

Abstract
Epithelial-mesenchymal transition (EMT) is a fundamental process that occurs during embryogenesis and tissue repair. However, EMT can be hijacked by malignant cells, where it may promote immune evasion and metastasis. Classically considered a dichotomous transition, EMT in cancer has recently been considered a plastic process whereby malignant cells display and interconvert among hybrid epithelial/mesenchymal (E/M) states. Epithelial-mesenchymal plasticity (EMP) and associated hybrid E/M states are divergent from classical EMT with unique immunomodulatory effects. Here, we review recent insights into the EMP-immune crosstalk, highlighting possible mechanisms of immune evasion conferred by hybrid E/M states and roles of immune cells in EMP.
Introduction
Approximately 90% of cancer-associated deaths are due to metastatic disease (1), and accordingly, 5-year disease-free survival in patients with metastatic disease compared to locoregional disease is greatly reduced across most cancer types (2). Metastatic cancer often displays resistance to systemic therapies including chemotherapy, immunotherapy, and targeted biologics (3). The advent of immunotherapy, namely immune checkpoint blockade (ICB) has dramatically advanced treatment of metastatic disease for a fraction of patients in select cancer types, including melanoma and lung cancer (4). Unfortunately, of the 43% of cancer patients eligible for ICB, only 14% are estimated to respond (5). Thus, a major focus of cancer research is addressing the low response rates of ICB by elucidating mechanisms underlying ICB resistance and identifying opportunities for combination therapies that may overcome resistance (1,4).
Epithelial-mesenchymal transition (EMT) has emerged as a potential contributor to immunotherapy resistance (6). EMT describes the transitional process of an epithelial cell becoming a mesenchymal cell and occurs in embryogenesis and development. However, cancer cells may hijack EMT to drive therapy resistance, invasion, metastasis, and thus, poor outcomes in cancer (7). Although classically described as a binary switch between epithelial and mesenchymal states, EMT in cancer is now understood to be a plastic process in which cells interconvert between hybrid epithelial/mesenchymal (E/M) states with varying degrees of both epithelial and mesenchymal features, referred to as epithelial–mesenchymal plasticity (EMP) (8). The hybrid E/M state has recently been found to modulate the immune system to support cancer invasion and metastasis (1,7), and attention has been given to the role of hybrid E/M states in ICB resistance (7,9). Understanding the hybrid E/M-immune crosstalk in cancer is now proving useful in the development of prognostic markers, therapeutic indications, and novel therapy combinations (7,9–11). Here, we review the relevant literature to highlight mechanisms of immune evasion conferred by hybrid E/M states, emphasize the role of the immune system in promoting the hybrid E/M phenotype, and propose areas of investigation that may advance our understanding of EMP in tumor immune evasion.
The Plasticity of EMT
Initial hypotheses addressing EMT were generated in the 1960s by Elizabeth Hay, and EMT has since been attributed important roles in embryogenesis, wound healing, and cancer (8,12–16). The plasticity of EMT, or EMP, and its intra-tumoral and inter-tumoral heterogeneity is a recent major discovery in oncology with clinical relevance (7–9,17–21). Indeed, hybrid E/M states are associated with poor prognosis in several cancers (22–25) and have been shown to promote therapy resistance (26) and local and distant metastases (27). Metastable hybrid E/M states exhibiting plasticity were predicted through computational modeling of EMT (9,28–33). RNA sequencing and histological staining of primary tumors corroborated these in silico findings, demonstrating hybrid E/M states in several cancers (10,11,17,22,23,27,28,34–46). Moreover, recent pan-cancer studies using single cell RNA sequencing (scRNA-seq) have highlighted that the EMP gene program is conserved across cancer types, although their constituent genes are variable and correlate with the tissue of origin (10,34,45,46).
While EMP induction is heterogeneous across tumors (10), some signals appear conserved. These include extracellular ligands such as transforming growth factor β (TGFβ), tumor necrosis factor α (TNFα), and vascular endothelial growth factor (VEGF), environmental factors like hypoxia, dysregulated intracellular signaling in MAPK or p53 pathways, biophysical properties of the extracellular matrix (ECM), genetic mutations, and epigenetic alterations (10,47–50) (Figure 1). Downstream canonical transcription factors (TFs) that mediate EMP include SNAIL1/2, TWIST1/2, and ZEB1/2 (51). However, others TFs such as JUNB, FOSB, and KLF4/6 are also associated with EMP (10). The inducing signals of EMP interface with a dynamic balance of intracellular inducing and inhibiting signals occurring epigenetically, transcriptionally, post-transcriptionally by microRNA (miRNA) and long non-coding RNA (lncRNA), and post-translationally by regulatory circuits and signaling pathways that dictate the relative expression of epithelial and mesenchymal features (9,17,32,47,52,53). A classic example of post-translational regulation is the balance between the interconnected miR-34/SNAIL negative feedback loop and miR-200/ZEB negative feedback loop, in which SNAIL and ZEB promote EMP while the miRNAs inhibit EMP (30,54,55).
Figure 1. Induction of EMP promotes tumor immune escape.
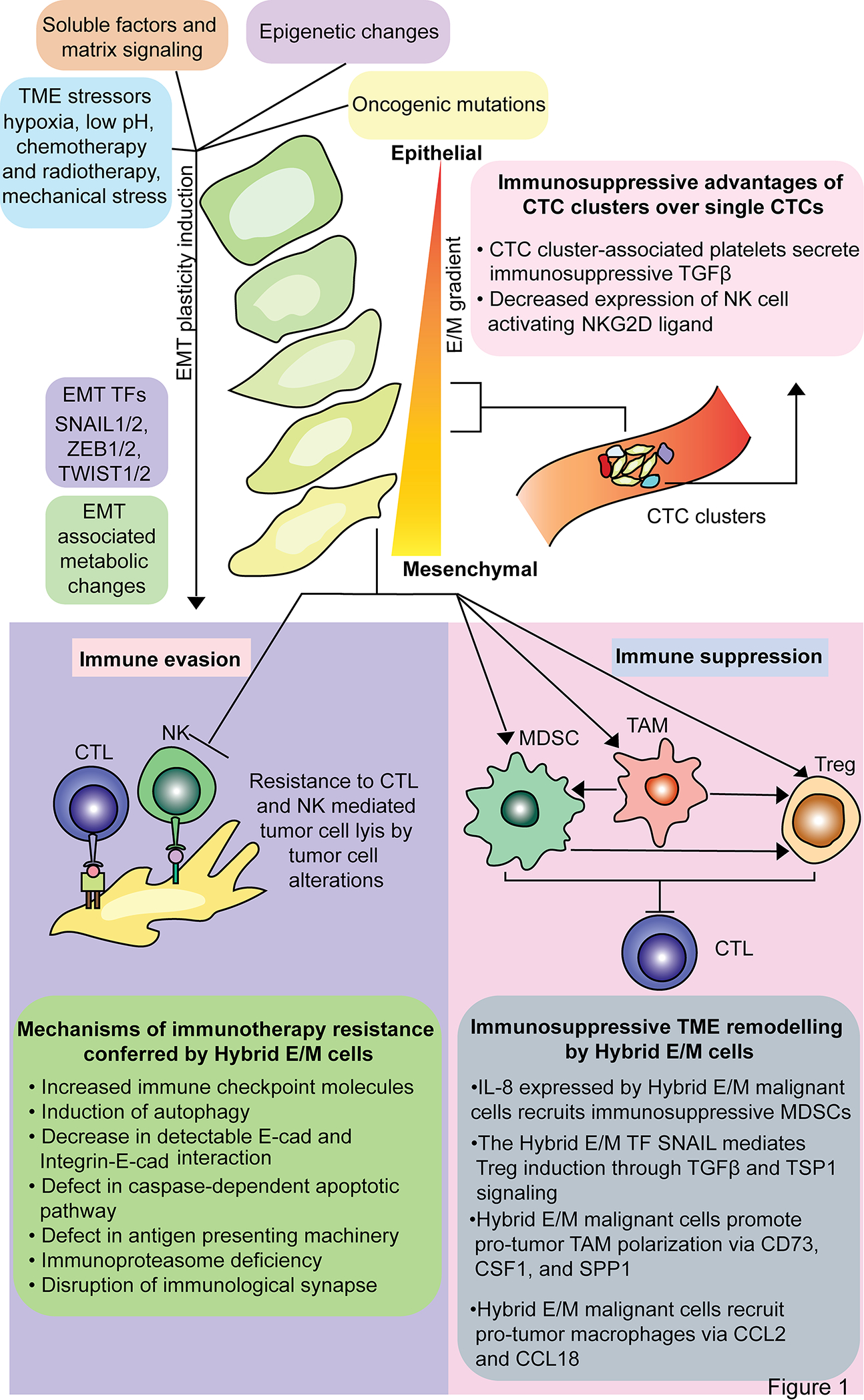
EMP is induced by environmental and genomic factors that canonically activate SNAIL, ZEB, and TWIST transcription factor families. Hybrid E/M states mediate tumor immune escape through evading immune cell cytotoxicity and polarizing immune cells to an immunosuppressive phenotype.
EMP in Tumor Immunology and Cancer Immunoediting
The dynamic relationship between the immune system and tumors is directly influenced by the cancer immunoediting hypothesis (56). Classically, cancer immunoediting is a three-stage model including elimination, equilibrium, and escape. In the elimination phase, cancer cells are recognized by the immune system as foreign due to expression of tumor-associated antigens (TAAs). TAAs are released to the extracellular environment following cancer cell death, and these antigens are taken up by antigen presenting cells (APCs) that present these neoantigens to a cytotoxic T lymphocyte (CTL) with a T cell receptor (TCR) specific to the TAA, triggering CTL activation. Although dendritic cells (DCs) are the prototypical APC, all nucleated cells – including cancer cells – express major histocompatibility complex (MHC) class I that serves to primarily present endogenous antigens to the immune system and activate CTLs. Activated CTLs home to the tumor, bind to a TAA presented on MHC class I, and lyse the cancer cell in combination with initiating a local immune response via inflammatory cytokines and chemokines (57). The cycle of immune recognition of cancer cells via MHC class I and their subsequent lysis results in either tumor elimination or an equilibrium phase. In the equilibrium phase, the persistent immune response acts as a selective pressure that, in combination with tumor heterogeneity and cancer cells’ DNA repair malfunctions, enriches tumor clones with an immune escape phenotype. The immune escape tumor phenotype is characterized by enrichment of immunosuppressive cell types such as regulatory T cells (Tregs) or myeloid-derived suppressor cells (MDSCs), expression of immune checkpoint proteins including programmed death-ligand 1 (PD-L1), defective antigen presentation, and/or dysregulated cytokine and metabolism regulation that promotes immune suppression (58). Tumor clones that enter the escape phase resist immune cell cytotoxicity and grow despite the anti-tumor immune response.
Spatial and Temporal Features of EMP-Tumor Immune Modulation
Advances in single cell technologies and the use of genetically engineered animal models have allowed for increased granularity in evaluating the roles of malignant, stromal, and immune cells of the tumor microenvironment (TME) in promoting EMP (23,27,38). Single cell sequencing has also helped to elucidate the heterogeneity in EMP, defining commonalities and differences in the EMT gene signature from tumor to tumor within and across primary cancer sites (10,34,45,59). Pastuschenoko et al. (2018) were among the first to demonstrate that malignant cells with varying expression of epithelial and mesenchymal markers existed within the same tumor and formed neighborhoods with the most mesenchymal cells in close proximity to a fibrovascular niche composed of endothelial cells and cancer-associated fibroblasts (CAFs) (23). Our group used scRNA-seq in head and neck squamous cell carcinoma (HSNCC) patient samples and found that cells enriched for a hybrid E/M signature localized to the leading edge of tumors adjacent to immune cells of the stroma and engaged with CAFs via TGFβ signaling (27). More importantly, we found that tumors enriched for this hybrid E/M signature were associated with worse clinical outcomes (22). Additionally, histological evidence of expression of classical EMT markers at the tumor-stroma interface has been associated with poor prognosis in several cancer types, such as breast, lung, prostate, pancreatic, colorectal, and HNSCC (27,40,60–63). The mechanism underlying this spatial localization is unresolved. However, a selective advantage of the hybrid E/M state in resisting stromal cytotoxic immune cells while coopting other stromal cells to promote EMP via soluble and contact signaling at the tumor-stroma interface likely contributes.
The hybrid E/M state is associated with immune evasion and suppression that may mediate such a selective advantage at the tumor leading edge. Immune evasion mechanisms include decreased expression of MHC class I and other antigen presentation proteins, which may be directly regulated by the hybrid E/M TF SNAIL2 and prevent recognition of hybrid E/M cells by CTLs (64–66) (Figure 1). Additionally, autophagy induction and the activity of EMT TFs brachyury and mucin 1 (MUC1) mediate resistance to natural killer (NK) and CTL-meditated cytotoxicity (67–69) (Figure 1). EMP is also associated with increased expression of immunosuppressive proteins, such as PD-L1, CD73, and CD276 (35,66) (Figure 1). The microRNA miR-200 has a dual role in both inhibiting EMP and PD-L1 expression; however, the EMT TF ZEB1 is activated in EMP, suppresses miR-200, and thereby increases PD-L1 expression (33,70–72). Interestingly, compared to epithelial cells, hybrid E/M cells and full mesenchymal cells upregulate PD-L1 to a similar extent, suggesting that hybrid E/M states adopt immunosuppressive features of a mesenchymal cell without losing the plasticity advantageous for metastasis (33). Due to increased immune checkpoint protein expression and alterations in adhesion proteins in hybrid E/M states, CTL activation is attenuated in the immunological synapse with an EMP-induced cell compared to a non-EMP-induced cell (73–75) (Figure 1).
