

Abstract
Branched-chain amino acid transaminase 1 (BCAT1) is upregulated selectively in human isocitrate dehydrogenase (IDH) wildtype (WT) but not mutant glioblastoma multiforme (GBM) and promotes IDHWT GBM growth. Through a metabolic synthetic lethal screen, we report here that α-ketoglutarate (AKG) kills IDHWT GBM cells when BCAT1 protein is lost, which is reversed by re-expression of BCAT1 or supplementation with branched-chain α-ketoacids (BCKAs), downstream metabolic products of BCAT1. In patient-derived IDHWT GBM tumors in vitro and in vivo, co-treatment of BCAT1 inhibitor gabapentin and AKG resulted in synthetic lethality. However, AKG failed to evoke a synthetic lethal effect with loss of BCAT2, BCKDHA, or GPT2 in IDHWT GBM cells. Mechanistically, loss of BCAT1 increased the NAD+/NADH ratio but impaired oxidative phosphorylation, mTORC1 activity, and nucleotide biosynthesis. These metabolic alterations were synergistically augmented by AKG treatment, thereby causing mitochondrial dysfunction and depletion of cellular building blocks, including ATP, nucleotides, and proteins. Partial restoration of ATP, nucleotides, proteins, and mTORC1 activity by BCKA supplementation prevented IDHWT GBM cell death conferred by the combination of BCAT1 loss and AKG. These findings define a targetable metabolic vulnerability in the most common subset of GBM that is currently incurable.
Keywords: Synthetic lethality, BCAT1, α-ketoglutarate, NADH, mitochondria, mTORC1
INTRODUCTION
Glioblastoma multiforme (GBM), which represents about 15% of all brain tumors, is a highly malignant grade IV brain tumor that dominantly expresses wildtype isocitrate dehydrogenase (IDHWT) (1). With current standard of treatment consisting of maximal surgical resection followed by radiotherapy and temozolomide chemotherapy, median survival of IDHWT GBM patients is only about 15 months (1), which highlights an unmet need for new therapies that can better treat this aggressive disease. Synthetic lethality was initially described in Drosophila melanogaster in 1922 and has received much enthusiasm for the treatment of cancer by selectively killing cancer cells with simultaneous loss of two interacting survival genes or pathways (2). Although a couple of synthetic lethal interactions with altered metabolism in mutant IDH (IDHMut) gliomas have been reported (3,4), it remains unknown whether metabolic synthetic lethality can be applied to treat IDHWT GBM.
Amino acids are one of the most fundamental bricks of cell structure and essential for synthesis of protein and nucleotide to support cell proliferation. Recent studies have revealed the significance of altered branched-chain amino acid (BCAA) metabolism in various human cancers including GBM, pancreatic cancer, lung cancer, breast cancer, leukemia, kidney cancer, ovarian cancer, and liver cancer (5). BCAAs (i.e., leucine, isoleucine, and valine) belong to a group of essential amino acids and are reversibly catabolized by cytosolic branched-chain aminotransferase (BCAT) 1 and mitochondrial BCAT2 into branched-chain α-ketoacids (BCKAs), including α-ketoisocaproate (KIC), α-ketoisovalerate (KIV), α-keto-β-methylvalerate (KMV) (5). Meanwhile, their amino groups are transferred to α-ketoglutarate (AKG) to produce glutamate, and thus BCAAs are the critical nitrogen donors for synthesis of nucleotides and non-essential amino acids in cancer cells (6). Studies from early pancreatic cancer, lung cancer, leukemia, and liver cancer showed that reductive metabolism of BCAAs is dominant in these cancer cells, leading to accumulation of BCAAs and subsequent mTORC1 activation (6–11). mTORC1 is a master regulator of protein synthesis and metabolic reprogramming and, upon activation, promotes cancer cell proliferation (12). In contrast, the BCAA catabolic pathway is highly active in leukemia stem cells and IDHWT GBM (13–15). High expression of BCAT1 increases glutamate production but reduces intracellular levels of AKG in leukemia cells, which causes activation of hypoxia-inducible factor 1α (HIF-1α) and DNA hypermethylation (15), two important oncogenic drivers of tumor growth. Interestingly, none or very little (1–3% in pancreatic cancer) of the final metabolic products in the BCAA metabolic pathway contributes to carbons for the tricarboxylic acid cycle in cancer cells (6–8), strongly arguing that the intermediates produced from BCAA metabolism may play a critical role in cancer cell proliferation. BCAT1 is upregulated in IDHWT GBM, which is controlled by its promoter hypomethylation and hypoxia (13,14), whereas both BCAT1 and BCAT2 are inactivated by R-2-hydroxyglutarate (HG) in IDHMut glioma (4). BCAT1 promotes growth of IDHWT GBM in vitro and in vivo (13,14), suggesting that BCAT1 is an intriguing target for the development of metabolic synthetic lethality in IDHWT GBM.
In the present study, we reported that genetic or pharmacological inhibition of BCAT1 (BCAT1i) in combination of AKG kills IDHWT GBM cells in vitro and in vivo. Supplementation with KIC prevents BCAT1i and AKG-induced mitochondrial dysfunction and depletion of ATP, nucleotides, and proteins, leading to IDHWT GBM cell survival. These findings define a novel metabolic vulnerability in the most common and currently incurable subset of GBM and develop an innovative and non-toxic therapeutic strategy.
MATERIALS AND METHODS
Plasmid constructs.
Full-length human BCAT1 cDNA was amplified by PCR and cloned into pcFUGW-3XFLAG vector. DNA oligonucleotides of the single guide RNA targeting human BCAT1, BCAT2, branched-chain α-keto acid dehydrogenase E1 subunit α (BCKDHA), or GPT2 (Supplemental Table 1) were annealed and ligated into BsmBI-linearized lentiCRISPRv2 vector (Addgene, #52961). All recombinant plasmids were verified by Sanger sequencing.
Cell culture.
U251MG (RRID: CVCL_0021), LN229 (RRID: CVCL_0393) (gifts from Sandeep Burma at UT Health San Antonio in 2015), U87MG (RRID: CVCL_0022, gift from Gregg L. Semenza at Johns Hopkins in 2014), T98G (RRID: CVCL_0556), and LN18 (RRID: CVCL_0392) (purchased from ATCC in 2017) cells were cultured in DMEM supplemented with 5 or 10% heat-inactivated fetal bovine serum (FBS) at 37°C in a 5% CO2/95% air incubator. Patient-derived IDHWT GBM C116 cells were generated by J.A.C. and cultured in DMEM/Ham’s F-12 supplemented with 1% non-essential amino acid, 1 nM T3, 8 ng/ml EGF, 5 μg/ml ITS, 5% iron-supplemented bovine calf serum, and 1% penicillin-streptomycin at 37°C in a 5% CO2/95% air incubator. Primary IDHWT GBM tumorspheres were cultured as described previously (13), which was approved by the Institutional Review Board at UT Southwestern Medical Center with written informed consent. Primary mouse astrocytes were isolated from the cortex of 1–4-day old C57BL/6J mice and cultured in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin at 37°C in a 5% CO2/95% air incubator. Microglia and oligodendrocyte precursor cells were removed by sequential shaking at 180 rpm for 30 mins and 240 rpm for 6 hours. Astrocytes at the second passage were used for the experiment. Hypoxia was conducted by exposure of cells to 1% O2/5% CO2/balanced N2 gas in a modular incubator chamber (Billups-Rothenberg). All KO cell lines were generated using the CRISPR/Cas9 technique as described (13). All cell lines used for experiments were replaced by thawing frozen cells every month. All cell lines and C116 cells were annually tested to be mycoplasma-free and authenticated by STR DNA profiling analysis during 2015–2017 (Supplemental Table 2).
Clonogenic assay.
Cells were seeded on a 48-well plate and treated with indicated chemicals for 7 or 12 days. Colonies were washed with phosphate-buffered saline (PBS), fixed with 100% methanol, and stained with 0.01% crystal violet. The crystal violet dye was dissolved in 10% acetic acid and measured at OD 570 nm in a Tecan Spark 10M plate reader.
Cell viability assay.
Cells (4,000 cells/well) were seeded on a 96-well plate and treated with indicated chemicals for 2 days. Cell viability was measured by the CellTiter 96® AQueous non-radioactive cell proliferation assay kit (Cat.#: G5421, Promega). For propidium iodide staining, cells were stained with 2 μM propidium iodide for 10 min and imaged under a Zeiss Axio Observer Z1 microscope.
Metabolomics assay.
Parental and BCAT1 KO#2 U251MG cells were seeded on 6-cm dishes and treated with vehicle or cell permeable AKG (10 mM) for 24 hours. After washing twice with cold PBS, cells were lysed by 1 ml of cold extraction buffer (80% methanol/20% water/0.2 μM internal heavy standard mix), vortexed for 30 sec, and centrifuged at 13,000 rpm for 15 minutes at 4°C. The supernatant containing intracellular metabolites was filtered using a PVDF syringe filter (Cat.#: F2504-6, ThermoFisher) and analyzed by liquid chromatography-tandem mass spectrometry (LC-MS). The pellet was dissolved in 0.1 N NaOH and their protein concentration was quantified by Bradford assay for metabolite normalization. The heatmap was generated by the statistical analysis module on the MetaboAnalyst website.
For nucleotide quantification, parental and BCAT1 KO#3 U251MG cells were treated with vehicle, cell permeable AKG (10 mM), KIC (1 mM), or both for 24 hours. After washing with ice-cold saline, cells were lysed by 1 ml of 80% acetonitrile mixed with 1 μmol/L Adenosine-15N5 5′-monophosphate disodium salt. Intracellular metabolites were extracted by repeated freeze-thaw cycles, followed by centrifugation at 20,160 g for 15 min. The supernatant containing nucleotides was filtered through a pre-treated SPE cartridge and analyzed by LC-MS. The pellet was dissolved in 0.1 N NaOH and their protein concentration was quantified by Bradford assay for nucleotide normalization.
Mitochondrial complex activity assay.
Parental and BCAT1 KO cells were harvested, washed once with ice-cold PBS, and resuspended in isolation buffer (250 mM sucrose, 10 mM KCl, 1.5 mM MgCl2, 1 mM Na2EDTA, 1 mM dithiothreitol, 1 mM PMSF, 10 mM Tris-HCl, pH 7.4, and protease inhibitor mixture). After incubation on ice for 30 min, cells were homogenized and subjected to centrifugation at 100 g for 5 min at 4 °C to remove cells that are not fractionated. The supernatant was collected and centrifuged twice at 700 g for 10 min at 4 °C to remove the nuclear fraction. The resulting supernatant was subjected to centrifugation at 12,000 g for 15 min at 4 °C and the pellet as the mitochondrial fraction was collected and washed once with isolation buffer. Mitochondria were resuspended in lysis buffer (30 mM Tris-HCl pH7.4, 200 mM KCl, 5 mM EDTA, 0.5% Triton X-100, 1 mM PMSF, and protease inhibitor mixture), and subjected to freeze in liquid nitrogen and thaw for three rounds to prepare mitochondrial extracts. The activity of mitochondrial complex I was determined through oxidation of NADH with ubiquinone as the electron acceptor. Mitochondrial extracts (20 μg) were mixed with the assay buffer (25 mM KHPO4, 25 mM K2HPO4, pH 7.2, 2.5 mg/ml BSA, 5 mM MgCl2, 2 mM KCN, 0.01 mg/ml antimycin A, 1.3 mM NADH, 0.01 mM decylubiquinone) pre-warmed for 5 min at 30°C. The absorbance at 340 nm wavelength was monitored every 30 sec for 10 min in a plate reader at 37 °C after the stable baseline was achieved. 0.25 mM Rotenone (10 μL) was added to stop the reaction and the absorbance at 340 nm was monitored for additional 5 min. The activity of mitochondrial complex V was determined through the NADH oxidation via conversion of phosphoenolpyruvate to lactate by two-step reaction. Mitochondrial extracts (20 μg) were mixed with 2.5 mM ATP in the assay buffer (40 mM Tris-HCO3, 2.5 M phosphoenolpyruvate, 0.2 mM NADH, 0.025 mg/ml antimycin A, 50 mM MgCl2, 0.5 mg/ml lactate dehydrogenase, 0.1 mg/ml pyruvate kinase) pre-warmed for 5 min at 30°C. The absorbance at 340 nm wavelength was monitored every 30 sec for 10 min in a plate reader at 37 °C after the stable baseline was achieved. Both activities of mitochondrial complex I and V were normalized to citrate synthase activity (16).
