard JA. Back to the future: recombinant polyclonal antibody therapeutics. Curr. Opin. Chem. Eng. 2013;2:405–415. doi: 10.1016/j.coche.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zwick Michael B, et al. Neutralization synergy of human immunodeficiency virus type 1 primary isolates by cocktails of broadly neutralizing antibodies. J. Virol. 2001;75:12198–12208. doi: 10.1128/JVI.75.24.12198-12208.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goulet DR, Atkins WM. Considerations for the design of antibody-based therapeutics. J. Pharm. Sci. 2020;109:74–103. doi: 10.1016/j.xphs.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y, Yang S. Multispecific drugs: the fourth wave of biopharmaceutical innovation. Signal Transduct. Target. Ther. 2020;5:86. doi: 10.1038/s41392-020-0201-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Labrijn AF, Janmaat ML, Reichert JM, Parren P. Bispecific antibodies: a mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019;18:585–608. doi: 10.1038/s41573-019-0028-1. [DOI] [PubMed] [Google Scholar]
- 21.Schuurman J, Parren PW. Editorial overview: special section: new concepts in antibody therapeutics: what’s in store for antibody therapy? Curr. Opin. Immunol. 2016;40:vii–xiii. doi: 10.1016/j.coi.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 22.Balocco R, De Sousa Guimaraes Koch S, Thorpe R, Weisser K, Malan S. New INN nomenclature for monoclonal antibodies. Lancet. 2022;399:24. doi: 10.1016/S0140-6736(21)02732-X. [DOI] [PubMed] [Google Scholar]
- 23.Karush F. Multivalent binding and functional affinity. Contemp. Top. Mol. Immunol. 1976;5:217–228. doi: 10.1007/978-1-4684-8142-6_8. [DOI] [PubMed] [Google Scholar]
- 24.Vorup-Jensen T. On the roles of polyvalent binding in immune recognition: perspectives in the nanoscience of immunology and the immune response to nanomedicines. Adv. Drug Deliv. Rev. 2012;64:1759–1781. doi: 10.1016/j.addr.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 25.Burnet FM, Keogh EV, Lush D. The immunological reactions of the filterable viruses. Aust. J. Exp. Biol. Med. Sci. 1937;15:227–368. doi: 10.1038/icb.1937.23. [DOI] [Google Scholar]
- 26.Bleeker WK, et al. Dual mode of action of a human anti-epidermal growth factor receptor monoclonal antibody for cancer therapy. J. Immunol. 2004;173:4699–4707. doi: 10.4049/jimmunol.173.7.4699. [DOI] [PubMed] [Google Scholar]
- 27.Rhoden JJ, Dyas GL, Wroblewski VJ. A modeling and experimental investigation of the effects of antigen density, binding affinity, and antigen expression ratio on bispecific antibody binding to cell surface targets. J. Biol. Chem. 2016;291:11337–11347. doi: 10.1074/jbc.M116.714287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang D, Kroe-Barrett R, Singh S, Roberts CJ, Laue TM. IgG cooperativity — is there allostery? Implications for antibody functions and therapeutic antibody development. mAbs. 2017;9:1231–1252. doi: 10.1080/19420862.2017.1367074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beurskens FJ, et al. Exhaustion of cytotoxic effector systems may limit monoclonal antibody-based immunotherapy in cancer patients. J. Immunol. 2012;188:3532–3541. doi: 10.4049/jimmunol.1103693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lilienthal G-M, et al. Potential of murine IgG1 and human IgG4 to inhibit the classical complement and Fcγ receptor activation pathways. Front. Immunol. 2018;9:958. doi: 10.3389/fimmu.2018.00958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prodjinotho UF, Hoerauf A, Adjobimey T. IgG4 antibodies from patients with asymptomatic bancroftian filariasis inhibit the binding of IgG1 and IgG2 to C1q in a Fc–Fc-dependent mechanism. Parasitol. Res. 2019;118:2957–2968. doi: 10.1007/s00436-019-06451-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dal Porto JM, et al. B cell antigen receptor signaling 101. Mol. Immunol. 2004;41:599–613. doi: 10.1016/j.molimm.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 33.Feng Y, Wang Y, Zhang S, Haneef K, Liu W. Structural and immunogenomic insights into B-cell receptor activation. J. Genet. Genom. 2020;47:27–35. doi: 10.1016/j.jgg.2019.12.003. [DOI] [PubMed] [Google Scholar]
- 34.Treanor B. B-cell receptor: from resting state to activate. Immunology. 2012;136:21–27. doi: 10.1111/j.1365-2567.2012.03564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peterson ML. Mechanisms controlling production of membrane and secreted immunoglobulin during B cell development. Immunol. Res. 2007;37:33–46. doi: 10.1007/BF02686094. [DOI] [PubMed] [Google Scholar]
- 36.Cattaneo A, Neuberger MS. Polymeric immunoglobulin M is secreted by transfectants of non-lymphoid cells in the absence of immunoglobulin J chain. EMBO J. 1987;6:2753–2758. doi: 10.1002/j.1460-2075.1987.tb02569.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hughey CT, Brewer JW, Colosia AD, Rosse WF, Corley RB. Production of IgM hexamers by normal and autoimmune B cells: implications for the physiologic role of hexameric IgM. J. Immunol. 1998;161:4091–4097. [PubMed] [Google Scholar]
- 38.Keyt BA, Baliga R, Sinclair AM, Carroll SF, Peterson MS. Structure, function, and therapeutic use of IgM antibodies. Antibodies. 2020;9:53. doi: 10.3390/antib9040053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Batista FD, Neuberger MS. Affinity dependence of the B cell response to antigen: a threshold, a ceiling, and the importance of off-rate. Immunity. 1998;8:751–759. doi: 10.1016/S1074-7613(00)80580-4. [DOI] [PubMed] [Google Scholar]
