

Abstract
Exchanging the ribose backbone of an oligonucleotide for a peptide can enhance its physiologic stability and nucleic acid binding affinity. Ordinarily, the eneamino nitrogen atom of a nucleobase is fused to the side chain of a polypeptide through a new C–N bond. The discovery of C–C linked nucleobases in the human transcriptome reveals new opportunities for engineering nucleopeptides that replace the traditional C–N bond with a non-classical C–C bond, liberating a captive nitrogen atom and promoting new hydrogen bonding and π-stacking interactions. We report the first late-stage synthesis of C–C linked carba-Nucleopeptides (cNPs) using aqueous Rhodamine B photoredox catalysis. We prepare brand-new cNPs in batch, in parallel, and in flow using three long-wavelength photochemical setups. We detail the mechanism of our reaction by experimental and computational studies and highlight the essential role of diisopropylethylamine as a bifurcated two-electron reductant.
Keywords: Nucleopeptides, Photoredox catalysis, Flow Synthesis, Parallel Synthesis, Rhodamine B
Graphical Abstract
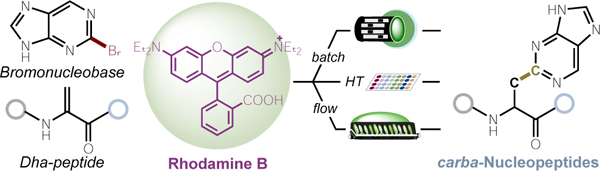
Exchanging the amino acid side chains of a polypeptide with C–N linked nucleobases forms oligonucleotide-like structures (aza-Nucleopeptides) with favorable physiochemical properties. Connecting the nucleobase to the peptide through a C–C bond can promote new, beneficial noncovalent interactions. A general method to make C–C linked carba-Nucleopeptides in batch, parallel, and flow using aqueous Rhodamine B photoredox catalysis is reported.
Introduction
Artificial DNA/RNA-mimetics can act as viable drugs, vehicles for understanding disease pathology, and as building blocks for new biomaterials and nanostructures.[1] One way to imitate endogenous oligonucleotides is to tether nucleobases to the side chains of a polypeptide via a C–N linkage (peptide nucleic acids).[2–4] These form stable hybrids with naturally occurring nucleic acid sequences and targets (e.g., duplexes, triplexes, G-quadraplexes, i-motifs, and aptamers)[2,5–7], and unlike classical oligonucleotides, cannot be degraded by nucleases.[8] In recent years, other strategies for connecting nucleobases to peptides have emerged, such as uniting the N1 (pyrimidine) or N9 (purine) nitrogen atoms of one or more nucleobases with alanine-based amino acids through sp3C–N bonds, referred to as aza-Nucleopeptides (aNPs).[9] These structures conserve the amide backbone of peptides enabling them to self-assemble and to recognize target oligonucleotides in ways akin to standard peptides. The discovery of the ‘fifth nucleotide’, pseudouridine,[10] unlocks new opportunities for nucleopeptides. Pseudouridine is linked to its ribose ring through an unusual C–C bond. This ubiquitous yet enigmatic feature allows pseudouridine to form an extra hydrogen bond and to enhance its π-stacking capabilities, which enables the proper folding of tRNAs and rRNAs and the efficient decoding and processing of mRNAs in the human ribosome, essential for protein synthesis. Pseudouridine in small nuclear RNAs also enhances spliceosomal RNA-pre-mRNA interactions, heavily regulating gene expression.[11,12] Hence, connecting a polypeptide to the carbon atom of even a single nucleobase, herein termed carba-Nucleopeptide (cNP), has the capacity to transform the function and chemical versatility of nucleopeptides, but no general method for assembling cNPs exists (Figure 1). To explore the untapped potential of carba-Nucleopeptides, a synthetic platform to readily access libraries of cNPs is needed.
Figure 1.
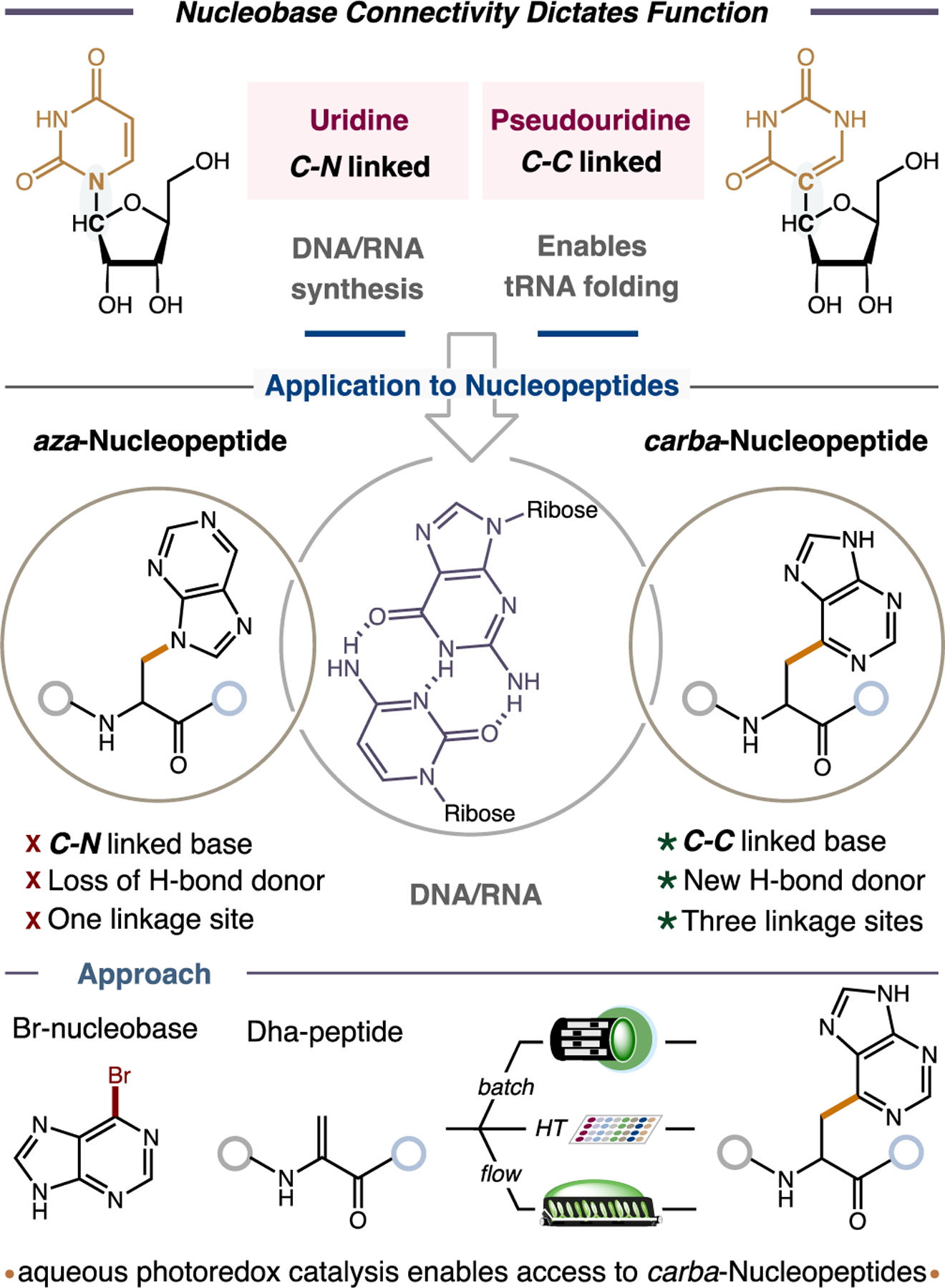
The connectivity of a nucleobase can dictate its role in biological systems as demonstrated by the endogenous nucleobase pseudouridine. Applying this concept to Nucleopeptides creates a brand new class of molecules called carba-Nucleopeptides with untapped functions that could be synthesized through radical-Dha coupling.
