

Abstract
Introduction:
Cystic fibrosis transmembrane conductance regulator (CFTR)-mediated chloride and bicarbonate secretion is integral to the pancreas’ ability to produce the alkaline pancreatic juice required for proper activation of enzymes for digestion. Disruption in this process increases the risk for pancreatitis.
Areas Covered:
Using original basic and clinical research, as well as clinical case reports and recent reviews indexed in PubMed, we discuss why patients with CFTR dysfunction are at risk for pancreatitis. We also discuss diagnostic modalities for assessing CFTR function, as well as new therapeutic advancements and the impact these are having on pancreatic function, pancreatitis in particular.
Expert Opinion:
CFTR-related pancreatitis occurs in the presence of monallelic or biallelic mutations and/or from toxin-mediated channel disruption. Research-based CFTR diagnostics have been expanded, yet all current methods rely on measuring CFTR chloride transport in non-pancreatic cells/tissue. Newer CFTR-directed therapies (“CFTR modulators”) are both improving pancreatitis (pancreatic-sufficient CF patients) and increasing the risk for pancreatitis (previously pancreatic-insufficient CF patients). Our experiences with these drugs are enlightening us on how CFTR modulation can affect pancreatitis risk across a wide spectrum of pancreatic disease, and represents an opportunity for therapeutic relief from pancreatitis in those without CF, but who suffer from CFTR-related pancreatitis.
Keywords: CF, cystic fibrosis, recurrent pancreatitis, chronic pancreatitis, hereditary pancreatitis CFTR modulators
1. Introduction
The cystic fibrosis transmembrane conductance regulator (CFTR) protein is expressed in various epithelial and non-epithelial tissues throughout the body, but plays a particularly prominent role in epithelial anion transport (especially chloride and/or bicarbonate). CFTR dysfunction is most closely linked with cystic fibrosis (CF), one of the most common life-shortening genetic diseases, which is caused by biallelic pathogenic mutations in CFTR. Over 2,000 mutations in the CFTR gene have bene identified with varying phenotypic properties. CF, and by extension non-CF CFTR dysfunction, is most commonly thought to affect the lungs where lung failure is the primary cause of mortality in CF, and CFTR mutations have been linked to chronic obstructive pulmonary disease and asthma.[1–3] However, CFTR is expressed throughout most of the gastrointestinal (GI) tract, liver, and pancreas.[4] It is highly expressed on the apical aspect of the pancreatic duct, making the pancreas particularly sensitive to CFTR mutations.[5, 6] CFTR-mediated chloride and bicarbonate secretion is integral to the pancreas’ ability to produce the alkaline pancreatic juice required for proper activation of enzymes for digestion and absorption of nutrients.[7, 8] The pancreatic disease resulting from CFTR dysfunction is in fact its defining phenotype of CF, named as such in 1938 when Dr. Dorothy Anderson termed the pancreatic changes she noted in autopsy findings of children who died from malnutrition as “cystic fibrosis of the pancreas”.[9, 10] Dr. Sydney Farber later described cases of pancreatic fibrosis associated with meconium ileus, chronic pneumonia and nutritional failure and suggested that these findings are the result of a systemic disease.[11] Di Sant’Agnese et al reported abnormal electrolyte concentrations in sweat in several patients with “cystic fibrosis of the pancreas” who were undergoing mild thermal stress in New York City in 1953. [12] Gibson and Cooke later measured electrolytes in sweat of patients diagnosed with “cystic fibrosis of the pancreas” by introducing pilocarpine into the skin using iontophoresis and found elevated sweat chloride in all patients.[13] These studies provided early evidence of a channelopathy resulting in phenotypic disease. Drs. Lap-Chee Tsui, Francis Collins, and their teams later discovered the first mutation in CFTR, thereby setting the stage for years of genotype-phenotype research aimed at improving the lives of people with CF and CFTR dysfunction.[14]
The purpose of this review is to outline pancreatitis as it relates to CFTR dysfunction, with special attention to the evolving data on the diagnosis of CFTR-related pancreatic dysfunction and emerging therapeutic opportunities for CFTR-related pancreatitis. We will also address remaining unmet needs and areas of research requiring special attention.
1.1. Background
Pancreatitis refers to “inflammation of the pancreas” and is characterized by both local and systemic inflammatory responses. It is in fact, the most common cause of hospitalization due to a gastrointestinal (GI) condition, with an average annual incidence of 34 per 100,000 person years in high-income countries, although this varies by age.[15–17] A recent review of pancreatitis in children suggested that while the incidence of pancreatitis in children is lower than adults, it remains an important cause of GI disease in children.[18] Chronic pancreatitis refers to the irreversible damage rendered in the pancreas, often as a consequence of repetitive episodes of pancreatic inflammation. Chronic pancreatitis is a devastating disease in both adults and children and frequently leads to intractable pain, impaired nutrition and significantly poorer quality of life. Despite this, treatment options remain limited.[19, 20] The association between CFTR mutations and the development of chronic hereditary pancreatitis was first highlighted in the late 1990s.[21, 22] Physiologically, the CFTR protein functions as a cyclic nucleotide-regulated ion channel that allows for anion movement, particularly chloride and bicarbonate, along their electrochemical gradient, contributing to fluid and electrolyte secretion. The exocrine pancreas is particularly susceptible to CFTR dysfunction.[23]
Animal studies further aid our understanding of the effect of CFTR dysfunction in pancreatic disease. They help to elucidate pathways of disease that may mirror human pathophysiology, and allow for further understanding of disease pathogeneisis and/or potential therapeutic targets to treat pancreatic disease. Myerholz et al examined pathological specimens of pigs with CF and reported ubiquitous pancreatic lesions. The authors reported that acinar cells were reduced in number with decreased cytoplasmic zymogen granules. In addition, the pancreatic ductal lumen was filled with mucous and altered zymogen material.[24] Abu-El-Haija et al also reported on similar pancreatic damage in fetal CF pigs with increased expression of proinflammatory and profibrotic genes, compared to non-CF pigs.[25] Mouse models of CF show similar findings.[26] Navis et al reported on the loss of pancreatic tissue in larval zebrafish with CFTR dysfunction, and exocrine pancreatic dysfunction is also noted in CF ferret models. [27, 28] Both animal and human studies support the finding that CFTR dysfunction can lead to both acute, and subsequently acute recurrent or chronic pancreatitis. Thus, it is valuable to gain a deep understanding of the pathophysiologic role that CFTR dysfunction plays in pancreatitis and the potential diagnostic value of measuring CFTR function in pancreatitis. Furthermore, therapeutic opportunities to target CFTR modulation will be increasingly important in ameliorating CFTR-related pancreatitis in children and adults with or without CF.
1.2. Diagnostic Techniques in CF and CFTR-Related Disorders
Because CFTR dysfunction may lead to pancreatic disease, testing for CF or a CFTR-related disorder should be part of the work up of patients with recurrent acute or chronic pancreatic disease. The diagnosis of CF and CFTR-related disorders was outlined in recent guidelines published by the Cystic Fibrosis Foundation.[29] The diagnosis of CF is not always straightforward. At times, even with comprehensive evaluation the diagnosis can remain elusive, especially in inidivuduals with single-organ manifestation of CF.[30] In general, the diagnosis of CF can be made with family history or clinical symptoms consistent with CF, and a sweat chloride measurement of >60 mmol/L.[29] Patients with indeterminate sweat chloride levels, defined as 30–59 mmol/L require further testing with CF genetic analysis and potentially alternative CFTR functional testing. Patients found to have 2 pathogenic mutations receive a CF diagnosis. Advancements in genetic testing have accelerated our ability to detect CFTR gene mutations. However, approaches to genetic testing are not all the same. Commonly, gene panels are utilized which contain a set number of frequently occurring CFTR mutations. These gene panels are generally highly effective at identifying CFTR mutations in non-Hispanic White individuals, where CF has been most widely characterized and the F508del mutation is common (~90% of CF individuals that are White). However, there is increasing appreciation that non-White ethnic groups may have CFTR mutations that are distinct and may not be present in these panels. For example, in California, which has an ethnically diverse population, up to 26% of new CF diagnoses have been found to have novel CFTR mutations.[31] As such, California integrates full CFTR gene sequencing (with deletions and duplications) into its newborn screening algorithm for CF. It is therefore recommended that when testing for possible CFTR mutations, full gene sequencing be considered, especially in non-White individuals.