- 40.Foote J, Eisen HN. Kinetic and affinity limits on antibodies produced during immune responses. Proc. Natl Acad. Sci. USA. 1995;92:1254–1256. doi: 10.1073/pnas.92.5.1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cambier JC, Gauld SB, Merrell KT, Vilen BJ. B-cell anergy: from transgenic models to naturally occurring anergic B cells? Nat. Rev. Immunol. 2007;7:633–643. doi: 10.1038/nri2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Daëron M. Fc receptors as adaptive immunoreceptors. Curr. Top. Microbiol. Immunol. 2014;382:131–164. doi: 10.1007/978-3-319-07911-0_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mazor Y, et al. Enhancement of immune effector functions by modulating IgG’s intrinsic affinity for target antigen. PLoS ONE. 2016;11:e0157788. doi: 10.1371/journal.pone.0157788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yamaguchi Y, et al. The Fab portion of immunoglobulin G has sites in the CL domain that interact with Fc gamma receptor IIIa. mAbs. 2022;14:2038531. doi: 10.1080/19420862.2022.2038531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang, et al. Molecular basis of assembly and activation of complement component C1 in complex with immunoglobulin G1 and antigen. Mol. Cell. 2016;63:135–145. doi: 10.1016/j.molcel.2016.05.016. [DOI] [PubMed] [Google Scholar]
- 46.Strasser J, et al. Unraveling the macromolecular pathways of IgG oligomerization and complement activation on antigenic surfaces. Nano Lett. 2019;19:4787–4796. doi: 10.1021/acs.nanolett.9b02220. [DOI] [PubMed] [Google Scholar]
- 47.Imkeller K, et al. Antihomotypic affinity maturation improves human B cell responses against a repetitive epitope. Science. 2018;360:1358–1362. doi: 10.1126/science.aar5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oyen D, et al. Cryo-EM structure of P. falciparum circumsporozoite protein with a vaccine-elicited antibody is stabilized by somatically mutated inter-Fab contacts. Sci. Adv. 2018;4:eaau8529. doi: 10.1126/sciadv.aau8529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Calarese DA, et al. Antibody domain exchange is an immunological solution to carbohydrate cluster recognition. Science. 2003;300:2065–2071. doi: 10.1126/science.1083182. [DOI] [PubMed] [Google Scholar]
- 50.Wu Y, et al. Structural basis for enhanced HIV-1 neutralization by a dimeric immunoglobulin G form of the glycan-recognizing antibody 2G12. Cell Rep. 2013;5:1443–1455. doi: 10.1016/j.celrep.2013.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huber M, et al. Very few substitutions in a germ line antibody are required to initiate significant domain exchange. J. Virol. 2010;84:10700–10707. doi: 10.1128/JVI.01111-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miller NL, Subramanian V, Clark T, Raman R, Sasisekharan R. Conserved topology of virus glycoepitopes presents novel targets for repurposing HIV antibody 2G12. Sci. Rep. 2022;12:2594. doi: 10.1038/s41598-022-06157-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Klein JS, et al. Examination of the contributions of size and avidity to the neutralization mechanisms of the anti-HIV antibodies b12 and 4E10. Proc. Natl Acad. Sci. USA. 2009;106:7385–7390. doi: 10.1073/pnas.0811427106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ofek G, et al. Structure and mechanistic analysis of the anti-human immunodeficiency virus type 1 antibody 2F5 in complex with its gp41 epitope. J. Virol. 2004;78:10724–10737. doi: 10.1128/JVI.78.19.10724-10737.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang MY, et al. Identification and characterization of a new cross-reactive human immunodeficiency virus type 1-neutralizing human monoclonal antibody. J. Virol. 2004;78:9233–9242. doi: 10.1128/JVI.78.17.9233-9242.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Klein JS, Bjorkman PJ. Few and far between: how HIV may be evading antibody avidity. PLoS Pathog. 2010;6:e1000908. doi: 10.1371/journal.ppat.1000908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schofield DJ, Stephenson JR, Dimmock NJ. Variations in the neutralizing and haemagglutination-inhibiting activities of five influenza A virus-specific IgGs and their antibody fragments. J. Gen. Virol. 1997;78:2431–2439. doi: 10.1099/0022-1317-78-10-2431. [DOI] [PubMed] [Google Scholar]
- 58.Wu H, et al. Ultra-potent antibodies against respiratory syncytial virus: effects of binding kinetics and binding valence on viral neutralization. J. Mol. Biol. 2005;350:126–144. doi: 10.1016/j.jmb.2005.04.049. [DOI] [PubMed] [Google Scholar]
- 59.Bournazos S, et al. Broadly neutralizing anti-HIV-1 antibodies require Fc effector functions for in vivo activity. Cell. 2014;158:1243–1253. doi: 10.1016/j.cell.2014.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hessell AJ, et al. Fc receptor but not complement binding is important in antibody protection against HIV. Nature. 2007;449:101–104. doi: 10.1038/nature06106. [DOI] [PubMed] [Google Scholar]
- 61.Gunn BM, et al. A Fc engineering approach to define functional humoral correlates of immunity against Ebola virus. Immunity. 2021;54:815–828. doi: 10.1016/j.immuni.2021.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Slon Campos JL, Mongkolsapaya J, Screaton GR. The immune response against flaviviruses. Nat. Immunol. 2018;19:1189–1198. doi: 10.1038/s41590-018-0210-3. [DOI] [PubMed] [Google Scholar]
- 63.Beltramello M, et al. The human immune response to Dengue virus is dominated by highly cross-reactive antibodies endowed with neutralizing and enhancing activity. Cell Host Microbe. 2010;8:271–283. doi: 10.1016/j.chom.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.van der Neut Kolfschoten M, et al. Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science. 2007;317:1554. doi: 10.1126/science.1144603. [DOI] [PubMed] [Google Scholar]
- 65.Labrijn AF, et al. Efficient generation of stable bispecific IgG1 by controlled Fab-arm exchange. Proc. Natl Acad. Sci. USA. 2013;110:5145–5150. doi: 10.1073/pnas.1220145110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Labrijn AF, et al. Species-specific determinants in the IgG CH3 domain enable Fab-arm exchange by affecting the noncovalent CH3–CH3 interaction strength. J. Immunol. 2011;187:3238. doi: 10.4049/jimmunol.1003336. [DOI] [PubMed] [Google Scholar]
- 67.Vergoossen DL, et al. Functional monovalency amplifies the pathogenicity of anti-MuSK IgG4 in myasthenia gravis. Proc. Natl Acad. Sci. USA. 2021;118:e2020635118. doi: 10.1073/pnas.2020635118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Melis JP, et al. Complement in therapy and disease: regulating the complement system with antibody-based therapeutics. Mol. Immunol. 2015;67:117–130. doi: 10.1016/j.molimm.2015.01.028. [DOI] [PubMed] [Google Scholar]
