

Abstract
Despite advances in breast cancer treatments and related 5-year survival outcomes, metastatic breast cancer cures remain elusive. The current standard of care includes a combination of surgery, radiation therapy and drug therapy. However, even the most advanced procedures and treatments do not prevent breast cancer recurrence and metastasis. Once metastasis occurs, patient prognosis is poor. Recent elucidation of the spatiotemporal transit of metastatic cancer cells from primary tumor sites to distant sites provide an opportunity to integrate knowledge of drug disposition in our effort to enhance drug localization and exposure in cancer laden tissues. Novel technologies have been developed, but could be further refined to facilitate the distribution of drugs to target cancer cells and tissues. The purpose of this review is to highlight the challenges in metastatic breast cancer treatment and focus on novel drug combination and nanotechnology approaches to overcome the challenges. With improved definition of metastatic tissue target, directed localization and retention of multiple, pharmacologically active drugs to tissues and cells of interest may overcome the limitations in breast cancer treatment that may lead to a cure for breast cancer.
Keywords: Metastatic breast Cancer, Nanoparticles, Combination therapy, Targeted therapy, Drug delivery
1. Introduction
In the United states, approximately 1 in 8 women develop breast cancer over the course of their lives (Rojas & Stuckey, 2016). There are many interventions available for breast cancer patients including surgery, radiation therapy and drug therapies (Moo, Sanford, Dang, & Morrow, 2018). Unfortunately, even though available treatment options are effective in early stages, ~30% of patients progress to metastatic disease (Redig & McAllister, 2013). Most patients at the time of diagnosis will undergo surgery to remove the primary breast tumor, but residual cancer cells can remain. Tumor recurrence from these foci likely reflects an inability of subsequent interventions (like radiation or targeted therapy) to eliminate residual cells in the mammary or lymphatic tissue, which results in distant metastasis (Franci, et al., 2013). Initial drug therapies for treating breast cancer, such as those targeted to inhibit hormone receptors or growth receptor kinases (which support rapid growth of cancer cells), can become ineffective after recurrence and lead to the spread of cancer cells due to the emergence of drug resistance characteristics. Once patients progress to the metastatic breast cancer (MBC; or cancer cells spread beyond breast tissue) stage, cytotoxic drug therapy—referred to as chemotherapy—remains as a viable, and perhaps the best, treatment option (Schneeweiss, Ruckhäberle, & Huober, 2015). Chemotherapy regimens used in the treatment of MBC are potent and remain effective in killing cancer cells. Unfortunately, they also exhibit dose-limiting toxicities from off-target exposure of drugs in the cells of healthy tissues. Thus, patients often do not complete the prescribed course of chemotherapies due to intolerable, severe side-effects. Additionally, chemotherapy is administered as multiple drugs in combination to maximize pharmacologic effects. Some drugs such as the anthracycline derivatives doxorubicin and daunorubicin exhibit a cumulative toxicity that may prevent patients from completing the required dose to eliminate residual cancer cells.
Combination regimens used in MBC are based on either an anthracycline (doxorubicin, epirubicin or daunorubicin) or taxane (paclitaxel, docetaxel) combined with other cytotoxic drugs. Each class of chemotherapy has their own specific toxicity profile. For example, anthracycline therapy damages cardiac tissue by irreversibly binding to cardiolipin enriched in the cells of heart tissues (Geisberg & Sawyer, 2010). Anthracycline cardiotoxicity is lifetime dose-limiting and is a major issue in late-stage disease where patients have already been exposed to numerous cycles of anthracycline therapy. In other words, when a patient has reached a life-time total dose, that patient can no longer receive any additional anthracycline therapy. Taxane based regimens (e.g., paclitaxel, docetaxel) on the other hand cause bone marrow toxicity, which is dose-limiting, but myelosuppression is generally reversible and can be managed clinically (Markman, 2003). Unlike anthracycline induced cumulative cardiotoxicity that prevents patients from receiving additional doses, taxane based therapy can be used in continuity after a period of rest to reverse myelotoxicity. Thus, while anthracycline and taxane based therapies have comparable therapeutic effects as single agents, the more manageable and reversible toxicity profile of taxane favors their usage in late-stage disease. Regardless of the regimen, the key to success for combination chemotherapy (as opposed to monotherapy) is to balance the effectiveness of killing cancer cells with manageable off-target toxicity to healthy organs.
Gemcitabine (antimetabolite, logP = −1.4) and paclitaxel (tubulin inhibitor, logP = 3) are used clinically as a taxane-based 2-drug combination chemotherapy regimen (GT, G=gemcitabine, T=Taxol=Paclitaxel) to improve outcomes of MBC (Colomer, 2005). Unlike anthracycline and paclitaxel combinations, the substitution of anthracycline with gemcitabine has overcome cumulative dose-limiting cardiotoxicity. Gemcitabine and paclitaxel are prescribed for patients who have progressed from initial therapies targeted to hormones, receptors and kinases that regulate breast cancer cell growth. However, gemcitabine and paclitaxel are given sequentially and separately in each drug formulation as two intravenous dosage forms. Two different formulations and two infusions are necessary due to the disparate physicochemical properties of GT. These physicochemical differences are also reflected in their distinctions in disposition, clearance, and plasma time course in vivo. If both GT drugs can simultaneously accumulate and persist in the same cancer cells following drug combination therapy, then antitumor effects could be maximized. In addition, the overall dose requirement could be lowered to minimize drug exposure to healthy organs. Unfortunately, the diverging clearance and disposition of GT soon after their IV administration makes this scenario unlikely for current injectable dosage forms (Gianni, et al., 1995; Reid, et al., 2004). Simultaneous, sustained IV infusion of both drugs may synchronize plasma levels of gemcitabine and paclitaxel, but injection site reactions of paclitaxel and incompatible (water soluble and insoluble) dosage forms of the two drugs make this approach impractical. Due to these factors, maintaining the concentrations of both drugs in tumors after sequential infusion without causing off-target, dose-limiting toxicities is nearly impossible.
To maintain drug levels at therapeutic concentrations in plasma and target cancer cells, a high dose regimen is typically employed in MBC treatment guidelines to compensate for the varying disposition and elimination of GT. As a result of high drug exposure to healthy organs, dose-limiting toxicities of chemotherapy are a major barrier in the management of metastatic disease and highlight a significant limitation in the treatment of MBC. There are other potent and effective drugs that are available and can be used in combination to treat MBC, but the lack of a consistent cure is due to off-target toxicity and inadequate exposure of combined drugs in target cancer cells. If one can develop a strategy to deliver a combination of chemotherapeutic agents to cancer cells and improve the cellular retention of drugs for enhancing antitumor effects, then off-target toxicity could be minimized and a novel treatment that leads to a cure for breast cancer could be envisioned.
Currently, researchers are investigating novel drug delivery systems intended to overcome dose limiting toxicities and improve target drug localization in breast cancer cells and tissues. In theory, drug delivery systems can be engineered with unique properties and characteristics including size, shape and surface chemistry, which helps determine their in vivo disposition. By optimizing these characteristics, drug delivery systems can improve the in vivo behavior of cytotoxic drugs by increasing retention in target cancer cells and reducing off-target toxicity (Moss & Siccardi, 2014). Unfortunately, the optimal drug delivery system to enable targeted delivery to cancer cells has yet to be developed, in part because current designs ahead of in vivo testing do not use a systems approach (Ho, et al., 2015).
In the context of MBC, a systems approach to developing drug delivery systems incorporates a fit-for-purpose approach and should work toward gathering systems information including: how specific particles distribute dynamically in the body vs in the tumor cells, how particles are cleared relative to tumor accumulation and drug stability within drug delivery systems. In addition, unforeseen toxicity of nanoparticulate compositions needs to be considered. Certain drug delivery systems have distinct toxicities related to off-target distribution that would not be observed in freely solubilized drug.
Specific excipients used in nanoparticle compositions, including the commonly used polymeric excipient polyethylene glycol (PEG), are also associated with adverse events (Kozma, Shimizu, Ishida, & Szebeni, 2020; P. Zhang, Sun, Liu, & Jiang, 2016). Toxicities related to off-target distribution and/or excipients used would preclude the clinical development of highly promising drug delivery strategies. For example, liposomes or lipid vesicles are a nanoparticle system that are well established in the literature, but unmodified liposomes greater than 100 nm in diameter are extensively taken up in the liver (Hu, van Rooijen, & Liu, 1996). The sequestration of liposomes with the inappropriate particle size in the liver prevents drug access to target cancer cells and limits drug effect. However, not all liposomes exhibit the same characteristics because the composition of lipids, manufacturing processes, size charge and fluidity of bilayers as well as multiplicity of bilayer concentric structures all contribute to how these particles are distributed, localized and cleared from the body after dosing in vivo (Kraft, Freeling, Wang, & Ho, 2014).
Similarly, not all polymeric nanoparticles are the same. Data accumulated to date suggest that polymeric nanoparticles smaller than 10 nm (such as cyclodextrin, polyamidoamine (PAMAM) dendrimers, etc.) undergo rapid renal filtration for elimination from the body in a particle size-dependent manner. In these conditions, a fraction of particles accumulate chronically in renal tissues. The accumulation of a significant number of particles in the kidney can then lead to toxicological concerns from both the free drug and polymeric particles. The high clearance and elimination by renal filtration (without reabsorption) may also prevent polymeric particles from accessing cancer cells and limit the effectiveness of these carriers.
Over the last few decades, much effort has been dedicated toward the engineering and development of nanoparticulate delivery systems for therapeutics, ranging from liposomes, polymer nanoparticles, micelles, dendrimers, and nanocrystals. Progress in nanotechnology for tumor-targeted delivery and reduced toxicity has resulted in a number of nanoparticle-based products that are currently approved by the Food and Drug Administration (FDA) for the clinical market, with more in the clinical trial pipeline (Fraguas-Sánchez, Martín-Sabroso, Fernández-Carballido, & Torres-Suárez, 2019; Gadekar, et al., 2021; Wicki, Witzigmann, Balasubramanian, & Huwyler, 2015b). However, several limitations remain with nanoparticle systems, from the complex and laborious manufacturing processes to the inability to formulate more than one compound in certain nano delivery systems (i.e. nanocrystals). For example, liposomal drug products require finely tuned salt and pH gradients to encapsulate drugs, while bioconjugation steps are required to link drugs to dendrimers. Complicated manufacturing processes on a large scale are difficult to control; manufacturing challenges have been reported even in production plants for Doxil, a broadly available liposomal drug product to treat breast cancer (Alven & Aderibigbe, 2020; Crommelin, van Hoogevest, & Storm, 2020; Gubernator, 2011).
In addition, a stable, established drug delivery system capable of carrying multiple, chemically diverse chemotherapeutic drug combination is not broadly available. Thus, a novel approach is required to enable the targeted delivery of chemically diverse chemotherapy combinations. The ideal system would facilitate the delivery of multiple drugs to cancer cells for an extended time course to achieve full antitumor efficacy, with a simple and scalable manufacturing process that reduces manufacturing challenges with nanoparticle delivery systems currently available.
The purpose of this review is to describe the pathophysiology of breast cancer and pathogenesis of metastasis to understand the physiological factors that affect the efficacy of MBC treatment. While a number of drugs and biotherapeutics are available and effective for treating early phases of breast cancer, there are limited treatment options that are effective and tolerable for those who progress to metastatic breast cancer or MBC. The key challenge is the rapid growth and spread of cancer from breast to multiple tissues through lymph and blood circulation, which often accumulate in lung capillaries as micro or macro-MBC nodules. Current pharmacologic approaches to treating breast cancer—particularly for MBC—as the basis for combination therapy will be reviewed and factors that allow breast cancer cells to survive treatment will be identified. Subsequently, pharmaceutical approaches that address mechanisms of breast cancer cell survival will be reviewed and a novel approach to improving cancer cell uptake and retention of clinically effective drugs will be presented.
2. Pathophysiology of Breast Cancer
In the simplest terms, breast cancer can be defined as the uncontrolled growth of cells in the breast. There are two main types of tissue in the breast: the milk or mammary ducts and the breast lobules. The tissue that breast cancer originates from determines the clinical progression and behavior of disease. Cancers originating from milk ducts, classified as ductal carcinoma, account for 40–75% of all diagnosed breast cancers (Bombonati & Sgroi, 2011). The defining characteristics of ductal carcinomas are mutations of ERBB2 (erythroblastic oncogene B2), EGFR (epidermal growth factor receptor) and tumor suppressor p53 with metastasis to the lungs, pleura, and central nervous system (Arpino, Bardou, Clark, & Elledge, 2004; Harris, et al., 1984). In contrast, cancer originating from breast lobules, classified as lobular carcinoma, are less prevalent, accounting for ~10–15% of diagnosed breast cancers (Bombonati & Sgroi, 2011). Lobular carcinomas are more likely to overexpress hormone receptors such as the estrogen receptor (ER) or progesterone receptor (PR) and metastasize to the peritoneum, ovary, and gastrointestinal system (Arpino, et al., 2004; Harris, et al., 1984). Although ductal and lobular carcinomas have different expression patterns for specific oncoproteins, treatment algorithms are not based on the tissue of origin but the molecular classification of breast cancer.
There is a strong association between the classification of breast cancer by molecular targets and survival/clinical prognosis. The two major molecular targets and classifications in breast cancer therapy are the estrogen/progesterone hormone receptors (ER/PR) and the erythroblastic oncogene B2 (ERBB2) receptor, which is also referred to as human epidermal growth factor receptor 2 (HER2). Breast cancers (regardless of tissue origin) are categorized as either ER/PR+ or ERBB2+ based upon their expression of these receptors. Breast cancers that do not express any of the three major receptors (ER, PR or ERBB2) are known as triple negative breast cancer (TNBC), and the molecular pathogenesis of TNBC is the least understood. In addition to these 3 major classifications, patients that carry mutations in breast cancer type 1 or type 2 susceptibility protein (BRCA1/BRCA2) are predisposed to breast cancer development and are strongly associated with TNBCs. In general, ER/PR+ breast cancer has the best prognosis (70% of all cases), followed by ERBB2+ cancers (~15–20% of all cases), and TNBC (~10%) which has the worst clinical prognosis and survival rate (Waks & Winer, 2019). The clinical outlook of different types of breast cancer is likely related to the available therapeutic agents. In the case of ER/PR+ breast cancer, drugs have been developed to target these receptors specifically via selective estrogen receptor modulators or aromatase inhibitors. For ERBB2+ breast cancer, targeted agents such as monoclonal antibodies or kinase inhibitors can be used. As TNBC does not express any clearly defined molecular targets, it is treated with broadly cytotoxic chemotherapy. Although chemotherapy is the primary treatment in TNBC, it is also used in ERBB2+ and ER/PR+ cancers. The individual agents used in each class will be discussed in later sections.