The C–C linkage of cNPs could be made via a fragment-based approach, combining a C-centered nucleobase radical with a dehydroalanine (Dha) side chain, derived from a single cysteine reside, in a peptide. This type of approach has been successfully used by our lab (with boronic acids) and others to incorporate unnatural amino acids and non-ribosomal side chains into peptides and proteins through photoredox catalysis.[13–14] Unfortunately, borono-nucleobases are unstable[15] and, therefore, have low commercial availability and challenging syntheses. Their high oxidation potential also makes them less apt to form radicals by photocatalyzed oxidative deborylation.[16] Thus, to leverage our established radical-to-Dha approach for cNP synthesis, we needed to identify a different radical precursor and a new photocatalyst. bromo-Nucleobases are a readily accessible class of nucleobase analogs for chemical synthesis. We imagined that single-electron transfer (SET) reduction of a bromonucleobase by an aqueous photocatalyst could cleave the C–Br bond and unveil a free C-centered nucleobase radical. To this end, fluorescent organic dyes are water soluble and strongly reducing in their photoexcited state. Their ability to absorb longer wavelengths (>500 nm) of light also enables them to mediate photochemical transformations at wavelengths more amenable to biomolecules.[17] (Our lab and others have found that some peptides are readily degraded with higher energy <450 nm light).[18] By operating through SET reduction, organic dyes are also less likely to cause undesirable side reactions on peptides, which generally have amino acids that can be oxidatively modified, including Tyr[19], Trp[20], Met[21,22], His[23,24], N-termini[25], and C-termini[26]. The discovery of an organic dye that can facilitate cNP synthesis would not only provide ready access to a novel biopharmaceutical modality but bolster the utility of fluorescent organic dyes for biomolecular synthesis.
Results and Discussion
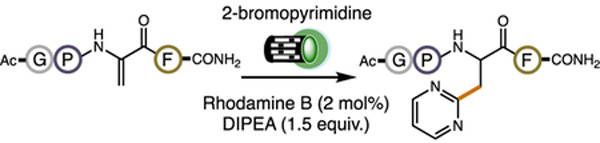
To identify a suitable photocatalyst for cNP synthesis, we surveyed several organic dyes and metal-based photocatalysts (10 mol%) using 2-bromopyridine (1 equiv.) as a surrogate for a bromonucleobase and water as solvent. (The single-electron reduction potential of 2-bromopyridine is similar to a bromonucleobase, and the 2-pyridyl radical has been shown to engage Dha residues by open-shell conjugate addition.[13a]) We also included 1-benzyl-3-carbamoylpyridinium bromide (reduced in situ) as a sacrificial reductant. Ac-Gly-Pro-Dha-Phe-CONH2 (2.3 μmol) was used as a test peptide and irradiation was provided by two 525 nm Kessil lamps (40 W each). We found Rhodamine B (Rh B), a standard dye for many biotechnology applications,[27–29] to be the best photocatalyst, delivering the desired cNP in 21% conversion. Using diisopropylethylamine (DIPEA) as reductant and decreasing photocatalyst loading to 2 mol% increased the conversion to 44%. These conditions translated well when the bromonucleobase, 2-bromopyrimidine, was used in place of 2-bromopyridine in our standard reaction (52% conversion). Including 2,2,2-trifluoroethanol (TFE) as a cosolvent (5% v/v) enhanced the efficiency of the reaction to 80% conversion, presumably by decreasing the reduction potential of 2-bromopyrimidine through hydrogen bonding.[30] Finally, adding a small amount of acetonitrile (5% v/v) increased the homogeneity of the reaction and provided an optimal 87% conversion to the desired cNP product. Common iridium- and ruthenium-based photocatalysts performed less admirably in our reaction with two 440 nm Kessil lamps (Table 1). Additionally, we found that our optimized reaction conditions with 2-bromopyrimidine could be successfully applied to the unprotected variant of our standard peptide, NH2-Gly-Pro-Dha-Phe-CO2H (44% conversion to cNP). See Supporting Information for full optimization details.
Table 1.
Reaction optimization parameters.
| ||
|---|---|---|
|
| ||
| Entry | Deviation from standard conditions[a] | Conversion [%][b] |
| 1 | none | 87 |
| 2 | no Rhodamine B | 0 |
| 3 | no light | 0 |
| 4 | no DIPEA | <5 |
| 5 | triphenylamine as amine | <5 |
| 6[c] | Ir(ppy)2(dtbby)PF6 as photocatalyst | 21 |
| 7[c] | Ru(bpy)3Cl2 as photocatalyst | 14 |
| 8 | 23 µmol scale (0.5 mol% photocatalyst) | >99 |
Reactions performed using 2.3 µmol of Ac-GPDhaF-CONH2, 5 equiv. of 2-bromopyrimidine, 2 mol% of Rhodamine B, and 1.5 equiv. of amine in a solution of 90:5:5 H20:ACN:TFE (3 mM overall concentration) with 15 h of Green light (525 nm) irridation.
Conversion [%] defined as the % area of the product peaks at 214 nm as compared to all other peptide peaks in the LC chromatogram.
Reaction performed with two Blue lights (440 nm). ppy — 2-phenylpyridine, dtbbpy — 4,4’-di-tert-butyl-2,2’-dipyridyl, bpy — 2,2’-bipyridine
We assessed the scope of bromopyrimidines that can be used for cNP synthesis (Scheme 1). (Note: The reaction was performed on a 23 μmol scale for ease of cNP product isolation. Interestingly, the reaction performed slightly better on scale and with lower catalyst loading (0.5 mol%). For example, 2-bromopyrimidine achieved >99% conversion to the desired cNP product with a 77% isolated yield, as a mixture of two epimers.) We found that our reaction tolerated a diverse array of substituents on the pyrimidine ring including several functional groups that are prone to reduction, such as exogenous halogen atoms[31] (compounds 3, 4, 5) and a nitrile[32] (compound 8). Unprotected aryl amines (prone to oxidation)[33] were also tolerated (compounds 7 and 8). For a tosyl protected alcohol, our reaction conditions not only afforded the desired cNP product 9, but also removed the tosyl protecting group in the same reaction. The ability to accomplish both transformations (alkylation and deprotection) on the same substrate is remarkable considering that only 0.5 mol% catalyst is used in the reaction. Finally, we found that our reaction could furnish cNP 10 having a decarbonyluracil side chain, an established group with herbicidal activity.[34] While uracil has traditionally been employed as a radical ‘acceptor’,[35] our reaction shows its competence as a radical ‘donor’. To our knowledge, this reactivity has not been demonstrated previously for uracil.
Scheme 1.
