

Abstract
Modification at the 5”-position of 4,5-disubstituted aminoglycoside antibiotics (AGAs) to circumvent inactivation by aminoglycoside modifying enzymes (AMEs) is well known. Such modifications, however, unpredictably impact activity and affect target selectivity thereby hindering drug development. A survey of 5”-modifications of the 4,5-AGAs and the related 5-O-furanosyl apramycin derivatives is presented. In the neomycin and the apralog series, all modifications were well-tolerated, but other 4,5-AGAs require a hydrogen bonding group at the 5”-position for maintenance of antibacterial activity. The 5”-amino modification resulted in parent-like activity, but reduced selectivity against the human cytosolic decoding A site rendering this modification unfavorable in paromomycin, propylamycin, and ribostamycin. Installation of a 5”-formamido group and, to a lesser degree, a 5”-ureido group resulted in parent-like activity without loss of selectivity. These lessons will aid the design of next-generation AGAs capable of circumventing AME action while maintaining high antibacterial activity and target selectivity.
Keywords: aminoglycoside modifying enzymes, antibacterial, antiribosomal, ototoxicity
Graphical Abstract
Multiple modifications (X) are tolerated at the ribofuranosyl 5-position in neomycin B and the apralogs, but only amine-based derivatives are active in the paromomycins, propylamycins and ribostamycins. This is discussed in terms of total amino group count and the ring 1 functionality, amino or hydroxy, with amino groups at the 6’- and 7’-positions in the neomycins and apralogs conferring greater flexibility
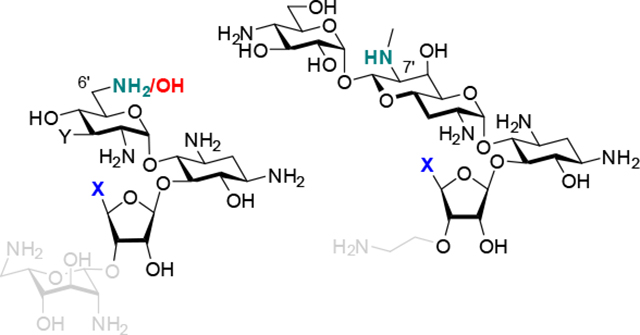
Introduction
The aminoglycoside phosphotransferases (APHs) are a class of aminoglycoside modifying enzymes (AMEs) that phosphorylate specific hydroxyl groups on aminoglycoside antibiotics (AGAs), thereby reducing their affinity for their biological target, the decoding A site on helix 44 in the 30S subunit of the bacterial ribosome, and resulting in AGA-resistant bacterial strains.[1] A case in point is the APH(3’) class, whose members phosphorylate the 3’-hydroxy group in ring I of both the 4,5- and 4,6-disubstituted classes of 2-deoxystreptamine-type AGAs (Figure 1).[1a] The action of the APH(3’)s at the 3’-hydroxy group can be thwarted by deoxygenation as illustrated by the clinical 4,6-class AGA tobramycin 2, which unlike the parent kanamycin B 1 is active in the presence of APH(3’)s. Thus, 3’-deoxygenation is a strategy widely employed both by nature and by chemists.[2]
Figure 1.

Structures of select 4,5- and 4,6-AGAs.
Some APH(3’) isozymes exhibit dual modes of reactivity at both the 3’- and 5”-hydroxy groups in the 4,5-AGAs, as was discovered by Courvalin and coworkers with their finding that certain APH(3’)s inactivate the 3’-deoxy-4,5-AGA lividomycin 5 in addition to the parent paromomycin 3.[3] Subsequently, in order to combat the full range of APH(3’)s acting on the 4,5-AGAs it became necessary to develop derivatives modified at both the 3’- and 5”-positions to prevent phosphorylation. Chemistry at the 5”-position is easier than at the 3’-position as it is one of only two possible primary alcohols in paromomycin 3, and the only one in neomycin 4 and in ribostamycin 6, but early modifications resulted in a reduction in activity and were thus not promising. For example, it was found that both 5”-deoxy- and 5”-amino-5”- deoxy-lividomycin A showed broadly reduced antibacterial activity over the parent lividomycin A.[4] Similarly, the 5”-chloro-5”-deoxy and 5”-fluoro-5”-deoxy analogues of lividomycin B as well as 5”-deoxy lividomycin B itself were markedly less active than the parent.[5] Although antibacterial activities were not reported, 5”-deoxyneomycin B was shown to have approximately 10-fold less affinity for a 27-mer RNA fragment modelling the decoding A site than neomycin B itself.[6] Attempts to block the APH(3’)s by the construction of cyclic AGAs with bridges spanning the 3’- and 5”-, or 2’- and 5”-positions were similarly fruitless, giving compounds with much reduced activity.[6–7] A series of 5”-O-monosaccharyl derivatives of neomycin B showed either comparable or reduced antibacterial activity.[8] In contrast to these mostly negative observations, it was reported that 5”-amino-5”-butirosin essentially retains the full spectrum of antibacterial activity when compared to butirosin 7 itself.[9] Likewise, 5”-amino- and 5”-guanidino derivatives of 4’,5”-dideoxybutirosin were shown to retain strong, parent-like antibacterial activity against multiple strains.[10] In comparison to the parent ribostamycin, 3’,4’,5”-trideoxyribostamycin was notably less active in all cases except for strains carrying AMEs acting at those positions.[11] The preparation of 5”-chloro-5”-deoxyribostamycin and 5”-deoxyribostamycin has been described in the patent literature but to our knowledge no antibacterial data for these compounds has been published.[12]
Particularly in the neomycin series, where the 5”-position carries the only primary hydroxy group making it easy to derivatize selectively, many more substantial modifications have been made beyond the simple aminations, deoxygenations, and halogenations listed above.[13] Often, however, these neomycin 5”-modifications were carried out with simultaneous modification at other positions, making it difficult to tease out the contribution to activity of the 5”-modification itself. Nevertheless, the fact that these highly modified derivatives sometimes retain high activity indicates that 5”-functionalization is broadly tolerated in neomycin. For example, a study by Chang and coworkers revealed that a series of 5”-deoxy-5”-triazolyl derivatives 8 and 9 retained significant activity against wild-type E. coli, and that several 5”-deoxy-5”-amido derivatives had comparable activity to the parent, with the optimal members being 5”-deoxy-5”-glycinamido neomycin derivatives 10 and 11, and 5”-deoxy-5”-palmitamido neomycin 12 (Figure 2).[14]
Figure 2.