2.1. The role of lymphatic tissue in breast cancer metastasis
The current major challenge in breast cancer treatment is not the primary tumor itself, but tumor recurrence and distant metastasis. When breast cancer is first detected, there are many options available for treatment including surgery, radiation therapy, hormone therapy, targeted therapy and chemotherapy. Surgical interventions consist of removing the primary tumor and, in some cases, removing the lymphatic architecture adjacent to the tumor. Radiation therapy is performed by using high-energy waves or particles to irradiate the area where the primary tumor was removed (also known as the tumor bed) to eliminate residual cancer cells. Drug therapy is then administered based on the molecular characteristics of the primary tumor to further inhibit residual cancer growth. These multi-modal treatments are excellent at extending survival but are not consistently curative so that disease progression (in the form of metastasis) is still prevalent.
Once metastasis occurs, treatment goals shift toward palliative care wherein treatment options are limited (typically due to the emergence of drug resistance). As primary breast cancer tumors grow, cancer cells can spread from the primary breast tumor site and invade systemic circulation and the draining lymphatic system (Figure 1) (Nathanson, et al., 2018; Rahman & Mohammed, 2015). As schematically presented in Figure 1, cancer cells remaining or newly detected in the sentinel lymph nodes adjacent to breast cancer tumors are a negative prognostic factor for clinical disease.
Figure 1.
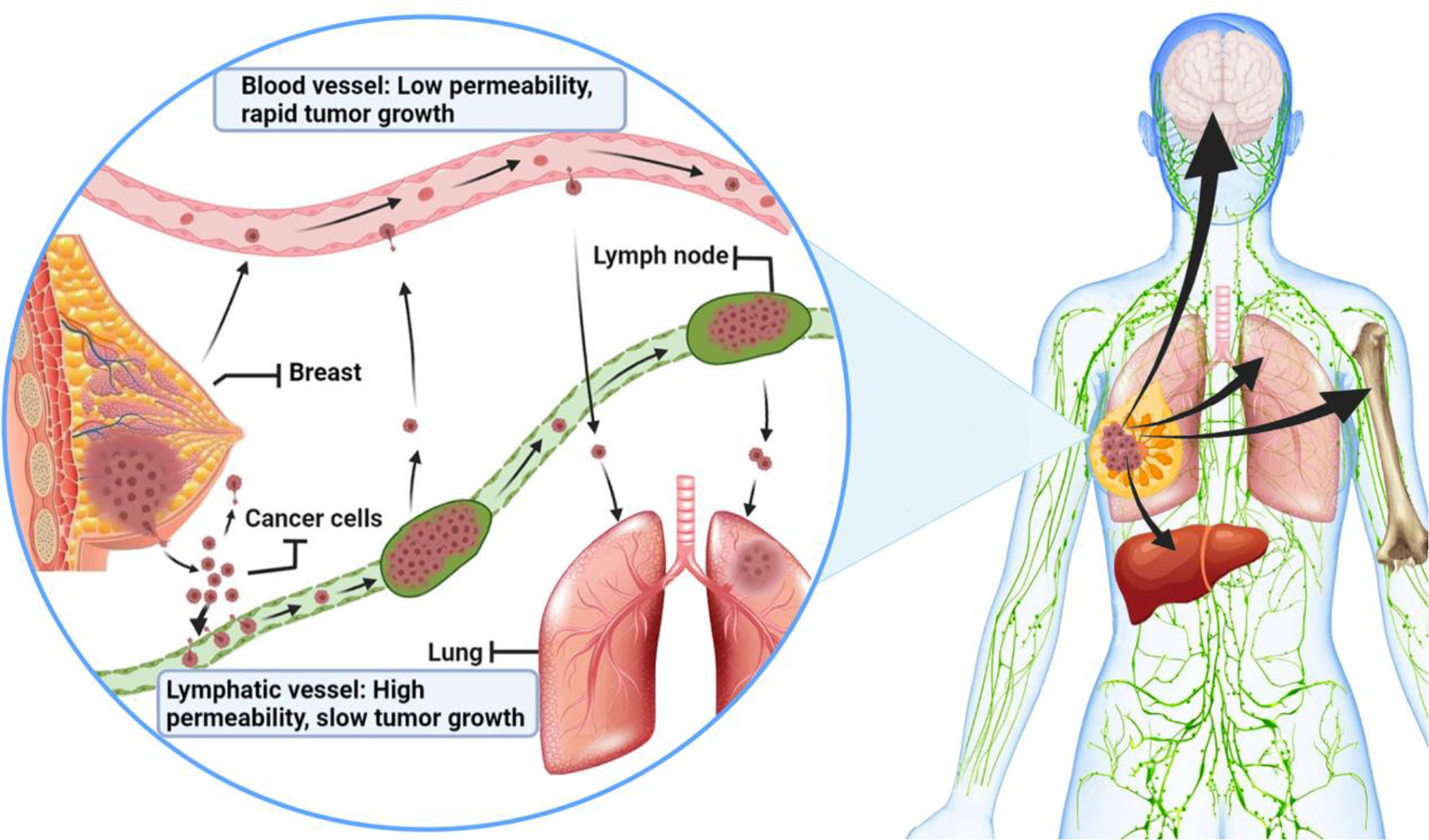
Metastasis of breast cancer cells and involvement of lymphatic and blood vessels. At metastatic stages, breast cancer cells are discovered in distant sites in humans: mostly bone, lung, brain, and liver (right). Recent discoveries in mouse models suggest that the local lymphatics and blood vessels are key players in the metastatic process (left) (Brown, et al., 2018; Pereira, et al., 2018). The cancer cells in tumor mass may first spread into adjacent lymphatic vessels and lymph nodes, which have high permeability for cancer cells but do not support rapid cancer growth. Cancer cells then spread into systemic blood circulation, which has low permeability but supports rapid cancer growth by delivering cells to distant sites to form nodules (such as metastatic lung nodules). Understanding this process helps the development of novel therapeutic strategies that can target cancer cells in lymphatics or in blood circulation.
Recently, studies have shown that lymphatics are also an important part of metastatic pathogenesis in breast cancer. Two reports from independent laboratories provided time and spatial insight into the metastatic spread of breast cancer cells from primary sites, which highlighted the importance of the lymph nodes and lymphatic system in metastatic pathogenesis (Brown, et al., 2018; Pereira, et al., 2018). These two studies used 4T1 metastatic mouse tumors as models to show that the removal of a primary tumor or the inoculation of a small number of cancer cells in the lymph node cortex invariably leads to rapidly growing metastatic nodules formed in the blood capillaries of the lungs. Regardless of where the cells are initially introduced (primary tumor followed by resection or directly into the lymph), cancer cells will penetrate into the blood to establish distant metastatic sites. The cited studies show that residual cancer cells will proliferate in the lymph and subsequently migrate into the blood. From there, the cells can transit to the lungs and colonize the pulmonary tissue as cancer nodules (Brown, et al., 2018). This time-course and spatial 4T1 tumor spread data highlights the lymph and blood as important compartments for supporting the growth and spread of MBC cancer cells. If cancer cells are not resected in the initial surgery and then come to reside in the lymph, those cells have the opportunity to penetrate back into the blood (where the circulation (flow) rate of blood is much higher than that of lymphatic system) and produce distant metastasis. Given the importance of the lymphatic and blood circulation systems in breast cancer metastasis, adequate concentrations of antitumor agents need to be maintained in both blood and lymphatic tissues to eliminate residual cells and prevent distant metastasis. The plasma (or blood) concentration time course of drugs used to treat breast cancer are well-characterized, but less is known about lymphatic drug concentrations over time. If there is poor penetration of chemotherapeutic agents into the lymph, this may explain persistent tumor recurrence and disease progression in breast cancer.
2.2. In vivo models used to study breast cancer growth
As researchers have developed a greater appreciation for the complexity of breast cancer pathophysiology, many preclinical models have been developed to mimic human breast cancer. Most of these models have been established in mice and are broadly categorized as (1) genetically engineered mouse models (GEMM), (2) (human) cell-derived xenografts (CDX), (3) patient derived xenografts (PDX), and (4) syngeneic mouse models (Gould, Junttila, & de Sauvage, 2015) (Table 1). As the name implies, GEMMs are mice that have been genetically modified to over express tumor inducing genes or under express tumor suppressing genes. This leads to the spontaneous formation of tumors that feature a native tumor microenvironment, native vascular and stromal cells and an intact immune system. However, these tumors lack the heterogeneity of naturally occurring human tumors as they are all driven by single point mutations. CDX models are immortalized human cancer cells that are transplanted into immune deficient mice. These models are well-characterized and reported in the literature. These CDX models with human cells implanted in immune-deficient mice can be used to study various human tumor types in mice, but lack the intact immune component, which may contribute to regulating breast cancer growth and suppression. PDX models are cancer cells directly harvested from a human patient and transplanted into an immunocompromised mouse. PDX models contain higher degrees of genetic heterogeneity compared to CDX and GEMM models and may be more representative of human tumors for evaluating therapeutic interventions, even if they lack the innate immune component of breast cancer growth in humans. Syngeneic mouse models use murine cancer cells that are transplanted into healthy mice of the same strain. This model allows for controlled tumors to grow in their native environment in the presence of an intact immune system but are entirely murine (both tumor and stroma). Tumor model selection to test treatment must take into consideration these aforementioned factors; Table 1 summarizes the general strengths and weaknesses of each model.
Table 1.
Examples of breast cancer models and distinctive characteristics.
| Model | Model Origin | Strengths | Weaknesses | References |
|---|---|---|---|---|
| Genetically engineered mouse model (GEMMs) | Genetic modification to promote spontaneous tumor development in mice |
|
|
(Gopinathan, Morton, Jodrell, & Sansom, 2015),(Kersten, de Visser, van Miltenburg, & Jonkers, 2017) |
| Cell-derived Xenograft (CDX) | Established immortalized human cancer cell lines transplanted into immunocompromised mice |
|
|
(Chavez, Garimella, & Lipkowitz, 2010) |
| Patient Derived Xenograft (PDX) | Human cancer cells harvested from patients and transplanted into immunocompromised mice |
|
|
(Murayama & Gotoh, 2019) (Whittle, Lewis, Lindeman, & Visvader, 2015) |
| Syngeneic | Established immortalized mouse cancer cell lines transplanted into immunocompetent mice |
|
|
(Park, Lee, & Lee, 2018), (Pulaski & Ostrand-Rosenberg, 2001) |
After selecting the appropriate cell line and mouse host strain (immunocompetent versus immunocompromised), the next consideration for mouse models is the method of transplantation (Rashid & Takabe, 2015). Ectopic transplantations (where the implantation tissue is different from the primary tumor tissue) are highly reproducible and consist of inoculating cells subcutaneously in the flank for breast cancer. Ectopic tumor growth can be precisely measured and tracked over time, but typically display slower growth rates and are present in an unnatural (non-breast tissue) stromal environment. Orthotopic transplantation is an alternative method where cancer cells are transplanted into their tissue of origin, typically the mammary fat pad for breast cancer. Orthotopic tumors grow in their native tissue and are often used to study spontaneous metastasis to distant organs. Finally, hematogenous models of breast cancer directly introduce cancer cells into the systemic circulation of mice by intravenous injection to study blood-based metastasis. Hematogenous models reproducibly form cancer nodules in the lungs and can recapitulate the transit of cancer cells through the systemic (blood) circulation. Intravenous administration of other types of cancer cells can also result in cancer nodule formation in the lungs due to the small size of the pulmonary capillary beds. However, hematogenous models are particularly useful for breast cancer due to the high prevalence of lung metastasis observed in the clinic. In general, a significant fraction—21 to 32%—of the secondary tumors observed in metastatic breast cancer patients form in the lungs (Medeiros & Allan, 2019). Thus, hematogenous models, which produce MBC growth as lung nodules, are well-suited to study treatment interventions for metastatic breast cancer.
With growing recognition that the lymphatic system is involved with breast cancer metastasis, many mouse models for breast cancer metastasis are also used to investigate lymphangiogenesis, or the growth of new lymph vessels. Cancer cells have been shown to secrete vascular endothelial growth factors (VEGF), which promote the development of new blood (VEGF-A) and lymph (VEGF-C/VEGF-D) vessels inside of primary tumors (Shibuya, 2011). The development of new lymph vessels is considered a crucial step for cancer cell entry into the lymph. Surprisingly, studies have shown that while intra-tumoral lymphatic vasculature can be collapsed and non-functional, peritumoral lymph vessels are sufficient to allow for metastasis to occur (Padera, et al., 2002). This observation is likely due to the dilation effect of VEGF-C/VEGF-D on collecting lymphatic vessels surrounding the sentinel lymph node (or primary draining lymph node from the tumor), which facilitates cell entry into the lymph (He, et al., 2005).
Mouse models of breast cancer metastasis are invaluable tools to study the growth and metastasis of breast cancer. These models help elucidate the underlying physiological changes caused by cancer. In doing so, these models further highlight the importance of the lymphatic system. For example, the rapid association of lymphatic vessels with cancerous tumors may provide a novel route for the delivery of pharmacologic agents. Chemotherapy used to treat breast cancer is associated with significant off-target, systemic toxicity. If chemotherapy can target the lymph, it may reduce the systemic, off-target toxicity and still achieve a cytotoxic effect in cancer cells. This manuscript will focus on a nanotechnology that enables the co-delivery two drugs, gemcitabine and paclitaxel, targeted to MBC to overcome some of the off-target toxicities linked to premature discontinuation of treatment cycles in MBC patients. We will also discuss the rationale for lymphatic-targeted drug combination delivery of specific nanoparticles called drug combination nanoparticles or DcNP. To appreciate how combined delivery and lymphatic targeting may benefit gemcitabine and paclitaxel efficacy, it is important to understand the role of these two drugs in the overall treatment of breast cancer. Thus, the following section will provide an overview of the current treatment approaches for breast cancer.
3. Principles of treating breast cancer
For breast cancers diagnosed in a localized region (~90% of all cases), the main goal of treatment is to completely remove the primary tumor by surgical intervention. During surgical resection, assessments of local tissue with the aid of biopsies can reveal if cancer cells have migrated to the draining lymph nodes and guide future treatment plans. Local therapy addressing cancer cells in regional lymph nodes can consist of surgical removal of the infiltrated lymph nodes, which is often followed by radiation therapy of the tumor bed. Systemic drug therapy is subsequently prescribed and intended to eliminate any residual cells. Breast cancer subtypes further guide follow-up therapies to prevent metastatic recurrence. However, just one or two residual cancer cells can lead to recurrence and spread to other tissues through the lymph and blood circulation systems. The following section will review the pharmacologic rationale behind specific drug classes used in each subtype of breast cancer and their role in overall treatment success.
