

Abstract
Recent studies have identified cancer-associated mutations in histone genes that lead to the expression of mutant versions of core histones called oncohistones. Many oncohistone mutations occur at Asp and Glu residues, two amino acids known to be ADP-ribosylated (ADPRylated) by PARP-1. We screened 25 Glu or Asp oncohistone mutants for their effects on cell growth in breast and ovarian cancer cells. Ectopic expression of six mutants of three different core histones (H2B, H3, and H4) altered cell growth in at least two different cell lines. Two of these sites, H2BD51 and H4-D68, were indeed sites of ADPRylation in wild-type (unmutated) histones, and mutation of these sites inhibited ADPRylation. Mutation of H2B-D51 dramatically altered chromatin accessibility at enhancers and promoters, as well as gene expression outcomes, whereas mutation of H4-D68 did not. Additional biochemical, cellular, proteomic, and genomic analyses demonstrated that ADPRylation of H2B-D51 inhibits p300-mediated acetylation of H2B at many Lys residues. In breast cancer cell xenografts in mice, H2B-D51A promoted tumor growth, but did not confer resistance to the cytotoxic effects of PARP inhibition. Collectively, these results demonstrate that functional Asp and Glu ADPRylation sites on histones are mutated in cancers, allowing cancer cells to escape the growth-regulating effects of post-translational modifications via distinct mechanisms.
Keywords: ADP-ribosylation, breast cancer, ovarian cancer, chromatin, chromatin accessibility, histones, histone acetylation, oncohistone, p300, poly(ADP-ribose) polymerase-1, (PARP-1), PARP inhibitor (PARPi), transcription, gene expression
Introduction
The functions of the core histones within chromatin are regulated by post-translational modifications (PTMs), such as methylation, phosphorylation, and acetylation (1,2). Recent studies have identified cancer-associated mutations in histone genes that lead to the expression of mutant versions of the core histones called “oncohistones,” some of which occur at sites of PTMs. Nacev and colleagues reported the prevalence of histone mutations in a broad array of tumor types (3). By integrating multiple public datasets, they identified more than 4,000 missense mutations in histone-encoding genes for all four core histones, occurring in about 4% of tumors (3). As we discuss below, many of the highest frequency oncohistone mutations across all cancer types occur in aspartate (Asp, D) and glutamate (Glu, E) residues, which are sites of modification by ADP-ribosylation (ADPRylation).
ADPRylation is a reversible post-translational modification of proteins resulting in the covalent attachment of a single ADP-ribose (ADPR) unit or polymers of ADPR units [i.e., poly(ADP-ribose), or PAR] derived from β-NAD+ on a variety of amino acid residues (e.g., Glu, Asp, Ser) (4,5). ADPRylation is catalyzed by members of the poly(ADP-ribose) polymerase (PARP) family of enzymes (5–7), which function as ADP-ribose “writers” that covalently attach ADP-ribose units on substrate proteins (8,9). The predominant nuclear PARP “polyenzymes” (e.g., PARPs 1 and 2) catalyze the addition of linear or branched chains of ADP-ribose (4,8). Although ADPRylation is an important regulatory PTM in many biological contexts, it is poorly understood compared to other PTMs. Interestingly, ADPRylation is highly associated with other PTMs, such as phosphorylation, across the proteome (10–12). Site-specific ADPRylation of proteins can (1) alter the biochemical or biophysical properties of the modified protein or (2) create new binding sites for ADPR binding domains that drive protein-protein interactions (4,8).
Initial studies of ADPRylation and PARPs were focused on the biochemistry and molecular biology of PARP-1 in DNA repair (13), but the mechanistic and functional understanding of the role of PARPs in different biological processes has grown considerably (7,8). A number of seminal studies on PARP-1-mediated ADPRylation over the past decade have shifted the focus to the regulation of gene expression (14,15). Nuclear PARPs modulate chromatin function through histone ADPRylation. All four core histones, as well as linker histone H1, have long been known to be substrates for ADPRylation (16) and PARP-mediated histone ADPRylation promotes chromatin decompaction (17–20). But, unlike other well characterized histone modifications (e.g., acetylation, methylation, phosphorylation), the specific sites of histone ADPRylation have historically been poorly characterized and their functions unknown. Recent advances in chemical biology and mass spectrometry have led to the identification of specific sites of ADPRylation on histones (10,11,21–23). These include: (1) Ser residues during genotoxic stress (12,24,25), (2) Glu and Asp residues in response to DNA damage (10,21,26), and (3) Glu and Asp residues under physiological conditions, such as adipogenesis (22).
In the studies described herein, we characterize functional sites of Asp and Glu ADPRylation in core histones. Moreover, we demonstrate that mutation of these sites as found in oncohistones allows cancer cells to escape the growth-regulating effects of ADPRylation via distinct mechanisms.
Materials and Methods
Additional details about the materials and methods can be found in the Supplementary Materials and Methods under the same headings listed here.
Cell culture
MCF-7, MDA-MB-231, OVCAR-3, and 293T cells were purchased from the American Type Culture Collection under standard conditions. Fresh cell stocks were regularly replenished from the original stocks, verified for cell type identity using the GenePrint 24 system (Promega, B1870), and confirmed as mycoplasma-free every three months using a commercial testing kit.
Cell treatments
Cells were grown until 70% confluence and treated with 5 μM A485 (Tocris Bioscience, 1104546-89-5), 5 to 20 μM Veliparib (Tocris Bioscience, 1104546-89-5), 10 μM Niraparib (MedChemExpress, HY-10619), or DMSO vehicle for 2 hours.
Antibodies
A detailed list of the antibodies used are provided in the Supplementary Materials and Methods.
Generation of lentiviral expression vectors for wild-type and site mutant histones
Double-stranded cDNAs encoding carboxyl-terminal FLAG epitope-tagged human wild-type and site-mutant histones H2B, H2A, H3, and H4 were synthesized as gene blocks (Integrated DNA Technologies), and then cloned individually into pCDH-EF1α-MCS-IRES-Puro lentiviral expression vector using Gibson assembly (NEB, E2621). Twenty-five individual glutamate (Glu, E) and aspartate (Asp, D) oncohistone sites (3) were mutated into alanine (Ala, A). H2B-D51 and H4-D68 were also mutated into asparagine (Asn, N).
Generation of stable ectopic protein expression cell lines
Lentiviruses were generated by transfecting the pCDH vectors described above into 293T cells, together with the expression vectors for the VSV-G envelope protein (pCMV-VSV-G, Addgene plasmid no. 8454), the expression vector for GAG-Pol-Rev (psPAX2, Addgene plasmid no. 12260), and a vector to aid with translation initiation (pAdVAntage, Promega) using Lipofectamine 3000 transfection reagent (Thermo Fisher Scientific, L3000015) according to the manufacturer’s instructions. Infected cells were selected with 2 μg/mL puromycin (Sigma-Aldrich, P9620), expanded, and frozen in aliquots for future use. Ectopic expression of the cognate proteins was confirmed by Western blotting.
Cell proliferation assays and cell migration assays
Cell proliferation assays were performed on fixed cells using a crystal violet staining assay. After washing to remove unincorporated stain, the crystal violet was extracted using 10% glacial acetic acid and the absorbance was read at 595 nm. Cell migration assays were performed in migration chambers formed from cell culture inserts (Corning, 353097). After 24 hours, the cells in the top chamber were removed. The chambers were stained with crystal violet (0.5% crystal violet in 20% methanol) and images of the cells at the bottom of the membrane were assessed by microscopy. All proliferation assays and migration assays were performed a minimum of three times to ensure reproducibility.
Preparation of cell extracts and Western blotting
Preparation of whole cell lysates.
Whole cell lysates were prepared as described previously (22).
Preparation of cell fractions.
Nuclear, chromatin and cytosolic fractions were prepared as described previously (22).
Determination of protein concentrations and Western blotting.
Protein concentrations were determined using a BCA protein assay (Pierce, 23225).
Nucleosome immunoprecipitation
MDA-MB-231 cells ectopically expressing FLAG-tagged wild-type or mutant histones by lentivirus-mediated delivery were seeded at ~5×106 cells per 15 cm diameter plate and cultured as described above. Nucleosome isolation was performed using micrococcal nuclease as described previously (22,27). The mononucleosomes were recovered from the beads by heating to 100°C for 5 minutes in 2 × SDS-PAGE loading buffer. The immunoprecipitated material was subjected to Western blotting as described above.
RNA-sequencing and data analysis
The following methods were used to generate, evaluate for QC, sequence, and analyze the RNA-seq data.
Generation and sequencing of RNA-seq libraries.
MDA-MB-231 cells expressing FLAG-tagged wild-type or mutant histones were seeded in 6-well plates. The cells were collected and total RNA was isolated using the RNeasy kit (Qiagen, 74136) according to the manufacturer’s instructions. The total RNA was then enriched for polyA+ RNA using Dynabeads Oligo(dT)25 (Life Technologies, 61002). The polyA+ RNA was then used to generate strand-specific RNA-seq libraries as described previously (28) and sequenced using an Illumina NextSeq 500 to an average depth of ~40 million reads total per condition. Two independent biological replicates with two sets of sequencing replicates were used.
Analysis of RNA-seq data.
The quality of RNA-seq datasets was assessed using the FastQC tool (http://www.bioinformatics.babraham.ac.uk/projects/fastqc) and the reads were then mapped to the human reference genome (hg38) using the spliced read aligner TopHat (v2.0.13) (29). Uniquely mapped reads were converted into bigwig files using the RSeQC tool (v2.6.4) (30) for visualization. Transcriptome assembly was performed using cufflinks (v2.2.1) with default parameters (31). An FDR cutoff of 0.05 and a fold change cutoff of 1.5 was used to determine significantly regulated genes in mutants compared to the WT H2B condition.
Gene ontology (GO) analyses.
Gene ontology analyses were performed on the differentially-expressed gene sets using DAVID (Database for Annotation, Visualization, and Integrated Discovery) (32).
ATAC- sequencing and data analysis
Generation and sequencing of ATAC-seq libraries.
ATAC-seq was performed using the Omni-ATAC protocol as previously described (33). The ATAC-seq libraries were subjected to QC analyses (i.e., the final library yield and the size distribution of the final library DNA fragments) and sequenced using an Illumina NextSeq 500 to an average depth of 57 million unique aligned reads per each biological replicate. Two independent biological replicates with three sets of sequencing replicates were used.
Analysis of ATAC-seq data.
The quality of the ATAC-seq datasets was assessed using the FastQC tool (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). The ATAC-seq reads were then aligned to the human genome (hg38) using BWAKit (v0.7.15) (34). For unique alignments, duplicate reads with quality score over 10 were filtered out using picard tools (v2.10.3) (http://broadinstitute.github.io/picard/). The resulting uniquely mapped reads were normalized to the same read depth across all samples and converted into bigWig files using the writeWiggle function from the groHMM package in R (35) for visualization in the UCSC Genome Browser. Peak calling was performed using MACS2 with default parameters (36,37). The read counts under the ATAC-seq peak calls were calculated using featureCounts and differentially enriched peaks were called using DESseq2 software (38) with an FDR cutoff of 0.05 and a fold change (FC) cutoff of ≥ 1.5.
Motif searching on the differentially enriched ATAC-seq peaks was performed using findMotifsGenome.pl program using HOMER (Heinz et al., 2010) motif discovery algorithm. To analyze footprinting signatures in ATAC-seq data the TOBIAS package was used (v0.12.10; available at https://github.com/loosolab/TOBIAS/) (39).
Native chromatin immunoprecipitation (ChIP), and ChIP-sequencing
Native chromatin immunoprecipitation.
Native ChIP was performed as previously described using 5×106 MDA-MB-231 cells (40). A spike-in strategy was used to normalize all ChIP-seq data to reduce the effects of technical variation and sample processing bias. Spike-In chromatin (Active Motif, 53083) and Spike-in antibody (Active Motif, 61686) were used according to manufacturer instructions.
ChIP-seq library preparation.
ChIP-seq libraries were prepared from 5 ng of ChIP’d DNA following the Illumina TruSeq protocol. The quality of the libraries was assessed using a D1000 ScreenTape on a 2200 TapeStation (Agilent) and quantified using a Qubit dsDNA HS Assay Kit (Thermo Fisher). Libraries with unique adaptor barcodes were multiplexed and sequenced on an Illumina NextSeq 500 (paired-end, 33 base pair reads). The ChIP-seq libraries were sequenced to an average depth of ~35 million unique aligned reads per condition. Two independent biological replicates were used.
Analysis of ChIP-seq data.
The quality of the ChIP-seq datasets was assessed using the FastQC tool (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). ChIP-seq raw reads were aligned separately to the human reference genome (hg38) and the spike-in Drosophila reference genome (dm3) using Bowtie2 (v2.3.2) (41). Only one alignment is reported for each read and duplicate reads were filtered using the Picard MarkDuplicates tool. Reads were converted into bigwig files using BEDTools (v2.29.0) genomecov function (42) for visualization in the UCSC Genome Browser. ChIP-seq peak calling was performed using the findPeaks function of HOMER (43) with an FDR cutoff of 0.001.
