

Abstract
Exatecan and deruxtecan are antineoplastic camptothecin derivatives in development as tumor-targeted-delivery warheads in various formulations including peptides, liposomes, polyethylene glycol (PEG) nanoparticles, and antibody-drug conjugates (ADCs). Here, we report the molecular pharmacology of exatecan compared to the clinically approved topoisomerase I (TOP1) inhibitors and preclinical models for validating biomarkers and the combination of exatecan with ATR inhibitors. Modeling exatecan binding at the interface of a TOP1 cleavage complex suggests two novel molecular interactions with the flanking DNA base and the TOP1 residue N352, in addition to the three known interactions of camptothecins with the TOP1 residues R364, D533 and N722. Accordingly, exatecan showed much stronger TOP1 trapping, higher DNA damage and apoptotic cell death than the classical TOP1 inhibitors used clinically. We demonstrate the value of SLFN11 expression and homologous recombination (HR)-deficiency (HRD) as predictive biomarkers of response to exatecan. We also show that exatecan kills cancer cells synergistically with the clinical ATR inhibitor ceralasertib (AZD6738). To establish the translational potential of this combination, we tested CBX-12, a clinically developed pH-sensitive peptide-exatecan conjugate that selectively targets cancer cells and is currently in clinical trials. The combination of CBX-12 with ceralasertib significantly suppressed tumor growth in mouse xenografts. Collectively, our results demonstrate the potency of exatecan as a TOP1 inhibitor and its clinical potential in combination with ATR inhibitors, using SLFN11 and HRD as predictive biomarkers.
Keywords: Exatecan, TOP1, SLFN11, Homologous Recombination, ceralasertib, LMP400, irinotecan, topotecan, SN-38, ATR inhibitors
Introduction
Camptothecin (CPT) and its derivatives trap topoisomerase 1 (TOP1)-DNA cleavage complexes (TOP1ccs) by binding at the interface of the cleaved DNA and TOP1, resulting in TOP1ccs that cause DNA damage leading to cell death and inhibition of TOP1-mediated DNA relaxation (1). Clinical CPT derivatives such as topotecan and irinotecan are ubiquitously used to treat a broad range of cancers (2,3). Exatecan (DX-8951f) was developed as a water-soluble CPT derivative with stronger inhibition of TOP1 activity and tumor suppression capability than the clinically approved CPT derivatives (4-6). However, its development as a single agent was ceased due to dose-limiting side effects, and the absence of therapeutic benefits in combination with gemcitabine compared with gemcitabine alone in clinical testing (7).
Exatecan is being re-evaluated in the context of tumor-targeted drug delivery approaches as TOP1-based antibody-drug conjugates (ADC) appear less toxic than pyrrolobenzodiazepine (PBD) DNA-crosslinking- and microtubule inhibitor derivative-based ADCs (8-14). Deruxtecan, a close derivative of exatecan is already successfully used as a cytotoxic payload (15) conjugated with a HER2 targeting antibody in Trastuzumab Deruxtecan (Enhertu®, T-DXd/DS-8201a), which has recently been clinically approved for the treatment of HER2-expressing solid tumors (16). A second TOP1 ADC, sacituzumab govitecan (Trodelvy®, IMMU-132) based on SN-38, the active metabolite of irinotecan (2) and targeting TROP2 (17) has also been recently approved for triple-negative breast cancers (18). Exatecan is being investigated as the cytotoxic payload on a novel HER2-targeting ADC (9). Mechanistically, targeting tumor antigens enables the release of the cytotoxic CPT derivative specifically to cancerous cells, stabilizing the drug in the bloodstream until its delivery to the tumor (8,19). Because clinical applications of ADC delivery are limited by subsets of cancers expressing high levels of antigen and off-target toxicity, the exatecan-based drug conjugate CBX-12 is being developed to improve drug delivery to a broad range of tumors while minimizing toxicity to normal tissues (14). CBX-12 is a pH-sensitive peptide conjugate that releases exatecan into tumor cells due to the acidic pH of the tumor microenvironment (14).
Clinical CPT derivatives are modified in the A and B rings of the CPT structure to increase their potency as TOP1cc poisons and their water solubility (Fig. 1A) (20). Exatecan bears an additional amino benzyl ring appended to the A and B rings over positions 7 and 9 and has additional substitutions with a methyl and a fluorine group at positions 10 and 11, respectively. However, the mechanistic and therapeutic benefits of these modifications have not been fully documented in comparison with SN-38, the active metabolite of irinotecan, which is the most active clinical derivative of CPT (20,21).
Figure 1. Structural insights into the potent trapping of TOP1ccs by exatecan.
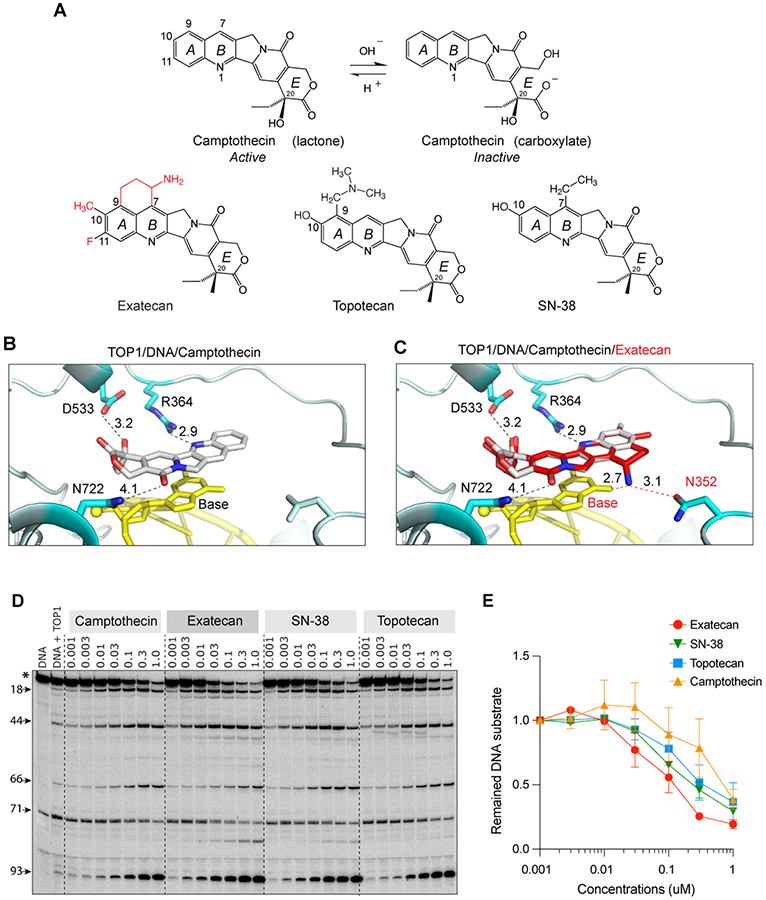
A. Chemical structures of CPT and its clinical derivatives (exatecan, topotecan, and SN-38). B. Representative view of CPT (light grey) bound to human TOP1 (cyan) and DNA (yellow) (PDB: 1T8I). In addition to base stacking, CPT makes 3 hydrogen bonds with TOP1 through D533, N722 and R364. The numbers indicate the distance of the bonds in Angstrom. C. Superposition of exatecan (red) and CPT (light grey) into the TOP1 (cyan)-DNA (yellow) structure. The 2 dotted lines (red) represent the potential additional hydrogen bonds of exatecan with the DNA base and N352 of TOP1. D. Comparative TOP1-mediated DNA cleavage (TOP1ccs) induced by exatecan and other TOP1 inhibitors. Recombinant TOP1 was incubated with 3'-end labeled 117 bp DNA oligo in the presence of the indicated drug concentrations. Fragmented DNA oligos were visualized on PAGE gel by using PhosphorImager (Molecular Dynamics). E. Quantitation of DNA substrates (*) as shown in panel D in duplicate experiments. The band intensity was determined by Image Quant software (Molecular Dynamics).
