

Analysis of >700 RAS structures helps define an expanded landscape of active, inactive, and druggable RAS conformations, the structural impact of common RAS mutations, and previously uncharacterized RAS inhibitor–binding modes.
Abstract
Mutations in RAS isoforms (KRAS, NRAS, and HRAS) are among the most frequent oncogenic alterations in many cancers, making these proteins high priority therapeutic targets. Effectively targeting RAS isoforms requires an exact understanding of their active, inactive, and druggable conformations. However, there is no structural catalog of RAS conformations to guide therapeutic targeting or examining the structural impact of RAS mutations. Here we present an expanded classification of RAS conformations based on analyses of the catalytic switch 1 (SW1) and switch 2 (SW2) loops. From 721 human KRAS, NRAS, and HRAS structures available in the Protein Data Bank (206 RAS–protein cocomplexes, 190 inhibitor-bound, and 325 unbound, including 204 WT and 517 mutated structures), we created a broad conformational classification based on the spatial positions of Y32 in SW1 and Y71 in SW2. Clustering all well-modeled SW1 and SW2 loops using a density-based machine learning algorithm defined additional conformational subsets, some previously undescribed. Three SW1 conformations and nine SW2 conformations were identified, each associated with different nucleotide states (GTP-bound, nucleotide-free, and GDP-bound) and specific bound proteins or inhibitor sites. The GTP-bound SW1 conformation could be further subdivided on the basis of the hydrogen bond type made between Y32 and the GTP γ-phosphate. Further analysis clarified the catalytic impact of G12D and G12V mutations and the inhibitor chemistries that bind to each druggable RAS conformation. Overall, this study has expanded our understanding of RAS structural biology, which could facilitate future RAS drug discovery.
Significance:
Analysis of >700 RAS structures helps define an expanded landscape of active, inactive, and druggable RAS conformations, the structural impact of common RAS mutations, and previously uncharacterized RAS inhibitor–binding modes.
Graphical Abstract

Introduction
Mutations in the RAS isoforms, KRAS (4A and 4B splice forms), NRAS, and HRAS, drive oncogenesis in approximately 20% of human cancers, and cause a variety of tumor predisposition syndromes, making these proteins high priority therapeutic targets (1). During the past 30 years, our molecular understanding of RAS mutations and our ability to drug these proteins has improved considerably, due, in part, to hundreds of structural studies examining wild-type (WT) and mutated RAS in complex with various signaling effector and regulatory proteins, or with small-molecule and designed protein inhibitors (2). However, our structural understanding of RAS mutations is incomplete, and, except for KRAS G12C, G12D, and G13C, all mutated RAS forms have not yet been selectively targeted by therapeutics (3).
The RAS proteins are molecular switches that modulate growth and other signaling pathways in almost all cells of the human body by conformationally cycling between GDP-bound (“OFF”) and GTP-bound (“ON”) states (4). In normal tissues, RAS conformational cycling is tightly regulated by the catalytic CDC25 domain of guanine exchange factors (GEFs; e.g., SOS1), which remove GDP allowing subsequent GTP rebinding (5), and GTPase activating proteins (GAPs; e.g., NF1), which catalyze the otherwise slow intrinsic rate of GTP hydrolysis to GDP (6). GEFs and GAPs interact with the conformationally dynamic switch 1 (SW1) and switch 2 (SW2) loops in RAS structures, which additionally provide binding interfaces for signal effector proteins (e.g., RAF1) and direct RAS inhibitors (7). RAS targeted therapies mostly bind to an SW1/SW2 pocket (called here “SP12”) to block RAS–protein interactions or an SW2 pocket (called here “SP2”) to lock RAS in an inactive, GDP-bound conformation. Overall, the configurations of SW1 and SW2 are essential for RAS function and the druggability of these proteins.
Most tumor-associated RAS mutations modify the conformational preferences and/or dynamics of SW1 and SW2 in ways that reduce the rate of intrinsic and GAP-mediated hydrolysis (residues 12, 13, and 61) and/or enhance the rate of GEF-mediated exchange (residues 13, 61, and 146; refs. 8–12). The net effect of these alterations is to increase the steady-state cellular concentration of active, GTP-bound RAS that is capable of stimulating signaling pathways via the following mechanisms: by binding to and activating RAS effectors; by binding to the allosteric REM domain of the GEF SOS1, which functions to accelerate GDP release at the CDC25 domain of SOS1 (13); and by promoting homodimerization of RAS monomers at their helices α4 and α5, which is required for activation of certain dimeric effectors, such as RAF1 (14, 15). However, the exact SW1 and SW2 conformations that can form each RAS complex and their potential druggability are unknown, making it difficult to design therapeutics that selectively block the activities of all WT and mutated RAS forms. Furthermore, for the most part, the described structural impact of many mutated RAS forms is underdetermined, either based on comparison of one or two mutated structures or extrapolations made while observing WT structures, necessitating further structural examination of RAS mutations.
While all published RAS structures have been made publicly available through the Protein Data Bank (PDB), this structural dataset has not been leveraged in a comprehensive way to improve our understanding of RAS conformations and mutations and to inform RAS drug discovery. Therefore, we analyzed the 721 human KRAS, NRAS, and HRAS structures available in the PDB to define a more comprehensive classification of active, inactive, and druggable RAS conformations and identify the structural consequence of common RAS mutations. We first annotated the molecular contents of each RAS structure, including their mutation status, nucleotide state and bound protein (e.g., effector, GAP, GEF) or inhibitor site (e.g., SP12, SP2). Second, we conformationally classified all RAS structures based on the spatial positions of residue Y32 in SW1 and residue Y71 in SW2 and by the conformations of their catalytic SW1 and SW2 loops, as expressed by their backbone dihedral angles. By associating the identified SW1 and SW2 conformations with the annotated molecular contents of each RAS structure, we were able to create a biologically and therapeutically informed map of the RAS conformational landscape and determine the structural impact of the two most common RAS mutations, G12D and G12V. Overall, our study expands our knowledge of RAS biology and provides a valuable resource for analyzing RAS structures in ways that will improve our understanding of RAS mutations and our ability to drug this family of proteins. The results of this work are presented in a continually updated database called Rascore (http://dunbrack.fccc.edu/rascore/).
Materials and Methods
Preparation of RAS structures
PDB entries containing human KRAS (all are the 4B splice form), NRAS, and HRAS were identified by SwissProt identifier in the pdbaa file (March 1 2022) on the PISCES webserver (http://dunbrack.fccc.edu/pisces/, RRID:SCR_022181; ref. 16). For each PDB entry, the asymmetric unit and all biological assemblies were downloaded and renumbered according to UniProt scheme using PDBrenum (http://dunbrack.fccc.edu/PDBrenum/, RRID:SCR_022179; ref. 17). Furthermore, electron density of individual atom (EDIA) scores (a measure of model quality per atom; ref. 18) were downloaded from the ProteinPlus webserver (https://proteins.plus, RRID:SCR_022178; ref. 19).
Because some PDB entries contain multiple RAS polypeptide chains, each RAS chain of the asymmetric unit (only the first model for NMR structures) were separated with their corresponding bound ligands and/or proteins. Ligands were labeled Nucleotide, Ion, Inhibitor, Chemical, Modification, or Membrane using a custom dictionary prepared in considering annotations from FireDB (https://firedb.bioinfo.cnio.es, RRID:SCR_007655; ref. 20). Since ligands are not always labeled by the RAS chain with which they interact, all ligands were reassigned based on the following criteria:
1. Nucleotide, Ion, Chemical, of Modification – Possess the same chain label.
2. Inhibitor – Have more than 5 residue contacts within 4 Å of the chain.
3. Membrane – Link the chain to a nanodisc (synthetic membrane).
Proteins were assigned to an RAS chain based on the rules below, with assignments checked against available biological assemblies, and discrepancies corrected:
1. If they had more than 5 Cβ contacts within 12 Å and 1 atom contact within 5 Å of the RAS chain.
2. If they had more than 5 atom contacts within 5 Å of the RAS chain.
3. If they were the bounding protein component of a nanodisc.
Each RAS chain was treated as a unique RAS structure in subsequent analyses. Finally, RAS structures were annotated by various molecular contents, many of which are not reported in PDB entries (details provided in Supplementary Materials and Methods): mutation status, nucleotide state, bound protein, inhibitor site, inhibitor chemistry, and homodimer status. In addition, KRAS-NF1, KRAS-RASA1, HRAS-NF1, and HRAS-RASA1 cocomplexes were modeled with the AlphaFold-Multimer software (21, 22) within the ColabFold framework (23) utilizing the RASK_HUMAN (P01116–2), RASH_HUMAN (P01116), NF1_HUMAN (P21359–1, residues 1235–1451, RasGAP domain), and RASA1_HUMAN (P20936–1, residues 748–942, RasGAP domain) sequences.
