

Abstract
All human diseases involve proteins, yet our current tools to characterize and quantify them are limited. To better elucidate proteins across space, time, and molecular composition, we provide a >10 years of projection for technologies to meet the challenges that protein biology presents. With a broad perspective, we discuss grand opportunities to transition the science of proteomics into a more propulsive enterprise. Extrapolating recent trends, we describe a next generation of approaches to define, quantify, and visualize the multiple dimensions of the proteome, thereby transforming our understanding and interactions with human disease in the coming decade.
Keywords: proteins, proteomics, single-cell biology, biotechnology, single-molecule sequencing
Abbreviations: ECM, extracellular matrix; HuBMAP, Human BioMolecular Atlas Program; MS, mass spectrometry; PTM, post-translational modification
Graphical Abstract
Highlight
-
•
How functional proteomics tools, methods, and datasets can be integrated to accelerate the development of comprehensive spatiomolecular tissue maps that generate new biological insights?
In Brief
Capturing the biology of proteins will require improved technologies to readout their composition in space and time. Developing these improved technologies presents a major opportunity for biomedical research. How might we proceed in the decades ahead?
The Proteome is Extremely Complex
Proteins are the primary conduit connecting our genes to complex traits including the diseases that afflict us. While the genome provides the “biological script,” proteins run and fill it with life. Given this critical role, the enormous complexity of the proteome, and the pace of technology development, we highlight strategies and opportunities for proteomics in the coming decades. Compared with the deep coverage of DNA in the genome through sequencing, our ability to detect proteins achieves only ∼0.1× coverage of the proteome (1). To fill this proteomic gap in the coming years, we argue for accelerated technology development, concerted efforts across laboratories, and large consortia with meaningful data integration. These advances will need to match the complexity of our biology, enabling the emergence of new enterprises to boost detection and develop interventions that will reliably extend our life spans and quality of life.
The immense complexity of the proteome arises from the fact that each gene produces several protein variants, termed “proteoforms,” far beyond what can be predicted from DNA sequence (Fig. 1). The proteoform paradigm describes the composition of protein molecules arising from ∼250 types of post-translational modifications (PTMs) (2), alternative transcriptions or translation start sites, splicing, or amino acid changes (Fig. 1). A single cell easily encompasses hundreds of thousands of different proteoforms reflecting the true chemical diversity of protein molecules (3, 4), each having the potential to alter structure, function, and interactions. While the number of theoretically possible proteoforms is extremely large (3, 4), existing methods detect fewer than 10,000 proteoforms in a typical cell sample.
Fig. 1.

From human genes (far left), diverse forms of mature endogenous protein molecules are expressed (far right). Technologies are needed to fully measure the dynamic set of ∼1 million distinct proteoforms expressed in each cell (see the study by Aebersold et al. (4)).
Given the complexity of proteins, it is unsurprising that RNA abundance or ribosome-bound mRNA is imperfect surrogate for protein abundance. RNA abundance ranges over ∼4 orders of magnitude in cells, whereas proteoforms range over ∼7 orders of magnitude and even over 10 in body fluids (5). Researchers use diverse proteomic technologies to identify and quantify proteins, and the analytical sensitivity, reproducibility, and specificity of platforms are extensive. These tools enable both discovery experiments (where emphasis is on identifying large numbers of proteins) and targeted measurements that focus on individual proteins, pathways, and/or certain classes such as membrane proteins. Ongoing innovation in spatial proteomics and multiplexing of compositional proteomics continue to improve our ability to probe the approximately half of the 20,000 genes expressed into detectable proteins in a given cell type. These developments have substantially advanced our functional and mechanistic insights of proteins (6).
Looking forward, one significant goal in the coming decades is to increase the efficiency of and access to different proteomic platforms while also decreasing cost. To do so, we need to prioritize technology development to deliver diverse data types of high value to the community. Efforts are underway; for example, mapping projects that use multiplexed protein measurements with single-cell resolution via antibody-based tissue imaging. The Human Protein Atlas (7) and the Human BioMolecular Atlas Program (HuBMAP) (8) are examples of such efforts. But, if proteomics are to capture biological complexity in its dynamics and relationships across scales, we need to upgrade our ability to sample proteoforms spatially, their complexes in organelles, and in regions of phase transition to make functional assertions with high confidence. Such effort will require creating technologies capable of molecular deconvolution from millimeters to nanometers and their temporal dynamics from seconds to the human life span.
Another significant goal will involve integration of such proteoform data with other multiomic data. Such “spatial multiomics data” will allow us to infer emergent properties of whole organisms, rather than investigating one aspect at a time. Integration is still challenging because of missing values, different properties of different data types, and the fact that defining precise proteoforms is challenging. Still, we believe that breakthroughs for these two goals—new technology combined with meaningful data integration—are achievable within the next 10 years.
PTMs are Major Drivers of Proteoform Diversity
Top–Down Proteomics
A first step toward comprehensively understanding proteome complexity is the efficient detection of proteoforms and their PTMs. PTMs are one of the major routes to produce proteoforms, and many proteins have multiple modifications, but it is still unclear which of these PTMs are functional and which are passengers or “bystanders.”
With “top–down” mass spectrometry (MS), intact proteoforms and their assemblies are analyzed, thereby directly informing on co-occurring events (i.e., PTMs, isoforms, or mutations). This approach has some advantages over peptide-centric “bottom–up” approaches described later, including (9) resolving the protein inference problem (10), identifying isoforms directly, and retaining both stoichiometry and the combinatorial occurrence of PTMs (Fig. 2) (9). Because so many questions about PTMs remain unanswered, expanding this approach will improve the efficiency of detection and assignment of function to proteoforms and their PTMs (i.e., single-cell measurements). However, our current ability to measure proteoforms at single-cell resolution is low and requires major leaps in technology development, that is, higher sensitivity and spatial as well as temporal resolution. Proteoform knowledge will also accelerate the development of disruptive technologies to improve biomarker discovery in translational proteomics by more accurately annotating the chemical diversity of protein variation in disease models and patient cohorts.
Fig. 2.
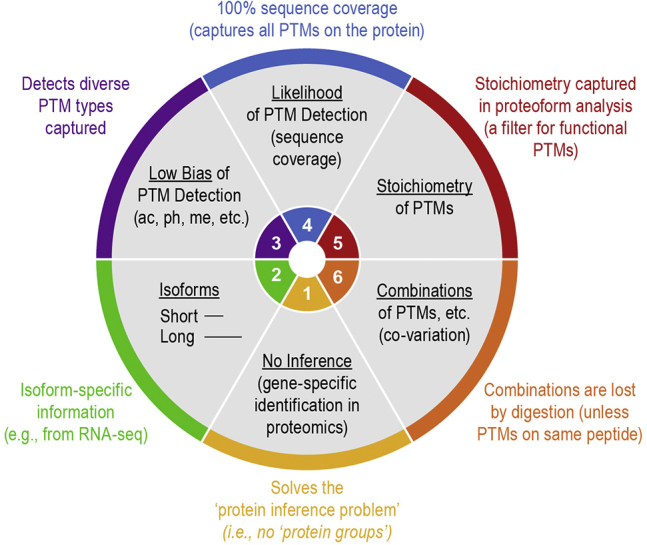
Proteoform measurement fills knowledge gaps. This figure details specific advantages of top–down measurements of intact proteins. Measuring proteoforms will improve molecular precision in asserting protein sequence and composition (i.e., primary structure). Such measurements should improve the efficiency of detection and assignment of function to all protein variants and their modifications in the coming decade.