3.1. Pharmacologic basis for the treatment of ER/PR+ breast cancer
Estrogen receptors (ER) are nuclear steroid receptors that act as ligand activated transcription factors. ERs exist as two isoforms: ERα and ERβ and both receptors play important roles in regular breast development. Activation of ERα occurs by binding its endogenous substrate, estradiol, and recruiting coactivators to gene promoters for the transcription of genes associated with cell proliferation, inhibition of cell death and new blood vessel formation (Thakkar & Mehta, 2011). This mechanism is a natural process in breast development and the dysregulation of this pathway causes developmental aberrations and carcinogenesis. As a result, the overexpression (or over activation) of ERα on cancer cells results in uncontrolled cell proliferation and leads to the development of breast cancer. Since this pathway is endogenous to all breast cell development, the dysregulation of this pathway accounts for the majority of breast cancers (70%). The progesterone receptor is also a nuclear steroid receptor that reflects ERα signaling (Daniel, Hagan, & Lange, 2011; Kariagina, Aupperlee, & Haslam, 2008). Interestingly, ERβ may act as a tumor suppressor gene and oppose the effects of ERα, but the role of ERβ in breast cancer development is still unclear (Rizza, et al., 2014). Early lobular breast cancer has been shown to express both ERα and ERβ receptors, but ERβ receptors are lost in late stage lobular cancer (Huang, et al., 2014). This may suggest that targeting the ERβ signaling pathway has a limited role in early-stage disease. Nevertheless, there is still uncertainty behind ERβ targeting and no ERβ agonists are used in the treatment of breast cancer. For that reason, this section will focus on ERα targeted drugs.
There are a number of pharmacologic agents that target the ERα signaling pathways to prevent breast cancer proliferation. They fall into 2 major categories: drugs that competitively inhibit ERα or drugs that deplete endogenous levels of estrogen. Selective estrogen receptor modulators (SERMs) are a group of molecules that can have both agonistic and antagonistic activity at the ERα in different tissues. In breast cancer cells, tamoxifen and raloxifene (two commonly used SERMs) compete with estradiol for binding to the ER (Frasor, et al., 2004). This competitive inhibition of ERα signaling reduces cancer cell proliferation and prevents disease progression. Fulvestrant, a selective estrogen down regulator (SERD), also binds to the ERα, similar to tamoxifen and raloxifene, but has a significantly greater binding affinity for ERα (~40-fold greater than tamoxifen) (Osborne, Wakeling, & Nicholson, 2004). The fulvestrant-ERα complex is unstable and results in ERα degradation, a mechanism not observed with SERMs (Nicholson, et al., 1995; Osborne, et al., 2004).
Aromatase inhibitors (AIs) target the ERα signaling pathway by interfering with the body’s ability to produce estrogen from circulating androgens. After menopause, estrogen produced by the ovaries decreases and local levels of estrogen are generated by the aromatase enzyme present in various tissues such as the breast. In proliferative breast cancer, aromatase activity can be increased, and local levels of estrogen near the primary tumor can be increased (Simpson & Davis, 2001). By interfering with the formation of estrogen, AIs deplete the endogenous ligands necessary for the ERα signaling pathway and inhibit cancer cell growth. Some examples of aromatase inhibitors used in breast cancer include: letrozole and anastrozole (reversible inhibitors) and exemestane (irreversible inhibitor). As a point of clarification, aromatase inhibitors are only effective at reducing levels of estrogen in women with post-menopausal ovaries. In pre-menopausal women, AIs can be used in women undergoing ovarian oblation or oophorectomy to reduce ovarian function but will not be effective in women with estrogen forming ovaries (Pistelli, Mora, Ballatore, & Berardi, 2018). In general, approximately 2/3 of women diagnosed with breast cancer are above post-menopausal age and most patients are eligible for AI therapy.
An important distinction in hormone receptor targeted therapy is the availability of oral dosage forms. Tablets and capsules are convenient for patients, but medication adherence is crucial to maximally suppress the ERα signaling pathway and prevent cancer growth. Even with good medication adherence, oral drug dosing is subject to fluctuations in GI physiology that can affect dissolution or absorption. Additionally, orally administered medications undergo first-pass metabolism and P glycoprotein-mediated drug efflux in the gut and liver. These factors lower the systemic drug bioavailability after oral dosing and, in turn, the fraction of drug that can enter effect sites, such as the breast. In breast cancer patients, surgical resection of the primary tumor and radiation therapy also disrupts the local vasculature of the breast. Thus, it is possible that fluctuations in the systemic drug concentrations of orally administered SERMs and AIs can result in momentary instances of autocrine signaling of estrogen in the breast and contribute to cancer cell survival.
3.2. Pharmacologic basis for the treatment of ERBB2+ breast cancer
ERBB2, also known as HER2, is a membrane bound tyrosine kinase that is overexpressed in several cancers including breast cancer. Overexpression of ERBB2 promotes homo- or heterodimerization with other ERBB (1 through 4) receptors and dimerization with other receptors in activating cell proliferation pathways (M. Tan & Yu, 2007). There are a few key factors that make the ERBB2 pathway particularly oncogenic: (1) ERBB2 has a relatively slow rate of endocytosis and can remain overexpressed on a cell surface for an extended period. (2) ERBB2 heterodimers have been shown to undergo recycling instead of degradation, and (3) activation of ERBB2 heterodimers persists due to the slow release of ligands from the complex (Harari & Yarden, 2000). These factors allow ERBB2 to stay present and active on the breast cancer cell surface while also making these proteins attractive pharmacologic targets for the treatment of breast cancer.
Different therapeutic modalities are used to target ERBB2 including monoclonal antibodies, small molecule tyrosine kinase inhibitors, and antibody drug conjugates. The monoclonal antibodies trastuzumab and pertuzumab bind to different extracellular domains of ERBB2 to mediate their effect. Trastuzumab binds to domain IV of ERBB2, which is not involved in heterodimerization (Fuentes, Scaltriti, Baselga, & Verma, 2011). Pertuzumab, on the other hand, binds to domain II (a region associated with dimerization) and may sterically hinder heterodimerization of other ERBB receptors. The net effect of either pertuzumab or trastuzumab binding to ERBB results in antibody dependent cell-mediated cytotoxicity and the inhibition of tumor growth (Agus, et al., 2002; Franklin, et al., 2004; Sliwkowski, et al., 1999). The different binding domains of trastuzumab and pertuzumab allow both agents to be used in combination to achieve synergistic clinical effects (J. Baselga, et al., 2012; Tolaney, 2014).
Targeting ERBB2 to treat breast cancer is not only achieved through monoclonal antibodies, but also through small molecule kinase inhibitors such as lapatinib and neratinib. Both neratinib and lapatinib bind to the intracellular regions of ERBB receptors and prevent autophosphorylation and downstream effects. The main difference between the two drugs are their mechanisms of inhibition and affinity for different members of the ERBB family. Lapatinib is a potent reversible inhibitor of ERBB1 (EGFR) and ERBB2 (HER2) and binding ERBB2 results in decreased signaling in the P13K (phosphoinositide 3-kinases) and MAPK (mitogen-activated protein kinase) pathways, pushing the cell towards apoptosis. Neratinib is an irreversible inhibitor of ERBB1 and ERBB2 but also ERBB4 and mediates the same inhibition of the P13K and MAPK signaling. Interestingly, lapatinib has been shown to stabilize HER2 expression on the cell surface and may synergize with trastuzumab therapy (Scaltriti, et al., 2009), while neratinib decreases HER2 cell surface expression without the need for additional monoclonal therapy (Collins, et al., 2017).
The final modalities used to target ERBB2 are antibody-drug conjugates where the monoclonal antibody, trastuzumab, is used as a delivery vehicle for highly cytotoxic agents such as emtansine and deruxtecan. Emtansine is a maytansine derivative that acts as a potent tubulin inhibitor. Early studies of maytansine showed that the compound was too toxic for use as a chemotherapy in its free form. To mitigate those toxicities, future derivatives were conjugated to monoclonal antibodies to target cancer cells only (Widdison, et al., 2006). Emtansine is conjugated to trastuzumab through a non-cleavable thioether linker, which prevents the release of emtansine in the systemic circulation. After trastuzumab binds to ERBB2 and receptor mediated endocytosis occurs, proteolytic degradation of trastuzumab releases emtansine and causes cell death (Barok, Joensuu, & Isola, 2014). In a similar fashion, deruxtecan is a novel camptothecin analog that inhibits topoisomerase 1 and is chemically conjugated to trastuzumab with an enzymatically cleavable peptide linker. The release of trastuzumab deruxtecan also occurs through receptor mediated endocytosis. However, trastuzumab deruxtecan carries 8 deruxtecan molecules for every antibody, compared to trastuzumab emtansine, which has a 3.5 to 1 drug to antibody ratio. This high loading capacity is thought to enable a bystander effect where cancer cells adjacent to the ERBB2+ cell will also experience cytotoxicity (Chen, et al., 2016; Nakada, Sugihara, Jikoh, Abe, & Agatsuma, 2019).
3.3. Pharmacologic basis for the treatment of triple negative breast cancer (TNBC)
While most breast cancer express druggable receptors responsive to current drug therapies, there are a small number of breast cancers that do not express these receptors, often referred to as triple negative breast cancer (TNBC) phenotypes. TNBC does not express druggable, well-defined molecular targets; consequently, currently available drugs that are targeted to specific receptors such as ER, PR or HER2 are not effective. While MBC may be associated with TNBC, not all TNBC may be MBC or vice versa. Chemotherapeutic agents that are not targeted to these molecular targets, but work through other pharmacological mechanisms such as those that can interfere with rapidly replicating cells are effective in TNBC. Therapeutic strategies using these agents are referred to as chemotherapy and it is often given in sequential, defined drug combinations that are key treatment options for patients presenting with TNBC. By targeting various points in cancer cell replication, combination chemotherapy can maximize pharmacologic effect and prevent drug resistance. These combination regimens are typically composed of either anthracyclines or taxanes in combination with other cytotoxic drugs. As an example, anthracyclines (doxorubicin, epirubicin, daunorubicin) are used in combination with alkylating agents (cyclophosphamide, ifosfamide) and taxanes (paclitaxel, docetaxel) as a preferred regimen for post-surgical TNBC. Anthracyclines target topoisomerase II to cause cell cycle arrest in the G1 phase, alkylating agents damage DNA to cause cell death and taxanes bind to microtubules to cause cell cycle arrest in the M phase. Alternative regimens can include the use of antimetabolites such as gemcitabine and capecitabine, which act by blocking DNA replication. By targeting different points in cancer cell replication, the likelihood of cancer cell death is high. However, the cumulative toxicity to healthy tissue limits the effectiveness of these agents.
Extended use of anthracyclines results in heart failure due to the irreversible binding of anthracyclines to cardiolipin, which is highly concentrated in slow growing heart tissues. The accumulation of cytotoxic anthracycline drug molecules eventually kills the cells of heart muscles leading to heart failure. This cardiotoxicity is lifetime dose-limiting and restricts the use of anthracyclines in late-stage disease. Alkylating agents also have a number of dose-limiting toxicities including GI, hematopoietic, gonadal and pulmonary toxicities, but these are more clinically manageable than the cardiotoxicity of anthracyclines (van der Wall, Beijnen, & Rodenhuis, 1995). Taxanes are associated with neurotoxicity, hypersensitivity reactions and myelosuppression, which can be dose-limiting but can also be managed clinically (Clemons, et al., 1997; Weiss, et al., 1990). Antimetabolites generally have the safest toxicity profile compared to other agents, but they have limited efficacy in monotherapy (Blackstein, et al., 2002; Feher, et al., 2005). All of these therapeutic agents are highly potent and have the ability to eliminate cancer cells. Unfortunately, these drugs broadly distribute into healthy tissue. Many of the adverse events listed above are the result of higher drug burdens in off-target tissues compared to cancer cells. If these agents could be directed towards cancer cells exclusively, breast cancer treatment could be improved but this is not achievable with the current clinical infusion protocols.
4. Current gaps in pharmacologic treatment of breast cancer: biological mechanisms for disease progression
The previous section outlines the various pharmacologic approaches used to target different subtypes of breast cancer. Despite the availability of these therapies, drug resistance and disease progression continue to persist as a major issue in breast cancer. Distant recurrent breast cancer is widely regarded as incurable and occurs in ~30% of all patients, despite the high overall 5-year survival rate of breast cancer (~90%). This suggests that pharmacologic treatment in breast cancer is effective at suppressing cancer cells, but that current treatments cannot eliminate them entirely. This gap in breast cancer treatment could be due to a number of factors including acquired drug resistance, poor drug exposure in tissues or cells of interest, or off-target toxicity associated with treatment. In the past few years, the FDA has approved several new drugs for metastatic breast cancer to address these gaps. These newly approved drugs are mostly used in combination with previously approved drugs and have shown promising results in MBC (Table 2). The following section will identify the major biological mechanisms within subtypes of breast cancer cells that lead to disease progression and drug combinations that can overcome these mechanisms.
Table 2.