TMRE assay.
Cells (2 × 105 cells/well) were seeded into a 12-well plate. Next day, cells were stained with 50 nM TMRE for 30 min at 37°C in 5% CO2 incubator. Fluorescence images were taken with a motorized Axio Observer Z1 microscope (Carl Zeiss). For negative control, cells were pretreated with 10 μM Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) 30 min before TMRE staining. Intensity of fluorescence was analyzed by Image J.
Reactive oxygen species (ROS) assay.
Cells (5 × 105 cells/well) were seeded into a 6-well plate. Next day, cells were treated with MitoSOX (2 μM) for 1 hour at 37°C in 5% CO2 incubator, dissociated with trypsin, washed once, and resuspended in 0.5 ml PBS. Flow cytometry was performed to quantify ROS levels.
NAD+/NADH assay.
The cellular NAD+ and NADH levels were measured using NAD/NADH-Glo™ assay (Cat.#: G9071, Promega). Cells (1 × 105 cells/well) were seeded into a 24-well plate. After 48 hours, cells were lysed by adding 300 μL/well of 0.1 M NaOH with 0.5% DTAB and subjected to measurement in a Tecan Spark 10M plate reader according to manufacturer’s instructions. The NAD+ and NADH levels were normalized to cellular protein concentrations quantified by Bradford assay (Bio-Rad).
ATP assay.
The cellular ATP levels were measured using an ATP determination kit (Cat.#: A22066, ThermoFisher). Cells (1 × 105 cells/well) were seeded into a 24-well plate. After 24 hours, cells were lysed by adding of 500 μL of 1% Triton X-100 and subjected to measurement in a Tecan Spark 10M plate reader according to manufacturer’s instructions. The ATP levels were normalized to cellular protein concentrations quantified by Bradford assay (Bio-Rad).
Seahorse assay.
The oxygen consumption rate (OCR) was measured in a Seahorse XF24e extracellular flux analyzer. Cells were seeded at 3–9 × 104/well into a V7 seahorse 24-well plate and next day subjected to Seahorse analysis. Oligomycin A (1 μM), FCCP (350 nM) and rotenone (2 μM) were added into different ports of the Seahorse cartridge. OCR was measured with a standard 8-min cycling program including mixture (3 min), waiting (2 min), and measurement (3 min). After Seahorse analysis, cells were lysed in 0.1 M NaOH and cellular protein concentrations were determined by Bradford assay (Bio-Rad) for data normalization.
AKG assay.
The cellular AKG levels were measured using a AKG colorimetric/fluorometric assay kit (Cat.#: K677-100, Biovision). 2 × 106 of cells were rapidly lysed with 150 μL of ice cold AKG assay buffer and then sonicated. After centrifugation at 13,000 rpm for 10 mins, supernatants were filtered using a 10 kDa spin column (Millipore) and subjected to measurement at 535/587 nm in a Tecan Spark 10M plate reader according to manufacturer’s instructions. The AKG levels were normalized to cellular protein concentrations quantified by Bradford assay (Bio-Rad).
Immunostaining assay.
Cells were seeded at 5 × 104 cells/well onto glass coverslips placed in a 12-well plate. Next day, cells were treated with cell permeable AKG (10 mM) or DMSO for 24 hours. After washing once with PBS, cells were fixed with 4% paraformaldehyde for 20 min at room temperature, permeabilized with 0.1% Triton X-100 in PBS for 15 min, and blocked with 5% BSA in PBS for 60 min. Cells then were incubated overnight with anti-Mitochondria antibody [MTC02] (1:50 dilution in PBS with 1% BSA, Cat.#: ab3298, Abcam, RRID: AB_303683) or anti-FLAG antibody (1:200 dilution in PBS with 1% BSA, RRID: AB_259529) in a 12-well plate at 4 °C, washed with PBST (PBS with 0.1% Tween-20) for 3 times, incubated for 90 min with Alexa488 donkey anti-mouse IgG (1:200 dilution in PBST with 1% BSA) or Cy3 donkey anti-mouse IgG (1:1,000 dilution in PBST with 1% BSA) in dark, washed again with PBST for 3 times, and incubated for 5 min with DAPI (1:1000 dilution in PBS) in dark. After washing 3 times, cells were mounted with anti-fade mounting medium. Mounted slides were observed with a Zeiss Axio Observer Z1 fluorescence microscope.
Transmission electron microscope assay.
Cells were fixed with 2.5% (v/v) glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.4) for 2 hours at room temperature. After washing with 0.1 M sodium cacodylate buffer (pH 7.4) five times, cells were post-fixed in 1% osmium tetroxide with 0.8% K3Fe(CN6) for 1.5 hours at room temperature. After rinsing with water five times, cells were stained with 4% uranyl acetate for 2 hours at room temperature in dark, dehydrated with increasing concentrations of ethanol ranging from 50%−95%, infiltrated with Embed-812 resin, and polymerized overnight in a 70°C oven. Samples were sectioned with a Leica UCT ultramicrotome, placed onto copper grids, and stained with 2% uranyl acetate. Images were acquired using a JEOL 1400 Plus transmission electron microscope equipped with an AMT CCD camera.
Immunoblot assay.
Cells were lysed in modified lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM β-mercaptoethanol, 1% Igepal, and protease inhibitor cocktail) as described previously (13). Equal amounts of lysates were fractionated by SDS-PAGE and subjected to immunoblot assay with the following antibodies: BCAT1 (Proteintech, Cat.#: 13640-1-AP, RRID: AB_2063569), BCAT2 (Proteintech, Cat.#: 16417-1-AP, RRID: AB_10792411), GPT2 (Proteintech, Cat.#: 16757-1-AP, RRID: AB_2112098), BCKDHA (Bethyl Laboratories, Cat. #A303-790A, RRID: AB_11218185), p-AMPK (Cell Signaling Technology, Cat.#: 2535, RRID: AB_331250), AMPK (Cell Signaling Technology, Cat.#: 2532S, RRID: AB_330331), p-P70S6K (Cell Signaling Technology, Cat.#: 9205S, RRID: AB_330944), P70S6K (Cell Signaling Technology, Cat.#: 9202S, RRID: AB_331676), or actin (Proteintech, Cat.#: 66009-1-Ig, RRID: AB_2687938).
Protein synthesis assay.
Cells were seeded at 50% confluency in a 6-well plate, and treated with indicated chemicals for 24–48 hrs. Before harvesting for lysis, cells were treated with 1 μM puromycin dihydrochloride (Cat.#: P8833, Sigma) for 30 min. Puromycin incorporation into proteins was detected by immunoblot assay with anti-puromycin antibody (1:5000 dilution, Cat.#: MABE343, Sigma, RRID: AB_2566826).
Animal studies.
Animal experiments were approved by the Animal Care and Use Committee at UT Southwestern Medical Center. IDHWT GBM patient-derived xenograft (PDX) tumors were obtained from the brain tumor PDX national resource at Mayo Clinic and approved by the Institutional Review Board at UT Southwestern Medical Center. PDX tumors were cut into 8 mm3 and implanted subcutaneously into the right flank of male NOD-SCIDIL2rγnull (NSG) mice. 100 μL Matrigel was injected into the area of implantation right after. Once the tumor volume reached 50–100 mm3, mice were randomly grouped and administered with saline, 150 mg/kg AKG disodium salt (prepared in saline solution), 60 mg/kg gabapentin (prepared in saline solution), or their combination by oral gavage daily for 16 days. Tumor growth was monitored with a caliper every 3 days and calculated with the formula: volume = 0.52 × length × height × width.
Immunohistochemistry assay.
Immunohistochemistry assays were performed by the Dako Autostainer Link 48 system as described (17). Briefly, the slides were baked at 60 °C for 20 minutes, deparaffinized and hydrated. After the antigen retrieval at optimized pH for 20 minutes in a Dako PT Link, the tissues were incubated with anti-Ki67 antibody (Proteintech, 1:1000, RRID: AB_2756525) for 20 minutes, and visualized using the EnVision FLEX visualization system (Dako). The percentage of Ki67-positive cells per field was counted manually.
Statistical analysis.
Statistical analysis was performed by two-tailed Student’s t test between two groups, and one-way or two-way ANOVA with multiple testing correction within multiple groups. p < 0.05 is considered significant. Data were expressed as mean ± SEM.
Data availability.
The data generated in this study are available within the article and its supplementary data file.
RESULTS
A metabolic screen identifies AKG and BCAT1i as a synthetic lethal approach in IDHWT GBM cells.
In order to perform a BCAT1-dependent synthetic lethal screening, we first generated two independent BCAT1 knockout (KO) U251MG cell lines using the CRISPR/Cas9 technique and their rescued cells by lentiviral transduction (Supplemental Figure 1, A and B). The levels of intracellular AKG, an amino group acceptor coupled with BCAT1-mediated transamination, were significantly elevated in BCAT1 KO#2 U251MG cells (Supplemental Figure 1C), whereas glutamate, the product of AKG, was significantly decreased in these KO cells (Supplemental Figure 1D). Re-expression of FLAG-BCAT1 in BCAT1 KO#2 U251MG cells reversed AKG levels comparable to those in parental U251MG cells (Supplemental Figure 1, A–C). These data indicate that BCAT1 KO U251MG cells we generated display the on-target loss-of-function effect on BCAA metabolism.
To identify metabolic synthetic lethal partners of BCAT1i in IDHWT GBM cells, we screened BCAA metabolism-related metabolites in BCAT1 KO#2 U251MG cells. The clonogenic assay demonstrated that dimethyl AKG (DMAKG, 10 mM), a cell-permeable precursor of AKG, almost completely inhibited growth of BCAT1 KO#2 cells but had a modest effect on parental U251MG cells (Figure 1A). In contrast, BCAAs (2 mM), dimethyl (DM)-glutamate (4 mM), DM-succinate (10 mM), or L-2-HG (5 mM) did not have a lethal effect on BCAT1 KO#2 cells (Figure 1A). Given the critical role of redox homeostasis and mitochondrial respiration in IDHWT GBM (18,19), we also included antioxidants and respiratory inhibitors N-acetyl cysteine (NAC, 1 mM), L-ascorbic acid (500 μM), MitoQ (250 nM), glutathione (3 mM), nicotinamide mononucleotide (NMN, 1 mM), rotenone (0.1 μM), and oligomycin (1 μM) in our screening. These compounds did not have a synergistic effect with BCAT1 KO on growth inhibition of U251MG cells (Figure 1A). The glycolysis-related inhibitor 2-deoxy-D-glucose (2-DG, 0.5 mM) also failed to enhance growth inhibition of BCAT1 KO#2 U251MG cells (Figure 1A). Likewise, the ketones, 3-hydroxybutyrate (10 mM) and acetoacetate (2 mM), had no effect on growth of parental and BCAT1 KO#2 U251MG cells (Figure 1A). Together, the metabolic screening reveals that cell permeable AKG induces a lethal vulnerability in BCAT1 KO IDHWT GBM cells and that the lethal phenotype is specifically caused by AKG but not metabolites from its oxidation and transamination.
Figure 1. Identification of AKG as a lethal metabolite in IDHWT GBM cells when combined with BCAT1i.