Bottom–up Proteomics
In detecting thousands of peptides and PTMs efficiently, bottom–up proteomics uses proteases to digest the proteome and is the current “to-go-to” platform for discovery proteomics. Unlike whole proteins, many peptides are easily fractionated, ionized, and fragmented via liquid chromatography–tandem MS methods. In addition, several options exist for collecting information on peptides and modified peptides and for infusing spatial information (Fig. 3, upper right) (11). However, data from bottom–up approaches remain limited in sequence coverage of individual proteins and coverage of the single-cell proteome. For example, because of the protein inference problem, biological signals are masked by combining peptides into a single protein quantity, losing information on combinatorial effects (12). Targeted MS assays, such as selected reaction monitoring, can precisely identify peptide sequences and specific PTMs to achieve absolute quantification and probe known proteoforms (13, 14). Future developments should continue by combining top–down with bottom–up strategies in an integrated approach for robust quantitation of isoforms and PTMs.
Fig. 3.

Multiple approaches for assessment of proteins in single cells. Antibody specificity dictates the protein detection specificity for technologies depicted in orange. bottom–up Mass Spectrometry, measures peptides; CODEX, codetection by antibody indexing; CyTOF, mass cytometry; nanoDESI, spatially resolved desorption by electrospray; PEA, proximity extension assay; PLAYR, proximity ligation assay for RNA; REAP, a rapid, efficient, and practical cell processing method; sc, single cell; top–down mass spectrometry, measures proteoforms.
Proteomics Goes Single Cell (and Single Molecule)
Single-Cell Proteomics
Tissues are heterogeneous mosaics of cells in different physiological stages, requiring single-cell analysis. However, given that we cannot amplify proteins and their concentrations cover a large dynamic range, single-cell proteomics is challenging. Some single-cell proteomics methods employing antibodies or other affinity reagents have existed for decades (Fig. 3, left). While these methods have improved significantly in power and sophistication (15), the newest developments via bottom–up MS have clearly expanded the horizon for single-cell proteomics (16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27).
Over the last 4 years, mass spectrometric methods have advanced from quantifying a few 100 proteins per cell to quantifying more than 1000 proteins per cell, with thousands of proteins quantified across datasets at a throughput of >200 single cells per day (Fig. 3, upper right) (28, 29). One enabling technology making such in-depth single-cell and “low-input” proteome profiling possible is multiplexed analysis of single cells labeled with isobaric mass tags and combined with an isobaric carrier (26) to enhance throughput and MS/MS peptide sequence identification (30). Miniaturization of sample preparation volumes to nanoliter or low-microliter levels has minimized surface losses and enhanced reaction kinetics (31, 32). In addition, miniaturization of liquid chromatography (25) and capillary electrophoresis (33) separations have boosted prospects, even adding peptide selectivity to the workflow (24, 27). However, future approaches still need to achieve higher coverage and throughput more comparable to those of single-cell RNA-Seq. We posit that this will be achievable given the rate of improvement through advances in different steps of the workflow. Integration of proteomics data with other single-cell modalities, such as small conditional RNA-Seq, have already provided initial results (16, 20, 28). We imagine that within the next 5 years, proteomics will cover >5000 proteins from thousands of single cells per day, with linked information on transcripts.
Single-Molecule Sequencing
Another promising area of technology development includes single-molecule protein sequencing and proteoform detection (34, 35, 36, 37). These approaches have the potential to be disruptive and to impact single-cell protein analysis, with sequencing demonstrated only on synthetic peptides thus far. Intriguingly, there have been major increases in private sector investments in this general area over the past year, creating a palpable sense that this could be the decade of breakthroughs in single-molecule protein measurement and sequencing.
Such technologies operative on single molecules will encounter the challenge of proteoform diversity expressed in human biology (Fig. 1). Therefore, they would benefit from better general compositional definition of the expressed proteome, prior to investigating the existence of different proteoforms in individual cells. Indeed, amassing a reference set of human proteoforms has been proposed (6, 38, 39) using both cell- and gene-based approaches (39). Using output of the Human Cell Atlas, HuBMAP, and other consortia, comprehensive maps of human cell types will allow the proteome of each cell type to be defined. Assuming, 5000 cell types and 1 million proteoforms in each cell type, such a human proteoform atlas would involve approximately 5 billion measurements, with perhaps approximately 50 million unique proteoforms needing to be asserted and mapped in the coming decade (39). Such efforts will only be possible based on the output of current atlasing efforts, providing reference sets of what proteoforms are actually present and where.
Single-Cell and Single-Molecule Immunoassays
Complementing methods for proteome-wide characterization, targeted assays using affinity reagents can accurately detect and quantify specific proteins. While often targeting fewer proteins, they achieve far higher throughput than nontargeted approaches. Furthermore, many proteins of interest in the human proteome are below the limit of detection of current MS methods and conventional immunoassays. One major group of targeted approaches includes immunoassays that are designed to detect known protein targets, with target identities informed by a priori discovery proteomics or driven by biological hypotheses. At the single-cell level, the formats are diverse and include immunohistochemistry, flow cytometry, mass cytometry, and various formats for multiplexed antibody-based imaging. Arrays of affinity reagents are also available for limited operation in discovery mode; use of these platforms is likely to increase in the coming decades.
Over the last 2 decades, miniaturization of single-cell immunoassays has improved to the point where proteins in thousands of samples can now be processed in parallel, while using far less time, sample, and reagents. Performance improvements stem from the underlying physics at miniature scale, and microfluidic approaches have enabled new assay designs (40). Microdevices afford scalable means to precisely compartmentalize individual cells, with the use of both droplets (41) and microwells being common strategies (42). Furthermore, ultrasensitive single-molecule technologies already access proteins at picomolar and femtomolar concentrations in the single-protein regime using digital immunoassays like SiMPull (43), SiMoAs (44), and droplet-based assays (45, 46). While not providing proteoform-level specificity, these single-cell frameworks are powerful for multimodal same-cell measurements (47, 48). Scrutinizing the protein content of lysates from thousands of individual organelles is viable today using microfluidic devices to overcome shortcomings of single capillary systems for single-cell (49, 50), subcellular (51), and multimodal (52, 53) immunoblotting.
Given new developments in droplet microfluidics, microfluidic imaging cytometry (41), and subcellular immunoblotting, we envision targeted single-cell proteomics to push two new frontiers: (1) monitoring protein expression in specific cellular compartments and (2) multimodal single-cell analyses that integrate proteomics with other omics data. While selective target detection and antibody availability remain an inherent limitation of immunoassays, we foresee a tremendous opportunity for immunoassays to become proteoform specific across diverse cells. We posit that clever immunoassay strategies that enhance analytical specificity (54, 55) will help translate proteoform knowledge into scalable single-molecule assays. We project that by 2030, innovations in this area will move such analyses to cost-effective benchtop instruments usable in diverse settings. Analysis of thousands of individual cells per assay, with specificity for hundreds of currently undiscernible proteoforms within each cell, is possible. Absolute, not simply relative, quantification of each proteoform beckons.