- 69.Reid KBM, Porter RR. Subunit composition and structure of subcomponent C1q of the first component of human complement. Biochem. J. 1976;155:19–23. doi: 10.1042/bj1550019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Burton DR. Antibody: the flexible adaptor molecule. Trends Biochem. Sci. 1990;15:64–69. doi: 10.1016/0968-0004(90)90178-E. [DOI] [PubMed] [Google Scholar]
- 71.Collins C, Tsui FWL, Shulman MJ. Differential activation of human and guinea pig complement by pentameric and hexameric IgM. Eur. J. Immunol. 2002;32:1802–1810. doi: 10.1002/1521-4141(200206)32:6<1802::AID-IMMU1802>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 72.Strasser J, et al. Weak fragment crystallizable (Fc) domain interactions drive the dynamic assembly of IgG oligomers upon antigen recognition. ACS Nano. 2020;14:2739–2750. doi: 10.1021/acsnano.9b08347. [DOI] [PubMed] [Google Scholar]
- 73.Meyer S, Leusen JHW, Boross P. Regulation of complement and modulation of its activity in monoclonal antibody therapy of cancer. mAbs. 2014;6:1133–1144. doi: 10.4161/mabs.29670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Alduaij W, et al. Novel type II anti-CD20 monoclonal antibody (GA101) evokes homotypic adhesion and actin-dependent, lysosome-mediated cell death in B-cell malignancies. Blood. 2011;117:4519–4529. doi: 10.1182/blood-2010-07-296913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Beers SA, Chan CH, French RR, Cragg MS, Glennie MJ. CD20 as a target for therapeutic type I and II monoclonal antibodies. Semin. Hematol. 2010;47:107–114. doi: 10.1053/j.seminhematol.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 76.Kumar A, Planchais C, Fronzes R, Mouquet H, Reyes N. Binding mechanisms of therapeutic antibodies to human CD20. Science. 2020;369:793–799. doi: 10.1126/science.abb8008. [DOI] [PubMed] [Google Scholar]
- 77.Oostindie SC, et al. CD20 and CD37 antibodies synergize to activate complement by Fc-mediated clustering. Haematologica. 2019;104:1841–1852. doi: 10.3324/haematol.2018.207266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dangl JL, et al. Segmental flexibility and complement fixation of genetically engineered chimeric human, rabbit and mouse antibodies. EMBO J. 1988;7:1989–1994. doi: 10.1002/j.1460-2075.1988.tb03037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tan LK, Shopes RJ, Oi VT, Morrison SL. Influence of the hinge region on complement activation, C1q binding, and segmental flexibility in chimeric human immunoglobulins. Proc. Natl Acad. Sci. USA. 1990;87:162–166. doi: 10.1073/pnas.87.1.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Coloma MJ, Trinh KR, Wims LA, Morrison SL. The hinge as a spacer contributes to covalent assembly and is required for function of IgG. J. Immunol. 1997;158:733. [PubMed] [Google Scholar]
- 81.Raju TS. Terminal sugars of Fc glycans influence antibody effector functions of IgGs. Curr. Opin. Immunol. 2008;20:471–478. doi: 10.1016/j.coi.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 82.van der Poel CE, Spaapen RM, van de Winkel JG, Leusen JH. Functional characteristics of the high affinity IgG receptor, FcγRI. J. Immunol. 2011;186:2699–2704. doi: 10.4049/jimmunol.1003526. [DOI] [PubMed] [Google Scholar]
- 83.Bruhns P, et al. Specificity and affinity of human Fcγ receptors and their polymorphic variants for human IgG subclasses. Blood. 2009;113:3716–3725. doi: 10.1182/blood-2008-09-179754. [DOI] [PubMed] [Google Scholar]
- 84.Ferrara C, et al. Unique carbohydrate–carbohydrate interactions are required for high affinity binding between FcγRIII and antibodies lacking core fucose. Proc. Natl Acad. Sci. USA. 2011;108:12669–12674. doi: 10.1073/pnas.1108455108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Larsen MD, et al. Afucosylated IgG characterizes enveloped viral responses and correlates with COVID-19 severity. Science. 2021;14:635. doi: 10.1126/science.abc8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang TT, et al. IgG antibodies to dengue enhanced for FcγRIIIA binding determine disease severity. Science. 2017;355:395–398. doi: 10.1126/science.aai8128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kapur R, et al. A prominent lack of IgG1-Fc fucosylation of platelet alloantibodies in pregnancy. Blood. 2014;123:471–480. doi: 10.1182/blood-2013-09-527978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Brandsma AM, et al. Potent Fc receptor signaling by IgA leads to superior killing of cancer cells by neutrophils compared to IgG. Front. Immunol. 2019;10:704. doi: 10.3389/fimmu.2019.00704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dechant M, et al. Effector mechanisms of recombinant IgA antibodies against epidermal growth factor receptor. J. Immunol. 2007;179:2936. doi: 10.4049/jimmunol.179.5.2936. [DOI] [PubMed] [Google Scholar]
- 90.Evers M, et al. Novel chimerized IgA CD20 antibodies: improving neutrophil activation against CD20-positive malignancies. mAbs. 2020;12:1795505. doi: 10.1080/19420862.2020.1795505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Otten MA, et al. Enhanced FcαRI-mediated neutrophil migration towards tumour colonies in the presence of endothelial cells. Eur. J. Immunol. 2012;42:1815–1821. doi: 10.1002/eji.201141982. [DOI] [PubMed] [Google Scholar]
- 92.van Egmond M, Bakema JE. Neutrophils as effector cells for antibody-based immunotherapy of cancer. Semin. Cancer Biol. 2013;23:190–199. doi: 10.1016/j.semcancer.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 93.Matlung HL, et al. Neutrophils kill antibody-opsonized cancer cells by trogoptosis. Cell Rep. 2018;23:3946–3959.e6. doi: 10.1016/j.celrep.2018.05.082. [DOI] [PubMed] [Google Scholar]
- 94.Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front. Immunol. 2014;5:520. doi: 10.3389/fimmu.2014.00520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Preiner J, et al. IgGs are made for walking on bacterial and viral surfaces. Nat. Commun. 2014;5:4394. doi: 10.1038/ncomms5394. [DOI] [PubMed] [Google Scholar]