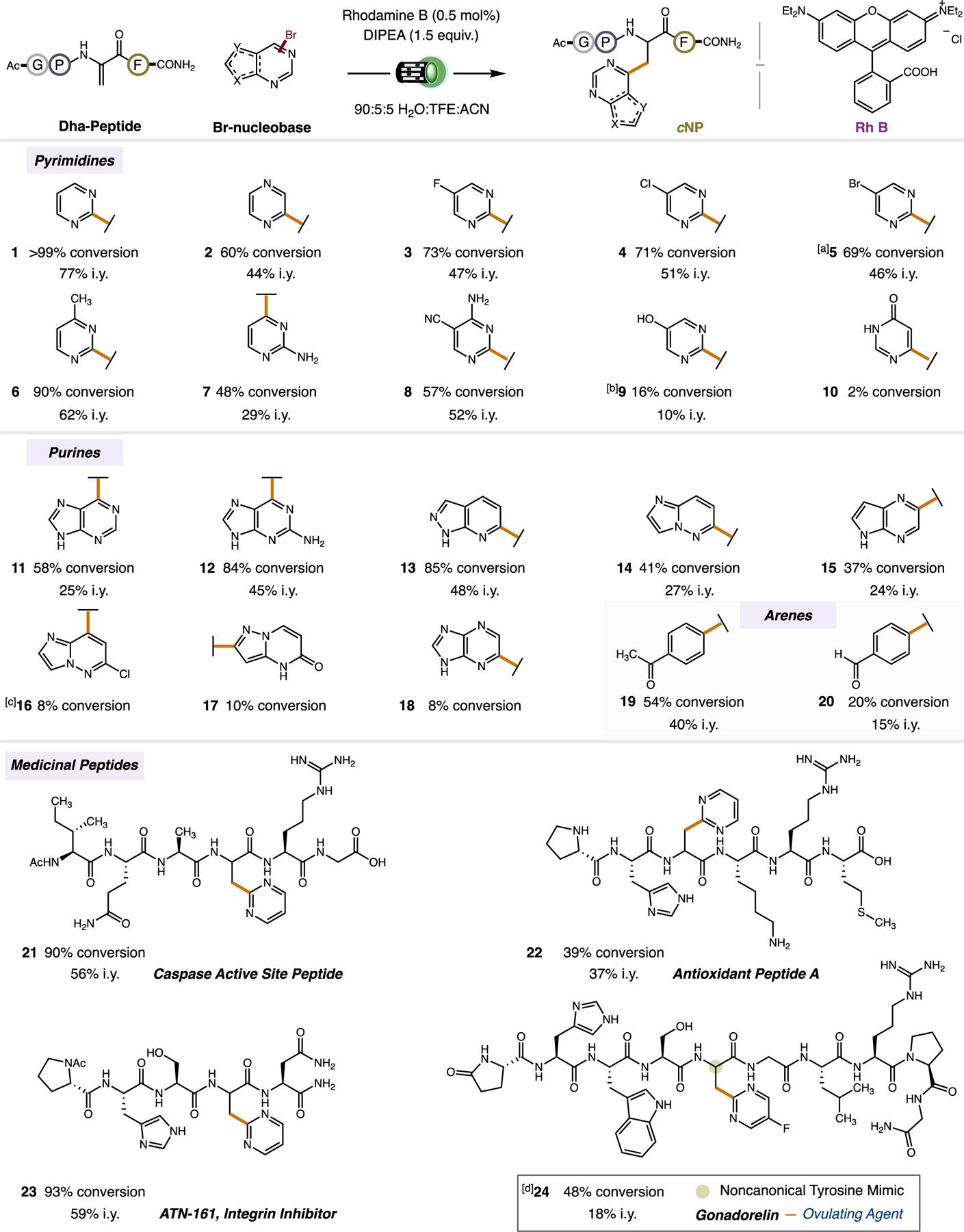
Scope of nucleobase bromides and medicinal peptides. All reactions were performed using 23 pmol of Ac-GPDhaF-CONH2 or other Dhapeptide, 5 equiv. of bromonucleobase, 0.5 mol% of Rhodamine B, and 1.5 equiv. of diisopropylethylamine in a solution of 90:5:5 H20:ACN:TFE (3 mM overall concentration) with 15 h of Green light (525 nm) irridation. [a] 5 yielded two cNP products giving a 13:1 selectivity for the C2-alkylated cNP vs. C5-alkylated cNP. [b] Reaction was performed with 5-OTs-2-bromopyrimidine. The Tosyl group deprotected under the reaction conditions to yield cNP 9. [c] Reaction performed with 4 mol% Rhodamine B. [d] Reaction performed on a 10 pmol scale. Conversion [%] is defined as the % area of the product peaks at 214 nm as compared to all other peptide peaks in the LC chromatQ4ram. i.y. = isolated yield.
We explored whether the position of the bromine atom in our standard pyrimidine affected the efficiency of cNP formation. 5-Bromopyrimidine failed to react to any significant extent (<1% conversion to cNP), and most of the bromide was recovered after the reaction. This suggests that reduction of the bromonucleobase at position-C5 of the pyrimidine ring is not favored. In contrast, bromides adjacent to electron withdrawing sp2-nitrogen atoms in the pyrimidine ring readily formed radicals. Taking advantage of this innate difference in reactivity, we performed our coupling reaction using 2,5-dibromopyrimidine. As expected, we obtained primarily the C2-alkylated cNP product (13:1 selectivity) determined by 1H NMR analysis (Figure S6). Our finding demonstrates that nucleobase radical formation is governed primarily by electronics, with more electron deficient C–Br sites being more susceptible to SET reduction. With this understanding, we strongly suspect nucleobases that are inherently electron rich (such as pseudouridine) and/or have activated C–Br sites could be problematic due to their higher reduction potentials. The inability to generate radicals at every conceivable position in a standard nucleobase poses a limitation to our methodology.