Availability and Validity of Affinity Reagents
Many assays in basic and clinical research rely on antibodies. However, there are proposed (56), but no widely accepted guidelines to determine the validity of these reagents. Furthermore, many recent publications have highlighted that commercial antibodies can fail to detect the intended target in diverse sample types. (57, 58) The utility in biological and biomedical research of affinity reagents for characterizing and quantifying proteins and peptides is central; however, it also should elevate the challenge in validating and reproducing many of the antibody-based assays published each year. For this purpose, increasing effort will be required to improve the rigor and reproducibility of antibody-based assays, (59), e.g. the antibody registries specific and unique identifiers for reagents. (60).
The proteomics field would greatly benefit from such antibody database and library, in particular if it is open source. Antibodies are expensive, and reducing their cost through a public database would allow researchers to devote more of their funding to other materials and maximize findings. To enhance reproducibility and operational efficiency, recombinant antibodies with known sequences would create a long-lasting and reproducible resource across laboratories (59). Increasingly, antibody sequencing and characterization using proteomics is on the rise (61, 62), making an open-source library of antibodies even more feasible in the coming decade.
Another important resource for proteomics of the future would be a proteoform-specific library of affinity reagents covering as many products of human genes as possible. Moving forward, we should promote gene-specific probes and create a generation of proteoform-aware PTM-recognizing affinity reagents (e.g., phosphorylation site–specific antibody reagents) (63). Creating such a resource will be a massive undertaking, but limited scope pilots in which antibodies to known proteoforms of key protein hubs in cellular regulatory networks (c-Myc, p53, NFF06BB, histones, etc) would be a very valuable resource and important next step. Construction of such a library of proteoform-specific antibodies, with known epitope-binding specificity, critically depends on the reproducibility, specificity, and scalability with which multiple antibodies can probe multiple epitopes and PTMs on proteoforms.
Thinking Outside the Cell: Extracellular Matrix, Body Fluids, and Extracellular Communication
Not only does one need to understand the single-cell proteome but also it is necessary to quantify the cellular environment and context. Quantification and defining extracellular communication and the cellular environment is required to infer tissue and organ function. The extracellular matrix (ECM) contains highly glycosylated and crosslinked proteins that form the scaffold of cellular anchorage and migration (64, 65). The ECM also liberates and sequesters secreted molecules such as growth factors (66). It plays critical roles in development, health, and disease via mechanical and chemical signal transduction. While recent proteomic methods tailored to biochemically unique ECM proteins have led to greater understanding of the ECM proteomes (67, 68, 69, 70, 71) in many tissues and organs, these methods cannot fully capture the broad dynamic range of the ECM from low-abundance growth factors to hyperabundant collagens (72). New imaging MS methods using collagenase digestions targeted to the ECM are beginning to address some spatial–temporal aspects (73, 74). To achieve near-complete coverage and single cell–level profiling of the matrisome and to capture its dynamics, our community will need to (1) expand methods to identify and isolate specific cell and tissue niches (e.g., laser-capture microdissection (75) and imaging MS), (2) develop reagents to enrich for ECM proteins or classes of ECM proteins (e.g., high-affinity antibodies against ECM-specific PTMs or splice variants), (3) use ECM-specific proteases such as elastase and collagenases instead of trypsin, and (4) apply adequate data analysis workflows (76, 77).
Beyond the ECM, the extracellular milieu contains protein-rich fluids (78, 79, 80) (e.g., tissue interstitial fluids, serum/plasma, urine, saliva, etc) that present unique challenges when it comes to proteomic profiling. Extracellular vesicles and their protein contents are key mediators of cell communications and regulators of cellular functions (81, 82), including those that can affect “in trans” chemoresistance and angiogenesis in cancer (83). An important activity for advancing proteomics will be to convert ultratrace analysis into a straightforward deep characterization of extracellular proteomes and to correlate the resulting information with their cells of origin.
The Future Lies in Integration Across Scales
Spatial Information
Be it through networks or other models, future efforts will have to integrate compositional, interactomics, and spatial information on proteoforms to reveal how the proteome drives human physiology. The proteomics toolkit offers a variety of methods for detecting protein–protein interactions that range from direct detection, such as affinity-purification MS, to less direct, such as correlational profiling. These approaches are relatively low throughput, whereas we need high-throughput methods for sensitive analysis of protein–protein interactions in primary tissues. We put forward a reductionist view in which enough spatial information—in conjunction with other omics data—can define the state, function, and trajectory of a cell within the tissue microenvironment. Approximately 20 spatial proteomics technologies are currently used within the proteomics field (54, 84, 85, 86, 87, 88, 89, 90, 91); these approaches can be targeted and multiplexed (e.g., using antibodies) or nontargeted (e.g., MS). Spatial technologies can measure specific protein–protein interactions, subcellular structures, individual cells, and tissue microenvironments within whole organs.
Imaging MS (85, 89, 92) assesses large numbers of glycans, lipids, and metabolites at spatial resolutions of 10 microns (85, 93, 94). At the proteomics level, imaging MS can currently identify >2000 proteins at spatial resolutions of 100 microns (89). Using different proteases and glycosidases, imaging MS also can be integrated with, for example, microscopy of the ECM to evaluate multiple analytes sequentially on the same tissue slice (73, 95). A future challenge will lie in applying these methods to resolve proteoforms down to 1 micron. To do so, major bottlenecks like low proteome coverage (especially for large and low abundance proteins) will need to be overcome.
Other imaging MS approaches we envision would combine artificial intelligence–powered image analysis with automated single-cell laser microdissection and ultrahigh sensitivity MS to link protein abundance to complex cellular or subcellular phenotypes while preserving spatial context. They have successfully identified rare cell states with distinct morphology (96). Spatial proteomics approaches are also commonly combined with magnetic resonance/computed tomography imaging data to assess anatomical features, stained microscopy to assess tissue morphology, multiplexed immunofluorescence approaches to differentiate cell types, and fluorescence in situ hybridization to observe DNA- and RNA-mediated influences. For example, multimodal imaging analyses of the murine spleen and pancreas have resolved pixel-level cellular populations and subregions (94, 97).
Another future challenge with imaging MS lies in tackling subcellular resolution. Exploiting fluorescence-based technologies, multiplexed protein imaging can now detail cell subtypes (Fig. 4). In the future, these protein imaging efforts could also include RNA information. We imagine that the future will bring a new kind of three-dimensional “AtomScope” capable of measuring every atom in a model cell, then reconstructing the structure of single molecules and their assemblies in a bottom–up process. As with single-cell and single-molecule proteomics, such aspirations will benefit from higher quality reference proteomes, structural predictors like AlphaFold, and a greatly improved baseline knowledge about proteoforms, their PTMs, and complexes. With meaningful data integration, models of the future will identify spatial dependencies of compositional data, correlate spatial patterns to specific outcomes, assess relationships between data types to enable predictions across them, and integrate data generated across bulk and single-cell scale. One early example lies in integration of protein imaging with protein–protein interaction data in the multiscale integrated cell model (98) that led to the identification of novel cellular systems.
Fig. 4.

Multiplexed spatial assays. Source: Ref. (8).
Meaningful Data Integration
Given the aforementioned developments, the way forward is to integrate different datasets in a biologically informed and statistically robust manner. Such integration will then detect emergent properties of the system as a whole (99) and consider the cell and its proteome as an “ecosystem” in which the sum is more than the parts. One popular method of integration exploits networks, with mathematical approaches available to understand and predict properties (Fig. 5A). Networks include both parts' lists and interactions among the parts. Constructing such networks will allow us to understand concepts such as the robustness of a system (the cell) to perturbations (i.e., mutations and loss of the protein) or efficiency and redundancy in signal transduction (i.e., through creation of short paths through a network that prioritize rapid response over versatility). Future work will accommodate different types of nodes (e.g., proteoforms) and different types of edges (e.g., kinase-target interactions or protein–protein interactions in a complex).