Homologous recombination deficiency (HRD), as observed in BRCA1 or BRCA2 mutant settings, has recently been shown to serve as predictive biomarkers for TOP1 inhibitors (22,23) in addition to their synthetic lethality with poly(ADP-ribose) polymerase (PARP) inhibitors (24). Another emerging biomarker of response to both TOP1 inhibitors and PARP inhibitors is the putative DNA/RNA helicase/nuclease, Schlafen 11 (SLFN11) (25,26). Cancer cells expressing SLFN11 are selectively more vulnerable to treatment with clinical TOP1 inhibitors including CPT and non-CPT derivatives (22,25). Yet, in the large proportion (approximately 50%) of cancer cells that do not express SLFN11, combination treatment with inhibitors of the replication checkpoint Ataxia Telangiectasia and Rad3-related kinase (ATR) has recently been shown to overcome resistance to TOP1 inhibitors (27,28).
In the present study, we elucidate the molecular mechanisms underlying the anticancer potency of exatecan. We demonstrate the selective susceptibility in HRD and SLFN11 expressing cancer cells to exatecan, and validate the synergy of the exatecan conjugate CBX-12 with the ATR inhibitor ceralasertib (AZD6738) in cell culture and human xenografts (29).
Materials and methods
Cell lines and reagents
DU145 (ATCC, HTB-81™) and DU145-SLFN11 KO cells were grown in DMEM medium with 10% FBS/1% penicillin-streptomycin. MOLT-4 (ATCC, CRL-1582™), MOLT-4-SLFN11 KO, CCRF-CEM (ATCC, CCL-119™), CCRF-CEM-SLFN11 KO, DMS114 (ATCC, CRL-2066™), DMS114-SLFN11 KO, and HCT-116 (ATCC, CCL-247™) cells were grown in RPMI1640 medium with 10% FBS/1% penicillin-streptomycin. DU145, MOLT-4, CCRF-CEM, DMS114, and HCT-116 were purchased from the ATCC. All SLFN11 KO cells were established in our laboratory (30). DT40, DT40-BRCA1 KO, and DT40-BRCA2 KO cells (gifts from Dr. S. Takeda, Kyoto University, Kyoto, Japan) were grown in RPMI1640 medium supplemented with 1% chicken serum, 10 nmol/L β-mercaptoethanol, 10% FBS, and 1% penicillin-streptomycin. UWB1.289 and UWB1.289+BRCA1 cells (gifts from Dr. J. Lee, NCI) were cultured in complete growth medium (50% RPMI-1640 medium, 50% MEGM medium with 10% FBS/1% penicillin-streptomycin. CPT, exatecan, topotecan, SN-38, LMP400, talazoparib and ceralasertib (AZD6738) were acquired from the Developmental Therapeutics Program (DCTD, NCI). CBX-12 is obtained from Cybrexa Therapeutics. All cell lines were passaged 15 times and examined by MycoAlert Mycoplasma Detection Kit (Lonza).
Modeling of exatecan in the TOP1cc
The structural coordinates of exatecan (PubChem: 151115), camptothecin (PubChem: 24360), SN-38 (PubChem: 443154), and topotecan (PubChem: 60700) were downloaded from the NCBI-PubChem compound (http://www.ncbi.nlm.nih.gov/pccompound) database in three-dimensional (3D) SDF-file format and then were converted to PDB format using PyMOL (ver.2.3.5., www.pymol.org). The spatial localization of TOP1-DNA-inhibitor (camptothecin/PDB: 1T8I (31) and topotecan/PDB: 1K4T (32)) within the 3D structures with the distances between molecules was graphically presented using PyMOL. Structural superposition modeling was carried out by overlapping exatecan to the bound camptothecin in the TOP1-DNA structure using CooT (33) and PyMOL. Docking simulations and estimated binding affinity of TOP1 inhibitors were performed using AutoDock Vina v.1.1.2.
TOP1-mediated DNA cleavage assay
A 3′-[32P]-labeled 117-bp DNA substrate oligonucleotide (34) was incubated with recombinant human TOP1 (purified from insect cells using a baculovirus construct for the full-length human TOP cDNA) (35) in 20 μl reaction buffer (10 mmol/L Tris-HCl, pH 7.5, 50 mmol/L KCl, 5 mmol/L MgCl2, 0.1 mmol/L EDTA, and 15 μg/ml BSA) at 30°C for 20 min in the presence of the indicated drug concentrations. Reactions were terminated by adding SDS (0.5% final concentration) followed by the addition of two volumes of loading dye (80% formamide, 10 mmol/L sodium hydroxide, 1 mmol/L sodium EDTA, 0.1% xylene cyanol, and 0.1% bromophenol blue). Aliquots of reaction mixtures were subjected to 16% denaturing PAGE. Gels were dried and visualized by using PhosphorImager and Image Quant software (Molecular Dynamics).
Detection of cellular TOP1ccs
DNA-trapped TOP1 was determined by a modified RADAR (rapid approach to DNA adduct recovery) assay (36). After treatment of TOP1 inhibitors, DU145 cells (1x106 cells/sample) were washed with 1x PBS and lysed with 600 μl of DNAzol (Invitrogen), followed by precipitation with 300 μl of 200 proof ethanol by centrifugation at 14,000 rpm. The nucleic acids were collected, washed with 75% ethanol, resuspended in 200 μl of TE buffer, and then heated at 65°C for 15 min, followed by shearing with sonication (40% output for 10 sec pulse and 10 sec rest for four times). The samples were centrifuged at 15,000 rpm for 5 min at 4°C, and the supernatant was collected. The sample (1 μl) was saved for spectrophotometric measurement of absorbance at 260 nm to quantitate DNA content (NanoDrop). Two μg of each sample was subjected to slot-blot for immunoblotting with anti-TOP1 antibody (#556597, BD Biosciences) or anti-dsDNA antibody (#3519, Abcam) as a loading control. The intensity of TOP1 and DNA was quantified by densitometric analysis using ImageJ.
Immunofluorescence microscopy
DU145 cells were plated in 6-well plates on sterilized coverslips and treated the next day with either DMSO as a vehicle or different concentrations (indicated in figure legends) of the TOP1 inhibitors exatecan and topotecan for 2 h. After washing with 1xPBS, cells were fixed with 4% paraformaldehyde in 1xPBS for 15 min at RT. Subsequent permeabilization was performed in 2.5% Triton-X/1xPBS for 15 min before 1xPBS washing and 1 h blocking in 5% BSA/1xPBS. Coverslips were then incubated for 1 h with mouse anti-phospho-Histone H2AX (Ser139) antibody (#05-636, Millipore), diluted 1:500 in 5% BSA/1xPBS. After three washes with 1xPBS, samples were incubated with secondary antibody (Alexa Fluor 488, goat anti-mouse, diluted 1:2000 in 5% BSA/1xPBS for 1 h in the dark. Coverslips were washed (3 × 5 min in 1xPBS), stained with DAPI, and mounted using VECTASHIELD (Vector Laboratories). Images were captured with a Zeiss LSM 880 super-resolution microscope with a 63x objective lens. The signal intensity was quantified by Image J software.