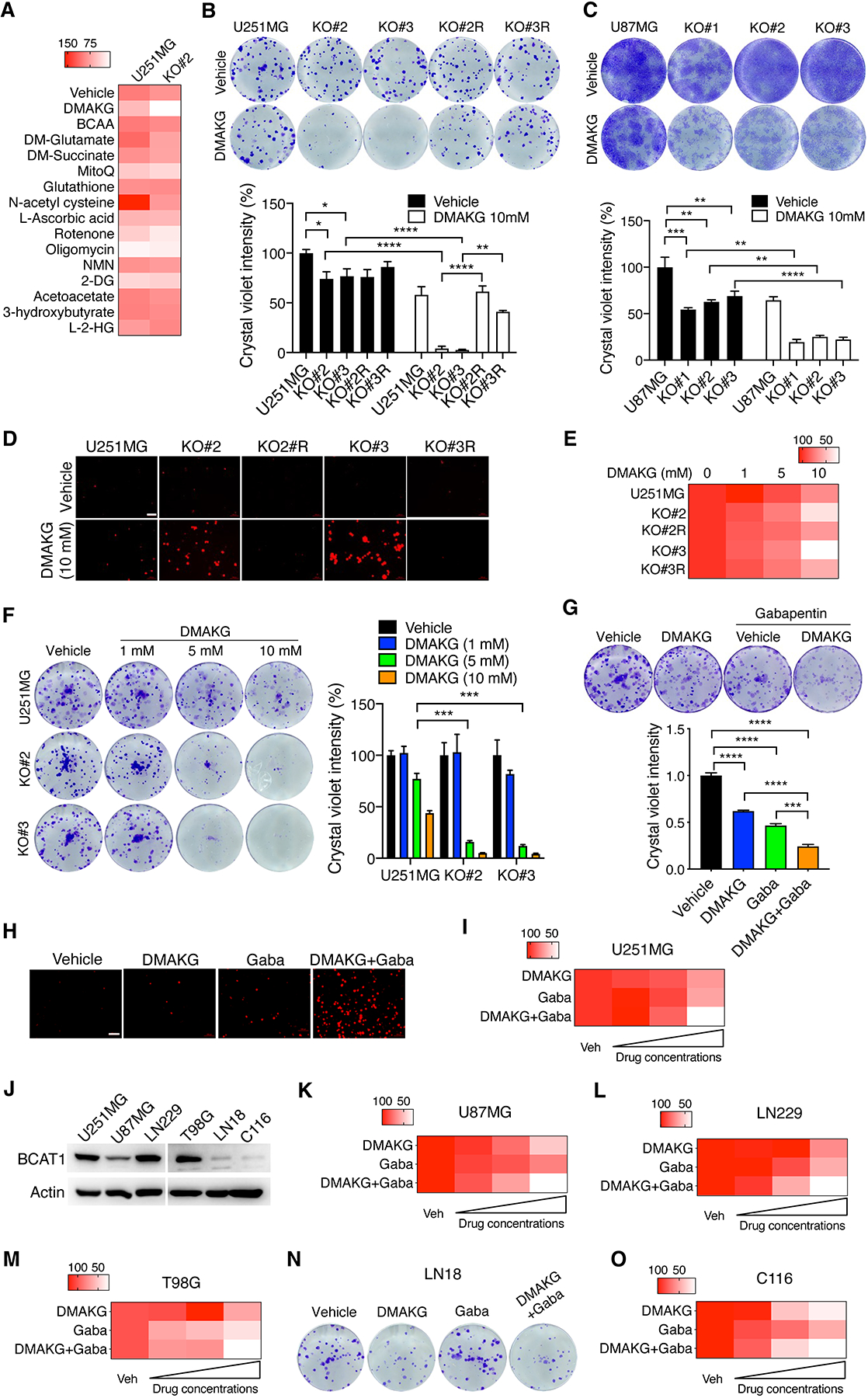
(A) A metabolic screen in parental and BCAT1 KO#2 U251MG cells. The mean percentage of colony growth was normalized to the U251MG-vehicle group and indicated by the gradient color (n = 3–9). (B) Clonogenic assay in parental, BCAT1 KO#2 or KO#3 and BCAT1 rescue (R) U251MG cells treated with vehicle or DMAKG for 7 days. Representative images are shown at the top. The intensity of crystal violet is quantified at the bottom (mean ± SEM, n = 3–6). *p < 0.05; **p < 0.01; ****p < 0.0001 by two-way ANOVA with Tukey’s test. (C) Clonogenic assay in parental and BCAT1 KO#1–3 U87MG cells treated with vehicle or DMAKG for 12 days. Representative images are shown at the top. The intensity of crystal violet is quantified at the bottom (mean ± SEM, n = 3). **p < 0.01; ***p < 0.001; ****p < 0.0001 by two-way ANOVA with Sidak’s test. (D) Parental, BCAT1 KO and BCAT1 rescue (R) U251MG cells treated with vehicle or DMAKG (10 mM) for 48 hours and stained propidium iodide for 10 min. Representative images from three independent experiments are shown. Scale bar, 100 μm. (E) Cell viability assay in parental, BCAT1 KO#2 or KO#3 and BCAT1 rescue (R) U251MG cells treated with vehicle or DMAKG for 48 hours. The mean percentage of cell viability was normalized to the U251MG-vehicle group and indicated by the gradient color (n = 3). (F) Clonogenic assay in parental and BCAT1 KO#2 or KO#3 U251MG cells treated with vehicle or DMAKG for 7 days. Representative images are shown at the left. The intensity of crystal violet is quantified and normalized to U251MG-vehicle at the right (mean ± SEM, n = 3). ***p < 0.001 by two-way ANOVA with Tukey’s test. (G) Clonogenic assay in U251MG cells treated with vehicle, DMAKG (10 mM), gabapentin (Gaba, 5 mM), or both for 7 days. Representative images are shown at the top. The intensity of crystal violet is quantified and normalized to vehicle at the bottom (mean ± SEM, n = 3). ***p < 0.001; ****p < 0.0001 by one-way ANOVA with Dunnett’s test. (H) U251MG cells were treated with vehicle, DMAKG (10 mM), Gaba (20 mM), or both for 48 hours and stained propidium iodide for 10 min. Representative images from three independent experiments are shown. Scale bar, 100 μm. (I) Cell viability assay in U251MG cells treated with vehicle, DMAKG (1, 5, 10 mM), Gaba (5, 10, 20 mM), or both for 48 hours. The mean percentage of cell viability was normalized to the vehicle group and indicated by the gradient color (n = 3). (J) Immunoblot analysis of BCAT1 in a panel of IDHWT GBM cell lines. (K-M, O) Cell viability assay in cells treated with vehicle, DMAKG (1, 5, 10 mM), Gaba (5, 10, 20 mM), or both for 48 hours. The mean percentage of cell viability was normalized to the vehicle group and indicated by the gradient color (n = 3). (N) Clonogenic assay in LN18 cells treated with vehicle, DMAKG (10 mM), Gaba (20 mM), or both for 7 days. Representative images from three independent experiments are shown.
To validate the screening data, we performed clonogenic assay in another BCAT1 KO#3 U251MG cell line and observed an identical synergistic growth inhibition of BCAT1 KO cells in the presence of cell permeable AKG (Figure 1B). A similar lethal phenotype was also observed in BCAT1 KO#1, KO#2, and KO#3 U87MG cells (Figure 1C). Cell death assay using propidium iodide staining confirmed that cell permeable AKG selectively killed BCAT1 KO#2 or KO#3 but not parental U251MG cells (Figure 1D). To determine whether cell permeable AKG specifically kills BCAT1 KO cells, we first performed the rescued experiment in U251MG cells. Ectopic expression of BCAT1 significantly restored survival and growth of BCAT1 KO#2 and KO#3 cells under conditions of cell permeable AKG treatment (Figure 1, B and D; Supplemental Figure 2A). We next found that cell permeable AKG had no inhibitory effect on BCAT2 KO U251MG cells (Supplemental Figure 3, A and B). Cell permeable AKG also failed to exert a combined lethal effect with KO of BCKDHA or another AKG-dependent transaminase GPT2 in U251MG cells (Supplemental Figure 3, C–G). Collectively, these results reveal the lethal effect of AKG specifically in BCAT1 KO IDHWT GBM cells in vitro.
The dose-response studies with increasing doses of cell permeable AKG showed that growth of BCAT1 KO#2 and KO#3 U251MG cells was gradually decreased (Figure 1, E and F). We next studied whether cell permeable AKG can synergize with a pharmacological BCAT1 inhibitor gabapentin to induce a lethal effect on IDHWT GBM cells. Co-treatment of cell permeable AKG (10 mM) and gabapentin (5 or 20 mM) caused significantly more U251MG cell death than their individual treatment (Figure 1, G and H; Supplemental Figure 2B). Moreover, U251MG cell death by this combined treatment was dose-dependent (Figure 1I). Similar synergistic cell death was also observed in many other IDHWT GBM U87MG, LN229, and T98G cells highly expressing BCAT1 (Figure 1, J–M). Importantly, cell permeable AKG alone but not gabapentin was sufficient to induce death of BCAT1-null LN18 and patient-derived IDHWT GBM C116 cells (Figure 1, N and O), indicating the specific inhibitory effect of gabapentin on BCAT1 in IDHWT GBM cells. Lastly, we found that the killing effect of gabapentin/AKG combination was not applicable to normal mouse astrocytes, and the combination instead increased astrocyte survival (Supplemental Figure 3H). Together, these findings from genetic and pharmacological studies indicate that BCAT1i combined with AKG induces a dose-dependent killing of IDHWT GBM cells but not normal astrocytes in vitro.
BCAT1i/AKG combination induces synthetic lethality in human patient-derived IDHWT GBM in vitro and in mice.
To study potential translational benefits of BCAT1i/AKG in IDHWT GBM patients, we treated patient-derived IDHWT GBM tumorspheres with gabapentin (5 mM), cell permeable AKG (10 mM), or their combination for 7 days. Treatment of gabapentin or cell permeable AKG alone had minimal effects on growth of tumorspheres in all four human IDHWT GBM models (Figure 2A). In contrast, gabapentin and AKG combination robustly inhibited growth of tumorspheres in vitro (Figure 2A), indicating that BCAT1i/AKG combination shows the killing effect in human IDHWT GBM in vitro. We next studied whether gabapentin and AKG combination induces killing of IDHWT GBM in mice. To this end, human patient-derived IDHWT GBM tumors were subcutaneously implanted into NSG mice and mice were administrated daily with gabapentin (60 mg/kg), AKG (150 mg/kg), or both by oral gavage once the tumor volume reached to 50–100 mm3. The combined treatment of gabapentin and AKG significantly inhibited growth of IDHWT GBM, whereas their individual treatment had no effect on tumor growth in mice (Figure 2B). This combination treatment did not show any toxicity as mouse body weight was not affected (Figure 2C). Analysis of Ki67 immunohistochemical staining revealed that gabapentin and AKG combination significantly reduced proliferating GBM tumor cells in PDX tumors as compared with saline, whereas individual treatment of gabapentin or AKG failed to do so (Figure 2D). Together, these in vivo results from patient-derived IDHWT GBM models indicate that BCAT1i/AKG combination reduces the rate of IDHWT GBM growth in mice.
Figure 2. BCAT1i and AKG induces synthetic lethality in patient-derived IDHWT GBM.