- 96.Tammen A, et al. Monoclonal antibodies against epidermal growth factor receptor acquire an ability to kill tumor cells through complement activation by mutations that selectively facilitate the hexamerization of IgG on opsonized cells. J. Immunol. 2017;198:1585–1594. doi: 10.4049/jimmunol.1601268. [DOI] [PubMed] [Google Scholar]
- 97.Ben M’Barek K, et al. Phagocytosis of immunoglobulin-coated emulsion droplets. Biomaterials. 2015;51:270–277. doi: 10.1016/j.biomaterials.2015.02.030. [DOI] [PubMed] [Google Scholar]
- 98.Grandal MM, et al. Simultaneous targeting of two distinct epitopes on MET effectively inhibits MET- and HGF-driven tumor growth by multiple mechanisms. Mol. Cancer Ther. 2017;16:2780–2791. doi: 10.1158/1535-7163.MCT-17-0374. [DOI] [PubMed] [Google Scholar]
- 99.Montagut C, et al. Efficacy of Sym004 in patients with metastatic colorectal cancer with acquired resistance to anti-EGFR therapy and molecularly selected by circulating tumor DNA analyses: a phase 2 randomized clinical trial. JAMA Oncol. 2018;4:e175245. doi: 10.1001/jamaoncol.2017.5245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pedersen MW, et al. Sym004: a novel synergistic anti-epidermal growth factor receptor antibody mixture with superior anticancer efficacy. Cancer Res. 2010;70:588–597. doi: 10.1158/0008-5472.CAN-09-1417. [DOI] [PubMed] [Google Scholar]
- 101.Jones S, et al. Targeting of EGFR by a combination of antibodies mediates unconventional EGFR trafficking and degradation. Sci. Rep. 2020;10:663. doi: 10.1038/s41598-019-57153-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Stefano JE, et al. A highly potent CD73 biparatopic antibody blocks organization of the enzyme active site through dual mechanisms. J. Biol. Chem. 2020;295:18379–18389. doi: 10.1074/jbc.RA120.012395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dechant M, et al. Complement-dependent tumor cell lysis triggered by combinations of epidermal growth factor receptor antibodies. Cancer Res. 2008;68:4998–5003. doi: 10.1158/0008-5472.CAN-07-6226. [DOI] [PubMed] [Google Scholar]
- 104.Klausz K, et al. Complement-mediated tumor-specific cell lysis by antibody combinations targeting epidermal growth factor receptor (EGFR) and its variant III (EGFRvIII) Cancer Sci. 2011;102:1761–1768. doi: 10.1111/j.1349-7006.2011.02019.x. [DOI] [PubMed] [Google Scholar]
- 105.Macor P, et al. Complement activated by chimeric anti-folate receptor antibodies is an efficient effector system to control ovarian carcinoma. Cancer Res. 2006;66:3876–3883. doi: 10.1158/0008-5472.CAN-05-3434. [DOI] [PubMed] [Google Scholar]
- 106.Oostindie SC, et al. DuoHexaBody-CD37®, a novel biparatopic CD37 antibody with enhanced Fc-mediated hexamerization as a potential therapy for B-cell malignancies. Blood Cancer J. 2020;10:30. doi: 10.1038/s41408-020-0292-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Peschiera Ilaria, et al. Structural basis for cooperativity of human monoclonal antibodies to meningococcal factor H-binding protein. Commun. Biol. 2019;2:241–241. doi: 10.1038/s42003-019-0493-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rijkers M, et al. Anti-HLA antibodies with complementary and synergistic interaction geometries promote classical complement activation on platelets. Haematologica. 2019;104:403–416. doi: 10.3324/haematol.2018.201665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Schütze K, et al. CD38-specific biparatopic heavy chain antibodies display potent complement-dependent cytotoxicity against multiple myeloma cells. Front. Immunol. 2018;9:2553. doi: 10.3389/fimmu.2018.02553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Steinhardt JJ, et al. Rational design of a trispecific antibody targeting the HIV-1 Env with elevated anti-viral activity. Nat. Commun. 2018;9:877. doi: 10.1038/s41467-018-03335-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Xu L, et al. Trispecific broadly neutralizing HIV antibodies mediate potent SHIV protection in macaques. Science. 2017;358:85–90. doi: 10.1126/science.aan8630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Huang S, et al. Structural and functional characterization of MBS301, an afucosylated bispecific anti-HER2 antibody. mAbs. 2018;10:864–875. doi: 10.1080/19420862.2018.1486946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wei H, et al. Structural basis of a novel heterodimeric Fc for bispecific antibody production. Oncotarget. 2017;8:51037–51049. doi: 10.18632/oncotarget.17558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sánchez-Martín FJ, et al. The first-in-class anti-EGFR antibody mixture Sym004 overcomes cetuximab resistance mediated by EGFR extracellular domain mutations in colorectal cancer. Clin. Cancer Res. 2016;22:3260–3267. doi: 10.1158/1078-0432.CCR-15-2400. [DOI] [PubMed] [Google Scholar]
- 115.Baum A, et al. REGN-COV2 antibodies prevent and treat SARS-CoV-2 infection in rhesus macaques and hamsters. Science. 2020;370:1110–1115. doi: 10.1126/science.abe2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hansen J, et al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science. 2020;369:1010. doi: 10.1126/science.abd0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cohen MS. Monoclonal antibodies to disrupt progression of early Covid-19 infection. N. Engl. J. Med. 2021;384:289–291. doi: 10.1056/NEJMe2034495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Baum A, et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science. 2020;369:1014. doi: 10.1126/science.abd0831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Takashita E, et al. Efficacy of antiviral agents against the SARS-CoV-2 Omicron subvariant BA.2. N. Engl. J. Med. 2022;386:1475–1477. doi: 10.1056/NEJMc2201933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.VanBlargan LA, et al. An infectious SARS-CoV-2 B.1.1.529 Omicron virus escapes neutralization by therapeutic monoclonal antibodies. Nat. Med. 2022;28:490–495. doi: 10.1038/s41591-021-01678-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zost SJ, et al. Potently neutralizing and protective human antibodies against SARS-CoV-2. Nature. 