We next examined bromopurines and purine-like nucleobases. To our delight, a wide array of bromopurines underwent successful fragment coupling with our standard Dha-peptide to afford new cNP products. In general, bromopurines where the bromide is adjacent to a sp2-nitrogen atom worked best in our reaction. Of significance, we found that non-coding purine mimetics, deaminoadenine (11) and decarbonylguanine (12) were coupled in good yields, replacing the endogenous amine and carbonyl groups of the parent nucleobases with a new C–C linkage, respectively. These two nucleobase mimetics are ideal substrates to probe the importance of hydrogen bonding in guanine and adenine nucleotides for base recognition and oligonucleotide folding, as they eliminate one hydrogen bond from each nucleobase that is normally used for base-pairing.[36,37] Aside non-coding nucleobases 11 and 12, we prepared the deaminoadenine analog (18) wherein N1 of the purine ring is transposed to the C6 position. Five additional non-coding nucleobase analogs were also successfully prepared, each having three nitrogen atoms scattered over the purine ring. As suspected, nucleobases 16 and 17 were less effective due to higher electron density at their C–Br sites. The corresponding bromide that makes cNP 18 is a highly colored material and likely presents its own challenges for photodriven Dha-diversification. The chemical/biological consequence of retaining the [5:6]-ring fusion of purines and redistributing the nitrogen atoms is largely unexplored, but we expect this will enable the purine to participate in unprecedented non-covalent interactions (e.g., new hydrogen bonding and π-stacking interactions), which could be useful for any number of applications, inter alia, medicine, chemical biology, and biomaterials. To conclude our survey, we examined simple bromoarenes. Without the nitrogen atom, we questioned whether our methodology would work at all. Gratifyingly, bromoarenes bearing a ketone 19 and an aldehyde 20 were viable substrates in our reaction. Thus, our method also has the potential to install non-heteroatom-containing arylbromides into Dha-peptides, including those with biorthogonal handles.[38]
We examined different peptide sequences for cNP synthesis. We prepared three medicinally relevant cysteine-containing peptides (caspase active site peptide,[39] antioxidant peptide A,[40] and ATN-161 integrin inhibitor[41]), converted their cysteine amino acids to Dha residues, and performed our standard coupling reaction with 2-bromopyrimidine. In each case, cNP products were isolated in good yields (compounds 21, 22, and 23). Considering the peptides we used, our reaction appears amenable to peptides having an unprotected C-termini (compounds 21 and 22), a free N-terminal proline and basic amino acid side chains (Arg and Lys, compound 22), and oxidatively sensitive amino acids (His[24] and Met[22], compounds 22 and 23). We also examined the hypothalamic decapeptide Gonadorelin, sequence: Pyr-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-CONH2. Residues His-2, Tyr-5, and Arg-8 of Gonadorelin form a tight triad that stabilizes the peptide in a conformation necessary for its biological activity.[42] Replacing the π-rich phenol ring of Tyr-5 with a more electron deficient imidazole ring increases affinity for the Gonadorelin receptor.[43] This suggested to us that substituting Tyr-5 with a dinitrogen-containing heteroaromatic ring that contains a bioisostere for a hydroxyl group might further improve activity. Accordingly, we used our newly minted cNP chemistry to exchange the phenol ring of Tyr-5 with an unprecedented 5-fluoropyrimidine side chain (compound 24). Its synthesis demonstrates clearly the unique opportunities of our side chain addition strategy (and reducing organophotocatalysts) for cNP synthesis in medicinal chemistry. Furthermore, the innate chemoselectivty of our reaction across four different Dha-containing peptides suggest that our synthetic approach could well translate to work on more complex peptides, including those with longer amino acid sequences, and potentially on proteins.
To detail the important biological applications of cNPs calls for strategies to simultaneously generate many numbers of disparate cNPs (for hit-to-lead peptide optimization) and to access larger quantities of the best peptide analogs for completing more advanced studies.[9b,44–46] In this vein, we sought to adopt our protocol for cNP synthesis to parallel and to flow technologies. The Lumidox® Generation II 527 nm reactor from Analytical Sales and Services Inc. is a two-in-one system that enables the user to transition between high-throughput and preparative (flow) synthesis. For parallel synthesis, a 96-well vial rack (containing unreacted samples) is placed atop an LED array with an integrated cooling base. All 96 samples are irradiated at once. For flow synthesis, the 96-well rack is exchanged for a pre-assembled unit containing flow-tubing of desired path length and transparency. An exogenous syringe pump is attached to the tubing inlet and controls the sample flow rate. We first optimized our reaction for parallel synthesis. We performed our reaction on a scale of 2.3 μmol (1 mg peptide) at 200 mW per well with Ac-Gly-Pro-Dha-Phe-CONH2 and 16 different bromides (9 pyrimidines, 5 purines, and 2 arenes) shown to work in batch at 23 μmol at 80 W per reaction. After 15 hours of irradiation, the light source was removed, and polymer-bound 2-mercaptoethylamine resin was added to each reaction (stirred for 5 hours at 40 °C). This resin sequesters any unreacted Ac-Gly- Pro-Dha-Phe-CONH2 and allows it to be conveniently filtered away. Unreacted bromide was removed by diethyl ether extraction. Finally, all products were purified in tandem by solid-phase extraction (SPE) with a Waters Oasis HLB sorbent plate (60 mg sorbent per well) using 5% aq. NaOH followed by 50% TFE/H2O as eluent. ~50% of our cNP products were obtained asa mixture of two diastereomers (epimers) in purities of ≥ 65%, with respect to all other components found in the final purified LC spectrum and amounts of ≥ 0.05 mg (Figure 2A). Our lab has shown that peptide mixtures meeting these two criteria are suitable for direct biochemical testing, with peptide mixtures giving comparable results to single diastereomer products.[14] For complete experimental details see Table S6.
Figure 2.
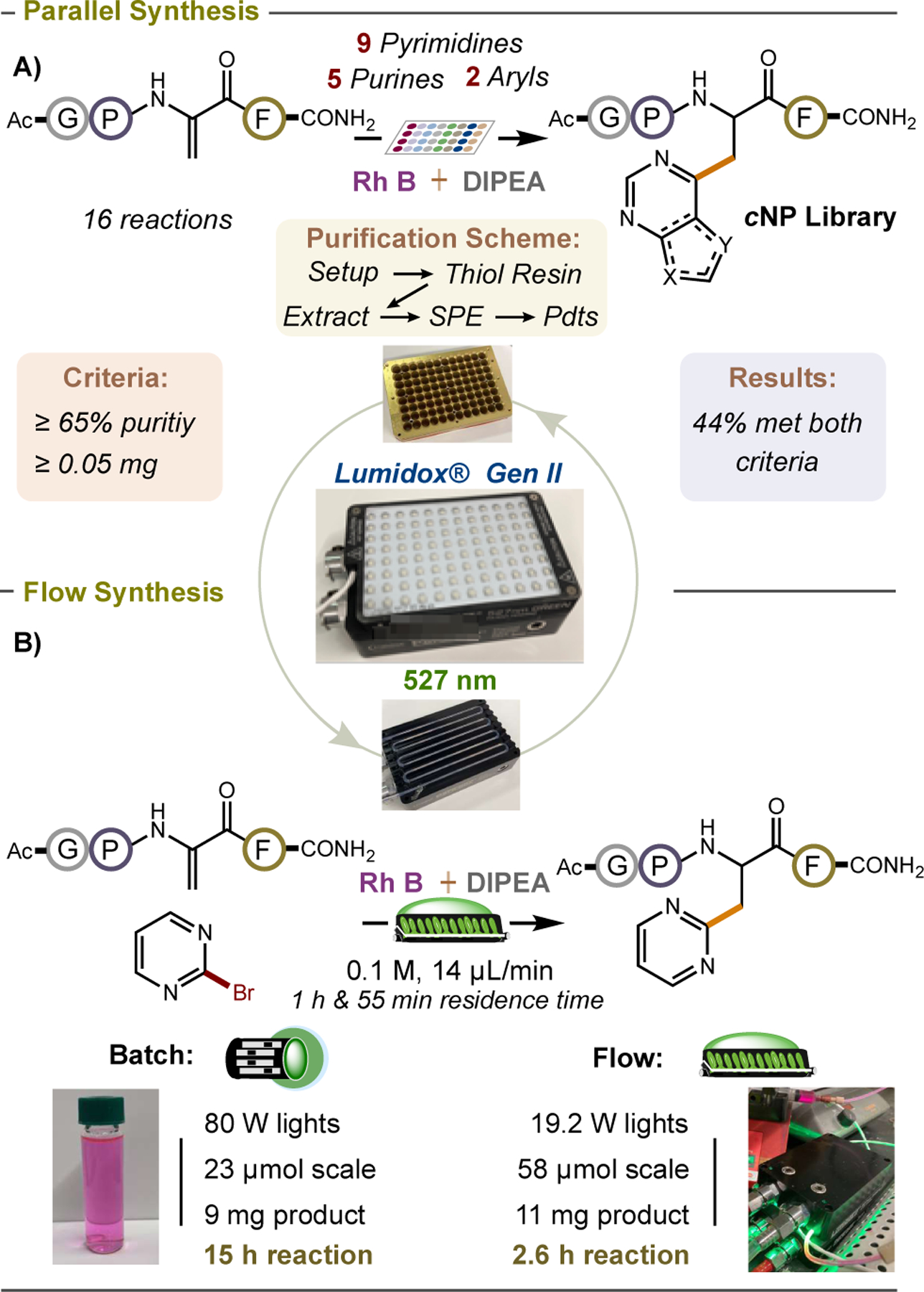
Merging cNP synthesis with the Lumidox® Gen II system for applications in A) parallel synthesis of cNP libraries and B) flow synthesis of cNPs.