In addition to immune evasion, hybrid E/M malignant cells trigger immune suppression through secreted factors upregulated in this state, such as TGFβ, IL-8, colony stimulating factor 1 (CSF1), and CCL18, and support immunosuppressive cell recruitment and polarization of Tregs, tumor associated macrophages (TAMs), and MDSCs (76–78) (Figure 2). Leveraging a multi-omics approach incorporating scRNA-seq, spatial transcriptomics, and multiplex ion beam imaging, Ji et al. (2020) identified a population of “tumor-specific keratinocytes” (TSKs) in human cutaneous squamous cell carcinoma with striking overlap with the hybrid E/M signature identified in HNSCC (27,35). These TSKs co-localized with CAFs, endothelial cells, macrophages, and Tregs. In addition, both TSKs and CAFs were found to express CD276 (B7-H3), an immune checkpoint protein that dampens the anti-tumor immune response (79). Notably, TSK cells expressed integrins ITGA3 and ITGB1, which serve as receptors for ligands expressed by TAMs and MDSCs (35). Hybrid E/M cells’ secreted factors can polarize TAMs and MDSCs to a pro-tumor phenotype that promotes immunosuppressive polarization of other immune cells as well as EMP induction, tumor growth, and invasion (62,80–83) (Figure 2). Tumor immune evasion and suppression mechanisms conferred by the hybrid E/M state are consistent with the observation that non-small cell lung cancer (NSCLC) and breast cancer tumors with more mesenchymal-like features show decreased infiltration of CTLs and increased immunosuppressive cell infiltration (66,84–86).
Figure 2. Crosstalk between hybrid E/M malignant cells and stromal cell populations.
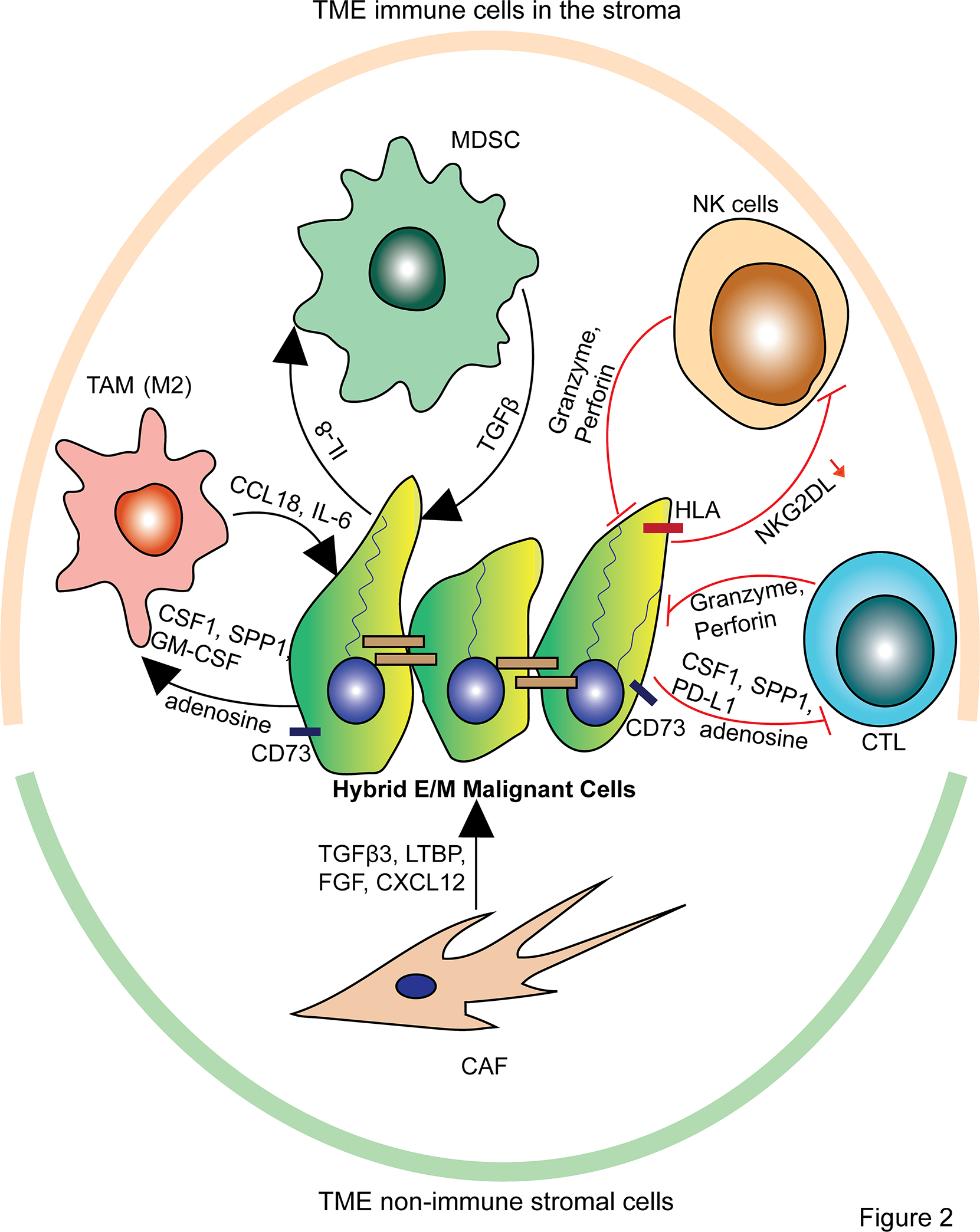
MDSCs and TAMs are recruited and polarized to a pro-tumor phenotype by secreted factors from hybrid E/M malignant cells, and in turn, the polarized MDSCs and TAMs secrete factors that promote EMP. Additionally, CAFs promote EMP through their secreted factors. At the same time, hybrid E/M malignant cells resist NK cell- and CTL-mediated cytotoxicity via membrane bound proteins and secreted factors.
The spatial localization of hybrid E/M cells to the tumor invasive front may also arise due to modulation of non-immune cells, including CAFs and normal epithelium. Indeed, CAFs have been shown to contribute to induction and maintenance of EMP in malignant cells via their secretion of latent TGFβ binding proteins (LTBPs), stromal cell-derived factor 1 (SDF1, or CXCL12), fibroblast growth factor (FGF), and TGFβ (27,85–87) (Figure 2). Additionally, hybrid E/M cells in HNSCC have been shown to promote lateral tumor invasion by secreting TGFβ and TNFα that acts on adjacent normal epithelium to induce EMT, which represents a selective advantage of hybrid E/M cells to invade that is permitted by their localization at the tumor-stroma interface (88).
The Role of EMP in Immune Evasion by Circulating Tumor Cells
Along with disrupting the basement membrane and local invasion of tissue, EMP is believed to confer circulating tumor cells (CTCs) with increased potential for distant metastasis (1). CTCs are considered a metastatic precursor that disseminate from the primary tumor mass, travel through the circulatory system, and seed distant sites to establish a metastatic niche that supports secondary tumors. EMP is implicated in CTC-mediated metastasis in several cancer types including breast cancer (89), NSCLC (90), prostate cancer (91), gastric cancer (92), and colorectal cancer (CRC) (93). Hybrid E/M cells travel as CTC clusters and decrease expression of NK cell activating ligands, which may shield CTC clusters from NK cell-mediated lysis when in circulation (94) (Figure 1). While in transit, CTCs of colon and breast cancer cell lines in vivo were shown to be coated in platelets that release TGFβ thereby promoting a mesenchymal phenotype, decreasing NK cell recognition, and increasing survival during transit to a metastatic site (1,95) (Figure 1). Upon seeding a metastatic site, a mesenchymal shift within the hybrid E/M state, or a mesenchymal-epithelial transition (MET), is essential for cancer cell seeding and outgrowth (47,96–101). The epithelial shift has been shown to be regulated by key TFs. For instance, Prrx1 promotes EMT and is suppressed to enable metastatic colonization (100), and the TFs OVOL1 and OVOL2 inhibit EMT and promote MET (102,103). The molecular mechanism of an epithelial shift at a metastatic site is likely distinct from the mechanism of a mesenchymal shift at the primary tumor site. Indeed, hysteresis regulation of EMP, in which the paths of epithelial and mesenchymal shifts are distinct, was found to have increased metastatic potential compared to non-hysteresis regulation in a breast cancer mouse model (104). For disseminated hybrid E/M malignant cells that seed a distant site, cell extrinsic factors likely contribute to the epithelial shift. For example, evidence in mouse models of breast cancer suggests that MDSCs facilitate a reversion to a more epithelial state, enhance survival of metastatic cells, and promote outgrowth (105). Thus, understanding the EMP-immune crosstalk in metastatic colonization may reveal immune interactions that are uniquely associated with MET and not observed at the primary tumor.
Cancer Stem Cells and Hybrid E/M Cells Similarly Modulate the Immune System
Cancer stem cells (CSCs) comprise a fraction of hybrid E/M cells and have the ability to differentiate into diverse cell types, self-renew, and both evade and suppress the immune system (106–108). Strikingly, cancer stemness is molecularly and functionally associated with EMP. For instance, the EMT TFs TWIST1 and ZEB1 increase cancer cell stemness, which has been shown to be associated with the enhanced metastatic potential of the hybrid E/M state (24,25,33,109–112). Indeed, a full EMT state in which a cell fully differentiates into a mesenchymal cell is unable to colonize a metastatic site due to loss of stemness (99,100). Thus, EMP, in which cells do not fully differentiate, is proposed to be compatible with maintenance of stemness, tumor initiating capacity, and metastatic seeding (24,98,108).
Similar to hybrid E/M states, CSCs also display immune evasion and suppression tactics (107,113,114). For instance, CSCs display MHC class I downregulation (115), and CSCs may evade NK cells through downregulation of the NK cell-activating ligand NKG2D (116). In addition, through secreted cytokines such as TGFβ, IL-10, and periostin, CSCs suppress and polarize local immune cells into a pro-tumor phenotype (117–119). For example, CSCs induce macrophages into a pro-tumor phenotype associated with secretion of cytokines that further promote stemness (120,121). However, in contrast to EMP, the transcriptional profiles characteristic of cancer stemness negatively correlate with PD-L1 expression in several cancer types (122). Although CSCs and hybrid E/M cells appear to share mechanisms of immune evasion and suppression, their similarity in underlying signaling remains incompletely understood; however, a more complete characterization of these cell states has the potential to reveal converging hubs suitable for pharmacologic inhibition.
Direct and Indirect EMP-Immune Crosstalk
EMP is involved in immunosuppression as result of soluble mediators that recruit and promote immunosuppressive immune cells as well as direct inhibition of immune cells (Figure 2). Many malignant tumors with an immunosuppressive phenotype also demonstrate an enrichment of EMP markers (84,123). These tumors feature decreased CTL infiltration and increased expression of immune checkpoint molecules, often portending a poor prognosis (124). In a study of lung cancer patients, a strong correlation between a “tumor EMT score” (based on the expression of 76 classical EMT signature genes) and expression of the immune checkpoint PD-L1 expression was identified (125). A similar result was found using a pan-cancer EMT signature spanning 11 different cancer types: Tumors with a high EMT score also had high expression of immune checkpoint proteins, including programmed cell death protein 1 (PD-1), PD-L1, cytotoxic T-Lymphocyte associated protein 4 (CTLA-4), and PD-L2. In patients with metastatic melanoma, an upregulation of E/M-related genes in the primary tumor was associated with primary resistance to anti-PD-1 therapy (126). A similar observation has been noted in CTCs, where classical EMT markers N-cadherin and vimentin were enriched in the CTCs of NSCLC patients with recurrence following anti-PD-L1 therapy (127). While these studies strongly suggest an association between EMP and markers of immunosuppression, mechanistic studies underlying the EMP-immune signaling axes are needed to better understand the link between hybrid E/M states and immune resistance.
Factors with Dual Roles in EMP Induction and Immunomodulation
Many of the soluble factors known to induce EMP also contribute to immune suppression in the TME (Figure 2). TGFβ is perhaps the most well-studied EMP-inducing soluble factor with additional immunomodulatory effects (128). TGFβ can be secreted by malignant, immune, and stromal cells. Induction of EMP by TGFβ is mediated by upregulation of key EMT TFs SNAIL (129), SLUG (15), and ZEB1/2 (130,131), as well as other TFs associated with EMP including HMGA2 (132) and ETS1 (133). In addition to modulating malignant cell phenotypes, TGFβ drives immune suppression in the TME by multiple mechanisms. For instance, EMP driven by TGFβ1 in hepatocellular carcinoma (HCC) cells resulted in increased expression of immune checkpoint proteins PD-L1 and B7-H3 (CD276), and the coordinate expression of TGFβ1 with PD-L1 and B7-H3 correlated with poor prognosis in HCC patients (134). In gastric carcinoma cell lines, EMP triggered by TGFβ1 resulted in increased expression of PD-L1 through activation of nuclear factor kappa B (NF-κB) (135). NF-κB is a known transcriptional regulator of several EMT TFs including TWIST1, SLUG, and ZEB2 in several cancer types, such as NSCLC and pancreatic cancer (136,137). Indeed, inhibition of NF-κB signaling has been shown to decrease the migratory capacity of gastric carcinoma cells and abrogate TGFβ1-mediated expression of PD-L1 (135).