2020;584:443–449. doi: 10.1038/s41586-020-2548-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Winkler ES, et al. Human neutralizing antibodies against SARS-CoV-2 require intact Fc effector functions for optimal therapeutic protection. Cell. 2021;184:1804–1820.e16. doi: 10.1016/j.cell.2021.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ullah I, et al. Live imaging of SARS-CoV-2 infection in mice reveals that neutralizing antibodies require Fc function for optimal efficacy. Immunity. 2021;54:2143–2158.e15. doi: 10.1016/j.immuni.2021.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Schäfer A, et al. Antibody potency, effector function, and combinations in protection and therapy for SARS-CoV-2 infection in vivo. J. Exp. Med. 2020;218:e20201993. doi: 10.1084/jem.20201993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Li D, et al. In vitro and in vivo functions of SARS-CoV-2 infection-enhancing and neutralizing antibodies. Cell. 2021;184:4203–4219.e32. doi: 10.1016/j.cell.2021.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mulangu S, et al. A randomized, controlled trial of Ebola virus disease therapeutics. N. Engl. J. Med. 2019;381:2293–2303. doi: 10.1056/NEJMoa1910993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Pascal KE, et al. Development of clinical-stage human monoclonal antibodies that treat advanced Ebola virus disease in nonhuman primates. J. Infect. Dis. 2018;218:S612–S626. doi: 10.1093/infdis/jiy285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wec AZ, et al. Development of a human antibody cocktail that deploys multiple functions to confer pan-Ebolavirus protection. Cell Host Microbe. 2019;25:39–48.e35. doi: 10.1016/j.chom.2018.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zeitlin L, et al. Enhanced potency of a fucose-free monoclonal antibody being developed as an Ebola virus immunoprotectant. Proc. Natl Acad. Sci. USA. 2011;108:20690. doi: 10.1073/pnas.1108360108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jacobsen HJ, et al. Pan-HER, an antibody mixture simultaneously targeting EGFR, HER2, and HER3, effectively overcomes tumor heterogeneity and plasticity. Clin. Cancer Res. 2015;21:4110–4122. doi: 10.1158/1078-0432.CCR-14-3312. [DOI] [PubMed] [Google Scholar]
- 131.Deshaies RJ. Multispecific drugs herald a new era of biopharmaceutical innovation. Nature. 2020;580:329–338. doi: 10.1038/s41586-020-2168-1. [DOI] [PubMed] [Google Scholar]
- 132.Harms BD, Kearns JD, Iadevaia S, Lugovskoy AA. Understanding the role of cross-arm binding efficiency in the activity of monoclonal and multispecific therapeutic antibodies. Methods. 2014;65:95–104. doi: 10.1016/j.ymeth.2013.07.017. [DOI] [PubMed] [Google Scholar]
- 133.Zheng S, et al. Cross-arm binding efficiency of an EGFR x c-Met bispecific antibody. mAbs. 2016;8:551–561. doi: 10.1080/19420862.2015.1136762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Clynes RA, Desjarlais JR. Redirected T cell cytotoxicity in cancer therapy. Annu. Rev. Med. 2019;70:437–450. doi: 10.1146/annurev-med-062617-035821. [DOI] [PubMed] [Google Scholar]
- 135.Grugan KD, et al. Fc-mediated activity of EGFR x c-Met bispecific antibody JNJ-61186372 enhanced killing of lung cancer cells. mAbs. 2017;9:114–126. doi: 10.1080/19420862.2016.1249079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Neijssen J, et al. Discovery of amivantamab (JNJ-61186372), a bispecific antibody targeting EGFR and MET. J. Biol. Chem. 2021;296:100641. doi: 10.1016/j.jbc.2021.100641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lameris R, et al. A single-domain bispecific antibody targeting CD1d and the NKT T-cell receptor induces a potent antitumor response. Nat. Cancer. 2020;1:1054–1065. doi: 10.1038/s43018-020-00111-6. [DOI] [PubMed] [Google Scholar]
- 138.Mouquet H, et al. Polyreactivity increases the apparent affinity of anti-HIV antibodies by heteroligation. Nature. 2010;467:591–595. doi: 10.1038/nature09385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Carravilla P, et al. Molecular recognition of the native HIV-1 MPER revealed by STED microscopy of single virions. Nat. Commun. 2019;10:78. doi: 10.1038/s41467-018-07962-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kügler M, et al. A recombinant trispecific single-chain Fv derivative directed against CD123 and CD33 mediates effective elimination of acute myeloid leukaemia cells by dual targeting. Br. J. Haematol. 2010;150:574–586. doi: 10.1111/j.1365-2141.2010.08300.x. [DOI] [PubMed] [Google Scholar]
- 141.Wu L, et al. Trispecific antibodies enhance the therapeutic efficacy of tumor-directed T cells through T cell receptor co-stimulation. Nat. Cancer. 2020;1:86–98. doi: 10.1038/s43018-019-0004-z. [DOI] [PubMed] [Google Scholar]
- 142.Shivange, et al. A single-agent dual-specificity targeting of FOLR1 and DR5 as an effective strategy for ovarian cancer. Cancer Cell. 2018;34:331–345.e11. doi: 10.1016/j.ccell.2018.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Goulet DR, Zwolak A, Williams JA, Chiu ML, Atkins WM. Design and characterization of novel dual Fc antibody with enhanced avidity for Fc receptors. Proteins. 2020;88:689–697. doi: 10.1002/prot.25853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Jain A, et al. Tumour antigen targeted monoclonal antibodies incorporating a novel multimerisation domain significantly enhance antibody dependent cellular cytotoxicity against colon cancer. Eur. J. Cancer. 2013;49:3344–3352. doi: 10.1016/j.ejca.2013.06.009. [DOI] [PubMed] [Google Scholar]
- 145.Rowley TF, et al. Engineered hexavalent Fc proteins with enhanced Fc-gamma receptor avidity provide insights into immune-complex interactions. Commun. Biol. 2018;1:146. doi: 10.1038/s42003-018-0149-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zhang X, et al. Anti-CD20 antibody with multimerized Fc domains: a novel strategy to deplete B cells and augment treatment of autoimmune disease. J. Immunol. 2016;196:1165–1176. doi: 10.4049/jimmunol.1501755. [DOI] [PubMed] [Google Scholar]