A further challenge with genetic testing can be how to interpet the mutations identified. With over 2,000 identified CFTR variants, it is a challenge to know what mutations are pathogenic vs. non-pathogenic, and for clinicians, what types of symptoms may be attributed to a set of mutations. Substantial advances in this area occurred initially with development of the CFTR mutation database at Sick Kids Hospital in Toronto, Canada, followed by assembly of an international research group called “the clinical and functional translation of CFTR (CFTR2) team” by the CF Foundation in the United States. The CFTR2 project has assembled data regarding CFTR mutations from national registries of patients with CF. The data sets are primarily from North America, Europe and Australia but also contain data from the Middle East, Asia and South America. Although not fully-encompassing, the CFTR2 repository of data allows for better phenotypic understanding of CFTR mutations using clinical, functional and population/penetrance criteria.[32]
If a diagnosis of CF is unclear, patients may receive a diagnosis of CF-screen positive, inconclusive diagnosis (CFSPID) or CFTR-related metabolic syndrome (CRMS).[33] Infants with CFSPID/CRMS are at increased risk to receive a diagnosis of CF in the future compared to other infants.[34] Recent guidelines defined CFSPID/CRMS as a symptomatic infant with a positive newborn screen who has either (1) normal sweat chloride testing and two CFTR variants with unclear significance or (2) borderline sweat chloride testing with one or zero CF causing variants.[33] Terlizzi et al analyzed the role of CFTR genotype as a predictor of disease evaluation in children with CFSPID/CRMS. They assessed 43 people with at least one D1152H variant mutation in CFTR and divided people into two groups. This variant is classified in the CFTR2 database as having varying clinical consequences with heterogenous clinical expression. The D1152H variant has been frequently observed in patients with CFSPID/CRMS. Group A contained participants with one mutation in D1152H and a known disease-causing mutation, while group B contained participants with one mutation in D1152H and one mutation of unknown clinical significance. Of interest is that 3/28 people in group A (11.5%) had episodes of pancreatitis, whereas no pancreatitis was noted in group B. Study results ultimately showed that patients in group A were more likely to develop episodes of pancreatitis, have isolation of pseudomonas aeruginosa, and respiratory exacerbations in the first year of life. These participants also had a higher risk of evolving to CF.[35]
In addition to sweat chloride analysis and genetic testing, other methodologies such as in vitro intestinal or nasal current measurements using fresh biopsies or cultured cell monolayers, in vivo nasal potential difference (NPD), or organoid swelling assays can be used to evaluate CFTR function and/or diagnose CF in inconclusive cases.[36, 37] Additionally, there are several other types of sweat gland research tests for CFTR analysis, including beta-adrenergic sweat test, evaporimeter sweat test, and the individual sweat gland bubble test.[38–40] Minso et al report use of NPD and rectal biopsy ICM measurements in 219 individuals with symptoms of CF but inconclusive sweat testing or CFTR genetic testing. NPD measures epithelial sodium channel (ENaC)-mediated sodium and CFTR-mediated chloride ion conductance in nasal respiratory epithelium, while intestinal current measurements target CFTR-mediated chloride secretory responses to agonists that increase intracellular cyclic adenosine monophosphate (cAMP) (in this instance fresh rectal biopsy tissue). Using these techniques, the authors diagnosed CF in 79% of patients with a CFTR genotype of unknown or variable clinical significance, and identified 27% of individuals with partially dysfunctional CFTR despite no identified CFTR mutation.[41] All of the above use non-pancreatic tissue to identify CFTR dysfunction. Given the invasive nature of obtaining human pancreas tissue, these studies have generally not been done with pancreas tissue. Pancreatic bicarbonate concentration from endoscopic pancreatic function testing has been used to diagnose chronic pancreatitis.[42] Beyond this modality, there are few methodologies that measure CFTR-dependent bicarbonate transport, despite evidence that some CFTR mutations preferentially affect bicarbonate transport.[43] It remains unclear if and/or how non-pancreatic CFTR chloride and/or bicarbonate measurements can inform risk stratification for pancreatitis.
1.3. The role of CFTR in Genetic Pancreatitis
The association between mutations in CFTR and pancreatitis have been well described. Traditionally, pancreatitis episodes were noted to occur only in patients with less-severe CFTR mutations due to the near-complete or complete lack of pancreatic enzymes in those with more severe mutations.[44] Ahmed et al outlined how pancreatic ductular secretion of chloride and bicarbonate was compromised in all patients with CFTR mutations classes, but that residual function remained in people with milder class mutations, predisposing to development of pancreatitis. The paper further highlighted the strong genotype-phenotype correlation of CF mutation class to exocrine pancreatic function.[5] CFTR has since been understood to play a role in genetic/hereditary pancreatitis. Many patients with chronic pancreatitis attributed to chronic idiopathic pancreatitis were later determined to have genetic factors that predisposed them to this disease. Cohn et al examined whether idiopathic pancreatitis was associated with CFTR mutations in patients who did not have CF lung disease. They found that 10/27 patients (37%) had at least one abnormal CFTR mutation.[21] This study helped to highlight a statistically significant association between mutations in the CFTR gene and pancreatitis. Sharer et al similarly drew attention to this association where they found that 18/134 (13.4%) patients with chronic pancreatitis had a CFTR mutation on one allele. No patients had mutations on both alleles, and no patients were diagnosed with CF, further showing that heterozygous mutations in CFTR predispose to development of chronic pancreatitis.[22] LaRusch et al screened 984 cases of pancreatitis and found several variants of CFTR mutations that were not associated with CF but were associated with pancreatitis. The findings introduced a new class of CFTR functional variants, those which may preferentially affect bicarbonate secretion over chloride secretion, which were more common in patients with pancreatitis than controls.[43] Ooi et al examined whether CFTR genotype status determined the risk of pancreatitis in patients with CF. They found that there appeared to be a gradation of risk of developing pancreatitis according to CFTR genotype, with increased pancreatitis risk in patients who had milder CF mutations. The authors proposed a mechanism by which pancreatitis occurs when a critical threshold is reached between the balance of pancreatic acinar cell function and pancreatic ductal obstruction. They termed this risk the Pancreatic Insufficiency Prevalence (PIP) score, which is determined from the ratio between pancreatic sufficient patients carrying a specific mutation and all pancreatic sufficient and insufficient patients carrying the mutation. The PIP score allowed for classification of exocrine pancreatic function in CF beyond the CF class mutation system. The authors also note that not all patients who carried mild CF mutations developed pancreatitis and highlighted that additional modifier factors (genetic, environmental) may play a role.[44] [45] Palermo et al conducted a retrospective review of 50 patients enrolled in a pancreatitis registry at a single institution. They found a higher rate of CFTR mutations in patients with chronic pancreatitis compared to acute recurrent pancreatitis.[46] These results are similar to a recent report from the INSPPIRE Consortium (International Study Group of Pediatric Pancreatitis: In Search of a CuRe) where they identified that genetic mutations were one of the most common risk factors for the development of acute recurrent or chronic pancreatitis, with CFTR mutations being the most commonly identified gene, accounting for 34% of the genetic causes of pancreatitis.[47] CFTR mutations that are co-inherited with other pancreatitis-causing mutations further increases the risk of chronic pancreatitis. Schneider et al screened patients with idiopathic chronic pancreatitis and found that pathogenic CFTR variants co-inherited with serine protease inhibitor Kazal-type 1 (SPINK1) mutations significantly increased the risk of chronic pancreatitis. [48] Ooi et al also reported an increase in pancreatitis risk in patients with both CFTR and SPINK1 mutations. [49] The compounded effects of CFTR mutation with other pathogenic mutations must be considered when assessing patient risk for genetic pancreatitis.