Conformational clustering
RAS structures were first subdivided into broad structural groups based on the spatial positions of residue Y32 in SW1 and residue Y71 in SW2. For Y32, two broad groups were empirically defined:
1. Y32in – Y32 “in” the active site; Y32(OH)-G12(CA) distance < 10.5 Å.
2. Y32out – Y32 “out” of the active site; Y32(OH)-G12(CA) distance > 10.5 Å.
For Y71, two broad groups were empirically defined:
1. Y71in – Y71 buried “in” the hydrophobic core; Y71(OH)-V9(CA) distance < 8.75 Å.
2. Y71out – Y71 facing “out” to the solvent; Y71(OH)-V9(CA) distance > 8.75 Å.
To identify conformational subsets within these spatial classes, for each nucleotide state, structures labeled Y32in and Y32out were clustered separately by the backbone configuration of their SW1 loop (residues 25–40), and similarly structures labeled Y71in and Y71out were clustered separately by the backbone configuration of their SW2 loop (residues 56–76).
In each conformational clustering, only completely modeled loop structures possessing carbonyl atom EDIA scores > 0.4 (indicating atoms well placed within the electron density) were clustered. The conformational clustering was performed using the Density-Based Spatial Clustering of Applications with Noise (DBSCAN) algorithm with a backbone dihedral-based distance metric (previously implemented in refs. 24–27). DBSCAN finds major clusters and removes outliers (28), which is ideal for clustering structural datasets since they usually contain several outliers that were poorly modeled or solved under rare experimental conditions. In this study, a distance metric was used that locates the maximum angular difference (d) upon pairwise comparison of the backbone dihedral angle values phi (φ), psi (ψ), and omega (ω) for residues 1 through n of compared loops i versus j, where d(θi, θj) = 2(1 – cos(θj – θi)):
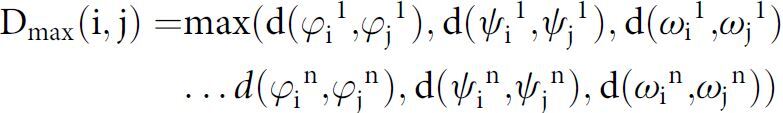 |
As in our previous study (24), DBSCAN was run across a grid of parameters and a set of quality control filters were applied to generate a robust consensus clustering (Supplementary Materials and Methods).
Detection of hydrogen bonds
Hydrogen (H)-bond cutoffs were based on a previous analysis of protein structures in the PDB (29):
i. H-bond – 2.0–3.2 Å donor–acceptor distance with 90–180° carbon–donor–acceptor and carbon–acceptor–donor angles.
- ii. Water-mediated (WM) H-bond
- a 2.0–3.0 Å donor–water and acceptor–water distances with 80–140° carbon–water–acceptor and carbon–water–donor angles.
- b 2.8–5.6 Å donor–acceptor distance (arrived at using the Law of Cosines with the previously specified cutoffs).
Data availability
All data are available as static tables in Supplementary Datasets S1-S4. In addition, our database, called Rascore, presents a continually updated dataset of all human RAS structures in the PDB annotated by their molecular contents and conformationally classified (http://dunbrack.fccc.edu/rascore/, RRID:SCR_022180). The Rascore database includes a page to conformationally classify user-uploaded structures. PyMOL sessions for all figures from the paper can be downloaded as well from the Rascore database. Open-source code pertaining to our conformational classification algorithm and the Rascore database can be found in GitHub (https://github.com/mitch-parker/rascore) and Code Ocean (https://codeocean.com/capsule/7782236/tree/v1).
Results
Conformationally classifying RAS structures
We identified 721 human KRAS (n = 436), HRAS (n = 275), and NRAS (n = 10) structures from 408 PDB entries (some entries contain multiple copies of the RAS protein, sometimes in different conformations). In all, there were 206 RAS-protein cocomplexes, 190 inhibitor-bound, and 325 unbound structures, comprising 204 WT and 517 mutated structures (Supplementary Dataset S1). Subsequently, we created an automated system for annotating RAS structures by various molecular contents, including their mutation status, nucleotide state (“3P” for GTP or any triphosphate analogue, “2P” for GDP, or “0P” for nucleotide-free), bound protein (effector, GAP, GEF CDC25 or REM domain, designed protein “binder,” or synthetic membrane “nanodisc”), small-molecule inhibitor site (SP12 or SP2), and whether the α4α5 homodimer is present in the protein crystal (only X-ray structures). In this work, we defined an expanded RAS conformational classification by identifying the observed SW1 and SW2 conformations within the prepared dataset of RAS structures and associated each conformation with the annotated molecular contents to gain novel insights into WT and mutated RAS function and inhibition.
Several RAS conformations have been previously described based on the commonly observed configurations of SW1 and SW2. For SW1, the conformations are named by their nucleotide state and include: GDP-bound, nucleotide-free, and GTP-bound “state 1” (inactive) and “state 2” (active; ref. 2); these conformations have been visually differentiated by the position of residue Y32 in SW1 relative to the active site (“in” or “out”; Fig. 1A). Of note, a noncanonical, GDP-bound SW1 conformation called “β’” or “Mg-Free” has been recently observed in WT as well as V14I- and A146T-mutated RAS structures, but it is uncertain if this conformation is biologically common (11, 30, 31). For SW2, two GTP-bound conformations have been characterized, including an inactive, “T state” [also called “off” or “ordered off” (32) and “state 2*” (33)] and an active, “R state” (also called “on”; ref. 32), which possess Y71 facing “out” to the solvent or buried “in” the hydrophobic core, respectively (Fig. 1B). Furthermore, other unnamed GDP-bound SW2 conformations have been differentiated on the basis of their ability to bind to certain small-molecule inhibitors (34–36). However, only a few RAS structures in the PDB have been visually classified into the previously named and unnamed SW1 and SW2 conformations, and there is no systematic method to differentiate all known RAS conformations from each other and from potentially unidentified ones.
Figure 1.

Broad structural classification of RAS structures. A and B, Previous conformational schemes based on the spatial position of, Y32 in SW1 (A) and Y71 in SW2 (B). C, Separation of available RAS structures in the Protein Data Bank by nucleotide states 0P (nucleotide-free), 2P (GDP-bound), and 3P (GTP or GTP analogue-bound). D and E, Distribution of distances by nucleotide state between the hydroxyl (OH) atom of residue Y32 and alpha carbon (CA) atom of residue G12 (D) and the OH atom of residue Y71 and CA atom of residue V9 (E); vertical dividing lines in plots indicate distance cutoffs for “in” versus “out” positions of Y32 (D) and Y71 (E), respectively. F–H, Structures classified Y32in (pink) and Y32out (cyan) within 0P (F), 2P (G), and 3P nucleotide states (H). I–K, Structures classified Y71in (purple) and Y71out (olive) within, 0P (I), 2P (J), and 3P nucleotide states (K).