Some established neomycin 5”-derivatives.
In our own laboratories we developed two series of 5-O-furanosyl apramycin derivatives, dubbed apralogs and advanced apralogs, carrying modifications at the primary position of the ribofuranosyl residue that is equivalent to the 5”-position in the 4,5-AGAs. We note that in apramycin the 5”-position is formally C5 in the terminal 4-aminoglucopyranosyl ring, such that the furanosyl ring in the apralogs and advanced apralogs should be numbered 1”’−5”’. However, to avoid confusion in this discussion, here we consistently designate positions in the furanose ring 1”−5” across all series of compounds We found that the 5-amino-5-deoxy modification in the ribose ring was optimal and afforded compounds (16 and 18) with excellent in vitro and in vivo antibacterial activity coupled with low ototoxicity in a cochlear explant model.[15] In contrast, the corresponding 5”-deoxy-5”-amino derivative 20 of the next-generation 4,5-AGA propylamycin 19, while retaining strong activity, was not as active as the 5”-deoxy-5”-formamido modification 21 (Figure 3).[16]
Figure 3.

Apralogs, advanced apralogs, and propylamycin derivatives.
In view of the somewhat disparate results reported by multiple groups for modifications at the 5”-position, and particularly of the differences between the propylamycin and apralog series in our own laboratories, we undertook a systematic study of a series of minimal modifications at the 5”-position in each of the neomycin, ribostamycin, paromomycin, propylamycin, apralog, and advanced apralog series of 4,5-AGAs with the aim of establishing structure activity relationships that would enable the informed choice of modification for use in the development of advanced AGAs circumventing the action of the APH(3’)s.
Results and Discussion
Synthesis.
Literature compound 23[17] was subjected to one-pot tritylation and acetylation followed by subsequent deprotection of the trityl group using FeCl3·6H2O.[18] The resulting primary alcohol was oxidized using BAIB and TEMPO[19] to give a 33% yield of carboxylic acid 24, which was immediately subjected to Barton decarboxylation[20] to generate intermediate 26 in 42% yield. Subsequent treatment with Mg(OMe)2 to selectively cleave the trifluoroacetamide groups followed by NaOH gave the 4”-des(hydroxymethyl) neomycin derivative 27. Final purification was achieved through chromatography over Sephadex C25 and lyophilization with acetic acid to generate the corresponding peracetate salt in 55% overall yield (Scheme 1).
Scheme 1.

Synthesis of 4”-des(hydroxymethyl)neomycin 27.
All other neomycin derivatives were prepared from common intermediate 29, which was generated through trisylation of literature compound 28[21] in 61% yield. Displacement of the trisyl group with sodium iodide and potassium phthalimide afforded intermediates 30 and 32 in 74% and 50% yield respectively. Hydrogenolysis of 30 followed by chromatographic purification through Sephadex C-25 and lyophilization with acetic acid afforded 5”-deoxyneomycin 31 in 33% yield. Cleavage of the phthalimide group of 32 with hydrazine hydrate gave the free amine 33 in 73% yield, which was subsequently subjected to either hydrogenolysis or acylation followed by saponification of any undesired esters and reduction of the azido groups. Each of the fully deprotected neomycin derivatives was passed through Sephadex C-25 and lyophilized with acetic acid to generate the corresponding peracetate salts (Scheme 2).
Scheme 2.

Synthesis of 5”-deoxy, 5”-deoxy-5”-amino, and 5”-deoxy-5”-amidoneomycin derivatives.
Turning to modifications at the 5”-position of paromomycin, literature compound 37[21] was subjected to one-pot tritylation and acetylation followed by trityl deprotection and oxidation of the resulting primary alcohol[18] to give carboxylic acid 38 in 36% overall yield. Decarboxylation[20] of 38 gave the intermediate 39 in 47% yield, which upon acidic hydrolysis of the benzylidene acetal and basic hydrolysis of the trifluoroacetamides and esters followed by purification by Sephadex C-25 chromatography and lyophilization with acetic acid generated the pentaacetate salt of des(hydroxymethyl) paromomycin 40 in 83% overall yield (Scheme 3).
Scheme 3.

Synthesis of 4”-des(hydroxymethyl)paromomycin.
Literature compounds 41[22] and 42[23] were treated with trisyl chloride to generate intermediates 43 and 44 in 53% and 58% yield respectively. Displacement of the trisyl group of 43 with sodium iodide gave alkyl iodide 45 in 68% yield, which was deprotected through acidic hydrolysis of the benzylidene acetal and hydrogenolysis, purified by Sephadex C-25 chromatography, and lyophilized with acetic acid to give 5”-deoxyparomomycin 46 in 54% overall yield. Displacement of the trisyl groups of 43 and 44 with sodium azide afforded 5”-azido derivatives 47 and 48 in 78% yield in each case. Intermediate 47 was subsequently subjected to hydrogenolysis, purification over Sephadex C-25, and lyophilization with acetic acid to give the hexaacetate salt of 49 in 57% overall yield. After subjection of 48 to Staudinger conditions to generate the 5”-amino derivative 50, acylation of the free amine followed by global deprotection, Sephadex C-25 purification, and lyophilization with acetic acid gave the peracetate salts of 5”-N-acyl derivatives 51–53 (Scheme 4).
Scheme 4.

Synthesis of 5”-deoxy, 5”-deoxy-5”-amino, and 5”-deoxy-5”-amidoparomomycin derivatives.
In the ribostamycin series, neamine derivative 54[21] was glycosylated with erythrosyl donor 55[15a] and boron trifluoride diethyl etherate as an activator to give 56 as a single isomer in quantitative yield. Subsequent basic hydrolysis of the benzoates, Staudinger reduction of the azides, and hydrogenolysis of the benzyl ethers generated the 4”-des(hydroxymethyl) ribostamycin derivative 57, which was purified over Sephadex C-25 and lyophilized with acetic acid to form the peracetate salt in 35% overall yield (Scheme 5). The anomeric configuration at the newly generated glycosidic linkage of 57 was assigned based on the 13C chemical shift of C1”’ (δ105.7) in accordance with well-established rules.[24]
Scheme 5.

Synthesis of 4”-des(hydroxymethyl)ribostamycin.
As with the paromomycin series, azide-protected 58, prepared in 57% yield from treatment of ribostamycin with triflyl azide and copper sulfate, and literature compound 59[11] were each subjected to sulfonation at the 5”-hydroxyl group to form derivatives 60 and 61 in 41% and 26% yield respectively. Subsequent displacement of the sulfonate with sodium azide afforded compounds 62 and 63 in 74% and 47% yield respectively. Hydrogenolysis of 62 followed by Sephadex C-25 purification and lyophilization with acetic acid generated the peracetate salt of 64 in 24% yield. Deprotection of the 5”-azido group of 63 under Staudinger conditions gave intermediate 65 in 77% yield, which was subsequently formylated and saponified or acetylated, hydrogenated, purified over Sephadex C-25, and lyophilized with acetic acid to give the peracetate salts of 66 and 67 in 26% and 46% yield respectively (Scheme 6).
Scheme 6.