1.4. Non-genetic causes of CFTR dysfunction leading to pancreatitis
In addition to genetic defects leading to CFTR dysfunction, environmental toxins can also cause CFTR downregulation and/or dysfunction, which can predispose to the development of pancreatitis. Perhaps the most common of these known toxins is cigarette smoke. Cantin et al examined the effects of cigarette smoke on CFTR expression and function in both smoker and non-smoker men who had no detectable CFTR mutations. They found that cigarette smoke decreased the expression of CFTR mRNA, protein, and function in vitro and that acquired CFTR deficiency occurred in the nasal respiratory epithelium of smokers.[50] Rasmussen et al identified that cigarette smoke induced prolonged calcium release into the cytoplasm which triggered removal of CFTR from the plasma membrane in human bronchial epithelial cultures, suggesting that calcium dysregulation may play an important role in acquired CFTR dysfunction.[51] The effect of smoking on CFTR does not appear to be limited to the airway; smoking is known to be an independent risk factor for pancreatitis.[52] Kadiyala et al compared pancreatic duct cell function in smokers and non-smokers by measurement of secretin-stimulated peak bicarbonate concentration in endoscopically collected pancreatic fluid. They found peak bicarbonate secretion to be significantly greater in the never smoker group when compared to the past or current smoker group. This suggests impairment of CFTR, the main protein responsible for bicarbonate secretion in the exocrine pancreas. Interestingly, the authors found lower bicarbonate secretion in both current and past smokers, with no significant difference between the groups, suggesting that cigarette smoke may have chronic and long-term effects on pancreatic function.[53] Furthermore, second-hand smoke exposure may also increase one’s risk of developing pancreatitis. Ballengee et al conducted a single-center, retrospective, observation study in which they examined patients aged 0–21 years with a diagnosis of acute pancreatitis. They demonstrated that children who were exposed to second-hand smoke experienced an increased number of acute pancreatitis episodes and increased length of hospital stay for their symptoms, compared to children with no cigarette smoke exposure.[54] Baker et al examined the role of tobacco smoke exposure in pediatric patients who were taking the CFTR modulator tezacaftor/ivacaftor. The investigators found that the therapeutic benefit of tezacaftor/ivacaftor, measured as the slope of change in lung function (FEV1), was nullified in patients who had tobacco smoke exposure.[55] This raises important questions about the effects of toxins from cigarette smoke on pancreatic function in patients on CF modulator therapies.
In addition to cigarette smoke, alcohol may play a role in CFTR dysfunction predisposing to pancreatitis. Excess consumption of ethanol may sensitize individuals to the development of acute and chronic pancreatitis.[56] One mechanistic explanation of the deleterious effects of alcohol consumption on pancreatitis is through the disruption of CFTR. Maleth et al measured CFTR activity based on sweat chloride concentrations in patients with CF, patients admitted to the emergency department due to excessive alcohol consumption, and healthy volunteers. They also measured CFTR levels and localization in patients with acute or chronic pancreatitis induced by alcohol. The authors found that low concentrations of ethanol stimulated CFTR and chloride/bicarbonate exchangers to increase bicarbonate and fluid, but high concentrations of ethanol inhibited these processes. Ethanol and its metabolites had deleterious effects on CFTR folding, expression, and membrane density.[57]
Thus, both genetic and environmental toxins that affect CFTR expression and/or function play an important role in presidposing to development of pancreatitis. Gaining a better understanding on toxin-mediated damage can help us to better highlight mechanistic pathways of pancreatic disease, thereby expanding treatment options to reduce disease burden.
1.5. CFTR Modulators and Pancreatitis
Pharmacologic treatments for pancreatitis were previously limited to lipid-lowering drugs for hypertriglyceridemia-induced pancreatitis. However, this changed with the Federal Drug Agency approval of ivacaftor in 2012 in the United States, followed by approval of lumacaftor/ivacaftor, tezacaftor/ivacaftor, and elexacaftor/tezacaftor/ivacaftor in 2015, 2018, and 2019, respectively. This group of CFTR correctors and potentiators are broadly known as “CFTR modulators” and were approved for CF patients with a defined set of CFTR mutations. Clinical experiences with CFTR modulator therapies has changed our landscape of knowledge with respect to CFTR dysfunction and pancreatitis. It was first suggested nearly a decade ago that the risk of pancreatitis may be altered by CFTR modulation.[44] Since the approval of ivafactor, multiple case reports have shown that CF children and adults with retained exocrine pancreatic function (“pancreatic sufficient” or “PS”) and a history of recurrent pancreatitis showed improvement in these episodes on CFTR modulator therapy. This literature is summarized in Table 1. As an example, Akshintala et al reported a series of fifteen adult patients with exocrine pancreatic sufficiency on CFTR modulator therapy. All patents had between 1–10 episodes of pancreatitis prior to modulator use. The study authors found that none of the patients on CFTR modulators had pancreatitis episodes while taking modulator therapy (Figure 1). [58] Furthermore, there have been multiple reports of patients with a history of exocrine pancreatic insufficiency who regained significant pancreatic function after initiation of CFTR modulators (Table 1). Emery et al assessed body mass index (BMI) and pancrealipase use in 28 patients with genetically confirmed CF and compared patients taking ivacaftor with those who were not. They found improved fecal elastase-1 (FE-1) levels, improved BMI, and decreased pancrealipase use in the patients taking ivacaftor vs. those not taking a CFTR modulator.[59] Thus, it is becoming increasingly clear that CFTR modulators can improve pancreatic health in CF patients. However, this improvement in exocrine pancreatic function can result in new pancreatic complications for some patients.
Table 1.
| Reference | Number of Patients | PI or PS | Age Range (Years) | CFTR Modulator(s) | Effect |
|---|---|---|---|---|---|
| Carrion et al | 6 | PS | 11.5 – 60 y | Ivacaftor | Reduced risk and frequency of ARP |
| Akshintla et al | 15 | PS | 27 – 76 y | Ivacaftor/Tezacaftor | Reduced risk and frequency of ARP |
| Hutchinson et al | 18 | PI | 1 – 11.4 y | Ivacaftor | 11/18 patients became PS |
| Munce et al | 3 | PI | 8 – 10 y | Ivacaftor | 2 patients became PS; one patient had improvement in ARP |
| Johns et al | 1 | PS | 24 y | Ivacaftor | Improvement in ARP |
ARP: acute recurrent pancreatitis; PI: pancreatic insufficient; PS: pancreatic sufficient; y: years
Figure 1. Improvement of acute pancreatitis frequency in CF patients treated with CFTR modulators.