Alkaline comet assay
DNA single- and double-stranded breaks were determined by the Alkaline CometAssay kit (Trevigen) according to the manufacturer’s protocol. Briefly, after treatment with the TOP1 inhibitors, cells were harvested, washed in 1xPBS, and combined at 3x105cells/ml with molten LMAagrose in a 1:10 (v/v) ratio. Fifty μl of the combined mixture was added onto the comet slide. After the gel was solidified at 4°C, slides were immersed in 4°C lysis solution for 30 min and subsequently incubated in alkaline unwinding solution for 20 min at RT in the dark. Alkaline electrophoresis was carried out at 1 V/cm and 300 mA for 40 min at 4°C. Slides were rinsed twice in deionized H2O for 5 min each, then in 70% ethanol for 5 min and air-dried overnight. DNA was stained with 100 μl SYBR Gold for 30 min, briefly rinsed in water, and allowed to air-dry. Fluorescent signals were visualized using fluorescence microscopy and quantified by using ImageJ plugin OpenComet (version 1.3)
Apoptotic cell death
Cells were seeded on 6-well plates (3x105cells/well) and treated with drugs for 24 h and 48 h. After harvest, cells were examined with the ApoDETECT Annexin V-FITC kit according to the manufacturer’s protocol (Invitrogen) and analyzed by flow cytometry using a FACS Canto (Becton Dickinson) and FlowJo software.
Western blotting
Cells were lysed with NETN300 buffer (1% NP40, 300 mM NaCl, 0.1 mM EDTA, and 50 mM Tris [pH 7.5]) supplemented with protease inhibitor cocktail (Cell Signaling). For TOP1 degradation, cells were incubated in alkaline lysis buffer (200 mM NaOH, 2 mM EDTA), and samples neutralized with neutralization buffer (1M HCl, 600 mM Tris, pH8.0). Subsequently, cell lysates were incubated in nuclease digestion buffer (5 mM CaCl2, 50 mM Tris-HCl, pH8.0). After adding SDS-PAGE sample buffer or 2X boiling lysis buffer (50 mM Tris-HCl pH6.8, 2% SDS, 850 mM β-mercaptoethanol), samples were resolved by SDS-PAGE gels (Novex Tris-Glycine Mini Gels, Invitrogen) and transferred onto PVDF membranes (Millipore). Membranes were immunoblotted with the following antibodies: TOP1 (#556597, BD Biosciences), GAPDH (GTX100118; GeneTex), PARP (9542; Cell signaling), and cleaved caspase-3 (9661; Cell signaling). After overnight incubation, membranes were incubated with the species-appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h. Protein signals were visualized by ChemiDoc MP Imaging System (Bio-Rad) with SuperSignal™ West Pico PLUS Chemiluminescent Substrate (Thermo Scientific). Signal intensity was quantified with the Image J software.
Cell viability
Cells were plated in 96-well white plates at a density of 2,000 cells/well in 100 μl complete growth medium. Cells were incubated with TOP1 inhibitors for 72 h. Cell viability was determined by CellTiter-Glo Luminescent Cell Viability Assay (Promega) according to the manufacturer’s protocol. Briefly, the reaction solution was added at 50 μl/well and the plates were kept in the dark for 10 min with mild shaking, and then luminescence was measured by Envision 2104 Multi-label Microplate Reader (Perkin Elmer).
Xenograft studies
For MDA-MB-231 xenografts, 3- to 4-week-old female athymic nude Foxnnu mice (HSD: Athymic nude-Foxn1nu, Envigo Labs) were inoculated subcutaneously with MDA-MB-231 tumor cells (1 x 106) combined 1:1 with Matrigel (Corning, 47743-716) in a total volume of 0.1 ml. Once the tumors reached a mean volume of 50-100 mm3, mice were randomized into treatment groups (n = ±10 mice/arm) and treated as indicated. CBX-12 (Cybrexa Therapeutics) doses were prepared by diluting DMSO stocks in 5% (w/v) mannitol in citrate vehicle, as described (14). CBX-12 doses were administered intraperitoneally at 10 mg/kg once daily for 4 days, repeated weekly for 3 weeks. Ceralasertib (AZD6738) doses were prepared by diluting DMSO stocks in 10% (w/v) 2-hydroxy-propyl-b-cyclodextrin (Sigma #H107) vehicle. Ceralasertib doses were then administered via oral gavage at 25 mg/kg once daily for 5 days, repeated weekly for 3 weeks. For HCT-116 xenografts, 6-week-old female athymic nude Foxnnu mice were obtained from Taconic Labs (Cat# NCRNU-F). Each mouse was inoculated subcutaneously with HCT-116 tumor cells (2.5 x 106) with Matrigel (1:1). After tumors had grown to a mean size of approximately 100-200 mm3, the mice were then split into groups (n = ±10 mice/arm) and treated as indicated. CBX-12 doses were administered intraperitoneally at 5 mg/kg once daily for 4 days, repeated weekly for 3 weeks. Ceralasertib doses were administered via oral gavage at 25 mg/kg once daily for 21 days. Mice were followed for the three weeks of treatment and throughout the subsequent three-week washout period. Tumor volumes were measured twice weekly with calipers and calculated according to the formula for ellipsoid volume: π/6 x (tumor length) x (tumor width)2. Mice with tumors exceeding 2 cm3 in volume or exhibiting significant weight loss or tumor ulceration were euthanized, in accordance with institutional protocols. All animal studies were approved by the Institutional Animal Use and Care Committee and performed by the Guide for the Care and Use of Laboratory Animals.
Statistical analyses
Statistical analysis was calculated using two-tailed unpaired Student's t-test, one-way ANOVA with using the GraphPad Prism 8 software (GraphPad Software, La Jolla, CA, USA).
Data availability
The data generated in this study are available within the article and its supplementary data files.
Results
TOP1cc trapping by exatecan
As interfacial inhibitors (1), CPT and its derivatives trap TOP1ccs by π∓π stacking with the base pairs flanking the DNA cleavage site and by 3 hydrogen bonds with TOP1 residues (R364, D533 and N722) (Fig. 1B and Supplementary Fig. S1A) (37). Because exatecan possesses an amino benzyl ring between the A and B rings (Fig. 1A), we hypothesized that these modifications may form additional interactions with the TOP1 and DNA complex. To test this hypothesis, we modeled and superimposed exatecan onto the structure of the TOP1-DNA-CPT complex (PDB: 1T8I) (Fig. 1C). We found that the amino group of the benzyl ring was positioned to make two additional hydrogen bonds with the +1 DNA base oxygen and the N352 residue of TOP1, implying enhanced binding affinity of exatecan compared to CPT (Supplementary Fig. S1B). These results show that exatecan can be readily modeled in the TOP1cc, and that it may form additional interaction that would stabilize its binding at the interface of TOP1-DNA complex to a greater extent than CPT, topotecan and SN-38.