Similar to TGFβ, IL-8 has a dual role in EMP and immune modulation by signaling through its G-protein coupled receptors, CXCR1 and CXCR2. The resulting acquisition of mesenchymal features, namely increased expression of vimentin, fibronectin, and classical EMT TFs, promotes cell migration and invasion (138). Interestingly, in addition to EMP induction, IL-8 has also been shown to drive immunosuppression in the TME by recruiting and enhancing the function of MDSCs (80,81) (Figure 2). MDSCs are known to play a critical role in facilitating tumor immune escape by inhibiting immune cells that are required for the anti-tumor immune response, such as CTLs, NK cells, and DCs (139). However, it is unclear whether the induction of EMP and immunosuppression by IL-8 are parallel pathways or interconnected. Most often, EMP displays a positive feedback loop whereby malignant cells exhibiting EMP produce the very factors that induce a further transition towards this state. In line with this positive feedback loop model, IL-8 has been shown to be upregulated upon induction of EMP by TNFα or TGFβ in colon cancer (140) and by epidermal growth factor (EGF) in breast cancer (141) (Figure 2). In turn, the transcription of IL-8 can be promoted by the EMT TFs SNAIL and brachyury. SNAIL was shown to induce IL-8 production in vivo in a NSCLC model (142) and directly regulate IL-8 production by binding to E-box regions within the IL-8 promoter in CRC cells (143). Additionally, IL-8 was one of the most upregulated secretory molecules in human carcinoma cells induced to a mesenchymal state by brachyury expression (144).
The tyrosine receptor kinase AXL is another well studied common inducer of both EMP and tumor immune evasion. In cancer, AXL signaling can become aberrantly activated due to membrane alterations of a mesenchymal-like cell that increase expression of AXL, EGFR, HER2, and Met (65). AXL expression positively correlates with expression of EMT genes in several cancers, including HER2+ breast cancer (145) and esophageal squamous cell carcinoma (146). Furthermore, in pancreatic cancer cells, AXL has been shown to regulate the expression of EMT TFs SNAIL, SLUG, and TWIST (147). AXL was also shown to mediate resistance to NK cell- and CTL-mediated killing through NF-κB and MAPK signaling in lung cancer clones derived from NSCLC patient tumors and induced to a mesenchymal phenotype by hypoxia (148). In keeping with the positive feedback cycle of EMP, the expression of AXL seems to be positively regulated by EMT TFs. For instance, in non-malignant human breast epithelial cells, stable expression of TWIST, ZEB2, SNAIL, or SLUG induced AXL expression along with an EMP phenotype (149). Thus, expression of the very inducers of EMP and tumor immune suppression, such as TGFβ, IL-8, and AXL, by malignant cells exhibiting EMP indicates their capacity to both sustain this EMP phenotype and further promote immune suppression.
Transcriptional Regulation of EMP-Mediated Immune Suppression
The EMT TFs SNAIL and ZEB1 play important roles in promoting tumor immune suppression. In murine and human melanoma cells, SNAIL transduction induced EMP, and these E/M-induced clones demonstrated immunosuppressive features both in cancer-immune cell co-culture experiments in vitro and experiments performed in vivo (150). In these hybrid E/M clones, SNAIL mediates the immunosuppressive function of Treg cells, resistance to CTL lysis, and decreases the function of DCs through TGFβ and thrombospondin 1 (TSP1) signaling (150). In an in vivo model of ovarian cancer, SNAIL was also found to have important immunomodulatory effects whereby SNAIL knockdown reduced tumor growth, increased CTL tumor infiltration, and decreased tumor-infiltrating MDSCs (77). Dongre et al. (2017) developed a model of EMP in mouse mammary tumor virus (MMTV) breast cancers using a SNAIL-driven yellow fluorescent protein (YFP) to identify SNAIL-high and SNAIL-low cells, which were labelled “quasi-mesenchymal” (akin to hybrid E/M) and epithelial, respectively (85). While the epithelial cells in this study were immunogenic, quasi-mesenchymal cells promoted an immunosuppressive TME associated with CTL exclusion from the tumor core, M2-like pro-tumor macrophage polarization, and decreased response to anti-CTLA-4 therapy. Interestingly, only a small percentage of quasi-mesenchymal cells (10%) in mixed tumors was needed to cross-protect the surrounding epithelial malignant cells from immune attack, suggesting a potent immunosuppressive effect of the hybrid E/M state. A subsequent study from the same group identified genes that were enriched in quasi-mesenchymal cells yet did not affect hybrid E/M status to identify downstream mediators of immune suppression in the hybrid E/M state (66). Among these genes, CD73, CSF1, secreted phosphoprotein 1 (SPP1), galectin-3, MBL associated serine protease 1 (MASP1), and CXCL12 all negatively regulated the number and function of CTLs (Figure 2). However, knockdown of only CD73, CSF1, or SPP1 could mobilize CTLs into the tumor core. Of these, CD73 was the only gene that upon knockdown led to tumor regression and decreased metastasis in anti-CTLA-4 therapy. This is one of the first studies establishing a causal role of the hybrid E/M state in immune modulation and resistance to ICB therapy (66).
Like SNAIL, ZEB1 has also been shown to confer immune resistance. In a K-Ras model of lung adenocarcinoma, ZEB1 expression specifically in invading cancer cells directly upregulated the expression of immune checkpoint proteins PD-L1 and CD47, a surface protein that promotes TAM polarization to a tumor-promoting phenotype (78). The resultant PD-L1 induction and CD47-dependent polarization of TAMs created a protective niche for invading cancer cells that was deficient in CTLs. This study demonstrates a direct link between EMP and immune resistance (78). Regulation of PD-L1 by ZEB1 was also found in an experiment implementing the K-ras LA1/+p53R172HΔg/+ (KP) mouse model of NSCLC that demonstrated ZEB1 increases PD-L1 expression by repressing miR-200, a miRNA that binds PD-L1 mRNA (125) (Figure 3).
Figure 3. The hybrid E/M state prevents immune recognition and anti-tumor cytotoxicity of cancer cells by CTLs.
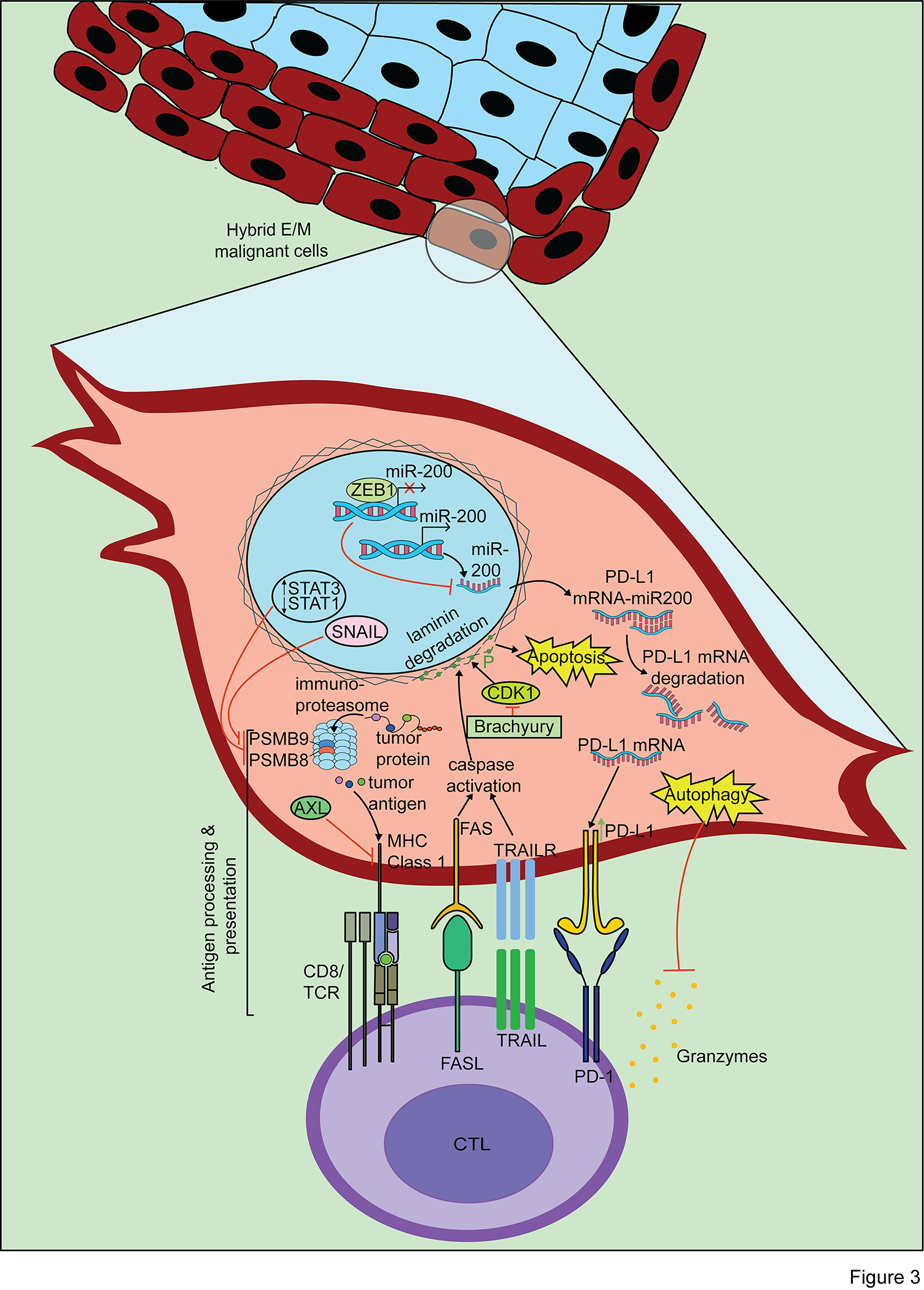
The hybrid E/M state prevents CTL recognition due to downregulation of the antigen presentation pathway. The tumor killing mechanisms of CTLs, including granzymes, FAS/FASL, and TRAIL/TRAILR, are blocked by hybrid E/M-associated signaling pathways including increased PD-L1 expression, autophagy induction, and inhibition of caspase-dependent cell death.
EMP and Macrophage Polarization
Macrophages play a key role in EMP, and in a mouse model of skin squamous cell carcinoma, macrophage depletion by anti-CSF1R and anti-CCL2 blocking antibodies decreased tumor formation and caused a shift of malignant cell states toward more epithelial-like phenotypes (135). Positive feedback loops between malignant cells and TAMs promoted the induction of EMP and ultimately facilitated intravasation. In a breast cancer mouse model, macrophages induced EMP via CCL18 while malignant cells with an EMP phenotype could induce macrophage secretion of CCL18 through GM-CSF along with environmental factors of the TME including lactate (62) (Figure 2). The expression of this GM-CSF and CCL18 positive feedback loop was associated with metastasis in mouse models and worse outcomes in patients. Similar to the GM-CSF and CCL18 positive feedback loop, macrophage-secreted IL-6 has been shown to modulate EMP through activating signal transducer and activator of transcription 3 (STAT3) signaling in cancer cells, which increases metastasis and macrophage recruitment through CCL2 (151) (Figure 2). In a separate study focusing on NSCLC, TAMs were found to create a pro-tumor niche, including induction of EMP and recruitment of Tregs in early tumorigenesis and seeding of a NSCLC cell line injected through the tail vein (152). These studies illustrate that the hybrid E/M phenotype promotes macrophage polarization into a phenotype that both sustains hybrid E/M phenotypes and the tumor-promoting macrophage phenotype itself, ultimately contributing to tumor progression.
Opposing Roles of NK Cells in EMP
Evidence for the role of EMP in regulating NK cells is controversial due to pleotropic, even conflicting, observations across studies. In melanoma cells, induction of EMP was found to increase resistance to NK cell cytotoxicity, which the authors attributed to increased surface expression of MHC class I – a suppressive signal to NK cells – along with decreased expression of ligands that activate NK cells (153) (Figure 2). Interestingly, no significant difference was observed in the surface expression of the NK cell activating ligand NKG2D upon EMP induction (153). Increased NK cell resistance was also shown in mesenchymal subclones from lung adenocarcinoma (compared to epithelial subclones) via sustained hypoxic stress of IGR-Heu cells established from primary NSCLC tumor samples (74). Increased resistance to NK cell cytotoxicity in the mesenchymal clones was associated with reduced tyrosine phosphorylation in contact areas of NK cells with mesenchymal subclones indicative of decreased NK cell activation. Hybrid E/M states facilitated this resistance – inhibiting EMP in the hypoxia-driven mesenchymal subclones increased their susceptibility to NK cell-mediated killing.