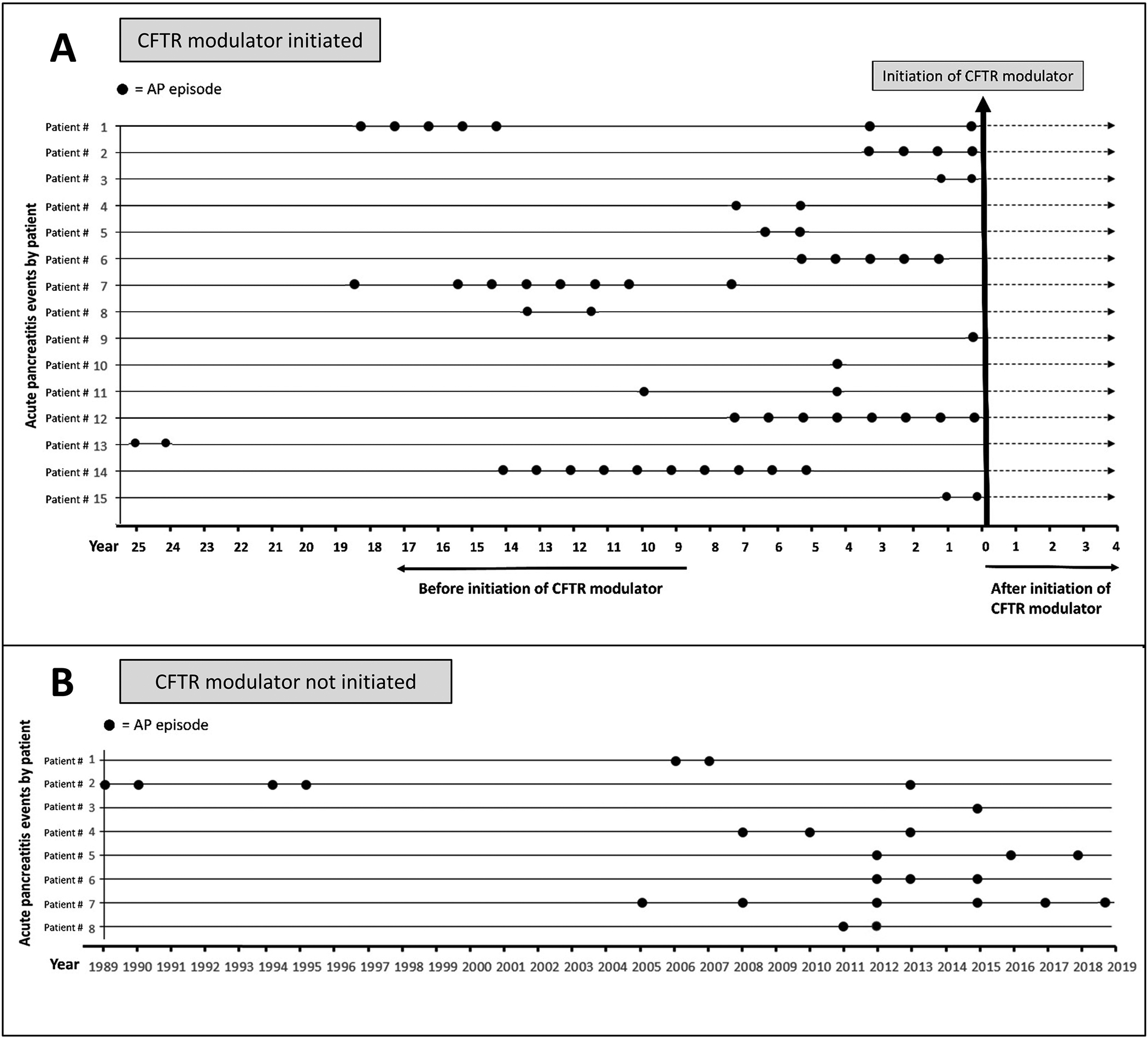
Retrospective review of AP episodes amongst 33 patients with CF and a history of acute recurrent pancreatitis before and after initiation of a CFTR modulator (A) or without any CFTR modulator treatment (B). Each row represents a single patient and every dot represents an episode of AP. Reproduced with permission from Akshintala et al.[55]
Mutations that have traditionally been associated with exocrine pancreatic insufficiency and little risk of development of pancreatitis have recently been associated with development of pancreatitis after treatment with modulators. Megalaa et al reported the case of a 6-year-old boy with CF (G542X/G551D) and documented exocrine pancreatic insufficiency who was started on ivacaftor. The patient presented to the emergency department four years into modulator therapy with abdominal pain and was found to have acute pancreatitis. Upon reassessment of his pancreatic function, he was found to have a normal fecal elastase and ultimately discontinued use of pancreatic enzyme replacement therapy (PERT).[60] Multiple case reports such as this have described patients who were previously documented as exocrine pancreatic insufficient gaining improved pancreas function on modulator therapy, thereby increasing the risk of acute pancreatitis episodes in these patients (Table 2). These studies have debunked the theory that exocrine pancreatic insufficiency in CF patients is irreversible, and also highlight the importance of continued vigilance in assessing current exocrine pancreatic function in those with CF and/or CFTR mutations with new exposures and/or new drug treatment regimens. Further highlighting this is a recent case report by Fortner et al in which they describe a false-negative CF newborn screen in a child born to a mother with CF who was taking CFTR modulator therapy during pregnancy.[61] After a period of infertility, the mother became pregnant 6 weeks after starting elexacaftor/tezacaftor/ivacaftor. She gave birth to a seemingly healthy infant at 39 weeks gestational age. The infant had a normal immunoreactive trypsinogen level, but due to family history of mother having CF and father being a carrier of one copy of F508del, CFTR mutation analysis was sent in the infant. She was found to be F508del homozygous and was diagnosed with CF. Her sweat chloride levels were elevated at 60 and 67 mmol/L, though noted to be well below the 25th percentile compared to patients with the same mutations. Interestingly, the infant was found to have normal fecal elastase levels (>500 micrograms/g) and normal growth. The fetal bioavailability of CFTR modulators is just beginning to be understood. Collins et al recently reported relatively high levels of CFTR modulators in cord blood, as well as low levels in breastmilk and infant blood, suggesting potential transfer of these compounds pre- and post-natally.[62] These data raise important questions about the mechanism of action of CFTR modulators and their utility in alleviating CFTR pathology in utero, particularly the pancreatic disease that infants with this mutation are typically born with.
Table 2.
| Reference | Number of Patients | PI or PS | Age Range (Years) | CFTR Modulator(s) | Effect |
|---|---|---|---|---|---|
| Megalaa et al | 1 | PI | 10 y | Ivacaftor | Developed AP |
| Gould et al | 1 | PI | 16 y | Lumacaftor/Ivacaftor | Developed AP |
| Petrocheilou et al | 1 | PI | 14 y | Ivacaftor | Developed AP |
| Redman et al | 1 | PI | 12 y | Lumacaftor/Ivacaftor | Developed AP |
AP: acute pancreatitis; PI: pancreatic insufficient; PS: pancreatic sufficient; y: years
The effect of CFTR modulators on the pancreas has also been noted in animal studies. A recent study by Zeng et al used a mouse model of pancreatic inflammation due to autoimmunity (non-obese diabetic (NOD) mice with 4 weeks of polyinosinic:polycytidylic acid) and found that treating the mice with the CFTR corrector C18 and potentiator VX-770 improved pancreatic ductal and gland function in mice.[63]
While more work and larger scale studies are needed in this area, it is important to note these changes challenge the traditional approach to pancreatitis and pancreatic function in CF. In a recent review, Sellers noted that pancreatic complications in CF were previously thought to be binary (ie CF-PI vs CF-PS) but given emerging evidence, they should instead be regarded as a spectrum of disease that takes into account several factors.[64]
1.6. Surgical Interventions for Pancreatitis Related to CFTR Dysfunction
Pancreatic surgery is considered for patients with intractable chronic pancreatitis with chronic pain and impaired quality of life. Traditional pancreatic surgeries have included longitudinal pancreaticojejunostomy (modified Peustow), Frey, Beger/Berne, or Whipple pancreaticoduodenectomy procedures. Over the last 40 years, total pancreatectomy with islet cell autotransplantation (TPIAT) has been shown to also be a safe and effective treatment for patients with intractable symptoms from pancreatic disease. A recent report outlined international consensus guidelines for TPIAT and suggests that TPIAT may improve quality of life, reduce pain and opiate use, and may reduce medical utilization in patients with chronic pancreatitis.[65] Therefore, TPIAT is increasingly being considered as the surgery of choice for those with genetic chronic pancreatitis who are unlikely to receive sustained benefit from traditional surgeries. Translational studies identifying that islets from CF patients may not themselves be impaired, but rather, are dysfunctional in CF due to dysfunctional ductal CFTR transport provide reassurance that transplanted islets may have preserved function.[66] Colling et al reviewed the role of TPIAT in patients with chronic pancreatitis due to CFTR mutations. They compared outcomes between patients who had undergone TPIAT for chronic pancreatitis for CF, CFTR heterozygotes, and those who had undergone TPIAT due to other etiologies. Baseline characteristics among the groups were similar. They found that post-TPIAT outcomes of pain relief and endocrine function were indistinguishable among patients with CF and those with CFTR heterozygosity, suggesting that TPIAT is a safe and effective treatment for carefully selected patients with CFTR mutations.[67] St Onge et al reported the case of a 4-year old who underwent TPIAT for chronic pancreatitis in the setting of exocrine pancreatic-sufficient CF. Her expanded 97-mutation genetic panel to test for CF was negative, but gene sequencing revealed a genotype of c.4111G>T (E1371X, clinically significant) and c.2657+2_2657+3insA (2789+2insA, clinical significance uncertain). She suffered from monthly episodes of pancreatitis and growth failure requiring intravenous nutrition. Post-TPIAT, she was able to be weaned off of intravenous nutrition and was not requiring insulin by 6 months post-surgery.[68]
These studies and cases suggest that TPIAT is a viable option in patients with CFTR-related pancreatitis who do not respond to medical therapy and is in fact the preferred surgical option for treatment of genetic pancreatitis. TPIAT can be considered even in young children. While long-term outcomes are continuing to be collected, data thus far are promising.[69]
2. Conclusion
CFTR-related pancreatitis is a significant contributor to genetic pancreatitis, whether it be in individuals with or without CF. As such, genetic testing for CFTR is an important component in working up the etiology of acute recurrent or chronic pancreatitis. Careful consideration of the method of genetic testing must be taken, especially in non-White individuals who may have non-classical CFTR mutations found in those not of European heritage. Pancreatitis is also important to consider as a cause of abdominal pain, nausea, anorexia in patients with CF, especially individuals who are on CFTR modulators, as increases in pancreatic function may bring new susceptibility of the pancreas to pancreatitis. The development of drugs that directly target CFTR expression and/or function are providing new insights in pancreatitis disease, as well as new opportunities for therapeutic relief to those who suffer from this painful, potentially debilitating disease. But, more work needs to be done to identify therapies for all patients with CFTR mutations (regardless of the specific mutation) and ensure equitable access to these potentially life altering therapies.