Next, we optimized our reaction in flow. In many cases, performing a reaction in flow requires that entirely new reaction conditions be identified so that the reaction is homogeneous, avoids the formation of precipitates, and can be concentrated. It is also highly desirable to use comparable (and low) stoichiometries between different reactants. Fortunately, our reaction already satisfies many of these criteria, and this allowed us to start our optimization using 1.0 equiv. of peptide (58 μmol), 1.5 equiv. of 2-bromopyrimidine, 1.5 equiv. of DIPEA, and 0.5 mol% Rh B, at 0.1 M. In flow (flow rate: 14 μL/min; residence time: 1 hour and 55 minutes), we achieved a 38% isolated yield of our desired cNP product. More importantly, we generated the same amount of product in this 2.6 h reaction that would normally take us 15 h to make in batch: ~ a 6-fold improvement in time (Figure 2B). This is remarkable if you consider that our flow system has ~ 4 x less wattage (80 W for batch vs. 19.2 W for flow). While S–S,[47,48] S–O,[49] S–C,[50,51] and C–N[52] bond forming reactions have been achieved on peptides in flow by photoredox catalysis, our work represents the first example of C–C bond formation.
As a final point of interest, we probed the mechanism of our reaction. SET reduction of 2-bromopyrimidine by photoexcited Rhodamine B could generate a C-centered nucleobase radical, which can be captured by radical trap TEMPO•. Indeed, adding TEMPO• to our standard reaction without Ac-Gly-Pro-Dha-Phe-CONH2 yielded a pyrimidine-TEMPO adduct in 3% conversion by LC-MS analysis (Figure 3A). While this observation is consistent with SET reduction by Rhodamine B, it does not indicate whether Rhodamine B reduces our bromonucleobase from its photoexcited (*RhB) or reduced (RhB•-) state, as our reaction contains the sacrificial amine reductant DIPEA (i). SET oxidation of DIPEA (0.68 V vs. SCE)[53] by photoexcited Rhodamine B (0.92 V vs. SCE)[54,55] is exergonic. Direct SET reduction of 2-bromopyrimidine (−1.86 vs. SCE)[56] by photoexcited Rhodamine B (−1.3 V vs. SCE)[54,55] is endergonic. Thus, reductive quenching of Rhodamine B to yield highly reducing RhB•- (more negative potential than −1.3 V vs. SCE) and DIPEA• (ii) appears most probable, and RhB•- converts our bromonucleobase to a C-centered radical (iii) via SET reduction. The nucleobase radical is free to engage our Dha-peptide (iv) by open-shell conjugate addition, producing a captodative α-carbonyl radical (v; ΔE = −45 kcalmol−1 at B3LYP/6–311+G**). DIPEA• reduces the α-carbonyl radical to an α-carbonyl anion by SET. The resulting iminium (vi) is hydrolyzed to diisopropylamine and acetaldehyde,[57] and the α-carbonyl anion is protonated by solvent to give the final cNP product (vii; ΔE = −53 kcalmol−1 at B3LYP/6–311+G**). It is critical to point out that the reduction of the α-carbonyl radical (v) by oxidized DIPEA (ii) is highly endergonic (+107 kcalmol−1). However, this SET process becomes favorable when the resulting iminium ion is hydrolyzed to diisopropylamine and acetaldehyde giving a net −53 kcalmol−1 driving force (Figure 3B). We find this to be an important result considering emerging reports on the promiscuity of α-amino radicals as halogen-atom transfer reagents for photoredox catalysis and as bifurcated SET reductants for photolytic reactions.[13b, 58–59] We now show that in addition to being modest reducing agents, α-amino radicals can help drive endergonic steps in photoredox catalyzed reactions via oxidative fragmentation. This mechanistic paradigm could find general applications for the design of other photocatalyzed transformations, especially those performed in aqueous solvent. In support of our proposed mechanistic steps: (1) HRMS analysis of our crude reaction mixture revealed the formation of diisopropylamine (102.13 m/z, Figure S5), (2) using D2O as solvent furnished the α-deuterated cNP product (>95% d-content, Figure 3A), and (3) when triphenylamine (1.09 vs. SCE)[60]–similar oxidation potential to DIPEA but lacks α-hydrogen atoms–was used as a sacrificial reductant, only a small amount of cNP was formed, highlighting the importance of an α-amino radical to cNP formation (Table 1). Our experimental results are consistent with the mechanism depicted in Figure 3B.
Figure 3.
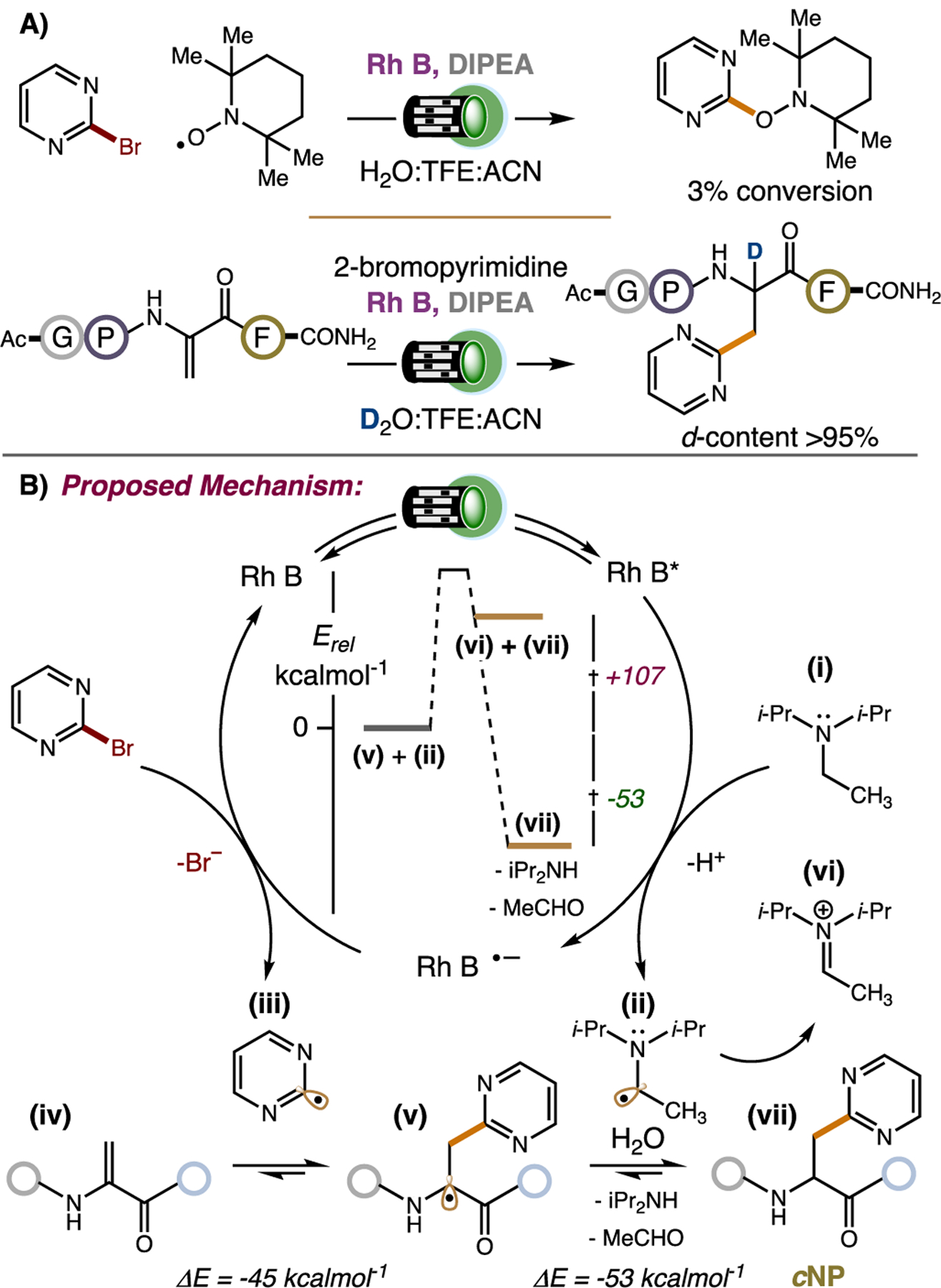
A) TEMPO trapping and a deuterium labeling experiment. B) Proposed mechansim for cNP synthesis.