While some studies show decreased susceptibility of malignant cells to NK cell killing associated with EMP (153), others show increased susceptibility of malignant cells to NK cell cytotoxicity upon induction of EMP. For instance, in human lung cancer cell lines, TGFβ-mediated EMP induction triggered the initiation of metastasis yet increased susceptibility to NK cell-mediated killing by both a human NK cell line and NK cells isolated from healthy donors, which acted as a rate limiting step in EMP-driven metastasis (154). In this study, spontaneous metastasis of lung cancer cells was observed upon NK cell depletion in RAG1−/− mice, which lack T and B cells but retain functional NK cells. In addition to the findings in lung cancer, this study showed a similar effect of increased NK cell susceptibility in EMP-induced estrogen receptor positive breast cancer cell lines (154). A potential mechanism for these findings is that EMP induces the expression of the NK cell activating ligand cell adhesion molecule 1 (CADM1), while downregulating type 1 E-cadherin ligand that inhibits NK cells (154). Interestingly, while EMP induction in this study was associated with increased expression of NKG2D ligands, namely UL16 Binding Protein 1 (ULBP1) and ULBP4, blocking NKG2D on NK cells did not affect NK cell-mediated lysis. However, another study has shown that inhibition of the NKG2D receptor with blocking antibodies decreased NK cell cytotoxicity against EMP-induced malignant cells (155). The contrasting findings of these studies may be due to differences in disease models and/or heterogeneity of EMP across cancer types. Further investigation of EMP and NK cell interactions will be critical to resolve these seemingly contradictory results and might reveal context-specific cues for these observations.
Although prior studies have provided limited insight into how malignant cells may leverage EMP to manipulate the surrounding immune cells, a recent study explored the role of NK cell-mediated cytotoxicity towards CTCs in a single cell form compared to CTCs in clusters using an in vivo mammary tumor model (94). Interestingly, CTC clusters had higher expression of an epithelial-related gene set (specifically genes which are suppressed by ZEB1 during EMT) and CDH1-related genes (whose low expression indicates a downregulation of E-cadherin). These more epithelial-like CTC clusters showed relatively decreased susceptibility to NK cell-mediated cytotoxicity relative to CTCs travelling as a single cell, which provided a metastatic advantage to the CTC clusters. Perturbation of EMP by the ZEB1 inhibitory miRNA, miR-200c, led to a more epithelial-like phenotype in the mammary cancer cells accompanied by a switch from single-cell dissemination to a collective dissemination as CTC clusters. Increased susceptibility of single cell CTCs to NK cell cytotoxicity upon induction of EMP was dominantly mediated by the NKG2D signaling pathway (Figure 2). Thus, this study suggests that EMP may confer mesenchymal features at the primary site to promote invasion while subsequently inducing epithelial features in circulation within CTC clusters to protect from NK cell cytotoxicity.
EMP and the Antigen Presentation Pathway
Signaling events associated with EMP contribute to decreased MHC class I presentation and result in an immune evasion phenotype (Figure 3). Peptides presented on MHC class I are generated in the proteasome or the immunoproteasome, the latter of which is the primary producer of MHC class I binding peptides and is induced by interferon gamma (IFNγ) (156). The immunoproteasome is defined by inclusion of catalytic subunits PSMB8, PSMB9, and PSMB10 (157). Increased expression of PSMB8 and PSMB9 has been associated with improved patient survival and slower disease progression in several cancers, including NSCLC (64) and melanoma (156). In melanoma, PSMB8 and PSMB9 expression was the most predictive marker of a response to anti-CTLA-4 or anti-PD-1 therapy, more so than an IFNγ gene signature, mutational load, or CTL infiltration (156). A clinical association between EMP and the immunoproteasome has been described in patient-derived samples of NSCLC, in which an intermediate mesenchymal phenotype (akin to hybrid E/M) with dual expression of epithelial and mesenchymal markers showed decreased immunoproteasome expression (64) (Figure 3). Similarly, EMP induced by TGFβ in NSCLC cell lines was associated with decreased protein levels of PSMB8 and PSMB9 (64) (Figure 3). These mesenchymal-like, hybrid E/M NSCLC cell lines showed increased levels of phospho-STAT3 and decreased levels of phospho-STAT1 relative to epithelial-like NSCLC cell lines. STAT1 is activated by IFNγ and is a main promoter of NLR family CARD domain containing 5 (NLRC5) protein, a key promoter of genes in the MHC class I antigen presentation pathway including the immunoproteasome subunits (158,159). Thus, as expected, treatment of mesenchymal NSCLC cell lines with IFNγ increased phospho-STAT1 levels, immunoproteasome subunit expression, and lysis by CTLs (64). However, a pathway connecting EMP explicitly with STAT1 and NLRC5 in their regulation of antigen presentation has yet to be well defined, representing an opportunity to uncover new pharmacologic targets.
The EMT inducer AXL is implicated in downregulating MHC class I presentation genes in several cancer types, including NSCLC and melanoma (160) (Figure 3). AXL signaling mechanistically links EMP to an immune evasion phenotype. In the MMTV breast cancer model, a radiation therapy-resistant cell line was found to exhibit an EMP state along with increased AXL expression and decreased MHC class I expression relative to a radiation-sensitive cell line (161). AXL knockdown promoted a more epithelial state, restored MHC class I expression, and promoted tumor elimination in vivo (161). A role of EMP in decreased MHC class I expression has also been noted in melanoma tumor samples, in which SNAIL was the highest expressed gene in HLA-low tumors (162). In this study, 7 out of the 16 melanoma tumor samples with anti-PD-1 resistance had downregulation of MHC class I, which was associated with an increase in EMP and its associated gene expression and pathway activation, including AXL, SNAIL, and TGFβ signaling.
Using the MMTV breast cancer model and a SNAIL-YFP reporter in malignant cells, Dongre et al. also identified a link between the hybrid E/M phenotype, SNAIL, and the antigen presentation machinery (66). In this study, cells in the quasi-mesenchymal state (akin to hybrid E/M) had lower expression of PSMB9 and STAT1 (Figure 3). Additionally, relative to epithelial-like cells, the quasi-mesenchymal cells showed downregulation of proteins in the MHC class I antigen presentation pathway, including transporter associated with antigen processing 1 (TAP1), TAP binding protein (TABP), and immunoproteasome subunit PSMB9. A chromatin immunoprecipitation sequencing (CHIP-seq) assay identified that SNAIL binds DNA in proximity to these MHC class I presentation genes, suggesting SNAIL may inhibit transcription of these genes (66). Collectively, these studies suggest that a more mesenchymal phenotype is associated with decreased MHC class I antigen presentation, which mechanistically may be explained by aberrant AXL signaling, STAT1/STAT3 signaling, and SNAIL activity.
The Immunologic Synapse and EMP
The immunological synapse is an organized clustering of membrane-bound signaling and adhesive proteins between a T cell and any cell presenting an antigen, including malignant cells. Immunologic synapse disruptions have been observed in vitro between CTLs and mesenchymal malignant cells, namely decreased TCR signaling and reduced CTL killing of malignant cells with mesenchymal features (73–75). Adhesive proteins in the interaction of a CTL with an APC, including malignant cells, play an important role in effecting the tumor lysis capacity of CTLs. The interaction of CD103 (ITGAE) on CTLs with E-cadherin on malignant cells is one such adhesive interaction (Figure 4). The CD103-E-cadherin interaction mediates polarization of cytolytic granules and subsequent exocytosis to the surrounding cancer cells that mediates cancer cell lysis (163). CD103 expression is upregulated in CTLs by TGFβ signaling, and CD103+ CTLs have increased anti-cancer activity compared to CD103− CTLs as well as increased secretion of TGFβ in the TME, which self-sustains their CD103 expression (164). Interestingly, the CD103+ population of CTLs has been shown to include the vast majority of tumor-specific CTL clones in several malignancies (164,165). Moreover, CD103+ CTLs exhibit localization to cancer cells expressing E-cadherin in lung cancer samples, and in vitro CTL migration toward cancer cell lines expressing E-cadherin was inhibited by an anti-CD103 blocking antibody (164). It stands to reason that hybrid E/M malignant cells are uniquely suited to induce CD103 expression on CTLs due to their increased TGFβ expression and their localization to the tumor-stroma border, while their mesenchymal features prevent CTL-mediated lysis. Furthermore, sustained E-cadherin expression in a hybrid E/M state may promote recruitment of CD103+ CTLs to hybrid E/M cells at the tumor-stroma border that in turn contributes to the induction of a hybrid E/M state via CD103+ CTL secretion of TGFβ, a major inducer of EMP (7) (Figure 4). As immune cells are often excluded from the tumor core and positioned at the tumor-stroma interface adjacent to hybrid E/M malignant cells, investigation of adhesive interactions and associated signaling between hybrid E/M malignant cells and CTLs may reveal determinants of immune exclusion from the tumor core.
Figure 4. Potential regulation of the immunological synapse by hybrid E/M cells.
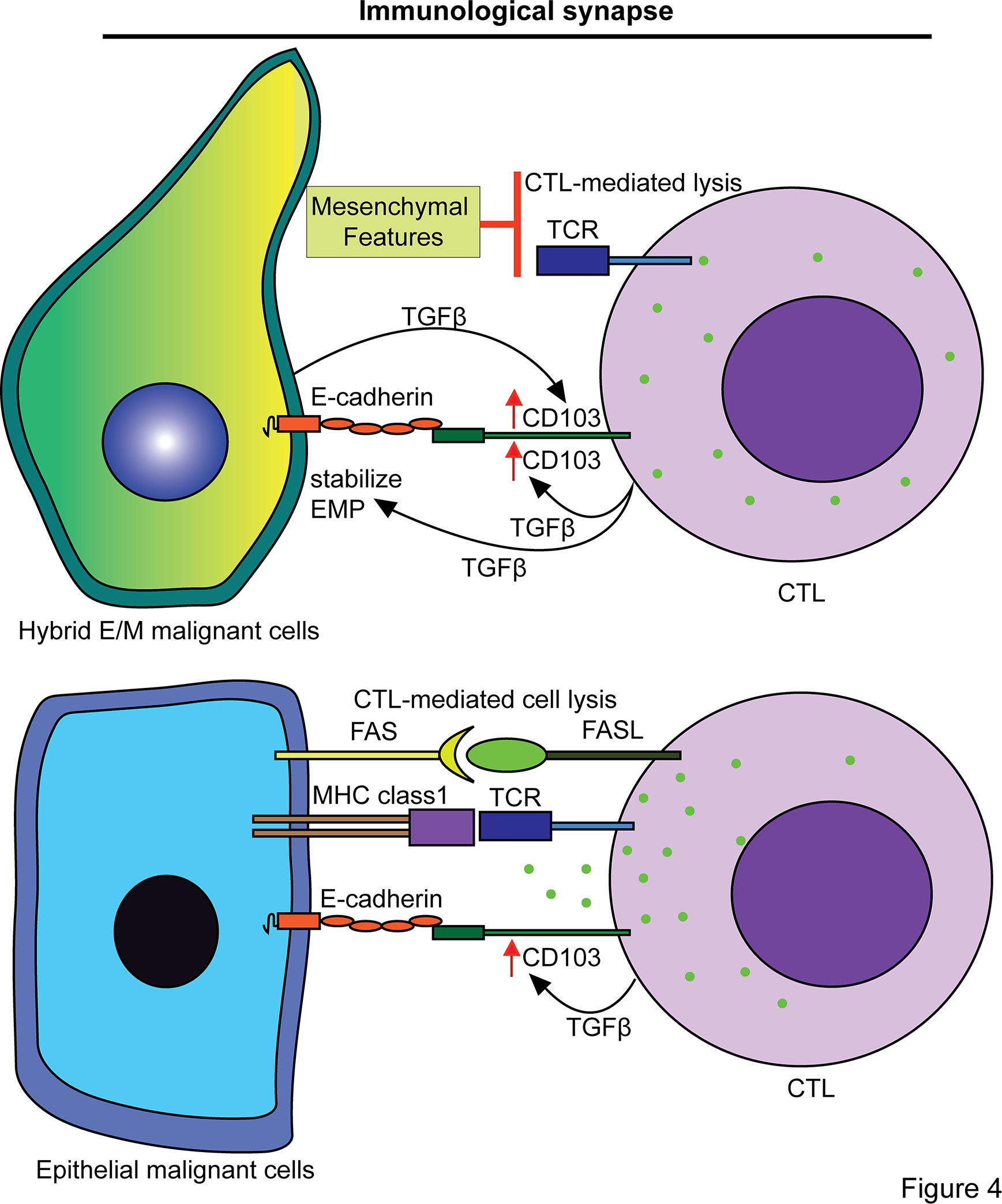
Hybrid E/M cells resist CTL-mediated cytotoxicity yet maintain adhesion proteins, such as E-cadherin, that are implemented in the immunological synapse with CD103+ CTLs, which secrete TGFβ and thereby may promote the hybrid E/M state.