3. Expert Opinion
Prior to the advent of CFTR modulators, treatment of CFTR-related pancreatitis had been limited to symptomatic management, similar to most of other genetic causes of pancreatitis. But now there is mounting anectdotal evidence that exocrine pancreatic sufficient CF individuals plagued by recurrent pancreatitis may experience relief from pancreatitis with these CFTR modulators.While this is tremendous and worthy of celebration, new areas of disparities are becoming apparent and it is important that these be addressed through education, research, and advocacy.
3.1. Improve access to CFTR modulators in CF patients with qualifying CFTR mutations.
In the United States there was relatively rapid FDA approval of CFTR modulator drugs, the first in 2012 and the most recent in 2019. Approximately half of qualifying CF patients in the US are on one of these CFTR modulators.[70] Some of these may be due to medical contraindications, however, there remains a signficiant number of individuals in the US who could benefit from these therapies, but are not. We should ensure all efforts are made to give patients access to these medications, especially the highly effective modulators ivacaftor and elexacaftor/tezacaftor/ivacaftor. Beyond the US, the disparities may be even larger, with some countries not having access to any modulators.
3.2. Expand access to non-CF individuals with CFTR-related diseases, such as pancreatitis, and qualifying CFTR mutattions.
As of the writing of this expert review, CFTR modulators are not FDA approved for the treatment of CFTR-related disorders without a CF diagnosis, meaning insurance providers generally will not approve them for the treatment of CFTR-related pancreatitis. The high cost of these drugs (estimated ~USD $300,000/year) precludes most individuals from paying out of pocket. A robust pharmaceutical industry with strong academic collaborations is vital to maintaining a pipeline of new therapies for CFTR-related pancreatitis. However, we must collaboratively identify strategies that put therapies into the hands of all who may benefit. With the increasing evidence on the benefit of CFTR modulators to relieve CFTR-related pancreatitis, these efforts should be prioritized in order to provide relief to these indivuals who suffer from the acute and chronic complications of pancreatitis. While more research is needed in this area, CFTR modulators could be a reasonable option for treatment of chronic pancreatitis prior to surgery such as TPIAT. Although TPIAT for CFTR-related chronic pancreatitis yields overall favorable outcomes, medical treatment would be worth consideration as it may provide benefit in a non-invasive manner.
3.3. Diversify the therapeutic portfolio of CFTR modulators, gene therapy approaches, and non-CFTR alternative targets.
Clinical trials and additional research efforts are already underway to target nonsense mutations and splice variants. These efforts are especially important because of their increased prevalence in non-White patients who currently have less access to CFTR modulators.[71] Further research and development of gene therapies are criticial to providing a cure and minimizing treatment disparities. These may be mutation-specific, or may be able to correct the entire CFTR gene so that all patients, regardless of their CFTR mutations, will benefit. [72, 73]
3.4. Increase awareness of CFTR mutations in non-White individuals and international research in countries where CF and/or CFTR mutations may be underappreciated.
There is an increasing awareness of the presence of CFTR mutations in populations beyond White individuals, however more work needs to be done, especially in native populations. For example, full CFTR sequencing programs to truly understand the epidemiology and characterization of CFTR variants in Asia and Africa is scarce, often due to faulty assumptions that CF and/or CFTR-related disease are not present in significant numbers.
Article Highlights:
Mutations in CFTR are a significant contributor to genetic pancreatitis in individuals with and without CF.
There is an expanding pool of diagnostic techniques beyond sweat chloride measurements to diagnose CFTR dysfunction.
CFTR modulator therapy in CF is challenging our traditional thoughts and approaches to pancreatitis and exocrine pancreatic function.
We suggest that expanded use of CFTR modulators to non-CF individuals with CFTR-related diseases, including chronic pancreatitis, will decrease morbidity and health care costs and increase quality of life in these individuals.
TPIAT can be considered as a surgical treatment for chronic pancreatitis due to CFTR dysfunction both with and without presence of CF.
Funding:
M Phadke and Z Sellers are both CF Foundation DIGEST Awardees (PHADKE19GE0, SELLER19GE0). Z Sellers is also supported by NIDDK (K08DK124684) and Cystic Fibrosis Foundation (SELLER20A0-KB).
Footnotes
Declaration of Interest:
Z Sellers has served as a paid consultant for Vertex Pharmaceuticals, AbbVie, Anionix, and Renexxion for CF-related gastrointestinal/liver complications, but has no financial interest in any of these companies. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
Papers of special note have been highlighted as either of interest (*) or of considerable interest (**) to readers.
- 1.Goetz D and Ren CL, Review of Cystic Fibrosis. Pediatr Ann, 2019. 48(4): p. e154–e161. [DOI] [PubMed] [Google Scholar]
- 2.Crespo-Lessmann A, et al. , Association of the CFTR gene with asthma and airway mucus hypersecretion. PLoS One, 2021. 16(6): p. e0251881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fernandez Fernandez E, et al. , CFTR dysfunction in cystic fibrosis and chronic obstructive pulmonary disease. Expert Rev Respir Med, 2018. 12(6): p. 483–492. [DOI] [PubMed] [Google Scholar]
- 4.Strong TV, Boehm K, and Collins FS, Localization of cystic fibrosis transmembrane conductance regulator mRNA in the human gastrointestinal tract by in situ hybridization. J Clin Invest, 1994. 93(1): p. 347–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahmed N, et al. , Molecular consequences of cystic fibrosis transmembrane regulator (CFTR) gene mutations in the exocrine pancreas. Gut, 2003. 52(8): p. 1159–64. [DOI] [PMC free article] [PubMed] [Google Scholar]; *This study assessed 742 patients with CF for whom genotype and clinica data were available. Mutations were identified on both alleles in 85.3% of patients, one allele in 12.8% of patients, and on neither allele in 1.9% of patients. Seventy six different mutations were identified. Over 95% of patietns with severe (Class I, II or III) mutations were pancreatic insufficient. Patients with milder (Class IV or V) mutations were consistently pancreatic sufficient. This study highlighted that predicted or known functional consequences of specific CFTR mutant alleles correlates with the severity of pancreatic disease in CF.