- 147.Miller A, Carr S, Rabbitts T, Ali H. Multimeric antibodies with increased valency surpassing functional affinity and potency thresholds using novel formats. mAbs. 2020;12:1752529. doi: 10.1080/19420862.2020.1752529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Smith RI, Coloma MJ, Morrison SL. Addition of a mu-tailpiece to IgG results in polymeric antibodies with enhanced effector functions including complement-mediated cytolysis by IgG4. J. Immunol. 1995;154:2226–2236. [PubMed] [Google Scholar]
- 149.Li F, Ravetch JV. Apoptotic and antitumor activity of death receptor antibodies require inhibitory Fcγ receptor engagement. Proc. Natl Acad. Sci. USA. 2012;109:10966–10971. doi: 10.1073/pnas.1208698109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.White AL, et al. Interaction with FcγRIIB is critical for the agonistic activity of anti-CD40 monoclonal antibody. J. Immunol. 2011;187:1754–1763. doi: 10.4049/jimmunol.1101135. [DOI] [PubMed] [Google Scholar]
- 151.Wilson NS, et al. An Fcγ receptor-dependent mechanism drives antibody-mediated target-receptor signaling in cancer cells. Cancer Cell. 2011;19:101–113. doi: 10.1016/j.ccr.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 152.Miller K, et al. Design, construction, and in vitro analyses of multivalent antibodies. J. Immunol. 2003;170:4854–4861. doi: 10.4049/jimmunol.170.9.4854. [DOI] [PubMed] [Google Scholar]
- 153.de Miguel D, Lemke J, Anel A, Walczak H, Martinez-Lostao L. Onto better TRAILs for cancer treatment. Cell Death Differ. 2016;23:733–747. doi: 10.1038/cdd.2015.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.von Karstedt S, Montinaro A, Walczak H. Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat. Rev. Cancer. 2017;17:352–366. doi: 10.1038/nrc.2017.28. [DOI] [PubMed] [Google Scholar]
- 155.Gieffers C, et al. APG350 induces superior clustering of TRAIL receptors and shows therapeutic antitumor efficacy independent of cross-linking via Fcγ receptors. Mol. Cancer Ther. 2013;12:2735–2747. doi: 10.1158/1535-7163.MCT-13-0323. [DOI] [PubMed] [Google Scholar]
- 156.Liu H, et al. Improvement of pharmacokinetic profile of TRAIL via trimer-tag enhances its antitumor activity in vivo. Sci. Rep. 2017;7:8953. doi: 10.1038/s41598-017-09518-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Piao X, et al. TRAIL-receptor 1 IgM antibodies strongly induce apoptosis in human cancer cells in vitro and in vivo. Oncoimmunology. 2016;5:e1131380. doi: 10.1080/2162402X.2015.1131380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Yang Y, et al. Tetravalent biepitopic targeting enables intrinsic antibody agonism of tumor necrosis factor receptor superfamily members. mAbs. 2019;11:996–1011. doi: 10.1080/19420862.2019.1625662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Merz C, et al. The hexavalent CD40 agonist HERA-CD40L induces T-cell-mediated antitumor immune response through activation of antigen-presenting cells. J. Immunother. 2018;41:385–398. doi: 10.1097/CJI.0000000000000246. [DOI] [PubMed] [Google Scholar]
- 160.Richards DM, et al. HERA-GITRL activates T cells and promotes anti-tumor efficacy independent of FcγR-binding functionality. J. Immunother. Cancer. 2019;7:191–191. doi: 10.1186/s40425-019-0671-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Thiemann M, et al. A single-chain-based hexavalent CD27 agonist enhances T cell activation and induces anti-tumor immunity. Front. Oncol. 2018;8:387. doi: 10.3389/fonc.2018.00387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Byrne KT, Leisenring NH, Bajor DL, Vonderheide RH. CSF-1R-dependent lethal hepatotoxicity when agonistic CD40 antibody is given before but not after chemotherapy. J. Immunol. 2016;197:179–187. doi: 10.4049/jimmunol.1600146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Vonderheide RH, Glennie MJ. Agonistic CD40 antibodies and cancer therapy. Clin. Cancer Res. 2013;19:1035–1043. doi: 10.1158/1078-0432.CCR-12-2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Richards DM, Sefrin JP, Gieffers C, Hill O, Merz C. Concepts for agonistic targeting of CD40 in immuno-oncology. Hum. Vaccin. Immunother. 2020;16:377–387. doi: 10.1080/21645515.2019.1653744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kedl RM, et al. CD40 stimulation accelerates deletion of tumor-specific CD8+ T cells in the absence of tumor-antigen vaccination. Proc. Natl Acad. Sci. USA. 2001;98:10811–10816. doi: 10.1073/pnas.191371898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Papadopoulos KP, et al. Unexpected hepatotoxicity in a phase I study of TAS266, a novel tetravalent agonistic Nanobody® targeting the DR5 receptor. Cancer Chemother. Pharmacol. 2015;75:887–895. doi: 10.1007/s00280-015-2712-0. [DOI] [PubMed] [Google Scholar]
- 167.Leusen JH. IgA as therapeutic antibody. Mol. Immunol. 2015;68:35–39. doi: 10.1016/j.molimm.2015.09.005. [DOI] [PubMed] [Google Scholar]
- 168.van Tetering G, Evers M, Chan C, Stip M, Leusen J. Fc engineering strategies to advance IgA antibodies as therapeutic agents. Antibodies. 2020;9:70. doi: 10.3390/antib9040070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Chen Y, et al. Targeted IgMs agonize ocular targets with extended vitreal exposure. mAbs. 2020;12:1818436. doi: 10.1080/19420862.2020.1818436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.de Jong RN, et al. A novel platform for the potentiation of therapeutic antibodies based on antigen-dependent formation of IgG hexamers at the cell surface. PLoS Biol. 2016;14:e1002344. doi: 10.1371/journal.pbio.1002344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Sopp JM, et al. On-target IgG hexamerisation driven by a C-terminal IgM tail-piece fusion variant confers augmented complement activation. Commun. Biol. 2021;4:1031. doi: 10.1038/s42003-021-02513-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Cook EM, et al. Antibodies that efficiently form hexamers upon antigen binding can induce complement-dependent cytotoxicity under complement-limiting conditions. J. Immunol. 2016;197:1762–1775. doi: 10.4049/jimmunol.1600648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Overdijk MB, et al. Dual epitope targeting and enhanced hexamerization by DR5 antibodies as a novel approach to induce potent antitumor activity through DR5 agonism. Mol. Cancer Ther. 2020;19:2126. doi: 10.1158/1535-7163.MCT-20-0044. [DOI] [PubMed] [Google Scholar]