Select drugs approved and indicated to treat metastatic breast cancer as mono or combination therapies in the past 5 years (2015–2020).*
| Common (Brand) name | MBC drug target (phenotype) | Drug platform Small or biologic molecules | Combination or mono-drugtherapy | Route | Pharmacological mechanisms | Efficacy (Primary outcome measure) | Main side effects | Ref |
|---|---|---|---|---|---|---|---|---|
| Approved in 2020 | ||||||||
| Margetuximab (Margenza) | HER2 (HER2+) | Monoclonal antibody (Biologics) | In combination with capecitabine, eribulin, gemcitabine, or vinorelbine | IV | An Fc-engineered chimeric anti-ERBB2 immunoglobulin G1 that has increased activation of antibody-dependent cellular cytotoxicity and natural killer cells, relative to trastuzumab. | Median PFS in the margetuximab group was 5.8 months vs 4.9 months in the control group | Infusion-related reactions, left ventricular dysfunction | (Rugo, et al., 2021) |
| Sacituzumab-govitecan (Trodelvy**) | Trop-2, topoisomerase-I (TNBC) | Antibody-drug conjugate (Biologic) | Mono-drug therapy | IV | This drug contains SN-38. The mAb delivers SN-38 into Trop-2+ TNBC cells. SN-38 is topoisomerase-I inhibitor. | Median PFS in sacituzumab govitecan group was 4.8 months vs 1.7 months in chemotherapy group | Neutropenia, hypersensitivity | (Bardia, et al., 2021) |
| Tucatinib (Tukysa) | HER2&3 kinase (HER2+) | Small Molecule | In combination with trastuzumab and capecitabine | PO | Tucatinib inhibits phosphorylation of HER2 and HER3, resulting in inhibition of downstream MAPK and AKT signaling and cell proliferation | Median PFS in tucatinib combination group was 7.8 months vs 5.6 months in placebo combination group | Elevated aminotransferase levels | (Murthy, et al., 2020) |
| Approved in 2019 | ||||||||
| Trastuzumab-deruxtecan (Enhertu) | HER2, Topoisomerase-I (HER2+) | Antibody-drug conjugate (Biologic) | Mono-drug therapy | IV | Trastuzumab-deruxtecan targets HER-2+ MBC. Deruxtecan(Dxd) is a topoisomerase I inhibitor. | The median response duration was 14.8 months and the median PFS was 16.4 months | Neutrophil count decreases | (Modi, et al., 2020) |
| Alpelisib (Piqray) | PI3K (HR+) | Small Molecule | In combination with fulvestrant | PO | Alpelisib selectively inhibits PI3K in the PI3K/AKT kinase signaling pathway, which results in inhibition of tumor cell proliferation. | Median PFS was 11.0 months in the alpelisib plus fulvestrant group vs 5.7 months in the placebo plus fulvestrant group | Hyperglycemia | (Andre, et al., 2019) |
| Approved in 2018 | ||||||||
| Talazoparib (Talzenna) | PARP (BRCA, HER2−) | Small Molecule | In combination with capecitabine, eribulin, gemcitabine, or vinorelbine | PO | Talazoparib binds to and inhibits PARP enzymatic activity, then increases formation of PARP-DNA complexes resulting in DNA damage in tumor cells. | Median PFS was 8.6 months in talazoparib group vs 5.6 months in the standard therapy group | Anemia, thrombocytopenia | (Litton, et al., 2018) |
| Approved in 2017 | ||||||||
| Abemaciclib (Verzenio) | CDK4/6 (HR+, HER2−) | Small Molecule | In combination with fulvestrant | PO | Abemaciclib inhibits CDK4 and CDK6, thus, it inhibits retinoblastoma protein phosphorylation in G1 phase resulting in arresting the cell cycle in the G1 phase | Median PFS was 16.4 in abemaciclib plus fulvestrant group vs 9.3 months in placebo group | Diarrhea, neutropenia | (George W. Sledge, et al., 2017) |
| Ribociclib (Kisqali) | CDK4/6 (HR+, HER2−) | Small Molecule | In combination with endocrine therapy, also received either as a nonsteroidal aromatase inhibitor or tamoxifen and goseline | PO | Ribociclib is an inhibitor of CDK 4 and 6 which interferes the phosphorylation of retinoblastoma protein and leads to arrest the cell cycle in the G1 phase. | Median PFS was 25.3 months in ribociclib plus letrozole group vs16.0 months in placebo plus letrozole group | Neutropenia, leucopenia | (Hortobagyi, et al., 2018) |
| Approved in 2015 | ||||||||
| Palbociclib (Ibrance) | CDK4/6 (HR+) | Small Molecule | In combination with letrozole | PO | Palbociclib is an inhibitor of CDK4 and CDK6 causing the prevention of Rb phosphorylation and E2F1 release, therefore leads to arrest the cell cycle in the G1 phase | Median PFS was 24.8 months in the palbociclib plus letrozole group vs 14.5 months in the placebo plus letrozole group | Neutropenia | (Finn, et al., 2016) |
Please see abbreviation list for the full names (PARP, CDK4/6, HR, HER2, E2F1, PFS).
Trodelvy received accelerated approval in April 2020 and regular approval in April 2021.
4.1. Biological mechanisms for disease progression in ER/PR+ cancer
As the understanding of ER action has evolved over the years, the complexity of ER signaling has shed light on the mechanisms of drug resistance. Breast cancer treated with anti-estrogen therapy can proliferate through alternate growth pathways or retain activity through the ER pathway in the presence of anti-estrogen therapy. For example, ER+ breast cancers that overexpress epidermal growth factor receptors (ERBB family) can undergo loss of ER expression and proliferate through the ERBB pathway causing tumors to become hormone independent, rendering hormone therapy ineffective (Arpino, Wiechmann, Osborne, & Schiff, 2008; Osborne, Shou, Massarweh, & Schiff, 2005; Schiff, et al., 2004). Alternatively, increased levels of specific coactivator proteins like AIB1 can shift the effect of the tamoxifen-ER complex from antagonism to agonism, leading to drug resistance (Osborne, et al., 2003; Smith, Nawaz, & O’Malley, 1997). This effect is due to the large role transcriptional co-regulators have on ER gene expression (McDonnell & Wardell, 2010). The activation of the ER pathway is dependent on the recruitment of coregulator proteins and differences in protein levels (such as AIB1) in the cell cytoplasm or nucleus can change the effects of ligand binding to ER. The shift from antagonism to agonism effects of tamoxifen as a function of co-regulator protein expression is likely what causes the tissue specific effects observed in tamoxifen therapy (i.e., breast vs ovary) (Hall & McDonnell, 2005). The ability of cancer cells to utilize other growth pathways and the sensitivity of the ER pathway to other co-regulatory proteins both serve as driving factors behind intrinsic and acquired drug resistance.
To overcome these mechanisms, combination therapy is often used. For example, a combination of the aromatase inhibitor, letrozole, with the selective estrogen receptor down regulator, fulvestrant, utilizes two independent mechanisms to target hormone responsive cancer cells (Johnston, Martin, Head, Smith, & Dowsett, 2005). Letrozole reduces the endogenous levels of estradiol required to bind to the ER complex and cause cell proliferation. Fulvestrant binds to the ER and reduces the absolute levels of the receptor. By using both drugs, the substrate and the receptor are inhibited, and cell proliferation through the ER pathway does not occur. Although this approach is effective, it does not prevent the activation of alternative growth pathways such that drug resistance still emerges (Bunone, Briand, Miksicek, & Picard, 1996). Currently, the most effective combination therapy is letrozole with palbociclib, a CDK4/6 inhibitor. CDK4/6 proteins are highly conserved in ER/PR+ breast cancers and are downstream from growth hormone signaling (Preusser, et al., 2018). Depletion of endogenous estradiol through letrozole and the inhibition of CDK4/6 prevent signaling through the ER pathway and promote cell death. Unfortunately, even with the best available options, cancer recurrence and progression remain a life-long issue (Pan, et al., 2017).
4.2. Biological mechanisms for disease progression in ERBB2+ cancer
Targeting ERBB2 has made significant improvements in the overall survival of ERBB2+ breast cancer patients. However, most patients will still have disease progression and metastasis to the brain (Pernas & Tolaney, 2019). Disease progression in ERBB2+ cancer cells is mediated through mechanisms similar to ER/PR+ resistance mechanisms. ERBB signaling can be activated downstream from the point of inhibition or an alternative growth pathway can be present. Specifically, the P13K pathways can be activated by the loss of PTEN expression, a signaling event downstream from ERBB2 dimerization (José Baselga, et al., 2016). The incomplete blockade of ERBB2 and compensatory mechanisms by receptors in the ERBB family (such as ERBB3) can also lead to resistance (Pernas & Tolaney, 2019). The activation of other signaling receptor tyrosine kinases such as IGF-1 has also been associated with ERBB2+ drug resistance (Lu, Zi, Zhao, Mascarenhas, & Pollak, 2001).
To overcome these mechanisms, several combination approaches are used. One approach has been to use both trastuzumab and pertuzumab in combination to maximize the ERBB2 blockade at the cell surface. In so doing, trastuzumab directly blocks the ERBB2 receptor while pertuzumab prevents heterodimerization with any compensatory family receptors such as ERBB3 (J. Baselga, et al., 2012). Alternatively, trastuzumab and lapatinib have been used concomitantly to inhibit ERBB2 on the cell membrane (trastuzumab) and intracellularly (lapatinib) (Blackwell, et al., 2012). There is some evidence to suggest that the irreversible binding of neratinib to ERBB2 can overcome trastuzumab resistance; however, resistance to neratanib will also develop (Breslin, Lowry, & O’Driscoll, 2017; Canonici, et al., 2013). It is important to note that ERBB2 targeted therapies are given in conjunction with conventional chemotherapy to preemptively counter the rapid emergence of drug resistance. Thus, antibody-drug conjugates with trastuzumab loaded with either emtansine or deruxtecan are in essence combination approaches wherein chemotherapy and monoclonal antibodies are administered as a single entity (Dhillon, 2014). Both trastuzumab emtansine and trastuzumab deruxtecan are only used in patients that have previously received ERBB2 targeted therapies with conventional chemotherapy.
4.3. Biological mechanisms for disease progression in TNBC
Due to the various pharmacologic agents used in TNBC (IE: alkylating agents, topoisomerase inhibitors, tubulin inhibitors and antimetabolites), the mechanisms of resistance are diverse. In general, the major cellular mechanisms that lead to drug resistance and disease progression are based on increased drug inactivation and increased drug efflux. In addition to the cellular mechanisms that can lead to drug resistance, off-target toxicity in patients undergoing chemotherapy can lead to disease progression as patients can no longer bear the burden of additional treatment.
Many of the drugs used in TNBC require metabolic activation in order to perform their pharmacologic function. For example, the nucleoside analogue gemcitabine is sequentially phosphorylated to gemcitabine-triphosphate (gem-TP) before being integrated into DNA and causing chain termination. The enzyme responsible for the first phosphorylation event of gemcitabine is deoxycytidine kinase (dCK) and dCK activity is generally accepted as the limiting factor in gemcitabine therapy. dCK has been shown to saturate when plasma levels of gemcitabine are around 15–20 μM and dCK activity is strongly correlated to gemcitabine sensitivity (Abbruzzese, et al., 1991; Grunewald, Abbruzzese, Tarassoff, & Plunkett, 1991; Kroep, et al., 2002). Interestingly, the competing inactivation of gemcitabine is also strongly correlated with gemcitabine effects. The majority of gemcitabine clearance is driven by metabolic inactivation to dFdU by cytidine deaminase (CDA). This clearance pathway accounts for 90% of the fraction metabolized for gemcitabine, with the remaining clearance occurring through renal filtration (Peters, et al., 2007). In comparison to the activation pathway of dCK, the metabolic inactivation of gemcitabine appears to saturate at much higher concentrations. In one pharmacokinetic study, the linear plasma pharmacokinetics of gemcitabine were observed at doses up to 4000 mg/m2; however, activated gem-TP levels peaked at 960 mg/m2 and higher dosing did not increase measured gem-TP levels (Peters, et al., 2007). Thus, any mutations that shift the relationship between gemcitabine activation and inactivation (i.e., dCK downregulation or CDA overexpression) can lead to drug resistance. Alternatively, reduced CDA expression is also associated with increased toxicity (Mercier, et al., 2007).
Another mechanism for TNBC drug resistance is the active efflux of chemotherapy from breast cancer cells. This phenomenon is mediated by ATP-Binding Cassette (ABC) transporters: specifically P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP). There are other ABC transporters that cause multi-drug resistance, such as multi-drug resistance associated protein 1 (MRP1) and lung resistance protein (LRP), but their expression has limited relevance in breast cancer. P-gp is an extensively studied ATP-dependent efflux pump that transports several drugs used to treat breast cancer including doxorubicin, paclitaxel, and vinblastine (Nanayakkara, et al., 2018). In healthy tissue, P-gp is expressed in the GI, kidneys, liver and blood brain barrier and plays an important role in governing the oral absorption of xenobiotics (B. Tan, Piwnica-Worms, & Ratner, 2000). More importantly, P-gp is expressed in ~40% of breast cancers and is shown to be inducible by radiation therapy (Ng, Lam, Ng, Kwong, & Sham, 1998; Trock, Leonessa, & Clarke, 1997). An increase in P-gp expression is associated with drug resistance and suggests that P-gp efflux can prevent chemotherapeutic agents from binding to their intracellular targets. BCRP is another transporter protein that acts in a similar manner to P-gp to cause drug resistance. BCRP can be upregulated in breast cancer cells and can efflux topoisomerase I inhibitors, methotrexate and anthracyclines under certain conditions (Austin Doyle & Ross, 2003).
In contrast to ER/PR or ERBB2 targeted agents, disease progression after chemotherapy is typically due to reduced drug exposure and residence time at the intracellular target leading to reduced effects. Off-target toxicity and systemic adverse events also limit the achievable concentration of drugs in cancer cells. As such, combination chemotherapy is utilized with the rationale that cancer cells are unlikely to have multiple resistance mechanisms to overcome two different drugs targets. By using combination therapy, lower individual drug dosing can be administered to achieve a greater effect (although the total dose is still high). For example, in the case of gemcitabine and paclitaxel combination therapy, upregulation of P-gp can enable increased efflux of paclitaxel that would likely cause drug resistance in monotherapy. However, it would be less likely for a cancer cell to have both increased efflux of paclitaxel (upregulated P-gp) and increased metabolism of gemcitabine (upregulated CDA) in the same cell. Based on this rationale, combination chemotherapy would likely overcome drug resistance. Unfortunately, drugs used in chemotherapy display varying dispositions, clearance mechanisms and plasma concentration time courses. Thus, the synchronization of combination drugs in target breast cancer cells after intravenous administration is unlikely. Additionally, the use of combination chemotherapy compounds places toxicological burdens on the body and adverse events may limit the effective doses that patients can receive.
4.4. Biological factors leading to disease progression and limitations of conventional combination chemotherapy
Across the various subtypes of cancer, drug resistance, off-target toxicity and disease progression continue to be major issues. In all forms of breast cancer, combination therapy has a role in addressing drug resistance, but cancer cells are incredibly adept at altering signaling pathways and developing intracellular mechanisms to prevent drugs from reaching their targets. Cancer cells can also develop resistance mechanisms to counteract the antitumor effects of drugs. For example, DNA repair processes can be upregulated in cancer cells to overcome the cytotoxic effects of DNA alkylating agents. Physiological factors can also play a role in causing drug resistance by limiting drug exposure in sites of effect. For example, breast cancer cells invade through the lymphatic and circulatory systems. Drug exposure in the systemic circulation is well established, but there is limited penetration of drugs into the lymph by conventional dosage forms (e.g., solubilized drugs) where cancer cells can grow. Off-target toxicity of these free drugs further limits the total dose that can be safely administered intravenously to patients and reduces the maximum achievable concentration of drugs in target tissues where cancer cells may reside. Under these conditions, the therapeutic levels required to kill cancer cells may not be achieved in tissues like the lymph, allowing persistent cancer cells to grow. Furthermore, existing breast cancer combination therapy regiments are based on sequential and separated administrations of drugs. The time windows for drug co-existence in plasma and tissues above therapeutic levels are usually brief (e.g., ~4 h for a gemcitabine/paclitaxel combination regimen). The asynchronized plasma and tissue pharmacokinetics of the drug pairs may promote drug resistance by cancer cells while causing toxicities to healthy tissues.