- 6. http://www.genet.sickkids.on.ca/cftr/app.
- 7.Gonska T, Genetic predisposition in pancreatitis. Curr Opin Pediatr, 2018. 30(5): p. 660–664. [DOI] [PubMed] [Google Scholar]; *The study outlines the evolving field of genetic pancreatitis. New pancreatitis-associated genes are continually emerging. New research findings and targeted therapy in pancreatitis are discussed. Acute recurrent and chronic pancreatitis are introduced as complex diseases caused by multiple genetic and non-geentic factors. The role of CFTR in development in pancreatitis is also reviewed.
- 8.Paranjape SM and Mogayzel PJ Jr., Cystic fibrosis. Pediatr Rev, 2014. 35(5): p. 194–205. [DOI] [PubMed] [Google Scholar]
- 9.Anderson DH, Cystic fibrosis of the panreas and its relation to celiac disease: A clinical and pathologic study Am J Dis Child, 1938. 56(2): p. 344–399. [Google Scholar]
- 10.Andersen DH and Hodges RG, Celiac syndrome; genetics of cystic fibrosis of the pancreas, with a consideration of etiology. Am J Dis Child, 1946. 72: p. 62–80. [DOI] [PubMed] [Google Scholar]
- 11.Farber S, The relation of pancreatic achylia to meconium ileus J. Pediat, 1944. 24: p. 387–392. [Google Scholar]
- 12.Di Sant’Agnese PA, et al. , Abnormal electrolyte composition of sweat in cystic fibrosis of the pancreas; clinical significance and relationship to the disease. Pediatrics, 1953. 12(5): p. 549–63. [PubMed] [Google Scholar]
- 13.Gibson LE and Cooke RE, A test for concentration of electrolytes in sweat in cystic fibrosis of the pancreas utilizing pilocarpine by iontophoresis. Pediatrics, 1959. 23(3): p. 545–9. [PubMed] [Google Scholar]
- 14.Marx JL, The cystic fibrosis gene is found. Science, 1989. 245(4921): p. 923–5. [DOI] [PubMed] [Google Scholar]
- 15.Xiao AY, et al. , Global incidence and mortality of pancreatic diseases: a systematic review, meta-analysis, and meta-regression of population-based cohort studies. Lancet Gastroenterol Hepatol, 2016. 1(1): p. 45–55. [DOI] [PubMed] [Google Scholar]
- 16.Boxhoorn L, et al. , Acute pancreatitis. Lancet, 2020. 396(10252): p. 726–734. [DOI] [PubMed] [Google Scholar]
- 17.Sellers ZM, et al. , Nationwide Trends in Acute and Chronic Pancreatitis Among Privately Insured Children and Non-Elderly Adults in the United States, 2007–2014. Gastroenterology, 2018. 155(2): p. 469–478. e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uc A and Husain SZ, Pancreatitis in Children. Gastroenterology, 2019. 156(7): p. 1969–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]; *This review outlines characteristics of acute, acute recurrent and chronic pancreatitic in children. Risk factors, disease burden, epidemiology, management and features unique to pancreatitic in children are discussed.
- 19.Beyer G, et al. , Chronic pancreatitis. Lancet, 2020. 396(10249): p. 499–512. [DOI] [PubMed] [Google Scholar]
- 20.Perito E, et al. , Complications of chronic pancreatitis in children. Curr Opin Gastroenterol, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cohn JA, et al. , Relation between mutations of the cystic fibrosis gene and idiopathic pancreatitis. N Engl J Med, 1998. 339(10): p. 653–8. [DOI] [PubMed] [Google Scholar]; **This studies investigated 27 patients with idiopathic pancreatitis. DNA was tested for 17 CFTR mutations. Ten patients (38%) had at least one 1 abnormal CFTR allele. Eight CFTR mutations were detected. There was a strong association between mutations in the CFTR gene and pancreatitis in patients with idiopathic pancreatitis. This in among the first studies to implicate CFTR gene mutations in the development of genetic pancreatitis.
- 22.Sharer N, et al. , Mutations of the cystic fibrosis gene in patients with chronic pancreatitis. N Engl J Med, 1998. 339(10): p. 645–52. [DOI] [PubMed] [Google Scholar]; **This study examined 134 patients with chronic pancreatitis. DNA was examined for 22 mutations in CFTR. They concluded that mutations of the CFTR gene and the 5T genotype are associated with chronic pancreatitis. This in among the first studies to implicate CFTR gene mutations in the development of genetic pancreatitis.
- 23.Weiss FU, Skube ME, and Lerch MM, Chronic pancreatitis: an update on genetic risk factors. Curr Opin Gastroenterol, 2018. 34(5): p. 322–329. [DOI] [PMC free article] [PubMed] [Google Scholar]; *This review outlines developments in discovery of genetic mutations within and outside the trypsin-dependent pathway that are associated with chronic pancreatitis.
- 24.Meyerholz DK, et al. , Pathology of gastrointestinal organs in a porcine model of cystic fibrosis. Am J Pathol, 2010. 176(3): p. 1377–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abu-El-Haija M, et al. , Pancreatic damage in fetal and newborn cystic fibrosis pigs involves the activation of inflammatory and remodeling pathways. Am J Pathol, 2012. 181(2): p. 499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Durie PR, et al. , Characteristic multiorgan pathology of cystic fibrosis in a long-living cystic fibrosis transmembrane regulator knockout murine model. Am J Pathol, 2004. 164(4): p. 1481–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Navis A and Bagnat M, Loss of cftr function leads to pancreatic destruction in larval zebrafish. Dev Biol, 2015. 399(2): p. 237–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olivier AK, et al. , Abnormal endocrine pancreas function at birth in cystic fibrosis ferrets. J Clin Invest, 2012. 122(10): p. 3755–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Farrell PM, et al. , Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. J Pediatr, 2017. 181s: p. S4–S15. e1. [DOI] [PubMed] [Google Scholar]
- 30.Ooi CY, et al. , Does extensive genotyping and nasal potential difference testing clarify the diagnosis of cystic fibrosis among patients with single-organ manifestations of cystic fibrosis? Thorax, 2014. 69(3): p. 254–60. [DOI] [PubMed] [Google Scholar]
- 31.Kharrazi M, et al. , Newborn Screening for Cystic Fibrosis in California. Pediatrics, 2015. 136(6): p. 1062–72. [DOI] [PubMed] [Google Scholar]
- 32.Sosnay PR, et al. , Applying Cystic Fibrosis Transmembrane Conductance Regulator Genetics and CFTR2 Data to Facilitate Diagnoses. J Pediatr, 2017. 181s: p. S27–S32. e1. [DOI] [PubMed] [Google Scholar]; **This paper discussed the significant advance in CF genetics with the development of durie the CFTR2 project. The project seeks to characterize CFTR mutations from patients with CF around the world. The project also established guidelines for the clinical, functional and penetrance cirteria that can be used to interpret mutations not yet inclusded in the CFTR2 database. The CFTR2 database provides valuable information to continue to characterize and understand phenotypic consequences of CF.
- 33.Barben J, et al. , Updated guidance on the management of children with cystic fibrosis transmembrane conductance regulator-related metabolic syndrome/cystic fibrosis screen positive, inconclusive diagnosis (CRMS/CFSPID). J Cyst Fibros, 2020. [DOI] [PubMed] [Google Scholar]; **This study used a consensus approach to designate and guide management of infants with inconclusive CF-related diagnoses.