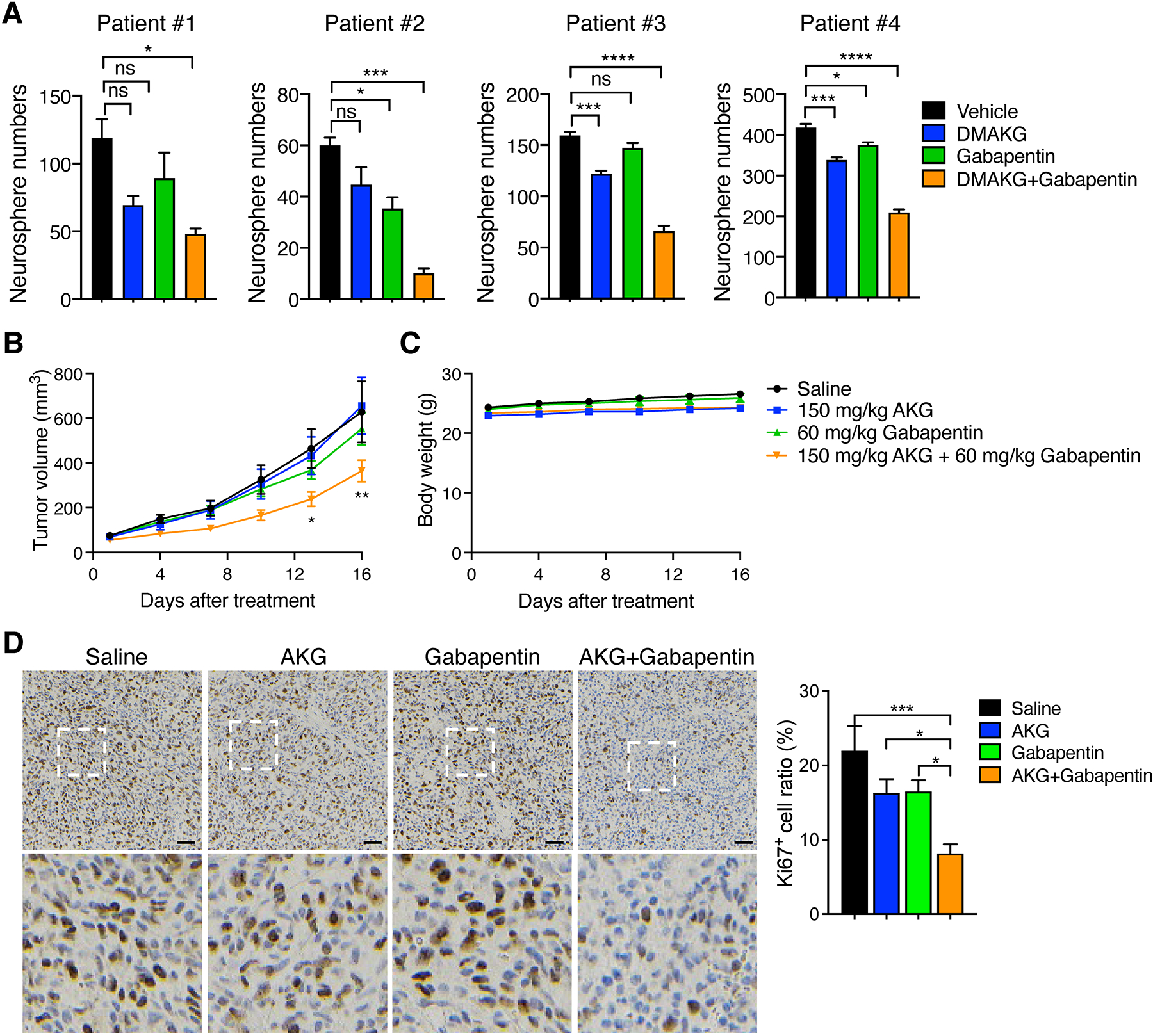
(A) Tumorsphere assay in patient-derived IDHWT GBM treated with vehicle, DMAKG (10 mM), gabapentin (5 mM), or both for 7 days. Tumorspheres with > 50 μm diameter are counted (mean ± SEM, n = 3). ns, not significant. *p < 0.05; ***p < 0.001; ****p < 0.0001 by one-way ANOVA with Dunnett’s test. (B and C) Patient-derived IDHWT GBM xenograft assay. Tumors were treated with saline, AKG, gabapentin, or both for 16 days. The tumor growth curve is shown in B (mean ± SEM, n = 7–9). The mouse body weight is shown in C (mean ± SEM, n = 7–9). *p < 0.05; **p < 0.01 by two-way ANOVA with Tukey’s test. (D) Ki67 immunohistochemical staining in patient-derived IDHWT GBM tumors harvested from xenograft assay. Representative images are shown at the top and magnified images of the boxed area are shown at the bottom. Scale bar, 50 μm. The percentage of Ki67+ cells is quantified at the right (mean ± SEM, n = 7–8). *p < 0.05; ***p < 0.001 by one-way ANOVA with Tukey’s test.
Multiple cell death inhibitors fail to prevent BCAT1i/AKG-induced IDHWT GBM cell death.
Next, we studied which cell death pathway is responsible for BCAT1i/AKG-induced IDHWT GBM cell death. Various cell death inhibitors including Z-Vad (10 μM, apoptosis inhibitor), ferrostatin-1 (Fer-1, 2 μM, ferroptosis inhibitor), necrostatin-1 (Nec-1, 20 μM, necroptosis inhibitor), 3-methyladenine (3-MA, 1 mM, autophagy inhibitor), or DPQ (30 μM, parthanatos inhibitor) were applied with or without cell permeable AKG into parental and BCAT1 KO#2 and KO#3 U251MG cells. Neither of these inhibitors rescued survival of BCAT1 KO U251MG cells in the presence of cell permeable AKG (Supplemental Figure 4, A–E). These findings indicate that the known cell death inhibitors cannot prevent BCAT1i/AKG-induced killing of IDHWT GBM cells.
Supplementation with BCKA prevents BCAT1i/AKG-induced IDHWT GBM cell death.
Next, we studied whether BCAT1-dependent metabolites can eliminate the synergy from BCAT1i/AKG combination to prevent IDHWT GBM cell death. BCAT1 reversibly converts BCAAs into BCKAs in the cell (Figure 3A). We first studied by clonogenic assay whether supplementation with BCAAs can prevent BCAT1i/AKG-induced cell death. BCAT1 KO cells were used for the following mechanistic studies because of their specificity (Supplemental Figure 1). BCAAs including Leu, Ile, and Val (2 mM) failed to restore survival of BCAT1 KO#2 and KO#3 U251MG cells under conditions of cell permeable AKG treatment (Figure 3B). Similarly, cell permeable glutamate and glutamine did not show the rescued effect (Supplemental Figure 5A). In contrast, supplementation with KIC (1 mM) significantly reversed death of BCAT1 KO U251MG or U87MG cells conferred by cell permeable AKG (Figure 3C; Supplemental Figure 5B). KIV (1 mM) and KMV (1 mM) also prevented AKG-induced death of BCAT1 KO#2 and KO#3 U251MG cells to a certain degree (Figure 3C). The individual BCKA itself had no obvious effect on parental and BCAT1 KO cell growth in the absence of cell permeable AKG (Figure 3C; Supplemental Figure 5B). Mass spectrometry analysis showed that all BCKAs were reduced in BCAT1 KO#2 and KO#3 U251MG cells (Figure 3D). Together, these results indicate that the depletion of BCKAs in BCAT1-null IDHWT GBM cells promotes a synergy with AKG treatment to induce cell death.
Figure 3. BCAT1i/AKG-induced IDHWT GBM cell death is rescued by BCKAs.
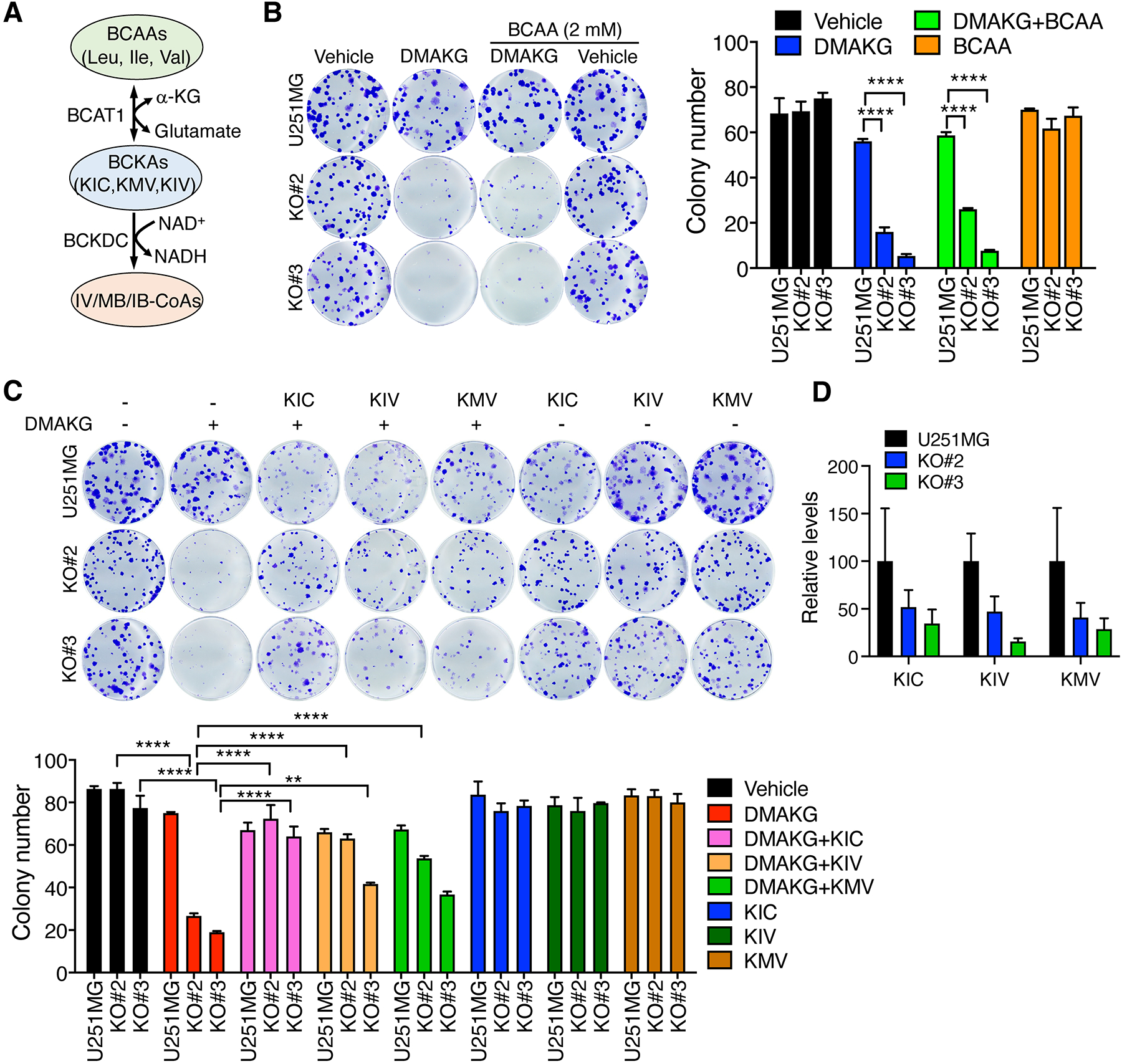
(A) The scheme of the BCAA metabolic pathway. (B) Clonogenic assay in parental and BCAT1 KO#2 or KO#3 U251MG cells treated with vehicle, DMAKG (10 mM), BCAA (2 mM), or both for 7 days. Representative images are shown at the left. The colonies are counted at the right (mean ± SEM, n = 3). (C) Clonogenic assay in parental and BCAT1 KO#2 or KO#3 U251MG cells treated with vehicle, DMAKG (10 mM), BCKA (1 mM), or both for 7 days. Representative images are shown at the top. The colonies are counted at the bottom (mean ± SEM, n = 3). (D) Mass spectrometry analysis of BCKA levels in parental and BCAT1 KO#2 or KO#3 U251MG cells (mean ± SEM, n = 3). **p < 0.01; ****p < 0.0001 by two-way ANOVA with Tukey’s test.
A previous study reported that dimethyl AKG induces HIF-1α protein expression in cancer cells (20). Consistently, both HIF-1α and HIF-2α protein levels were elevated in parental and BCAT1 KO#2 or KO#3 U251MG cells treated with cell permeable AKG for 24 hr (Supplemental Figure 6A). To determine the role of HIF in BCAT1i/AKG-induced IDHWT GBM cell death, we performed clonogenic assays under 20% or 1% O2 and found that hypoxia had no obvious effect on viability of parental and BCAT1 KO#2 or KO#3 U251MG cells with or without cell permeable AKG treatment (Supplemental Figure 6, B and C), which excludes a possible role of HIF signaling in BCAT1i/AKG-induced IDHWT GBM cell death.