EMP and its Modulation of CTL Motility
Structural proteins within the ECM may be specifically secreted or regulated by hybrid E/M malignant cells, representing an additional layer of interaction with the immune system. For example, laminin-332 is a structural protein enriched in the hybrid E/M state and serves as an archetypal example, contributing to decreased CTL migration and effector functions (166). This heterotrimeric protein consists of laminin α3, laminin β3, and laminin γ2 and can be secreted to the apical surface to promote migration via interactions with α3β1 and α6β1 integrin receptors located at the apical surface of cancer cells (167,168). Laminin-332 subunit enrichment at the leading edge has been observed in several cancer types, such as NSCLC, HCC, lung adenocarcinoma, and esophageal squamous cell carcinoma (27,166,169,170). Additionally, in three studies characterizing hybrid E/M states in primary samples or cancer cell lines, laminin-332 subunits were specifically enriched in the hybrid E/M state (27,35,171). Laminin γ2 expression can be induced by TGFβ through JNK/AP1 signaling (166). Importantly, the laminin γ2 subunit of laminin-332 has been shown to decrease T cell chemotaxis, downregulate TCR expression, and be a predictor of poor response to anti-PD-1 therapy in NSCLC and esophageal squamous cell carcinoma patients and cancer models (166). Although the precise effect of laminins expressed by cancer cells on immune cell functions is not well understood, given that immune cells express a variety of integrins capable of binding laminins (172), the laminin-immune interactions appear to be an important source of immune modulation by hybrid E/M malignant cells.
A major limitation in existing knowledge related to EMP and immune interactions is the use of approaches that merely capture a “snapshot” of in vitro CTL-hybrid E/M malignant cell interactions. In vivo studies of CTL-tumor interactions have found that CTLs remain motile while on a target cell, have heterogenous killing rates ranging from zero to twelve target cells per day, and that malignant cells are more likely to die with multiple CTL interactions (173–175). Thus, assessing CTL killing in vitro as a snapshot of functional synapses as has been done in EMP and CTL interaction studies to date does not capture the complex cellular dynamics essential to tumor elimination. Indeed, CTL migration and scanning for antigens across the tumor-stroma border is important for CTL-mediated lysis: A study of CD44-knockout CTLs showed that these CTLs were less efficient in tumor killing due to decreased ability to migrate across the tumor despite retaining antigen-TCR interactions (176). Given that CTL motility across a tumor is crucial for tumor elimination and hybrid E/M malignant cells are localized at the tumor edge, investigation into the relation of hybrid E/M states and CTL interactions may reveal insights into mechanisms of tumor immune evasion that are associated with EMP.
EMP Confers Resistance to CTL Cytotoxicity
Cancer cells that retain MHC class I expression and antigen presentation for recognition by CTLs have mechanisms related to EMP that may prevent CTL-mediated lysis, including resistance to caspase- or mitochondrial-dependent apoptosis and the induction of autophagy. The EMP-associated TFs brachyury and MUC1 mediate CTL resistance (67,68,177) (Figure 3). Death ligands expressed on CTLs, such as Fas ligand (FASL) and TNF-related apoptosis inducing ligand (TRAIL), bind death receptors on target cells to induce intracellular signaling pathways in the target cell that lead to cell death (178). CTL cytotoxicity is also mediated by perforins that perforate the membrane and serine proteases called granzymes that enter the target cell (178). Brachyury has been shown to prevent caspase-mediated nuclear laminin degradation through CDK1 in the death receptor/death ligand pathway of CTL cytotoxicity in pancreatic, breast, lung, and colon cancer cell lines (177) (Figure 3). In CRC cell lines, brachyury has been shown to stabilize MUC1 mRNA, which in turn conferred increased resistance to mitochondrial-induced apoptosis and perforin- and granzyme-mediated lysis (67) (Figure 3). While EMP can support an anti-apoptosis phenotype in malignant cells, the anti-apoptotic proteins can also, in turn, induce EMP. For instance, the anti-apoptotic Bcl-2 family of proteins are involved in the induction of E/M states in cancer while also functioning to prevent granzyme-mediated apoptosis in malignant cells (179–181). Additionally, though classically described as anti-tumor interactions, the FAS/FASL interaction and the TRAIL-R1 and -R2 receptors that are implemented in CTL tumor killing also play important roles in modulating EMP in malignant cells (182,183).
Autophagy is a mechanism of CTL resistance that acts by neutralizing granzymes as seen in breast cancer cell lines exposed to hypoxia, a known inducer of hybrid E/M states (184). The breast cancer cell line MCF-7 transfected with the EMP TF SNAIL displayed greater resistance to CTL lysis than the parental, epithelial-like MCF-7 cells due to autophagy induction (73). A role for autophagy induction in CTL resistance was also identified in a CRISPR screen using several mouse and cancer cell lines involving 19,069 protein-coding genes (185). In both the CRISPR screen and SNAIL transfection studies, pharmacologic inhibition of autophagy increased CTL-mediated lysis of cancer cells. While EMP and autophagy are associated with CTL resistance, the mechanistic links between these two processes and signaling pathways are still undetermined. For instance, while the induction of autophagy resists CTL-mediated lysis in hybrid E/M-induced MCF-7 cells, the induction of autophagy can also inhibit EMP through destabilization of EMT TFs (69). As the autophagy pathways involve stabilization or degradation of EMT TF proteins, we suspect there may be relevant post-translational interactions between the autophagy pathway and EMT TFs contributing to broader EMP regulation and CTL resistance.
Clinical Trials Coupling Immune Checkpoint Blockade with Agents Targeting EMP
As studies elucidate the critical signals mediating EMP and immune escape, there has been a corresponding increase in clinical trials addressing these pathways. Targeting EMP immune escape via inhibition of TGFβ, AXL, or IL-8 in combination with ICB is being evaluated in 88 clinical trials ranging from phase I to III in several solid cancer types. Most notably due the prominent role of TGFβ in EMP, 70 clinical trials – including phase I to phase III – feature TGFβ inhibition combined with ICB. These anti-TGFβ agents include small molecule inhibitors of TGFβ signaling including vactosertib (NCT03724851, NCT03732274, and NCT04064190) and LY3200882 (NCT02937272 and NCT04158700), the anti-PD-L1/TGFβ decoy receptor fusion proteins bintrafusp alfa (e.g., NCT04220775) and SHR-1701 (e.g., NCT04856774), and anti-TGFβ antibodies including BCA101 (NCT04429542), NIS793 (NCT04390763 and NCT02947165), and SAR439459 (NCT04524871). AXL inhibition combined with ICB is in 13 clinical trials ranging from phase I to phase III. AXL inhibitors include multi-tyrosine kinase inhibitors that inhibit AXL including cabozantinib (e.g., NCT04471428), INCB081776 (NCT03522142) and sitravatinib (NCT03941873 and NCT03666143), the AXL-specific inhibitor bemcentinib (NCT03654833 and NCT03184571), and the decoy receptor batiraxcept (NCT04004442 and NCT04019288) that binds the AXL ligand Gas6. Inhibitors of IL-8 being used in combination with ICB include the anti-IL-8 antibody BMS-986253 (NCT03689699 and NCT04572451) and SX-682 (NCT04599140, NCT04477343, and NCT03161431), which inhibits the IL-8 receptors CXCR1 and CXCR2.
The results of these clinical trials and mechanistic study of these combination therapies have yet to be completed. As such, the efficacy and mechanisms related to targeting the hybrid E/M cell-immune crosstalk from these combination therapies remain uncertain but will provide critical insight into the role of targeting EMP to improve ICB efficacy and patient outcomes. Given that even a small percentage (10%) of hybrid E/M cells can provide immune evasion to a tumor, identifying these mechanisms of immune evasion may reveal novel therapeutic targets to enhance ICB (85). Indeed, Dongre et al. (2021) found that CD73 expressed specifically in hybrid E/M cells supported tumor immune evasion, and upon knockdown, increased efficacy of ICB in a pre-clinical model of breast cancer (66). Future research that focuses on identifying hybrid E/M-specific mechanisms of tumor immune evasion may similarly define potential novel targets to increase ICB efficacy.
Conclusion
The study of EMP and immune interactions is an exciting field due to expanding evidence of a spectrum of hybrid E/M states with unique effects on the anti-tumor immune response and therapeutic interventions. The spatial localization of malignant cells with a hybrid E/M phenotype at the tumor-stroma interface and in CTCs physically positions hybrid E/M cells in proximity to immune cells. EMP confers resistance to cytotoxicity from the anti-tumor immune response, while capitalizing on immune cell plasticity to polarize immune cells to an immunosuppressive phenotype that further promotes EMP. Indeed, this arrangement supports both soluble and contact signaling in the crosstalk between EMP and immune cells, which in general leads to immune suppression. Moreover, the spatial proximity to immune cells and the immunomodulatory effects of hybrid E/M malignant cells indicates that targeting the hybrid E/M state may overcome tumor immune evasion, especially in combination with ICB. Many studies to date have focused on classical EMT pathways in immune evasion and have identified associations between EMT and immune resistance. The distinction between a complete EMT and a hybrid E/M state and their presence in patient tumors are not always clear in prior studies. We believe that future investigations focused on establishing the mechanistic, causal roles of more subtle and distinct hybrid E/M states and their comparison to full mesenchymal states on immune evasion will be critical for advancing our understanding of tumor immunology and improving cancer treatment.
Acknowledgements
This work was supported by V Foundation V Scholars Award, Cancer Research Institute Technology Impact Award, Cancer Research Foundation Young Investigator Award, Siteman Cancer Center and Barnes Jewish Foundation, American Cancer Society Institutional Research Grant, K08CA237732 (NIH/NCI), R21DE031072 (NIH/NIDCR), R21DE31366 (NIH/NIDCR) (S.V.P.) and T32DC000022 (NIH/NIDCD) (T.F.B.).