Synthesis of 5”-deoxy-5”-amino and 5”-deoxy-5”-amidoribostamycin derivatives.
Finally, apramycin derivatives 75, 76, and 79 were synthesized to enable comparison between the apralogs, advanced apralogs and the 4,5-AGAs. Glycosylation of acceptor 68 and either donor 70 or donor 71 with boron trifluoride diethyl etherate as an activator gave exclusively the β-anomers of 72 and 73 in 76% and 45% yield respectively. Saponification of the esters (and cleavage of the phthalimide with sodium borohydride in the case of 72) followed by formylation with 74[25] and Staudinger reduction of azides generated final compounds 75 and 76, each of which was purified by Sephadex C-25 chromatography and lyophilized in acetic acid to give the peracetate salts in 75% and 57% yield respectively. Glycosylation at room temperature of acceptor 69 with commercial donor 77 using boron trifluoride diethyl etherate afforded 78 in 27% isolated yield (α:β= 0.3:1), which was subsequently subjected to one-pot ester hydrolysis and Staudinger reduction, followed by subsequent purification via Sephadex C-25 and lyophilization with acetic acid to give the peracetate salt of 79 in 57% yield (Scheme 7). As above, the anomeric configurations at the newly generated glycosidic linkages of 72, 73, and 78 were assigned based on the 13C chemical shifts of C1”’ (δ106.9, 106.7, and 105.5 respectively) in accordance with well-established rules.[24]
Scheme 7.

Synthesis of apralog and advanced apralog derivatives.
Antiribosomal Activity and Selectivity.
To determine the influence of 5”-modification at the level of the drug target, all compounds (Figure 4) were screened in cell-free translation assays for their ability to inhibit luciferase production by wild type Mycobacterium smegmatis bacterial ribosomes. In parallel and as a measure of selectivity for binding to the bacterial ribosome, the more active compounds were also subjected to cell-free translation assays using engineered M. smegmatis ribosomes containing the human drug binding pocket, namely the mitochondrial decoding A site and its A1555G mutant, and the human cytosolic ribosome decoding A site (Figure 5, Table 1).[26] Lack of selectivity in binding of the AGA to the bacterial ribosome over the human mitochondrial ribosome is an established predictor of ototoxicity, as AGA-induced ototoxicity has been linked to translational inhibition of mitochondrial ribosomes in cochlear hair cells.[26b, 27] Moreover, hypersusceptibility to AGA-induced ototoxicity has been linked to translational inhibition of the A1555G mutant in genetically predisposed patients.[26d, 28] Lack of selective binding of the drug to bacterial ribosomes over human cytosolic ribosomes is an indicator of systemic toxicity.
Figure 4.

Set of parent compounds and derivatives screened.
Figure 5.