To test this possibility further, we measured the trapping of TOP1ccs by exatecan by performing DNA cleavage assays with recombinant TOP1 and 32P-labeled DNA oligonucleotides. As shown in Fig. 1D, 1E, and Supplementary S1C, exatecan induced DNA cleavage more effectively than the other clinical TOP1 inhibitors tested: CPT, SN-38, and topotecan. Together, these results establish that exatecan is a potent TOP1 poison, acting at low nanomolar concentrations.
We also confirmed that CBX-12, which is a pH-sensitive peptide-exatecan conjugate (14), acts as a prodrug. As shown in Supplementary Fig. S1D, CBX-12 was at least 100 times less active than exatecan in the induction of DNA cleavage by recombinant human TOP1.
Exatecan induces cellular TOP1ccs and induces TOP1 degradation at nanomolar concentrations
To confirm the potency of exatecan as a TOP1 poison, we examined the level of DNA-trapped TOP1 in comparison with clinical TOP1 inhibitors topotecan, SN-38, and CPT using a modified RADAR assay. Exatecan was the most potent drug and induced TOP1ccs at a lower concentration (0.03 μM) than the other TOP1 inhibitors (Fig. 2A, 2B, and Supplementary S2A and S2B).
Figure 2. Exatecan leads to greater TOP1-DNA trapping than topotecan, SN-38 and CPT.

A. Detection of DNA-trapped TOP1 by exatecan and other TOP1 inhibitors. DU145 cells were treated with the indicated drug concentrations for 30 min. TOP1ccs were isolated by RADAR assay. B. Quantitation of TOP1ccs from panel A in a single experiment. The intensity of TOP1 was analyzed by ImageJ software and normalized to DNA loading. Data are plotted with GraphPad Prism 8. C. TOP1 degradation induced by exatecan. DU145 cells were incubated with the indicated TOP1 inhibitors for 2 h. Following TOP1 reversal for 30 min without inhibitors, TOP1 levels were determined by Western blotting. D. Quantification of total cellular TOP1 bands in duplicate experiments. Band intensity was analyzed using the ImageJ software and normalized to GAPDH used as a loading control.
Given that DNA-trapped TOP1 is rapidly removed from DNA and degraded by the ubiquitin-proteasome pathway (38), we hypothesized that exatecan may produce faster cellular TOP1 degradation than the other TOP1 inhibitors. To demonstrate this, we first treated cells with the TOP1 inhibitors for 2 h and then allowed the cells to grow without drugs for 30 min to allow the reversal of TOP1ccs and TOP1 degradation. As expected, exatecan induced TOP1 degradation in a dose-dependent manner and appeared markedly more effective than the other clinical TOP1 inhibitors, SN-38 and topotecan (Fig. 2C and 2D).
Collectively, these results demonstrate that exatecan is a potent TOP1 inhibitor, inducing cytotoxic TOP1ccs leading to the degradation of TOP1 at nanomolar concentrations.
Exatecan causes cellular DNA breakage and apoptotic cell death
Given that TOP1 inhibitor-mediated TOP1ccs lead to DNA damage (20), we determined the induction of phosphorylated H2AX (γH2AX), a sensitive biomarker for DNA double-strand breaks (DSBs), in cells treated with exatecan in comparison with topotecan. As shown in Fig. 3A-B and Supplementary Fig. S3A, γH2AX was induced by exatecan at 10 nM drug concentration and increased in a dose-dependent manner. The induction of γH2AX by topotecan was significantly less than that of exatecan.
Figure 3. DNA damage and cell death induced by exatecan.
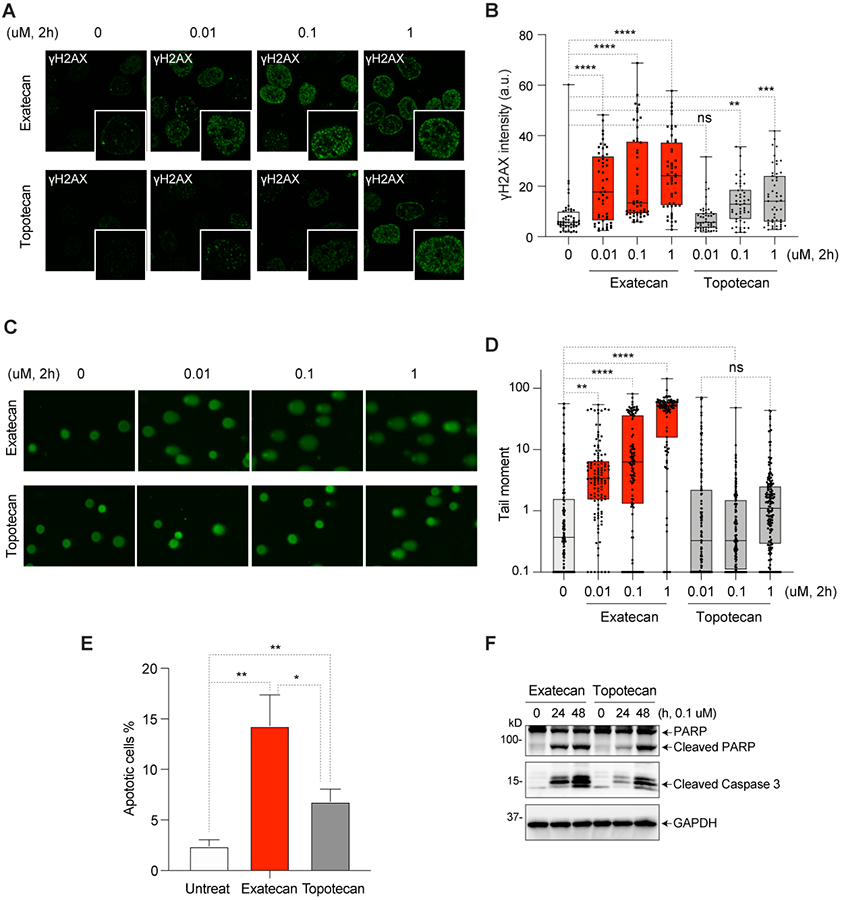
A. Representative immunofluorescence images of γH2AX (green) in exatecan- or topotecan-treated DU145 cells. B. Intensity of γH2AX fluorescence (average per cell) for the experiment depicted in panels A (mean ± SEM, N = 50/each) ** p-value <0.002, ***p-value <0.0004, ****p-value <0.0001. a.u., arbitrary units. C. Representative images of comet analysis in DU145 cells treated with exatecan and topotecan. D. Quantitation of tail moments of experiments depicted in panel B (mean ± SEM, N = 100/each) are quantified with the Open Comet/ImageJ program. ** p-value <0.006, ****p-value <0.0001. E. Apoptotic cell death induced by exatecan and topotecan and measured by Annexin V/PI staining. * p-value <0.01, **p-value <0.005. F. Cleavage of PARP1 and caspase-3 in exatecan and topotecan treated cells measured by Western blotting.
Next, DNA break induction by exatecan was examined using the comet assay. Exatecan produced DNA breaks in a dose-dependent manner and was significantly more effective than topotecan (Supplementary Fig. 3C and 3D).
To determine whether the TOP1-induced DNA breaks generated by exatecan result in cell death, we measured apoptosis using Annexin V-FITC staining. As shown in Fig. 3E and Supplementary Fig. S3B, exatecan-treated cells exhibited higher apoptotic responses than the topotecan-treated cells. These findings were confirmed by detecting cleaved PARP and caspase 3 (Fig. 3F).