Considering the various pharmacologic targets in breast cancer and the resistance mechanisms that occur from treatment, there is a clear distinction between targeted (ER/PR, ERBB2) and untargeted (chemotherapeutic) drug resistance. The pharmacology of targeted agents can change as cancer cells shift from one signaling pathway to another. When resistance to ER/PR or ERBB2 targeted agents occurs, drugs can still engage their target receptors, but the cancer cells can still proliferate through other pathways. In contrast, the pharmacology of chemotherapy is relatively stable over time and drug binding to target proteins will generally retain cytotoxic effects. Disease progression from chemotherapy is due to resistance mechanisms preventing drug molecules from reaching their intracellular targets (e.g., efflux drug transporters or pumps such as P-gp, MRP, BCRP) or from patients suffering too many adverse events. In principle, we can overcome the limitations in chemotherapy by using rationally designed vehicles to deliver combination chemotherapy specifically to target cancer cells instead of healthy tissue. Advanced delivery approaches will also be able to deliver drugs in combination regimens into the body at the same time, therefore synchronizing the pharmacokinetics. Another advantage with a specific carrier is that the drugs can be delivered specifically to lymphatics where cancer cells metastasize. In the following sections, we will explore advanced delivery systems for metastatic breast cancer combination chemotherapy in both preclinical and clinical investigations.
5. Strategies to overcome drug insufficiencies and improve cancer cell drug exposure
The intent of targeted drug delivery systems is to concentrate drugs in tissues and cells of interest while sparing healthy organs from unnecessary exposure. If designed with appropriate fit-for-purpose intent, targeted drug delivery systems can overcome drug insufficiencies. For a targeted drug delivery system to transport drugs from an injection site to specific cancer tissues and produce pharmacologic action, a number of criteria must be considered and achieved. The carriers should be engineered to have precise characteristics such as size, shape, surface charge and composition to produce specific effects such as defined distribution, exposure and clearance profiles in vivo. Due to these additional characteristic requirements and demands, the formulation of drug delivery systems is more complex than conventional dosage forms and requires more process control. Despite these challenges, some drug delivery systems have been developed and have proven useful as strategies to overcome drug insufficiency with acceptable safety profiles (Figure 2). The purpose of the following section will be to summarize the current pharmaceutical approaches for drug targeting of chemotherapy based on drug delivery systems to treat breast cancer. The challenges and benefits associated with each drug delivery system will be discussed before providing the rationale for the need to develop a novel drug delivery system that is targeted and capable of simultaneously delivering multiple chemotherapeutic agents to tissues and cells at higher concentrations and longer duration.
Figure 2.

Examples of existing nanoscale drug carriers for treatment of metastatic breast cancer (MBC) and introduction of an innovative lipid-based drug combination nanoparticle (DcNP). Liposomes (A), polymeric nanoparticles (B), dendrimers (C) and antibody-drug conjugates (D) are among the most investigated in clinical settings. Drugs or their combinations, depending on their hydrophobicity/water solubility, are loaded either in aqueous cavities of these vehicles or hydrophobic regions of lipophilic structures. The drugs are either non-covalently encapsulated inside the nanoparticles, or covalently conjugated to the inner or outer areas of these carriers. Each of the existing nanocarriers has their own advantages and disadvantages comprehensively summarized in the literature. The DcNP (E) is a new type of nanocarrier for anti-MBC drug combination delivery (Mu, et al., 2020; Jesse Yu, 2020; J. Yu, Mu, et al., 2020). DcNPs possess several key advantages over the existing nanocarriers bullet pointed in the figure. Their clinical efficacy and adverse effects are worth in-depth investigations.
5.1. Clinically used nanoplatforms for single drug delivery and limitations
Modification of drug disposition and clearance through pharmaceutical carriers is an important strategy in delivering therapeutic agents to breast cancer cells versus healthy cells. Pharmaceutical carriers are typically nano- to micro-meter sized particles that can circulate in the systemic circulation and distribute into cancer tissue from the leaky vasculature formed around tumors (a phenomena known as the enhanced permeation and retention effect or EPR) (Golombek, et al., 2018). As mentioned above, examples of these carriers include liposomes, polymeric nanoparticles, dendrimers and antibody drug conjugates (Figure 2). In general, the rationale for associating drugs to particles is that particle-bound drugs will have prolonged residence in systemic circulation with a limited free fraction of drugs. The reduced free fraction of drugs from particle binding thus results in less off-target toxicity. As particle-bound drugs continues to circulate in the blood, accumulation in tumors can be achieved through different approaches such as active targeting or EPR. Besides the clinically used platforms, many other nanoparticle approaches have investigated for either single or combination drug delivery such as quantum dots (Probst, Zrazhevskiy, Bagalkot, & Gao, 2013), iron oxide nanoparticles (Vangijzegem, Stanicki, & Laurent, 2019), ceramic nanoparticles (Zang, et al., 2019), carbon nanoparticles (Attia, et al.), metal-organic framework (C. Y. Sun, Qin, Wang, & Su, 2013), etc. Although many of these approaches are materially novel, they are not scalable, well understood, pose safety concerns (such as high liver/spleen accumulations), or are not suitable for drug combination formulation and thus not discussed in detail here (Maurer-Jones, Bantz, Love, Marquis, & Haynes, 2009; Paliwal, Babu, & Palakurthi, 2014; Wicki, Witzigmann, Balasubramanian, & Huwyler, 2015a). In the following, we discuss a few of most promising and clinically used nanoplatforms that are used for treatment of breast cancer or metastatic breast cancer.
5.1.1. Liposomes or lipid membrane vesicles
Liposomes are spherical vesicles composed of phospholipids with a bilayer structure similar to cell membranes. Due to their aqueous core and lipid shell, liposomes have been used to incorporate both hydrophilic and hydrophobic drugs. In drug delivery, liposomes are the most studied and clinically successful nanoparticle system. For example, the pegylated liposome, Doxil, encapsulates the anthracycline doxorubicin to target various types of solid tumors. The major limitation in anthracycline therapy is the irreversible cardiotoxicity caused by binding to cardiolipin in the heart. Although the lifetime dose limitation for liposomal doxorubicin is the same as conventional doxorubicin, there is evidence to suggest that encapsulation reduces the cardiotoxicity by limiting the exposure of free doxorubicin to cardiac tissue (O’Brien, et al., 2004). In addition, liposomal doxorubicin has been shown to localize in Kaposi’s Sarcoma lesions 10-fold greater than conventional doxorubicin (Northfelt, et al., 1996). By enabling a higher concentration of doxorubicin in the sarcoma lesions at the same dose as conventional doxorubicin, Doxil may have a greater pharmacologic effect against those cells and overcome drug resistance.
Liposomes are one of the most well-studied drug carrier systems and have demonstrated applications in the clinic (Saraf, et al., 2020). However, there are also limitations to this drug delivery system. Depending upon the lipid composition, liposomes can be associated with extensive hepatic and splenic uptake (Kraft, et al., 2014). For example, large (378 nm) and small (113 nm) liposomes were intravenously administered to mice and, after 4 hours, 93% of large liposomes and 67% of small liposomes were recovered in the liver and spleen (Hu, et al., 1996). Pegylated liposomes can reduce the splenic and hepatic uptake of liposomes for a single dose, but accelerated blood clearance is observed after multiple dosing (Ishida, et al., 2005). Many studies have incorporated active targeting ligands to the surface of liposomes, but this approach has yet to be tested in the clinic. The hepatic/splenic uptake of liposomes remains as a major barrier to liposomal targeted chemotherapy delivery and further study is required to optimize these drug delivery systems.
5.1.2. Polymeric Nanoparticles
Another set of drug carriers used to enable cancer targeting are polymeric nanoparticles made of amphiphilic copolymers. When placed in aqueous solution, polymers can self-assemble to form a hydrophobic core to stabilize water insoluble drugs. Polymeric nanoparticles can be calibrated based upon the length of the monomers while polymer residues can be used as anchor points for covalent conjugation to improve stability. For example, CT-2103 is a polymer-drug nanoparticle that conjugates hydrophobic paclitaxel to water soluble, poly-L-glutamic acid polymers. Poly-L-glutamic acid is biodegradable and is broken down to glutamic acid. Chemical conjugation of paclitaxel occurs at the 2’-hydroxyl position, which is necessary for tubulin inhibition, and assures that conjugated paclitaxel will not have off-target effects (Singer, et al., 2005). Preclinical studies show that polymer-bound paclitaxel produces a 5-fold greater tumor AUC compared to conventional paclitaxel (Li, et al., 2000). Additionally, polymer-bound paclitaxel significantly increases aqueous solubility of the paclitaxel dosage form. Conventional paclitaxel is solubilized in a Cremophor EL/Ethanol co-solvent that is associated with significant hypersensitivity reactions70. By circumventing the need for these excipients, polymer-bound paclitaxel also reduces the toxicity of paclitaxel treatment and allows a higher dose to be administered. By administering a higher dose of drug, drug resistant cells can be eliminated without causing undue burden to the rest of the body. In addition to CT-2103, many other polymeric nanoparticles have advanced into clinical studies and show great promise (Afsharzadeh, Hashemi, Mokhtarzadeh, Abnous, & Ramezani, 2018; Wicki, et al., 2015b).
Many polymeric nanoparticles have advanced to clinical studies but face similar biological barriers as liposomes. For example, a mass balance study of CT-2103 administered IV produced an 8-fold greater paclitaxel AUC in the liver compared to the primary tumor in mice (Li, et al., 2000). In addition to hepatic and splenic uptake, very small polymeric particles (<10 nm) can be passively filtered through the glomerulus which may present an additional barrier to tumor targeting. Surprisingly, a recent study demonstrated that even large polymeric particles (~400 nm) composed of PLGA-PEG could accumulate in the kidney cells and tissues through active endocytosis by peritubular endothelial cells (Williams, et al., 2015). This effect was not related to the surface charge of the particle and highlights the importance of understanding the biodistribution of drug carriers to enable tumor targeting. If renal accumulation were to occur, it could pose a potential toxicological effect from both the free drug and the polymeric particles.
5.1.3. Dendrimers
Dendrimers are macromolecules that are synthesized by sequential chemical reactions to form “generations” (referred as G0, G1, G2) of branches originating from a central core. Each sequential addition of a generation produces a radiating, well-defined, spherical morphology branching from the central core. Dendrimer size can be precisely controlled, and the surface properties of dendrimers are easily functionalized to produce highly monodisperse particles. The precision with which dendrimers can be modified allows novel approaches to targeting such as covalent attachment of monoclonal antibodies (Marcinkowska, et al., 2018). Dendrimer synthesis is generally achieved through either a diverging method or a converging method (Abbasi, et al., 2014). The diverging method begins with a central core to which sequential arms are added in a step-wise fashion. The converging method pre-assembles the exterior arms of the dendrimer and covalently attaches them to the core as a final step. The flexibility with which dendrimers can be synthesized has led to the development of diverse structures such as block dendrimers, heterobifunctional dendrimers, and core-functionalized dendrimers (Sowinska & Urbanczyk-Lipkowska, 2014). Although there is great potential for dendrimers, the ideal linkers, size and in vivo characteristics of dendrimer drug conjugates are still being identified in preclinical species with only a few candidates currently approved for early phase clinical trials (Afsharzadeh, et al., 2018; Alven & Aderibigbe, 2020; Kojima, 2015; Longmire, Ogawa, Choyke, & Kobayashi, 2011; Luo, et al., 2012). Early biodistribution studies have shown that dendrimers can distribute into the kidney, liver and spleen. Kidney accumulation of dendrimers is thought to occur through electrostatic interactions between the cationic exterior of dendrimers and the negative charge of the glomerular basement membrane (Mager, et al., 2012; Okuda, Kawakami, Akimoto, et al., 2006; Okuda, Kawakami, Maeie, et al., 2006). Pegylation of dendrimers can minimize this interaction and reduce renal accumulation (Okuda, Kawakami, Akimoto, et al., 2006). Dendrimers are also known to distribute into pancreas to the same extent as the liver and spleen, which may represent a toxicological risk. Dendrimers enter cells through endocytic pathways and some data suggests that excess endocytosis of dendrimers may lead to hepatic toxicity (Duncan & Izzo, 2005; Roberts, Bhalgat, & Zera, 1996). Dendrimers are a relatively new drug delivery system and the specific physical properties that affect biodistribution and clearance of these macromolecules are still being defined. Once these properties are optimized, dendrimers may be a relevant strategy for drug delivery but the current biodistribution profile (accumulation in kidney, liver, spleen and pancreas) and off-target toxicity risk limits their application in clinical treatment.
5.1.4. Anti-body Drug Conjugates
Although previously addressed in the ERBB2+ targeted therapy section, antibody conjugates (ADC) also constitute a form of targeted drug delivery. Antibodies are nano-biological materials with ~5–10 nm in diameter, thus they are considered a type of nanocarrier in this review. These nanosized molecules with distinct characteristics can provide high receptor specificity,affinity and long circulation in plasma. Due to these attributes, drugs are conjugated onto antibodies or antibody fragments to target various receptors. By covalently linking cytotoxic drugs to antibodies, ADCs reduce distribution to healthy tissue and selectively target cancer cells. After binding to target receptors and undergoing receptor mediated endocytosis, drugs can be released intracellularly for effect. Recently, clinical success with monoclonal ADCs for the treatment of various cancers (such as Kadcyla for ERBB2+ breast cancer and Adcetris for CD30+ lymphomas) has demonstrated the value of this approach in targeted drug delivery. This clinical success was achieved through significant effort and optimization. For example, the earliest generations of ADCs had limitations such as immunogenicity and relatively low in vivo stability. Gemtuzumab ozogamicin (GO) is an ADC designed to deliver highly toxic calicheamicin to CD33+ leukemia cells. Although effective and capable of achieving complete remission in 15–20% of patients with relapsed disease, GO is associated with major toxicities including myelosuppression and hepatoxicity (Giles, 2002). These toxicities are likely related to the (relatively) unstable linker between calicheamicin and gemtuzumab, with almost 50% of the drug being released after 48 hours in mice (Doronina, et al., 2003; Ho & Chien, 2014). As ADC technology has matured, the trend has shifted towards utilizing more stable linkers that can carry more potent payloads (such as those used in Kadcyla) (Bargh, Isidro-Llobet, Parker, & Spring, 2019; Doronina, et al., 2003). A focus on several key factors such as target selectivity, drug-antibody linkers, product homogeneity and finding the right patient population has resulted in clinical success and approval of multiple ADCs for the treatment of cancer.
ADCs can overcome the limitations of broadly toxic chemotherapy by using specific monoclonal antibodies targeted to receptors on cancer cells to deliver potent, cytotoxic drugs preferentially. In breast cancer, well-defined signaling pathways such as ERBB2 present a validated molecular target for ADCs (as used in Kadcyla). Unfortunately, the most aggressive form of breast cancer, TNBC, does not have clear molecular targets. The pathogenesis of TNBC is the least understood and although certain biomarkers have been identified (PD-L1, BRCA, PARP), there is not a single receptor that is unilaterally overexpressed in this breast cancer subtype. Thus, the development of ADCs in TNBC could benefit from the identification of ubiquitous molecular targets in TNBC that have not yet been discovered.