Decoding A sites of prokaryotic and eukaryotic ribosomes. The bacterial AGA binding pocket is boxed. The bacterial numbering scheme is illustrated for the AGA binding pocket. Changes from the bacterial ribosome binding pocket are colored green. The A1555G mutant conferring hypersusceptibility to AGA ototoxicity is colored red.
Table 1.
Antiribosomal activities and selectivities of 4,5-AGA derivatives[a]
| IC50 (μM) | Selectivity | |||||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||
| Compound | 4”-Substituent | wt | Mit13 | A1555G | Cyt14 | Mit13 | A1555G | Cyt14 |
|
|
|
|||||||
| Neomycin series (6 basic amines in parent) | ||||||||
| Neomycin, 4 | CH2OH | 0.034 | 3.6 | 0.40 | 34 | 106 | 12 | 1000 |
| 27 | H | 0.038 | 5.0 | 0.077 | 32 | 132 | 2 | 865 |
| 31 | CH3 | 0.039 | 10.7 | 0.945 | 32 | 274 | 24 | 821 |
| 34 | CH2NH2 | 0.025 | 14 | 0.470 | 32 | 560 | 19 | 1391 |
| 35 | CH2NHCHO | 0.048 | 15 | 1.09 | 56 | 313 | 23 | 1167 |
| 36 | CH2NHAc | 0.038 | 18 | 1.2 | 38 | 474 | 32 | 1000 |
| Paromomycin series (5 basic amines in parent) | ||||||||
| Paromomycin, 3 | CH2OH | 0.033 | 131 | 12 | 37 | 3970 | 364 | 1121 |
| 40 | H | 1.059 | - | - | - | - | - | - |
| 46 | CH3 | 1.092 | - | - | - | - | - | - |
| 49 | CH2NH2 | 0.090 | 36 | 6.3 | 6.6 | 400 | 70 | 73 |
| 51 | CH2NHCHO | 0.040 | 166 | 27 | 114 | 4140 | 675 | 2850 |
| 52 | CH2NHAc | 0.513 | - | - | - | - | - | - |
| 53 | CH2NHCONH2 | 0.096 | 114 | 45 | 184 | 1188 | 469 | 1917 |
| Propylamycin series (5 basic amines in parent) | ||||||||
| Propylamycin, 19 | CH2OH | 0.022 | 150 | 56 | 61 | 6818 | 2545 | 2773 |
| 20 | CH2NH2 | 0.14 | 62 | 38 | 9.0 | 443 | 271 | 64 |
| 21 | CH2NHCHO | 0.034 | 505 | 118 | 175 | 14853 | 3471 | 5147 |
| 22 | CH2NHCONH2 | 0.078 | 318 | 189 | 194 | 4077 | 2423 | 2487 |
| Ribostamycin series (4 basic amines in parent) | ||||||||
| Ribostamycin, 6 | CH2OH | 0.089 | 461 | 86 | 416 | 5180 | 966 | 4674 |
| 57 | H | 1.612 | - | - | - | - | - | - |
| 64 | CH2NH2 | 0.088 | 76 | 34 | 50 | 861 | 386 | 570 |
| 66 | CH2NHCHO | 0.686 | - | - | - | - | - | - |
| 67 | CH2NHAc | 3.542 | - | - | - | - | - | - |
| Apralog series (5 basic amines in parent) | ||||||||
| 14 | CH2OH | 0.16 | 439 | 272 | 475 | 2815 | 1745 | 3045 |
| 15 | H | 0.128 | 332 | 211 | 444 | 2594 | 1648 | 3469 |
| 79 | CH3 | 0.28 | 562 | 323 | 462 | 2022 | 1162 | 1662 |
| 16 | CH2NH2 | 0.12 | 113 | 81 | 111 | 941 | 675 | 925 |
| 75 | CH2NHCHO | 0.31 | 572 | 232 | 567 | 1863 | 755 | 1847 |
| Advanced Apralog series (6 basic amines in parent) | ||||||||
| 17 | CH2OH | 0.071 | 68 | 13 | 190 | 957 | 183 | 2676 |
| 18 | CH2NH2 | 0.030 | 42 | 20 | 38 | 1400 | 667 | 1267 |
| 76 | CH2NHCHO | 0.085 | 93 | 11 | 142 | 1096 | 130 | 1669 |
All values were determined in at least duplicate using 2.5-fold dilution series
It is apparent from Table 1 that modification of the 5”-position in neomycin has no significant impact on the ability of the drug to inhibit wild type bacterial ribosomes, consistent with the reported use of the neomycin 5”-position as attachment point for the preparation of multiple derivatives with retention of antibacterial activity.[13–14] In the paromomycin series, on the other hand, an approximately 3-fold loss of activity is seen on conversion of the 5”-hydroxy group to an amino group (49). Functionalization of the amino group in 49 in the form of a formamide (51) restores the lost activity while acetylation (52) is highly detrimental. The urea derivative 53, which can be considered isosteric with the acetamide 52 but with greater hydrogen-bonding capabilities, in part regains activity. The complete removal of hydrogen-bonding capability at the 5”-position as in both 40 and 46 results in an approximately 30-fold loss of activity. In the propylamycin series no attempt was made to prepare the 5”-deoxy, 4”-des(hydroxymethyl), and 5”-acetamido-5”-deoxy derivatives in view of the detrimental nature of these modifications to paromomycin: essentially the same trend in activity against the bacterial ribosome as in the paromomycin series was seen with the 5”-amino 20, 5”-formamido 21 and 5”-ureido 22 modifications to propylamycin with the formamide 21 retaining the greatest level of activity. In the ribostamycin series a different pattern was observed with the 5”-amino-5”-deoxy modification 64 having essentially the same activity as the parent, the formamide 66 with approximately 8-fold lower activity, and the des(hydroxymethyl) and acetamide derivatives 57 and 67 showing 20 and 40-fold losses of activity relative to the parent.
Moving to the apralog and advanced apralog series of compounds, replacement of the 5”-hydroxy group by an amino group (16 and 18) results in a minor increase in antiribosomal activity, which disappears on conversion to the corresponding formamido derivatives (75 and 76). In so far as the formamido derivatives are less active than the amino derivatives, this pattern in the apralogs resembles that seen in the neomycin series. The complete removal of functionality at the apralog 5”-position as in the deoxy derivative 79 or the des(hydroxymethyl) derivative 15 similarly has little impact on selectivity, as seen in the neomycin series.
Overall, in the neomycin series with its six basic amines, mostly protonated at physiological pH[29] and providing a strong affinity for the negatively charged ribosomal decoding A site,[30] all modifications studied at the 5”-position are tolerated. In all other series except the apralogs the complete removal of functionality from the ribosyl side chain, as in the deoxy and des(hydroxymethyl) modifications results in a significant loss of activity indicating the need for a hydrogen bonding capable group (donating and/or accepting) at that position that is only offset by the presence of six basic amines. The amino modification is effective in all series resulting in compounds with only minor losses or minor gains in activity. The formamido modification is similarly effective in all series except the ribostamycins, where the parent is inherently less tolerant of modification. Whenever studied, except in the tightly bound neomycin series, the acetamido modification is detrimental, which we ascribe to the presence of the hydrophobic methyl group as activity is largely recovered with the isosteric but hydrophilic ureido group. While formamido groups are known to populate the Z-conformation to a much greater extent than acetamides,[31] and indeed are seen to do so in the NMR spectra of 35, 51, 66, 21, 75, and 76 in free solution, we see no reason to invoke preferential binding of the formamides through this conformation in view of the activity of the ureido derivatives.
Turning to the hybrid ribosomes carrying the human mitochondrial ribosome (Mit13), with the exception of the tightly bound neomycin series where all modifications result in a minor reduction in activity, the amino modification stands out in causing a modest increase in activity, which in the paromomycin, propylamycin, and ribostamycin series results in a reduction in selectivity compared to the parent. In the A1555G mutant mitochondrial hybrid ribosomes a comparable pattern is seen.
In the Cyt14 series of hybrid ribosomes carrying the human cytosolic decoding A site, which is characterized by a C1409•A1491 mismatch at the base of the binding pocket as well as an A1409G substitution at the site of interaction with positions 4’ and 6’ of the drug, all substitutions at the 5”-position of the drug are similarly accommodated in the tightly bound neomycin B framework. In the other series of 4,5-compounds, the amino modification affords noticeably higher activity than any other changes made, which is attributable to the stronger H bond between the protonated 5”-amino group and N7 of A1491, such that the 5”-amino-5”-deoxy compounds have noticeably lower selectivity for the bacterial ribosome over the cytosolic variant. The observation that 5”-amino modified 4,5-AGAs show less selectivity for the bacterial over the A1555G mutant mitochondrial and cytoplasmic ribosomes than the parents suggests that compounds carrying this modification will suffer from increased toxicity over the parent compounds and will not be good candidates for development as antibacterial agents. The pattern of the relatively high activity of the 5”-amino modified compounds against the cytoplasmic ribosome accords with the status of the 5”-aminoribostamycin derivative ELX-02 as a candidate drug for the treatment of genetic diseases arising from the replacement of an amino acid codon by a premature stop codon (Figure 6).[32] For the apralogs, the 5”-amino modification affects drug selectivity to a significantly lower extent. Notably, the 5”-amino-3”-O-(2-aminoethyl)apralog 18 shows increased selectivity over the mitochondrial and A1555G mutant mitochondrial ribosomes predictive of lower ototoxicity and as borne out previously by toxicity studies with mouse cochlear explants. Inspection of the data reveals that this apparent anomaly is not the result of breakdown of the interaction with the humanized hybrid ribosomes but is due to a larger increase in activity for the bacterial ribosome.
Figure 6.

Structure of the Experimental Drug ELX-02.
It is widely accepted that the affinity of AGAs for the decoding A site is dependent on the number of basic amines and their protonation state,[30, 33] begging the question of why installation of a further basic amine in the 5”-amino derivatives (to a total of seven in 18 and 34, six in 16, 20, and 49, and five in 64) does not lead to a larger increase in activity over the parents. For this, we turn to the prototypical X-ray structure of the 4,5-AGA paromomycin in the drug binding pocket (Figure 7)[34] and the hydrogen bonding interactions involving the 5”-substituent, particularly the intramolecular hydrogen bond between the protonated N2’ in ring I and the 5”-hydroxy group in the ribofuranose ring.
Figure 7.