Exatecan is the most potent cytotoxic inhibitor among clinical TOP1 inhibitors
Because of the higher levels of DNA damage and apoptosis induced by exatecan compared to topotecan, we tested the cytotoxicity of exatecan in comparison with the other clinical TOP1 inhibitors, topotecan, SN-38, and LMP400 (indotecan) in 4 different human cancer cell lines: acute leukemia MOLT-4 and CCRF-CEM, prostate cancer DU145, and small cell lung cancer DMS114. As shown in Fig. 4, exatecan stood out as being significantly more active than the 3 other TOP1 inhibitors. IC50 values of exatecan were in the picomolar range against the four tested cancer cell lines and showed over 10 to 50 times higher potency of exatecan compared to the next best TOP1 inhibitor, SN-38 (Fig. 4E).
Figure 4. Exatecan is the most potent TOP1 inhibitor.

A~D. Cytotoxicity of clinical TOP1 inhibitors (Exatecan, SN-38, Topotecan, and LMP400) in MOLT-4, CCRF-CEM, DU145 and DMS114 cells. Cells were treated as indicated for 72 h and cell viability was measured by CellTiter-Glo assay. Error bars represent standard deviations in the triplicate. Statistical values were calculated using one-way ANOVA with Dunnett's multiple comparisons test. * p-value<0.03. E. IC50 values of the TOP1 inhibitors calculated by GraphPad Prism 8. The IC50 values represent the mean (nM) obtained from triplicate experiments in MOLT-4, CCRF-CEM, DMS114, and DU145 cells. CI, 95% confidence interval. Ratios indicate comparative IC50 values between exatecan and the other TOP1 inhibitors.
Susceptibility to exatecan is selectively increased in cancer cells expressing SLFN11 and with defective homology-directed recombination (HR)
Given that SLFN11 expression is a dominant biomarker of response to TOP1cc-targeting chemotherapeutic agents, which kills cancer cells under replicative stress (26), we compared the activity of exatecan in 4 pairs of SLFN11-KO isogenic cancer cell lines: prostate DU145, acute leukemia CCRF-CEM, acute leukemia MOLT-4, and small cell lung cancer DMS114. As expected, SLFN11 positive cancer cells were consistently more sensitive to exatecan than their isogenic SLFN11-negative cells counterpart (Fig. 5A-D), which confirms the potential value of SLFN11 expression as a predictive biomarker for exatecan-based therapies.
Figure 5. SLFN11-proficient and HR-deficient cells are preferentially vulnerable to exatecan.

A~D. Cytotoxicity of exatecan in the isogenic DU145, CCRF-CEM, MOLT-4, DMS114 and paired SLFN11 knock-out (KO) cells. Cells were treated as indicated for 72 h and cell viability was measured by CellTiter-Glo assay. E. Cytotoxicity of exatecan in the isogenic DT40 chicken B-cells and paired BRCA1/2 KO cells. Cells were treated as indicated for 72 h and cell viability was measured by CellTiter-Glo assay. F. Cytotoxicity of exatecan in UWB1.289 (carrying a BRCA1 mutation, BRCA1-null) and UWB1.289+BRCA1 cells. Cells were treated as indicated for 72 h, and cell viability was measured by e CellTiter-Glo assay. Error bars represent standard deviations in the triplicate.
In addition to SLFN11, HR deficiency increases susceptibility to TOP1 inhibitors because of defective DNA repair (22,23). To determine whether HR-deficiency enhances chemosensitivity to exatecan, we tested cell viability in BRCA1-KO and BRCA2-KO genetically altered DT40 cell lines, which are derived from chicken B-cell lymphoma. As expected, exatecan was more potent than topotecan and SN-38 in DT40 cells (Supplementary Fig. S4A-C), as observed in the human cancer cells (Fig. 4). We also confirmed that cell killing in BRCA1-KO and BRCA2-KO DT40 cells by exatecan was significantly higher than in DT40 parental (WT) cells (Fig. 5E). These results were further validated in the BRCA1-null human ovarian cancer cell line UWB1.289, in which complementation with wild-type BRCA1 partially reversed chemosensitivity to exatecan (Fig. 5F), indicating that HR status is a potential predictive biomarker for the clinical use of exatecan.
Exatecan synergizes with ATR inhibitor
To our knowledge, no studies of exatecan with other clinical chemotherapeutic drugs have been reported since the unsuccessful combination clinical trial with gemcitabine (7). Given that topotecan and CPT show consistent synergy with ATR inhibitors (27,28,39), we studied the cytotoxicity of combination treatments of exatecan with the ATR inhibitor ceralasertib (AZD6738), which is being developed in various clinical trials (40,41). To do so, we tested exatecan with minimally toxic doses of ceralasertib (0.5 and 1 μM) as a single treatment. As shown in Fig. 6A and 6B, low doses of combination with ceralasertib enhanced the cytotoxicity of exatecan in human MDA-MB-231 breast cancer and HCT-116 colon cancer cells, which are both SLFN11-negative (http://discover.nci.nih.gov/cellminercdb). Combination index (CI) computation showed strong synergistic effects of ceralasertib for a range of concentrations of exatecan, implying that combination with low doses of ATR inhibitors could be utilized for cancer treatment in the clinic (Fig. 6C and 6D).
Figure 6. Exatecan synergizes with the ATR inhibitor ceralasertib.

A-B. Cytotoxicity of combination treatments of exatecan with ATR inhibitor. Human breast cancer MDA-MB-231 and colon adenocarcinoma HCT116 cells were treated with the indicated concentrations of exatecan without or with ceralasertib (0.5 and 1 μM) for 72 h, and cell viability was measured by CellTiter-Glo assays. Error bars represent standard deviations in the triplicate. C-D. Combination index (CI) plots for the combinations exatecan and ceralasertib from data obtained from panels A and B. The CI values were calculated by using CompuSyn. Additive combination: 0.5<CI<1, and synergistic combinations: 0 <CI<0.5.
Antitumor activity of CBX-12, a pH-sensitive peptide-exatecan conjugate, as a single agent and with the ATR inhibitor ceralasertib in mouse xenograft models
CBX-12 is a pH-sensitive alphalex™-exatecan conjugate currently being tested in early-phase clinical trials (14). Alphalex™ is a tumor-targeting technology consisting of a unique variant of a family of pH-Low Insertion Peptides (pHLIP®) that enables the targeting of acidic cell surfaces (42). In CBX-12, it makes exatecan specifically target the surface of cancer cells while avoiding exposure to the active payload in the vascular system and reducing cytotoxicity to normal cells. To validate our previous results with CBX-12 in pre-clinical settings, we tested the antitumor activity of CBX-12 alone and in combination with ceralasertib in MDA-MB-231 and HCT116 xenograft (Fig. 7A-D). Combination treatment significantly inhibited tumor growth without significant toxicity in both mouse xenografts compared with CBX-12 and ceralasertib monotherapy without significant toxicity (Fig. 7A and 7B). Overall survival of mice treated with the combination was also better than that of CBX-12 alone and ceralasertib single-treatment (Fig. 7C and 7D). No significant body weight loss was noted in the single-agent or combination groups, highlighting a lack of combination toxicity due to tumor-selective delivery of exatecan by CBX-12 (Supplementary Fig. S5A and S5B). The dose of ceralasertib chosen is in the range of that used frequently in the preclinical settings. Preclinical efficacy of ceralasertib in combination with chemotherapy appears to translate in the clinical setting, as recent early phase 1 data demonstrates preliminary signs of efficacy (NCT02630199). Thus, these results suggest that combining CBX-12 and ceralasertib may be a potentially new treatment approach that warrants further investigation.