Fig. 5.

Capturing the multidimensional biology of our proteome.A, from a parts lists to interaction mapping and proteoform networks underlying cell-based biology. B, capturing time-resolved human biology at the protein level. Top, depiction of multiple spatiomolecular proteoform images collected over time. Bottom, temporal dynamics vary widely across disease progression (years) across cell differentiation (days) in human bone marrow and blood or through embryogenesis (months).
We propose that in the coming decades, “filling this network with life” will involve rethinking commonly used terms, for example, that of protein function. While functional descriptions are standardized in a controlled vocabulary such as Gene Ontology (100), we will need to identify more quantifiable definitions (e.g., by the profile of the genetic, physical, or regulatory interactions of a protein). Such definitions would also allow us to measure similarity and changes to proteoform function. For example, once a transcript is spliced, rearranged, and modified, the resulting different proteoforms are expected to mediate divergent functions or behave as alloforms, rather than retain similar functions, or acting as isoforms (101).
Other proteomics integration efforts will need to venture toward other molecule types, such as RNA, or metabolites that are already being captured with spatial resolution (Fig. 5). Existing efforts have already shown the power of such an approach to infer regulatory mechanisms and most tightly connected and therefore potentially most functionally important relationships. For example, integrating complementary RNA, protein, and metabolite in the Kidney Precision Medicine Project has generated many new hypotheses (102). More generalized efforts for multiomics data integration across space and time are exemplified by the Human Tissue Atlas Network (103).
Finally, we propose that the future of proteomics lies not only in integrative network approaches and redefining entities to render them quantifiable but also will include consideration of the combinatorial nature of biological processes. Traditionally, we study one protein or pathway at a time as the cell is otherwise too complex. Including combinatorial action of proteoforms under the same conditions, attacking the same problem (e.g., through aforementioned network approaches) will allow us to truly understand cells and functional tissue units as systems driving life processes such as those shown in Figure 5B (bottom). While constructing networks, quantifying protein activity, defining proteoforms, and considering combinatorial actions are being addressed in specific areas, one future of proteomics requires simultaneous implementation of all these approaches to develop a comprehensive predictive framework of cell and whole tissues for the first time.
Meeting the Scale of the Challenge
Given the vast challenge of capturing protein-level biology (depicted as a hypercube in Figs. 5B and 6A) and the state of measurement, we argue for an increased rate of progress in proteomics. Combinations of new accessible technologies need to provide synergistic data streams to move us beyond census taking and tenuous inferences to far greater understanding of how protein function and how they go awry. The future of proteomics clearly lies in expanding its “dimensions” and building stronger connections across the spatial, temporal, and compositional domains. To achieve major outcomes in biomedicine (Fig. 6C), we should move beyond partial and siloed information on proteins, to an increasingly integrated field (104) providing functional information on a bounded defined proteome.
Fig. 6.

Collaborative and big science models for approaching the human proteome in the decade ahead. Scope of the solution needs to match scope of the challenge, if we are to make interactions with the human proteome more deterministic for the goals of biomedical research, including regenerative medicine, more efficient drug development, and early detection of all types of human disease.
Science is inherently a multiscale endeavor (Fig. 6B). In addition to ground-breaking work in individual research laboratories, there are many initiatives building on and maximizing information gain at a larger scale, including the Human Cell Atlas (105), the Human Proteoform Project (39), the Human Proteome Project (6), the Human Protein Atlas (7), and HuBMAP (8), and many organ- or disease-specific consortia such as Kidney Precision Medicine Project (106), Human Tissue Atlas Network (103), and so on. Increasingly, consortia are elevating and integrating proteomics with related fields. Consortia have demonstrated science at a scale not possible in typical single-investigator laboratories, convening multidisciplinary communities and building resources in close collaboration with the research community. Currently, even large teams are only able to generate a small portion of the data needed to comprehensively capture the enigmatic proteome (Fig. 6A). Democratic access to adjacent atlasing projects is essential as well as a balance between building a multiscale scientific approach and implementation of new technologies, including respective training and adoption. However, many consortia are responding with agile management and investigator buy-ins to achieve these goals.
All human diseases involve proteins, and there is an exciting range of new tools providing new insights in protein composition, spatial patterning, and temporal dynamics. A multitude of technologies and multidisciplinary expertise are required (104), and extensive interactions between computational, statistical, clinical, and biology experts will be more important than ever. Proteomics needs to be deeply crossdisciplinary, providing the data and connections to start capturing the complexity of the cell, functional tissue units, and complex traits of an entire person. To accomplish this in proteomics, we need hardened platforms capable of powering quantitative models to reliably connect protein information to disease and unlock precise decision making (Fig. 6C). These hardened platforms will be enabled through a collection of tools, techniques, and statistical approaches. Tremendous advances are possible within the next decade but only with increased level of focus and intention: a “proteomic moonshot” will be needed that can reach this “criticality” with the human proteome.
The National Institutes of Health Office of Strategic Coordination convened a virtual meeting to foster discussion among experts on existing gaps and opportunities within the functional proteomics field. Organized by the National Institutes of Health Office of Strategic Coordination, in collaboration with Workshop Co-Chairs (K. E. B.-J. and N. L. K.) and HuBMAP-funded principal investigators, this meeting engaged proteomics and atlasing experts in discussions designed to (1) identify how functional proteomics tools, methods, and datasets can be integrated to accelerate the development of comprehensive spatiomolecular tissue maps that generate new biological insights and (2) develop a prospective publication that articulates a vision of the field, including state-of-the-art and near-future tools and methods, a roadmap for integrating multiple proteomics data types (e.g., spatial proteomics, single-cell proteomics), and an articulation of the most important knowledge gaps.
Conflict of interest
T. P. C. is a Thermo Fisher Scientific Advisory Board member and receives research funding from AbbVie, United States. A. E. H. receives research support from Thermo Fisher Scientific, United States. J. E. V. E. is a Bruker Software Development SAB member and receives support from Thermo Fisher Scientific, Sciex, Canada, Agilent, United States, CellenONE, and Neoteryx. Many authors are involved with private sector endeavors and actively manage competing financial interests. All the other authors declare no competing interests. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Acknowledgments
We thank Nathan Johnson (Pacific Northwest National Laboratory) and Michael Mullowney for assistance in generating figures. The opinions expressed in this article are the authors’ own and do not necessarily reflect the views of the National Institute of Diabetes and Digestive and Kidney Diseases.
Author contributions
K. E. B.-J., T. P. C., R. R. D., A. E. H., R. I., R. T. K., E. L., M. J. M., A. N., G. P. N., P. A. P., K. D. R., S. S., N. S., J. M. S., J. E. V. E., M. V., C. V., D. R. W., and N. L. H. conceptualization; K. E. B.-J., T. P. C., R. R. D., A. E. H., R. I., R. T. K., E. L., M. J. M., A. N., G. P. N., P. A. P., K. D. R., S. S., N. S., J. M. S., J. E. V. E., M. V., C. V., D. R. W., and N. L. H. writing–original draft.