- 174.Zhang D, et al. Functional optimization of agonistic antibodies to OX40 receptor with novel Fc mutations to promote antibody multimerization. mAbs. 2017;9:1129–1142. doi: 10.1080/19420862.2017.1358838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zhang D, Goldberg MV, Chiu ML. Fc engineering approaches to enhance the agonism and effector functions of an anti-OX40 antibody. J. Biol. Chem. 2016;291:27134–27146. doi: 10.1074/jbc.M116.757773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Natsume A, et al. Engineered antibodies of IgG1/IgG3 mixed isotype with enhanced cytotoxic activities. Cancer Res. 2008;68:3863–3872. doi: 10.1158/0008-5472.CAN-07-6297. [DOI] [PubMed] [Google Scholar]
- 177.Natsume A, Shimizu-Yokoyama Y, Satoh M, Shitara K, Niwa R. Engineered anti-CD20 antibodies with enhanced complement-activating capacity mediate potent anti-lymphoma activity. Cancer Sci. 2009;100:2411–2418. doi: 10.1111/j.1349-7006.2009.01327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Sensel MG, Kane LM, Morrison SL. Amino acid differences in the N-terminus of CH2 influence the relative abilities of IgG2 and IgG3 to activate complement. Mol. Immunol. 1997;34:1019–1029. doi: 10.1016/S0161-5890(97)00112-0. [DOI] [PubMed] [Google Scholar]
- 179.Xu Y, Oomen R, Klein MH. Residue at position 331 in the IgG1 and IgG4 CH2 domains contributes to their differential ability to bind and activate complement. J. Biol. Chem. 1994;269:3469–3474. doi: 10.1016/S0021-9258(17)41886-2. [DOI] [PubMed] [Google Scholar]
- 180.Dall’Acqua WF, Cook KE, Damschroder MM, Woods RM, Wu H. Modulation of the effector functions of a human IgG1 through engineering of its hinge region. J. Immunol. 2006;177:1129. doi: 10.4049/jimmunol.177.2.1129. [DOI] [PubMed] [Google Scholar]
- 181.Idusogie EE, et al. Mapping of the C1q binding site on rituxan, a chimeric antibody with a human IgG1 Fc. J. Immunol. 2000;164:4178–4184. doi: 10.4049/jimmunol.164.8.4178. [DOI] [PubMed] [Google Scholar]
- 182.Idusogie EE, et al. Engineered antibodies with increased activity to recruit complement. J. Immunol. 2001;166:2571–2575. doi: 10.4049/jimmunol.166.4.2571. [DOI] [PubMed] [Google Scholar]
- 183.Michaelsen TE, Sandlie I, Bratlie DB, Sandin RH, Ihle O. Structural difference in the complement activation site of human IgG1 and IgG3. Scand. J. Immunol. 2009;70:553–564. doi: 10.1111/j.1365-3083.2009.02338.x. [DOI] [PubMed] [Google Scholar]
- 184.Moore GL, Chen H, Karki S, Lazar GA. Engineered Fc variant antibodies with enhanced ability to recruit complement and mediate effector functions. mAbs. 2010;2:181–189. doi: 10.4161/mabs.2.2.11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Lazar GA, et al. Engineered antibody Fc variants with enhanced effector function. Proc. Natl Acad. Sci. USA. 2006;103:4005–4010. doi: 10.1073/pnas.0508123103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Richards JO, et al. Optimization of antibody binding to FcγRIIa enhances macrophage phagocytosis of tumor cells. Mol. Cancer Ther. 2008;7:2517–2527. doi: 10.1158/1535-7163.MCT-08-0201. [DOI] [PubMed] [Google Scholar]
- 187.Shields RL, et al. High resolution mapping of the binding site on human IgG1 for FcγRI, FcγRII, FcγRIII, and FcRn and design of IgG1 variants with improved binding to the FcγR. J. Biol. Chem. 2001;276:6591–6604. doi: 10.1074/jbc.M009483200. [DOI] [PubMed] [Google Scholar]
- 188.Chu SY, et al. Inhibition of B cell receptor-mediated activation of primary human B cells by coengagement of CD19 and FcγRIIb with Fc-engineered antibodies. Mol. Immunol. 2008;45:3926–3933. doi: 10.1016/j.molimm.2008.06.027. [DOI] [PubMed] [Google Scholar]
- 189.Mimoto F, et al. Novel asymmetrically engineered antibody Fc variant with superior FcγR binding affinity and specificity compared with afucosylated Fc variant. mAbs. 2013;5:229–236. doi: 10.4161/mabs.23452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Mimoto F, et al. Engineered antibody Fc variant with selectively enhanced FcγRIIb binding over both FcγRIIa(R131) and FcγRIIa(H131) Protein Eng. Des. Sel. 2013;26:589–598. doi: 10.1093/protein/gzt022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Horton HM, et al. Antibody-mediated coengagement of FcγRIIb and B cell receptor complex suppresses humoral immunity in systemic lupus erythematosus. J. Immunol. 2011;186:4223–4233. doi: 10.4049/jimmunol.1003412. [DOI] [PubMed] [Google Scholar]
- 192.Iwayanagi Y, et al. Inhibitory FcγRIIb-mediated soluble antigen clearance from plasma by a pH-dependent antigen-binding antibody and its enhancement by Fc engineering. J. Immunol. 2015;195:3198–3205. doi: 10.4049/jimmunol.1401470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Nordstrom JL, et al. Anti-tumor activity and toxicokinetics analysis of MGAH22, an anti-HER2 monoclonal antibody with enhanced Fcγ receptor binding properties. Breast Cancer Res. 2011;13:R123. doi: 10.1186/bcr3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Rugo HS, et al. Efficacy of margetuximab vs trastuzumab in patients with pretreated ERBB2-positive advanced breast cancer: a phase 3 randomized clinical trial. JAMA Oncol. 2021;7:573–584. doi: 10.1001/jamaoncol.2020.7932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Niwa R, et al. Defucosylated chimeric anti-CC chemokine receptor 4 IgG1 with enhanced antibody-dependent cellular cytotoxicity shows potent therapeutic activity to T-cell leukemia and lymphoma. Cancer Res. 2004;64:2127–2133. doi: 10.1158/0008-5472.CAN-03-2068. [DOI] [PubMed] [Google Scholar]