A synergistic increase in NAD+/NADH by BCAT1i/AKG combination causes IDHWT GBM cell death.
BCKA oxidation by branched-chain α-keto acid dehydrogenase is coupled with NAD+ reduction into NADH in mitochondria, which controls electron transfer in the mitochondrial complex I to regulate mitochondrial respiration. We next studied whether BCAT1i increases the NAD+/NADH ratio and whether this altered ratio is augmented by AKG treatment to block mitochondrial respiration leading to IDHWT GBM cell death. Our biochemical studies showed that cellular NAD+ levels were significantly increased but cellular NADH levels were decreased in BCAT1 KO#2 and KO#3 U251MG cells (Supplemental Figure 7, A and B). As a result, the ratio of NAD+/NADH was significantly increased in BCAT1 KO U251MG cells (Supplemental Figure 7C). Consistently, BCAT1 KO#2 and KO#3 significantly reduced the activity of the mitochondrial complex I in U251MG cells (Supplemental Figure 7D). Imbalance of NAD+/NADH ratio is associated with redox homeostasis in mitochondria (21). As shown by MitoSOX-based flow cytometry, the mitochondrial ROS levels were significantly increased in BCAT1 KO#2 and KO#3 U251MG cells as compared with parental cells (Supplemental Figure 7E). Seahorse analysis demonstrated that BCAT1 KO#3 inhibited the basal OCR as well as maximal respiratory capacity in mitochondria (Supplemental Figure 7F). The OCR was also inhibited when BCKDHA was knocked out in U251MG cells (Supplemental Figure 7G), which supports the idea that reduced BCKA oxidation increases the NAD+/NADH ratio to inhibit oxidative phosphorylation in BCAT1 KO cells. Consistently, the cellular ATP levels were significantly decreased in BCAT1 KO#2 and KO#3 U251MG cells (Supplemental Figure 7H). TMRE staining revealed that both BCAT1 KO#2 and KO#3 significantly reduced mitochondrial membrane potential in U251MG cells (Supplemental Figure 7, I and J), which further support the role of BCAT1 in mitochondrial ATP production. However, the activity of the mitochondrial complex V ATP synthase was not altered in BCAT1 KO#2 and KO#3 U251MG cells (Supplemental Figure 7K). Collectively, these results indicate that BCAT1 loss inhibits NADH/NAD+ ratio and mitochondrial complex I activity to impair mitochondrial respiration in IDHWT GBM cells.
Distinct to BCAT1 KO (Supplemental Figure 7C), BCAT2 KO significantly decreased the ratio of NAD+/NADH in U251MG cells (Supplemental Figure 8A). Consistently, BCAT2 KO modestly increased the basal OCR and intracellular ATP levels, although it had no effect on maximal respiratory capacity and mitochondrial membrane potential in U251MG cells (Supplemental Figure 8, B–E). These results indicate that BCAT1 and BCAT2 have a distinct role in mitochondrial respiration in IDHWT GBM cells.
Remarkably, cell permeable AKG synergistically increased the ratio of NAD+/NADH in BCAT1 KO#1 U87MG cells and BCAT1 KO#2 and KO#3 U251MG cells (Figure 4, A and B). Supplementation with KIC (1 mM) significantly reversed a synergistic increase in NAD+/NADH ratio conferred by BCAT1 KO/AKG in U87MG cells (Figure 4A). Similar results were also observed in cell permeable AKG-treated BCAT1 KO#3 U251MG cells when expression of BCAT1 was rescued (Figure 4B). To determine whether NAD+/NADH imbalance causes BCAT1i/AKG-induced IDHWT GBM cell death, we treated parental and BCAT1 KO#2 U251MG cells with 1 mM NADH in the presence or absence of cell permeable AKG as previous studies showed that NADH can be transported into the cell (22). Supplementation with NADH partially but significantly rescued NAD+/NADH homeostasis in cell permeable AKG-treated BCAT1 KO#2 U251MG cells (Figure 4C). Under this condition, survival of BCAT1 KO#2 U251MG cells was restored (Figure 4D). In contrast, NAD+ precursors nicotinamide (1 mM) and NMN (1 mM) failed to restore growth of BCAT1 KO#2 U251MG cells under conditions of cell permeable AKG treatment (Supplemental Figure 9, A–D). We further found that NMN treatment did not replenish intracellular NADH levels in BCAT1 KO#3 U251MG cells, although it increased intracellular NAD+ levels in parental and BCAT1 KO#3 U251MG cells as well as intracellular NADH levels in U251MG cells (Supplemental Figure 9, E and F). Collectively, these results indicate that blockade of NADH regeneration from NAD+ during BCKA oxidation sensitizes IDHWT GBM cells to AKG treatment in vitro.
Figure 4. BCAT1i/AKG causes mitochondrial dysfunction and energy depletion in IDHWT GBM cells.
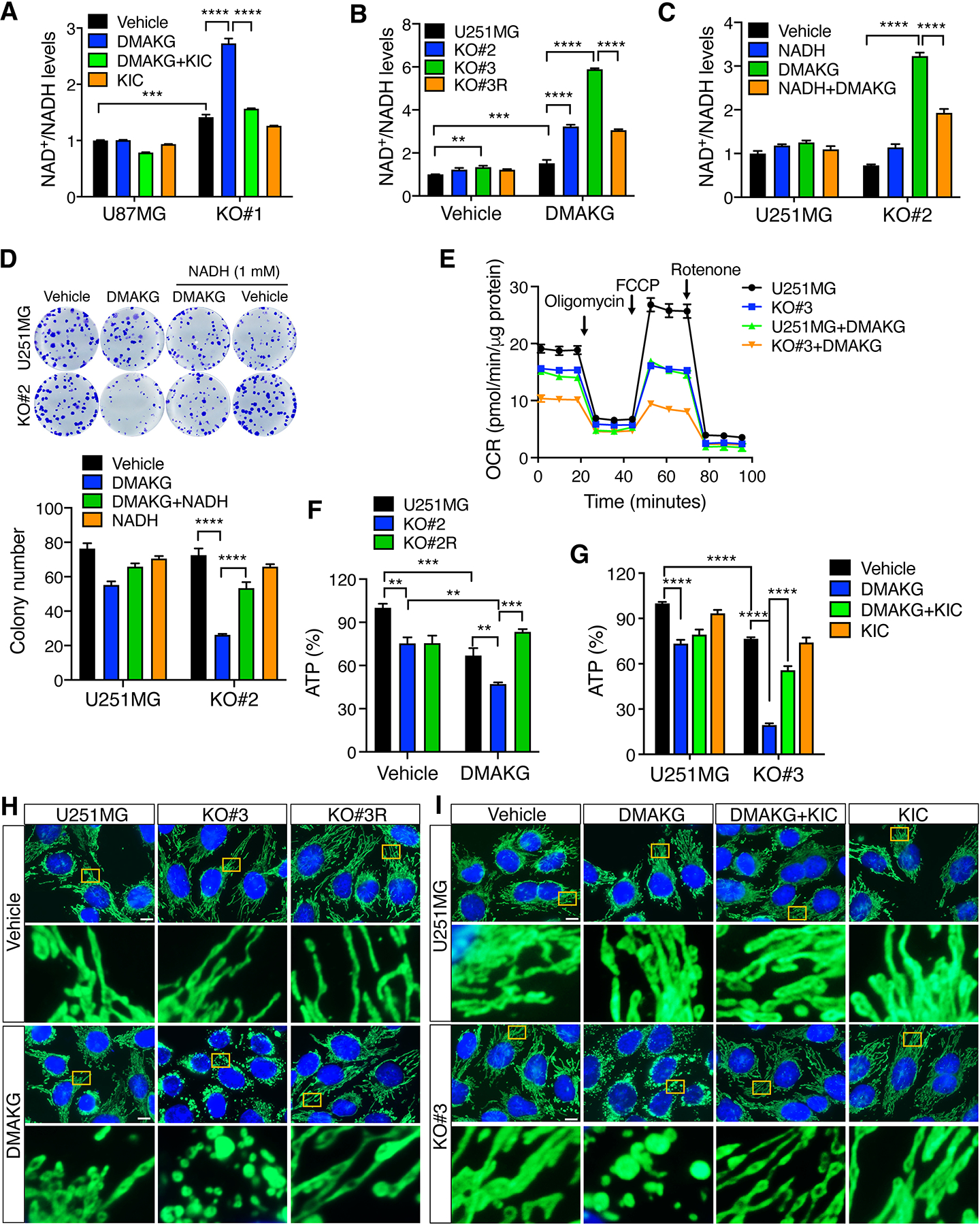
(A) Analysis of NAD+ and NADH levels in parental and BCAT1 KO#1 U87MG cells treated with vehicle, DMAKG (10 mM), KIC (1 mM), or both for 24 hours (mean ± SEM, n = 3). (B) Analysis of NAD+ and NADH levels in parental, BCAT1 KO#2 or KO#3, and BCAT1 rescue (R) U251MG cells treated with vehicle or DMAKG (10 mM) for 24 hours (mean ± SEM, n = 3–17). (C) Analysis of NAD+ and NADH levels in parental and BCAT1 KO#2 U251MG cells treated with vehicle, DMAKG (10 mM), NADH (1 mM), or both for 24 hours (mean ± SEM, n = 3). (D) Clonogenic assay in parental and BCAT1 KO#2 U251MG cells treated with vehicle, DMAKG (10 mM), NADH (1 mM), or both for 7 days. Representative images are shown on the top. The colonies are counted at the bottom (mean ± SEM, n = 5–6). (E) OCR analysis in parental and BCAT1 KO#3 U251MG cells treated with vehicle or DMAKG (10 mM) for 24 hours (mean ± SEM, n = 4). (F) Analysis of cellular ATP levels in parental, BCAT1 KO#2, and BCAT1 rescue (R) U251MG cells treated with vehicle or DMAKG (10 mM) for 24 hours (mean ± SEM, n = 3). (G) Analysis of cellular ATP levels in parental and BCAT1 KO#3 U251MG cells treated with vehicle, DMAKG (10 mM), KIC (1 mM), or both for 24 hours (mean ± SEM, n = 3). (H) Analysis of mitochondrial morphology in parental, BCAT1 KO#3, and BCAT1 rescue (R) U251MG cells treated with vehicle or DMAKG (10 mM) for 24 hours. Representative images from three independent experiments are shown at the top. Magnified images of the boxed area are shown at the bottom. Scale bar, 10 μm. (I) Analysis of mitochondrial morphology in parental and BCAT1 KO#3 U251MG cells treated with vehicle, DMAKG (10 mM), KIC (1 mM), or both for 24 hours. Representative images from three independent experiments are shown at the top. Magnified images of the boxed area are shown at the bottom. Scale bar, 10 μm. **p < 0.01; ***p < 0.001; ****p < 0.0001 by two-way ANOVA with Tukey’s test.
In line with the altered NAD+/NADH ratio, the OCR was significantly attenuated by cell permeable AKG treatment in BCAT1 KO#3 U251MG cells, which was much more severe than that in parental U251MG cells treated with cell permeable AKG or BCAT1 KO#3 U251MG cells treated with vehicle (Figure 4E). Consistently, ATP production was synergistically decreased by cell permeable AKG treatment in BCAT1 KO#2 U251MG cells, but not in BCAT1 rescued cells (Figure 4F). Supplementation with KIC (1 mM) also restored ATP production in cell permeable AKG-treated BCAT1 KO U251MG or U87MG cells (Figure 4G; Supplemental Figure 10A). Notably, mitochondria became massive swollen 24 hrs after cell permeable AKG treatment in BCAT1 KO#3 U251MG cells and BCAT1 KO#2 U87MG cells (Figure 4H; Supplemental Figure 10B). In contrast, BCAT1 KO or cell permeable AKG alone had little effect on mitochondrial morphology in IDHWT GBM cells (Figure 4H; Supplemental Figure 10B). Re-expression of BCAT1 rescued mitochondrial morphological changes caused by BCAT1 KO and AKG combination (Figure 4H). A similar rescued effect was also observed when KIC (1 mM) was added into BCAT1 KO#3 U251MG cells or BCAT1 KO#2 U87MG cells treated with cell permeable AKG (Figure 4I; Supplemental Figure 10B). Electron microscope images further confirmed mitochondrial swelling and loss of cristae in BCAT1 KO#3 U251MG cells treated with cell permeable AKG, which were partially rescued by re-expression of BCAT1 or supplementation with KIC (Supplemental Figure 10C). Mitochondrial swelling is an indicator of opening of the mitochondrial permeability transition pore and a hallmark of mitochondrial dysfunction (23). Thus, altered mitochondrial morphology confirms disruption of mitochondrial respiration and ATP production by BCAT1 KO/AKG combination in IDHWT GBM cells. Together, these results support the notion that blockade of NADH regeneration by BCAT1i/AKG combination causes death of IDHWT GBM cells possibly through mitochondrial dysfunction and energy depletion.