Partial crystal structure of paromomycin bound to the decoding A site of Thermus thermophilus (PDB ID 1FJG), with dashed blue lines denoting hydrogen bonds.
This same interaction has been established to pre-organize 4,5-AGAs in free solution for binding to the target.[35] Clearly, in the 5”-amino derivatives it is not possible to fully protonate both N2’ and N5” at the same time as this would result in a strongly repulsive electrostatic interaction between two proximal ammonium ions, hence the breakdown in the rule of thumb relating activity to the number of basic amines. Rather we suggest that N2’ and N5” share a hydrogen bonded proton thereby maintaining the overall geometry apparent for the parent in Figure 7 and resulting in no net increase in positive charge. According to this hypothesis, N5” in the amino series nevertheless carries a partial positive charge making it a strong hydrogen bond donor in its interaction with N7 of G1491 (Figure 8 a).
Figure 8.

a) Proposed hydrogen bonding scheme between N2’, N5”, and G1491, and the ring I A1408 pseudo-base pair in the 5”-deoxy-5”-amino of the 6’amino (X = NH2+) and 6’hydroxy AGAs (X = O), and b) Proposed hydrogen bonding scheme in the 5”-amino apralog interaction with the target.
The importance of the hydrogen bond between the protonated N2’ in ring I and the 5”-substituent is borne out by the limited range of modifications possible at the 5”-position, where only groups capable of hydrogen bonding are tolerated in all but the tightly bound neomycin series. The importance of this hydrogen bond in all but the neomycin series is further reflected in earlier studies on the modification of N2’ in which it was shown that the retention of a hydrogen bonding group was essential.[30a]
Finally in this section, we return to the differences in relative inhibitory activities of the various amino and formamido derivatives for the bacterial ribosome. Thus, in the neomycin and apralog series the amines are better inhibitors than the formamides. Conversely, in the paromomycin and propylamycin series the formamides are better inhibitors than the amines. We hypothesize that this difference in behavior is again related to the interaction between N2’ and the 5”-amines, and that this interaction is modulated by the functionality at the 6’-position and in the case of the apralogs the 7’-position. Thus, the paromomycin and propylamycin series of compounds carry a hydroxy group at the 6’-position (Figure 8a, X = O), whereas neomycin is a 6’-amine, whose protonated ammonium form interacts with A1408 in the decoding A site (Figure 8a, X = NH2+). Protonation of the 6’-amino group in neomycin necessarily inductively reduces the ability of the 2’-amino group to accept a hydrogen bond from a protonated amine at the 5”-position and so increases the ability of the latter to hydrogen bond with G1491. In contrast, hydrogen bond donation from the 6-hydroxy group of the paromomycin and propylamycin series of compounds to A1408 in the interaction with the target will tend to increase the basicity of N2’ and hence its interaction with a protonated amine at the 5”-position. The apralogs are 6’-hydroxy AGAs and so are nominally most closely related to the paromomycin and propylamycin series, but they also carry a secondary amino group at the 7’-position. This secondary amine does not contact the ribosome directly,[36] but is critical for activity[37] from which it follows that its protonated form donates a hydrogen bond to the adjacent 6’-hydroxy group, which in turn inductively modulates the hydrogen bond accepting ability of N2’ (Figure 8b): in this manner the apralogs are functionally analogs to neomycin.
Antibacterial Activity Against Wild Type Bacterial Strains.
All compounds were screened for activity against a panel of Gram-negative pathogens (Escherichia coli, Enterobacter cloacae, Klebsiella pneumoniae, Acinetobacter baumannii, and Pseudomonas aeruginosa) as well as the Gram-positive methicillin-resistant Staphylococcus aureus (MRSA), all of which were obtained from the Diagnostic Department of the Institute of Medical Microbiology at the University of Zurich (Table 2).
Table 2.
Antibacterial Activity Against Relevant Pathogens (MIC, μg/mL)[a]
| Strain |
AG212 | AG215 | AG290 | AG225 | AG220 | AG038 | |
|---|---|---|---|---|---|---|---|
| Compound | 4”-Substituent | E. coli | K. pneumoniae | E. cloacae | A. baumanii | P. aeruginosa | MRSA |
|
| |||||||
| Neomycin series (6 basic amines in parent) | |||||||
| Neomycin, 4 | CH2OH | 1 | 0.5 | 1 | 1–2 | 32–64 | 1 |
| 27 | H | 1–2 | 0.5–1 | 1–2 | 2–4 | >64 | 1 |
| 31 | CH3 | 2 | 1–2 | 1–2 | 2–4 | >64 | 1–2 |
| 34 | CH2NH2 | 2–4 | 2 | 2–4 | 8 | 16–32 | 1–2 |
| 35 | CH2NHCHO | 1–2 | 0.5–1 | 1 | 2–4 | 64 | 1–2 |
| 36 | CH2NHAc | 1–2 | 0.5–1 | 1–2 | 2–4 | >64 | 1 |
| Paromomycin series (5 basic amines in parent) | |||||||
| Paromomycin, 3 | CH2OH | 2–4 | 1 | 2 | 2–4 | >64 | 2 |
| 40 | H | 16 | 4–8 | 8–16 | 16 | >64 | 8 |
| 46 | CH3 | 32 | 16 | 32 | 64 | >64 | 16–32 |
| 49 | CH2NH2 | 4 | 2–4 | 4 | 4 | >64 | 2–4 |
| 51 | CH2NHCHO | 2–4 | 1 | 2 | 2–4 | >64 | 2–4 |
| 52 | CH2NHAc | 16 | 8 | 8–16 | 8–16 | >64 | 8 |
| 53 | CH2NHCONH2 | 4–8 | 2 | 2–4 | 4–8 | >64 | 2–4 |
| Propylamycin series (5 basic amines in parent) | |||||||
| Propylamycin, 19 | CH2OH | 1 | 0.25–0.5 | 0.5 | 1–2 | 8 | 1–2 |
| 20 | CH2NH2 | 2–4 | 1 | 1 | 2 | 4 | 2 |
| 21 | CH2NHCHO | 1–2 | 1 | 1 | 2 | 8 | 1–2 |
| 22 | CH2NHCONH2 | 4 | 2–4 | 2–4 | 2–4 | >64 | 2–4 |