After separating RAS structures by 0P (n = 67), 2P (n = 262), and 3P (n = 392) nucleotide states (Fig. 1C), we broadly classified these structures into the known conformational scheme based on the spatial positions of residue Y32 relative to the active site (“Y32in” or “Y32out”) in SW1 and residue Y71 relative to the hydrophobic core (“Y71in” or “Y71out”) in SW2. By examining the distribution of distances between the Y32 hydroxyl (OH) atom and carbon alpha (CA) atom of residue G12 in the active site (Fig. 1D; Supplementary Dataset S2), we determined that almost all 0P (96.8%; n = 61 of 63 classified) and 2P (98.1%; n = 207 of 211 classified) structures are Y32out, as defined by an empirically determined distance cutoff of 10.5 Å, while 3P structures contain a mix of Y32in (86.0%; n = 296 of 344 classified; includes structures labeled state 2) and Y32out (14.0%; n = 48 of 344 classified; includes structures labeled state 1; Supplementary Table S1). The distribution of distances between the Y71 OH atom and CA atom of V9 in the hydrophobic core demonstrate that each nucleotide state contains a mix of Y71in and Y71out structures (defined by an empirically determined distance cutoff of 8.75 Å), with 3P structures preferring Y71in and 0P and 2P structures preferring Y71out (Fig. 1E; Supplementary Dataset S2); the Y71in.3P and Y71out.3P structures include those classified as the R state and T state, respectively. In all, we were able to spatially classify 85.7% (n = 618 of 721) of structures by the positions of Y32 and 94.2% (n = 679 of 721) by the position of Y71 (Supplementary Table S1).
Since considerable conformational variability was observed for SW1 and SW2 within the defined Y32 and Y71 spatial groups by nucleotide state (Fig. 1F–K), we sought to identify further conformational subsets than previously described. To do so, we analyzed the RAS structures by their SW1 and SW2 configurations based on the backbone dihedral angle values of these loops: φ (phi), ψ (psi), and ω (omega). Because the RAS structures in the PDB displayed the most dihedral variability on the Ramachandran map (φ vs. ψ plot; ref. 37) in residues 25–40 (SW1) and residues 56–76 (SW2), we selected these residue ranges to analyze (Supplementary Fig. S1). After removing loop structures with incomplete modeling or poor electron density, we arrived at 542 SW1 (75.2% of 721 structures) and 423 SW2 (58.7% of 721 structures) loop structures for conformational clustering. In our analysis, we used the Density-Based Spatial Clustering of Applications with Noise (DBSCAN) algorithm (28), which clusters points with sufficient numbers of near neighbors and classifies the remainder as outliers. We employed a distance metric that locates the maximum backbone dihedral difference upon pairwise comparison of loop residues (previously implemented in (24–27)). We first separated RAS structures by nucleotide state (0P, 2P, and 3P) and spatial class (Y32in/out for SW1 and Y71in/out for SW2) and subsequently clustered the conformations of SW1 and SW2 within each group using DBSCAN. We then assigned a small number of poorly or incompletely modeled loops to the clusters obtained from DBSCAN through a nearest neighbors (NN) approach.
Overall, we were able to conformationally cluster 69.2% (n = 375 out of 542) of SW1 and 56.7% (n = 240 of 423) of SW2 loops that passed completeness and electron density checks. In addition, we assigned conformational labels using our NN approach to 85 SW1 and 51 SW2 loop structures that were not included in the original clustering due to removal by quality filters. Finally, we labeled any unclassified loops “outlier” if they were completely modeled or “disordered” if they were incompletely modeled. In all, we identified three SW1 and nine SW2 conformations, each of which were found across multiple RAS isoforms, PDB entries, and crystal forms (entries with the same space group and similar unit cell dimensions and angles; Supplementary Table S2, including the mean dihedral distance and loop α carbon atom root-mean-square deviation for each conformation), indicating that these configurations are conserved within the residue interactions of the protein structure and are not solely the product of crystal contacts.
For clarity and brevity in our expanded RAS conformational classification, we named each SW1 and SW2 conformation by its spatial class, nucleotide state, and a conformational label (written in all-capital letters). Correlated counts of the SW1 and SW2 conformations are provided in Table 1 and reported throughout the text. The SW1 conformations are labeled Y32in.3P-ON (GTP-bound state 2), Y32out.2P-OFF (GDP-bound), and Y32out.0P-GEF (nucleotide-free; Fig. 2A). There was no structurally uniform cluster within the Y32out.3P structures that could be called the GTP-bound state 1 conformation, nor a cluster within the Y32out.2P structures that could be called the GDP-bound β’/Mg-Free conformation. For SW2, the only nucleotide-free conformation was Y32out.0P-GEF (Fig. 2B). The four GDP-bound SW2 conformations are all named for their predominant binding partners, which consist of SP2 and SP12 inhibitors and protein binders: Y71out.2P-SP2-A, Y71out.2P-SP2-B, Y71in.2P-SP12, and Y71out.2P-BINDER (Fig. 2C). GTP-bound SW2 conformations included Y71in.3P-R (R state) and Y71out.3P-T (T state), and two previously unclassified druggable conformations associated with inhibitors at the SP12 site, which we named Y71in.3P-SP12-A and Y71in.3P-SP12-B (Fig. 2D). Except for Y71out.2P-SP2-A, the other clusters labeled with SP12 or SP2 include structures with and without bound inhibitors (Supplementary Dataset S1). In Fig. 2E and F, the results for our SW1 and SW2 conformational clustering (with NN assignments added) are displayed as Ramachandran maps per residue of each cluster.
Table 1.
Correlation of SW1 and SW2 conformational clusters.
| Conformation label | Y32out.0P-GEF | Y32out.2P-OFF | Y32in.3P-ON | SW1 Outlier | SW1 Disordered | All |
|---|---|---|---|---|---|---|
| Y71out.0P-GEF | 23 | 29 | 52 | |||
| Y71out.2P-SP2-A | 26 | 1 | 27 | |||
| Y71in.2P-SP12 | 20 | 20 | ||||
| Y71out.2P-SP2-B | 19 | 19 | ||||
| Y71out.2P-BINDER | 12 | 1 | 13 | |||
| Y71in.3P-R | 94 | 3 | 97 | |||
| Y71in.3P-SP12-A | 30 | 1 | 31 | |||
| Y71out.3P-T | 20 | 20 | ||||
| Y71in.3P-SP12-B | 12 | 12 | ||||
| SW2 Outlier | 59 | 53 | 85 | 53 | 250 | |
| SW2 Disordered | 32 | 60 | 25 | 63 | 180 | |
| All | 23 | 168 | 269 | 145 | 116 | 721 |
Figure 2.

SW1 and SW2 conformational clusters. A, SW1 conformations. In the Y32out.0P-GEF conformation, the central Y32 residue in SW1 is ∼12–13 Å from the active site. In the Y32out.2P-OFF conformation, SW1 is “closed” and interacts with the nucleotide through the backbone atoms of residues 28–32. In the Y32in.3P-ON conformation, further interactions are made with the nucleotide involving the side chains of residues Y32 and T35. B–D, SW2 conformations within 0P (B), 2P (C), and 3P states (D). In the Y71out.0P-GEF conformation, residues 58–60 of SW2 are pulled towards the nucleotide site, and the side chains of residues Q61 and Y71 form an intra- SW2 hydrogen bond (not displayed), which is not seen in other SW2 conformations. In all 2P SW2 conformations, except for Y71in.2P-SP12, Y71 is exposed to the solvent; the opposite trend is observed in all 3P SW2 conformations where Y71 is buried in the hydrophobic core of the protein, except in Y71out.3P-T where it is exposed. E and F, Ramachandran maps (φ versus ψ backbone dihedrals) for SW1 (E) and SW2 (F) conformational clusters. Lighter points in E and F correspond to loop structures, with one or more residues belonging to different regional subdivisions of the Ramachandran map relative to the overall cluster that they belong to.
SW1 and SW2 conformations found in RAS–protein cocomplexes
Previously, we did not know the combination of SW1 and SW2 conformations involved in each RAS interaction. Therefore, we analyzed which RAS conformations are found in the 206 RAS–protein cocomplexes currently available in the PDB, except for nanodisc-linked structures, which almost all were classified as outliers or disordered (Table 2).
Table 2.