KIC partially prevents BCAT1i/AKG-induced nucleotide depletion.
Our results about selective killing of BCAT1 KO but not BCKDHA KO U251MG cells by cell permeable AKG (Figure 1D; Supplemental Figure S3G) suggest that metabolic mechanisms other than NADH depletion may also contribute to BCAT1i/AKG-induced IDHWT GBM cell death. To investigate these underlying mechanisms in greater detail, we conducted the metabolomics profiling by LC-MS in parental and BCAT1 KO#2 U251MG cells treated with or without cell permeable AKG for 24 hours. Consistent with biochemical analysis (Supplemental Figure 7, A–C), NAD+/NADH was increased in BCAT1 KO#2 U251MG cells (Figure 5A), which validated our assay. BCAT1 KO upregulated 10 intracellular metabolites but downregulated 33 intracellular metabolites in U251MG cells (p < 0.05 and 1.3-fold change as cutoffs, Figure 5A). Of 33 downregulated metabolites, 17 are nucleotides and nucleotide derivatives and precursors (Figure 5A). The levels of intracellular aspartate, which is involved in de novo synthesis of purines and pyrimidines, were also reduced in BCAT1 KO#2 U251MG cells (Figure 5A). Furthermore, 6-phosphate-D-gluconate and 3-phospho-serine from the respective pentose phosphate pathway and serine-glycine biosynthesis pathway that control the flux from glucose towards purine biosynthesis were downregulated in BCAT1 KO#2 U251MG cells (Figure 5A). Together, these results indicate that BCAT1 controls nucleotide biosynthesis in IDHWT GBM cells. Treatment of cell permeable AKG upregulated 16 and 29 metabolic pathways but downregulated 34 and 19 metabolic pathways (FDR < 0.05) in parental and BCAT1 KO#2 U251MG cells, respectively (Figure 5, B and C). Notably, in addition to NAD+/NADH, nucleotides were synergistically reduced in BCAT1 KO#2 U251MG cells treated with cell permeable AKG (Figure 5A). Purine metabolism was the sole pathway downregulated by BCAT1 KO, AKG, and both (Figure 5B; Supplemental Table 3). No upregulated metabolic pathways were overlapped among these groups (Figure 5C; Supplemental Table 4). Importantly, supplementation with KIC (1 mM) partially rescued ATP, GTP, and CTP in AKG-treated BCAT1 KO#2 and KO#3 U251MG cells (Figure 5D). Supplementation with nucleosides (30 μM), however, failed to rescue BCAT1 KO#2 and KO#3 U251MG cells after cell permeable AKG treatment (Supplemental Figure 9, G and H). Nucleotide is one of the fundamental building blocks required for cancer cell proliferation. Thus, these results suggest that nucleotides are depleted by BCAT1i/AKG combination, which may be one of mechanisms underlying BCAT1i/AKG-induced IDHWT GBM cell death.
Figure 5. BCAT1i/AKG causes nucleotide depletion in IDHWT GBM cells.
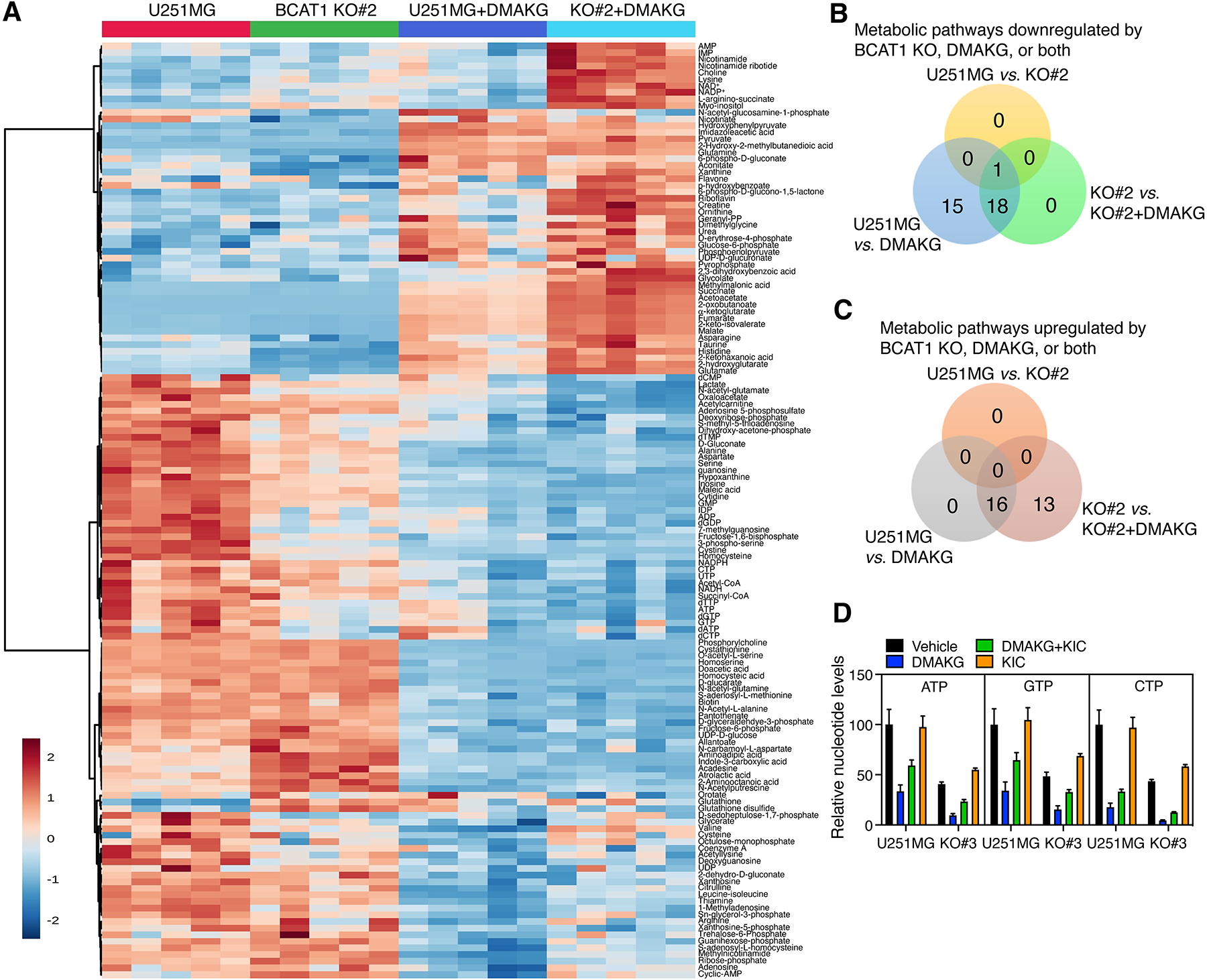
(A) The heatmap of cellular metabolites in parental and BCAT1 KO#2 U251MG cells treated with vehicle or DMAKG (10 mM) for 24 hours (n = 5). (B and C) Analysis of the enriched metabolic pathways in parental and BCAT1 KO#2 U251MG cells treated with vehicle or DMAKG (10 mM) for 24 hours. (D) Mass spectrometry analysis of nucleotides in parental and BCAT1 KO#3 U251MG cells treated with vehicle, DMAKG (10 mM), KIC (1 mM), or both for 24 hours (mean ± SEM, n = 4).
KIC partially prevents BCAT1i/AKG-induced mTORC1 inactivation and protein depletion.
Energy depletion triggers AMP-dependent protein kinase (AMPK) activation to block mTORC1 activity in the cell (12). We next studied the role of BCAT1i and/or AKG in the AMPK-mTORC1 signaling pathway in IDHWT GBM cells. Our metabolomics study showed that the AMP/ATP ratio was modestly increased by BCAT1 KO#2 (Figure 6A). The levels of phosphorylated AMPK, but not total AMPK, were increased in BCAT1 KO#2 and KO#3 U251MG cells (Supplemental Figure 11A), indicating that BCAT1 KO leads to AMPK activation in IDHWT GBM cells. In contrast, we found that BCAT1 KO caused mTORC1 inactivation as the putative mTORC1 substrate p70S6 kinase (p70S6K) was less phosphorylated at Threonine 389 in three individual BCAT1 KO U251MG cell lines (Supplemental Figure 11B, left). Similar results were also observed in U87MG cells (Supplemental Figure 11B, right). Expression of FLAG-BCAT1 in BCAT1 KO#2 U251MG cells restored phosphorylation of p70S6K (Supplemental Figure 11C), indicating specific regulation of mTORC1 by BCAT1 in IDHWT GBM cells. In line with genetic KO of BCAT1 (Supplemental Figure 11, B and C), treatment of gabapentin increased AMPK phosphorylation but decreased p70S6K phosphorylation in U251MG and U87MG cells (Supplemental Figure 11D). Previous studies showed that leucine and glutamate activate mTORC1 in cancer cells (24,25). Supplementation with leucine, glutamate, or both in BCAT1 KO#2 and KO#3 U87MG cells failed to rescue mTORC1 activity (Supplemental Figure 11E). Together, these findings indicate that BCAT1 inhibits AMPK activation leading to mTORC1 activation in IDHWT GBM cells.
Figure 6. BCAT1i/AKG causes mTORC1 inactivation and protein depletion in IDHWT GBM cells.