5.1.5. Limited synchronization of combination drugs from single drug nanocarriers
The targeted drug delivery approaches mentioned above highlight the potential to improve chemotherapy in breast cancer treatment. By associating with nanoparticles, cytotoxic drugs can overcome toxicity and solubility limitations and may even partially avoid MDR efflux by undergoing receptor mediated endocytosis (Li, et al., 2000). However, most pharmaceutical drug carriers in clinical studies are single-drug delivery systems and face significant biological barriers before reaching target tumors. If a single drug delivery system avoided sequestration in healthy organs and accumulated in tumors, it may improve the efficacy (or decrease toxicity) of that drug, but combination therapy would require a separate infusion. Unfortunately, sequential infusions will likely limit the synchronicity of multiple drugs in target cells. An ideal pharmaceutical carrier would target and deliver a clinically relevant combination of chemotherapeutic drugs to target cells to maximize cytotoxic effect. Given this limitation, there is an active interest in developing combination nanoparticles that can deliver multiple active drugs with a single pharmaceutical carrier (Figure 2E) (Mu, et al., 2018).
5.2. Drug combination nanoparticles in the treatment of cancer
Currently, there are no clinically approved drug combination nanoparticles for BC or MBC; however, there is one clinically approved drug combination nanoparticle for the treatment of acute myeloid leukemia (AML). Vyxeos, which is composed of two encapsulated liposomal drugs, expands on the liposomal approach and contains both cytarabine and daunorubicin. Both drugs are shown to circulate in plasma at a fixed ratio for an extended period of time. The liposomal properties of Vyxeos enables a targeted effect of both drugs to important (bone marrow, spleen) tissues, which are associated with AML. This improved distribution into the bone marrow and spleen produces a greater effect than the conventional cytarabine/daunorubicin regimen (Mayer, Tardi, & Louie, 2019). Although effective against liquid tumors in blood and bone, Vyxeos has not been tested in solid tumors such as breast cancer. Based on tissue distribution of the liposome, the combination liposomal approach demonstrated by Vyxeos may not be as effective in targeting solid tumors. For AML, bone marrow accumulation of Vyxeos is beneficial because bone marrow is the site of effect. However, the site of effect in breast cancer is the solid tumor or individual cancer foci present in the breast or lymph. In this scenario, extensive accumulation in bone marrow can be a risk for myelosuppression (untoward off target effects) with no therapeutic advantage. Extensive distribution into the spleen would also limit the fraction of doses available for targeting to solid tumors. Furthermore, the manufacturing process required to make Vyxeos is complex and specifically optimized for daunorubicin and cytarabine (Tardi, et al., 2009). This manufacturing process may not directly apply to other drug combination regimens, especially considering the chemically diverse drugs used in breast cancer. The ability to stabilize and associate hydrophilic and hydrophobic drugs in a single particle remains elusive. Despite these challenges in formulation and scaleup as a product, the clinical development of Vyxeos clearly highlights the potential for combination nanoparticles to address the current limitations in cancer treatment by co-delivering chemotherapy to tissues of interest.
The combination nanoparticle approach has also gained attention in the treatment of solid cancers (such as breast cancer, Table 3), but not all combination regimens are appropriate in the treatment of late-stage metastatic breast cancer from pharmaceutical development or pharmacology perspectives. For example, the most common approaches for drug combination nanoparticles are only suitable for drugs with similar hydrophobicity, and the platforms either possess large particle size or require complex manufacturing processes or chemical conjugations. Some pH or redox-sensitive systems may be unstable in the in vivo environment with dilution and plasma protein interference. As mentioned earlier, anthracyclines suffer from life-time dose limiting cardiotoxicity and even encapsulation in liposomes (as seen with Doxil) does not completely eliminate that toxicity. Consequently, taxane-based regimens like gemcitabine and paclitaxel have a greater role in late-stage disease because they do not have any lifetime dose-limitations like anthracyclines. Based on the pharmacokinetic properties of gemcitabine and paclitaxel, this combination regimen will likely benefit from a combination nanoparticle approach. Gemcitabine is a potent antimetabolite that undergoes rapid metabolism by cytidine deaminase, an enzyme expressed ubiquitously in healthy tissue. Rapid metabolism may limit the amount of active gemcitabine that can reach cancer cells before inactivation. Paclitaxel is a potent taxane but is a substrate for P-gp efflux, which may reduce the intracellular concentration of paclitaxel in cancer cells. Following a standard clinical infusion protocol, the time interval during which both drugs are present above their minimum inhibitory concentration in plasma is estimated to be only about 2 hours (Rowinsky, Jiroutek, Bonomi, Johnson, & Baker, 1999; Wang, Liu, Huang, & Xu, 2007). Thus, the time interval where GT is synchronized in cancer cells will potentially be even smaller than that of plasma. By binding both drugs to targeted drug combination nanoparticles, GT can have greater synchronized exposure in cancer cells. With both drugs available intracellularly, the individual pharmacologic effects of gemcitabine (DNA chain termination) and paclitaxel (tubulin inhibitor) can be potentiated and the likelihood of cancer cells escaping cell death will decrease. If GT nanoparticles can distribute into tissues of interest (such as the tissue where MBC cells spread into lymph and blood) while avoiding healthy tissue, then this approach could also reduce the off-target toxicities observed with free drug. A schematic representation of this concept is presented in Figure 1.1.
Table 3.
Examples of nanoparticle-based combination chemotherapy for metastatic breast cancer and formulation analysis.a
| Delivery platform | Drug combination (solubilityb) | Formulation properties | Reference | ||
|---|---|---|---|---|---|
| Drug 1 | Drug 2 | Distinctions | Potential limitations for pharmaceutical development | ||
| Lipid drug combination nanoparticles (DcNP) | Gemcitabine (S) | Paclitaxel (I) | Co-loading of soluble/insoluble drugs, high drug association with good stability in vitro and in vivo, unique elongated shape, FDA-approved excipients, simple and safe manufacturing, high scalability | Fixed-drug ratio optimum, maximum drug loads need to be investigated | (Mu, et al., 2020; J. Yu, Mu, et al., 2020) |
| Liposome | Doxorubicin (I) | Paclitaxel (I) | dual-drug multilamellar vesicles with crosslinking | Large particle size (d~220 nm), instability of liposomes (in vivo) | (Y. R. Liu, Fang, Joo, Wong, & Wang, 2014) |
| Polymers | Oleanolic acid (I) | Paclitaxel (I) | Drug (oleanolic acid) participates in the formation of nanoparticles | Large particle size (d~260 nm), harsh solvent needed in formulation | (Bao, et al., 2020) |
| Everolimus (I) | Paclitaxel (I) | Conjugation of anti-HER2 and anti-EGFR antibody Fab fragments for tumor targeting | Complex manufacturing process, instability of antibody fragments, chemical conjugation (new drug per FDA perspective) | (Houdaihed, Evans, & Allen, 2019) | |
| Gemcitabine (S) | Paclitaxel (I) | Co-loading of soluble/insoluble drugs with high drug association | Micelle particle structures may not be stable and subject to dissociation in diluting conditions and plasma protein interference | (Dong, et al., 2018) | |
| Dasatinib (I) | Doxorubicin (I) | redox-responsive polymeric micelles | Complex manufacturing, potential premature release in oxidative environment such as high GSH in liver, large particle size (d~200 nm), chemical conjugation (new drug per FDA perspective) | (J. J. Sun, et al., 2017) | |
| Paclitaxel (I) | Rapamycin (I) | Loading of two hydrophobic drugs with small NP size | Micelle particle structures may not be stable and subject to dissociation in diluting conditions and plasma protein interference | (Blanco, et al., 2014) | |
| Inorganic/lipid complex | Cisplatin (S) | Tolfenamic acid (I) | Co-loading of soluble/insoluble drugs, two drugs were conjugated into a coordination prodrug | Chemical conjugation (new drug per FDA perspective), drug ratio not tunable (1:1 fixed) | (Yang, et al., 2020) |
| Combretastatin-A4 (I) | Doxorubicin (I) | Fe3O4 as nanoparticle core pay provide multiple functions, DOX conjugated to lipid to form prodrug | Drug loading on surface only, chemical conjugation (new drug per FDA perspective), drug 1 is not FDA-approved | (H. M. Liu, et al., 2016) | |
| Inorganic/polymer complex | Quercetin (I) | Topotecan (I) | Mesoporous silica nanoparticle, pores loaded with topotecan, surface conjugated with quercetin | Complex manufacturing, chemical conjugation (new drug per FDA perspective), degradation issue of silica in vivo | (Murugan, et al., 2016) |
Literature selected are based on dual-drug loaded nanoparticle-based combination chemotherapy. All examples selected have been evaluated in animal models and have improved PK and/or tumor inhibition properties compared to the free drugs. The administration route was all IV, thus not shown in the table.
Water solubility of drugs is shown in brackets. I, insoluble; S, soluble.
The coordinated delivery of GT as a single drug combination nanoparticle to eliminate metastatic cancer cells is a compelling project to pursue. However, the co-formulation of water soluble (gemcitabine, LogP= −1.4) and water insoluble (paclitaxel, LogP=3) drugs together is challenging. To our knowledge, there are only a few published reports that have achieved the co-formulation of GT for breast cancer (Aryal, Hu, & Zhang, 2010; Dong, et al., 2018; Lei, et al., 2019; Meng, et al., 2015; Noh, et al., 2015; J. Zhang, et al., 2018). In those studies, GT are chemically conjugated to polymers, encapsulated in monophosphate form or self-assembled to achieve drug loading. Each study notes an improved effect of GT combination nanoparticles versus free GT in models of cancer and highlights the potential for GT co-delivery. Unfortunately, many of the processes used to synthesize these particles would be difficult to translate into a clinical product. For example, lipid-calcium-phosphate particles were used to co-formulate gemcitabine and paclitaxel in a 16-step process. These particles were very effective at inhibiting tumor growth, but the complex synthesis process and extensive use of organic solvents/surfactants (cyclohexane, THF, IGEPAL Co-520) limits the large-scale manufacturing of this product. Thus, a new strategy is needed to make drug combination nanoparticles that can be easily scaled for clinical development.
5.3. A novel approach to drug combination nanoparticles: Targeting chemotherapy to metastatic breast cancer cells.
Recently, we have developed a novel method to stabilize hydrophobic and hydrophilic cancer drugs—gemcitabine and paclitaxel—together in lipid nanoparticles (Figures 2E&3, Table 4). Chemically distinct drugs and lipid excipients are combined to form drug combination nanoparticles (DcNPs). This DcNP technology platform is a simple 3-step manufacturing process to produce injectable drug-combinations in suspension. The manufacturing process is intended for scalability and includes controlled solvent removal of drug and lipid excipients in solution to trap drugs with lipid excipients in the powder form (J. Yu, Yu, Lane, McConnachie, & Ho, 2020); subsequently the DcNP powder can be suspended in an aqueous buffer to yield uniform particles after subjecting them to a size reduction processes. No unbound drug removal is required in producing injectable DcNPs in suspension. This concept has been validated with multiple combination drug formulations in which each composition is composed of both hydrophilic and hydrophobic HIV drugs including protease inhibitors (lopinavir, ritonavir and atazanavir) and reverse transcriptase inhibitors (efavirenz, tenofovir and lamivudine). The drug combination in each formulation is stabilized in lipid excipients and tested subcutaneously in non-human primates (NHPs). In early iterations, DcNPs were thought to be structurally similar to liposomal products; however, DcNPs have been demonstrated to exhibit unique in vivo characteristics that are not typically associated with standard liposomes. Subcutaneous dosing of DcNPs formulated in drug combination were demonstrated to target the mononuclear cells of lymph nodes in the lymphatic tissue as well as mononuclear lymphocyte cells in the blood of NHPs while producing long-acting plasma circulation (Freeling, Koehn, Shu, Sun, & Ho, 2015; Koehn, et al., 2018; Kraft, et al., 2017; McConnachie, et al., 2018; Perazzolo, et al., 2018). The higher intracellular drug concentrations in blood mononuclear cells compared to plasma demonstrated the cell targeting ability of DcNP associated HIV drugs. These studies also demonstrated that the reverse transcriptase inhibitor tenofovir is accessible to cellular phosphorylation for conversion to the active metabolite, tenofovir-diphosphate, at similar ranges as that delivered by oral dosage form in HIV host cells (Kraft, et al., 2017; McConnachie, et al., 2018). Additionally, DcNP technology appears to be compatible with a wide range of constituent drugs, including tenofovir (logP = −1.6), lopinavir (logP = 4.7), atazanavir (logP = 4.5) and ritonavir (logP = 5.2). As the development and optimization of DcNPs continued, it became evident that the particles are very different from other lipid-based formulations (such as liposomes). Structural analysis revealed that DcNPs have a discoid morphology with no evidence of membrane bilayers (J. Yu, Mu, et al., 2020). The unique structure of DcNPs likely facilitates the binding of chemically distinct compounds together and enables stable, long-acting circulation in vivo. DcNP manufacturing is also relatively simple and easy to scale, which has enabled sizable studies in NHPs. These results highlight the potential for novel DcNP technology to extend beyond what has been accomplished with current delivery approaches.
Figure 3.

The DcNP approach provides opportunities for effective and safe treatment of MBC. A. Schematic representation of how GT DcNP enhances tumor tissue and cell selectivity and likely mechanisms that lead to enhanced exposure and extension of effective drug concentrations in metastatic breast cancer cells (Jesse Yu, 2020). B. Association of GT to DcNPs increases the concentration of GT in plasma over time compared to CrEl control suspension (J. Yu, Mu, et al., 2020). Doses: 50/5 mg/kg G/T. C. Enhancement of GT fixed-dose combination treatment on MBC in the lung by DcNP (Mu, et al., 2020). Mice with IV inoculation of 4T1-luc to form lung metastasis were administered with a single IV bolus dose of 50/5 mg/kg GT combination in DcNP or CrEL control formulation. On day 14, the total tumor burden was examined by bioluminescence of luciferase transfected cancer cells. Contents in this figure were reproduced with permission of publishers.
Table 4.
A comparison of drug combination nanoparticles Vyxeos and GT-DcNP with respect to therapeutic purpose, physical characteristics and manufacturing process.
| Vyxeosa | GT-DcNPb | |
|---|---|---|
| Disease indication | Acute myeloid leukemia | Metastatic breast cancer, pancreatic cancer |
| Target cell locations | Blood and bone marrow | Mammary grand or pancreas, blood and lymphatics, distant sites of the body |
| Physical properties | ||
| Characteristics | Liposomes (lipid bilayer vesicles) | Drug combination particles (Lipid excipient stabilized drug particles) |
| Hydrodynamic size | 100 ± 20 nm | ~60 nm |
| Excipients (Function) | DSPC, DSPG, cholesterol (bilayer enclosure) | DSPC, DSPE-mPEG2000 (serving as a bridge for two drugs-G & T; no bilayer structure) |
| Drugs and solubility (LogP) | Daunorubicin (LogP =1.83) Cytarabine (LogP=−2.8) | Paclitaxel (T) (LogP=3.0) Gemcitabine (GT) (LogP=−1.4) |
| Drug ratios (molar)c | 1:5 | 1:1 to 1:30 |
| Manufacturing process | ||
| Manufacturing stepsd |
|
|
Vyxeos® (or CPX-351) is an approved liposomal formulation consisting of daunorubicin and cytarabine with fixed ratio for treatment of AML.