Conclusion
In conclusion, we have developed a general method to access carba-Nucleopeptides having new pyrimidine and purine side chains. We demonstrate the compatibility of our system for parallel synthesis and for on-scale synthesis in flow using a brand-new photochemical setup. Finally, we detail the mechanism of our reaction, demonstrating the feasibility of organic dyes (such as Rhodamine B) to perform reductive transformations under biocompatible conditions, and for α-amino radicals to offset endergonic reactions through oxidative fragmentation.
Supplementary Material
Acknowledgements
This work was performed in the Department of Medicinal Chemistry at the University of Kansas, and supported by the School of Pharmacy, University of Kansas, and the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health under award number P20GM113117. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supporting information for this article is given via a link at the end of the document
Contributor Information
Jacob R. Immel, Department of Medicinal Chemistry, University of Kansas, Lawrence, Kansas 66045
Steven Bloom, Department of Medicinal Chemistry, University of Kansas, Lawrence, Kansas 66045.
References
- [1].a) Veedu RN, Wengel J, Chem. Biodivers 2010, 7, 536–542; [DOI] [PubMed] [Google Scholar]; b) Ziach K, Chollet C, Parissi V, Prabhakaran P, Marchivie M, Corvaglia V, Bose PP, Laxmi-Reddy K, Godde F, Schmitter J-M, Chaignepain S, Pourquier P, Huc I, Nat. Chem 2018, 10, 511–518; [DOI] [PubMed] [Google Scholar]; c) Yang H, Xi W, Polymers 2017, 9, 666; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Swenson CS, Velusamy A, Argueta-Gonzalez HS, Heemstra JM, J. Am. Chem. Soc 2019, 141, 19038–19047; [DOI] [PubMed] [Google Scholar]; e) Del Prado A, González-Rodríguez D, Wu Y-L, ChemistryOpen 2020, 9, 409–430; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Wang F, Liu B, Huang P-JJ, Liu J, Anal. Chem 2013, 85, 12144–12151; [DOI] [PubMed] [Google Scholar]; g) Baumann V, Winkler J, Future Med. Chem 2014, 6, 1967–1984; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Kulkarni JA, Witzigmann D, Thomson SB, Chen S, Leavitt BR, Cullis PR, van der Meel R, Nat. Nanotechnol 2021, 16, 630–643. [DOI] [PubMed] [Google Scholar]
- [2].Pellestor F, Paulasova P, Eur. J. Hum. Genet 2004, 12, 694–700. [DOI] [PubMed] [Google Scholar]
- [3].Singh KR, Sridevi P, Singh RP, Eng. Rep 2020, 2, e12238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Das A, Pradhan B, Chem. Biol. Drug Des 2021, 97, 865–892. [DOI] [PubMed] [Google Scholar]
- [5].Goux E, Lespinasse Q, Guieu V, Perrier S, Ravelet C, Fiore E, Peyrin E, Methods 2016, 97, 69–74. [DOI] [PubMed] [Google Scholar]
- [6].Modi S, Wani AH, Krishnan Y, Nucleic Acids Res 2006, 34, 4354–4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Panyutin IG, Onyshchenko MI, Englund EA, Appella DH, Neumann RD, Curr. Pharm. Des 2012, 18, 1984–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Patel R, Sarma S, Shukla A, Parmar P, Goswami D, Saraf M, Mol. Biol. Rep 2020, 47, 8113–8131. [DOI] [PubMed] [Google Scholar]
- [9].a) Takahashi T, Hamasaki K, Kumagai I, Ueno A, Mihara H, Chem. Commun 2000, 349–350; [DOI] [PubMed]; b) Pomplun S, Gates ZP, Zhang G, Quartararo AJ, Pentelute BL, J. Am. Chem. Soc 2020, 142, 19642–19651; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Chaltin P, Lescrinier E, Lescrinier T, Rozenski J, Hendrix C, Rosemeyer H, Busson RHC, van Aerschot A, Herdewijn P, Helv. Chim. Acta 2002, 85, 2258–2283; [Google Scholar]; d) Miyanishi H, Takahashi T, Mihara H, Bioconjug. Chem 2004, 15, 694–698; [DOI] [PubMed] [Google Scholar]; e) Takahashi T, Hamasaki K, Ueno A, Mihara H, Bioorg. Med. Chem 2001, 9, 991–1000; [DOI] [PubMed] [Google Scholar]; f) Du X, Zhou J, Li X, Xu B, Interface Focus 2017, 7, 20160116; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Tomassi S, Ieranò C, Mercurio ME, Nigro E, Daniele A, Russo R, Chambery A, Baglivo I, Pedone PV, Rea G, Napolitano M, Scala S, Cosconati S, Marinelli L, Novellino E, Messere A, Di Maro S, Bioorg. Med. Chem 2018, 26, 2539–2550; [DOI] [PubMed] [Google Scholar]; h) Yuan D, Du X, Shi J, Zhou N, Zhou J, Xu B, Angew. Chem. Int. Ed 2015, 54, 5705–5708; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Li X, Kuang Y, Lin H-C, Gao Y, Shi J, Xu B, Angew. Chem. Int. Ed 2011, 50, 9365–9369; [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Roviello GN, Musumeci D, Bucci EM, Pedone C, Int. J. Pharm 2011, 415, 206–210; [DOI] [PubMed] [Google Scholar]; k) Roviello GN, Musumeci D, RSC Adv 2016, 6, 63578–63585; [DOI] [PMC free article] [PubMed] [Google Scholar]; l) Murai K, Inagaki K, Hiraoka C, Minoshima S, Kinoshita T, Nagata K, Higuchi M, CrystEngComm 2019, 21, 3557–3567; [Google Scholar]; m) Musumeci D, Mokhir A, Roviello GN, Bioorganic Chem 2020, 100, 103862; [DOI] [PubMed] [Google Scholar]; n) Scognamiglio PL, Platella C, Napolitano E, Musumeci D, Roviello GN, Molecules 2021, 26, 3558; [DOI] [PMC free article] [PubMed] [Google Scholar]; o) Boback K, Bacchi K, O’Neill S, Brown S, Dorsainvil J, Smith-Carpenter JE, Molecules 2020, 25, 5493; [DOI] [PMC free article] [PubMed] [Google Scholar]; p) Kumar Bandela Anil, Nathaniel Wagner, Hava Sadihov, Sara Morales-Reina, Agata Chotera-Ouda, Kingshuk Basu, Rivka Cohen-Luria, Andrés de la Escosura, Gonen Ashkenasy, Proc. Natl. Acad. Sci 2021, 118, e2015285118;33622789 [Google Scholar]; q) Roviello GN, Musumeci D, Bucci EM, Pedone C, Mol. Biosyst 2011, 7, 1073–1080; [DOI] [PubMed] [Google Scholar]; r) Roviello G, Vicidomini C, Di Gaetano S, Capasso D, Musumeci D, Roviello V, RSC Adv 2016, 6, 14140–14148; [DOI] [PMC free article] [PubMed] [Google Scholar]; s) Musumeci D, Roviello V, Roviello GN, Int. J. Nanomedicine 2018, 13, 2613–2629; [DOI] [PMC free article] [PubMed] [Google Scholar]; t) Tomassi S, Montalban FF, Russo R, Novellino E, Messere A, Di Maro S, Symmetry 2019, 11, 567. [Google Scholar]