- 34.Ooi CY, et al. , Immunoreactive trypsinogen levels in newborn screened infants with an inconclusive diagnosis of cystic fibrosis. BMC Pediatr, 2019. 19(1): p. 369. [DOI] [PMC free article] [PubMed] [Google Scholar]; **This study discussed newborn screening practices for CF with a focus on those with an uncertain diagnosis of CF. The authors hypothesized that immunoreactive trypsinogen (IRT) levels may reflect the degree of CFTR dysfunction and help to identify those with CRMS/CFSPID who are are increased risk of development of CF. They discovered that infants who are ultimately diagnosed with CF have higher NBS IRT levels that those with CRMS/CFSPID, and that NBS IRT levels increase in a graded fasion.
- 35.Terlizzi V, et al. , CRMS/CFSPID Subjects Carrying D1152H CFTR Variant: Can the Second Variant Be a Predictor of Disease Development? Diagnostics (Basel), 2020. 10(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vonk AM, et al. , Protocol for Application, Standardization and Validation of the Forskolin-Induced Swelling Assay in Cystic Fibrosis Human Colon Organoids. STAR Protoc, 2020. 1(1): p. 100019. [DOI] [PMC free article] [PubMed] [Google Scholar]; **This article describes the isolation, handling, culture of and experiments with human colon stem cell organoids in the setting of CF. Procedures and validation experiemnts are described for several human colon organoid lines. CFTR genotypes are tested for baseline CFTR function and response to CFTR-modulating drugs. This methodology is promising for future research in CF, particularly for therapeutic targets.
- 37.Derichs N, et al. , Intestinal current measurement for diagnostic classification of patients with questionable cystic fibrosis: validation and reference data. Thorax, 2010. 65(7): p. 594–9. [DOI] [PubMed] [Google Scholar]
- 38.Salinas DB, et al. , Low Beta-Adrenergic Sweat Responses in Cystic Fibrosis and Cystic Fibrosis Transmembrane Conductance Regulator-Related Metabolic Syndrome Children. Pediatr Allergy Immunol Pulmonol, 2017. 30(1): p. 2–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Quinton P, et al. , β-adrenergic sweat secretion as a diagnostic test for cystic fibrosis. Am J Respir Crit Care Med, 2012. 186(8): p. 732–9. [DOI] [PubMed] [Google Scholar]
- 40.Kim J, et al. , Evaporimeter and Bubble-Imaging Measures of Sweat Gland Secretion Rates. PLoS One, 2016. 11(10): p. e0165254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Minso R, et al. , Intestinal current measurement and nasal potential difference to make a diagnosis of cases with inconclusive CFTR genetics and sweat test. BMJ Open Respir Res, 2020. 7(1). [DOI] [PMC free article] [PubMed] [Google Scholar]; **NPD and ICM are CF biomarkers recommended to make a disgnoaiss in patients with inconclusice CF genetics or sweat testing. The study assessed 219 individuals by NPD or ICM who had been referred due to symptoms of CF but with inconclusive sweat test and CFTR genetics. CF or CFTR-trlated disorder was diagnosed in 76% of patients with a CFTR genotype of unknown or variable clinical significance. This study suggests that NPD or ICM should be used when there is an inconclusive CF or CF-related diagnosis.
- 42.Hart PA, et al. , Endoscopic Pancreas Fluid Collection: Methods and Relevance for Clinical Care and Translational Science. Am J Gastroenterol, 2016. 111(9): p. 1258–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.LaRusch J, et al. , Mechanisms of CFTR functional variants that impair regulated bicarbonate permeation and increase risk for pancreatitis but not for cystic fibrosis. PLoS Genet, 2014. 10(7): p. e1004376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ooi CY, et al. , Type of CFTR mutation determines risk of pancreatitis in patients with cystic fibrosis. Gastroenterology, 2011. 140(1): p. 153–61. [DOI] [PubMed] [Google Scholar]; **This study investigated the effect of CFTR genotype on risk of pancreatitis. Two hunderd seventy seven patients’ genotypes and clinical characteristics were analyzed. Loss of pancreatic function associated with each CFTR genotype was determined based on pancreatic insufficiecny prevalence (PIP) score. Patients with pancreatitis were more likely to have genotypes associated with mild PIP scores. This study highlighted that specific CFTR genotypes are more significantly associated with pancreatitis.
- 45.Ooi CY and Durie PR, Cystic fibrosis transmembrane conductance regulator (CFTR) gene mutations in pancreatitis. J Cyst Fibros, 2012. 11(5): p. 355–62. [DOI] [PubMed] [Google Scholar]
- 46.Palermo JJ, et al. , Genophenotypic Analysis of Pediatric Patients With Acute Recurrent and Chronic Pancreatitis. Pancreas, 2016. 45(9): p. 1347–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kumar S, et al. , Risk Factors Associated With Pediatric Acute Recurrent and Chronic Pancreatitis: Lessons From INSPPIRE. JAMA Pediatr, 2016. 170(6): p. 562–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schneider A, et al. , Combined bicarbonate conductance-impairing variants in CFTR and SPINK1 variants are associated with chronic pancreatitis in patients without cystic fibrosis. Gastroenterology, 2011. 140(1): p. 162–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ooi CY, D. R, Keenan K, et al. , SPINK1 mutation N34S increases risk of pancreatitis in patients with CF & CFTR-related disorders. Pediatric Pulmonology 2011: p. 390. [Google Scholar]
- 50.Cantin AM, et al. , Cystic fibrosis transmembrane conductance regulator function is suppressed in cigarette smokers. Am J Respir Crit Care Med, 2006. 173(10): p. 1139–44. [DOI] [PubMed] [Google Scholar]; *This study examined the effects of cigarette smoke exposure on Calu-3 and T84 cell CFTR expression. Nasal potential difference (NPD) was also measured in 26 men who had no detectable CFTR mutations. Cigarette smoke decreased CFTR expression and function in Calu-3 and T84 cells. NPDs of cigarette smokers showed a pattern typical of CFTR deficiency compared to nonsmokers
- 51.Rasmussen JE, et al. , Cigarette smoke-induced Ca2+ release leads to cystic fibrosis transmembrane conductance regulator (CFTR) dysfunction. J Biol Chem, 2014. 289(11): p. 7671–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alexandre M, et al. , The emerging role of smoking in the development of pancreatitis. Pancreatology, 2011. 11(5): p. 469–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kadiyala V, et al. , Cigarette smoking impairs pancreatic duct cell bicarbonate secretion. Jop, 2013. 14(1): p. 31–8. [DOI] [PMC free article] [PubMed] [Google Scholar]; *This study examined pancreatic function in smokers and nonsmokers. Pancreatic fluid collection was conducted during endoscopy. Measurement of pancreatic fluid bicarbonate reveled that both past and current cigarette smoking is an independent risk factor for pancreatic duct cell secretory dysfunction.
- 54.Ballengee CR, et al. , Effects of Second-Hand Smoke on Pancreatitis in Children. Pancreas, 2019. 48(5): p. 706–710. [DOI] [PubMed] [Google Scholar]
- 55.Baker E, et al. , Tobacco smoke exposure limits the therapeutic benefit of tezacaftor/ivacaftor in pediatric patients with cystic fibrosis. J Cyst Fibros, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Samokhvalov AV, Rehm J, and Roerecke M, Alcohol Consumption as a Risk Factor for Acute and Chronic Pancreatitis: A Systematic Review and a Series of Meta-analyses. EBioMedicine, 2015. 2(12): p. 1996–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maléth J, et al. , Alcohol disrupts levels and function of the cystic fibrosis transmembrane conductance regulator to promote development of pancreatitis. Gastroenterology, 2015. 148(2): p. 427–39. e16. [DOI] [PMC free article] [PubMed] [Google Scholar]; *This study examined the effect of ethanol on pancreatic function. CFTR activity was measured in sweat chloride in patients with CF, patients admitted to the emergency department with excessive alcohol consumption, and helathy volunteers. Decreased sweat chloride levels were found in patients who acutely abused alcohol but not in healthy volunteers. The effects of ethanol were also assessed in pancreatic cell lines and tissue from guinea pics. Administration of ethanol reduced expression of CFTR mRNA, reduced the stability of CFTR at the cell surface, and disrupted foling of CFTR at the endoplasmic reticulum.