Distribution of SW1 and SW2 conformations by bound proteins.
| Conformation label | Effector | GAP | GEF.CDC25 | GEF.REM | Binder | Nanodisc | Other | None | All |
|---|---|---|---|---|---|---|---|---|---|
| Y32out.0P-GEF | 23 | 23 | |||||||
| Y32out.2P-OFF | 13 | 1 | 154 | 168 | |||||
| Y32in.3P-ON | 17 | 6 | 38 | 10 | 3 | 3 | 192 | 269 | |
| SW1 Outlier | 5 | 1 | 32 | 11 | 10 | 3 | 83 | 145 | |
| SW1 Disordered | 6 | 22 | 2 | 86 | 116 | ||||
| Y71out.0P-GEF | 52 | 52 | |||||||
| Y71out.2P-SP2-A | 27 | 27 | |||||||
| Y71in.2P-SP12 | 20 | 20 | |||||||
| Y71out.2P-SP2-B | 1 | 18 | 19 | ||||||
| Y71out.2P-BINDER | 11 | 2 | 13 | ||||||
| Y71in.3P-R | 12 | 6 | 34 | 4 | 2 | 39 | 97 | ||
| Y71in.3P-SP12-A | 31 | 31 | |||||||
| Y71out.3P-T | 20 | 20 | |||||||
| Y71in.3P-SP12-B | 12 | 12 | |||||||
| SW2 Outlier | 8 | 1 | 9 | 4 | 40 | 11 | 4 | 173 | 250 |
| SW2 Disordered | 2 | 1 | 2 | 2 | 173 | 180 | |||
| All | 22 | 7 | 61 | 38 | 56 | 13 | 9 | 515 | 721 |
As expected, the SW1 conformation, Y32out.0P-GEF, and SW2 conformation, Y71out.0P-GEF, only exist in structures bound to the GEF.CDC25 domain of SOS1, which has SW1 held open by a SOS1 region called the “helical-hairpin” (Fig. 3A and B; ref. 5). As a new finding, we identified that the SW1 conformation, Y32in.3P-ON, and the SW2 conformation, Y71in.3P-R, bind to the GEF.REM domain of SOS1 (Fig. 3C and D), effectors (Fig. 3E and F, including RAF1, PI3K, RALGDS, and PLCε, among others), and the GAP NF1 (Fig. 3G and H). Originally, Y32in.3P-ON and Y71in.3P-R were not suggested to bind to GAPs (rather an unidentified GTP-bound “state 3” conformation; refs. 38, 39). Furthermore, we found that slight positional variations of Y32 in the Y32in.3P-ON structures influence whether the catalytic GAP “arginine (R)-finger” (6) of NF1 can enter the RAS active site: when Y32 is within 4.5 Å of the GTP γ-phosphate [Fig. 3G, left; called “Tyr32in” (40) or “Ground State” (9) by others], the R-finger is excluded from the active site and, when it moves ∼2 Å further from the γ-phosphate [Fig. 3G, right; called “Tyr32out” (40) or “Transition State” (9) by others], the R-finger can enter the active site to potentially interact with GTP. Modeling KRAS–NF1, KRAS–RASA1, HRAS–NF1, and HRAS–RASA1 cocomplexes using the AlphaFold-Multimer software (21, 22), we found that RAS in these cocomplexes is predicted to be Y32in.3P-ON and Y71in.3P-R with Y32 slightly shifted outward, allowing the R-finger to enter the active site and directly interact with GTP, as observed in a previously published HRAS–RASA1 transition state structure (PDB: 1WQ1; Supplementary Fig. S2; ref. 6). Similar variability in the Y32 position is present in effector-bound structures (Fig. 3E). Later, we examine the functional significance of this Y32 positional variability as it relates to intrinsic and GAP-mediated hydrolysis and the structural impact of RAS mutations.
Figure 3.

SW1 and SW2 conformations associated with bound proteins. SW1 and SW2 conformations bound to the GEF.CDC25 (catalytic) domain of SOS (A and B), the GEF.REM (allosteric) domain of SOS1 (C and D), effectors (E and F), the GAP NF1 (G and H), 3P targeting designed protein “binders” (I and J), and 2P targeting binders (K and L). M and N, structures forming the α4α5 homodimer. A, the “helical hairpin” of SOS1 opening SW1 of RAS. G, Comparison of the catalytic “arginine (R) finger” position for the GAP NF1 with Y32 within 4.5 Å of the GTP γ-phosphate (left) and ∼2 Å further away from the γ-phosphate (right, PDB: identical to the transition state stabilized in 1WQ1).
Besides the RAS cocomplexes with signaling molecules described above, many RAS structures are found in the PDB bound to designed protein inhibitors (called here “binders”; Table 2). In analyzing these cocomplexes, we found that binders target three major sites on RAS structures: the SW1/SW2 pocket (SP12), the SW2 pocket (SP2), and the α4α5 interface (Fig. 3I–L). In 3P structures, we identified that anti-RAS antibodies, the monobodies 12VC1 and 12VC3, and the DARPin K55 preferentially bind at the SP12 site to the SW1 conformation, Y32in.3P-ON, and the SW2 conformation, Y71in.3P-R. Because these loop conformations are found in effector-bound structures as well (Fig. 3I and J), it is no surprise that the listed binders are effective inhibitors of RAS–effector interactions (41–44). Next, we found that binders with preference for targeting 2P structures bind to multiple RAS interfaces; these include the Affimers K3 (SP2 site), K6 (SP12 site), and K69 (α4α5 interface) as we all the DARPin K27 (SP12 site). Of the 2P-interacting binders, Affimer K3 and DARPin K27, which both function to block nucleotide exchange (43, 45), bind to the SW1 conformation, Y32in.2P-OFF, and the SW2 conformation, Y71out.2P-BINDER (hence the chosen conformational label; Fig. 3K and L). Moving forward, the identified binder-interacting conformations can be used as structural templates in creating and optimizing additional protein inhibitors of RAS–effector interactions and nucleotide exchange.
SW1 and SW2 conformations found in α4α5 homodimer complexes
Homodimerization of GTP-bound RAS monomers at their α4 and α5 helices is required in some cases for signal effector activation (e.g., RAF1; refs. 14, 15), and has been identified across numerous KRAS, NRAS, and HRAS crystal structures in the PDB (15, 46). However, the conformations that can homodimerize are entirely unknown. We found the α4α5 homodimer in 144 HRAS (n = 115), KRAS (n = 28), and NRAS (n = 1) structures (31% of the X-ray experiments; 119 PDB entries and 19 crystal forms; Supplementary Table S3). The functional relevance of the α4α5 homodimer is further supported by the observance of α4α5 homodimers in cocrystal complexes with the signaling effectors RAF1 (n = 5), PLCε1 (n = 2), and RASSF1 (n = 1) as well as the GEF, GRP4 (n = 6; Supplementary Dataset S1).
From the SW1 perspective, Y32in.3P-ON structures most commonly form the α4α5 homodimer (52.1%; n = 75 of 144 dimers), with Y32out.2P-OFF forming this complex but less commonly (13.9%; n = 20 of 144 dimers), and the remainder found in outlier or disordered structures (34.0%; n = 49 of 144 dimers; Fig. 3M). Surprisingly, we found that both Y71in.3P-R (active) and Y71out.3P-T (inactive) are the most common SW2 conformations (at approximately equal rates) that form the α4α5 homodimer (Fig. 3N), contrary to the expectation that only active RAS would form this complex.
SW1 and SW2 conformations involved in small-molecule inhibitor binding
RAS proteins are notoriously difficult to drug, because of their conformational variability and lack of deep surface pockets (3). Therefore, we sought to associate the presence of small-molecule inhibitor–bound and unbound sites with the set of identified SW1 and SW2 configurations, with the goal of cataloguing all potentially druggable RAS conformations.
Using the Fpocket software (47), we first obtained pocket descriptors for inhibitor-bound sites observed on RAS structures, including their pocket volumes and druggability scores. Of the 721 RAS structures, 190 were bound to small-molecule inhibitors: 44.7% at the SW1/SW2 pocket (SP12) site (n = 85 of 190), 48.9% at the SW2 pocket (SP2) site (n = 93 of 190), and the remaining 6.3% involving other or multiple sites (n = 12 of 190; Table 3). Subsequently, we focused our analysis on the most targeted pockets, SP12 and SP2. Overall, we were able to detect and calculate pocket descriptors for 92.9% of SP12 (n = 79 of 85) and 91.4% of SP2 (n = 85 of 93) inhibitor-bound sites with Fpocket (Fig. 4A and B). We then used Fpocket to predict potentially druggable pockets in unbound structures and classified which were found at the SP12 or SP2 sites based on the similarity of their residue contacts to observed inhibitor-bound sites. In summary, we identified 208 SP12 and 222 SP2 unbound sites, which translated to about 70% of these sites existing in the absence of inhibitor binding.