Integration, analysis, and visualization of ChIP-seq, ATAC-seq, and RNA-seq data
The up- or downregulated ATAC-seq peaks nearest to the transcription start sites (TSSs) of up- or downregulated regulated genes (from RNA-seq) were obtained using BEDTools closest function (42). The distance between the TSS and the nearest regulated ATAC-seq peak was represented as a histogram using the hist function in R. The metaplots of ATAC-seq and ChIP-seq data at the TSSs and nearest regulated ATAC-seq peaks were generated using the metagene function in the groHMM software package (35).
Crosslinked ChIP and ChIP-qPCR
Crosslinked ChIP.
Formaldehyde crosslinked ChIP was performed as described previously (44,45).
ChIP-qPCR.
ChIP-qPCR was performed as described previously (46) in triplicate using a LightCycler® 480 Instrument II system and Power SYBR Green PCR master mix. The qPCR primer sequences used in this study are listed in the Supplementary Materials and Methods.
Expression and purification of recombinant proteins
Purification of PARP-1 expressed in Sf9 insect cells.
Sf9 insect cells, cultured in SF-II 900 medium (Invitrogen), were transfected with 1 μg of bacmid driving expression of FLAG-tagged human PARP-1 using Cellfectin transfection reagent (Invitrogen) as described by the manufacturer. PARP-1-expressing Sf9 cells were collected by centrifugation, flash frozen in liquid N2, and stored at −80°C. PARP-1 was purified from the cells as described (22). Eluted proteins were aliquoted, flash frozen in liquid N2, and stored at −80°C.
Purification of p300 expressed in Sf9 insect cells.
FLAG-tagged human p300 was expressed in Sf9 cells at the Protein and Monoclonal Antibody Production Shared Resource at Baylor College of Medicine. The Sf9 cells were treated for 3 hours prior to harvesting with the following p300 inhibitors: 10 μM SGC-CBP30 (Sigma-Aldrich, SML1133), 25 μM C646 (Sigma-Aldrich, SML0002), and 10 μM of A-485 (Tocris, 6387). p300 protein was purified from the cells as described (47). Eluted proteins were aliquoted, flash frozen in liquid N2, and stored at −80°C.
Purification of histone H2B expressed in E. coli.
Wild-type and mutant human histone H2B cDNAs were cloned into pRUTH5 to yield an N-terminal His6-TEV tagged version of the cDNA. Transformed bacteria were grown in LB containing ampicillin at 37°C and the expression of recombinant protein was induced by the addition of 1 mM IPTG for 4 h at 37°C. The recombinant histones were purified using Ni-NTA chromatography. Eluted and dialyzed proteins were quantified using a Bradford protein assay (Bio-Rad), aliquoted, flash-frozen in liquid N2, and stored at −80°C.
Purification of NMNAT-1 expressed in E. coli.
6x His-tagged human NMNAT-1 (UniProt entry Q9EPA7) was expressed in E. coli strain BL21(DE3) using a pET19b-based bacterial expression vector and purified as described previously (22). Eluted and dialyzed proteins were quantified using a Bradford protein assay (Bio-Rad), aliquoted, flash-frozen in liquid N2, and stored at −80 °C.
In vitro PARylation assays
In vitro PARylation assays were performed essentially as described previously (22,48,49). One μg of purified human H2B protein or 500 nM of assembled human mononucleosomes (EpiCypher, 16-0009) was mixed with 0.1 μM purified human PARP-1, 1 μM purified human NMNAT-1, 100 μM NAD+, and 100 μM ATP in Reaction Buffer (50 mM Tris-HCl pH 7.4, 10 mM MgCl2, 5% glycerol, and 1 mM DTT). The PARylation reactions were initiated by adding 25-mer DNA, incubated at room temperature for 20 minutes, and then stopped by the addition of 4x SDS-PAGE Loading Buffer followed by heating to 100°C for 5 minutes, or by the addition of PJ34. The reaction products were then subjected to Western blotting as described above, or were subjected to additional in vitro HAT assays with p300 as described below.
In vitro HAT assays
One μg of purified human H2B protein or 500 nM of assembled mononucleosomes (EpiCypher, 16-0009) were incubated with purified p300 protein (50 ng) in Reaction Buffer (50 mM Tris-HCl pH 7.4, 10 mM MgCl2, 5% glycerol, and 1 mM DTT) in the presence of 25 μM acetyl-CoA. The HAT reactions were incubated at 30 °C for 30 min and stopped by the addition of one third of a reaction volume of 4x SDS-PAGE Loading Buffer (200 mM Tris•HCl, pH 6.8, 8% SDS, 40% glycerol, 4% β-mercaptoethanol, 50 mM EDTA, 0.08% bromophenol blue), followed by heating to 100°C for 5 minutes. The reaction products were then subjected to Western blotting as described above.
Identification of histone PTMs by mass spectrometry
MDA-MB-231 cells expressing FLAG-tagged wild-type or mutant histone H2B were seeded at ~5×106 cells per 15 cm diameter plate and cultured as described above. Nucleosome immunoprecipitation was performed as described above. The immunoprecipitated material was electrophoresed on a precast SDS-PAGE gel and the regions containing histone proteins were excised from the gel. The proteins were digested with trypsin and processed for mass spectrometry as described previously (22). The resulting peptides were injected onto an Orbitrap Fusion Lumos mass spectrometer (Thermo Electron) coupled to an Ultimate 3000 RSLC-Nano liquid chromatography system (Dionex). The samples were eluted with a gradient from 1–28% Buffer B over 90 minutes. Buffer A contained 2% (v/v) ACN and 0.1% formic acid in water, and Buffer B contained 80% (v/v) ACN, 10% (v/v) trifluoroethanol, and 0.1% formic acid in water. MS scans were acquired at a 120,000 resolution in the Orbitrap and up to 10 MS/MS spectra were obtained in the ion trap for each full spectrum acquired using higher-energy collisional dissociation (HCD) for ions with charges 2–7. Raw MS data files were analyzed using Proteome Discoverer v2.2 (Thermo), with peptide identification performed using Sequest HT searching against the Homo sapiens database from UniProt.
Cell-derived xenograft experiments in mice
All animal use was performed with oversight from UT Southwestern’s Institutional Animal Care and Use Committee under an approved animal use protocol. Female NOD scid gamma (NSG) mice at 6–8 weeks of age were used (The Jackson Laboratory, 005557) to establish breast cancer xenografts. Five million MDA-MB-231 cells engineered for ectopic expression of FLAG-tagged histone H2B (WT or D51A mutant) were injected subcutaneously into the flank of the mice. Mice carrying ~100 mm3 subcutaneous tumors were randomized to receive 25 mg/kg of Niraparib (MedChemExpress, HY-10619) or vehicle intraperitoneally, 5 days a week (2 days off) for 4 weeks. Tumor volumes were calculated using a modified ellipsoid formula: Tumor volume = ½ (length × width × height). The experiment was terminated and the mice were euthanized when the average tumor diameter from any group exceeded 20 mm. The tumor tissues were homogenized to make extracts used for Western blotting.
Quantification and statistical analyses
All sequencing-based genomic experiments were performed a minimum of two times with independent biological samples. Statistical analyses for the genomic experiments were performed using standard genomic statistical tests as described above. All cell proliferation, cell migration, and qPCR-based gene-specific experiments were performed a minimum of three times with independent biological samples. All Western blotting experiments with quantification were performed a minimum of three times with independent biological samples and analyzed by Image Lab 6.0.
Data availability
The high throughput sequencing data from this study are available from the NCBI’s Gene Expression Omnibus repository (GEO; http://www.ncbi.nlm.nih.gov/geo/) under the super series accession number GSE180463. The ATAC-seq, ChIP-seq, and RNA-seq datasets can be found under the subseries GSE180458, GSE180459, and GSE180461 respectively.
Results
Identification of functional sites of ADPRylation that are mutated in oncohistones
Nacev and colleagues previously reported a total of 4205 missense mutations for all four core histones, identified from a total of 3143 sequenced samples from 3074 unique patients across 183 specific tumor types (3). In our further analysis, we found that among the 20 most prevalent somatic missense mutations for each core histone, 60% of H2B mutations are Glu/Asp mutations occurring on 8 different residues, 50% of H3 mutations are Glu/Asp mutations occurring on 8 different residues, 40% of H4 mutations are Glu/Asp mutations occurring on 5 different residues, and 35% of H2A mutations are Glu/Asp mutations occurring on 4 different residues (Fig. 1A; Supplementary Table S1). Overall, 46% of the oncohistone mutations considered occur on 25 different Glu or Asp residues (35% on Glu, and 11% on Asp) for all four core histones (Fig. 1B; Supplementary Table S1). Notably, among these top 25 unique Glu or Asp oncohistone mutation sites, 20 have been identified previously as sites of ADPRylation by PARP-1 in wild-type histones (Fig. 1C; Supplementary Data S1) (10,21,22,50,51), suggesting that these mutations may interrupt the regulatory effects of ADPRylation on growth of cancer cells. We confirmed nearly half of these ADPRylation sites in our own mass spectrometry analyses using MCF-7 cells, including H2B-D51 (Supplementary Fig. S1 and Supplementary Data S1), although this screen was not saturated.
Fig. 1. Oncohistone mutations occur at sites of Asp and Glu ADPRylation.
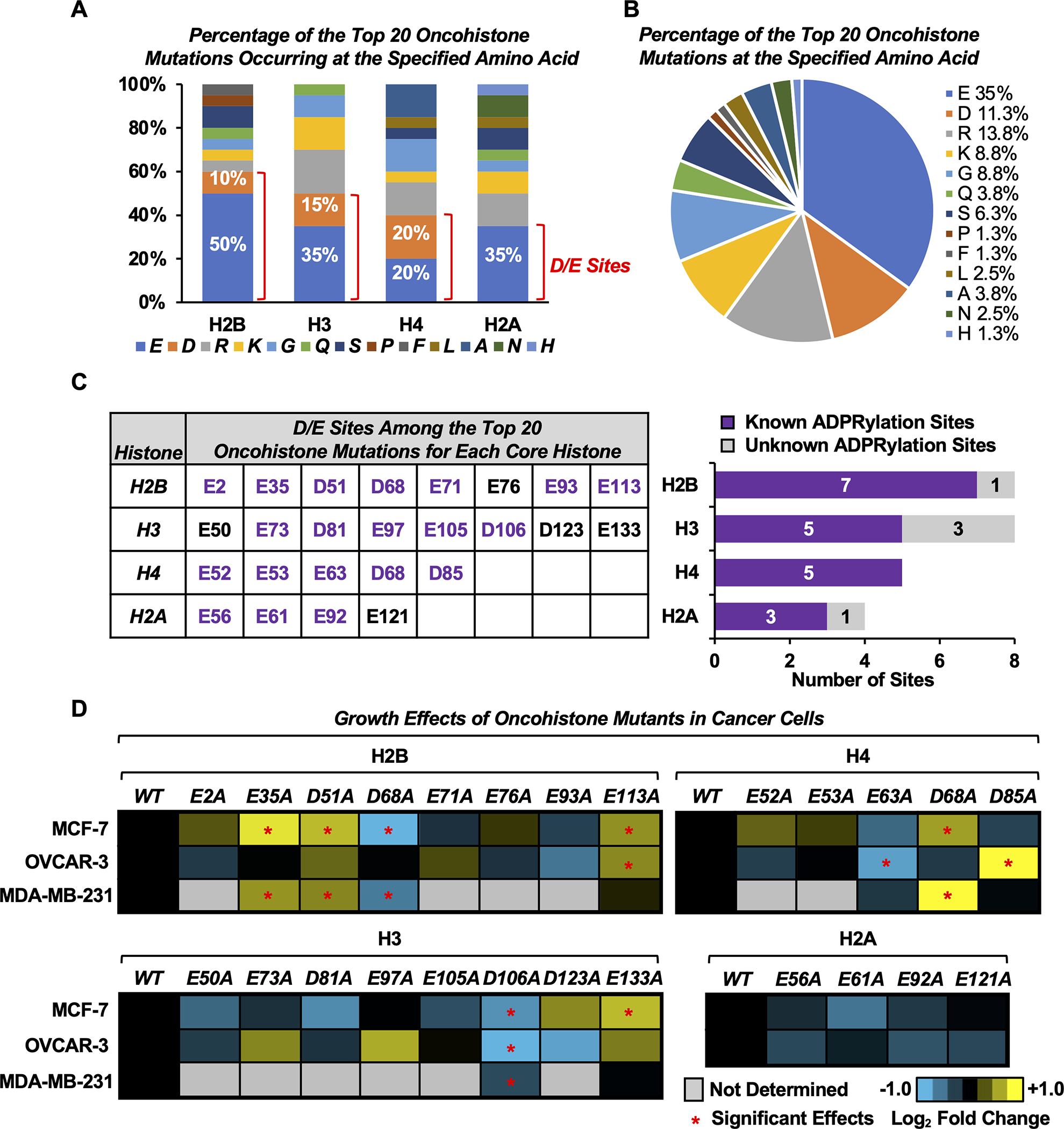
A) Bar graph showing the percentage of the top 20 oncohistone mutations for each histone occurring at the specified amino acid. Those occurring at Asp (D) and Glu (E) sites are indicated.