Funding and additional information
C. V. was supported by awards R35GM127089, 75N93019C00052, and the Chan Zuckerberg Initiative; K. E. B.-J. was supported by award UH3 CA255132-03; N. L. K. by UH3 CA246635-02; G. P. N. by U54 HG010426-03; and J. M. S. U54 DK120058-03, all of which are part of the Human Biomolecular Atlas Program. A. N. was supported by R01 CA232517 and Catalyst Award C-088 from the Chicago Biomedical Consortium, United States. A. E. H. was supported by a National Institutes of Health Cancer Moonshot, United States grant (grant no.: R33CA225296) and as a Chan Zuckerberg Biohub Investigator. E. L. was supported by the KAW Foundation (grant nos.: 2016.0204 and 2018.0172), the Swedish Research Council, Sweden (grant no.: 2017-05327), and the Chan Zuckerberg Initiative, United States (grant no.: CZF2019-002448). N. S. was supported by DP2GM123497 and CZF2019-002424, and N. S. and N. L. K. were supported by Allen Distinguished Investigator awards through the Paul G. Allen Frontiers Group. R. T. K. was supported by R01 GM138931.
Footnotes
The authors were participants in an NIH Workshop on Functional and Integrative Proteomics.
Contributor Information
Kristin E. Burnum-Johnson, Email: kristin.burnum-johnson@pnnl.gov.
Neil L. Kelleher, Email: n-kelleher@northwestern.edu.
References
- 1.Ruggles K.V., Tang Z., Wang X., Grover H., Askenazi M., Teubl J., et al. An analysis of the sensitivity of proteogenomic mapping of somatic mutations and novel splicing events in cancer. Mol. Cell Proteomics. 2016;15:1060–1071. doi: 10.1074/mcp.M115.056226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Montecchi-Palazzi L., Beavis R., Binz P.A., Chalkley R.J., Cottrell J., Creasy D., et al. The PSI-MOD community standard for representation of protein modification data. Nat. Biotechnol. 2008;26:864–866. doi: 10.1038/nbt0808-864. [DOI] [PubMed] [Google Scholar]
- 3.Smith L.M., Kelleher N.L., Consortium for Top Down, P Proteoform: a single term describing protein complexity. Nat. Met. 2013;10:186–187. doi: 10.1038/nmeth.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aebersold R., Agar J.N., Amster I.J., Baker M.S., Bertozzi C.R., Boja E.S., et al. How many human proteoforms are there? Nat. Chem. Biol. 2018;14:206–214. doi: 10.1038/nchembio.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zubarev R.A. The challenge of the proteome dynamic range and its implications for in-depth proteomics. Proteomics. 2013;13:723–726. doi: 10.1002/pmic.201200451. [DOI] [PubMed] [Google Scholar]
- 6.Adhikari S., Nice E.C., Deutsch E.W., Lane L., Omenn G.S., Pennington S.R., et al. A high-stringency blueprint of the human proteome. Nat. Commun. 2020;11:5301. doi: 10.1038/s41467-020-19045-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thul P.J., Akesson L., Wiking M., Mahdessian D., Geladaki A., Ait Blal H., et al. A subcellular map of the human proteome. Science. 2017;356 doi: 10.1126/science.aal3321. [DOI] [PubMed] [Google Scholar]
- 8.Consortium H. The human body at cellular resolution: the NIH human biomolecular atlas program. Nature. 2019;574:187–192. doi: 10.1038/s41586-019-1629-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith L.M., Kelleher N.L. Proteoforms as the next proteomics currency. Science. 2018;359:1106–1107. doi: 10.1126/science.aat1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nesvizhskii A.I., Aebersold R. Interpretation of shotgun proteomic data: the protein inference problem. Mol. Cell Proteomics. 2005;4:1419–1440. doi: 10.1074/mcp.R500012-MCP200. [DOI] [PubMed] [Google Scholar]
- 11.Branon T.C., Bosch J.A., Sanchez A.D., Udeshi N.D., Svinkina T., Carr S.A., et al. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 2018;36:880–887. doi: 10.1038/nbt.4201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Plubell D.L., Kall L., Webb-Robertson B.J., Bramer L.M., Ives A., Kelleher N.L., et al. Putting humpty dumpty back together again: what does protein quantification mean in bottom-up proteomics? J. Proteome Res. 2022;21:891–898. doi: 10.1021/acs.jproteome.1c00894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang B., Whiteaker J.R., Hoofnagle A.N., Baird G.S., Rodland K.D., Paulovich A.G. Clinical potential of mass spectrometry-based proteogenomics. Nat. Rev. Clin. Oncol. 2019;16:256–268. doi: 10.1038/s41571-018-0135-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kennedy J.J., Yan P., Zhao L., Ivey R.G., Voytovich U.J., Moore H.D., et al. Immobilized metal affinity chromatography coupled to multiple reaction monitoring enables reproducible quantification of phospho-signaling. Mol. Cell Proteomics. 2016;15:726–739. doi: 10.1074/mcp.O115.054940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levy E., Slavov N. Single cell protein analysis for systems biology. Essays Biochem. 2018;62:595–605. doi: 10.1042/EBC20180014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu Y., Scheibinger M., Ellwanger D.C., Krey J.F., Choi D., Kelly R.T., et al. Single-cell proteomics reveals changes in expression during hair-cell development. Elife. 2019;8 doi: 10.7554/eLife.50777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu Y., Clair G., Chrisler W.B., Shen Y., Zhao R., Shukla A.K., et al. Proteomic analysis of single mammalian cells enabled by microfluidic nanodroplet sample preparation and ultrasensitive NanoLC-MS. Angew. Chem. Int. Ed. Engl. 2018;57:12370–12374. doi: 10.1002/anie.201802843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Williams S.M., Liyu A.V., Tsai C.F., Moore R.J., Orton D.J., Chrisler W.B., et al. Automated coupling of nanodroplet sample preparation with liquid chromatography-mass spectrometry for high-throughput single-cell proteomics. Anal. Chem. 2020;92:10588–10596. doi: 10.1021/acs.analchem.0c01551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsai C.F., Zhao R., Williams S.M., Moore R.J., Schultz K., Chrisler W.B., et al. An improved boosting to amplify signal with isobaric labeling (iBASIL) strategy for precise quantitative single-cell proteomics. Mol. Cell Proteomics. 2020;19:828–838. doi: 10.1074/mcp.RA119.001857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Specht H., Emmott E., Petelski A.A., Huffman R.G., Perlman D.H., Serra M., et al. Single-cell proteomic and transcriptomic analysis of macrophage heterogeneity using SCoPE2. Genome Biol. 2021;22:50. doi: 10.1186/s13059-021-02267-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schoof E.M., Furtwangler B., Uresin N., Rapin N., Savickas S., Gentil C., et al. Quantitative single-cell proteomics as a tool to characterize cellular hierarchies. Nat. Commun. 2021;12:3341. doi: 10.1038/s41467-021-23667-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Z.Y., Huang M., Wang X.K., Zhu Y., Li J.S., Wong C.C.L., et al. Nanoliter-scale oil-air-droplet chip-based single cell proteomic analysis. Anal. Chem. 2018;90:5430–5438. doi: 10.1021/acs.analchem.8b00661. [DOI] [PubMed] [Google Scholar]