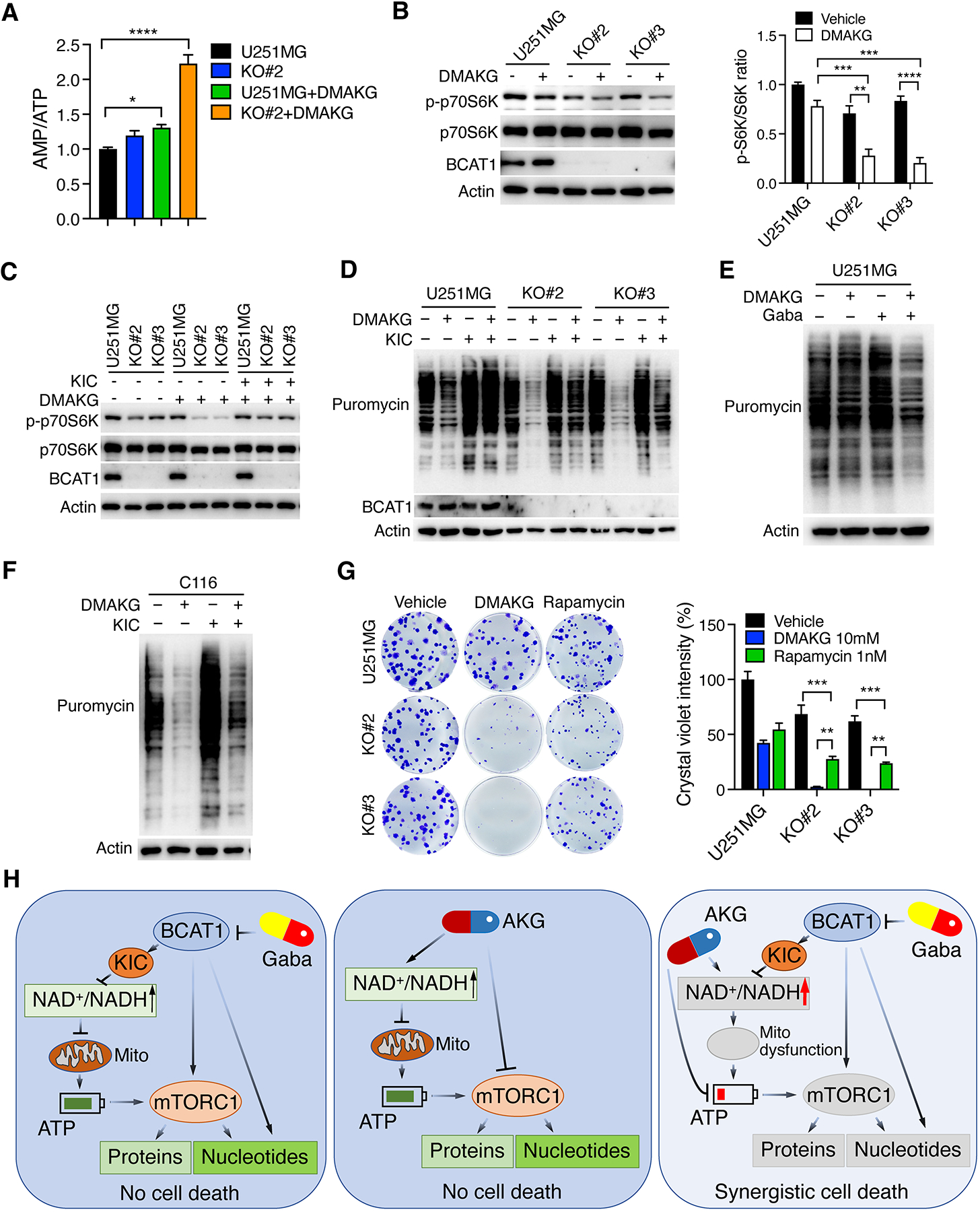
(A) Mass spectrometry analysis of AMP/ATP ratio in parental and BCAT1 KO#2 U251MG cells treated with vehicle or DMAKG (10 mM) for 24 hours (mean ± SEM, n = 5). *p < 0.05; ****p < 0.0001 by one-way ANOVA with Dunnett’s test. (B) Immunoblot analysis of mTORC1 activation in parental and BCAT1 KO#2 or KO#3 U251MG cells treated with vehicle (-) or DMAKG (10 mM) for 24 hours. Representative blots are shown in the left. Intensities of p-p70S6K and p70S6K bands are quantified and their ratio is shown in the right (mean ± SEM, n = 3). **p < 0.01; ***p < 0.001; ****p < 0.0001 by two-way ANOVA with Tukey’s test. (C) Immunoblot analysis of mTORC1 activation in parental and BCAT1 KO#2 or KO#3 U251MG cells treated with vehicle (-), DMAKG (10 mM), KIC (1 mM), or both for 24 hours. (D) Analysis of protein synthesis in parental and BCAT1 KO#2 or KO#3 U251MG cells treated with vehicle (-), DMAKG (10 mM), KIC (1 mM), or both for 24 hours. (E) Analysis of protein synthesis in U251MG cells treated with vehicle (-), DMAKG (10 mM), gabapentin (Gaba, 20 mM), or both for 48 hours. (F) Analysis of protein synthesis in C116 cells treated with vehicle (-), DMAKG (10 mM), KIC (1 mM), or both for 24 hours. (G) Clonogenic assay in parental and BCAT1 KO#2 or KO#3 U251MG cells treated with vehicle, DMAKG (10 mM), or Rapamycin (1 nM) for 7 days. Representative images are shown at the left. The intensity of crystal violet is quantified at the right (mean ± SEM, n = 3). **p < 0.01; ***p < 0.001 by two-way ANOVA with Tukey’s test. (H) A proposed model of BCAT1i/AKG-induced synthetic lethality in IDHWT GBM. BCAT1 inhibition causes a modest reduction of NADH, ATP, nucleotides, and proteins through loss of BCKAs, nitrogen donor glutamate, and/or mTORC1 activity in IDHWT GBM cells. Likewise, AKG modestly reduces nucleotide biosynthesis and protein synthesis through mTORC1 inhibition, inhibits ATP production, and increases NADH oxidation into NAD+. While BCAT1 loss or AKG alone-induced metabolic alterations are not sufficient to kill cells, BCAT1i/AKG combination has a significant impact on mitochondrial dysfunction and depletion of ATP, nucleotides, and proteins, leading to IDHWT GBM cell death. Gaba, gabapentin. Mito, mitochondrial. All immunoblot data are shown from one of three independent experiments.
Similar to BCAT1 loss, the treatment of cell permeable AKG modestly increased the AMP/ATP ratio and inhibited mTORC1 activity in U251MG cells, which were synergistically augmented in BCAT1 KO#2 and KO#3 U251MG cells (Figure 6, A and B). The loss of mTORC1 activity was partially restored by supplementation with KIC (1 mM) in U251MG cells (Figure 6C), suggesting that BCKA depletion promotes the synergistic effect of BCAT1i/AKG combination on mTORC1 activation, similar to its effect on NAD+/NADH ratio, ATP, and nucleotides (Figure 4, A and G, and 5D). In line with altered phospho-p70S6K, global protein synthesis was synergistically inhibited in BCAT1 KO#2 and KO#3 U251MG cells treated with cell permeable AKG (Figure 6D). Similar effects were also observed when U251MG cells were co-treated with gabapentin and cell permeable AKG (Figure 6E). Again, synergistic inhibition of protein synthesis by BCAT1 KO/AKG combination was partially rescued by supplementation with KIC (1 mM) in U251MG, U87MG, and patient-derived C116 cells (Figure 6, D and F; Supplemental Figure 11F). Finally, we found that mTORC1 inhibitor Rapamycin (1 nM) phenocopied cell permeable AKG but exhibited a less synergistic effect on death of BCAT1 KO#2 and KO#3 U251MG cells (Figure 6G). Collectively, these results suggest that the AMPK-mTORC1-protein synthesis axis is synergistically inhibited by BCAT1i/AKG combination partially contributing to IDHWT GBM cell death.
DISCUSSION
In the present study, we defined BCAT1i/AKG as an innovative metabolic synthetic lethal approach to treat IDHWT GBM and revealed that multiple metabolic events including mitochondrial dysfunction, mTORC1 inactivation, and depletion of ATP, nucleotides, and proteins may contribute to BCAT1i/AKG-induced synthetic lethality. The intracellular BCKAs are the key determinants of BCAT1i/AKG-induced IDHWT GBM cell death.
We showed that BCAT1 is highly expressed and controls metabolic rewiring of BCAA metabolism toward mitochondrial respiration in IDHWT GBM cells, whereas BCAT2 has a minimal effect on oxidative phosphorylation, indicating that BCAT1 is an important regulator of the oxidative phosphorylation phenotype in IDHWT GBM cells. Previous studies elucidated a critical role of BCAT2 in oxidative phosphorylation in pancreatic cancer cells (7,8). Thus, our results and others reveal a context-dependent role of BCAT1 vs. BCAT2 in mitochondrial metabolism in human cancers. Our studies suggest that NAD+/NADH homeostasis during BCKA oxidation regulates oxidative phosphorylation in IDHWT GBM cells. We also found that KIC but not Leu or Glu is required for mTORC1 activation in IDHWT GBM cells. The final metabolites in the BCAA metabolic pathway are unlikely to contribute to carbons in cancer cells (6–8). Collectively, these findings indicate that the intermediates rather than the final metabolites generated from BCAA metabolism contribute to cellular building blocks and also control the cell survival signaling pathways, leading to cancer cell proliferation.
Since Otto Warburg’s seminal work in 1920s, aerobic glycolysis was thought to play a dominant role in cancer cell proliferation (26). Our studies reveal that mitochondrial dysfunction by BCAT1i/AKG combination leads to ATP depletion in IDHWT GBM cells, which further blocks the AMPK-mTORC1-nucleotide/protein signaling pathway. Interestingly, genetic KO of BCAT2 failed to sensitize AKG-induced IDHWT GBM cell death. BCAT1 and BCAT2 have a distinct role in oxidative phosphorylation in IDHWT GBM cells, highlighting the key contribution of mitochondrial dysfunction to BCAT1i/AKG-induced IDHWT GBM cell death. The distinct biochemical property of BCAT1 and BCAT2 in BCAA metabolism was also reported in pancreatic tumors, where BCAT1 controls BCAA catabolism to produce BCKAs in cancer-associated fibroblasts, while BCAT2 converts BCKAs secreted from cancer-associated fibroblasts into BCAAs in pancreatic cancer cells (8). Such an interactive function between BCAT1 and BCAT2 in mitochondrial metabolism remains unknown in IDHWT GBM tumors and needs to be defined in future. A recent report showed that 36% of IDHWT GBM tumors heavily rely on oxidative phosphorylation for their growth (18). Thus, targeting mitochondria by BCAT1i/AKG combination will have a strong clinical significance in IDHWT GBM patients. This notion is also supported from studies in lung cancer, leukemia, and lymphoma that mitochondrion is an essential cellular organelle for cancer cell survival and proliferation and can be a therapeutic target to treat cancers (27–29).
NADH is an electron donor in the mitochondrial complex I where it transfers electrons after oxidizing into NAD+ to initiate the mitochondrial respiratory chain. We showed that BCAT1 loss increases the NAD+/NADH ratio but blocks oxidative phosphorylation in IDHWT GBM cells, which is synergized by AKG treatment. The NAD+/NADH ratio and ATP production can be partially reversed by supplementation with KIC, suggesting that NADH regeneration from NAD+ during KIC oxidation in mitochondria is crucial for oxidative phosphorylation. In agreement, we showed that BCKDHA loss robustly inhibits oxidative phosphorylation in U251MG cells. However, AKG fails to kill BCKDHA KO U251MG cells, which is most likely due to that accumulated BCKAs maintains mTORC1 activity in these KO cells. AKG can be converted to L-2-HG by lactate dehydrogenase A and malate dehydrogenases and, meanwhile, NADH is oxidized into NAD+ (30,31). Our metabolomics study showed that 2-HG is dramatically increased but lactate is reduced in U251MG cells after AKG treatment. These results suggest that AKG may hijack lactate dehydrogenase A for its metabolism into L-2-HG along with NADH oxidation to NAD+, which leads to increase in NAD+/NADH ratio in IDHWT GBM cells. This hypothesis requires future investigation. Nevertheless, our studies reveal a molecular mechanism of depletion of the NADH pool inhibiting oxidative phosphorylation in IDHWT GBM. NAD+ and NADH are critical for the function of many cellular enzymes in human health and diseases. Previous studies have shown that NAD+ depletion is associated with neurodegeneration, heart diseases, and inflammation (32). Thus, it will be important to understand how cellular NAD+ and NADH levels are precisely manipulated to develop a potent and safe therapeutic window for IDHWT GBM treatment.
mTORC1 is a well-known regulator of synthesis of nucleotides and proteins, which are essential for cell survival and proliferation (33,34). We showed that BCAT1 loss causes inhibition of mTORC1, which is further augmented by AKG treatment. The synergistic inhibition of mTORC1 is largely due to ATP and BCKA depletion by BCAT1i/AKG combination in IDHWT GBM cells. Leucine is a known potent activator of mTORC1 (24). Our current findings about mTORC1 activation by BCAA-derived KIC reveal another layer of regulatory mechanism of mTORC1 activation. In addition, AKG itself can inhibit mTORC1 in IDHWT GBM cells, which is consistent with a previous report showing that AKG blocks the ATP synthase activity in the mitochondrial complex V to inhibit mTOR (35). Although BCAT1 controls nitrogen donation from BCAAs for nucleotide biosynthesis (6,7), severe inactivation of mTORC1 by BCAT1i/AKG combination at least partially leads to nucleotide depletion in IDHWT GBM cells. Therefore, multiple layers of mechanisms contribute to BCAT1i/AKG-induced disruption of cellular building blocks leading to IDHWT GBM cell death (Figure 6H). It is also worth noting that a mTORC1 inhibitor rapamycin has a less synthetic lethal effect than AKG, which further supports the multifunctional role of AKG in metabolic synthetic lethality in IDHWT GBM. To date, mTOR inhibitors do not show any clinical benefits in GBM patients (36). Our findings strongly argue the necessity of targeting multiple metabolic pathways to treat IDHWT GBM. Further studies are needed to investigate whether alterations in other metabolic pathways we identified in metabolomics profiling studies also contribute to BCAT1i/AKG-induced IDHWT GBM cell death.