- 58.Akshintala VS, et al. , Cystic fibrosis transmembrane conductance regulator modulators reduce the risk of recurrent acute pancreatitis among adult patients with pancreas sufficient cystic fibrosis. Pancreatology, 2019. 19(8): p. 1023–1026. [DOI] [PubMed] [Google Scholar]; *This retrospective study examined patients with PS-CF, history of acute pancreatitis and initiation of CFTR modulator therapy. Fifteen patients were identified. It eas found that CFTR modulators, alone or in combination, substantially reduced the risk of recurrent pancreatitis over a 3-year period. this stidu highlight the potential therapeutic benefit with regar to pancreatitis in patients with PS-CF.
- 59.Emery J, M. D, Chroinin MN. 2019, The effects of ivacaftor on pancreatic function in paediatric patients with cystic fibrosis gating mutations. Arch Dis Child. Suppl 3. A149–150 [Google Scholar]
- 60.Megalaa R, et al. , Time for a gut check: Pancreatic sufficiency resulting from CFTR modulator use. Pediatr Pulmonol, 2019. 54(8): p. E16–e18. [DOI] [PubMed] [Google Scholar]
- 61.Fortner CN, Seguin JM, and Kay DM, Normal pancreatic function and false-negative CF newborn screen in a child born to a mother taking CFTR modulator therapy during pregnancy. J Cyst Fibros, 2021. [DOI] [PubMed] [Google Scholar]; **This case report outlines the case of a female infant born to a mother with CF (F508del/F508del) who was taking elexacaftor/tezacaftor/ivacaftor. The infant had a false-negative newborn immunoreactive trypsinogen screen but was foung to have CF on CFTR mutation analysis (F508del/F508del). The infant had a normal fecal elastase level and maintained normal growth wihtout PERT. while the infant’s sweat chloride levels were elevated, they were well below the 25th percentile for her mutation. this case report raises important questions about the effect of CFTR modulator therapy during fetal development.
- 62.Collins B, et al. , Drug exposure to infants born to mothers taking Elexacaftor, Tezacaftor, and Ivacaftor. J Cyst Fibros, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zeng M, et al. , Restoration of CFTR Activity in Ducts Rescues Acinar Cell Function and Reduces Inflammation in Pancreatic and Salivary Glands of Mice. Gastroenterology, 2017. 153(4): p. 1148–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]; **This review highlights recent research in exocrine pancreatic function in the era of CFTR modulator therapy. New research regarding pancreatic endocrine-exocrine interplay and imaging advances are also reviewed.
- 64.Sellers ZM, Pancreatic complications in children with cystic fibrosis. Curr Opin Pediatr, 2020. 32(5): p. 661–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Abu-El-Haija M, et al. , The role of total pancreatectomy with islet autotransplantation in the treatment of chronic pancreatitis: A report from the International Consensus Guidelines in chronic pancreatitis. Pancreatology, 2020. 20(4): p. 762–771. [DOI] [PubMed] [Google Scholar]
- 66.Shik Mun K, et al. , Patient-derived pancreas-on-a-chip to model cystic fibrosis-related disorders. Nat Commun, 2019. 10(1): p. 3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Colling KP, et al. , Total Pancreatectomy With Intraportal Islet Autotransplantation as a Treatment of Chronic Pancreatitis in Patients With CFTR Mutations. Pancreas, 2018. 47(2): p. 238–244. [DOI] [PubMed] [Google Scholar]; **This retrospective study examined all TPIATs performed at a single institution between 2002–2014. Outcomes after TPIAT associated with CFTR mutations and CP not due to CFTR mutations were compared. Twenty CFTR homozygotes, 19 CFTR heterozygotes and 20 ane/sex matched controls were identified. Postoperative HBA1C, C-peptide levels, postoperative complications, an dislet yield were similar between groups. TPIAT is a safe and effective treatment option for chronic pancreatitis due to CFTR dysfunction.
- 68.St Onge I, et al. , Total pancreatectomy with islet autotransplantation in a pancreatic-sufficient cystic fibrosis patient. J Cyst Fibros, 2019. 18(5): p. e53–e55. [DOI] [PubMed] [Google Scholar]
- 69.Bellin MD, et al. , A multicenter study of total pancreatectomy with islet autotransplantation (TPIAT): POST (Prospective Observational Study of TPIAT). Pancreatology, 2018. 18(3): p. 286–290. [DOI] [PMC free article] [PubMed] [Google Scholar]; *This study outlines the fact that large, multi-center studies to guide decisions about seleting timing and indication for TPIAT are lacking. Several centers are prospectively enrolling patients undergoing TPIAT for chronic pancreatitis in the POST study with several outcome measures including participant questionnaires, visual analong pain scale, pain interference scores, opoid requirements, insuling requirements, insluin graft function, and HBA1C. A biorepository is also being established for future studies. This large-scale multicenter study will help us better understand outcomes of TPIAT
- 70.Cystic Fibrosis Foundation Patient Registry. Annual Report 2019. [Google Scholar]
- 71.McGarry ME and McColley SA, Cystic fibrosis patients of minority race and ethnicity less likely eligible for CFTR modulators based on CFTR genotype. Pediatr Pulmonol, 2021. 56(6): p. 1496–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vaidyanathan S, et al. , High-Efficiency, Selection-free Gene Repair in Airway Stem Cells from Cystic Fibrosis Patients Rescues CFTR Function in Differentiated Epithelia. Cell Stem Cell, 2020. 26(2): p. 161–171. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vaidyanathan S, et al. , Targeted replacement of full-length CFTR in human airway stem cells by CRISPR-Cas9 for pan-mutation correction in the endogenous locus. Mol Ther, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Carrion A, et al. , Reduction of Recurrence Risk of Pancreatitis in Cystic Fibrosis With Ivacaftor: Case Series. J Pediatr Gastroenterol Nutr, 2018. 66(3): p. 451–454. [DOI] [PubMed] [Google Scholar]; *This study was a multi-center retrospective study of patients with CF and a history of recurrent pancreatitis taking ivacaftor. The use of ivacaftor was associated with a reduced frequency and recurrence of pancreatitis episodes.
- 75.Hutchinson I and McNally P, Appearance of Pancreatic Sufficiency and Discontinuation of Pancreatic Enzyme Replacement Therapy in Children with Cystic Fibrosis on Ivacaftor. Ann Am Thorac Soc, 2021. 18(1): p. 182–183. [DOI] [PubMed] [Google Scholar]; *This study examines 3 pediatric patients with CF and PI who showed restored exocrine pancreatic function and biochemical parameters on ivacaftor.
- 76.Munce D, Lim M, and Akong K, Persistent recovery of pancreatic function in patients with cystic fibrosis after ivacaftor. Pediatr Pulmonol, 2020. 55(12): p. 3381–3383. [DOI] [PubMed] [Google Scholar]
- 77.Johns JD and Rowe SM, The effect of CFTR modulators on a cystic fibrosis patient presenting with recurrent pancreatitis in the absence of respiratory symptoms: a case report. BMC Gastroenterol, 2019. 19(1): p. 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gould MJ, R. F, Gonska T, Pancreatitis in a previously pancreatic insufficient patient with cystic fibrosis after treatment with lumacaftor/ivacaftor combination therapy J Can Assoc Gastroenterol, 2019. 2: p. 361–362. [Google Scholar]
- 79.Petrocheilou A, Kaditis AG, and Loukou I, Pancreatitis in A Patient with Cystic Fibrosis Taking Ivacaftor. Children (Basel), 2020. 7(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Redman AY, Michelle; Freswick P; Thompson K, Acute Pancreatitis in a Previously Exocrine Pancreatic Insufficient Cystic Fibrosis Patient Who Had Improved Pancreatic Function After Being Treated With Lumacaftor/Ivacaftor. JPGN Reports 2021. 2(3): p. e096. [DOI] [PMC free article] [PubMed] [Google Scholar]