- 196.Ollila TA, Sahin I, Olszewski AJ. Mogamulizumab: a new tool for management of cutaneous T-cell lymphoma. Onco Targets Ther. 2019;12:1085–1094. doi: 10.2147/OTT.S165615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Tobinai K, Klein C, Oya N, Fingerle-Rowson G. A review of obinutuzumab (GA101), a novel type II anti-CD20 monoclonal antibody, for the treatment of patients with B-cell malignancies. Adv. Ther. 2017;34:324–356. doi: 10.1007/s12325-016-0451-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Ghazi A, Trikha A, Calhoun WJ. Benralizumab — a humanized mAb to IL-5Rα with enhanced antibody-dependent cell-mediated cytotoxicity — a novel approach for the treatment of asthma. Expert Opin. Biol. Ther. 2012;12:113–118. doi: 10.1517/14712598.2012.642359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Kolbeck R, et al. MEDI-563, a humanized anti-IL-5 receptor alpha mAb with enhanced antibody-dependent cell-mediated cytotoxicity function. J. Allergy Clin. Immunol. 2010;125:1344–1353.e2. doi: 10.1016/j.jaci.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 200.Vijayaraghavan S, et al. Amivantamab (JNJ-61186372), an Fc enhanced EGFR/cMet bispecific antibody, induces receptor downmodulation and antitumor activity by monocyte/macrophage trogocytosis. Mol. Cancer Ther. 2020;19:2044–2056. doi: 10.1158/1535-7163.MCT-20-0071. [DOI] [PubMed] [Google Scholar]
- 201.Bournazos S, Corti D, Virgin HW, Ravetch JV. Fc-optimized antibodies elicit CD8 immunity to viral respiratory infection. Nature. 2020;588:485–490. doi: 10.1038/s41586-020-2838-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Junghans RP, Anderson CL. The protection receptor for IgG catabolism is the β2-microglobulin-containing neonatal intestinal transport receptor. Proc. Natl Acad. Sci. USA. 1996;93:5512–5516. doi: 10.1073/pnas.93.11.5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Pyzik M, et al. The neonatal Fc receptor (FcRn): a misnomer? Front. Immunol. 2019;10:1540. doi: 10.3389/fimmu.2019.01540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Müller R, et al. High-resolution structures of the IgM Fc domains reveal principles of its hexamer formation. Proc. Natl Acad. Sci. USA. 2013;110:10183. doi: 10.1073/pnas.1300547110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Sharp TH, et al. Insights into IgM-mediated complement activation based on in situ structures of IgM-C1-C4b. Proc. Natl Acad. Sci. USA. 2019;116:11900. doi: 10.1073/pnas.1901841116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Michaelsen TE, et al. Human secretory IgM antibodies activate human complement and offer protection at mucosal surface. Scand. J. Immunol. 2017;85:43–50. doi: 10.1111/sji.12508. [DOI] [PubMed] [Google Scholar]
- 207.Redpath S, Michaelsen TE, Sandlie I, Clark MR. The influence of the hinge region length in binding of human IgG to human Fcγ receptors. Hum. Immunol. 1998;59:720–727. doi: 10.1016/S0198-8859(98)00075-5. [DOI] [PubMed] [Google Scholar]
- 208.Hobeika E, Maity PC, Jumaa H. Control of B cell responsiveness by isotype and structural elements of the antigen receptor. Trends Immunol. 2016;37:310–320. doi: 10.1016/j.it.2016.03.004. [DOI] [PubMed] [Google Scholar]
- 209.Übelhart R, et al. Responsiveness of B cells is regulated by the hinge region of IgD. Nat. Immunol. 2015;16:534–543. doi: 10.1038/ni.3141. [DOI] [PubMed] [Google Scholar]
- 210.Kerr MA. The structure and function of human IgA. Biochem. J. 1990;271:285–296. doi: 10.1042/bj2710285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Breedveld A, van Egmond M. IgA and FcαRI: pathological roles and therapeutic opportunities. Front. Immunol. 2019;10:553–553. doi: 10.3389/fimmu.2019.00553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Sutton BJ, Davies AM, Bax HJ, Karagiannis SN. IgE antibodies: from structure to function and clinical translation. Antibodies. 2019;8:19. doi: 10.3390/antib8010019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Shade KT, et al. A single glycan on IgE is indispensable for initiation of anaphylaxis. J. Exp. Med. 2015;212:457–467. doi: 10.1084/jem.20142182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Kubo S, Nakayama T, Matsuoka K, Yonekawa H, Karasuyama H. Long term maintenance of IgE-mediated memory in mast cells in the absence of detectable serum IgE. J. Immunol. 2003;170:775–780. doi: 10.4049/jimmunol.170.2.775. [DOI] [PubMed] [Google Scholar]
- 215.Bruhns P, Jönsson F. Mouse and human FcR effector functions. Immunol. Rev. 2015;268:25–51. doi: 10.1111/imr.12350. [DOI] [PubMed] [Google Scholar]
- 216.Dhodapkar KM, Dhodapkar MV. Recruiting dendritic cells to improve antibody therapy of cancer. Proc. Natl Acad. Sci. USA. 2005;102:6243. doi: 10.1073/pnas.0502547102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Dhodapkar MV, et al. A reversible defect in natural killer T cell function characterizes the progression of premalignant to malignant multiple myeloma. J. Exp. Med. 2003;197:1667–1676. doi: 10.1084/jem.20021650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Groh V, et al. Efficient cross-priming of tumor antigen-specific T cells by dendritic cells sensitized with diverse anti-MICA opsonized tumor cells. Proc. Natl Acad. Sci. USA. 2005;102:6461. doi: 10.1073/pnas.0501953102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Boruchov AM, et al. Activating and inhibitory IgG Fc receptors on human DCs mediate opposing functions. J. Clin. Invest. 2005;115:2914–2923. doi: 10.1172/JCI24772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.DiLillo DJ, Ravetch JV. Differential Fc-receptor engagement drives an anti-tumor vaccinal effect. Cell. 2015;161:1035–1045. doi: 10.1016/j.cell.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Holgado MP, Sananez I, Raiden S, Geffner JR, Arruvito L. CD32 ligation promotes the activation of CD4+ T cells. Front. Immunol. 2018;9:2814. doi: 10.3389/fimmu.2018.02814. [DOI] [PMC free article] [PubMed] [Google Scholar]