The synthetic lethal role of AKG in combination with BCAT1i in IDHWT GBM is unexpected since AKG is a glutamate-derived metabolite involved in energy production in the cell. Accumulating studies have uncovered several non-canonical functions of AKG in hypoxia signaling, gene regulation, cell death, cell differentiation, and immune response (37–41). The metabolic synthetic lethality is emerging in the field of cancer research given that metabolic alterations are the hallmarks of human cancers (42). Recently, several groups reported a similar role of AKG in vulnerability of cancer cells with altered cellular metabolism (38–40). Our work and others showing diverse mechanisms of AKG-mediated cancer cell death suggest a therapeutic potential of AKG in a broad range of human cancers.
In conclusion, we identified BCAT1i/AKG combination as a new synthetic lethal approach in IDHWT GBM. BCAT1i/AKG combination causes mitochondrial dysfunction and depletes the fundamental cellular building blocks including ATP, nucleotides, and proteins, leading to death of IDHWT GBM cells (Figure 6H). Gabapentin is an FDA-approved anti-epileptic drug. Our studies reveal its new potential application for a combination therapy with AKG in combating IDHWT GBM. This combined approach shows the killing effect specifically on IDHWT GBM cells but not normal astrocytes, suggesting that BCAT1i/AKG combination is a non-toxic therapeutic strategy that can be translated into the clinical investigation in patients with IDHWT GBM.
Supplementary Material
Significance:
Metabolic synthetic lethal screening in IDHWT glioblastoma defines a vulnerability to α-ketoglutarate following BCAT1 loss, uncovering a therapeutic strategy to improve glioblastoma treatment.
Acknowledgments
We thank the UTSW Cancer Center Tissue Resource for assistance in immunohistochemistry, which was supported by NCI Cancer Center Grant P30CA142543, the UTSW Electron Microscopy Core for assistance in sample preparation, which was supported by NIH shared instrumentation award 1S10OD021685-01A1, and the UTSW Metabolomics Core for metabolite analysis. We also thank the brain tumor PDX national resource at Mayo Clinic for providing IDHWT GBM PDX tumor. This work was supported by grants from the CPRIT (RR140036, RP220178), the NIH (R01CA222393), and the Welch Foundation (I-1903) to W.L., and the NIH (R35GM124693 and R01AG066166) to Y.W.. W.L. is a CPRIT Scholar in Cancer Research.
Footnotes
Conflict of interest
R.J.D. is an advisor for Agios Pharmaceuticals. Other authors declare that they have no conflict of interest.
REFERENCES
- 1.Ohgaki H, Kleihues P. The definition of primary and secondary glioblastoma. Clin Cancer Res 2013;19:764–72 [DOI] [PubMed] [Google Scholar]
- 2.Setton J, Zinda M, Riaz N, Durocher D, Zimmermann M, Koehler M, et al. Synthetic lethality in cancer therapeutics: the next generation. Cancer Discov 2021;11:1626–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karpel-Massler G, Ishida CT, Bianchetti E, Zhang Y, Shu C, Tsujiuchi T, et al. Induction of synthetic lethality in IDH1-mutated gliomas through inhibition of Bcl-xL. Nat Commun 2017;8:1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McBrayer SK, Mayers JR, DiNatale GJ, Shi DD, Khanal J, Chakraborty AA, et al. Transaminase inhibition by 2-hydroxyglutarate impairs glutamate biosynthesis and redox homeostasis in glioma. Cell 2018;175:101–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peng H, Wang Y, Luo W. Multifaceted role of branched-chain amino acid metabolism in cancer. Oncogene 2020;39:6747–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mayers JR, Torrence ME, Danai LV, Papagiannakopoulos T, Davidson SM, Bauer MR, et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science 2016;353:1161–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li JT, Yin M, Wang D, Wang J, Lei MZ, Zhang Y, et al. BCAT2-mediated BCAA catabolism is critical for development of pancreatic ductal adenocarcinoma. Nat Cell Biol 2020;22:167–74 [DOI] [PubMed] [Google Scholar]
- 8.Zhu Z, Achreja A, Meurs N, Animasahun O, Owen S, Mittal A, et al. Tumour-reprogrammed stromal BCAT1 fuels branched-chain ketoacid dependency in stromal-rich PDAC tumours. Nat Metab 2020;2:775–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gu Z, Liu Y, Cai F, Patrick M, Zmajkovic J, Cao H, et al. Loss of EZH2 reprograms BCAA metabolism to drive leukemic transformation. Cancer Discov 2019;9:1228–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hattori A, Tsunoda M, Konuma T, Kobayashi M, Nagy T, Glushka J, et al. Cancer progression by reprogrammed BCAA metabolism in myeloid leukaemia. Nature 2017;545:500–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ericksen RE, Lim SL, McDonnell E, Shuen WH, Vadiveloo M, White PJ, et al. Loss of BCAA catabolism during carcinogenesis enhances mTORC1 activity and promotes tumor development and progression. Cell Metab 2019;29:1151–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Valvezan AJ, Manning BD. Molecular logic of mTORC1 signalling as a metabolic rheostat. Nat Metab 2019;1:321–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang B, Chen Y, Shi X, Zhou M, Bao L, Hatanpaa KJ, et al. Regulation of branched-chain amino acid metabolism by hypoxia-inducible factor in glioblastoma. Cell Mol Life Sci 2021;78:195–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tonjes M, Barbus S, Park YJ, Wang W, Schlotter M, Lindroth AM, et al. BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild-type IDH1. Nat Med 2013;19:901–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Raffel S, Falcone M, Kneisel N, Hansson J, Wang W, Lutz C, et al. BCAT1 restricts alphaKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature 2017;551:384–8 [DOI] [PubMed] [Google Scholar]
- 16.Kirby DM, Thorburn DR, Turnbull DM, Taylor RW. Biochemical assays of respiratory chain complex activity. Methods Cell Biol 2007;80:93–119 [DOI] [PubMed] [Google Scholar]
- 17.Chen Y, Zhang B, Bao L, Jin L, Yang M, Peng Y, et al. ZMYND8 acetylation mediates HIF-dependent breast cancer progression and metastasis. J Clin Invest 2018;128:1937–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garofano L, Migliozzi S, Oh YT, D’Angelo F, Najac RD, Ko A, et al. Pathway-based classification of glioblastoma uncovers a mitochondrial subtype with therapeutic vulnerabilities. Nat Cancer 2021;2:141–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng X, Geng F, Pan M, Wu X, Zhong Y, Wang C, et al. Targeting DGAT1 ameliorates glioblastoma by increasing fat catabolism and oxidative stress. Cell Metab 2020;32:229–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hou P, Kuo CY, Cheng CT, Liou JP, Ann DK, Chen Q. Intermediary metabolite precursor dimethyl-2-ketoglutarate stabilizes hypoxia-inducible factor-1α by inhibiting prolyl-4-hydroxylase PHD2. PLoS One 2014;9:e113865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiao W, Wang RS, Handy DE, Loscalzo J. NAD(H) and NADP(H) redox couples and cellular energy metabolism. Antioxid Redox Signal 2018;28:251–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu H, Burns D, Garnier P, Wei G, Zhu K, Ying W. P2X7 receptors mediate NADH transport across the plasma membranes of astrocytes. Biochem Biophys Res Commun 2007;362:946–50 [DOI] [PubMed] [Google Scholar]
- 23.Zoratti M, Szabo I. The mitochondrial permeability transition. Biochim Biophys Acta 1995;1241:139–76 [DOI] [PubMed] [Google Scholar]
- 24.Jewell JL, Kim YC, Russell RC, Yu FX, Park HW, Plouffe SW, et al. Differential regulation of mTORC1 by leucine and glutamine. Science 2015;347:194–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dyachok J, Earnest S, Iturraran EN, Cobb MH, Ross EM. Amino acids regulate mTORC1 by an obligate two-step mechanism. J Biol Chem 2016;291:22414–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Warburg O On the origin of cancer cells. Science 1956;123:309–14 [DOI] [PubMed] [Google Scholar]
- 27.Deribe YL, Sun Y, Terranova C, Khan F, Martinez-Ledesma J, Gay J, et al. Mutations in the SWI/SNF complex induce a targetable dependence on oxidative phosphorylation in lung cancer. Nat Med 2018;24:1047–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med 2018;24:1036–46 [DOI] [PubMed] [Google Scholar]
- 29.Zhang L, Yao Y, Zhang S, Liu Y, Guo H, Ahmed M, et al. Metabolic reprogramming toward oxidative phosphorylation identifies a therapeutic target for mantle cell lymphoma. Sci Transl Med 2019;11:eaau1167. [DOI] [PubMed] [Google Scholar]
- 30.Oldham WM, Clish CB, Yang Y, Loscalzo J. Hypoxia-mediated increases in L-2-hydroxyglutarate coordinate the metabolic response to reductive stress. Cell Metab 2015;22:291–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Intlekofer AM, Dematteo RG, Venneti S, Finley LW, Lu C, Judkins AR, et al. Hypoxia induces production of L-2-hydroxyglutarate. Cell Metab 2015;22:304–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katsyuba E, Romani M, Hofer D, Auwerx J. NAD+ homeostasis in health and disease. Nat Metab 2020;2:9–31 [DOI] [PubMed] [Google Scholar]
- 33.Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 2013;339:1323–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ben-Sahra I, Hoxhaj G, Ricoult SJH, Asara JM, Manning BD. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 2016;351:728–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chin RM, Fu X, Pai MY, Vergnes L, Hwang H, Deng G, et al. The metabolite alpha-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature 2014;510:397–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kreisl TN, Lassman AB, Mischel PS, Rosen N, Scher HI, Teruya-Feldstein J, et al. A pilot study of everolimus and gefitinib in the treatment of recurrent glioblastoma (GBM). J Neurooncol 2009;92:99–105 [DOI] [PubMed] [Google Scholar]
- 37.Tran TQ, Hanse EA, Habowski AN, Li H, Gabra MBI, Yang Y, et al. alpha-Ketoglutarate attenuates Wnt signaling and drives differentiation in colorectal cancer. Nat Cancer 2020;1:345–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sica V, Bravo-San Pedro JM, Izzo V, Pol J, Pierredon S, Enot D, et al. Lethal poisoning of cancer cells by respiratory chain inhibition plus dimethyl alpha-ketoglutarate. Cell Rep 2019;27:820–34 e9 [DOI] [PubMed] [Google Scholar]
- 39.Madala HR, Helenius IT, Zhou W, Mills E, Zhang Y, Liu Y, et al. Nitrogen trapping as a therapeutic strategy in tumors with mitochondrial dysfunction. Cancer Res 2020;80:3492–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang JY, Zhou B, Sun RY, Ai YL, Cheng K, Li FN, et al. The metabolite alpha-KG induces GSDMC-dependent pyroptosis through death receptor 6-activated caspase-8. Cell Res 2021;31:980–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gyanwali B, Lim ZX, Soh J, Lim C, Guan SP, Goh J, et al. Alpha-Ketoglutarate dietary supplementation to improve health in humans. Trends in endocrinology and metabolism: TEM 2022;33:136–46 [DOI] [PubMed] [Google Scholar]
- 42.Zecchini V, Frezza C. Metabolic synthetic lethality in cancer therapy. Biochim Biophys Acta Bioenerg 2017;1858:723–31 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available within the article and its supplementary data file.