| Ribostamycin series (4 basic amines in parent) | |||||||
| Ribostamycin, 6 | CH2OH | 4 | 2 | 2–4 | 4 | >128 | 4 |
| 57 | H | 64 | 32 | 64 | 64 | >128 | 32 |
| 64 | CH2NH2 | 8 | 4 | 4 | 4 | >32 | 8 |
| 66 | CH2NHCHO | 16 | 8 | 8–16 | 16 | >128 | 16–32 |
| 67 | CH2NHAc | 64–128 | 32 | 64 | 128 | >128 | 64–128 |
| Apralog series (5 basic amines in parent) | |||||||
| 14 | CH2OH | 4–8 | 1–2 | 2–4 | 8–16 | 32–64 | 4–8 |
| 15 | H | 8 | 2–4 | 2–4 | 8 | 16–32 | 2–4 |
| 79 | CH3 | 8–16 | 8 | 8–16 | 16 | 64 | 4 |
| 16 | CH2NH2 | 4 | 1–2 | 2 | 8 | 4–8 | 2 |
| 75 | CH2NHCHO | 8 | 2–4 | 4–8 | 16 | >64 | 8–16 |
| Advanced Apralog series (6 basic amines in parent) | |||||||
| 17 | CH2OH | 2 | 1–2 | 1–2 | 4–8 | 16 | 2–4 |
| 18 | CH2NH2 | 1–2 | 0.5–1 | 1 | 2 | 2 | 1–2 |
| 76 | CH2NHCHO | 2–4 | 1 | 1–2 | 4–8 | 16–32 | 4 |
All values were determined in at least duplicate using 2-fold dilution series
On the whole, the observed antibacterial activities against wild type bacteria were consistent with the antiribosomal activities; thus, the 4”des(hydroxymethyl), 5”-deoxy, and 5”-acetamido derivatives of all parent compounds studied displayed decreased antibacterial activity, though to a significantly lesser degree in the case of neomycin. Also consistent with the antiribosomal activity data is the decreased antibacterial activity resulting from 5”s-NH2 installation in neomycin, paromomycin, ribostamycin, and propylamycin, which mirrors previous observations from the Hanessian[38] and Baasov[32c] groups.
Antibacterial Activity against Resistant Bacterial Strains.
All compounds were additionally screened for activity against both engineered strains and clinical isolates of Escherichia coli expressing various APH(3’) isozymes (Table 3). All compounds in the ribostamycin, paromomycin, and neomycin series remained susceptible to APH(3’) isozymes, an unsurprising result in view of the free hydroxyl group at the 3’-position in each case. In the case of propylamycin, installation of a 5”-amino, 5”-formamido, and 5”-ureido groups resulted in significantly reduced susceptibility to the APH(3’)-I isoforms which act on the 5”-position. The apralogs, lacking a 3’-hydroxy group and so retaining activity in the presence by APH(3’)s acting only at that position, showed minimal susceptibility to the APH(3’) isozymes acting at the 5”-position.
Table 3.
MIC assays on engineered (EC) and clinical (AG) strains of E. coli bearing specific resistance determinants (MIC, μg/mL) [a]
| Engineered (EC) Strains | Clinical (AG) Strains | ||||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|||||||||
| EC026 | EC189 | EC191 | EC125 | EC141 | AG212 | AG163 | AG166 | ||
| Compound | 4”-Substituent | wt | APH(3’)-Ia | APH(3’)-IIa | APH(3’)-IIb | APH(3’)-VI | wt | APH(3’)-I | APH(3’)-II |
|
| |||||||||
| Neomycin series (6 basic amines in parent) | |||||||||
| Neomycin, 4 | CH2OH | 1 | 32–64 | 16–32 | 16–32 | 8–16 | 1 | 64–128 | 64–128 |
| 27 | H | 1 | 128 | ≥128 | 128 | 64–128 | 1–2 | >128 | >128 |
| 31 | CH3 | 1 | ≥128 | >128 | 128 | 64–128 | 2 | >128 | >128 |
| 34 | CH2NH2 | 1–2 | 64–128 | 32 | 32 | 16–32 | 2–4 | 128 | 128 |
| 35 | CH2NHCHO | 1 | 8–16 | 16 | 8–16 | 16 | 1–2 | 32–64 | 64 |
| 36 | CH2NHAc | 0.5–1 | ≥128 | ≥128 | 128 | 128 | 1–2 | >128 | >128 |
| Paromomycin series (5 basic amines in parent) | |||||||||
| Paromomycin, 3 | CH2OH | 1 | >128 | 32–64 | 64–128 | 64 | 2–4 | >128 | >128 |
| 40 | H | 4 | >64 | >64 | >64 | >64 | 16 | >128 | >128 |
| 46 | CH3 | 16 | >128 | >128 | >128 | >128 | 32 | >128 | >128 |
| 49 | CH2NH2 | 4 | 16–32 | 16–32 | 32 | 16–32 | 4 | >64 | >64 |
| 51 | CH2NHCHO | 1 | 8–16 | 32 | 64 | 32–64 | 2–4 | 64 | >128 |
| 52 | CH2NHAc | 4–8 | >128 | >128 | >128 | >128 | 16 | >128 | >128 |
| 53 | CH2NHCONH2 | 2 | 32 | 32–64 | >32 | >32 | 4–8 | >128 | >128 |
| Propylamycin series (5 basic amines in parent) | |||||||||
| Propylamycin, 19 | CH2OH | 0.25–0.5 | 64–128 | 0.5–1 | 0.5–1 | 0.5 | 1 | >128 | 1–2 |
| 20 | CH2NH2 | 0.5–1 | 2–4 | 1–2 | 0.5–1 | 0.5–1 | 2–4 | 16 | 2–4 |
| 21 | CH2NHCHO | 0.5–1 | 2 | 0.5–1 | 0.5 | 0.25 | 1–2 | 4–8 | 2–4 |
| 22 | CH2NHCONH2 | 1 | 8 | 2–4 | 2 | 0.5–1 | 4 | 32 | 16–32 |
| Ribostamycin series (4 basic amines in parent) | |||||||||
| Ribostamycin, 6 | CH2OH | 1–2 | >128 | >128 | >128 | >128 | 4 | >128 | >128 |
| 57 | H | 16 | >128 | >128 | >128 | >128 | 64 | >128 | >128 |
| 64 | CH2NH2 | - | - | - | - | - | 8 | >128 | >128 |
| 66 | CH2NHCHO | 4 | >128 | >128 | >128 | >128 | 16 | >128 | >128 |
| 67 | CH2NHAc | 16–32 | >128 | >128 | >128 | >128 | 64–128 | >128 | >128 |
| Apralog series (5 basic amines in parent) | |||||||||
| 14 | CH2OH | 1–2 | 1–2 | 1 | 1 | 1 | 4–8 | 8 | 4–8 |
| 15 | H | 2 | 1–2 | 1 | 1 | 1 | 8 | 8 | 8 |
| 79 | CH3 | 2–4 | 4–8 | 2–4 | 2–4 | 2–4 | 8–16 | 8–16 | 8–16 |
| 16 | CH2NH2 | 1–2 | 1 | 1 | 1 | 1 | 4 | 2 | 8 |
| 75 | CH2NHCHO | 2–4 | - | - | - | - | 8 | 16 | 8–16 |
| Advanced Apralog series (6 basic amines in parent) | |||||||||
| 17 | CH2OH | 0.5–1 | 0.5–1 | 0.5 | 0.5 | 0.5 | 2 | 4 | 2 |
| 18 | CH2NH2 | 0.25–0.5 | 0.5–1 | 0.5 | 0.25 | 0.5 | 1–2 | 1–2 | 2 |
| 76 | CH2NHCHO | 0.5–1 | 0.5–1 | 0.25–0.5 | 0.25 | 0.25–0.5 | 2–4 | 2–4 | 2–4 |
All values were determined in at least duplicate using 2-fold dilution series
Conclusion