B) Pie chart showing the percentage of the top 20 oncohistone mutations for all histones occurring at the specified amino acid.
C) Most of the top oncohistone mutations occurring at D and E sites have previously been identified by mass spectrometry as ADPRylation sites (“known sites”). Left, D/E sites among the top 20 oncohistone mutations for each core histone. Purple text indicates known ADPRylation sites. Right, the number of previously known and unknown histone ADPRylation sites from the collection in the left panel.
D) Heatmaps showing proliferation of MCF-7, OVCAR-3 and MDA-MB-231 cells expressing FLAG-tagged wild-type (WT) or alanine (A) mutants of D/E oncohistone mutation sites for individual core histones, assayed by crystal violet staining. Results represent the mean fold changes of cell proliferation for cells expressing the histone mutants versus cells expressing the cognate WT histone at day 7 (n = 3–5). Asterisks indicate significant differences from the WT (two-way ANOVA followed by Dunnett’s multiple comparisons test; * p < 0.05). The corresponding cell growth curves and Western blots for ectopic FLAG-tagged histone expression are shown in Supplementary Figs. S2–4.
[See also Supplementary Figs. S1–S5, Supplementary Table S1, and Supplementary Data S1]
To test this hypothesis, we first individually mutated the top 25 Glu/Asp oncohistone mutation sites into alanine (Ala, A), which cannot be ADPRylated (Fig. 1C, left). We ectopically expressed FLAG-tagged versions of the wild-type (WT) and Glu/Asp mutant histones in three different human cancer cell lines: MCF-7 [estrogen receptor (ER)-positive luminal breast cancer], MDA-MB-231 (triple-negative/basal breast cancer), and OVCAR-3 (high-grade serous ovarian adenocarcinoma) (Supplementary Figs. S2–S4). We then screened for proliferation in cells expressing the WT or mutant histones. Altogether, we found that ectopic expression of six mutants of three different core histones (H2B, H3, and H4) altered cell proliferation in at least two different cell lines (Fig. 1D; Supplementary Figs. S2–S5). Among them, four mutants including H2B-E35, H2B-D51, H2B-E113, and H4-D68 enhanced cell growth, while two mutants including H2B-D68 and H3-D106 inhibited cell growth (Fig. 1D; Supplementary Figs. S2–S4). We then chose two known ADPRylation sites, H2B-D51 and H4-D68, whose mutation resulted in enhanced cell proliferation in both MCF-7 and MDA-MB-231 breast cancer cell lines, for further characterization, with a focus on the transcription regulatory effects of H2B-D51.
To investigate the functions of H2B-D51 mutants as oncohistones, we mutated the Asp51 residue to two non-ADPRylatable residues: Ala, as used in our cell proliferation screening assays, and asparagine (Asn, N), which is a naturally-occurring alteration found in cancers (3). We ectopically expressed FLAG-tagged WT and mutant versions of H2B (D51A and D51N) in MDA-MB-231 cells. Both of the H2B-D51 mutants (Ala and Asn) enhanced cell proliferation and cell migration compared to WT H2B (Fig. 2, A and B). Similar results were not observed in two “normal” cell lines: FT-194 (a normal fallopian tube cell line representing a suspected source of ovarian cancer progenitors) and MCF-10A (commonly used to represent normal mammary epithelial cells) (Supplementary Fig. S6). Thus, the H2B mutants drive the enhanced growth phenotype in cancer cells, but not in untransformed cells.
Fig. 2. Mutation of the H2B-D51 ADPRylation site enhances cell proliferation and migration in MDA-MB-231 cells through gene regulation.
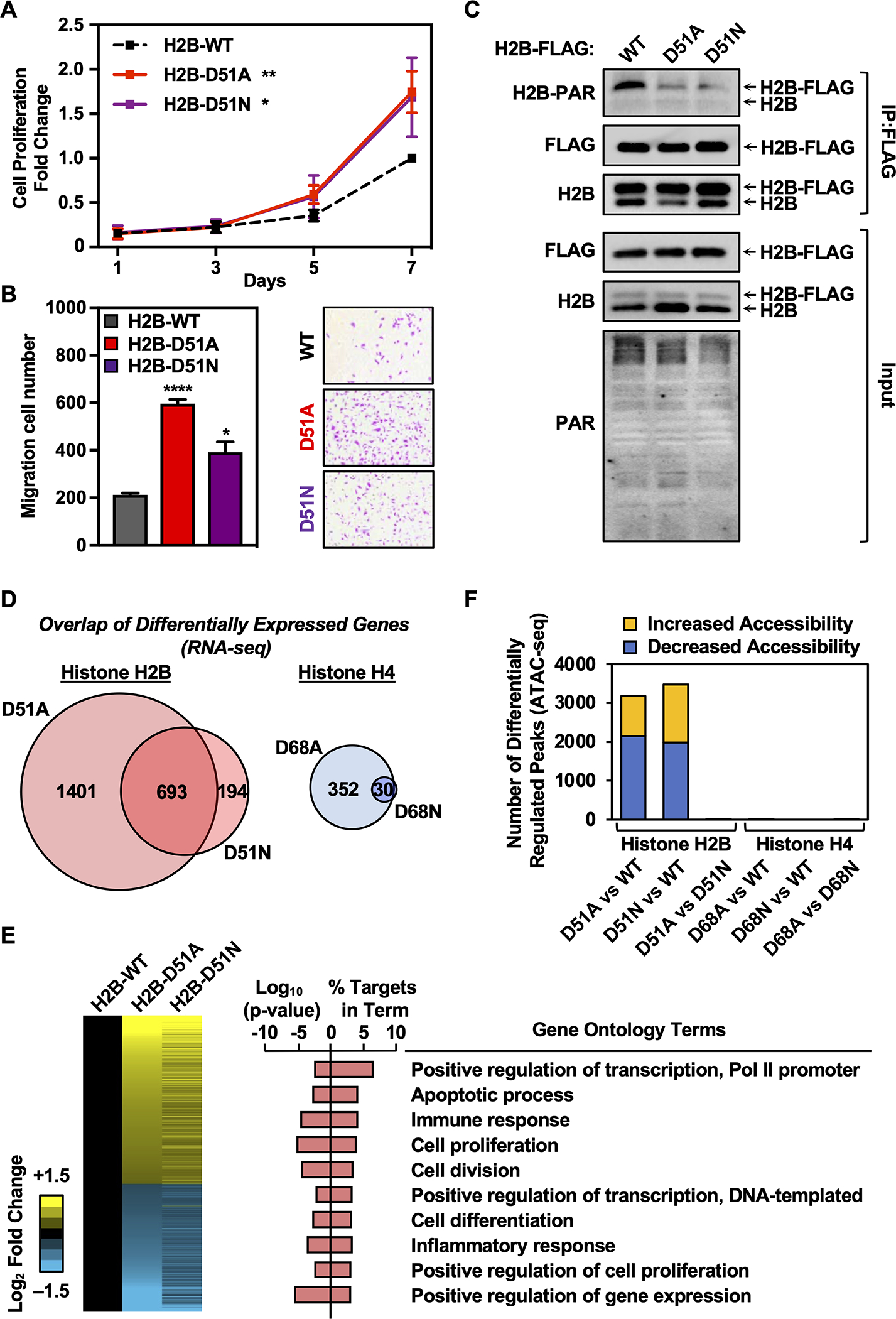
A) Growth curves of MDA-MB-231 cells ectopically expressing FLAG-tagged wild-type (WT), D51A, or D51N H2B assayed by crystal violet staining. Each point represents the mean ± SEM, n = 3. Asterisks indicate significant differences between the mutant and WT at day 7 (two-way ANOVA followed by Dunnett’s multiple comparisons test; * p < 0.05, and ** p < 0.01).
B) Cell migration assays for MDA-MB-231 cells ectopically expressing FLAG-tagged WT, D51A, or D51N H2B. Left, Quantification of cell migration numbers. Each bar represents the mean ± SEM, n = 3. Asterisks indicate significant differences between the mutant and WT (Student’s t-test; * p < 0.05, and **** p < 0.0001). Right, Representative images.
C) Western blots showing the levels of ADPRylation on H2B in MDA-MB-231 cells ectopically expressing FLAG-tagged WT, D51A, or D51N H2B after immunoprecipitation of FLAG-tagged nucleosomes.
D) Comparison of gene expression alterations mediated by H2B-D51 and H4-D68 oncohistone mutants in MDA-MB-231 cells as assessed by RNA-seq. Left, Venn diagram depicting the overlap of differentially expressed genes in cells ectopically expressing H2B-D51A and D51N mutants versus WT H2B. Right, Venn diagram depicting the overlap of differentially expressed genes in cells ectopically expressing H4-D68A and D68N mutants versus WT H4.
E) Regulation of gene expression programs in breast cancer cells by H2B-D51 oncohistone mutations. Left, Heatmaps representing the fold change of common differentially expressed genes from RNA-seq performed in MDA-MB-231 cells ectopically expressing FLAG-tagged WT, D51A, or D51N H2B. Right, Gene ontology terms enriched for the common differentially expressed genes in MDA-MB-231 cells ectopically expressing the H2B-D51 mutant versus cells expressing WT H2B.
F) Comparison of chromatin accessibility alterations mediated by H2B-D51 and H4-D68 mutants in MDA-MB-231 cells as assessed by ATAC-seq performed in cells ectopically expressing WT, D51A, or D51N H2B, or WT, D68A, or D68N H4. Yellow, differentially upregulated ATAC-seq peaks; Blue, differentially downregulated ATAC-seq peaks. Minimum counts 10, FDR < 0.05, fold-change cutoff 1.5.
[See also Supplementary Figs. S6–S8]
To characterize the expression and ADPRylation of the ectopically expressed WT and mutant H2B proteins, we performed a series of Western blotting assays comparing input (i.e., extract) to MNase-digested mononucleosomes that were enriched for ectopically-expressed H2B by FLAG immunoprecipitation. The use of mononucleosomes eliminated potential contributions from unincorporated H2B. In these assays, the addition of the FLAG epitope allowed us to distinguish between the endogenous and ectopically expressed histones (Fig. 2C). The total levels of ectopically expressed H2B (WT or D51 mutant) was less than 20% of the endogenous H2B (Fig. 2C; see the condition labeled “H2B” in the “Input”). For immunoprecipitated nucleosomes (“IP:FLAG”), only those with one or two copies of ectopically-expressed FLAG-tagged H2B, but not those with two copies of the endogenous histone, should be enriched. Thus, we would expect an enrichment of FLAG-tagged H2B in the immunoprecipitated material, as seen in Fig. 2C. Moreover, both H2B mutants exhibited reduced levels of PARylation as demonstrated by Western blotting of the FLAG-immunoprecipitated nucleosomes for PAR (Fig. 2C). Importantly, the reduction in PARylation was only observed on ectopically-expressed H2B harboring the D51 mutations, not endogenous H2B (Supplementary Fig. S7).
Similar results for the expression and ADPRylation of ectopically expressed WT and D68 mutant H4 were observed (Supplementary Fig. S8). Overall, these results demonstrate that sites of oncohistone mutation can be functional sites of ADPRylation, with direct impact on cellular phenotypes often attributed to oncogenesis, such as proliferation.
H2B-D51 oncohistone mutants promote alterations in chromatin accessibility and gene expression
Histone ADPRylation has been linked to both gene regulation and DNA damage repair (16). To determine whether loss of ADPRylation on H2B-D51 or H4-D68 could impact gene expression in MDA-MB-231 cells, we performed RNA-seq. We observed strikingly different effects of H2B-D51 and H4-D68 mutations on gene expression. We found that exogenous expression of the H2B-D51 mutants resulted in dramatic alterations in gene expression compared to WT H2B, with an overlap of 693 genes differentially regulated between D51A and D51N mutants (Fig. 2D). In contrast, exogenous expression of the H4 mutants resulted in little change in gene expression compared to WT H4, with only 30 commonly dysregulated genes observed between the D68A and D68N mutants (Fig. 2D). Moreover, ectopic expression of the H2B D51A and D51N mutants resulted in similarly altered patterns of gene expression (Fig. 2E, left). Gene ontology (GO) analyses indicated that the common set of genes whose expression was altered by the H2B-D51 mutants was enriched in GO terms including regulation of transcription, gene expression, and cell proliferation (Fig. 2E, right), in line with the growth advantage we observed in culture after ectopic expression of the H2B-D51 mutants.
A recent study showed that sites of oncohistone mutation in the globular domains of core histones, especially in H2A and H2B, can destabilize nucleosomes and enhance chromatin remodeling (52). To further explore whether the oncohistone mutations in H2B-D51 or H4-D68 could impact chromatin accessibility, we performed ATAC-seq, which provides a genomic readout of chromatin accessibility at distinct loci. Consistent with the effects on gene expression, the H2B-D51 mutants, but not the H4-D68 mutants, promoted dramatic changes in chromatin accessibility, with both H2B-D51A and H2B-D51N showing remarkably similar patterns (Fig. 2F). Collectively, these results suggest that H2B-D51 oncohistone mutations enhance the oncogenic properties of breast cancer cells by altering transcriptional regulation.