- 23.Dou M., Clair G., Tsai C.F., Xu K., Chrisler W.B., Sontag R.L., et al. High-throughput single cell proteomics enabled by multiplex isobaric labeling in a nanodroplet sample preparation platform. Anal. Chem. 2019;91:13119–13127. doi: 10.1021/acs.analchem.9b03349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cong Y., Motamedchaboki K., Misal S.A., Liang Y., Guise A.J., Truong T., et al. Ultrasensitive single-cell proteomics workflow identifies >1000 protein groups per mammalian cell. Chem. Sci. 2021;12:1001–1006. doi: 10.1039/d0sc03636f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cong Y., Liang Y., Motamedchaboki K., Huguet R., Truong T., Zhao R., et al. Improved single-cell proteome coverage using narrow-bore packed NanoLC columns and ultrasensitive mass spectrometry. Anal. Chem. 2020;92:2665–2671. doi: 10.1021/acs.analchem.9b04631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Budnik B., Levy E., Harmange G., Slavov N. SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol. 2018;19:161. doi: 10.1186/s13059-018-1547-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brunner A.D., Thielert M., Vasilopoulou C., Ammar C., Coscia F., Mund A., et al. Ultra-high sensitivity mass spectrometry quantifies single-cell proteome changes upon perturbation. Mol. Syst. Biol. 2022;18 doi: 10.15252/msb.202110798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Slavov N. Single-cell protein analysis by mass spectrometry. Curr. Opin. Chem. Biol. 2020;60:1–9. doi: 10.1016/j.cbpa.2020.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kelly R.T. Single-cell proteomics: progress and prospects. Mol. Cell Proteomics. 2020;19:1739–1748. doi: 10.1074/mcp.R120.002234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen A.T., Franks A., Slavov N. DART-ID increases single-cell proteome coverage. PLoS Comput. Biol. 2019;15 doi: 10.1371/journal.pcbi.1007082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu Y., Piehowski P.D., Zhao R., Chen J., Shen Y., Moore R.J., et al. Nanodroplet processing platform for deep and quantitative proteome profiling of 10-100 mammalian cells. Nat. Commun. 2018;9:882. doi: 10.1038/s41467-018-03367-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shao X., Wang X., Guan S., Lin H., Yan G., Gao M., et al. Integrated proteome analysis device for fast single-cell protein profiling. Anal. Chem. 2018;90:14003–14010. doi: 10.1021/acs.analchem.8b03692. [DOI] [PubMed] [Google Scholar]
- 33.Lombard-Banek C., Moody S.A., Manzini M.C., Nemes P. Microsampling capillary electrophoresis mass spectrometry enables single-cell proteomics in complex tissues: developing cell clones in live Xenopus laevis and Zebrafish embryos. Anal. Chem. 2019;91:4797–4805. doi: 10.1021/acs.analchem.9b00345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alfaro J.A., Bohlander P., Dai M., Filius M., Howard C.J., van Kooten X.F., et al. The emerging landscape of single-molecule protein sequencing technologies. Nat. Met. 2021;18:604–617. doi: 10.1038/s41592-021-01143-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Callahan N., Tullman J., Kelman Z., Marino J. Strategies for development of a next-generation protein sequencing platform. Trends Biochem. Sci. 2020;45:76–89. doi: 10.1016/j.tibs.2019.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Restrepo-Perez L., Joo C., Dekker C. Paving the way to single-molecule protein sequencing. Nat. Nanotechnol. 2018;13:786–796. doi: 10.1038/s41565-018-0236-6. [DOI] [PubMed] [Google Scholar]
- 37.Swaminathan J., Boulgakov A.A., Hernandez E.T., Bardo A.M., Bachman J.L., Marotta J., et al. Highly parallel single-molecule identification of proteins in zeptomole-scale mixtures. Nat. Biotechnol. 2018;36:1076–1082. doi: 10.1038/nbt.4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kelleher N.L. A cell-based approach to the human proteome project. J. Am. Soc. Mass Spectrom. 2012;23:1617–1624. doi: 10.1007/s13361-012-0469-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith L.M., Agar J.N., Chamot-Rooke J., Danis P.O., Ge Y., Loo J.A., et al. The human proteoform project: defining the human proteome. Sci. Adv. 2021;7 doi: 10.1126/sciadv.abk0734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duncombe T.A., Tentori A.M., Herr A.E. Microfluidics: Reframing biological enquiry. Nat. Rev. Mol. Cell Biol. 2015;16:554–567. doi: 10.1038/nrm4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holzner G., Mateescu B., van Leeuwen D., Cereghetti G., Dechant R., Stavrakis S., et al. High-throughput multiparametric imaging flow cytometry: toward diffraction-limited sub-cellular detection and monitoring of sub-cellular processes. Cell Rep. 2021;34 doi: 10.1016/j.celrep.2021.108824. [DOI] [PubMed] [Google Scholar]
- 42.Liu L., Chen D., Wang J., Chen J. Advances of single-cell protein analysis. Cells. 2020;9:1271. doi: 10.3390/cells9051271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jain A., Liu R., Xiang Y.K., Ha T. Single-molecule pull-down for studying protein interactions. Nat. Protoc. 2012;7:445–452. doi: 10.1038/nprot.2011.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rissin D.M., Kan C.W., Campbell T.G., Howes S.C., Fournier D.R., Song L., et al. Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat. Biotechnol. 2010;28:595–599. doi: 10.1038/nbt.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yelleswarapu V., Buser J.R., Haber M., Baron J., Inapuri E., Issadore D. Mobile platform for rapid sub-picogram-per-milliliter, multiplexed, digital droplet detection of proteins. Proc. Natl. Acad. Sci. U. S. A. 2019;116:4489–4495. doi: 10.1073/pnas.1814110116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cohen L., Cui N., Cai Y., Garden P.M., Li X., Weitz D.A., et al. Single molecule protein detection with attomolar sensitivity using droplet digital enzyme-linked immunosorbent assay. ACS Nano. 2020;14:9491–9501. doi: 10.1021/acsnano.0c02378. [DOI] [PubMed] [Google Scholar]
- 47.Stoeckius M., Hafemeister C., Stephenson W., Houck-Loomis B., Chattopadhyay P.K., Swerdlow H., et al. Simultaneous epitope and transcriptome measurement in single cells. Nat. Met. 2017;14:865–868. doi: 10.1038/nmeth.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peterson V.M., Zhang K.X., Kumar N., Wong J., Li L., Wilson D.C., et al. Multiplexed quantification of proteins and transcripts in single cells. Nat. Biotechnol. 2017;35:936–939. doi: 10.1038/nbt.3973. [DOI] [PubMed] [Google Scholar]
- 49.Kang C.C., Yamauchi K.A., Vlassakis J., Sinkala E., Duncombe T.A., Herr A.E. Single cell-resolution western blotting. Nat. Protoc. 2016;11:1508–1530. doi: 10.1038/nprot.2016.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hughes A.J., Spelke D.P., Xu Z., Kang C.C., Schaffer D.V., Herr A.E. Single-cell western blotting. Nat. Met. 2014;11:749–755. doi: 10.1038/nmeth.2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamauchi K.A., Herr A.E. Subcellular western blotting of single cells. Microsyst. Nanoeng. 2017;3:16079. doi: 10.1038/micronano.2016.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rosas-Canyelles E., Modzelewski A.J., Geldert A., He L., Herr A.E. Assessing heterogeneity among single embryos and single blastomeres using open microfluidic design. Sci. Adv. 2020;6 doi: 10.1126/sciadv.aay1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rosas-Canyelles E., Modzelewski A.J., Geldert A., He L., Herr A.E. Multimodal detection of protein isoforms and nucleic acids from mouse pre-implantation embryos. Nat. Protoc. 2021;16:1062–1088. doi: 10.1038/s41596-020-00449-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lundberg E., Borner G.H.H. Spatial proteomics: a powerful discovery tool for cell biology. Nat. Rev. Mol. Cell Biol. 2019;20:285–302. doi: 10.1038/s41580-018-0094-y. [DOI] [PubMed] [Google Scholar]