A systematic study of the impact of 5”-modifications on activity and selectivity at the target level in the neomycin, paromomycin, propylamycin ribostamycin, and the related 5-O-ribofuranosyl apramycin derivatives series of AGAs has been conducted. In the neomycin and apralog series, modifications at the 5”-position were well-tolerated, as any potential destabilizing interactions are outweighed by the significant Coulombic stabilization of the AGA-ribosome complex from the high number of protonated amines. In 4,5-AGAs with fewer basic amines, we find that a hydrogen bonding-capable group at the 5”-position is critical for maintenance of comparable antibacterial activity to the parent. Though antibacterial activity is maintained in the 5”-amino derivatives of paromomycin, ribostamycin, and propylamycin, reduced selectivity against the human cytosolic ribosome renders this modification generally unfavorable. No such reduction of selectivity is observed, however, for the comparably active 5”-formamido and 5”-ureido modifications. In contrast, for the apralog series all 5”-amino modifications affect selectivity minimally indicating ample room for further modification. In this respect the apralogs resemble neomycin but come with a much-improved selectivity profile. These lessons will inform the design of next-generation antibiotics exhibiting reduced toxicity, greater antibacterial activity, and reduced susceptibility to the aminoglycoside modifying enzymes.
Supplementary Material
Acknowledgements
We thank the NIH (AI123352) for support of this work. Graphics were generated using UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311.
Footnotes
Conflict of Interest
AV, ECB, SNH, and DC are cofounders of and equity holders in Juvabis AG, a biotech start-up developing aminoglycoside antibiotics.
Supporting information for this article is given via a link at the end of the document.
References
- [1]a).Wright GD, Thompson PR, Front. Biosci. 1999, 4, d9–21; [DOI] [PubMed] [Google Scholar]; b) Shakya T, Wright GD, in Aminoglycoside Antibiotics: From Chemical Biology to Drug Discovery (Ed.: Arya DP), Wiley, Hoboken, 2007, pp. 119–140; [Google Scholar]; c) Yang L, Ye XS, Curr. Top. Med. Chem. 2010, 10, 1898–1926; [DOI] [PubMed] [Google Scholar]; d) Garneau-Tsodikova S, Labby KJ, Med. Chem. Commun. 2016, 7, 11–27; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Armstrong ES, Kostrub CF, Cass RT, Moser HE, Serio AW, Miller GH, in Antibiotic Discovery and Development (Eds.: Dougherty TJ, Pucci MJ), Springer Science+Business Media, New York, 2012, pp. 229–269; [Google Scholar]; f) Davies J, Davies D, Microbiol. Mol. Biol. Rev. 2010, 74, 417–433; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Chang H-H, Cohen T, Grad YH, Hanage WP, O’Brien TF, Lipsitch M, Microbiol. Mol. Biol. Rev. 2015, 79, 101–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2]a).Chandrika NT, Shrestha SK, Ranjan N, Sharma A, Arya DP, Garneau-Tsodikova S, ACS Infect. Dis. 2018, 4, 196–207; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zárate SG, De la Cruz Claure ML, Benito-Arenas R, Revuelta R, Santana AG, Bastida A, Molecules 2018, 23, 284, doi: 210.3390/molecules23020284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Courvalin P, Davies J, Antimicrob. Agents Chemother. 1977, 11, 619–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Yamamoto H, Kondo S, Maeda K, Umezawa H, Antibiotics J 1982, 25, 487–488. [DOI] [PubMed] [Google Scholar]
- [5].Torii T, Tsuchiya T, Umezawa S, Umezwa H, Bull. Chem. Soc. Jpn. 1983, 56, 1522–1526. [Google Scholar]
- [6].Bastida A, Hidalgo A, Chiara JL, Torrado M, Corzana F, Pérez-Cañadillas JM, Groves P, Garcia-Junceda E, Gonzalez C, Jimenez-Barbero J, Asensio JL, J. Am. Chem. Soc. 2006, 128, 100–116. [DOI] [PubMed] [Google Scholar]
- [7]a).Asako T, Yoshioka K, Mabuchi H, Hiraga K, Heterocycles 1978, 11, 197–2002; [Google Scholar]; b) Blount KF, Zhao F, Hermann T, Tor Y, J. Am. Chem. Soc. 2005, 127, 9818–9829; [DOI] [PubMed] [Google Scholar]; c) Barbieri CM, Kaul M, Bozza-Hingos M, Zhao F, Tor Y, Hermann T, Pilch DS, Antimicrob. Agents Chemother. 2007, 51, 1760–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Fridman M, Belakhov V, Yaron S, Baasov T, Org. Lett. 2003, 5, 3575–3578. [DOI] [PubMed] [Google Scholar]
- [9].Hayashi T, Saeki H, Takeda N, Ohki E, Antibiotics J 1979, 32, 1280–1287. [DOI] [PubMed] [Google Scholar]
- [10].Narita Y, Masuyoshi S, Yamasaki T, Naito T, Kawaguchi H, J. Antibiotics 1991, 44, 86–92. [DOI] [PubMed] [Google Scholar]
- [11].Umezawa S, Tsuchiya T, Ikeda D, Umezawa H, J. Antibiotics 1972, 25, 613–616. [DOI] [PubMed] [Google Scholar]
- [12].Hiraga K, Okutani T, Yoshioka K, Asako T, Takeda, USA Patent Appl. 1977, 4,029,883. [Google Scholar]
- [13].Chandrika NT, Garneau-Tsodikova S, Chem. Soc. Rev. 2018, 47, 1189–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhang J, Chiang F-I, Wu L, Czyryca PG, Li D, Chang C-WT, J. Med. Chem. 2008, 51, 7563–7573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15]a).Quirke JCK, Rajasekaran P, Sarpe VA, Sonousi A, Osinnii I, Gysin M, Haldimann K, Fang Q-J, Shcherbakov D, Hobbie SN, Sha S-H, Schacht J, Vasella A, Böttger EC, Crich D, J. Am. Chem. Soc. 2020, 142, 530–544; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sonousi A, Quirke JCK, Waduge P, Janusic T, Gysin M, Haldimann K, Xu S, Hobbie SN, Sha S-H, Schacht J, Chow CS, Vasella A, Böttger EC, Crich D, Chem. Med. Chem. 2021, 16, 335–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lubriks D, Zogota R, Sarpe VA, Matsushita T, Sati GC, Haldimann K, Gysin M, Böttger EC, Vasella A, Suna E, Hobbie SN, Crich D, ACS Infect. Dis. 2021, 7, 2413–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kiviniemi A, Virta P, Lönnberg H, Bioconj. Chem. 2010, 21, 1890–1901. [DOI] [PubMed] [Google Scholar]