References
- 1.Lambert AW, Pattabiraman DR, Weinberg RA. Emerging Biological Principles of Metastasis. Cell. 2017. Feb 9;168(4):670–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steeg PS. Targeting metastasis. Nat Rev Cancer. 2016. Apr;16(4):201–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ganesh K, Massagué J. Targeting metastatic cancer. Nat Med. 2021. Jan;27(1):34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sharma P, Allison JP. Immune Checkpoint Targeting in Cancer Therapy: Toward Combination Strategies with Curative Potential. Cell. 2015. Apr 9;161(2):205–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haslam A, Prasad V. Estimation of the Percentage of US Patients With Cancer Who Are Eligible for and Respond to Checkpoint Inhibitor Immunotherapy Drugs. JAMA Netw Open. 2019. May 3;2(5):e192535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Soundararajan R, Fradette JJ, Konen JM, Moulder S, Zhang X, Gibbons DL, et al. Targeting the Interplay between Epithelial-to-Mesenchymal-Transition and the Immune System for Effective Immunotherapy. Cancers. 2019. May 24;11(5):714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dongre A, Weinberg RA. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. 2019. Feb;20(2):69–84. [DOI] [PubMed] [Google Scholar]
- 8.Yang J, Antin P, Berx G, Blanpain C, Brabletz T, Bronner M, et al. Guidelines and definitions for research on epithelial–mesenchymal transition. Nat Rev Mol Cell Biol. 2020. Jun;21(6):341–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nieto MA, Huang RY-J, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016. Jun 30;166(1):21–45. [DOI] [PubMed] [Google Scholar]
- 10.Cook DP, Vanderhyden BC. Transcriptional census of epithelial-mesenchymal plasticity in cancer. Sci Adv. 2022/01/05 ed. 2022. Jan 7;8(1):eabi7640–eabi7640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Puram SV, Parikh AS, Tirosh I. Single cell RNA-seq highlights a role for a partial EMT in head and neck cancer. Mol Cell Oncol. 2018;5(3):e1448244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greenburg G, Hay ED. Epithelia suspended in collagen gels can lose polarity and express characteristics of migrating mesenchymal cells. J Cell Biol. 1982. Oct;95(1):333–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thiery JP, Duband JL, Rutishauser U, Edelman GM. Cell adhesion molecules in early chicken embryogenesis. Proc Natl Acad Sci U S A. 1982. Nov;79(21):6737–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vallés AM, Boyer B, Badet J, Tucker GC, Barritault D, Thiery JP. Acidic fibroblast growth factor is a modulator of epithelial plasticity in a rat bladder carcinoma cell line. Proc Natl Acad Sci U S A. 1990. Feb;87(3):1124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Savagner P, Yamada KM, Thiery JP. The zinc-finger protein slug causes desmosome dissociation, an initial and necessary step for growth factor-induced epithelial-mesenchymal transition. J Cell Biol. 1997. Jun 16;137(6):1403–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sexén L III. Epithelial-mesenchymal interactions: 18th Hahnemann Symposium. R. Fleischmajer and R. E. Billingham eds. Williams and Wilkins Co., Baltimore. 326 pp. 1968. Teratology. 1970. Feb 1;3(1):100–1. [Google Scholar]
- 17.Pal A, Barrett TF, Paolini R, Parikh A, Puram SV. Partial EMT in head and neck cancer biology: a spectrum instead of a switch. Oncogene. 2021. Aug;40(32):5049–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y, Weinberg RA. Epithelial-to-mesenchymal transition in cancer: complexity and opportunities. Front Med. 2018. Aug;12(4):361–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lambert AW, Weinberg RA. Linking EMT programmes to normal and neoplastic epithelial stem cells. Nat Rev Cancer. 2021. May;21(5):325–38. [DOI] [PubMed] [Google Scholar]
- 20.Thiery JP. Epithelial–mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002. Jun;2(6):442–54. [DOI] [PubMed] [Google Scholar]
- 21.Jolly MK, Somarelli JA, Sheth M, Biddle A, Tripathi SC, Armstrong AJ, et al. Hybrid epithelial/mesenchymal phenotypes promote metastasis and therapy resistance across carcinomas. Pharmacol Ther. 2019. Feb 1;194:161–84. [DOI] [PubMed] [Google Scholar]
- 22.Parikh AS, Puram SV, Faquin WC, Richmon JD, Emerick KS, Deschler DG, et al. Immunohistochemical quantification of partial-EMT in oral cavity squamous cell carcinoma primary tumors is associated with nodal metastasis. Oral Oncol. 2019;99:104458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pastushenko I, Brisebarre A, Sifrim A, Fioramonti M, Revenco T, Boumahdi S, et al. Identification of the tumour transition states occurring during EMT. Nature. 2018. Apr;556(7702):463–8. [DOI] [PubMed] [Google Scholar]
- 24.Kröger C, Afeyan A, Mraz J, Eaton EN, Reinhardt F, Khodor YL, et al. Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proc Natl Acad Sci. 2019. Apr 9;116(15):7353–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bierie B, Pierce SE, Kroeger C, Stover DG, Pattabiraman DR, Thiru P, et al. Integrin-β4 identifies cancer stem cell-enriched populations of partially mesenchymal carcinoma cells. Proc Natl Acad Sci. 2017. Mar 21;114(12):E2337–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017. Oct;14(10):611–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell. 2017. Dec 14;171(7):1611–1624.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jolly MK, Murphy RJ, Bhatia S, Whitfield HJ, Redfern A, Davis MJ, et al. Measuring and Modelling the Epithelial- Mesenchymal Hybrid State in Cancer: Clinical Implications. Cells Tissues Organs. 2022;211(2):110–33. [DOI] [PubMed] [Google Scholar]
- 29.Hong T, Watanabe K, Ta CH, Villarreal-Ponce A, Nie Q, Dai X. An Ovol2-Zeb1 Mutual Inhibitory Circuit Governs Bidirectional and Multi-step Transition between Epithelial and Mesenchymal States. PLOS Comput Biol 2015. Nov 10;11(11):e1004569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jolly MK, Boareto M, Huang B, Jia D, Lu M, Ben-Jacob E, et al. Implications of the Hybrid Epithelial/Mesenchymal Phenotype in Metastasis. Front Oncol 2015;5:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jolly MK, Tripathi SC, Jia D, Mooney SM, Celiktas M, Hanash SM, et al. Stability of the hybrid epithelial/mesenchymal phenotype. Oncotarget. 2016. Mar 17;7(19):27067–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med. 2013. Nov;19(11):1438–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sahoo S, Nayak SP, Hari K, Purkait P, Mandal S, Kishore A, et al. Immunosuppressive Traits of the Hybrid Epithelial/Mesenchymal Phenotype. Front Immunol. 2021;12:797261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tyler M, Tirosh I. Decoupling epithelial-mesenchymal transitions from stromal profiles by integrative expression analysis. Nat Commun. 2021. May 10;12(1):2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ji AL, Rubin AJ, Thrane K, Jiang S, Reynolds DL, Meyers RM, et al. Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma. Cell. 2020. Jul 23;182(2):497–514.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Navas T, Kinders RJ, Lawrence SM, Ferry-Galow KV, Borgel S, Hollingshead MG, et al. Clinical Evolution of Epithelial–Mesenchymal Transition in Human Carcinomas. Cancer Res. 2020. Jan 15;80(2):304–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meyer SN, Galván JA, Zahnd S, Sokol L, Dawson H, Lugli A, et al. Co-expression of cytokeratin and vimentin in colorectal cancer highlights a subset of tumor buds and an atypical cancer-associated stroma. Hum Pathol. 2019. May 1;87:18–27. [DOI] [PubMed] [Google Scholar]
- 38.Qi Z, Barrett T, Parikh AS, Tirosh I, Puram SV. Single-cell sequencing and its applications in head and neck cancer. Oral Oncol. 2019. Dec 1;99:104441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kinker GS, Greenwald AC, Tal R, Orlova Z, Cuoco MS, McFarland JM, et al. Pan-cancer single-cell RNA-seq identifies recurring programs of cellular heterogeneity. Nat Genet. 2020. Nov;52(11):1208–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kohler I, Bronsert P, Timme S, Werner M, Brabletz T, Hopt UT, et al. Detailed analysis of epithelial-mesenchymal transition and tumor budding identifies predictors of long-term survival in pancreatic ductal adenocarcinoma. J Gastroenterol Hepatol. 2015. Mar;30 Suppl 1:78–84. [DOI] [PubMed] [Google Scholar]
- 41.Bronsert P, Enderle-Ammour K, Bader M, Timme S, Kuehs M, Csanadi A, et al. Cancer cell invasion and EMT marker expression: a three-dimensional study of the human cancer–host interface. J Pathol. 2014;234(3):410–22. [DOI] [PubMed] [Google Scholar]
- 42.Godin L, Balsat C, Van Eycke Y-R, Allard J, Royer C, Remmelink M, et al. A Novel Approach for Quantifying Cancer Cells Showing Hybrid Epithelial/Mesenchymal States in Large Series of Tissue Samples: Towards a New Prognostic Marker. Cancers. 2020. Apr 8;12(4):E906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Giannelli G, Bergamini C, Fransvea E, Sgarra C, Antonaci S. Laminin-5 With Transforming Growth Factor-β1 Induces Epithelial to Mesenchymal Transition in Hepatocellular Carcinoma. Gastroenterology. 2005. Nov 1;129(5):1375–83. [DOI] [PubMed] [Google Scholar]
- 44.Børretzen A, Gravdal K, Haukaas SA, Beisland C, Akslen LA, Halvorsen OJ. FOXC2 expression and epithelial–mesenchymal phenotypes are associated with castration resistance, metastasis and survival in prostate cancer. J Pathol Clin Res. 2019;5(4):272–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cook DP, Vanderhyden BC. Context specificity of the EMT transcriptional response. Nat Commun. 2020. May 1;11(1):2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gavish A, Tyler M, Simkin D, Kovarsky D, Gonzalez Castro LN, Halder D, et al. The transcriptional hallmarks of intra-tumor heterogeneity across a thousand tumors. bioRxiv. 2021. Jan 1;2021.12.19.473368. [Google Scholar]
- 47.De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013. Feb;13(2):97–110. [DOI] [PubMed] [Google Scholar]
- 48.Gonzalez DM, Medici D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci Signal. 2014. Sep 23;7(344):re8–re8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tripathi S, Levine H, Jolly MK. The Physics of Cellular Decision Making During Epithelial–Mesenchymal Transition. Annu Rev Biophys. 2020. May 6;49(1):1–18. [DOI] [PubMed] [Google Scholar]
- 50.McFaline-Figueroa JL, Hill AJ, Qiu X, Jackson D, Shendure J, Trapnell C. A pooled single-cell genetic screen identifies regulatory checkpoints in the continuum of the epithelial-to-mesenchymal transition. Nat Genet. 2019. Sep;51(9):1389–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial–mesenchymal transition. Nat Rev Mol Cell Biol. 2014. Mar;15(3):178–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xiong H-G, Li H, Xiao Y, Yang Q-C, Yang L-L, Chen L, et al. Long noncoding RNA MYOSLID promotes invasion and metastasis by modulating the partial epithelial-mesenchymal transition program in head and neck squamous cell carcinoma. J Exp Clin Cancer Res. 2019. Jun 25;38(1):278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Latil M, Nassar D, Beck B, Boumahdi S, Wang L, Brisebarre A, et al. Cell-Type-Specific Chromatin States Differentially Prime Squamous Cell Carcinoma Tumor-Initiating Cells for Epithelial to Mesenchymal Transition. Cell Stem Cell. 2017. Feb 2;20(2):191–204.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Park S-M, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008. Apr 1;22(7):894–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim NH, Kim HS, Li X-Y, Lee I, Choi H-S, Kang SE, et al. A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial-mesenchymal transition. J Cell Biol. 2011/10/24 ed. 2011. Oct 31;195(3):417–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schreiber RD, Old LJ, Smyth MJ. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science. 2011. Mar 25;331(6024):1565–70. [DOI] [PubMed] [Google Scholar]
- 57.Chen DS, Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity. 2013. Jul 25;39(1):1–10. [DOI] [PubMed] [Google Scholar]
- 58.O’Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. 2019. Mar;16(3):151–67. [DOI] [PubMed] [Google Scholar]
- 59.Qi Z, Liu Y, Mints M, Mullins R, Sample R, Law T, et al. Single-Cell Deconvolution of Head and Neck Squamous Cell Carcinoma. Cancers. 2021. Jan;13(6):1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Spaderna S, Schmalhofer O, Hlubek F, Berx G, Eger A, Merkel S, et al. A Transient, EMT-Linked Loss of Basement Membranes Indicates Metastasis and Poor Survival in Colorectal Cancer. Gastroenterology. 2006. Sep 1;131(3):830–40. [DOI] [PubMed] [Google Scholar]
- 61.Mahmood MQ, Ward C, Muller HK, Sohal SS, Walters EH. Epithelial mesenchymal transition (EMT) and non-small cell lung cancer (NSCLC): a mutual association with airway disease. Med Oncol 2017. Feb 14;34(3):45. [DOI] [PubMed] [Google Scholar]
- 62.Su S, Liu Q, Chen J, Chen J, Chen F, He C, et al. A Positive Feedback Loop between Mesenchymal-like Cancer Cells and Macrophages Is Essential to Breast Cancer Metastasis. Cancer Cell. 2014. May 12;25(5):605–20. [DOI] [PubMed] [Google Scholar]
- 63.Cheaito KA, Bahmad HF, Hadadeh O, Saleh E, Dagher C, Hammoud MS, et al. EMT Markers in Locally-Advanced Prostate Cancer: Predicting Recurrence? Front Oncol 2019. Mar 11;9:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tripathi SC, Peters HL, Taguchi A, Katayama H, Wang H, Momin A, et al. Immunoproteasome deficiency is a feature of non-small cell lung cancer with a mesenchymal phenotype and is associated with a poor outcome. Proc Natl Acad Sci U S A. 2016. Mar 15;113(11):E1555–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Antony J, Huang RY-J. AXL-Driven EMT State as a Targetable Conduit in Cancer. Cancer Res. 2017. Jul 15;77(14):3725–32. [DOI] [PubMed] [Google Scholar]