H2B-D51 oncohistone mutations enhance H2B acetylation
We next explored the potential molecular mechanisms by which expression of H2B-D51 mutants influence transcriptional regulation in cancer cells. Previous studies have established that modification of histones at one site can either promote or inhibit modification at other sites, termed histone modification crosstalk (22,40,53). To identify other histone PTMs that might be altered in the H2B-D51 mutants, we isolated FLAG-tagged nucleosomes by immunoprecipitation after MNase digestion of chromatin from MDA-MB-231 cells expressing WT H2B, H2B-D51A, or H2B-D51N, and subjected them to mass spectrometry (Supplementary Data Set 2). Both H2B-D51A and H2B-D51N resulted in similar increases in many different histone acetylation marks on H2B, including K12ac, K20ac, and K120ac, histone PTMs that are enriched at promoters and enhancers (54) (Fig. 3, A and B; Supplementary Fig. S9A). We confirmed these results by Western blotting (Fig. 3C). Increased acetylation was specific to H2A and H2B; other sites of acetylation, such as H3K27, showed little consistent global change in cells expressing H2B-D51A or H2B-D51N (Fig. 3, A and B; Supplementary Fig. S9B). Interestingly, the effects of the H2B-D51 mutants on H2B acetylation occurred in both cis and trans, since enhanced acetylation was observed for both exogenous H2B (containing H2B-D51 mutations; cis) and endogenous H2B (no H2B-D51 mutations; trans) (Fig. 3C; see H2BK12ac, two bands).
Fig. 3. Site-specific H2B acetylation is elevated in MDA-MB-231 cells expressing H2B-D51 oncohistone mutants.
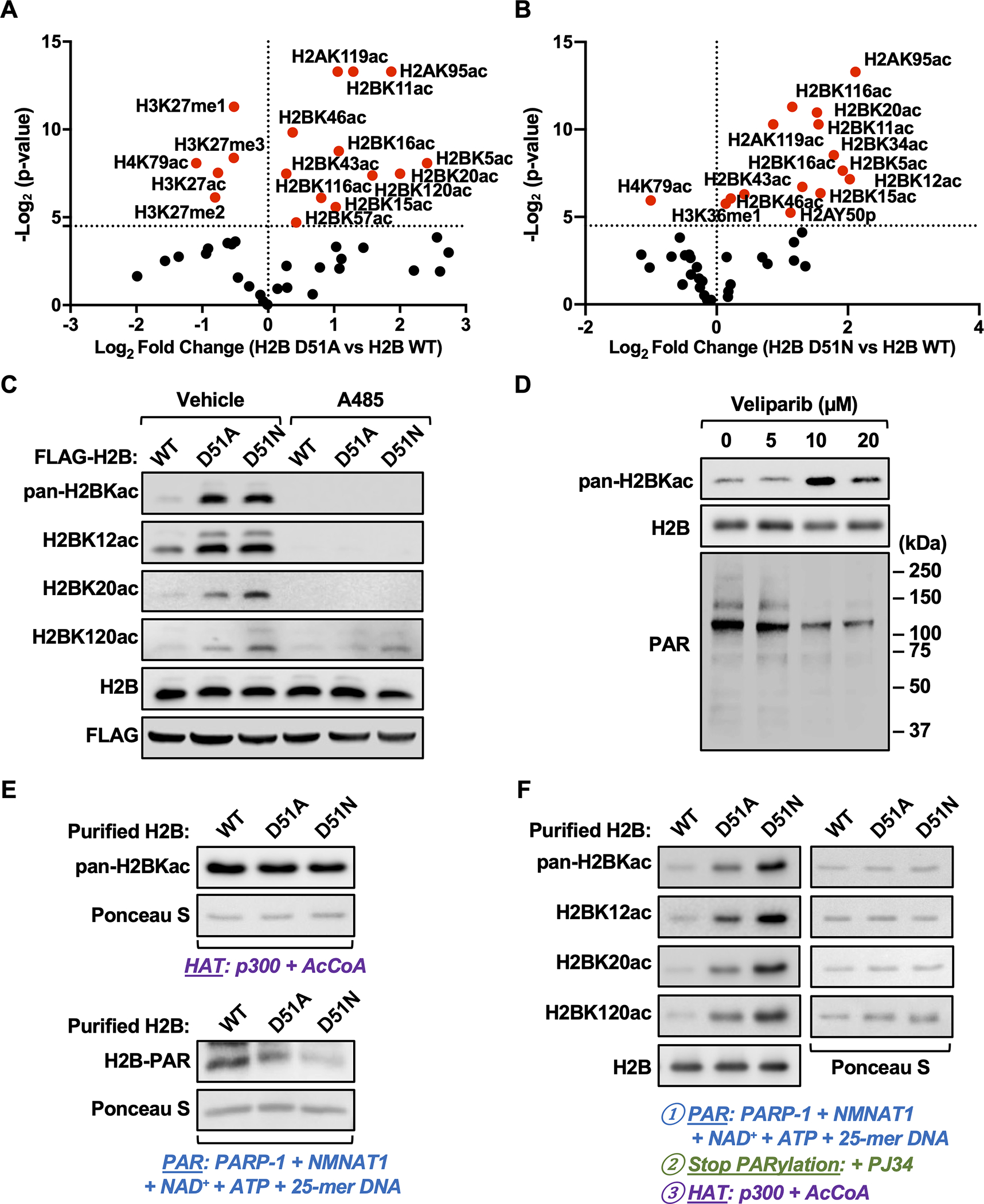
A and B) Quantification of histone modifications by mass spectrometry in MDA-MB-231 cells ectopically expressing FLAG-tagged WT, D51A, or D51N H2B. Volcano plots showing statistical significance (p-value) plotted versus the abundance of modified peptides (fold change of H2B D51 mutant versus H2B WT; D51A, panel A; D51N, panel B) for individual site-specific histone modifications. Points plotted in red indicate histone modifications significantly different in the H2B mutants versus H2B WT (p < 0.05). The data are from three biological replicates of FLAG-tagged nucleosomes from MDA-MB-231 cells enriched by immunoprecipitation after MNase digestion of chromatin analyzed by LC-MS/MS.
C) Western blots showing the levels of pan-H2BKac, H2BK12ac, H2BK20ac, and H2BK120ac in MDA-MB-231 cells ectopically expressing FLAG-tagged WT, D51A, or D51N H2B ± treatment with A485 (5 μM, 2 hours) or vehicle (DMSO).
D) Western blots showing the levels of pan-H2BKac and PAR in MDA-MB-231 cells ± treatment with the PARPi Veliparib (5–20 μM) for 2 hours.
E) In vitro HAT assays were performed using purified H2B (WT, D51A and D51N), p300, and acetyl-CoA (upper panels). In vitro PARylation assays were performed using purified H2B (WT, D51A and D51N), PARP-1, NMNAT-1, NAD+, ATP, and 25-mer DNA (lower panels). Western blots showing the levels of pan-lysine acetylation (upper panels) and PAR (lower panels) on H2B. Ponceau S staining of H2B was used to assess equal loading of material.
F) In vitro PARylation assays with purified H2B (WT, D51A and D51N) were performed as described in (E) and stopped by the addition of the PARPi PJ34, followed by HAT assays with the addition of p300 and acetyl-CoA. Western blots showing the levels of pan-H2BKac, H2BK12ac, H2BK20ac, and H2BK120ac. Blots for H2B (left bottom panel) and Ponceau S staining of H2B (right panels) were used to assess equal loading of material.
[See also Supplementary Fig. S9 and Supplementary Data S2]
The histone acetyltransferase CBP/p300 is responsible for a broad array of histone acetylation events on histone proteins (55). Treatment with A485, an inhibitor of CBP/p300, resulted in global reduction of H2B acetylation in both wild-type and mutant expressing cells (Fig. 3C). Moreover, treatment of wild-type cells with Veliparib, a PARP inhibitor (PARPi), dramatically increased H2B acetylation, similar to the effects of the H2B-D51 mutants (Fig. 3D). To explore this interplay in more detail, we reconstituted PARP-1-mediated PARylation and p300-mediated acetylation of H2B in biochemical assays using purified proteins, with both free H2B and recombinant nucleosomes (22,40,47). First, we determined that mutation of H2B had no effect on H2B-directed p300 activity in vitro (Fig. 3E, upper panels). The H2B-D51A and H2B-D51N mutants, however, exhibited reduced PARylation by PARP-1 versus WT H2B protein, confirming H2B-D51 as a PARylation site (Fig. 3E, lower panels). Furthermore, we found that PARP-1-mediated PARylation on D51 blocked subsequent H2B acetylation, as both the H2B-D51A and H2B-D51N mutants exhibited enhanced pan-acetylation, as well as acetylation on specific lysine sites (K12, K20 and K120) on H2B (Fig. 3F). Finally, we observed similar results with recombinant nucleosomes, namely that PARP-1-mediated PARylation inhibited p300-mediated acetylation of H2B (Supplementary Fig. S9C). Collectively, these results suggest that H2B-D51 PARylation is an important regulatory mechanism to suppress p300-mediated H2B acetylation, thus impacting gene expression and cancer cell properties.
Loss of H2B-D51 ADPRylation promotes H2BK12 acetylation genome-wide
To further explore the relationship between H2B-D51 ADPRylation and p300-mediated H2B acetylation at regulatory elements genome-wide, we performed chromatin immunoprecipitation-sequencing (ChIP-seq) for H2BK12ac in MDA-MB-231 cells expressing WT H2B, H2B-D51A, or H2B-D51N with or without A485 treatment. Compared to WT H2B, we observed that both H2B-D51A and H2B-D51N caused a significant increase in H2BK12ac levels at intergenic regions and promoters genome-wide, which was inhibited by treatment with A485 (Fig. 4, A–C). Increased p300-mediated H2BK12 acetylation in cells expressing H2B-D51 mutants was confirmed in an independent ChIP-qPCR experiment (Supplementary Fig. S10).
Fig. 4. H2B-D51 oncohistone mutations promote genome-wide H2BK12 acetylation.
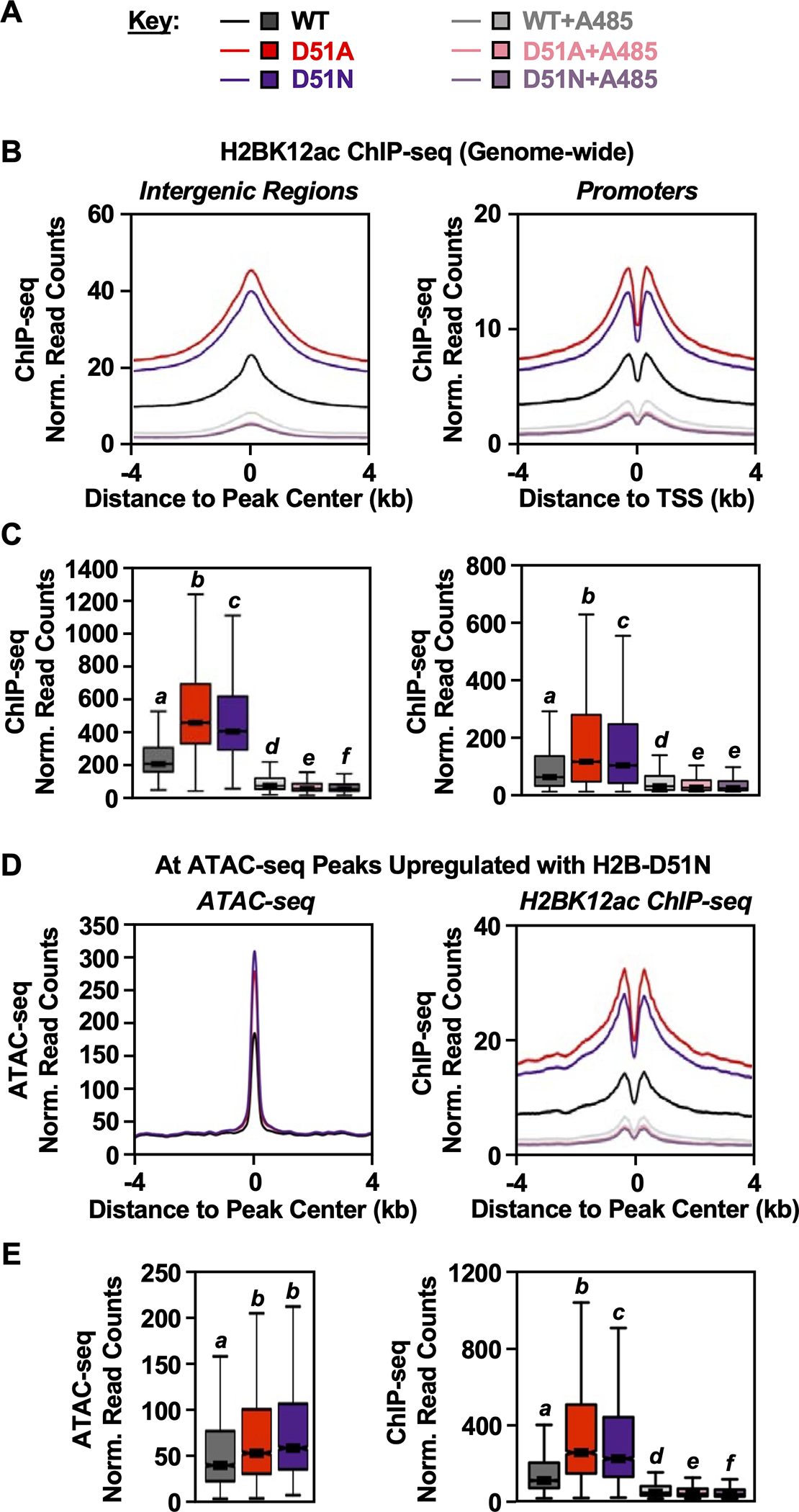
A) Key for the conditions used in the other panels of this figure. H2BK12ac ChIP-seq and ATAC-seq were performed in MDA-MB-231 cells ectopically expressing FLAG-tagged WT, D51A, or D51N H2B ± treatment with A485 (5 μM, 2 hours) or vehicle (DMSO).