- [18].Ding X, Wang W, Kong F, Carbohydr. Res. 1997, 303, 445–448. [Google Scholar]
- [19].Epp JB, Widlanski TS, J. Org. Chem. 1999, 64, 293–295. [DOI] [PubMed] [Google Scholar]
- [20].Barton DHR, Crich D, Motherwell WB, J. Chem. Soc., Chem. Commun. 1983, 939–941. [Google Scholar]
- [21].Greenberg WA, Priestley ES, Sears PS, Alper PB, Rosenbohm C, Hendrix M, Hung S-C, Wong C-H, J. Am. Chem. Soc. 1999, 121, 6527–6541. [Google Scholar]
- [22].Pathak R, Böttger EC, Vasella A, Helv. Chim. Acta 2005, 88, 2967–2985. [Google Scholar]
- [23].Hanessian S, Takamoto T, Massé R, Patil G, Can. J. Chem. 1978, 56, 1482–1491. [Google Scholar]
- [24].Taha HA, Richards MR, Lowary TL, Chem. Rev. 2013, 113, 1851–1876. [DOI] [PubMed] [Google Scholar]
- [25].Akikusa N, Mitsui K, Sakamoto T, Kikugawa Y, Synthesis 1992, 1992, 1058–1060. [Google Scholar]
- [26]a).Hobbie SN, Kalapala SK, Akshay S, Bruell C, Schmidt S, Dabow S, Vasella A, Sander P, Böttger EC, Nucleic Acids Res. 2007, 35, 6086–6093; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hobbie SN, Akshay S, Kalapala SK, Bruell CM, Shcherbakov D, Böttger EC, Proc. Natl. Acad. Sci. USA 2008, 105, 20888–20893; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Hobbie SN, Kaiser M, Schmidt S, Shcherbakov D, Janusic T, Brun R, Böttger EC, PLOS Neglected Trop. Dis. 2011, 5, e1161; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Hobbie SN, Bruell CM, Akshay S, Kalapala SK, Shcherbakov D, Böttger EC, Proc. Natl. Acad. Sci. USA 2008, 105, 3244–3249; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Perez-Fernandez D, Shcherbakov D, Matt T, Leong NC, Kudyba I, Duscha S, Boukari H, Patak R, Dubbaka SR, Lang K, Meyer M, Akbergenov R, Freihofer P, Vaddi S, Thommes P, Ramakrishnan V, Vasella A, Böttger EC, Nat. Commun. 2014, 5, 3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27]a).Böttger EC, Schacht J, Hear. Res. 2013, 303, 12–19; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Duscha S, Boukari H, Shcherbakov D, Salian S, Silva S, Kendall A, Kato T, Akbergenov R, Perez-Fernandez D, Bernet B, Vaddi S, Thommes P, Schacht J, Crich D, Vasella A, Bottger EC, mBio 2014, 5, e01827–01814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28]a).Qian Y, Guan M-X, Antimicrob. Agents Chemother. 2009, 53, 4612–4618; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Prezant TR, Agapian JV, Bohlman MC, Bu X, Öztas S, Qiu W-Q, Arnos KS, Cortopassi GA, Jaber L, Rotter JI, Shohat M, Fischel-Ghodsian N, Nat. Genet. 1993, 4, 289–294. [DOI] [PubMed] [Google Scholar]
- [29].Alkhzem AH, Woodman TJ, Blagbrough IS, ACS Omega 2020, 5, 21094–21103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30]a).Sati GC, Sarpe VA, Furukawa T, Mondal S, Mantovani M, Hobbie SN, Vasella A, Böttger EC, Crich D, ACS Infect. Dis. 2019, 5, 1718–1730; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) François B, Russell RJM, Murray JB, Aboul-ela F, Masquida B, Vicens Q, Westhof E, Nucleic Acids Res. 2005, 33, 5677–5690; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kaul M, Pilch DS, Biochemistry 2002, 41, 7695–7706; [DOI] [PubMed] [Google Scholar]; d) Kaul M, Barbieri CM, Kerrigan JE, Pilch DS, J. Mol. Biol. 2003, 326, 1373–1387. [DOI] [PubMed] [Google Scholar]
- [31]a).Hu X, Zhang W, Carmichael I, Serianni AS, J. Am. Chem. Soc. 2010, 132, 4641–4652; [DOI] [PubMed] [Google Scholar]; b) LaPlanche LA, Rogers MT, J. Am. Chem. Soc. 1964, 86, 337–341; [Google Scholar]; c) Pawar DM, Khalil AA, Hooks DR, Collins K, Elliott T, Stafford J, Smith L, Noe EA, J. Am. Chem. Soc. 1998, 120, 2108–2112. [Google Scholar]
- [32]a).Sabbavarapu NM, Pienko T, Zalman B-H, Trylska J, Baasov T, Med. Chem. Commun. 2018, 9, 503–508; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Crawford DK, Alroy I, Sharpe N, Goddeeris MM, Williams G, J. Pharmacol. Expt. Therap 2020, 374, 264–272; [DOI] [PubMed] [Google Scholar]; c) Shalev M, Baasov T, Med. Chem. Commun. 2014, 5, 1092–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Becker K, Cao S, Nilsson A, Erlandsson M, Hotop S-K, Kuka J, Hansen J, Haldimann K, Grinberga S, Fernández TB, Huseby DL, Shariatgorji R, Lindmark E, Platzack B, Böttger EC, Crich D, Friberg LE, Vinsbo Lundberg C, Hughes D, Brönstrup M, Andrén PE, Liepinsh E, Hobbie SN, EBioMedicine 2021, 73, 103652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Carter AP, Clemons WM, Brodersen DE, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V, Nature 2000, 407, 340–348. [DOI] [PubMed] [Google Scholar]
- [35]a).Corzana F, Cuesta I, Freire F, Revuelta J, Torrado M, Bastida A, Jiménez-Barbero J, Asensio JL, J. Am. Chem. Soc. 2007, 129, 2849–2865; [DOI] [PubMed] [Google Scholar]; b) Fourmy D, Recht MI, Blanchard SC, Puglisi JD, Science 1996, 274, 1367–1371; [DOI] [PubMed] [Google Scholar]; c) Herzog IM, Louzoun Zada S, Fridman M, J. Med. Chem. 2016, 59, 8008–8018. [DOI] [PubMed] [Google Scholar]
- [36].Matt T, Ng CL, Lang K, Sha S-H, Akbergenov R, Shcherbakov D, Meyer M, Duscha S, Xie J, Dubbaka SR, Perez-Fernandez D, Vasella A, Ramakrishnan V, Schacht J, Böttger EC, Proc. Natl. Acad. Sci. 2012, 109, 10984–10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Mandhapati AR, Shcherbakov D, Duscha S, Vasella A, Böottger EC, Crich D, ChemMedChem 2014, 9, 2074–2083. [DOI] [PubMed] [Google Scholar]
- [38].Hanessian S, Massé R, Capmeau ML, J. Antibiot. 1977, 30, 893–896. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.