Figure 7. Antitumor activity of CBX-12 in human breast cancer and colon cancer xenografts and synergy with the ATR inhibitor ceralasertib.

A-B. Tumor suppression by CBX-12 without and with ceralasertib (AZD6738) in MDA-MB-231 and HCT-116 xenografts. MDA-MB-231 xenografts (A) were treated with CBX-12 intraperitoneally at 10 mg/kg once daily for 4 days, repeated weekly for 3 weeks. Ceralasertib was administered via oral gavage at 25 mg/kg once daily for 5 days, repeated weekly for 3 weeks. HCT-116 xenografts (B) were treated with CBX-12 intraperitoneally at 5 mg/kg once daily for 4 days, repeated weekly for 3 weeks. Ceralasertib doses were then administered via oral gavage at 25 mg/kg once daily for 21 days. Tumor volumes are shown as mean ± SEM (N = 10 mice for each group). Statistical values were calculated using one-way ANOVA with Dunnett's multiple comparisons test. * p-value<0.05, *** p-value<0.001, **** p-value<0.0001. C-D. Cell survival after drug treatments for the MDA-MB-231 (C) and HCT-116 (D) xenografts. E. Proposed model for potent TOP1cc trapping by exatecan. F. Therapeutic strategy and predictive biomarkers for targeted exatecan delivery. ATRi: ATR inhibitor, SLFN11: Schlafen 11 expression, HR: homologous recombination including BRCA1/2.
Discussion
In this study, we report the molecular pharmacology and outstanding potency of exatecan in comparison with the classical TOP1 inhibitors, CPT, topotecan and SN-38 (the active metabolite of irinotecan), and provide proof-of-concept pharmacodynamic (γH2AX) and clinical biomarkers (SLFN11 and HRD) to support the ongoing clinical investigations of exatecan as payload for targeted drug delivery systems (2,14,19,43). Consistent with our data, exatecan initially showed much stronger antitumor activity in multiple pre-clinical studies, promising better therapeutic benefits than the other clinical TOP1-targeted drugs (44-46). However, the high potency of exatecan as a free drug led to dose-limiting cytotoxicity and hindered its development as a new clinically applicable TOP1 poison (14). Here, we studied the underlying molecular pharmacology of exatecan. We also provide preclinical evidence of combination strategies with CBX-12, a peptide-exatecan conjugate, and ATR inhibitors as a strategy to overcome the limitations of free exatecan.
We demonstrate that exatecan leads to stronger TOP1 trapping than SN-38, topotecan or CPT in biochemical and cellular assays and that CBX-12 acts as a prodrug for exatecan. We propose that this enhanced trapping may result from additional molecular interactions with DNA and TOP1 (Fig. 7E). By docking simulation, we find that the amino group on the 6th amino benzyl ring of exatecan can form two additional molecular interactions with the oxygen of the +1 DNA base and the TOP1 residue (N352) in addition to three known interactions of CPT derivatives with the three TOP1 residues R364, D533, and N722 at the interface of the TOP1-DNA complex (31). Consistently, cells with a TOP1 mutation N352A have been reported to be resistant to a CPT derivative with a 10-OH substitution on the CPT A-ring. Additionally, by analyzing TCGA with cBioPortal, we found that a lung adenocarcinoma patient harbored a mutation at this same E352 residue (E352K) (Supplementary Fig. S5C), implying its potential clinical relevance for drug resistance. Furthermore, a mutant (E418K) of TOP1 at the TOP1cc interface and near the N352 residue has been detected in a patient with triple-negative breast cancer resistant to sacituzumab govitecan (47). These observations suggest the potential value of sequencing the TOP1 gene in patients treated with exatecan to analyze potential drug resistance.
Although TOP1 inhibitors are widely used in the clinic as the first-line chemotherapy, predictive biomarkers are not a current focus of attention. Although TOP1 overexpression and TDP1 deficiency have been proposed as potential predictive biomarkers (48), they are not systematically analyzed. Alternatively, SLFN11 has emerged as a dominant prediction biomarker for TOP1 inhibitors (25,26). The results presented here are consistent with this possibility, as we show in four different isogenic cancer cell line models with SLFN11-WT and -KO, that SLFN11-expressing cancer cells are selectively sensitive to exatecan, as observed with other clinical TOP1 inhibitors (25,49). Thus, evaluation of SLFN11 expression should be considered as a correlative factor for patient response to exatecan-based treatment, such as CBX-12. Ultimately, SLFN11 expression could be considered to select patients who may derive therapeutic benefits from exatecan-based cancer therapy. Here we also provide evidence that the relative resistance of SLFN11-negative cancer cells such as the breast cancer cell line MDA-MB-231 and the colon cancer cell line HCT-116 (50) (http://discover.nci.nih.gov/cellminercdb) (see Fig. 6 & 7), can be overcome by combination with the ATR inhibitor ceralasertib (Fig. 7F). In addition to SLFN11 expression, we confirmed that cancer cells with HRD are also selectively vulnerable to the exatecan (23). Thus, therapeutic strategies taking into account SLFN11 expression and HRD should enable the use of exatecan to aim accurately precision medicine (2) (Fig. 7F).
Recent attempts to combine a carrier and the cytotoxic warhead exatecan have provided new opportunities to overcome the therapeutic limitations of exatecan in the clinic, which are due to exatecan’s very high potency as a TOP1 poison (19,43). Cancer cell specific targeting with the exatecan derivative Deruxtecan has shown remarkable results in multiple clinical trials, which led to the FDA approval of Trastuzumab Deruxtecan (Enhertu®) (16). These results illustrate the possibility that exatecan can be re-formulated to harness its antineoplastic properties while limiting the toxicity (9,44). In particular, the newly developed pH-sensitive peptide-exatecan conjugate, CBX-12, showed that it can target most cancer cells regardless of limited oncogenic antigen expression observed in the antibody conjugation (14). CBX-12 selectively delivers exatecan to cancer cells in their low pH environment while sparing normal tissues and thereby shows better antitumor activity compared with non-conjugated exatecan itself in pre-clinical models. In two different mouse xenograft models, we show that CBX-12 significantly suppresses tumor growth as monotherapy and even more efficiently in combination with the ATR inhibitor ceralasertib, in line with previous data showing a synergistic effect with CBX12 and the PARP inhibitor talazoparib (14).
In conclusion, our data uncover mechanistic insight into the nature of exatecan-induced TOP1cc as the most potent TOP1 poison, and a therapeutic rationale for exatecan-based therapy via a drug-targeted system in combination with clinical ATR inhibitors. We also provide evidence for predictive clinical (SLFN11 and HRD) and pharmacodynamic (γH2AX) biomarkers that may improve the efficacy of exatecan.
Supplementary Material
Acknowledgments
Our studies are funded by the Center for Cancer Research, the intramural program of the National Cancer Institute (Z01-BCC006150). JCV is funded in part by the Robert Wood Johnson Harold Amos Medical Faculty Development Program and the Fund to Retain Clinical Scientists at Yale, sponsored by the Doris Duke Charitable Foundation award #2015216, and the Yale Center for Clinical Investigation.
Footnotes
Conflict of Interest: R.S.B. is a co-founder and consultant for Cybrexa Therapeutics. UJ, YM, KA, YS, LKS, XY, YA, SG, KJ, VP, RKS, JVD, JCV, WSC, and YP declare no potential conflicts of interest.