- 66.Dongre A, Rashidian M, Eaton EN, Reinhardt F, Thiru P, Zagorulya M, et al. Direct and Indirect Regulators of Epithelial–Mesenchymal Transition–Mediated Immunosuppression in Breast Carcinomas. Cancer Discov. 2021. May 1;11(5):1286–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.David JM, Hamilton DH, Palena C. MUC1 upregulation promotes immune resistance in tumor cells undergoing brachyury-mediated epithelial-mesenchymal transition. Oncoimmunology. 2016. Apr;5(4):e1117738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rajabi H, Kufe D. MUC1-C Oncoprotein Integrates a Program of EMT, Epigenetic Reprogramming and Immune Evasion in Human Carcinomas. Biochim Biophys Acta. 2017. Aug;1868(1):117–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gugnoni M, Sancisi V, Manzotti G, Gandolfi G, Ciarrocchi A. Autophagy and epithelial–mesenchymal transition: an intricate interplay in cancer. Cell Death Dis. 2016. Dec;7(12):e2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008. May;10(5):593–601. [DOI] [PubMed] [Google Scholar]
- 71.Noman MZ, Janji B, Abdou A, Hasmim M, Terry S, Tan TZ, et al. The immune checkpoint ligand PD-L1 is upregulated in EMT-activated human breast cancer cells by a mechanism involving ZEB-1 and miR-200. OncoImmunology. 2017. Jan 2;6(1):e1263412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn Y-H, Byers LA, et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun. 2014. Oct 28;5(1):5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Akalay I, Janji B, Hasmim M, Noman MZ, André F, Cremoux PD, et al. Epithelial-to-Mesenchymal Transition and Autophagy Induction in Breast Carcinoma Promote Escape from T-cell–Mediated Lysis. Cancer Res. 2013. Apr 15;73(8):2418–27. [DOI] [PubMed] [Google Scholar]
- 74.Terry S, Buart S, Tan TZ, Gros G, Noman MZ, Lorens JB, et al. Acquisition of tumor cell phenotypic diversity along the EMT spectrum under hypoxic pressure: Consequences on susceptibility to cell-mediated cytotoxicity. OncoImmunology. 2017. Feb 1;6(2):e1271858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Akalay I, Tan TZ, Kumar P, Janji B, Mami-Chouaib F, Charpy C, et al. Targeting WNT1-inducible signaling pathway protein 2 alters human breast cancer cell susceptibility to specific lysis through regulation of KLF-4 and miR-7 expression. Oncogene. 2015. Apr;34(17):2261–71. [DOI] [PubMed] [Google Scholar]
- 76.Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018. Feb;554(7693):544–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Taki M, Abiko K, Baba T, Hamanishi J, Yamaguchi K, Murakami R, et al. Snail promotes ovarian cancer progression by recruiting myeloid-derived suppressor cells via CXCR2 ligand upregulation. Nat Commun. 2018. Apr 27;9(1):1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Guo Y, Lu X, Chen Y, Rendon B, Mitchell RA, Cuatrecasas M, et al. Zeb1 induces immune checkpoints to form an immunosuppressive envelope around invading cancer cells. Sci Adv. 2021. May 21;7(21):eabd7455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Picarda E, Ohaegbulam KC, Zang X. Molecular Pathways: Targeting B7-H3 (CD276) for Human Cancer Immunotherapy. Clin Cancer Res. 2016. Jul 15;22(14):3425–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Alfaro C, Teijeira A, Oñate C, Pérez G, Sanmamed MF, Andueza MP, et al. Tumor-Produced Interleukin-8 Attracts Human Myeloid-Derived Suppressor Cells and Elicits Extrusion of Neutrophil Extracellular Traps (NETs). Clin Cancer Res. 2016. Aug 1;22(15):3924–36. [DOI] [PubMed] [Google Scholar]
- 81.Tobin RP, Jordan KR, Kapoor P, Spongberg E, Davis D, Vorwald VM, et al. IL-6 and IL-8 Are Linked With Myeloid-Derived Suppressor Cell Accumulation and Correlate With Poor Clinical Outcomes in Melanoma Patients. Front Oncol. 2019;9:1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017. Jul;14(7):399–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Condamine T, Ramachandran I, Youn J-I, Gabrilovich DI. Regulation of Tumor Metastasis by Myeloid-derived Suppressor Cells. Annu Rev Med 2015. Jan 14;66:97–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chae YK, Chang S, Ko T, Anker J, Agte S, Iams W, et al. Epithelial-mesenchymal transition (EMT) signature is inversely associated with T-cell infiltration in non-small cell lung cancer (NSCLC). Sci Rep. 2018. Feb 13;8(1):2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dongre A, Rashidian M, Reinhardt F, Bagnato A, Keckesova Z, Ploegh HL, et al. Epithelial-to-Mesenchymal Transition Contributes to Immunosuppression in Breast Carcinomas. Cancer Res. 2017. Aug 1;77(15):3982–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bagati A, Kumar S, Jiang P, Pyrdol J, Zou AE, Godicelj A, et al. Integrin αvβ6–TGFβ–SOX4 Pathway Drives Immune Evasion in Triple-Negative Breast Cancer. Cancer Cell. 2021. Jan 11;39(1):54–67.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Aggarwal V, Montoya CA, Donnenberg VS, Sant S. Interplay between tumor microenvironment and partial EMT as the driver of tumor progression. iScience. 2021. Feb 19;24(2):102113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Singh P, Banerjee R, Piao S, Costa de Medeiros M, Bellile E, Liu M, et al. Squamous cell carcinoma subverts adjacent histologically normal epithelium to promote lateral invasion. J Exp Med. 2021. Jun 7;218(6):e20200944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science. 2013. Feb 1;339(6119):580–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lecharpentier A, Vielh P, Perez-Moreno P, Planchard D, Soria JC, Farace F. Detection of circulating tumour cells with a hybrid (epithelial/mesenchymal) phenotype in patients with metastatic non-small cell lung cancer. Br J Cancer. 2011. Oct 25;105(9):1338–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Armstrong AJ, Marengo MS, Oltean S, Kemeny G, Bitting RL, Turnbull JD, et al. Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. Mol Cancer Res MCR. 2011. Aug;9(8):997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhao R, Cai Z, Li S, Cheng Y, Gao H, Liu F, et al. Expression and clinical relevance of epithelial and mesenchymal markers in circulating tumor cells from colorectal cancer. Oncotarget. 2017. Feb 7;8(6):9293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li T-T, Liu H, Li F-P, Hu Y-F, Mou T-Y, Lin T, et al. Evaluation of epithelial-mesenchymal transitioned circulating tumor cells in patients with resectable gastric cancer: Relevance to therapy response. World J Gastroenterol. 2015. Dec 21;21(47):13259–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lo HC, Xu Z, Kim IS, Pingel B, Aguirre S, Kodali S, et al. Resistance to natural killer cell immunosurveillance confers a selective advantage to polyclonal metastasis. Nat Cancer. 2020. Jul;1(7):709–22. [DOI] [PubMed] [Google Scholar]
- 95.Labelle M, Begum S, Hynes RO. Direct Signaling Between Platelets and Cancer Cells Induces an Epithelial-Mesenchymal-Like Transition and Promotes Metastasis. Cancer Cell. 2011. Nov 15;20(5):576–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Aiello NM, Maddipati R, Norgard RJ, Balli D, Li J, Yuan S, et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev Cell. 2018. Jun 18;45(6):681–695.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.del Pozo Martin Y, Park D, Ramachandran A, Ombrato L, Calvo F, Chakravarty P, et al. Mesenchymal Cancer Cell-Stroma Crosstalk Promotes Niche Activation, Epithelial Reversion, and Metastatic Colonization. Cell Rep 2015. Dec 6;13(11):2456–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Brabletz T, Hlubek F, Spaderna S, Schmalhofer O, Hiendlmeyer E, Jung A, et al. Invasion and Metastasis in Colorectal Cancer: Epithelial-Mesenchymal Transition, Mesenchymal-Epithelial Transition, Stem Cells and β-Catenin. Cells Tissues Organs 2005;179(1–2):56–65. [DOI] [PubMed] [Google Scholar]
- 99.Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell. 2012. Dec 11;22(6):725–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ocaña OH, Córcoles R, Fabra Á, Moreno-Bueno G, Acloque H, Vega S, et al. Metastatic Colonization Requires the Repression of the Epithelial-Mesenchymal Transition Inducer Prrx1. Cancer Cell. 2012. Dec 11;22(6):709–24. [DOI] [PubMed] [Google Scholar]
- 101.Brabletz T. To differentiate or not — routes towards metastasis. Nat Rev Cancer. 2012. Jun 1;12(6):425–36. [DOI] [PubMed] [Google Scholar]
- 102.Watanabe K, Villarreal-Ponce A, Sun P, Salmans ML, Fallahi M, Andersen B, et al. Mammary Morphogenesis and Regeneration Require the Inhibition of EMT at Terminal End Buds by Ovol2 Transcriptional Repressor. Dev Cell. 2014. Apr 14;29(1):59–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Roca H, Hernandez J, Weidner S, McEachin RC, Fuller D, Sud S, et al. Transcription Factors OVOL1 and OVOL2 Induce the Mesenchymal to Epithelial Transition in Human Cancer. PLoS ONE. 2013. Oct 4;8(10):e76773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Celià-Terrassa T, Bastian C, Liu DD, Ell B, Aiello NM, Wei Y, et al. Hysteresis control of epithelial-mesenchymal transition dynamics conveys a distinct program with enhanced metastatic ability. Nat Commun. 2018. Nov 27;9(1):5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ouzounova M, Lee E, Piranlioglu R, El Andaloussi A, Kolhe R, Demirci MF, et al. Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nat Commun. 2017. Apr 6;8:14979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dalerba P, Cho RW, Clarke MF. Cancer Stem Cells: Models and Concepts. Annu Rev Med. 2007. Feb 1;58(1):267–84. [DOI] [PubMed] [Google Scholar]
- 107.Prager BC, Xie Q, Bao S, Rich JN. Cancer Stem Cells: The Architects of the Tumor Ecosystem. Cell Stem Cell. 2019. Jan 3;24(1):41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. 2017. Oct;23(10):1124–34. [DOI] [PubMed] [Google Scholar]
- 109.Scheel C, Weinberg RA. Cancer stem cells and epithelial–mesenchymal transition: Concepts and molecular links. Semin Cancer Biol. 2012. Oct;22(5–6):396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ruscetti M, Quach B, Dadashian EL, Mulholland DJ, Wu H. Tracking and Functional Characterization of Epithelial–Mesenchymal Transition and Mesenchymal Tumor Cells during Prostate Cancer Metastasis. Cancer Res. 2015. Jul 1;75(13):2749–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mani SA, Guo W, Liao M-J, Eaton ENg, Ayyanan A, Zhou AY, et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell. 2008. May 16;133(4):704–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Morel A-P, Lièvre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of Breast Cancer Stem Cells through Epithelial-Mesenchymal Transition. PLOS ONE. 2008. Aug 6;3(8):e2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Maccalli C, Volontè A, Cimminiello C, Parmiani G. Immunology of cancer stem cells in solid tumours. A review. Eur J Cancer. 2014. Feb 1;50(3):649–55. [DOI] [PubMed] [Google Scholar]
- 114.Heft Neal ME, Brenner JC, Prince MEP, Chinn SB. Advancement in Cancer Stem Cell Biology and Precision Medicine-Review Article Head and Neck Cancer Stem Cell Plasticity and the Tumor Microenvironment. Front Cell Dev Biol. 2021;9:660210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Schatton T, Schütte U, Frank NY, Zhan Q, Hoerning A, Robles SC, et al. Modulation of T-Cell Activation by Malignant Melanoma Initiating Cells. Cancer Res. 2010. Jan 15;70(2):697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Malladi S, Macalinao DG, Jin X, He L, Basnet H, Zou Y, et al. Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT. Cell. 2016. Mar 24;165(1):45–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nappo G, Handle F, Santer FR, McNeill RV, Seed RI, Collins AT, et al. The immunosuppressive cytokine interleukin-4 increases the clonogenic potential of prostate stem-like cells by activation of STAT6 signalling. Oncogenesis. 2017. May;6(5):e342–e342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol. 2015. Feb;17(2):170–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Schatton T, Frank MH. Antitumor Immunity and Cancer Stem Cells. Ann N Y Acad Sci. 2009;1176(1):154–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fan Q-M, Jing Y-Y, Yu G-F, Kou X-R, Ye F, Gao L, et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial–mesenchymal transition in hepatocellular carcinoma. Cancer Lett. 2014. Oct 1;352(2):160–8. [DOI] [PubMed] [Google Scholar]
- 121.Jinushi M, Chiba S, Yoshiyama H, Masutomi K, Kinoshita I, Dosaka-Akita H, et al. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc Natl Acad Sci. 2011. Jul 26;108(30):12425–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Malta TM, Sokolov A, Gentles AJ, Burzykowski T, Poisson L, Weinstein JN, et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell. 2018. Apr 5;173(2):338–354.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Oki E, Saeki H, Tsutsumi S, Nakashima Y, Nakanishi R, Sugiyama M, et al. Association of epithelial-mesenchymal transition with an immunosuppressive tumor microenvironment with elevated levels of PD-L1 in esophageal carcinoma. J Clin Oncol. 2017. May 20;35(15_suppl):e15585–e15585. [Google Scholar]
- 124.Romeo E, Caserta CA, Rumio C, Marcucci F. The Vicious Cross-Talk between Tumor Cells with an EMT Phenotype and Cells of the Immune System. Cells. 2019. May 15;8(5):460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn Y-H, Byers LA, et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun. 2014;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2016. Mar 24;165(1):35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Raimondi C, Carpino G, Nicolazzo C, Gradilone A, Gianni W, Gelibter A, et al. PD-L1 and epithelial-mesenchymal transition in circulating tumor cells from non-small cell lung cancer patients: A molecular shield to evade immune system? OncoImmunology. 2017. Dec 2;6(12):e1315488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.David CJ, Massagué J. Contextual determinants of TGFβ action in development, immunity and cancer. Nat Rev Mol Cell Biol. 2018. Jul;19(7):419–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Cano A, Pérez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, et al. The transcription factor Snail controls epithelial–mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000. Feb;2(2):76–83. [DOI] [PubMed] [Google Scholar]