B and C) Metaplots (B) and box plots (C) of H2BK12ac ChIP-seq data at intergenic regions (left) and promoter regions (right). Bars marked with different letters are significantly different from each other (Wilcoxon rank sum test, p < 1 ×10−13).
D and E) Metaplots (D) and box plots (E) of ATAC-seq (left) and H2BK12ac ChIP-seq (right) data at ATAC-seq peaks upregulated in cells expressing the H2B-D51N oncohistone mutant versus cells expressing WT H2B. Bars marked with different letters are significantly different from each other (Wilcoxon rank sum test, p < 1 ×10−13).
[See also Supplementary Figs. S10–S11]
To assess potential relationships between H2B-D51 ADPRylation, chromatin accessibility, and enrichment of H2BK12ac, we integrated ATAC-seq and ChIP-seq datasets in MDA-MB-231 cells expressing WT H2B, H2B-D51A, or H2B-D51N. We focused on ATAC-seq peaks with increased accessibility (Fig. 4, D and E, left panels) or decreased accessibility (Supplementary Fig. 11; see left panels in B and C) in cells expressing the H2B mutants versus WT H2B. By ChIP-seq, we observed an enrichment of H2BK12ac at the upregulated ATAC-seq peaks (Fig. 4, D and E, right panels). Surprisingly, we also observed an enrichment of H2BK12ac at the downregulated ATAC-seq peaks (Supplementary Fig. 11; see right panels in B and C), suggesting that loss of H2B-D51 ADPRylation promotes H2BK12ac regardless of the chromatin accessibility state promoted by the H2B-D51A and H2B-D51N mutants.
H2B-D51 oncohistone mutations link PARP-1-mediated ADPRylation and p300-mediated acetylation to chromatin accessibility and gene expression
To assess potential relationships between H2B-D51 ADPRylation, chromatin accessibility, enrichment of H2BK12ac, and gene expression, we integrated all of the genomic data sets that we generated from MDA-MB-231 cells expressing WT H2B, H2B-D51A, or H2B-D51N (i.e., RNA-seq, ATAC-seq, H2BK12ac ChIP-seq). Based on chromatin accessibility (i.e., ATAC-seq) and gene expression (i.e., RNA-seq) that was up- or downregulated by expression of the H2B-D51N mutant (versus WT H2B), we categorized the ATAC-seq peaks into four groups: ATAC-seq peaks upregulated (Group A) or downregulated (Group B) with the H2B-D51N mutant located nearest to an upregulated gene, and ATAC-seq peaks upregulated (Group C) or downregulated (Group D) with the H2B-D51N mutant located nearest to a downregulated gene (Fig. 5A). We observed that upregulated ATAC-seq peaks were more enriched than downregulated ATAC-seq peaks nearest to the upregulated genes (Group A/60% versus Group B/40%, respectively) (Fig. 5B). In contrast, downregulated ATAC-seq peaks were more enriched than upregulated ATAC-seq peaks nearest to the downregulated genes (Group D/65% versus Group C/35%, respectively) (Fig. 5B). In addition, we observed that upregulated ATAC-seq peaks (Group A) are located closer to the TSSs of the nearest upregulated genes than the downregulated ATAC-seq peaks (Group B) (Fig. 5C, left panel; Supplementary Fig. S12A). A similar relationship between the Group C and Group D peaks was not observed for the downregulated genes (Fig. 5C, right panel; Supplementary Fig. S12B). These results suggest that loss of H2B-D51 ADPRylation in the oncohistone mutants promotes the upregulation of gene expression, at least in part, by increasing chromatin accessibility.
Fig. 5. H2B-D51 oncohistone mutations promote the upregulation of gene expression by increasing chromatin accessibility.
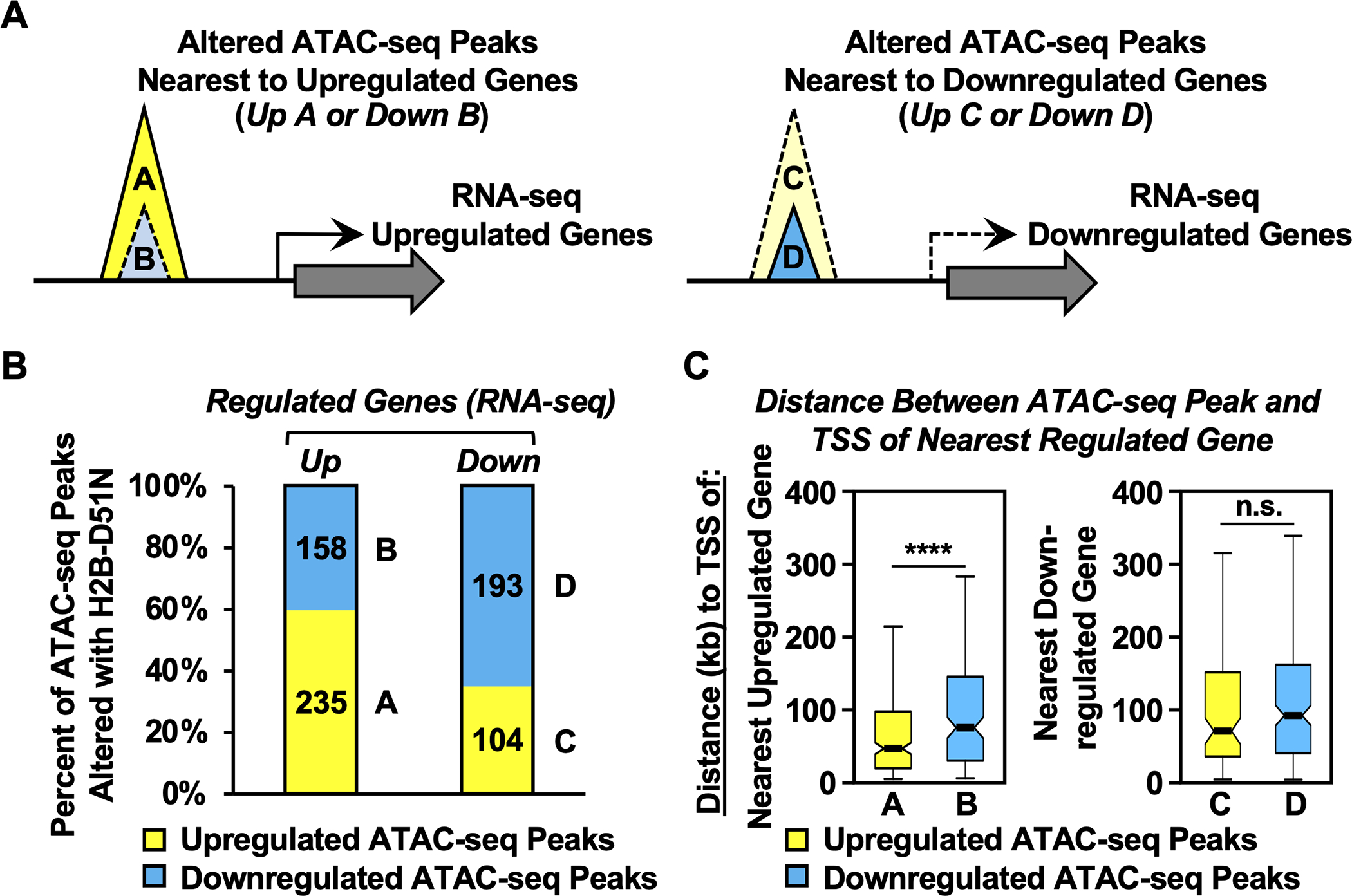
A) Schematic diagram showing the integration of ATAC-seq data with RNA-seq data to link chromatin accessibility to changes in gene expression in MDA-MB-231 cells expressing the H2B-D51N oncohistone mutant. Based on chromatin accessibility (i.e., ATAC-seq) and gene expression (i.e., RNA-seq) that was up- or downregulated by expression of the H2B-D51N mutant (versus WT H2B), the ATAC-seq peaks were categorized into four groups: ATAC-seq peaks upregulated (Group A) or downregulated (Group B) with the H2B-D51N mutant located nearest to an upregulated gene, and ATAC-seq peaks upregulated (Group C) or downregulated (Group D) with the H2B-D51N mutant located nearest to a downregulated gene.
B) Percent of upregulated or downregulated ATAC-seq peaks altered by expression of the H2B-D51N mutant located nearest to an upregulated (left: n = 393) or downregulated (right: n = 297) gene based on RNA-seq. The A, B, C, D labeling refers to the groups shown in panel A.
C) Box plots showing the average distance (in kb) between the center of the altered ATAC-seq peaks to the TSSs of the nearest upregulated or downregulated genes, as indicated. Asterisks indicate significant differences, p = 9.86 ×10−5. The A, B, C, D labeling refers to the groups shown in panel A.
[See also Supplementary Fig. S12]
To explore these genomic relationships further, we focused on the putative enhancers and the nearest neighboring genes associated with Group A ATAC-seq peaks (i.e., upregulated with the H2B-D51 mutants) (Fig. 6, A and B, left panels), representing a more accessible chromatin state associated with active gene transcription. At these ATAC-seq peaks, H2BK12ac was significantly enriched in the presence of H2B-D51A or H2B-D51N (Fig. 6, A and B, right panels). In addition, we observed that H2BK12ac was also enriched at the TSSs of the nearest neighboring genes upregulated with the H2B-D51 mutants (Fig. 6, C–E). These observations are represented with browser tracks for a single gene locus (i.e., LEFTY1) (Fig. 6F). We observed a similar enrichment of H2BK12ac at downregulated ATAC-seq peaks and the TSSs of the nearest neighboring downregulated genes (Supplementary Fig. S13). These results reaffirm the relationships observed in Fig. 5 among the H2B-D51 mutants, chromatin accessibility, and gene expression. They also suggest, again, that loss of H2B-D51 ADPRylation promotes H2BK12ac regardless of the chromatin accessibility state promoted by the H2B-D51 mutants.
Fig. 6. H2B-D51 oncohistone mutations link site-specific histone ADPRylation, chromatin accessibility, H2BK12 acetylation, and gene expression outcomes.