Table 3.
Distribution of SW1 and SW2 conformations by inhibitor site.
| SP12 | SP2 | Other | Multiple | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Conformation label | Bound | Unbound | Bound | Unbound | Bound | Unbound | Bound | Unbound | All |
| Y32out.0P-GEF | 23 | 23 | |||||||
| Y32out.2P-OFF | 16 | 4 | 64 | 37 | 24 | 2 | 21 | 168 | |
| Y32in.3P-ON | 53 | 97 | 1 | 24 | 65 | 3 | 26 | 269 | |
| SW1 Outlier | 9 | 25 | 13 | 50 | 4 | 31 | 13 | 145 | |
| SW1 Disordered | 7 | 12 | 15 | 18 | 3 | 51 | 10 | 116 | |
| Y71out.0P-GEF | 1 | 48 | 3 | 52 | |||||
| Y71out.2P-SP2-A | 26 | 1 | 27 | ||||||
| Y71in.2P-SP12 | 8 | 12 | 20 | ||||||
| Y71out.2P-SP2-B | 6 | 10 | 2 | 1 | 19 | ||||
| Y71out.2P-BINDER | 1 | 8 | 4 | 13 | |||||
| Y71in.3P-R | 11 | 37 | 7 | 34 | 8 | 97 | |||
| Y71in.3P-SP12-A | 14 | 11 | 2 | 4 | 31 | ||||
| Y71out.3P-T | 11 | 1 | 6 | 2 | 20 | ||||
| Y71in.3P-SP12-B | 7 | 3 | 2 | 12 | |||||
| SW2 Outlier | 15 | 47 | 50 | 47 | 1 | 65 | 25 | 250 | |
| SW2 Disordered | 29 | 28 | 11 | 29 | 3 | 58 | 4 | 18 | 180 |
| All | 85 | 138 | 93 | 152 | 7 | 171 | 5 | 70 | 721 |
Figure 4.

SW1 and SW2 conformations associated with inhibitor sites. A and B, Observed and predicted, SW1/SW2 pockets (SP12; A) and SW2 pockets (SP2; B) across RAS structures in the PDB. C, Pocket volumes and druggability scores across inhibitor-bound and unbound SP2, SP12, or other sites. D and E, Y71 positions in SP12 (D) and SP2 (E) inhibitor–bound structures. F–I, SW1 and SW2 conformations in RAS structures with an inhibitor-bound SP12 site (F and G) and an inhibitor-bound SP2 site (H and I). J–M, Percent of each SW1 and SW2 conformation bound to inhibitors with different chemistries at the SP12 site (J and K) and the SP2 site (L and M). J–M are colored by the same scheme, with gray indicating structures labeled outlier or disordered (F–I).
Examining the calculated results for the SP12 and SP2 sites (Supplementary Dataset S3), we found that inhibitor-bound pockets correlated with higher druggability scores (>0.5) than inhibitor-unbound pockets (Fig. 4C). Other unbound sites were detected but with lower pocket volumes (<500 Å3) and druggability scores (<0.5) than the SP12 and SP2 sites. Importantly, we found that the structures bound to SP12 inhibitors were mainly Y71in (90.2%; n = 77 of 85), possessing an exposed SP12 site but an SP2 site occluded by the buried Y71 (Fig. 4D), while the structures bound to SP2 inhibitors by contrast were mainly Y71out (92.3%; n = 86 of 93), with an exposed SP2 site but an SP12 site occluded by the exposed Y71 (Fig. 4E)
Further analyzing the conformational preferences of inhibitor-bound and unbound structures, we found that SP12 inhibitors preferentially bind to the SW1 conformation, Y32in.3P-ON, and the SW2 conformations, Y71in.3P-R, Y71in.3P-SP12-A, Y71in.3P-SP12-B, and Y71in.2P-SP12 (Fig. 4F and G). SP2 drugs alternatively bind to structures with the SW1 conformation, Y32out.2P-OFF, and the SW2 conformations, Y71out.2P-SP2-A and Y71out.2P-SP2-B (Fig. 4H and I). For both SP12 and SP2 sites, the inhibitor-bound and unbound structures had similar distributions of SW1 and SW2 conformations (Table 3). Notably, several SW1 or SW2 conformation that were found in both inhibitor-bound and unbound structures correlated with the highest druggability scores (Supplementary Fig. S3), indicating that these inhibitor-binding conformations may be the most optimal targets for small molecule inhibition. In further support, no outlier conformations were found within the set of SP2 unbound sites with druggability scores >0.5, and only two structures were identified within the set of SP12 unbound sites with scores greater than this cutoff (PDB: 1XCM and 4EFN, which are classified by some as GTP-bound state 1; ref. 48).
Recently, SP2 inhibitors with divergent core chemistries were found to bind to different SW2 configurations (34, 35), but the conformational binding preferences of other SP12 and SP2 inhibitor chemistries are entirely unknown. Therefore, we subdivided the SP12 and SP2 inhibitors by chemistries, focusing on inhibitor classes discussed repeatedly in the literature, and examined to which SW1 and SW2 conformations each inhibitor chemistry binds to (Supplementary Table S4). The inhibitor chemistries analyzed included indole, benzodioxane, and biphenyl for SP12 inhibitors (49–54) and acrylamide and sulfonamide for SP2 inhibitors (36, 55). SP12.Indole compounds preferred binding to structures with the SW1 conformation, Y32in.3P-ON and Y32out.2P-OFF, and the SW2 conformations, Y71in.3P-R, Y71in.2P-SP12, or Y71in.3P-SP12-A (Fig. 4J and K); these SP12.Indole compounds included RAS-SOS1 inhibitors (e.g., DCAI; refs. 56, 57) and those that block multiple key RAS interactions (e.g., BI-2852, Cmpd2; ref. 58). Most SP12.Indole compounds were found targeting KRAS G12D–mutated structures in the PDB (Supplementary Dataset S1). In contrast, SP12.Benzodioxane and SP12.Biphenyl compound, which block key RAS-effector interactions (e.g., PPIN-1, PPIN-2; ref. 50), were found to preferentially bind to structures with the SW1 conformation, Y32in.3P-ON, and the SW2 conformations, Y71in.3P-SP12-A or Y71in.3P-SP12-B (Fig. 4J and K); these inhibitors were mostly found targeting KRAS Q61H structures in the PDB (Supplementary Dataset S1). Lastly, SP2.Acrylamide compounds, which include the well-known KRAS G12C (covalent) inhibitors (e.g., sotorasib/AMG 510, adagrasib/MRTX849; refs. 36, 55), were found to preferentially bind to structures with the SW1 conformation, Y32out.2P-OFF, and the SW2 conformations, Y71out.2P-SP2-A or Y71out.2P-SP2-B (Fig. 4L and M). SP2.Sulfonamide compounds, which are another class of KRAS G12C (covalent) inhibitors, bound only to outlier or disordered labeled structures.
Structural impact of G12D and G12V mutations on intrinsic and GAP-mediated hydrolysis
Currently, there are at least 10 structures available in the PDB for each of the G12D, G12V, G12C, G13D, and Q61H mutated forms (Supplementary Dataset S1). Here, we leverage our prepared dataset of RAS structures to elucidate a proposed hypothesis regarding the active site configuration required for intrinsic and GAP-mediated hydrolysis and the structural consequence of the two most common RAS mutations, G12D and G12V, on these activities.