- [10].Li X, Ma S, Yi C, Curr. Opin. Chem. Biol 2016, 33, 108–116. [DOI] [PubMed] [Google Scholar]
- [11].Zhao Y, Dunker W, Yu Y-T, Karijolich J, Front. Bioeng. Bio.technol 2018, 6, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Charette M, Gray MW, IUBMB Life 2000, 49, 341–351. [DOI] [PubMed] [Google Scholar]
- [13].a) Aycock RA, Vogt DB, Jui NT, Chem. Sci 2017, 8, 7998–8003; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Constantin T, Zanini M, Regni A, Sheikh NS, Juliá F, Leonori D, Science 2020, 367, 1021–1026; [DOI] [PubMed] [Google Scholar]; c) de Bruijn AD, Roelfes G, Chem. Eur. J 2018, 24, 11314–11318; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Bottecchia C, Noël T, Chem. Eur. J 2019, 25, 26–42; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Faraggi TM, Rouget-Virbel C, Rincón JA, Barberis M, Mateos C, García-Cerrada S, Agejas J, de Frutos O, MacMillan DWC, Org. Process Res. Dev 2021, 25, 1966–1973; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Zhang O, Schubert JW, J. Org. Chem 2020, 85, 6225–6232; [DOI] [PubMed] [Google Scholar]; g) Wang J, Shao Z, Tan K, Tang R, Zhou Q, Xu M, Li YM, Shen Y, J. Org. Chem 2020, 85, 9944–9954; [DOI] [PubMed] [Google Scholar]; h) Seo H, Katcher MH, Jamison TF, Nat. Chem 2017, 9, 453–456; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Shatskiy A, Axelsson A, Stepanova EV, Liu J-Q, Temerdashev AZ, Kore BP, Blomkvist B, Gardner JM, Dinér P, Kärkäs MD, Chem. Sci 2021, 12, 5430–5437; [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Yin H, Zheng M, Chen H, Wang S, Zhou Q, Zhang Q, Wang P, J. Am. Chem. Soc 2020, 142, 14201–14209; [DOI] [PMC free article] [PubMed] [Google Scholar]; k) Ji P, Zhang Y, Dong Y, Huang H, Wei Y, Wang W, Org. Lett 2020, 22, 1557–1562; [DOI] [PMC free article] [PubMed] [Google Scholar]; l) Shah AA, Kelly MJ, Perkins JJ, Org. Lett 2020, 22, 2196–2200; [DOI] [PubMed] [Google Scholar]; m) Reich D, Trowbridge A, Gaunt MJ, Angew. Chem. Int. Ed 2020, 59, 2256–2261; [DOI] [PubMed] [Google Scholar]; n) Trowbridge A, Reich D, Gaunt MJ, Nature 2018, 561, 522–527; [DOI] [PubMed] [Google Scholar]; o) Merkens K, Aguilar Troyano FJ, Djossou J, Gómez-Suárez A, Adv. Synth. Catal 2020, 362, 2354–2359; [Google Scholar]; p) Sim J, Campbell MW, Molander GA, ACS Catal 2019, 9, 1558–1563; [DOI] [PMC free article] [PubMed] [Google Scholar]; q) Rossolini T, Ferko B, Dixon DJ, Org. Lett 2019, 21, 6668–6673; [DOI] [PubMed] [Google Scholar]; r) Rossolini T, Leitch JA, Grainger R, Dixon DJ, Org. Lett 2018, 20, 6794–6798; [DOI] [PubMed] [Google Scholar]; s) Aycock RA, Pratt CJ, Jui NT, ACS Catal 2018, 8, 9115–9119; [Google Scholar]; t) van Lier RCW, de Bruijn AD, Roelfes G, Chem. Eur. J 2021, 27, 1430–1437; [DOI] [PMC free article] [PubMed] [Google Scholar]; u) Josephson B, Fehl C, Isenegger PG, Nadal S, Wright TH, Poh AWJ, Bower BJ, Giltrap AM, Chen L, Batchelor-McAuley C, Roper G, Arisa O, Sap JBI, Kawamura A, Baldwin AJ, Mohammed S, Compton RG, Gouverneur V, Davis BG, Nature 2020, 585, 530–537; [DOI] [PubMed] [Google Scholar]; v) Brandhofer T, Mancheño OG, ChemCatChem 2019, 11, 3797–3801; [Google Scholar]; w) Yamamoto H, Wu A, Synfacts 2020, 16, 1372. [Google Scholar]
- [14].Immel JR, Chilamari M, Bloom S, Chem. Sci 2021, 12, 10083–10091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Knapp DM, Gillis EP, Burke MD, J. Am. Chem. Soc 2009, 131, 6961–6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chilamari M, Immel JR, Bloom S, ACS Catal 2020, 10, 12727–12737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wang H, Li W-G, Zeng K, Wu Y-J, Zhang Y, Xu T-L, Chen Y, Angew. Chem. Int. Ed 2019, 58, 561–565. [DOI] [PubMed] [Google Scholar]
- [18].Zhang L, Floyd BM, Chilamari M, Mapes J, Swaminathan J, Bloom S, Marcotte EM, Anslyn EV, ACS Chem. Biol 2021, 16, 2595–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Li BX, Kim DK, Bloom S, Huang RY-C, Qiao JX, Ewing WR, Oblinsky DG, Scholes GD, MacMillan DWC, Nat. Chem 2021, 13, 902–908. [DOI] [PubMed] [Google Scholar]
- [20].Tower SJ, Hetcher WJ, Myers TE, Kuehl NJ, Taylor MT, J. Am. Chem. Soc 2020, 142, 9112–9118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Taylor MT, Nelson JE, Suero MG, Gaunt MJ, Nature 2018, 562, 563–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kim J, Li BX, Huang RY-C, Qiao JX, Ewing WR, MacMillan DWC, J. Am. Chem. Soc 2020, 142, 21260–21266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Chen X, Ye F, Luo X, Liu X, Zhao J, Wang S, Zhou Q, Chen G, Wang P, J. Am. Chem. Soc 2019, 141, 18230–18237. [DOI] [PubMed] [Google Scholar]