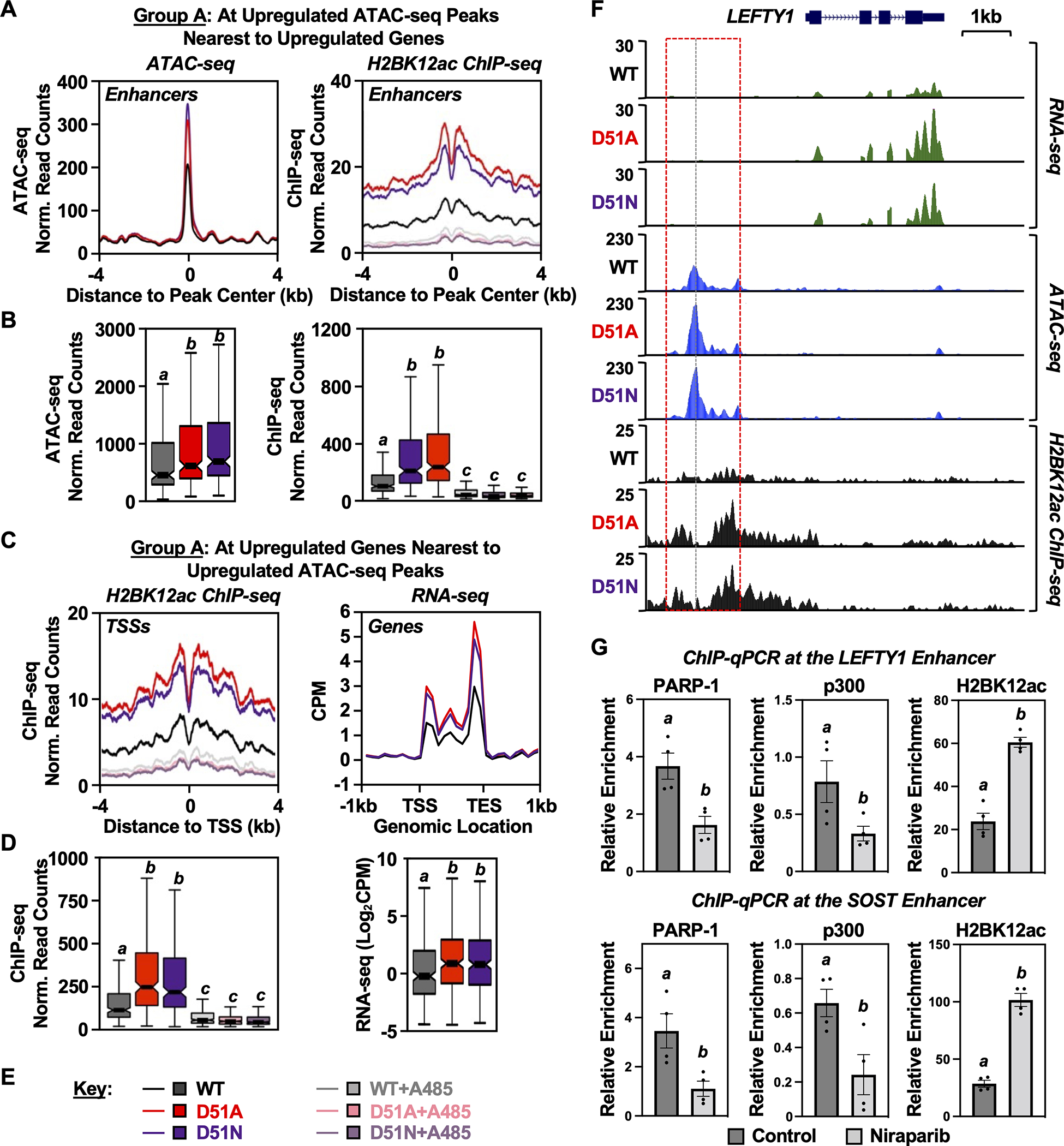
A and B) Metaplots (A) and box plots (B) of ATAC-seq (left) and H2BK12ac ChIP-seq (right) data at the Group A ATAC-seq peaks described in Fig. 5A (i.e., H2B-D51-upregulated ATAC-seq peaks located nearest to upregulated genes). Bars marked with different letters are significantly different from each other (Wilcoxon rank sum test, p < 8 ×10−7).
C and D) Metaplots (C) and box plots (D) of H2BK12ac ChIP-seq (left) and RNA-seq (right) data at the TSSs of upregulated genes nearest to the Group A ATAC-seq peaks described in Fig. 5A. Bars marked with different letters are significantly different from each other (Wilcoxon rank sum test, p < 8 ×10−7 for ChIP-seq data and p < 3 ×10−4 for RNA-seq data).
E) Key for the conditions used in the other panels of this figure. Genomic assays were performed in MDA-MB-231 cells ectopically expressing FLAG-tagged WT, D51A, or D51N H2B ± treatment with A485 (5 μM, 2 hours) or vehicle (DMSO).
F) Genome browser tracks representing the RNA-seq, ATAC-seq, and H2BK12ac ChIP-seq data at genomic locus (LEFTY1) containing a gene upregulated upon expression of the H2B-D51 oncohistone mutants in MDA-MB-231 cells.
G) ChIP-qPCR assays for PARP-1, p300, and H2BK12ac in MDA-MB-231 cells treated with vehicle or the PARPi Niraparib (10 μM, 2 hours) as indicated. PARP-1, p300, and H2BK12ac enrichment was assessed at the enhancers of LEFTY1 and SOST genes using gene-specific primers. These sites exhibit increased H2BK12ac by ChIP-seq in the presence of the H2B-D51 ADPRylation site mutants. Each column represents the mean ± SEM (n = 4). Bars marked with different letters are significantly different from each other (Student’s t-test; p < 0.05).
[See also Supplementary Figs. S13–S16]
To explore in more detail how ADPRylation of H2B-D51 might modulate p300-mediated acetylation of H2B, we performed locus-specific ChIP-qPCR assays. Two models are that (1) ADPRylation of H2B-D51 interferes with p300 binding to nucleosomes and, thus, reduces acetylation of H2B and (2) ADPRylation of H2B-D51 inhibits p300 activity without affecting its binding to nucleosomes. To test these models, we treated MDA-MB-231 cells ± the PARPi Niraparib, then performed ChIP-qPCR for PARP-1, p300, and H2BK12ac at a set of loci where the H2B-D51 ADPRylation site mutants promoted increased H2BK12 acetylation in the ChIP-seq assays. As expected, treatment with Niraparib caused a significant increase in H2BK12ac at 4 out of 5 of the loci tested (Fig. 6G; Supplementary Fig. S14). In addition, treatment with Niraparib caused a significant reduction in PARP-1 binding and, in most but not all cases, a concomitant reduction in p300 binding (Fig. 6G; Supplementary Fig. S14). Thus, reductions in PARylation and PARP-1 binding were not associated with increased p300 binding, even though we observed an increase in H2BK12 acetylation. These results suggest that p300 catalytic activity is released from inhibition as nucleosomal PAR levels are reduced [Model (2) above].
Potential mechanisms controlling chromatin accessibility at loci enriched in H2BK12ac
Interestingly, we observed that regions of accessible chromatin (from ATAC-seq), whether up or downregulated with the H2B-D51 mutants, were located within the troughs of H2BK12ac-enriched regions (Fig. 6F; Supplementary Fig. S13F). These results suggest that H2BK12ac, which is elevated with the H2B oncohistone mutants, is enriched around accessible loci and may provide context-specific cues for chromatin remodeling events that regulate accessibility. To explore this possibility further, we analyzed the ATAC-seq peaks up- or down-regulated with the H2B-D51N mutant to identify transcription factors (TF) enriched in differentially accessible regions. De novo motif analyses using HOMER (43) showed that loci with increased accessibility were significantly enriched in motifs for TFs contributing to tumor progression, such as RUNX, TEAD, AP-1, and KLF5 (Supplementary Fig. S15A). In contrast, loci with decreased accessibility were significantly enriched in motifs for other TFs, such as BATF, CTCF, C/EBPα, and ETS1 (Supplementary Fig. S15A).
To investigate further which TFs might drive the altered chromatin accessibility that was observed with the H2B-D51 mutants, we performed footprinting analysis on differentially accessible regions using TOBIAS (Transcription factor Occupancy prediction By Investigation of ATAC-seq Signal) (39). Consistent with the HOMER analysis, motifs for RUNX (i.e., RUNX1 and RUNX2) were enriched with the H2B-D51N mutant, whereas a motif for BATF was enriched with WT H2B (Supplementary Fig. S15B). In breast cancer, RUNX2 acts as a tumor-driving TF that acts to promote tumor growth and bone metastasis (56,57). In contrast, BATF has been reported to bind to closed chromatin and mediate the recruitment of CTCF (58), which can suppress cell proliferation and migration in breast cancer through transcriptional regulation of NF-κB and Nm23-H1 (59–61). These results are bolstered by ChIP-seq analyses in MDA-MB-231 cells showing enrichment of “activating” histone PTMs (e.g., H2K4me1, H3K27ac, and H3K9ac) at ATAC-seq peaks upregulated by H2B-D51N and reduced levels of these same histone PTMs at ATAC-seq peaks downregulated by H2B-D51N (Supplementary Fig. S16). These results suggest that loss of H2B-D51 ADPRylation in the oncohistone mutants promotes breast cancer progression by altering chromatin accessibility to enhance or block the binding of specific TFs to regulate gene expression.
The H2B-D51A ADPRylation site oncohistone mutant promotes the growth of cell-derived xenograft tumors in mice
To determine the effects of H2B-D51 ADPRylation on cancer cell growth in vivo, we performed xenograft tumor growth assays using MDA-MB-231 cells ectopically expressing FLAG-tagged H2B WT and D51A (Supplementary Fig. S17). We found that ectopic expression of H2B-D51A led to a significant increase in tumor growth versus H2B WT after four weeks (Figure 7, A and B). Interestingly, these effects were inhibited by treatment with PARPi (i.e., Niraparib), indicating that the presence of the H2B-D51A mutant alone does not confer resistance to PARP inhibition. Given the broad array of effects of PARPi in cells, each with distinct underlying mechanisms (62), this is perhaps not surprising. These results demonstrate that an amino acid residue in a core histone that functions as a site of ADPRylation can act to promote tumor growth when subject to oncohistone mutation.
Fig. 7. H2B-D51 oncohistone mutants promote tumor growth in vivo.
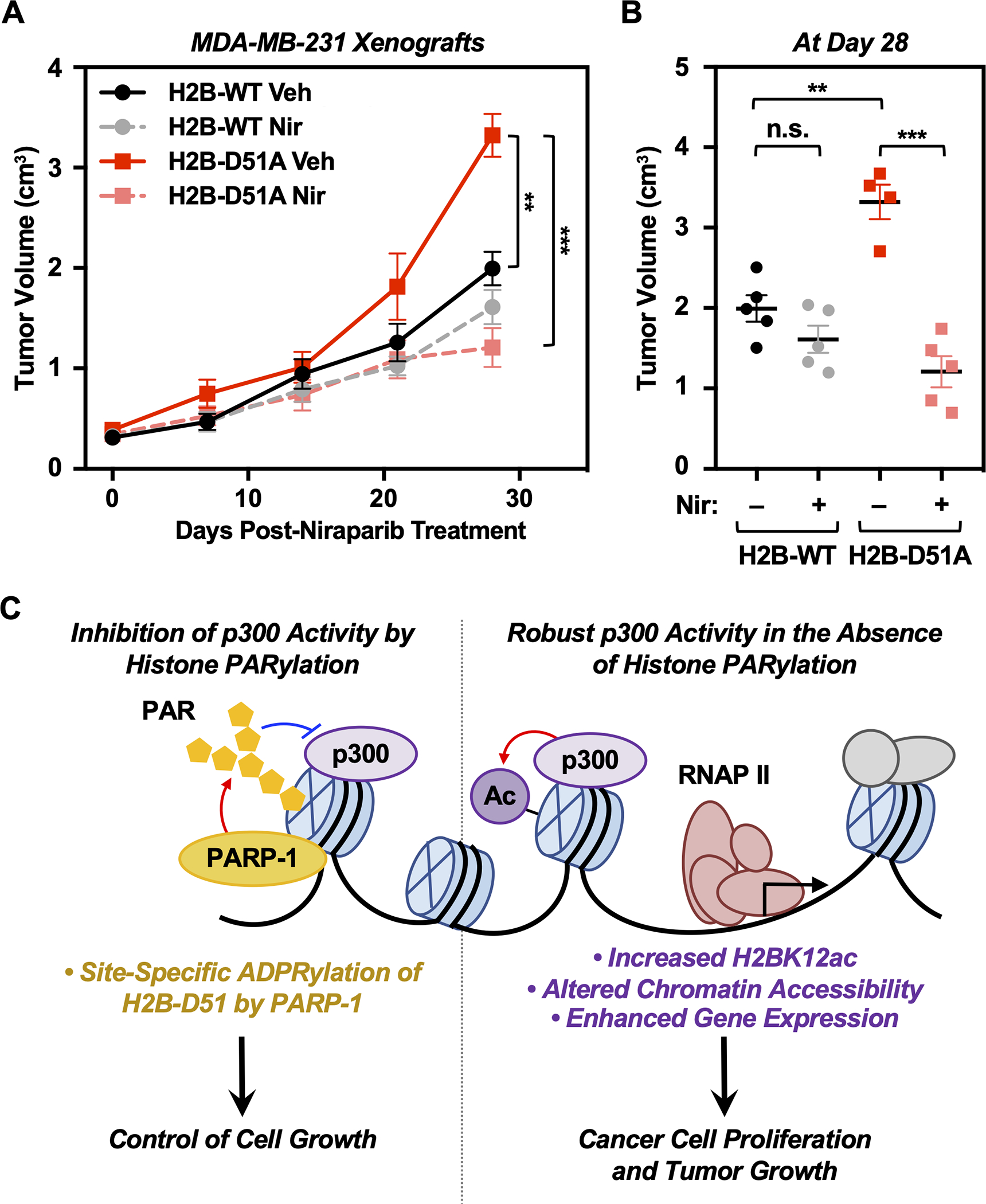
A) Growth of xenograft tumors formed from MDA-MB-231 cells engineered for ectopic expression of FLAG-tagged WT or D51A mutant H2B in NOD scid gamma (NSG) mice, treated with vehicle or 25 mg/kg Niraparib intraperitoneally (i.p.) 5 days a week over 4 weeks. Each point represents the mean ± SEM (n = 5). Asterisks indicate significant differences at day 28 as indicated (Student’s t-test; ** p < 0.01, *** p < 0.001).
B) Comparison of xenograft tumor volumes at day 28 among different groups as indicated. Each cluster in the plot represents the mean ± SEM (n = 5). Asterisks indicate significant differences as indicated (Student’s t-test; ** p < 0.01, *** p < 0.001, and n.s. not significant).
C) Schematic representation of the mechanism by which PARP-1-mediated ADPRylation of H2B-D51 inhibits p300-mediated H2B lysine acetylation and alters chromatin accessibility. Reversal of these inhibitory effects leads to enhanced p300-mediated histone acetylation and a gene expression program that favors oncogenic cell proliferation and tumor progression.