Previously, Mattos and colleagues proposed that the G12D and G12V mutations may alter intrinsic hydrolysis by shifting the equilibrium between GTP-bound substates defined by the H-bond type made between the hydroxyl (OH) atom of Y32 and the closest γ-phosphate oxygen (called here O1G) atom of GTP or GTP analogues: one within direct hydrogen (H)-bonding distance, which they theorized to be catalytically incompetent, and another within water-mediated (WM) H-bonding distance, which they theorized to be catalytically competent (59–61). Given the previous observation of multiple Y32 positions in RAS–effector (Fig. 3E) and RAS–GAP (Fig. 3G) cocomplexes, we wondered if there are possibly two or more hydrolytically relevant substates within the GTP-bound equivalent (i.e., 3P) structure. Examining the distribution of distances between the Y32(OH) and 3P(O1G) atoms (Supplementary Dataset S4), we found three peaks at distances of 3, 4.5, and 7 Å, which we associated with the observance of WM, direct, and no H-bonds, respectively (Fig. 5A).
Figure 5.

Structural impact of G12D and G12V mutations on GTP-bound substate preference. A, Distance distribution within 3P structure between the hydroxyl (OH) atom of residue Y32 and closest γ-phosphate (called here O1G) atom of GTP or GTP analogues, which was used to define hydrogen (H)-bonding subtypes: water-mediated (WM) H-bond, direct H-bond, and no H-bond. B–D, 3P substate preference within WT (B), G12D (C), and G12V (D) structures.
Next, we compared the 3P substate preferences of the WT, G12D, and G12V structures (included KRAS and HRAS isoforms) with the SW1 conformation, Y32in.3P-ON, and the SW2 conformation, Y71in.3P-R, since these loop configurations define the active form of RAS proteins that occurs immediately before their GTP to GDP transition. While WT structures (n = 23) had a nearly even distribution of 3P substates (Fig. 5B), 94% of G12D structures (n = 17 of 18) were observed in the WM H-bond substate (Fig. 5C) and 100% of G12V structures (n = 10 of 10) were found in the direct H-bond substate (Fig. 5D). The preference of G12V and G12D mutated structures for the WM H-bond (catalytically competent) and direct H-bond (catalytically incompetent) substates, respectively, may explain why G12V mutations severely impair intrinsic hydrolysis while G12D mutations only slightly dampen the hydrolytic reaction (8). Of note, we found that Y32 is stabilized in the WM H-bond substate in G12D-mutated structures by residue D12 replacing the location of the water molecule (Fig. 5C), suggesting that intrinsic hydrolysis can still be preserved if Y32 is held within WM H-bonding distance of the GTP γ-phosphate. Relating the identified 3P substates to GAP-mediated hydrolysis, we propose that the preference of G12D- and G12V-mutated structures for substates other than no H-bond is the likely reason why GAP-mediated hydrolysis is almost nonexistent in the context of these mutations (4).
Discussion
Ever since the first HRAS structures were experimentally solved in 1990 (62, 63), researchers have focused on characterizing the RAS conformational landscape by examining the possible structural configurations of their catalytic SW1 and SW2 loops. In this study, we used an extended dataset (721 human RAS structures, 67% of which were solved within the past 5 years), and an approach that differs from previous studies (64, 65), to create a data-driven RAS conformational classification, identifying three SW1 and nine SW2 RAS conformations. Our approach can be used to automatically conformationally classify and annotate the molecular contents of additional RAS structures as they are experimentally solved, providing a clear and consistent method for comparing WT and mutated structures across various biological and inhibitory contexts. To facilitate future RAS structural analyses, we have created a database, called Rascore, presenting the results of this study, which includes a page to conformationally classify user-uploaded RAS structures (http://dunbrack.fccc.edu/rascore/).
An uncertainty faced in defining our RAS conformational classification was identifying which GTP-bound SW1 conformations are state 1 and state 2. These SW1 conformations were discovered in the early 2000s with the observation of two peaks in 31P NMR spectra for GTP α and γ phosphates (66). Later studies found that mutations in residues Y32 and T35, as well as the common G12V mutation, cause a shift to the state 1-associated peaks, while other mutations, such as G12D, and the presence of the signaling effector, RAF1, cause a shift to state 2 (67–69). Subsequently, other researchers experimentally solved a potential state 1 structure using a T35S mutant construct (70), and following for WT, G12V, Q61L, and other mutations (48, 71). However, the previously labeled state 1 structures, which are found within our classified set of Y32out.3P structures, occurred too infrequently in this analysis to unambiguously call them the biological state 1 conformation. Moreover, the identified H-bonding substates within 3P structures (WM, direct, and no H-bond), could also explain the split state 1 and state 2 peaks in NMR spectra. Considering the NMR studies described above, and that we found that the G12D and G12V mutated structures prefer the WM and direct H-bond substates, respectively, we propose that the WM H-bond substate is most consistent with state 2 and that state 1 is either the direct or no H-bond substates, some part of Y32out.3P, or some mixture of these structural configurations.
In contrast to other studies (64, 65), we associated each SW1 and SW2 conformation with RAS interactions involving bound proteins and small molecular inhibitors. Overall, our analysis confirmed several previously held hypotheses regarding RAS conformations in a large experimental dataset and helped us uncover some new hidden trends. For example, it has been hypothesized that RAS preferentially binds to signaling effectors and the GEF.REM domain of SOS1 when its SW1 conformation is GTP-bound “state 2” (our Y32in.3P-ON) and its SW2 conformation is in the “R state” (our Y71in.3P-R; refs. 13, 32, 33). We confirmed these observations and discovered that these same conformations bind to GAPs as well. In addition, we validated that both GTP-bound and GDP-bound RAS structures can form the α4α5 homodimer, as was previously shown through NMR experiments of KRAS (14). Furthermore, we found that both the active, Y71in.3P-R and inactive, Y71out.3P-T SW2 conformations can α4α5 homodimerize, which was an observation not previously reported.
Another major value of this work is that it defined a comprehensive set of RAS conformations that are known targets for small-molecule or designed protein inhibitors. Six out of seven druggable SW2 conformations are newly characterized in this study (all except for Y71in.3P-R); these include: GTP-bound Y71in.3P-SP12-A and Y71in.3P-SP12-B, and GDP-bound Y71in.2P-SP12, Y71out.2P-SP2-A, Y71out.2P-SP2-B, and Y71out.2P-BINDER. We associated these druggable conformations with their preference for binding small-molecule inhibitors with certain chemical substructures, which is information researchers can use to select appropriate structural templates during structure-based drug design. One general finding from our analysis was that all the identified druggable RAS conformations except one (Y71out.2P-SP2-A) exist in the absence of inhibitor-binding, indicating that these structural configurations may naturally occur within a biological context and are not solely the product of an induced fit model. In addition, we found that the SP2 inhibitor site is mainly present in structures with Y71 exposed to the solvent (Y71out), while the SP12 inhibitor site appears in structures with Y71 buried into the protein core (Y71in). The consistency of this finding among many inhibitor-bound structures suggests it is an essential determinant of SP12 and SP2 druggability.
While this study has expanded our understanding of RAS structural biology, it only marks the beginning of characterizing the RAS conformational landscape. We hope that our RAS conformational classification system will be paired with structure-activity relationship data to create machine learning models for RAS drug discovery. In addition, the pharmaceutical industry has over six times as many inhibitor-bound RAS structures as there are available in the PDB (72), and analysis of these structures using our conformational classification approach can help identify further druggable RAS conformations. Most importantly, having all the RAS structures in the PDB consistently annotated and conformationally classified will enable simple use of this growing structural dataset for informing RAS drug discovery and studies of RAS mutations in human cancers and other diseases.
Supplementary Material
Acknowledgments
The authors thank Bulat Faezov for providing the program PDBrenum in advance of publication, Vivek Modi for sharing scripts for calculating dihedral angles, and Simon Kelow for guidance in developing the conformational clustering algorithm. This work was funded by NIH NIGMS Grants F30 GM142263 (to M.I. Parker) and R35 GM122517 (to R.L. Dunbrack), Colon Cancer Alliance Funding (to J.E. Meyer), and the NIH NCI Core Grant P30 CA006927 (to Fox Chase Cancer Center).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors' Disclosures
J.E. Meyer reports grants from NCI, grants from Colorectal Cancer Alliance, grants and personal fees from Varian Medical Systems, and other support from Quantigic Genomics outside the submitted work. No disclosures were reported by the other authors.