GT-DcNP is a lipid nanoparticle formulation consisting of gemcitabine and paclitaxel with fixed ratio for treatment of metastatic breast cancer and pancreatic cancer.
Drug ratios in these formulations are molar ratios of daunorubicin:cytarabine and paclitaxel:gemcitabine, respectively.
These steps indicate key procedures in manufacturing of Vyxeos and GT-DcNP.
In recent publications, we have evaluated whether DcNP technology can stabilize chemically distinct drugs for MBC. G & T, which are in clinical use, can be produced in a single GT-drug combination nanoparticle for use as an injectable dosage form (Figures 2E&3, Table 3). We have also evaluated whether a GT-DcNP dosage form may allow targeting of both drugs in MBC nodules found in the lung capillaries in a 4T1 mouse MBC model. The overall goal is to determine whether GT-DcNP may be able to overcome the current limitations of asynchronous infusion of the two drugs to MBC and dose-limiting toxicities due to wide distribution of GT to off-target non-cancer cells. In the 4T1 MBC mouse model, we found that, with a yet to be optimized composition, GT DcNPs can synchronize both drugs—gemcitabine and paclitaxel—in target cancer cells in the lungs, and maintain their concentrations above therapeutic levels for an extended period of time (J. Yu, Mu, et al., 2020).
In a pharmacokinetic time-course and drug disposition comparison between GT formulated in DcNP and freely soluble dosage forms, we found that DcNP extends plasma time course detected as an increased apparent half-life for both gemcitabine and paclitaxel. In addition, the in vivo stability of GT-DcNP as drug combination particles was evident in the greatly reduced metabolic inactivation to dFdU by cytidine deaminase (CDA), which is a major metabolic clearance route of gemcitabine when given by injection in the soluble dosage form. Analysis of 4T1 MBC nodule laden lungs also indicated a significantly higher degree of GT accumulation attributed to the DcNP formulation. The ratio of GT in the 4T1 lung tissues also exhibited a ratio similar to that of the original composition ratio of 10:1 GT (w/w). Collectively these data suggest that in the yet to be optimized GT-DcNP dosage form, a simple process can be used with lipid excipients to produce DcNPs. These GT DcNPs in suspension are able to facilitate long-acting GT drug combination therapy that is stable in mice and leads to the enhanced co-localization of both drugs (G+T) in target MBC nodules in lung tissues (J. Yu, Mu, et al., 2020).
Leveraging on enhanced tissue and the MBC targeting effect of DcNP loaded with GT, the therapeutic effects in 4T1 MBC in mouse were evaluated. We found that DcNP enhanced the potency of both drugs (GT) by inhibiting 4T1 MBC nodules in the lung at a significantly lower administered dose compared to that of free drug equivalent. While a 20/2 mg/kg G/T single IV injection in DcNP could completely inhibit 4T1 MBC nodules, such effects could not be achieved by free GT combination even at a 5-fold higher dose (100/10 mg/kg G/T). Moreover, we found that based on the gross side-effect profile (measured as weight reduction over 14-day study), a therapeutic index of ~15.8 is achievable (Mu, et al., 2020). With additional optimization of drug ratio and retention in the MBC nodules, this approach may be widely applicable for the treatment of MBC and other metastatic cancers. In addition, if the safety and tolerability can be improved significantly, it could be considered for wider use in other stages of breast cancer such as TNBCs with limited treatment options.
6. Concluding remarks and future perspective
With public awareness of early diagnosis and interventions, breast cancer survival has increased and benefited from a number of pharmacological treatment options that can inhibit disease progression. However, a cure for breast cancer is elusive and remains an important goal. The current drug therapies meaningfully increase the time of survival and extend the duration of disease remission. On average, one can expect about 17 years of disease-free status (Dent, et al., 2007). However, cancer progression to metastatic stage (even with active treatment) is a constant concern. People with a triple receptor negative phenotype carry even higher risks of metastatic disease as the recurrence rate is estimated to be within 1–4 years (Dent, et al., 2007; Liedtke, et al., 2008).
Advances have been made to develop oral dosage forms of molecularly targeted therapies to inhibit specific proteins (including checkpoints for cell growth, survival and other processes) and receptors that promote growth and motility (for metastasis). Some of these molecularly targeted drugs have enhanced selectivity and the benefit of oral dosing to improve patient acceptance and tolerability. The growing number of monoclonal antibody therapeutics for cancer may provide additional utility for targeting cancers with specific antigens expressed on their cell surface. Unfortunately, not all cancer types express distinct markers or those markers can be down-regulated over the treatment course. It is likely for these reasons, none of the current therapeutic modalities (either alone or used in combination) has been shown to provide a cure for breast cancer patients who have progressed to metastatic disease. Highly potent (but often intolerably cytotoxic) chemotherapy has been shown to be partially effective in progressive metastatic breast cancer. These chemotherapeutic agents do not depend on cell surface receptors nor growth regulating target expression. With decades of clinical and experimental experience with highly potent, chemotherapeutic agents that target crucial cell growth and replication mechanisms, it is clear that off-target toxicity (which relates to tolerability) continue to be a key barrier that limits their potential impact. The adverse events associated with combination chemotherapy such as gemcitabine and paclitaxel or doxorubicin with paclitaxel can reduce patient quality of life and lead to sub-optimal dosing. Untoward events due to dose-limiting toxicities may explain some of the higher early recurrence rates in TNBC and MBC patients. While cumulative anthracycline cardiotoxicity is dose-limiting, gemcitabine and paclitaxel combination therapy can overcome cardiotoxicity related to anthracyclines such as doxorubicin therapies. Even with improvements in clinical use of liposome encapsulated doxorubicin to reduce cardiotoxicity, doxorubicin nanomedicine still faces cardiotoxicity risk with its chronic use.
The combination of gemcitabine and paclitaxel is prescribed for metastatic breast cancer and not subjected to cardiotoxicity from chronic use. The gemcitabine-paclitaxel combination is effective but also faces dose-limiting toxicity at higher doses (although not lifetime dose limiting). With two different drugs given sequentially, it is not only inconvenient for both patients and clinicians but also limits their coordinated exposure in the same cancer cells for maximum effect. Ideally, both compounds are available to cancer cells at the same time with enough exposure to provide optimal impact on rapidly growing and mobile metastatic cancer cells. Unfortunately, due to their disparate physical (i.e., water solubility) properties, gemcitabine and paclitaxel combination are incompatible in one injectable dosage form; thus, the two drugs are given asynchronously. The sequential infusion would not optimally inhibit both DNA synthesis for cell replication and microtubule formation required for cell-division to knock out MBC completely. In addition, even if one can infuse these two drugs together, the drugs infused into the blood must penetrate and diffuse into cells, which typically would result in lower drug levels than what could achieved in blood.
While liposomes (or lipid membrane vesicles), polymeric carriers and drug conjugates have been evaluated to carry highly potent chemotherapeutic combinations, many of these require complex manufacturing process including separation of unbound drugs. Many struggle to develop a scalable strategy to ensure long enough shelf life for commercial development. As stated previously, nanoparticle systems can also struggle from premature release or rapid mononuclear phagocytosis (sometime referred to as cells of the reticuloendothelial system or RES) leading to rapid clearance of nanoparticles into liver and spleen. Recent discovery of a platform technology that uses biodegradable and biocompatible lipid excipients to stabilize the unlikely partners, water-soluble gemcitabine and water- insoluble paclitaxel may overcome these challenges. This technology (generally referred to DcNP in this review) is able to bring together the physically diverging drugs, gemcitabine and paclitaxel, which are “glued” together in GT-DcNPs through three steps-(1) co-solubilization in an alcoholic solution and produce drug powder, (2) suspension in buffer, and (3) size reduction by homogenization. This process produces a final product that does not require the need for removing unbound drug. Leveraging this simple and scalable drug-combination DcNP technology, we have enabled synchronous delivery and localization of GT into metastatic breast cancer cells in a metastatic mouse model. In addition to its storage and serum stability, DcNPs are reported to reduce the metabolic liability of gemcitabine in vivo and extend the plasma time course of both gemcitabine and paclitaxel. Using a combination of experimental and PK modeling approach, both gemcitabine and paclitaxel were shown to be stably associated to DcNP in GT-DcNP, leading to GT-DcNP’s ability to completely clear the 4T1 MBC nodules in mouse models. It is likely that the combination of serum stability of GT-DcNPs and enhanced drug exposure in metastatic cancer cells (as opposed to bystander host cells) facilitates a greater estimated therapeutic index value (15.8) when compared to freely solubilized drug combinations. These dose-ranging studies suggest improve safety and tumor clearing effect. With some modification and optimization, such as the adjustment of the gemcitabine to paclitaxel drug ratio and other composition variables of formulation, this DcNP approach may be useful for transforming cytotoxic drug combinations (which are effective but intolerable) into tissue and cell targeted drug combination therapies for metastatic breast cancer. Having a more specific, safe and highly potent chemotherapeutic drug combination that can eliminate aggressive, mobile and invasive tumor cells (independent of their surface marker expression) may allow us to envision a cure for breast cancer.
Key challenges of MBC treatments.
Chronic use of kinase inhibitors eventually lead to kinase-specific resistance as breast cancer progress to metastatic stage, and even combining 2 kinase inhibitors have not increase long-term survival.
Hormone receptor targeted drugs such as aromatase inhibitors also cause eventual drug resistance.
Highly potent chemo-drug combinations are frequently used in MBC; however, toxicity and intolerability lead to treatment stoppage; disparate drug properties and in vivo dispositions require frequent infusion visit.
Possible solutions.
Long-acting therapeutic agents with sustained drug coverage for less frequent dosing. Formulation challenges for development of long-acting drug combination in a single dosage must be addressed with enabling nanotechnology that capable to stabilizing highly potent drug cobinaiotn in a nanoparticle platform which is stable (in storage and in vivo) and scalable.
Recently, a novel platform DcNP technology that enables physical association of water soluble and insoluble drugs without modifications was shown to enhanced MBC localization and clear MBC in the lung. The DcNP has low liver/spleen/kidney uptake and less toxicity, thus likely increase safety.
Acknowledgements:
We thank Matthew Hartman for his editorial assistance.
Financial support:
This work was supported in part by NIH grants UM1 AI120176, R61 AI149665, U01 AI1448055 and T32 GM007750 Pharmacological training for J Yu, and by the University of Washington Royalty Research Fund for Q Mu (68-3104 RRF_MU).
Abbreviations
- ADC
Anti-body drug conjugates
- AIs
Aromatase inhibitors
- AML
Acute myeloid leukemia
- BCRP
Breast cancer resistance protein
- CDA
Cytidine deaminase
- CDK4/6
Cyclin-dependent kinases 4 and 6
- CDX
Cell-derived xenografts
- dCK
Deoxycytidine kinase
- DcNP
Drug combination nanoparticles
- Dxd
Deruxtecan
- ER
Estrogen receptor
- ERBB2
Erythroblastic oncogene B2
- G
Gemcitabine
- GEMM
Genetically engineered mouse models
- gem-TP
Gemcitabine-triphosphate
- GT
Gemcitabine and paclitaxel combination
- HER2
Human epidermal growth factor receptor 2
- MAPK
Mitogen-activated protein kinase
- MBC
Metastatic breast cancer
- MRP
Multi-drug resistance associated protein
- NHP
Non-human primates
- PARP
Poly-ADP ribose polymerase
- PDX
Patient derived xenografts
- PFS
Progression-free survival
- P-gp
P-glycoprotein
- PI3K
Phosphoinositide 3-kinases
- PK
Pharmacokinetics
- PR
Progesterone receptor
- SERM
Selective estrogen receptor modulators
- T
Paclitaxel
- TNBC
Triple negative breast cancer
- VEGF
Vascular endothelial growth factors
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement:
The authors declare no conflict of interest.