[See also Supplementary Figs. S17–S19]
H4-D68 ADPRylation site oncohistone mutants promote DNA damage responses
Our results demonstrate a role for H2B-D51 in cancers as both a site of ADPRylation and of oncohistone mutation, as well as a regulator of gene expression. In contrast, H4-D68, which is also a site of ADPRylation (Fig. 1C; Supplementary Fig. S8B), had little discernable effect on gene expression and chromatin accessibility when mutated (Fig. 2, D–F). Given the minimal effects of the H4-D68 mutants in these assays, we explored possible effects on other nuclear processes. To examine whether loss of ADPRylation on H4-D68 could impact DNA damage responses in MDA-MB-231 cells, we assayed γH2AX levels by immunofluorescence and Western blotting in cells treated with or without the PARPi, Niraparib. Interestingly, both H4-D68A and H4-D68N promoted the accumulation of γH2AX foci, especially upon treatment with Niraparib, suggesting that the H4-D68 oncohistone mutation sensitizes cells to PARPi (Supplementary Fig. S18, A and B). The H4-D68 mutants also increased the levels of phosphorylated p53 in MDA-MB-231 cells treated with Niraparib (Supplementary Fig. S18B). These effects were not observed for the H2B-D51 mutants (Supplementary Fig. S18, C and D). Similar results were observed for the H4-D68 mutants in MDA-MB-468 cells, with the effects of the mutants even more pronounced upon treatment with H2O2 (Supplementary Fig. S19). Collectively, these results suggest that H4-D68 mutations reduce the efficiency of DNA repair after damage, whereas H2B-D51 mutations enhance the oncogenic properties of breast cancer cells through transcriptional regulation.
Discussion
Although recent studies have identified mutations in histone genes as a class of oncogenic drivers in a broad array of cancer types (3,63), the regulatory mechanisms underlying oncohistone mutations and the selective advantages that they provide remain elusive. Herein, we identified site-specific ADPRylation of histones on Glu or Asp residues as an important regulator of cancer cell growth. Mutation of histone Glu/Asp ADPRylation sites to residues commonly found in oncohistones in human cancers promoted cell proliferation in breast and ovarian cancer cells. Specific oncohistone mutations that ablate ADPRylation sites contribute to cancer cell proliferation through distinct molecular mechanisms. For example, mutation of histone H2B-D51 enhanced global p300-mediated H2B lysine acetylation and altered chromatin accessibility, leading to a gene expression program that favors oncogenic cell proliferation. Collectively, our results have revealed functional consequences and diverse roles for the loss of regulatory Glu/Asp ADPRylation at sites of oncohistone mutation in tumorigenesis.
Oncohistone mutations occur at functional sites of ADPRylation to inhibit the proper regulation of cancer cell proliferation
Recent advances in chemical biology and mass spectrometry have helped to understand the role of site-specific histone ADPRylation in some cellular processes, including ADPRylation of H3-Ser10 and H2B-Glu18/Glu19 in DNA damage responses (26,64), and H2B-Glu35 in adipogenesis (22). We screened 25 Glu/Asp oncohistone mutation sites - 20 of which were previously identified as sites of ADPRylation - for their effects on proliferation in three different cancer cells lines. We found that six of them enhanced cell proliferation in at least one cell line when ectopically expressed, suggesting that mutation of those sites interferes with the regulatory effects of ADPRylation on cell proliferation. The link between ADPRylation and cell growth was confirmed for two sites, H2B-D51 and H4-D68, which exhibited a loss of ADPRylation and enhanced cell growth when changed to residues found in common oncohistone mutants. These results suggest that oncohistone mutations occur at functional sites of ADPRylation to inhibit the regulation of cancer cell proliferation.
Different molecular effects of oncohistone mutations occurring at functional sites of ADPRylation
To examine the molecular effects of ADPRylation site mutations in cancer cells, we focused on two oncohistone mutations (i.e., H2B-D51 and H4-D68). Mutation of these sites to Ala, as well as Asn which occurs naturally in human cancers, reduced the ADPRylation levels compared to the cognate wild-type histones, verifying that H2B-D51 and H4-D68 are sites of ADPRylation. We observed pronounced changes in the chromatin landscape and gene expression with the H2B-D51 mutant, but not with the H4-D68, compared to the cognate wild-type histones. Notably, the chromatin at enhancers associated with the genes upregulated in the presence of the H2B-D51 mutants were more likely to be open and accessible, enriched for activating histone PTMs (e.g., H3K4me1, H3K27ac, and H3K9ac), and enriched for DNA motifs for cancer-promoting transcription factors (e.g., RUNX, TEAD, AP-1, and KLF5). In addition, treatment with Niraparib caused a significant reduction in PARP-1 binding and, in most but not all cases, a concomitant reduction in p300 binding. Thus, reductions in PARylation and PARP-1 binding were not associated with increased p300 binding, even though we observed an increase in H2BK12 acetylation. These results suggest a model in which p300 catalytic activity is released from inhibition as nucleosomal PAR levels are reduced (Fig. 7C). The mechanisms of PARP-1/PAR-dependent inhibition of p300-mediated histone acetylation may involve steric inhibition of p300 catalytic activity or p300-histone interactions, or possibly even enhanced interactions of histone deacetylases with chromatin.
Unlike H2B-D51, mutations in H4-D68 had very little effect on either chromatin accessibility or gene expression [although Bagert et al. have reported a modest effect of H4-D68 mutations in dimerization exchange (52)]. Instead, we observed an increase in DNA damage with the H4-D68A and H4-D68N mutants under basal growth conditions, which was dramatically increased in cells treated with the PARPi Niraparib. One caveat from the studies with H4 is that D68 is buried in the nucleosome structure and faces inward, possibly making it inaccessible to PARP-1 in the context of the nucleosome. If so, then H4-D68 might only be ADPRylated as a free histone or in the H3/H4 tetramer, perhaps preventing nucleosome formation in cells (although our nucleosome immunoprecipitation assays argue against this). This would impact the potential mechanisms by which the H4-D68 oncohistone mutants affect DNA damage responses. This will be explored in more detail in future studies. Taken together, our results provide insights into the molecular mechanisms by which oncohistone mutations may regulate distinct cellular processes by inhibiting site-specific histone ADPRylation.
An important point to consider is that if oncohistone mutations are indeed drivers of cancer phenotypes, then they are likely selected by nature in the absence of a therapy, such as PARPi. In this case, the “goals” for the mutations would be to (1) subvert regulatory processes that attenuate tumor growth (such as histone ADPRylation that prevents growth-promoting gene expression, as seen with the H2B-D51 mutants) or (2) subvert regulatory processes that maintain genome integrity, thereby increasing DNA damage to promote a cancer-driving pro-mutagenic state (such as histone ADPRylation that helps maintain genome integrity, as seen with the H4-D68 mutants). Once selected and exposed to PARPi, the consequences could be varied - in some cases the mutants might sensitize cells to PARPi and in other cases make the cells resistant to PARPi. Future considerations for the therapeutic use of PARPi may need to include an understanding of the oncohistone mutation status of the cancers.
Functional links between histone ADPRylation and other modifications occurring at distinct sites
A growing body of evidence from patient samples and cell lines have shown that oncohistone mutations, such as those that occur in histone H3, alter the epigenomic landscape and interfere with the control of gene expression. This may involve crosstalk between distinct sites of histone modification. For example, the H3K27M oncohistone mutation inhibits the enzymatic activity of PRC2, leading to global reduction of H3K27me2/3 and an increase of H3K27ac (65–67). In addition, the H3K36M oncohistone mutation causes a global loss of H3K36 methylation and a gain of H3K27 methylation, which is involved in aberrant gene activation (68,69). These studies suggest that oncohistone mutations drive global changes in the abundance of other histone PTMs in human cancers.
ADPRylation has also been linked to other histone PTMs in gene regulation, on histones, transcription factors, and histone-modifying enzymes (22,45,47). Our previous work has revealed a functional link between site-specific Asp/Glu ADPRylation and nearby phosphorylation: (1) ADPRylation of histone H2B-Glu35 inhibits H2B-Ser36 phosphorylation, regulating adipogenic gene expression (22) and (2) ADPRylation of STAT1α-Asp721 in its transactivation domain is required for Ser727 phosphorylation, associated with transcriptional activation in macrophages (47). Other studies have connected histone ADPRylation and acetylation. For example, H3-Ser10 ADPRylation and H3-Lys9 acetylation are mutually exclusive in DNA damage responses (64). Herein, we observed dramatic increases in CBP/p300-mediated H2B acetylation with the H2B-D51 mutants on a broad array of lysine residues, without any appreciable impact on acetylation of H3K27, in both biochemical and genomic assays. The effects of H2B-D51 oncohistone mutation and ADPRylation on H2B acetylation indicate that both cis and inter-histone trans effects occur within the nucleosome, as described previously for other histone PTMs (40).
H2B acetylation, such as K12ac, K20ac, and K120ac, are enriched at promoters and enhancers (54). Thus, we surmised that enhanced H2B acetylation mediated the effects of the H2B-D51 mutants on chromatin accessibility and gene regulation. Indeed, ChIP-seq showed a global enrichment of H2BK12ac at enhancer and promoter regions in cells expressing the H2B-D51 mutants. Interestingly, the accessible chromatin regions from ATAC-seq, whether up or downregulated with the H2B-D51 mutants, were located within the troughs of H2BK12ac-enriched regions. These results suggest context-specific cues mediated by H2BK12 acetylation that drive chromatin remodeling complexes to regulate accessibility. Indeed, as noted above, motifs for specific types of transcription factors were enriched around the accessible loci.
Limitations of the study
In this study, we used a set of methods and approaches currently used by the field to investigate the effects of histone PTMs on chromatin-dependent events. These include ectopic expression of epitope-tagged histones and the use of histone ADPRylation site mutants. Although these approaches have been validated, more direct approaches, including CRISPR/Cas9-mediated genome engineering of endogenous histone loci (70), would simplify future analyses. The mutagenesis approach, however, has required us to make inferences about the functions of site-specific ADPRylation through loss-of-function studies, which may reveal effects of the mutations that are independent of ADPRylation. Although methods for the site-specific ADPRylation of histones in vitro are available (71), they are not applicable to studies in cells. This is a difficult limitation to overcome.
Other tools commonly used in the histone PTM field, but not available for studies of histone ADPRylation, are site-specific antibodies to modified histones. To date, the generation of site-specific histone Glu/Asp-ADPR antibodies has not been possible. Thus, experiments such as ChIP-seq localization of ADPRylated H2B-D51, which would aid the molecular analyses of p300-mediated H2B acetylation, are not possible. This has left some of our genomic analyses and mechanistic conclusions correlative. We have proposed a model for the interplay between PARP-1-mediated H2B-D51 ADPRylation and p300-mediated H2B acetylation, but same aspects of the model have not been tested directly. Moreover, although we have suggested that different classes of TFs may allow increased H2BK12 acetylation to be associated both with regions of enhanced and reduced chromatin accessibility, we have not tested this experimentally. Finally, we have not defined the mechanisms whereby H4-D68 mutations increase the levels of γH2AX. In spite of these limitations, our results illustrate how histone ADPRylation, which is tuned to the nuclear NAD+ environment of the cell (22,72), can serve as an upstream regulator of other histone modifications to control gene expression and downstream biological responses.
Supplementary Material
Significance.
This study identifies cancer-driving mutations in histones as sites of PARP-1-mediated ADP-ribosylation in breast and ovarian cancers, providing a molecular pathway by which cancers may subvert the growth-regulating effects of PARP-1.
Acknowledgements
We would like to thank members of Kraus lab for continued input and feedback on this project. We thank members of Dr. Gary Hon’s laboratory for technical help with ATAC-seq. We also thank Dr. Lianying Jiao for the structure-based alignment of the oncohistone residues on the nucleosome structure using PyMOL software. We acknowledge and thank the following UT Southwestern core facilities: the Next Generation Sequencing Core for deep sequencing services (Dr. Ralf Kittler and Vanessa Schmid), the Proteomics Core Facility for mass spectrometry services (Dr. Andrew Lemoff), and the Live Cell Imaging Core for microscopy support (Dr. Katherine Luby-Phelps). We also thank the Protein and Monoclonal Antibody Production Shared Resource at Baylor College of Medicine.
Funding:
This work was supported by a grant from the NIH/NIDDK (R01 DK069710), a grant from CPRIT (RP220325), and funds from the Cecil H. and Ida Green Center for Reproductive Biology Sciences Endowment to W.L.K.; a grant from the NIH/NIGMS (R35 GM124958) to L.A.B.; and fellowship support from the UT Southwestern Medical Center Hamon Center for Regenerative Science and Medicine to S.M.
Disclosures:
W.L.K. is a founder and stockholder of Ribon Therapeutics, Inc., and a founder, SAB member, BOD member, stockholder, and recipient of a research service agreement from Arase Therapeutics, Inc. He is also a co-holder of U.S. Patent 9,599,606 covering the ADP-ribose detection reagents used herein, which have been licensed to and is sold by EMD Millipore.
Footnotes
Competing Interests
All other authors declare no potential conflicts of interest.