Authors' Contributions
M.I. Parker: Conceptualization, resources, data curation, software, formal analysis, funding acquisition, validation, investigation, visualization, methodology, writing–original draft, project administration, writing–review and editing. J.E. Meyer: Supervision, funding acquisition, writing–review and editing. E.A. Golemis: Supervision, funding acquisition, writing–review and editing. R.L. Dunbrack: Conceptualization, resources, supervision, funding acquisition, validation, investigation, methodology, writing–original draft, project administration, writing–review and editing.
References
- 1. Prior IA, Hood FE, Hartley JL. The frequency of Ras mutations in cancer. Cancer Res 2020;80:2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lu S, Jang H, Muratcioglu S, Gursoy A, Keskin O, Nussinov R, et al. Ras conformational ensembles, allostery, and signaling. Chem Rev 2016;116:6607–65. [DOI] [PubMed] [Google Scholar]
- 3. Hofmann MH, Gerlach D, Misale S, Petronczki M, Kraut N. Expanding the reach of precision oncology by drugging all KRAS mutants. Cancer Discov 2022;12:924–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Simanshu DK, Nissley DV, McCormick F. RAS proteins and their regulators in human disease. Cell 2017;170:17–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boriack-Sjodin PA, Margarit SM, Bar-Sagi D, Kuriyan J. The structural basis of the activation of Ras by Sos. Nature 1998;394:337–43. [DOI] [PubMed] [Google Scholar]
- 6. Scheffzek K, Ahmadian MR, Kabsch W, Wiesmüller L, Lautwein A, Schmitz F, et al. The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science 1997;277:333–8. [DOI] [PubMed] [Google Scholar]
- 7. Kessler D, Bergner A, Böttcher J, Fischer G, Döbel S, Hinkel M, et al. Drugging all RAS isoforms with one pocket. Future Med Chem 2020;12:1911–23. [DOI] [PubMed] [Google Scholar]
- 8. Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, Westover KD. Biochemical and structural analysis of common cancer-associated KRAS mutations. Mol Cancer Res 2015;13:1325. [DOI] [PubMed] [Google Scholar]
- 9. Rabara D, Tran TH, Dharmaiah S, Stephens RM, McCormick F, Simanshu DK, et al. KRAS G13D sensitivity to neurofibromin-mediated GTP hydrolysis. Proc Natl Acad Sci U S A 2019;116:22122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Johnson CW, Lin Y-J, Reid D, Parker J, Pavlopoulos S, Dischinger P, et al. Isoform-specific destabilization of the active site reveals a molecular mechanism of intrinsic activation of KRas G13D. Cell Rep 2019;28:1538–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Poulin EJ, Bera AK, Lu J, Lin YJ, Strasser SD, Paulo JA, et al. Tissue-specific oncogenic activity of KRAS(A146T). Cancer Discov 2019;9:738–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Buhrman G, Wink G, Mattos C. Transformation efficiency of RasQ61 mutants linked to structural features of the switch regions in the presence of Raf. Structure 2007;15:1618–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Margarit SM, Sondermann H, Hall BE, Nagar B, Hoelz A, Pirruccello M, et al. Structural evidence for feedback activation by Ras.GTP of the Ras-specific nucleotide exchange factor SOS. Cell 2003;112:685–95. [DOI] [PubMed] [Google Scholar]
- 14. Lee KY, Fang Z, Enomoto M, Gasmi-Seabrook G, Zheng L, Koide S, et al. Two distinct structures of membrane-associated homodimers of GTP- and GDP-bound KRAS4B revealed by paramagnetic relaxation enhancement. Angew Chem Int Ed Engl 2020;59:11037–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Spencer-Smith R, Koide A, Zhou Y, Eguchi RR, Sha F, Gajwani P, et al. Inhibition of RAS function through targeting an allosteric regulatory site. Nat Chem Biol 2017;13:62–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang G, Dunbrack RL Jr. PISCES: a protein sequence culling server. Bioinformatics 2003;19:1589–91. [DOI] [PubMed] [Google Scholar]
- 17. Faezov B, Dunbrack RL Jr. PDBrenum: A webserver and program providing Protein Data Bank files renumbered according to their UniProt sequences. PLoS One 2021;16:e0253411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Meyder A, Nittinger E, Lange G, Klein R, Rarey M. Estimating electron density support for individual atoms and molecular fragments in X-ray structures. J Chem Inf Model 2017;57:2437–47. [DOI] [PubMed] [Google Scholar]
- 19. Fährrolfes R, Bietz S, Flachsenberg F, Meyder A, Nittinger E, Otto T, et al. ProteinsPlus: a web portal for structure analysis of macromolecules. Nucleic Acids Res 2017;45:W337–W43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Maietta P, Lopez G, Carro A, Pingilley BJ, Leon LG, Valencia A, et al. FireDB: a compendium of biological and pharmacologically relevant ligands. Nucleic Acids Res 2014;42:D267–D72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021;596:583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Evans R, O'Neill M, Pritzel A, Antropova N, Senior A, Green T, et al. Protein complex prediction with AlphaFold-Multimer. bioRxiv 2021. [Google Scholar]
- 23. Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, Steinegger M. ColabFold - Making protein folding accessible to all. bioRxiv 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kelow SP, Adolf-Bryfogle J, Dunbrack RL. Hiding in plain sight: structure and sequence analysis reveals the importance of the antibody DE loop for antibody-antigen binding. MAbs 2020;12:1840005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Modi V, Dunbrack RL. Defining a new nomenclature for the structures of active and inactive kinases. Proc Natl Acad Sci U S A 2019;116:6818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shapovalov M, Vucetic S, Dunbrack RL Jr. A new clustering and nomenclature for beta turns derived from high-resolution protein structures. PLoS Comput Biol 2019;15:e1006844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. North B, Lehmann A, Dunbrack RL Jr. A new clustering of antibody CDR loop conformations. J Mol Biol 2011;406:228–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ester M, Kriegel H-P, Sander J, Xu X. A density-based algorithm for discovering clusters in large spatial databases with noise. Proceedings of the Second International Conference on Knowledge Discovery and Data Mining. Portland, Oregon: AAAI Press; 1996. p226–31. [Google Scholar]
- 29. Jiang L, Kuhlman B, Kortemme T, Baker D. A “solvated rotamer” approach to modeling water-mediated hydrogen bonds at protein-protein interfaces. Proteins 2005;58:893–904. [DOI] [PubMed] [Google Scholar]
- 30. Bera AK, Lu J, Wales TE, Gondi S, Gurbani D, Nelson A, et al. Structural basis of the atypical activation mechanism of KRAS(V14I). J Biol Chem 2019;294:13964–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dharmaiah S, Tran TH, Messing S, Agamasu C, Gillette WK, Yan W, et al. Structures of N-terminally processed KRAS provide insight into the role of N-acetylation. Sci Rep 2019;9:10512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Holzapfel G, Buhrman G, Mattos C. Shift in the equilibrium between on and off states of the allosteric switch in Ras-GppNHp affected by small molecules and bulk solvent composition. Biochemistry 2012;51:6114–26. [DOI] [PubMed] [Google Scholar]
- 33. Matsumoto S, Miyano N, Baba S, Liao J, Kawamura T, Tsuda C, et al. Molecular mechanism for conformational dynamics of Ras·GTP elucidated from in-situ structural transition in crystal. Sci Rep 2016;6:25931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gentile DR, Rathinaswamy MK, Jenkins ML, Moss SM, Siempelkamp BD, Renslo AR, et al. Ras binder induces a modified switch-II pocket in GTP and GDP states. Cell Chem Biol 2017;24:1455–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zeng M, Lu J, Li L, Feru F, Quan C, Gero TW, et al. Potent and selective covalent quinazoline inhibitors of KRAS G12C. Cell Chem Bio 2017;24:1005–16. [DOI] [PubMed] [Google Scholar]
- 36. Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013;503:548–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ramachandran GN, Ramakrishnan C, Sasisekharan V. Stereochemistry of polypeptide chain configurations. J Mol Biol 1963;7:95–9. [DOI] [PubMed] [Google Scholar]