- 55.Assarsson E., Lundberg M., Holmquist G., Bjorkesten J., Thorsen S.B., Ekman D., et al. Homogenous 96-plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS One. 2014;9 doi: 10.1371/journal.pone.0095192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Uhlen M., Bandrowski A., Carr S., Edwards A., Ellenberg J., Lundberg E., et al. A proposal for validation of antibodies. Nat. Met. 2016;13:823–827. doi: 10.1038/nmeth.3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baker M. When antibodies mislead: the quest for validation. Nature. 2020;585:313–314. doi: 10.1038/d41586-020-02549-1. [DOI] [PubMed] [Google Scholar]
- 58.Baker M. Reproducibility crisis: blame it on the antibodies. Nature. 2015;521:274–276. doi: 10.1038/521274a. [DOI] [PubMed] [Google Scholar]
- 59.Bradbury A., Pluckthun A. Reproducibility: standardize antibodies used in research. Nature. 2015;518:27–29. doi: 10.1038/518027a. [DOI] [PubMed] [Google Scholar]
- 60.Bandrowski A., Brush M., Grethe J.S., Haendel M.A., Kennedy D.N., Hill S., et al. The resource identification initiative: a cultural shift in publishing. J. Comp. Neurol. 2016;524:8–22. doi: 10.1002/cne.23913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Safonova Y., Pevzner P.A. De novo inference of diversity genes and analysis of non-canonical V(DD)J recombination in immunoglobulins. Front. Immunol. 2019;10:987. doi: 10.3389/fimmu.2019.00987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Safonova Y., Bonissone S., Kurpilyansky E., Starostina E., Lapidus A., Stinson J., et al. IgRepertoireConstructor: a novel algorithm for antibody repertoire construction and immunoproteogenomics analysis. Bioinformatics. 2015;31:i53–61. doi: 10.1093/bioinformatics/btv238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhou X.X., Bracken C.J., Zhang K., Zhou J., Mou Y., Wang L., et al. Targeting phosphotyrosine in native proteins with conditional, bispecific antibody traps. J. Am. Chem. Soc. 2020;142:17703–17713. doi: 10.1021/jacs.0c08458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yamada K.M., Collins J.W., Cruz Walma D.A., Doyle A.D., Morales S.G., Lu J., et al. Extracellular matrix dynamics in cell migration, invasion and tissue morphogenesis. Int. J. Exp. Pathol. 2019;100:144–152. doi: 10.1111/iep.12329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Karamanos N.K., Theocharis A.D., Piperigkou Z., Manou D., Passi A., Skandalis S.S., et al. A guide to the composition and functions of the extracellular matrix. FEBS J. 2021;288:6850–6912. doi: 10.1111/febs.15776. [DOI] [PubMed] [Google Scholar]
- 66.Hynes R.O. The extracellular matrix: not just pretty fibrils. Science. 2009;326:1216–1219. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Taha I.N., Naba A. Exploring the extracellular matrix in health and disease using proteomics. Essays Biochem. 2019;63:417–432. doi: 10.1042/EBC20190001. [DOI] [PubMed] [Google Scholar]
- 68.Shao X., Taha I.N., Clauser K.R., Gao Y.T., Naba A. MatrisomeDB: the ECM-protein knowledge database. Nucl. Acids Res. 2020;48:D1136–D1144. doi: 10.1093/nar/gkz849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Raghunathan R., Sethi M.K., Klein J.A., Zaia J. Proteomics, glycomics, and glycoproteomics of matrisome molecules. Mol. Cell Proteomics. 2019;18:2138–2148. doi: 10.1074/mcp.R119.001543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Naba A., Clauser K.R., Hoersch S., Liu H., Carr S.A., Hynes R.O. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell Proteomics. 2012;11 doi: 10.1074/mcp.M111.014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Naba A., Clauser K.R., Ding H., Whittaker C.A., Carr S.A., Hynes R.O. The extracellular matrix: tools and insights for the "omics" era. Matrix Biol. 2016;49:10–24. doi: 10.1016/j.matbio.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bingham G.C., Lee F., Naba A., Barker T.H. Spatial-omics: novel approaches to probe cell heterogeneity and extracellular matrix biology. Matrix Biol. 2020;91-92:152–166. doi: 10.1016/j.matbio.2020.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Angel P.M., Schwamborn K., Comte-Walters S., Clift C.L., Ball L.E., Mehta A.S., et al. Extracellular matrix imaging of breast tissue pathologies by MALDI-imaging mass spectrometry. Proteomics Clin. Appl. 2019;13 doi: 10.1002/prca.201700152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Angel P.M., Comte-Walters S., Ball L.E., Talbot K., Mehta A., Brockbank K.G.M., et al. Mapping extracellular matrix proteins in formalin-fixed, paraffin-embedded tissues by MALDI imaging mass spectrometry. J. Proteome Res. 2018;17:635–646. doi: 10.1021/acs.jproteome.7b00713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hobeika L., Barati M.T., Caster D.J., McLeish K.R., Merchant M.L. Characterization of glomerular extracellular matrix by proteomic analysis of laser-captured microdissected glomeruli. Kidney Int. 2017;91:501–511. doi: 10.1016/j.kint.2016.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Merl-Pham J., Basak T., Knuppel L., Ramanujam D., Athanason M., Behr J., et al. Quantitative proteomic profiling of extracellular matrix and site-specific collagen post-translational modifications in an in vitro model of lung fibrosis. Matrix Biol. Plus. 2019;1 doi: 10.1016/j.mbplus.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Basak T., Vega-Montoto L., Zimmerman L.J., Tabb D.L., Hudson B.G., Vanacore R.M. Comprehensive characterization of glycosylation and hydroxylation of basement membrane collagen IV by high-resolution mass spectrometry. J. Proteome Res. 2016;15:245–258. doi: 10.1021/acs.jproteome.5b00767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tran B.Q., Miller P.R., Taylor R.M., Boyd G., Mach P.M., Rosenzweig C.N., et al. Proteomic characterization of dermal interstitial fluid extracted using a novel microneedle-assisted technique. J. Proteome Res. 2018;17:479–485. doi: 10.1021/acs.jproteome.7b00642. [DOI] [PubMed] [Google Scholar]
- 79.Hsu C.W., Chang K.P., Huang Y., Liu H.P., Hsueh P.C., Gu P.W., et al. Proteomic profiling of paired interstitial fluids reveals dysregulated pathways and salivary NID1 as a biomarker of oral cavity squamous cell carcinoma. Mol. Cell Proteomics. 2019;18:1939–1949. doi: 10.1074/mcp.RA119.001654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Geyer P.E., Kulak N.A., Pichler G., Holdt L.M., Teupser D., Mann M. Plasma proteome profiling to assess human health and disease. Cell Syst. 2016;2:185–195. doi: 10.1016/j.cels.2016.02.015. [DOI] [PubMed] [Google Scholar]