Supplementary Materials
This paper includes one Supplementary Table, 19 Supplementary figures, 2 Supplementary data sets, Supplementary materials and methods, and Supplementary references.
References
- 1.Zhao Z, Shilatifard A. Epigenetic modifications of histones in cancer. Genome Biol 2019;20:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Audia JE, Campbell RM. Histone modifications and cancer. Cold Spring Harb Perspect Biol 2016;8:a019521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nacev BA, Feng L, Bagert JD, Lemiesz AE, Gao J, Soshnev AA, et al. The expanding landscape of ‘oncohistone’ mutations in human cancers. Nature 2019;567:473–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gibson BA, Kraus WL. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nature reviews Molecular cell biology 2012;13:411–24 [DOI] [PubMed] [Google Scholar]
- 5.Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nature reviews Molecular cell biology 2006;7:517–28 [DOI] [PubMed] [Google Scholar]
- 6.Ame JC, Spenlehauer C, de Murcia G. The PARP superfamily. BioEssays : news and reviews in molecular, cellular and developmental biology 2004;26:882–93 [DOI] [PubMed] [Google Scholar]
- 7.Vyas S, Chesarone-Cataldo M, Todorova T, Huang YH, Chang P. A systematic analysis of the PARP protein family identifies new functions critical for cell physiology. Nat Commun 2013;4:2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gupte R, Liu Z, Kraus WL. PARPs and ADP-ribosylation: Recent advances linking molecular functions to biological outcomes. Genes Dev 2017;31:101–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hottiger MO. SnapShot: ADP-Ribosylation Signaling. Mol Cell 2015;58:1134– e1 [DOI] [PubMed] [Google Scholar]
- 10.Gibson BA, Zhang Y, Jiang H, Hussey KM, Shrimp JH, Lin H, et al. Chemical genetic discovery of PARP targets reveals a role for PARP-1 in transcription elongation. Science 2016;353:45–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hendriks IA, Larsen SC, Nielsen ML. An advanced strategy for comprehensive profiling of ADP-ribosylation sites using mass spectrometry-based proteomics. Mol Cell Proteomics 2019;18:1010–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Larsen SC, Hendriks IA, Lyon D, Jensen LJ, Nielsen ML. Systems-wide analysis of serine ADP-ribosylation reveals widespread occurrence and site-specific overlap with phosphorylation. Cell Rep 2018;24:2493–505 e4 [DOI] [PubMed] [Google Scholar]
- 13.Beck C, Robert I, Reina-San-Martin B, Schreiber V, Dantzer F. Poly(ADP-ribose) polymerases in double-strand break repair: focus on PARP1, PARP2 and PARP3. Exp Cell Res 2014;329:18–25 [DOI] [PubMed] [Google Scholar]
- 14.Kraus WL, Hottiger MO. PARP-1 and gene regulation: progress and puzzles. Molecular aspects of medicine 2013;34:1109–23 [DOI] [PubMed] [Google Scholar]
- 15.Krishnakumar R, Kraus WL. The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol Cell 2010;39:8–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Messner S, Hottiger MO. Histone ADP-ribosylation in DNA repair, replication and transcription. Trends Cell Biol 2011;21:534–42 [DOI] [PubMed] [Google Scholar]
- 17.de Murcia G, Huletsky A, Lamarre D, Gaudreau A, Pouyet J, Daune M, et al. Modulation of chromatin superstructure induced by poly(ADP-ribose) synthesis and degradation. J Biol Chem 1986;261:7011–7 [PubMed] [Google Scholar]
- 18.Poirier GG, de Murcia G, Jongstra-Bilen J, Niedergang C, Mandel P. Poly(ADP-ribosyl)ation of polynucleosomes causes relaxation of chromatin structure. Proc Natl Acad Sci U S A 1982;79:3423–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim MY, Hsiao SJ, Kraus WL. A role for coactivators and histone acetylation in estrogen receptor alpha-mediated transcription initiation. EMBO J 2001;20:6084–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tulin A, Spradling A. Chromatin loosening by poly(ADP)-ribose polymerase (PARP) at Drosophila puff loci. Science 2003;299:560–2 [DOI] [PubMed] [Google Scholar]
- 21.Karch KR, Langelier MF, Pascal JM, Garcia BA. The nucleosomal surface is the main target of histone ADP-ribosylation in response to DNA damage. Mol Biosyst 2017;13:2660–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang D, Camacho CV, Setlem R, Ryu KW, Parameswaran B, Gupta RK, et al. Functional interplay between histone H2B ADP-ribosylation and phosphorylation controls adipogenesis. Mol Cell 2020;79:934–49 e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hottiger MO. ADP-ribosylation of histones by ARTD1: an additional module of the histone code? FEBS Lett 2011;585:1595–9 [DOI] [PubMed] [Google Scholar]
- 24.Leidecker O, Bonfiglio JJ, Colby T, Zhang Q, Atanassov I, Zaja R, et al. Serine is a new target residue for endogenous ADP-ribosylation on histones. Nat Chem Biol 2016;12:998–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Palazzo L, Leidecker O, Prokhorova E, Dauben H, Matic I, Ahel I. Serine is the major residue for ADP-ribosylation upon DNA damage. Elife 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rakhimova A, Ura S, Hsu DW, Wang HY, Pears CJ, Lakin ND. Site-specific ADP-ribosylation of histone H2B in response to DNA double strand breaks. Sci Rep 2017;7:43750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jayaram H, Hoelper D, Jain SU, Cantone N, Lundgren SM, Poy F, et al. S-adenosyl methionine is necessary for inhibition of the methyltransferase G9a by the lysine 9 to methionine mutation on histone H3. Proc Natl Acad Sci U S A 2016;113:6182–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhong S, Joung JG, Zheng Y, Chen YR, Liu B, Shao Y, et al. High-throughput illumina strand-specific RNA sequencing library preparation. Cold Spring Harb Protoc 2011;2011:940–9 [DOI] [PubMed] [Google Scholar]
- 29.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 2013;14:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang L, Wang S, Li W. RSeQC: quality control of RNA-seq experiments. Bioinformatics 2012;28:2184–5 [DOI] [PubMed] [Google Scholar]
- 31.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010;28:511–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009;4:44–57 [DOI] [PubMed] [Google Scholar]
- 33.Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 2017;14:959–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chae M, Danko CG, Kraus WL. groHMM: a computational tool for identifying unannotated and cell type-specific transcription units from global run-on sequencing data. BMC Bioinformatics 2015;16:222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 2008;9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feng J, Liu T, Qin B, Zhang Y, Liu XS. Identifying ChIP-seq enrichment using MACS. Nat Protoc 2012;7:1728–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bentsen M, Goymann P, Schultheis H, Klee K, Petrova A, Wiegandt R, et al. ATAC-seq footprinting unravels kinetics of transcription factor binding during zygotic genome activation. Nat Commun 2020;11:4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martire S, Gogate AA, Whitmill A, Tafessu A, Nguyen J, Teng YC, et al. Phosphorylation of histone H3.3 at serine 31 promotes p300 activity and enhancer acetylation. Nat Genet 2019;51:941–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012;9:357–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010;26:841–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 2010;38:576–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krishnakumar R, Gamble MJ, Frizzell KM, Berrocal JG, Kininis M, Kraus WL. Reciprocal binding of PARP-1 and histone H1 at promoters specifies transcriptional outcomes. Science 2008;319:819–21 [DOI] [PubMed] [Google Scholar]
- 45.Krishnakumar R, Kraus WL. PARP-1 regulates chromatin structure and transcription through a KDM5B-dependent pathway. Mol Cell 2010;39:736–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martire S, Nguyen J, Sundaresan A, Banaszynski LA. Differential contribution of p300 and CBP to regulatory element acetylation in mESCs. BMC Mol Cell Biol 2020;21:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gupte R, Nandu T, Kraus WL. Nuclear ADP-ribosylation drives IFNgamma-dependent STAT1alpha enhancer formation in macrophages. Nat Commun 2021;12:3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin KY, Huang D, Kraus WL. Generating protein-linked and protein-free mono-, oligo-, and poly(ADP-ribose) in vitro. Methods Mol Biol 2018;1813:91–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang T, Berrocal JG, Yao J, DuMond ME, Krishnakumar R, Ruhl DD, et al. Regulation of poly(ADP-ribose) polymerase-1-dependent gene expression through promoter-directed recruitment of a nuclear NAD+ synthase. J Biol Chem 2012;287:12405–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rosenthal F, Nanni P, Barkow-Oesterreicher S, Hottiger MO. Optimization of LTQ-orbitrap mass spectrometer parameters for the identification of ADP-ribosylation sites. J Proteome Res 2015;14:4072–9 [DOI] [PubMed] [Google Scholar]
- 51.Zhang Y, Wang J, Ding M, Yu Y. Site-specific characterization of the Asp- and Glu-ADP-ribosylated proteome. Nat Methods 2013;10:981–4 [DOI] [PubMed] [Google Scholar]
- 52.Bagert JD, Mitchener MM, Patriotis AL, Dul BE, Wojcik F, Nacev BA, et al. Oncohistone mutations enhance chromatin remodeling and alter cell fates. Nat Chem Biol 2021;17:403–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee JS, Smith E, Shilatifard A. The language of histone crosstalk. Cell 2010;142:682–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet 2008;40:897–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weinert BT, Narita T, Satpathy S, Srinivasan B, Hansen BK, Scholz C, et al. Time-Resolved Analysis Reveals Rapid Dynamics and Broad Scope of the CBP/p300 Acetylome. Cell 2018;174:231–44 e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fritz AJ, Hong D, Boyd J, Kost J, Finstaad KH, Fitzgerald MP, et al. RUNX1 and RUNX2 transcription factors function in opposing roles to regulate breast cancer stem cells. J Cell Physiol 2020;235:7261–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shore P A role for Runx2 in normal mammary gland and breast cancer bone metastasis. J Cell Biochem 2005;96:484–9 [DOI] [PubMed] [Google Scholar]
- 58.Pham D, Moseley CE, Gao M, Savic D, Winstead CJ, Sun M, et al. Batf pioneers the reorganization of chromatin in developing effector T cells via Ets1-dependent recruitment of Ctcf. Cell Rep 2019;29:1203–20 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oh S, Oh C, Yoo KH. Functional roles of CTCF in breast cancer. BMB Rep 2017;50:445–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wong KM, Song J, Wong YH. CTCF and EGR1 suppress breast cancer cell migration through transcriptional control of Nm23-H1. Sci Rep 2021;11:491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu J, Li PC, Pang JY, Liu GY, Xie XM, Li JY, et al. CCCTC-binding factor inhibits breast cancer cell proliferation and metastasis via inactivation of the nuclear factor-kappaB pathway. Oncotarget 2017;8:93516–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim DS, Camacho CV, Kraus WL. Alternate therapeutic pathways for PARP inhibitors and potential mechanisms of resistance. Exp Mol Med 2021;53:42–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bennett RL, Bele A, Small EC, Will CM, Nabet B, Oyer JA, et al. A mutation in histone H2B represents a new class of oncogenic driver. Cancer Discov 2019;9:1438–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bartlett E, Bonfiglio JJ, Prokhorova E, Colby T, Zobel F, Ahel I, et al. Interplay of histone marks with serine ADP-ribosylation. Cell Rep 2018;24:3488–502 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013;340:857–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mohammad F, Weissmann S, Leblanc B, Pandey DP, Hojfeldt JW, Comet I, et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat Med 2017;23:483–92 [DOI] [PubMed] [Google Scholar]
- 67.Brien GL, Bressan RB, Monger C, Gannon D, Lagan E, Doherty AM, et al. Simultaneous disruption of PRC2 and enhancer function underlies histone H3.3-K27M oncogenic activity in human hindbrain neural stem cells. Nat Genet 2021 [DOI] [PubMed] [Google Scholar]
- 68.Lu C, Jain SU, Hoelper D, Bechet D, Molden RC, Ran L, et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 2016;352:844–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang S, Zheng X, Lu C, Li GM, Allis CD, Li H. Molecular basis for oncohistone H3 recognition by SETD2 methyltransferase. Genes Dev 2016;30:1611–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kang TZE, Zhu L, Yang D, Ding D, Zhu X, Wan YCE, et al. The elevated transcription of ADAM19 by the oncohistone H2BE76K contributes to oncogenic properties in breast cancer. J Biol Chem 2021;296:100374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hananya N, Daley SK, Bagert JD, Muir TW. Synthesis of ADP-ribosylated histones reveals site-specific impacts on chromatin structure and function. J Am Chem Soc 2021;143:10847–52 [DOI] [PubMed] [Google Scholar]
- 72.Ryu KW, Nandu T, Kim J, Challa S, DeBerardinis RJ, Kraus WL. Metabolic regulation of transcription through compartmentalized NAD(+) biosynthesis. Science 2018;360. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The high throughput sequencing data from this study are available from the NCBI’s Gene Expression Omnibus repository (GEO; http://www.ncbi.nlm.nih.gov/geo/) under the super series accession number GSE180463. The ATAC-seq, ChIP-seq, and RNA-seq datasets can be found under the subseries GSE180458, GSE180459, and GSE180461 respectively.