- 130.Eger A, Aigner K, Sonderegger S, Dampier B, Oehler S, Schreiber M, et al. DeltaEF1 is a transcriptional repressor of E-cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene. 2005. Mar;24(14):2375–85. [DOI] [PubMed] [Google Scholar]
- 131.Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, et al. The Two-Handed E Box Binding Zinc Finger Protein SIP1 Downregulates E-Cadherin and Induces Invasion. Mol Cell. 2001. Jun 1;7(6):1267–78. [DOI] [PubMed] [Google Scholar]
- 132.Thuault S, Valcourt U, Petersen M, Manfioletti G, Heldin C-H, Moustakas A. Transforming growth factor-β employs HMGA2 to elicit epithelial–mesenchymal transition. J Cell Biol. 2006. Jul 10;174(2):175–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Shirakihara T, Saitoh M, Miyazono K. Differential Regulation of Epithelial and Mesenchymal Markers by δEF1 Proteins in Epithelial–Mesenchymal Transition Induced by TGF-β. Mol Biol Cell. 2007. Sep 1;18(9):3533–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Shrestha R, Bridle KR, Crawford DHG, Jayachandran A. Immune checkpoint molecules are regulated by transforming growth factor (TGF)-β1-induced epithelial-to-mesenchymal transition in hepatocellular carcinoma. Int J Med Sci 2021. Apr 22;18(12):2466–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Xu D, Li J, Li R-Y, Lan T, Xiao C, Gong P. PD-L1 Expression Is Regulated By NF-κB During EMT Signaling In Gastric Carcinoma. OncoTargets Ther 2019. Nov 25;12:10099–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kumar M, Allison DF, Baranova NN, Wamsley JJ, Katz AJ, Bekiranov S, et al. NF-κB Regulates Mesenchymal Transition for the Induction of Non-Small Cell Lung Cancer Initiating Cells. PLOS ONE. 2013. Jul 30;8(7):e68597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Nomura A, Majumder K, Giri B, Dauer P, Dudeja V, Roy S, et al. Inhibition of NF-kappa B pathway leads to deregulation of epithelial–mesenchymal transition and neural invasion in pancreatic cancer. Lab Invest. 2016. Dec;96(12):1268–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.David JM, Dominguez C, Hamilton DH, Palena C. The IL-8/IL-8R Axis: A Double Agent in Tumor Immune Resistance. Vaccines. 2016. Sep;4(3):22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Li K, Shi H, Zhang B, Ou X, Ma Q, Chen Y, et al. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduct Target Ther. 2021. Oct 7;6(1):1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bates RC, DeLeo MJ, Mercurio AM. The epithelial-mesenchymal transition of colon carcinoma involves expression of IL-8 and CXCR-1-mediated chemotaxis. Exp Cell Res. 2004. Oct 1;299(2):315–24. [DOI] [PubMed] [Google Scholar]
- 141.Suarez-Carmona M, Bourcy M, Lesage J, Leroi N, Syne L, Blacher S, et al. Soluble factors regulated by epithelial-mesenchymal transition mediate tumour angiogenesis and myeloid cell recruitment. J Pathol. 2015. Aug;236(4):491–504. [DOI] [PubMed] [Google Scholar]
- 142.Yanagawa J, Walser TC, Zhu LX, Hong L, Fishbein MC, Mah V, et al. Snail promotes CXCR2 ligand dependent tumor progression in NSCLC. Clin Cancer Res Off J Am Assoc Cancer Res. 2009. Nov 15;15(22):6820–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hwang W, Yang M, Tsai M, Lan H, Su S, Chang S, et al. SNAIL Regulates Interleukin-8 Expression, Stem Cell–Like Activity, and Tumorigenicity of Human Colorectal Carcinoma Cells. Gastroenterology. 2011. Jul 1;141(1):279–291.e5. [DOI] [PubMed] [Google Scholar]
- 144.Fernando RI, Castillo MD, Litzinger M, Hamilton DH, Palena C. IL-8 signaling plays a critical role in the epithelial-mesenchymal transition of human carcinoma cells. Cancer Res. 2011. Aug 1;71(15):5296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Goyette M-A, Duhamel S, Aubert L, Pelletier A, Savage P, Thibault M-P, et al. The Receptor Tyrosine Kinase AXL Is Required at Multiple Steps of the Metastatic Cascade during HER2-Positive Breast Cancer Progression. Cell Rep 2018. May 1;23(5):1476–90. [DOI] [PubMed] [Google Scholar]
- 146.Zhang G, Kong X, Wang M, Zhao H, Han S, Hu R, et al. AXL is a marker for epithelial-mesenchymal transition in esophageal squamous cell carcinoma. Oncol Lett. 2018. Feb;15(2):1900–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Koorstra J-BM, Karikari CA, Feldmann G, Bisht S, Rojas PL, Offerhaus GJA, et al. The Axl receptor tyrosine kinase confers an adverse prognostic influence in pancreatic cancer and represents a new therapeutic target. Cancer Biol Ther. 2009. Apr;8(7):618–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Terry S, Abdou A, Engelsen AST, Buart S, Dessen P, Corgnac S, et al. AXL Targeting Overcomes Human Lung Cancer Cell Resistance to NK- and CTL-Mediated Cytotoxicity. Cancer Immunol Res. 2019. Nov 1;7(11):1789–802. [DOI] [PubMed] [Google Scholar]
- 149.Gjerdrum C, Tiron C, Høiby T, Stefansson I, Haugen H, Sandal T, et al. Axl is an essential epithelial-to-mesenchymal transition-induced regulator of breast cancer metastasis and patient survival. Proc Natl Acad Sci. 2010. Jan 19;107(3):1124–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kudo-Saito C, Shirako H, Takeuchi T, Kawakami Y. Cancer Metastasis Is Accelerated through Immunosuppression during Snail-Induced EMT of Cancer Cells. Cancer Cell. 2009. Mar 3;15(3):195–206. [DOI] [PubMed] [Google Scholar]
- 151.Wei C, Yang C, Wang S, Shi D, Zhang C, Lin X, et al. Crosstalk between cancer cells and tumor associated macrophages is required for mesenchymal circulating tumor cell-mediated colorectal cancer metastasis. Mol Cancer. 2019. Mar 30;18(1):64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Casanova-Acebes M, Dalla E, Leader AM, LeBerichel J, Nikolic J, Morales BM, et al. Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nature. 2021. Jul;595(7868):578–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Huergo-Zapico L, Parodi M, Cantoni C, Lavarello C, Fernández-Martínez JL, Petretto A, et al. NK-cell Editing Mediates Epithelial-to-Mesenchymal Transition via Phenotypic and Proteomic Changes in Melanoma Cell Lines. Cancer Res. 2018. Jul 15;78(14):3913–25. [DOI] [PubMed] [Google Scholar]
- 154.Chockley PJ, Chen J, Chen G, Beer DG, Standiford TJ, Keshamouni VG. Epithelial-mesenchymal transition leads to NK cell–mediated metastasis-specific immunosurveillance in lung cancer. J Clin Invest. 2021. Nov 5;128(4):1384–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.López-Soto A, Huergo-Zapico L, Galván JA, Rodrigo L, Herreros AG de, Astudillo A, et al. Epithelial–Mesenchymal Transition Induces an Antitumor Immune Response Mediated by NKG2 D Receptor. J Immunol. 2013. Apr 15;190(8):4408–19. [DOI] [PubMed] [Google Scholar]
- 156.Kalaora S, Lee JS, Barnea E, Levy R, Greenberg P, Alon M, et al. Immunoproteasome expression is associated with better prognosis and response to checkpoint therapies in melanoma. Nat Commun. 2020. Feb 14;11(1):896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ferrington DA, Gregerson DS. Immunoproteasomes: Structure, Function, and Antigen Presentation. Prog Mol Biol Transl Sci. 2012;109:75–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kobayashi KS, van den Elsen PJ. NLRC5: a key regulator of MHC class I-dependent immune responses. Nat Rev Immunol. 2012. Dec;12(12):813–20. [DOI] [PubMed] [Google Scholar]
- 159.Yoshihama S, Roszik J, Downs I, Meissner TB, Vijayan S, Chapuy B, et al. NLRC5/MHC class I transactivator is a target for immune evasion in cancer. Proc Natl Acad Sci. 2016. May 24;113(21):5999–6004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Aguilera TA, Giaccia AJ. Molecular Pathways: Oncologic Pathways and Their Role in T-cell Exclusion and Immune Evasion—A New Role for the AXL Receptor Tyrosine Kinase. Clin Cancer Res. 2017. Jun 15;23(12):2928–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Aguilera TA, Rafat M, Castellini L, Shehade H, Kariolis MS, Hui AB-Y, et al. Reprogramming the immunological microenvironment through radiation and targeting Axl. Nat Commun. 2016. Dec 23;7(1):13898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lee JH, Shklovskaya E, Lim SY, Carlino MS, Menzies AM, Stewart A, et al. Transcriptional downregulation of MHC class I and melanoma de- differentiation in resistance to PD-1 inhibition. Nat Commun. 2020. Apr 20;11(1):1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Floc’h AL, Jalil A, Vergnon I, Chansac BLM, Lazar V, Bismuth G, et al. αEβ7 integrin interaction with E-cadherin promotes antitumor CTL activity by triggering lytic granule polarization and exocytosis. J Exp Med. 2007. Feb 26;204(3):559–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hamid MA, Colin-York H, Khalid-Alham N, Browne M, Cerundolo L, Chen J-L, et al. Self-Maintaining CD103+ Cancer-Specific T Cells Are Highly Energetic with Rapid Cytotoxic and Effector Responses. Cancer Immunol Res. 2020. Feb 1;8(2):203–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Duhen T, Duhen R, Montler R, Moses J, Moudgil T, de Miranda NF, et al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat Commun. 2018. Jul 13;9(1):2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Li L, Wei J-R, Dong J, Lin Q-G, Tang H, Jia Y-X, et al. Laminin γ2–mediating T cell exclusion attenuates response to anti–PD-1 therapy. Sci Adv. 2021. Feb 1;7(6):eabc8346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kariya Y, Miyazaki K. The basement membrane protein laminin-5 acts as a soluble cell motility factor. Exp Cell Res. 2004. Jul 15;297(2):508–20. [DOI] [PubMed] [Google Scholar]
- 168.Margadant C, Raymond K, Kreft M, Sachs N, Janssen H, Sonnenberg A. Integrin α3β1 inhibits directional migration and wound re-epithelialization in the skin. J Cell Sci. 2009. Jan 15;122(2):278–88. [DOI] [PubMed] [Google Scholar]
- 169.Yasuda H, Nakagawa M, Kiyokawa H, Yoshida E, Yoshimura T, Koshikawa N, et al. Unique Biological Activity and Potential Role of Monomeric Laminin-γ2 as a Novel Biomarker for Hepatocellular Carcinoma: A Review. Int J Mol Sci. 2019. Jan;20(1):226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Moon YW, Rao G, Kim JJ, Shim H-S, Park K-S, An SS, et al. LAMC2 enhances the metastatic potential of lung adenocarcinoma. Cell Death Differ. 2015. Aug;22(8):1341–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Deshmukh AP, Vasaikar SV, Tomczak K, Tripathi S, den Hollander P, Arslan E, et al. Identification of EMT signaling cross-talk and gene regulatory networks by single-cell RNA sequencing. Proc Natl Acad Sci. 2021. May 11;118(19):e2102050118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Simon T, Bromberg JS. Regulation of the Immune System by Laminins. Trends Immunol. 2017. Nov;38(11):858–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Halle S, Halle O, Förster R. Mechanisms and Dynamics of T Cell-Mediated Cytotoxicity In Vivo. Trends Immunol. 2017. Jun 1;38(6):432–43. [DOI] [PubMed] [Google Scholar]
- 174.Purbhoo MA, Irvine DJ, Huppa JB, Davis MM. T cell killing does not require the formation of a stable mature immunological synapse. Nat Immunol. 2004. May;5(5):524–30. [DOI] [PubMed] [Google Scholar]
- 175.Vasconcelos Z, Müller S, Guipouy D, Yu W, Christophe C, Gadat S, et al. Individual Human Cytotoxic T Lymphocytes Exhibit Intraclonal Heterogeneity during Sustained Killing. Cell Rep. 2015. Jun 9;11(9):1474–85. [DOI] [PubMed] [Google Scholar]
- 176.Mrass P, Kinjyo I, Ng LG, Reiner SL, Puré E, Weninger W. CD44 Mediates Successful Interstitial Navigation by Killer T Cells and Enables Efficient Antitumor Immunity. Immunity. 2008. Dec 19;29(6):971–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Hamilton DH, Huang B, Fernando RI, Tsang K-Y, Palena C. WEE1 Inhibition Alleviates Resistance to Immune Attack of Tumor Cells Undergoing Epithelial–Mesenchymal Transition. Cancer Res. 2014. May 1;74(9):2510–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Martínez-Lostao L, Anel A, Pardo J. How Do Cytotoxic Lymphocytes Kill Cancer Cells? Clin Cancer Res. 2015. Nov 15;21(22):5047–56. [DOI] [PubMed] [Google Scholar]
- 179.Zuo J, Ishikawa T, Boutros S, Xiao Z, Humtsoe JO, Kramer RH. Bcl-2 Overexpression Induces a Partial Epithelial to Mesenchymal Transition and Promotes Squamous Carcinoma Cell Invasion and Metastasis. Mol Cancer Res. 2010. Feb 1;8(2):170–82. [DOI] [PubMed] [Google Scholar]
- 180.Voskoboinik I, Whisstock JC, Trapani JA. Perforin and granzymes: function, dysfunction and human pathology. Nat Rev Immunol. 2015. Jun;15(6):388–400. [DOI] [PubMed] [Google Scholar]
- 181.Wang Y, Xiao H, Wang C, Wu H, He H, Yao C, et al. M-phase phosphoprotein 8 promotes gastric cancer growth and metastasis via p53/Bcl-2 and EMT-related signaling pathways. J Cell Biochem. 2020. Mar;121(3):2330–42. [DOI] [PubMed] [Google Scholar]
- 182.Peyre L, Meyer M, Hofman P, Roux J. TRAIL receptor-induced features of epithelial-to-mesenchymal transition increase tumour phenotypic heterogeneity: potential cell survival mechanisms. Br J Cancer. 2021. Jan;124(1):91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Peter ME, Hadji A, Murmann AE, Brockway S, Putzbach W, Pattanayak A, et al. The role of CD95 and CD95 ligand in cancer. Cell Death Differ. 2015. Apr;22(4):549–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Baginska J, Viry E, Berchem G, Poli A, Noman MZ, van Moer K, et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc Natl Acad Sci U S A. 2013. Oct 22;110(43):17450–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Lawson KA, Sousa CM, Zhang X, Kim E, Akthar R, Caumanns JJ, et al. Functional genomic landscape of cancer-intrinsic evasion of killing by T cells. Nature. 2020. Oct;586(7827):120–6. [DOI] [PMC free article] [PubMed] [Google Scholar]