References
- 1.Pommier Y, Marchand C. Interfacial inhibitors: targeting macromolecular complexes. Nat Rev Drug Discov 2011;11(1):25–36 doi 10.1038/nrd3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thomas A, Pommier Y. Targeting Topoisomerase I in the Era of Precision Medicine. Clin Cancer Res 2019;25(22):6581–9 doi 10.1158/1078-0432.CCR-19-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kopetz S, Guthrie KA, Morris VK, Lenz HJ, Magliocco AM, Maru D, et al. Randomized Trial of Irinotecan and Cetuximab With or Without Vemurafenib in BRAF-Mutant Metastatic Colorectal Cancer (SWOG S1406). J Clin Oncol 2021;39(4):285–94 doi 10.1200/JCO.20.01994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mitsui I, Kumazawa E, Hirota Y, Aonuma M, Sugimori M, Ohsuki S, et al. A new water-soluble camptothecin derivative, DX-8951f, exhibits potent antitumor activity against human tumors in vitro and in vivo. Jpn J Cancer Res 1995;86(8):776–82 doi 10.1111/j.1349-7006.1995.tb02468.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Joto N, Ishii M, Minami M, Kuga H, Mitsui I, Tohgo A. DX-8951f, a water-soluble camptothecin analog, exhibits potent antitumor activity against a human lung cancer cell line and its SN-38-resistant variant. Int J Cancer 1997;72(4):680–6 doi . [DOI] [PubMed] [Google Scholar]
- 6.Vey N, Giles FJ, Kantarjian H, Smith TL, Beran M, Jeha S. The topoisomerase I inhibitor DX-8951f is active in a severe combined immunodeficient mouse model of human acute myelogenous leukemia. Clin Cancer Res 2000;6(2):731–6. [PubMed] [Google Scholar]
- 7.Abou-Alfa GK, Letourneau R, Harker G, Modiano M, Hurwitz H, Tchekmedyian NS, et al. Randomized phase III study of exatecan and gemcitabine compared with gemcitabine alone in untreated advanced pancreatic cancer. J Clin Oncol 2006;24(27):4441–7 doi 10.1200/JCO.2006.07.0201. [DOI] [PubMed] [Google Scholar]
- 8.Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov 2017;16(5):315–37 doi 10.1038/nrd.2016.268. [DOI] [PubMed] [Google Scholar]
- 9.Conilh L, Fournet G, Fourmaux E, Murcia A, Matera EL, Joseph B, et al. Exatecan Antibody Drug Conjugates Based on a Hydrophilic Polysarcosine Drug-Linker Platform. Pharmaceuticals (Basel) 2021;14(3) doi 10.3390/ph14030247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takegawa N, Nonagase Y, Yonesaka K, Sakai K, Maenishi O, Ogitani Y, et al. DS-8201a, a new HER2-targeting antibody-drug conjugate incorporating a novel DNA topoisomerase I inhibitor, overcomes HER2-positive gastric cancer T-DM1 resistance. Int J Cancer 2017;141(8):1682–9 doi 10.1002/ijc.30870. [DOI] [PubMed] [Google Scholar]
- 11.Iwata TN, Ishii C, Ishida S, Ogitani Y, Wada T, Agatsuma T. A HER2-Targeting Antibody-Drug Conjugate, Trastuzumab Deruxtecan (DS-8201a), Enhances Antitumor Immunity in a Mouse Model. Mol Cancer Ther 2018;17(7):1494–503 doi 10.1158/1535-7163.MCT-17-0749. [DOI] [PubMed] [Google Scholar]
- 12.Haratani K, Yonesaka K, Takamura S, Maenishi O, Kato R, Takegawa N, et al. U3-1402 sensitizes HER3-expressing tumors to PD-1 blockade by immune activation. J Clin Invest 2020;130(1):374–88 doi 10.1172/JCI126598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kotani D, Shitara K. Trastuzumab deruxtecan for the treatment of patients with HER2-positive gastric cancer. Ther Adv Med Oncol 2021;13:1758835920986518 doi 10.1177/1758835920986518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gayle S, Aiello R, Leelatian N, Beckta JM, Bechtold J, Bourassa P, et al. Tumor-selective, antigen-independent delivery of a pH sensitive peptide-topoisomerase inhibitor conjugate suppresses tumor growth without systemic toxicity. NAR Cancer 2021;3(2):zcab021 doi 10.1093/narcan/zcab021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li W, Veale KH, Qiu Q, Sinkevicius KW, Maloney EK, Costoplus JA, et al. Synthesis and Evaluation of Camptothecin Antibody-Drug Conjugates. ACS Med Chem Lett 2019;10(10):1386–92 doi 10.1021/acsmedchemlett.9b00301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keam SJ. Trastuzumab Deruxtecan: First Approval. Drugs 2020;80(5):501–8 doi 10.1007/s40265-020-01281-4. [DOI] [PubMed] [Google Scholar]
- 17.Goldenberg DM, Stein R, Sharkey RM. The emergence of trophoblast cell-surface antigen 2 (TROP-2) as a novel cancer target. Oncotarget 2018;9(48):28989–9006 doi 10.18632/oncotarget.25615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wahby S, Fashoyin-Aje L, Osgood CL, Cheng J, Fiero MH, Zhang L, et al. FDA Approval Summary: Accelerated Approval of Sacituzumab Govitecan-hziy for Third-line Treatment of Metastatic Triple-negative Breast Cancer. Clin Cancer Res 2021;27(7):1850–4 doi 10.1158/1078-0432.CCR-20-3119. [DOI] [PubMed] [Google Scholar]
- 19.Hafeez U, Parakh S, Gan HK, Scott AM. Antibody-Drug Conjugates for Cancer Therapy. Molecules 2020;25(20) doi 10.3390/molecules25204764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pommier Y DNA topoisomerase I inhibitors: chemistry, biology, and interfacial inhibition. Chem Rev 2009;109(7):2894–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tanizawa A, Fujimori A, Fujimori Y, Pommier Y. Comparison of topoisomerase I inhibition, DNA damage, and cytotoxicity of camptothecin derivatives presently in clinical trials. J Natl Cancer Inst 1994;86:836–42. [DOI] [PubMed] [Google Scholar]
- 22.Marzi L, Szabova L, Gordon M, Weaver Ohler Z, Sharan SK, Beshiri ML, et al. The Indenoisoquinoline TOP1 Inhibitors Selectively Target Homologous Recombination-Deficient and Schlafen 11-Positive Cancer Cells and Synergize with Olaparib. Clin Cancer Res 2019;25(20):6206–16 doi 10.1158/1078-0432.CCR-19-0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coussy F, El-Botty R, Chateau-Joubert S, Dahmani A, Montaudon E, Leboucher S, et al. BRCAness, SLFN11, and RB1 loss predict response to topoisomerase I inhibitors in triple-negative breast cancers. Sci Transl Med 2020;12(531) doi 10.1126/scitranslmed.aax2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lord CJ, Ashworth A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017;355(6330):1152–8 doi 10.1126/science.aam7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murai J, Thomas A, Miettinen M, Pommier Y. Schlafen 11 (SLFN11), a restriction factor for replicative stress induced by DNA-targeting anti-cancer therapies. Pharmacol Ther 2019;201:94–102 doi 10.1016/j.pharmthera.2019.