- 81.Wang Y.T., Shi T., Srivastava S., Kagan J., Liu T., Rodland K.D. Proteomic analysis of exosomes for discovery of protein biomarkers for prostate and bladder cancer. Cancers (Basel) 2020;12:2335. doi: 10.3390/cancers12092335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mathew B., Mansuri M.S., Williams K.R., Nairn A.C. Exosomes as emerging biomarker tools in neurodegenerative and neuropsychiatric disorders-A proteomics perspective. Brain Sci. 2021;11:258. doi: 10.3390/brainsci11020258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xu R., Rai A., Chen M., Suwakulsiri W., Greening D.W., Simpson R.J. Extracellular vesicles in cancer - implications for future improvements in cancer care. Nat. Rev. Clin. Oncol. 2018;15:617–638. doi: 10.1038/s41571-018-0036-9. [DOI] [PubMed] [Google Scholar]
- 84.Zhu Y., Piehowski P.D., Kelly R.T., Qian W.J. Nanoproteomics comes of age. Expert Rev. Proteomics. 2018;15:865–871. doi: 10.1080/14789450.2018.1537787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Spraggins J.M., Rizzo D.G., Moore J.L., Noto M.J., Skaar E.P., Caprioli R.M. Next-generation technologies for spatial proteomics: integrating ultra-high speed MALDI-TOF and high mass resolution MALDI FTICR imaging mass spectrometry for protein analysis. Proteomics. 2016;16:1678–1689. doi: 10.1002/pmic.201600003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schueder F., Unterauer E.M., Ganji M., Jungmann R. DNA-barcoded fluorescence microscopy for spatial omics. Proteomics. 2020;20 doi: 10.1002/pmic.201900368. [DOI] [PubMed] [Google Scholar]
- 87.Ryan D.J., Spraggins J.M., Caprioli R.M. Protein identification strategies in MALDI imaging mass spectrometry: a brief review. Curr. Opin. Chem. Biol. 2019;48:64–72. doi: 10.1016/j.cbpa.2018.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ryan D.J., Patterson N.H., Putnam N.E., Wilde A.D., Weiss A., Perry W.J., et al. MicroLESA: integrating autofluorescence microscopy, in situ micro-digestions, and liquid extraction surface analysis for high spatial resolution targeted proteomic studies. Anal. Chem. 2019;91:7578–7585. doi: 10.1021/acs.analchem.8b05889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Piehowski P.D., Zhu Y., Bramer L.M., Stratton K.G., Zhao R., Orton D.J., et al. Automated mass spectrometry imaging of over 2000 proteins from tissue sections at 100-mum spatial resolution. Nat. Commun. 2020;11:8. doi: 10.1038/s41467-019-13858-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pankow S., Martinez-Bartolome S., Bamberger C., Yates J.R. Understanding molecular mechanisms of disease through spatial proteomics. Curr. Opin. Chem. Biol. 2019;48:19–25. doi: 10.1016/j.cbpa.2018.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Baharlou H., Canete N.P., Cunningham A.L., Harman A.N., Patrick E. Mass cytometry imaging for the study of human diseases-applications and data analysis strategies. Front. Immunol. 2019;10:2657. doi: 10.3389/fimmu.2019.02657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Spraggins J.M., Rizzo D.G., Moore J.L., Rose K.L., Hammer N.D., Skaar E.P., et al. MALDI FTICR IMS of intact proteins: using mass accuracy to link protein images with proteomics data. J. Am. Soc. Mass Spectrom. 2015;26:974–985. doi: 10.1007/s13361-015-1147-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yin R., Burnum-Johnson K.E., Sun X., Dey S.K., Laskin J. High spatial resolution imaging of biological tissues using nanospray desorption electrospray ionization mass spectrometry. Nat. Protoc. 2019;14:3445–3470. doi: 10.1038/s41596-019-0237-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Van de Plas R., Yang J., Spraggins J., Caprioli R.M. Image fusion of mass spectrometry and microscopy: a multimodality paradigm for molecular tissue mapping. Nat. Met. 2015;12:366–372. doi: 10.1038/nmeth.3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Clift C.L., Drake R.R., Mehta A., Angel P.M. Multiplexed imaging mass spectrometry of the extracellular matrix using serial enzyme digests from formalin-fixed paraffin-embedded tissue sections. Anal. Bioanal. Chem. 2021;413:2709–2719. doi: 10.1007/s00216-020-03047-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mund A., Coscia F., Hollandi R., Kovács F., Kriston A., Brunner A.D., et al. AI-driven Deep Visual Proteomics defines cell identity and heterogeneity. bioRxiv. 2021 doi: 10.1101/2021.01.25.427969. [preprint] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jones M.A., Cho S.H., Patterson N.H., Van de Plas R., Spraggins J.M., Boothby M.R., et al. Discovering new lipidomic features using cell type specific fluorophore expression to provide spatial and biological specificity in a multimodal workflow with MALDI imaging mass spectrometry. Anal. Chem. 2020;92:7079–7086. doi: 10.1021/acs.analchem.0c00446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Qin Y., Huttlin E.L., Winsnes C.F., Gosztyla M.L., Wacheul L., Kelly M.R., et al. A multi-scale map of cell structure fusing protein images and interactions. Nature. 2021;600:536–542. doi: 10.1038/s41586-021-04115-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bhalla U.S., Iyengar R. Emergent properties of networks of biological signaling pathways. Science. 1999;283:381–387. doi: 10.1126/science.283.5400.381. [DOI] [PubMed] [Google Scholar]
- 100.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., et al. Gene ontology: tool for the unification of biology. The gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yang X., Coulombe-Huntington J., Kang S., Sheynkman G.M., Hao T., Richardson A., et al. Widespread expansion of protein interaction capabilities by alternative splicing. Cell. 2016;164:805–817. doi: 10.1016/j.cell.2016.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jens Hansen R.S., Menon R., Eadon M.T., Lake B.B., Steck B., Dobi D., et al. Towards building a smart Kidney atlas: network-based integration of multimodal transcriptomic, proteomic, metabolomic and imaging data in the Kidney precision medicine project. bioRxiv. 2020 doi: 10.1101/2020.07.23.216507. [preprint] [DOI] [Google Scholar]
- 103.Rozenblatt-Rosen O., Regev A., Oberdoerffer P., Nawy T., Hupalowska A., Rood J.E., et al. The human tumor atlas network: charting tumor transitions across space and time at single-cell resolution. Cell. 2020;181:236–249. doi: 10.1016/j.cell.2020.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Vitrinel B., Koh H.W.L., Mujgan Kar F., Maity S., Rendleman J., Choi H., et al. Exploiting interdata relationships in next-generation proteomics analysis. Mol. Cell Proteomics. 2019;18:S5–S14. doi: 10.1074/mcp.MR118.001246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rozenblatt-Rosen O., Shin J.W., Rood J.E., Hupalowska A., Human Cell Atlas S., Technology Working G., et al. Building a high-quality human cell atlas. Nat. Biotechnol. 2021;39:149–153. doi: 10.1038/s41587-020-00812-4. [DOI] [PubMed] [Google Scholar]
- 106.de Boer I.H., Alpers C.E., Azeloglu E.U., Balis U.G.J., Barasch J.M., Barisoni L., et al. Rationale and design of the Kidney precision medicine project. Kidney Int. 2021;99:498–510. doi: 10.1016/j.kint.2020.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]