- [24].Huvaere K, Skibsted LH, J. Am. Chem. Soc 2009, 131, 8049–8060. [DOI] [PubMed] [Google Scholar]
- [25].Wang S, Zhou Q, Zhang X, Wang P, Angew. Chem. Int. Ed 2022, 61, e202111388. [DOI] [PubMed] [Google Scholar]
- [26].Bloom S, Liu C, Kölmel DK, Qiao JX, Zhang Y, Poss MA, Ewing WR, MacMillan DWC, Nat. Chem 2018, 10, 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Forster S, Thumser AE, Hood SR, Plant N, PLOS ONE 2012, 7, e33253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chen YY, Wood AW, Bioelectromagnetics 2009, 30, 583–590. [DOI] [PubMed] [Google Scholar]
- [29].Oyekanmi AA, Ahmad A, Hossain K, Rafatullah M, J. Mol. Liq 2019, 281, 48–58. [Google Scholar]
- [30].Seath CP, Vogt DB, Xu Z, Boyington AJ, Jui NT, J. Am. Chem. Soc 2018, 140, 15525–15534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zeman CJ, Kim S, Zhang F, Schanze KS, J. Am. Chem. Soc 2020, 142, 2204–2207. [DOI] [PubMed] [Google Scholar]
- [32].Szostak M, Sautier B, Spain M, Procter DJ, Org. Lett 2014, 16, 1092–1095. [DOI] [PubMed] [Google Scholar]
- [33].Capperucci A, Tanini D, Chemistry 2022, 4, 77–97. [Google Scholar]
- [34].Kozak A; Shapiro I Novel non-coding heterocyclic amino acids (nchaa) and their use as herbicides, 2020, WO/2020/250223.
- [35].Zhang W, Xiang X-X, Chen J, Yang C, Pan Y-L, Cheng J-P, Meng Q, Li X, Nat. Commun 2020, 11, 638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Reha-Krantz LJ, Hariharan C, Subuddhi U, Xia S, Zhao C, Beckman J, Christian T, Konigsberg W, Biochemistry 2011, 50, 10136–10149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Leconte AM, Hwang GT, Matsuda S, Capek P, Hari Y, Romesberg FE, J. Am. Chem. Soc 2008, 130, 2336–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Xu Y, Wang Y, Liu P, Chu G-C, Xu H, Li Y-M, Wang J, Shi J, Org. Biomol. Chem 2018, 16, 7036–7040. [DOI] [PubMed] [Google Scholar]
- [39].Troy CM, Stefanis L, Prochiantz A, Greene LA, Shelanski ML, Proc. Natl. Acad. Sci 1996, 93, 5635–5640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kalmodia S, Vandhana S, Tejaswini Rama BR, Jayashree B, Sreenivasan Seethalakshmi T, Umashankar V, Yang W, Barrow CJ, Krishnakumar S, Elchuri SV, Cancer Nanotechnol 2016, 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Stoeltzing O, Liu W, Reinmuth N, Fan F, Parry GC, Parikh AA, McCarty MF, Bucana CD, Mazar AP, Ellis LM, Int. J. Cancer 2003, 104, 496–503. [DOI] [PubMed] [Google Scholar]
- [42].Shinitzky M, Fridkin M, Biochim. Biophys. Acta 1976, 434, 137–143. [DOI] [PubMed] [Google Scholar]
- [43].Millar RP, Flanagan CA, Milton RC, King JA, J. Biol. Chem 1989, 264, 21007–21013. [PubMed] [Google Scholar]
- [44].Porta R, Benaglia M, Puglisi A, Org. Process Res. Dev 2016, 20, 2–25. [Google Scholar]
- [45].Pastre JC, Browne DL, Ley SV, Chem. Soc. Rev 2013, 42, 8849–8869. [DOI] [PubMed] [Google Scholar]
- [46].Quartararo AJ, Gates ZP, Somsen BA, Hartrampf N, Ye X, Shimada A, Kajihara Y, Ottmann C, Pentelute BL, Nat. Commun 2020, 11, 3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Bottecchia C, Erdmann N, Tijssen PMA, Milroy L-G, Brunsveld L, Hessel V, Noël T, ChemSusChem 2016, 9, 1781–1785. [DOI] [PubMed] [Google Scholar]
- [48].Talla A, Driessen B, Straathof NJW, Milroy L-G, Brunsveld L, Hessel V, Noël T, Adv. Synth. Catal 2015, 357, 2180–2186. [Google Scholar]
- [49].Emmanuel N, Mendoza C, Winter M, Horn CR, Vizza A, Dreesen L, Heinrichs B, Monbaliu J-CM, Org. Process Res. Dev 2017, 21, 1435–1438. [Google Scholar]
- [50].Bottecchia C, Rubens M, Gunnoo SB, Hessel V, Madder A, Noël T, Angew. Chem. Int. Ed 2017, 56, 12702–12707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Vara BA, Li X, Berritt S, Walters CR, Petersson EJ, Molander GA, Chem. Sci 2018, 9, 336–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Su J, Mo J-N, Chen X, Umanzor A, Zhang Z, Houk KN, Zhao J, Angew. Chem. Int. Ed 2022, 61, e202112668. [DOI] [PubMed] [Google Scholar]
- [53].Pischel U, Zhang X, Hellrung B, Haselbach E, Muller P-A, Nau WM, J. Am. Chem. Soc 2000, 122, 2027–2034. [Google Scholar]
- [54].Yoshioka E, Kohtani S, Jichu T, Fukazawa T, Nagai T, Kawashima A, Takemoto Y, Miyabe H, J. Org. Chem 2016, 81, 7217–7229. [DOI] [PubMed] [Google Scholar]
- [55].Montalti M, Credi A, Prodi L, & Gandolfi MT Handbook of Photochemistry (3rd ed.). 2006, CRC Press. [Google Scholar]
- [56].O’Reilly JE, Elving PJ, J. Electroanal. Chem 1977, 75, 507–522. [Google Scholar]
- [57].DeLaive PJ, Foreman TK, Giannotti C, Whitten DG, J. Am. Chem. Soc 1980, 102, 5627–5631. [Google Scholar]
- [58].Nicastri MC, Lehnherr D, Lam Y, DiRocco DA, Rovis T, J. Am. Chem. Soc 2020, 142, 987–998. [DOI] [PubMed] [Google Scholar]
- [59].Wang M, Wang C, Huo Y, Dang X, Xue H, Liu L, Chai H, Xie X, Li Z, Lu D, Xu Z, Nat. Commun 2021, 12, 6873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Yurchenko O, Freytag D, zur Borg L, Zentel R, Heinze J, Ludwigs S, J. Phys. Chem. B 2012, 116, 30–39. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.