- 38. Girard E, Lopes P, Spoerner M, Dhaussy A-C, Prangé T, Kalbitzer HR, et al. Equilibria between conformational states of the Ras oncogene protein revealed by high pressure crystallography. Chem Sci 2022;13:2001–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kalbitzer HR, Rosnizeck IC, Munte CE, Narayanan SP, Kropf V, Spoerner M. Intrinsic allosteric inhibition of signaling proteins by targeting rare interaction states detected by high-pressure NMR spectroscopy. Angew Chem Int Ed 2013;52:14242–6. [DOI] [PubMed] [Google Scholar]
- 40. Li Y, Zhang Y, Großerüschkamp F, Stephan S, Cui Q, Kötting C, et al. Specific substates of ras to interact with GAPs and effectors: Revealed by theoretical simulations and FTIR experiments. J Phys Chem Lett 2018;9:1312–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Teng KW, Tsai ST, Hattori T, Fedele C, Koide A, Yang C, et al. Selective and noncovalent targeting of RAS mutants for inhibition and degradation. Nat Commun 2021;12:2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bery N, Legg S, Debreczeni J, Breed J, Embrey K, Stubbs C, et al. KRAS-specific inhibition using a DARPin binding to a site in the allosteric lobe. Nat Commun 2019;10:2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Guillard S, Kolasinska-Zwierz P, Debreczeni J, Breed J, Zhang J, Bery N, et al. Structural and functional characterization of a DARPin which inhibits Ras nucleotide exchange. Nat Commun 2017;8:16111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tanaka T, Williams RL, Rabbitts TH. Tumour prevention by a single antibody domain targeting the interaction of signal transduction proteins with RAS. EMBO J 2007;26:3250–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Haza KZ, Martin HL, Rao A, Turner AL, Saunders SE, Petersen B, et al. RAS-inhibiting biologics identify and probe druggable pockets including an SII-α3 allosteric site. Nat Commun 2021;12:4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xu Q, Dunbrack RL. ProtCID: a data resource for structural information on protein interactions. Nat Commun 2020;11:711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Le Guilloux V, Schmidtke P, Tuffery P. Fpocket: an open source platform for ligand pocket detection. BMC Bioinf 2009;10:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Muraoka S, Shima F, Araki M, Inoue T, Yoshimoto A, Ijiri Y, et al. Crystal structures of the state 1 conformations of the GTP-bound H-Ras protein and its oncogenic G12V and Q61L mutants. FEBS Lett 2012;586:1715–8. [DOI] [PubMed] [Google Scholar]
- 49. Cruz-Migoni A, Canning P, Quevedo CE, Bataille CJR, Bery N, Miller A, et al. Structure-based development of new RAS-effector inhibitors from a combination of active and inactive RAS-binding compounds. Proc Natl Acad Sci U S A 2019;116:2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Quevedo CE, Cruz-Migoni A, Bery N, Miller A, Tanaka T, Petch D, et al. Small molecule inhibitors of RAS-effector protein interactions derived using an intracellular antibody fragment. Nat Commun 2018;9:3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bery N, Cruz-Migoni A, Bataille CJR, Quevedo CE, Tulmin H, Miller A, et al. BRET-based RAS biosensors that show a novel small molecule is an inhibitor of RAS-effector protein-protein interactions. Elife 2018;7:e37122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen F-Y, Li X, Zhu H-P, Huang W. Regulation of the Ras-related signaling pathway by small molecules containing an indole core scaffold: a potential antitumor therapy. Front Pharmacol 2020;11:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Burns MC, Sun Q, Daniels RN, Camper D, Kennedy JP, Phan J, et al. Approach for targeting Ras with small molecules that activate SOS-mediated nucleotide exchange. Proc Natl Acad Sci U S A 2014;111:3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Abbott JR, Hodges TR, Daniels RN, Patel PA, Kennedy JP, Howes JE, et al. Discovery of aminopiperidine indoles that activate the guanine nucleotide exchange factor SOS1 and modulate RAS signaling. J Med Chem 2018;61:6002–17. [DOI] [PubMed] [Google Scholar]
- 55. Ostrem JM, Shokat KM. Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat Rev Drug Discov 2016;15:771–85. [DOI] [PubMed] [Google Scholar]
- 56. Sun Q, Burke JP, Phan J, Burns MC, Olejniczak ET, Waterson AG, et al. Discovery of small molecules that bind to K-Ras and inhibit Sos-mediated activation. Angew Chem Int Ed Engl 2012;51:6140–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Maurer T, Garrenton LS, Oh A, Pitts K, Anderson DJ, Skelton NJ, et al. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc Natl Acad Sci U S A 2012;109:5299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kessler D, Gmachl M, Mantoulidis A, Martin LJ, Zoephel A, Mayer M, et al. Drugging an undruggable pocket on KRAS. Proc Natl Acad Sci U S A 2019;116:15823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Johnson CW, Mattos C. Chapter Three - The allosteric switch and conformational states in Ras GTPase affected by small molecules. In: Tamanoi F., editor. The enzymes: Academic Press; 2013. p41–67. [DOI] [PubMed] [Google Scholar]
- 60. Buhrman G, Holzapfel G, Fetics S, Mattos C. Allosteric modulation of Ras positions Q61 for a direct role in catalysis. Proc Natl Acad Sci U S A 2010;107:4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Buhrman G, Kumar VSS, Cirit M, Haugh JM, Mattos C. Allosteric modulation of Ras-GTP is linked to signal transduction through RAF kinase*. J Biol Chem 2011;286:3323–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Pai EF, Krengel U, Petsko GA, Goody RS, Kabsch W, Wittinghofer A. Refined crystal structure of the triphosphate conformation of H-ras p21 at 1.35 A resolution: implications for the mechanism of GTP hydrolysis. Embo j 1990;9:2351–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Milburn MV, Tong L, deVos AM, Brunger A, Yamaizumi Z, Nishimura S, et al. Molecular switch for signal transduction: structural differences between active and inactive forms of protooncogenic ras proteins. Science 1990;247:939. [DOI] [PubMed] [Google Scholar]
- 64. Li H, Yao X-Q, Grant BJ. Comparative structural dynamic analysis of GTPases. PLoS Comput Biol 2018;14:e1006364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gorfe AA, Grant BJ, McCammon JA. Mapping the nucleotide and isoform-dependent structural and dynamical features of Ras proteins. Structure 2008;16:885–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Spoerner M, Herrmann C, Vetter IR, Kalbitzer HR, Wittinghofer A. Dynamic properties of the Ras switch I region and its importance for binding to effectors. Proc Natl Acad Sci U S A 2001;98:4944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Spoerner M, Wittinghofer A, Kalbitzer HR. Perturbation of the conformational equilibria in Ras by selective mutations as studied by 31P NMR spectroscopy. FEBS Lett 2004;578:305–10. [DOI] [PubMed] [Google Scholar]
- 68. Smith MJ, Neel BG, Ikura M. NMR-based functional profiling of RASopathies and oncogenic RAS mutations. Proc Natl Acad Sci U S A 2013;110:4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Parker JA, Volmar AY, Pavlopoulos S, Mattos C. K-Ras Populates conformational states differently from its isoform H-ras and oncogenic mutant K-RasG12D. Structure 2018;26:810–20. [DOI] [PubMed] [Google Scholar]
- 70. Shima F, Ijiri Y, Muraoka S, Liao J, Ye M, Araki M, et al. Structural basis for conformational dynamics of GTP-bound Ras protein. J Biol Chem 2010;285:22696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lu J, Bera AK, Gondi S, Westover KD. KRAS switch mutants D33E and A59G crystallize in the state 1 conformation. Biochemistry 2018;57:324–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mullard A. What does AlphaFold mean for drug discovery? Nat Rev Drug Discov 2021;20:725–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available as static tables in Supplementary Datasets S1-S4. In addition, our database, called Rascore, presents a continually updated dataset of all human RAS structures in the PDB annotated by their molecular contents and conformationally classified (http://dunbrack.fccc.edu/rascore/, RRID:SCR_022180). The Rascore database includes a page to conformationally classify user-uploaded structures. PyMOL sessions for all figures from the paper can be downloaded as well from the Rascore database. Open-source code pertaining to our conformational classification algorithm and the Rascore database can be found in GitHub (https://github.com/mitch-parker/rascore) and Code Ocean (https://codeocean.com/capsule/7782236/tree/v1).