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jo U, Murai Y, Takebe N, Thomas A, Pommier Y. Precision Oncology with Drugs Targeting the Replication Stress, ATR, and Schlafen 11. Cancers (Basel) 2021;13(18) doi 10.3390/cancers13184601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jo U, Senatorov IS, Zimmermann A, Saha LK, Murai Y, Kim SH, et al. Novel and Highly Potent ATR Inhibitor M4344 Kills Cancer Cells With Replication Stress, and Enhances the Chemotherapeutic Activity of Widely Used DNA Damaging Agents. Mol Cancer Ther 2021;20(8):1431–41 doi 10.1158/1535-7163.MCT-20-1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thomas A, Takahashi N, Rajapakse VN, Zhang X, Sun Y, Ceribelli M, et al. Therapeutic targeting of ATR yields durable regressions in small cell lung cancers with high replication stress. Cancer Cell 2021;39(4):566–79.e7 doi 10.1016/j.ccell.2021.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yap TA, Krebs MG, Postel-Vinay S, El-Khouiery A, Soria JC, Lopez J, et al. Ceralasertib (AZD6738), an Oral ATR Kinase Inhibitor, in Combination with Carboplatin in Patients with Advanced Solid Tumors: A Phase I Study. Clin Cancer Res 2021. doi 10.1158/1078-0432.CCR-21-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murai J, Feng Y, Yu GK, Ru Y, Tang SW, Shen Y, et al. Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget 2016;7(47):76534–50 doi 10.18632/oncotarget.12266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Staker BL, Feese MD, Cushman M, Pommier Y, Zembower D, Stewart L, et al. Structures of three classes of anticancer agents bound to the human topoisomerase I-DNA covalent complex. J Med Chem 2005;48(7):2336–45 doi 10.1021/jm049146p. [DOI] [PubMed] [Google Scholar]
- 32.Staker BL, Hjerrild K, Feese MD, Behnke CA, Burgin AB, Jr., Stewart L. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc Natl Acad Sci U S A 2002;99(24):15387–92 doi 10.1073/pnas.242259599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 2010;66(Pt 4):486–501 doi 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dexheimer TS, Pommier Y. DNA cleavage assay for the identification of topoisomerase I inhibitors. Nat Protoc 2008;3(11):1736–50 doi 10.1038/nprot.2008.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laco GS, Du W, Kohlhagen G, Sayer JM, Jerina DM, Burke TG, et al. Analysis of human topoisomerase I inhibition and interaction with the cleavage site +1 deoxyguanosine, via in vitro experiments and molecular modeling studies. Bioorg Med Chem 2004;12(19):5225–35 doi 10.1016/j.bmc.2004.06.046. [DOI] [PubMed] [Google Scholar]
- 36.Kiianitsa K, Maizels N. A rapid and sensitive assay for DNA-protein covalent complexes in living cells. Nucleic Acids Res 2013;41(9):e104 doi 10.1093/nar/gkt171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pommier Y, Cushman M. The indenoisoquinoline noncamptothecin topoisomerase I inhibitors: update and perspectives. Mol Cancer Ther 2009;8(5):1008–14 doi 10.1158/1535-7163.MCT-08-0706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun Y, Saha LK, Saha S, Jo U, Pommier Y. Debulking of topoisomerase DNA-protein crosslinks (TOP-DPC) by the proteasome, non-proteasomal and non-proteolytic pathways. DNA Repair (Amst) 2020;94:102926 doi 10.1016/j.dnarep.2020.102926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bradbury A, Hall S, Curtin N, Drew Y. Targeting ATR as Cancer Therapy: A new era for synthetic lethality and synergistic combinations? Pharmacol Ther 2020;207:107450 doi 10.1016/j.pharmthera.2019.107450. [DOI] [PubMed] [Google Scholar]
- 40.Yap TA, Krebs MG, Postel-Vinay S, El-Khouiery A, Soria JC, Lopez J, et al. Ceralasertib (AZD6738), an oral ATR kinase inhibitor, in combination with carboplatin in patients with advanced solid tumors: a Phase I study. Clin Cancer Res 2021;27(19):5213–24 doi 10.1158/1078-0432.CCR-21-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim ST, Smith SA, Mortimer P, Loembe AB, Cho H, Kim KM, et al. Phase I Study of Ceralasertib (AZD6738), a Novel DNA Damage Repair Agent, in Combination with Weekly Paclitaxel in Refractory Cancer. Clin Cancer Res 2021. doi 10.1158/1078-0432.CCR-21-0251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wyatt LC, Lewis JS, Andreev OA, Reshetnyak YK, Engelman DM. Applications of pHLIP Technology for Cancer Imaging and Therapy. Trends Biotechnol 2017;35(7):653–64 doi 10.1016/j.tibtech.2017.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khongorzul P, Ling CJ, Khan FU, Ihsan AU, Zhang J. Antibody-Drug Conjugates: A Comprehensive Review. Mol Cancer Res 2020;18(1):3–19 doi 10.1158/1541-7786.MCR-19-0582. [DOI] [PubMed] [Google Scholar]
- 44.Royce ME, Hoff PM, Dumas P, Lassere Y, Lee JJ, Coyle J, et al. Phase I and pharmacokinetic study of exatecan mesylate (DX-8951f): a novel camptothecin analog. J Clin Oncol 2001;19(5):1493–500 doi 10.1200/JCO.2001.19.5.1493. [DOI] [PubMed] [Google Scholar]
- 45.Esteva FJ, Rivera E, Cristofanilli M, Valero V, Royce M, Duggal A, et al. A Phase II study of intravenous exatecan mesylate (DX-8951f) administered daily for 5 days every 3 weeks to patients with metastatic breast carcinoma. Cancer 2003;98(5):900–7 doi 10.1002/cncr.11557. [DOI] [PubMed] [Google Scholar]
- 46.Sun FX, Tohgo A, Bouvet M, Yagi S, Nassirpour R, Moossa AR, et al. Efficacy of camptothecin analog DX-8951f (Exatecan Mesylate) on human pancreatic cancer in an orthotopic metastatic model. Cancer Res 2003;63(1):80–5. [PubMed] [Google Scholar]
- 47.Coates JT, Sun S, Leshchiner I, Thimmiah N, Martin EE, McLoughlin D, et al. Parallel genomic alterations of antigen and payload targets mediate polyclonal acquired clinical resistance to sacituzumab govitecan in triple-negative breast cancer. Cancer Discov 2021. doi 10.1158/2159-8290.CD-21-0702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gilbert DC, Chalmers AJ, El-Khamisy SF. Topoisomerase I inhibition in colorectal cancer: biomarkers and therapeutic targets. Br J Cancer 2012;106(1):18–24 doi bjc2011498 [pii] 10.1038/bjc.2011.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang B, Ramkumar K, Cardnell RJ, Gay CM, Stewart CA, Wang WL, et al. A wake-up call for cancer DNA damage: the role of Schlafen 11 (SLFN11) across multiple cancers. Br J Cancer 2021. doi 10.1038/s41416-021-01476-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zoppoli G, Regairaz M, Leo E, Reinhold WC, Varma S, Ballestrero A, et al. Putative DNA/RNA helicase Schlafen-11 (SLFN11) sensitizes cancer cells to DNA-damaging agents. Proc Natl Acad Sci U S A 2012;109(37):15030–5 doi 10.1073/pnas.1205943109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available within the article and its supplementary data files.