References
- Abbasi E, Aval SF, Akbarzadeh A, Milani M, Nasrabadi HT, Joo SW, Hanifehpour Y, Nejati-Koshki K, & Pashaei-Asl R (2014). Dendrimers: synthesis, applications, and properties. Nanoscale Res Lett, 9, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbruzzese JL, Grunewald R, Weeks EA, Gravel D, Adams T, Nowak B, Mineishi S, Tarassoff P, Satterlee W, Raber MN, & et al. (1991). A phase I clinical, plasma, and cellular pharmacology study of gemcitabine. J Clin Oncol, 9, 491–498. [DOI] [PubMed] [Google Scholar]
- Afsharzadeh M, Hashemi M, Mokhtarzadeh A, Abnous K, & Ramezani M (2018). Recent advances in co-delivery systems based on polymeric nanoparticle for cancer treatment. Artificial Cells, Nanomedicine, and Biotechnology, 46, 1095–1110. [DOI] [PubMed] [Google Scholar]
- Agus DB, Akita RW, Fox WD, Lewis GD, Higgins B, Pisacane PI, Lofgren JA, Tindell C, Evans DP, Maiese K, Scher HI, & Sliwkowski MX (2002). Targeting ligand-activated ErbB2 signaling inhibits breast and prostate tumor growth. Cancer Cell, 2, 127–137. [DOI] [PubMed] [Google Scholar]
- Alven S, & Aderibigbe BA (2020). The Therapeutic Efficacy of Dendrimer and Micelle Formulations for Breast Cancer Treatment. Pharmaceutics, 12, 1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andre F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, Iwata H, Conte P, Mayer IA, Kaufman B, Yamashita T, Lu YS, Inoue K, Takahashi M, Papai Z, Longin AS, Mills D, Wilke C, Hirawat S, Juric D, & Group S-S (2019). Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N Engl J Med, 380, 1929–1940. [DOI] [PubMed] [Google Scholar]
- Arpino G, Bardou VJ, Clark GM, & Elledge RM (2004). Infiltrating lobular carcinoma of the breast: tumor characteristics and clinical outcome. Breast Cancer Res, 6, R149–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arpino G, Wiechmann L, Osborne C, & Schiff R (2008). Crosstalk between the estrogen receptor and the HER tyrosine kinase receptor family: molecular mechanism and clinical implications for endocrine therapy resistance. Endocrine reviews, 29 2, 217–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aryal S, Hu C-MJ, & Zhang L (2010). Combinatorial Drug Conjugation Enables Nanoparticle Dual-Drug Delivery. Small, 6, 1442–1448. [DOI] [PubMed] [Google Scholar]
- Attia MS, Hassaballah MY, Abdelqawy MA, Emad-Eldin M, Farag AK, Negida A, Ghaith H, & Emam SE An updated review of mesoporous carbon as a novel drug delivery system. DRUG DEVELOPMENT AND INDUSTRIAL PHARMACY. [DOI] [PubMed] [Google Scholar]
- Austin Doyle L, & Ross DD (2003). Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2). Oncogene, 22, 7340–7358. [DOI] [PubMed] [Google Scholar]
- Bao Y, Zhang S, Chen Z, Chen AT, Ma J, Deng G, Xu W, Zhou J, Yu Z-Q, Yao G, & Chen J (2020). Synergistic Chemotherapy for Breast Cancer and Breast Cancer Brain Metastases via Paclitaxel-Loaded Oleanolic Acid Nanoparticles. Molecular Pharmaceutics, 17, 1343–1351. [DOI] [PubMed] [Google Scholar]
- Bardia A, Hurvitz SA, Tolaney SM, Loirat D, Punie K, Oliveira M, Brufsky A, Sardesai SD, Kalinsky K, Zelnak AB, Weaver R, Traina T, Dalenc F, Aftimos P, Lynce F, Diab S, Cortes J, O’Shaughnessy J, Dieras V, Ferrario C, Schmid P, Carey LA, Gianni L, Piccart MJ, Loibl S, Goldenberg DM, Hong Q, Olivo MS, Itri LM, Rugo HS, & Investigators ACT (2021). Sacituzumab Govitecan in Metastatic Triple-Negative Breast Cancer. N Engl J Med, 384, 1529–1541. [DOI] [PubMed] [Google Scholar]
- Bargh JD, Isidro-Llobet A, Parker JS, & Spring DR (2019). Cleavable linkers in antibody–drug conjugates. Chemical Society Reviews, 48, 4361–4374. [DOI] [PubMed] [Google Scholar]
- Barok M, Joensuu H, & Isola J (2014). Trastuzumab emtansine: mechanisms of action and drug resistance. Breast Cancer Res, 16, 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baselga J, Cortes J, Kim SB, Im SA, Hegg R, Im YH, Roman L, Pedrini JL, Pienkowski T, Knott A, Clark E, Benyunes MC, Ross G, Swain SM, & Group CS (2012). Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med, 366, 109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baselga J, Lewis Phillips GD, Verma S, Ro J, Huober J, Guardino AE, Samant MK, Olsen S, de Haas SL, & Pegram MD (2016). Relationship between Tumor Biomarkers and Efficacy in EMILIA, a Phase III Study of Trastuzumab Emtansine in HER2-Positive Metastatic Breast Cancer. Clinical Cancer Research, 22, 3755–3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackstein M, Vogel CL, Ambinder R, Cowan J, Iglesias J, & Melemed A (2002). Gemcitabine as first-line therapy in patients with metastatic breast cancer: a phase II trial. Oncology, 62, 2–8. [DOI] [PubMed] [Google Scholar]
- Blackwell KL, Burstein HJ, Storniolo AM, Rugo HS, Sledge G, Aktan G, Ellis C, Florance A, Vukelja S, Bischoff J, Baselga J, & O’Shaughnessy J (2012). Overall survival benefit with lapatinib in combination with trastuzumab for patients with human epidermal growth factor receptor 2-positive metastatic breast cancer: final results from the EGF104900 Study. J Clin Oncol, 30, 2585–2592. [DOI] [PubMed] [Google Scholar]
- Blanco E, Sangai T, Wu SH, Hsiao A, Ruiz-Esparza GU, Gonzalez-Delgado CA, Cara FE, Granados-Principal S, Evans KW, Akcakanat A, Wang Y, Do KA, Meric-Bernstam F, & Ferrari M (2014). Colocalized Delivery of Rapamycin and Paclitaxel to Tumors Enhances Synergistic Targeting of the PI3K/Akt/mTOR Pathway. Molecular Therapy, 22, 1310–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bombonati A, & Sgroi DC (2011). The molecular pathology of breast cancer progression. J Pathol, 223, 307–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslin S, Lowry MC, & O’Driscoll L (2017). Neratinib resistance and cross-resistance to other HER2-targeted drugs due to increased activity of metabolism enzyme cytochrome P4503A4. Br J Cancer, 116, 620–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M, Assen FP, Leithner A, Abe J, Schachner H, Asfour G, Bago-Horvath Z, Stein JV, Uhrin P, Sixt M, & Kerjaschki D (2018). Lymph node blood vessels provide exit routes for metastatic tumor cell dissemination in mice. Science, 359, 1408–1411. [DOI] [PubMed] [Google Scholar]
- Bunone G, Briand PA, Miksicek RJ, & Picard D (1996). Activation of the unliganded estrogen receptor by EGF involves the MAP kinase pathway and direct phosphorylation. EMBO J, 15, 2174–2183. [PMC free article] [PubMed] [Google Scholar]
- Canonici A, Gijsen M, Mullooly M, Bennett R, Bouguern N, Pedersen K, O’Brien NA, Roxanis I, Li JL, Bridge E, Finn R, Siamon D, McGowan P, Duffy MJ, O’Donovan N, Crown J, & Kong A (2013). Neratinib overcomes trastuzumab resistance in HER2 amplified breast cancer. Oncotarget, 4, 1592–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez KJ, Garimella SV, & Lipkowitz S (2010). Triple negative breast cancer cell lines: one tool in the search for better treatment of triple negative breast cancer. Breast Dis, 32, 35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Wang L, Shion H, Yu C, Yu YQ, Zhu L, Li M, Chen W, & Gao K (2016). In-depth structural characterization of Kadcyla(R) (ado-trastuzumab emtansine) and its biosimilar candidate. MAbs, 8, 1210–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemons M, Leahy M, Valle J, Jayson G, Ranson M, & Howell A (1997). Review of recent trials of chemotherapy for advanced breast cancer: the taxanes. European Journal of Cancer, 33, 2183–2193. [DOI] [PubMed] [Google Scholar]
- Collins DM, Gately K, Hughes C, Edwards C, Davies A, Madden SF, O’Byrne KJ, O’Donovan N, & Crown J (2017). Tyrosine kinase inhibitors as modulators of trastuzumab-mediated antibody-dependent cell-mediated cytotoxicity in breast cancer cell lines. Cell Immunol, 319, 35–42. [DOI] [PubMed] [Google Scholar]
- Colomer R (2005). Gemcitabine in combination with paclitaxel for the treatment of metastatic breast cancer. Womens Health (Lond), 1, 323–329. [DOI] [PubMed] [Google Scholar]
- Crommelin DJA, van Hoogevest P, & Storm G (2020). The role of liposomes in clinical nanomedicine development. What now? Now what? Journal of Controlled Release, 318, 256–263. [DOI] [PubMed] [Google Scholar]
- Daniel AR, Hagan CR, & Lange CA (2011). Progesterone receptor action: defining a role in breast cancer. Expert Rev Endocrinol Metab, 6, 359–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P, & Narod SA (2007). Triple-Negative Breast Cancer: Clinical Features and Patterns of Recurrence. Clinical Cancer Research, 13, 4429. [DOI] [PubMed] [Google Scholar]
- Dhillon S (2014). Trastuzumab Emtansine: A Review of Its Use in Patients with HER2-Positive Advanced Breast Cancer Previously Treated with Trastuzumab-Based Therapy. Drugs, 74, 675–686. [DOI] [PubMed] [Google Scholar]
- Dong S, Guo Y, Duan Y, Li Z, Wang C, Niu L, Wang N, Ma M, Shi Y, & Zhang M (2018). Co-delivery of paclitaxel and gemcitabine by methoxy poly(ethylene glycol)-poly(lactide-coglycolide)-polypeptide nanoparticles for effective breast cancer therapy. Anticancer Drugs, 29, 637–645. [DOI] [PubMed] [Google Scholar]
- Doronina SO, Toki BE, Torgov MY, Mendelsohn BA, Cerveny CG, Chace DF, DeBlanc RL, Gearing RP, Bovee TD, Siegall CB, Francisco JA, Wahl AF, Meyer DL, & Senter PD (2003). Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nature Biotechnology, 21, 778–784. [DOI] [PubMed] [Google Scholar]
- Duncan R, & Izzo L (2005). Dendrimer biocompatibility and toxicity. Adv Drug Deliv Rev, 57, 2215–2237. [DOI] [PubMed] [Google Scholar]
- Feher O, Vodvarka P, Jassem J, Morack G, Advani SH, Khoo KS, Doval DC, Ermisch S, Roychowdhury D, Miller MA, & von Minckwitz G (2005). First-line gemcitabine versus epirubicin in postmenopausal women aged 60 or older with metastatic breast cancer: a multicenter, randomized, phase III study. Ann Oncol, 16, 899–908. [DOI] [PubMed] [Google Scholar]
- Finn RS, Martin M, Rugo HS, Jones S, Im SA, Gelmon K, Harbeck N, Lipatov ON, Walshe JM, Moulder S, Gauthier E, Lu DR, Randolph S, Dieras V, & Slamon DJ (2016). Palbociclib and Letrozole in Advanced Breast Cancer. N Engl J Med, 375, 1925–1936. [DOI] [PubMed] [Google Scholar]
- Fraguas-Sánchez AI, Martín-Sabroso C, Fernández-Carballido A, & Torres-Suárez AI (2019). Current status of nanomedicine in the chemotherapy of breast cancer. Cancer Chemotherapy and Pharmacology, 84, 689–706. [DOI] [PubMed] [Google Scholar]
- Franci C, Zhou J, Jiang Z, Modrusan Z, Good Z, Jackson E, & Kouros-Mehr H (2013). Biomarkers of residual disease, disseminated tumor cells, and metastases in the MMTV-PyMT breast cancer model. PLoS One, 8, e58183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin MC, Carey KD, Vajdos FF, Leahy DJ, de Vos AM, & Sliwkowski MX (2004). Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell, 5, 317–328. [DOI] [PubMed] [Google Scholar]
- Frasor J, Stossi F, Danes JM, Komm B, Lyttle CR, & Katzenellenbogen BS (2004). Selective estrogen receptor modulators: discrimination of agonistic versus antagonistic activities by gene expression profiling in breast cancer cells. Cancer Res, 64, 1522–1533. [DOI] [PubMed] [Google Scholar]
- Freeling JP, Koehn J, Shu C, Sun J, & Ho RJ (2015). Anti-HIV drug-combination nanoparticles enhance plasma drug exposure duration as well as triple-drug combination levels in cells within lymph nodes and blood in primates. AIDS Res Hum Retroviruses, 31, 107–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentes G, Scaltriti M, Baselga J, & Verma CS (2011). Synergy between trastuzumab and pertuzumab for human epidermal growth factor 2 (Her2) from colocalization: an in silico based mechanism. Breast Cancer Res, 13, R54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadekar V, Borade Y, Kannaujia S, Rajpoot K, Anup N, Tambe V, Kalia K, & Tekade RK (2021). Nanomedicines accessible in the market for clinical interventions. Journal of Controlled Release, 330, 372–397. [DOI] [PubMed] [Google Scholar]
- Geisberg CA, & Sawyer DB (2010). Mechanisms of anthracycline cardiotoxicity and strategies to decrease cardiac damage. Curr Hypertens Rep, 12, 404–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sledge George W. J., Toi M, Neven P, Sohn J, Inoue K, Pivot X, Burdaeva O, Okera M, Masuda N, Kaufman PA, Koh H, Grischke E-M, Frenzel M, Lin Y, Barriga S, Smith IC, Bourayou N, & Llombart-Cussac A (2017). MONARCH 2: Abemaciclib in Combination With Fulvestrant in Women With HR+/HER2− Advanced Breast Cancer Who Had Progressed While Receiving Endocrine Therapy. Journal of Clinical Oncology, 35, 2875–2884. [DOI] [PubMed] [Google Scholar]
- Gianni L, Kearns CM, Giani A, Capri G, Viganó L, Lacatelli A, Bonadonna G, & Egorin MJ (1995). Nonlinear pharmacokinetics and metabolism of paclitaxel and its pharmacokinetic/pharmacodynamic relationships in humans. J Clin Oncol, 13, 180–190. [DOI] [PubMed] [Google Scholar]
- Giles FJ (2002). Gemtuzumab ozogamicin: promise and challenge in patients with acute myeloid leukemia. Expert Review of Anticancer Therapy, 2, 630–640. [DOI] [PubMed] [Google Scholar]
- Golombek SK, May JN, Theek B, Appold L, Drude N, Kiessling F, & Lammers T (2018). Tumor targeting via EPR: Strategies to enhance patient responses. Adv Drug Deliv Rev, 130, 17–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopinathan A, Morton JP, Jodrell DI, & Sansom OJ (2015). GEMMs as preclinical models for testing pancreatic cancer therapies. Dis Model Mech, 8, 1185–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould SE, Junttila MR, & de Sauvage FJ (2015). Translational value of mouse models in oncology drug development. Nature Medicine, 21, 431–439. [DOI] [PubMed] [Google Scholar]
- Grunewald R, Abbruzzese JL, Tarassoff P, & Plunkett W (1991). Saturation of 2’,2’-difluorodeoxycytidine 5’-triphosphate accumulation by mononuclear cells during a phase I trial of gemcitabine. Cancer Chemother Pharmacol, 27, 258–262. [DOI] [PubMed] [Google Scholar]
- Gubernator J (2011). Active methods of drug loading into liposomes: recent strategies for stable drug entrapment and increased in vivo activity. Expert Opinion on Drug Delivery, 8, 565–580. [DOI] [PubMed] [Google Scholar]
- Hall JM, & McDonnell DP (2005). Coregulators in nuclear estrogen receptor action: from concept to therapeutic targeting. Mol Interv, 5, 343–357. [DOI] [PubMed] [Google Scholar]
- Harari D, & Yarden Y (2000). Molecular mechanisms underlying ErbB2/HER2 action in breast cancer. Oncogene, 19, 6102–6114. [DOI] [PubMed] [Google Scholar]
- Harris M, Howell A, Chrissohou M, Swindell RI, Hudson M, & Sellwood RA (1984). A comparison of the metastatic pattern of infiltrating lobular carcinoma and infiltrating duct carcinoma of the breast. Br J Cancer, 50, 23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Rajantie I, Pajusola K, Jeltsch M, Holopainen T, Yla-Herttuala S, Harding T, Jooss K, Takahashi T, & Alitalo K (2005). Vascular Endothelial Cell Growth Factor Receptor 3–Mediated Activation of Lymphatic Endothelium Is Crucial for Tumor Cell Entry and Spread via Lymphatic Vessels. Cancer Research, 65, 4739. [DOI] [PubMed] [Google Scholar]
- Ho RJ, & Chien J (2014). Trends in translational medicine and drug targeting and delivery: new insights on an old concept-targeted drug delivery with antibody-drug conjugates for cancers. J Pharm Sci, 103, 71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho RJ, Yu J, Li B, Kraft JC, Freeling JP, Koehn J, & Shao J (2015). Systems Approach to targeted and long-acting HIV/AIDS therapy. Drug Deliv Transl Res, 5, 531–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hortobagyi GN, Stemmer SM, Burris HA, Yap YS, Sonke GS, Paluch-Shimon S, Campone M, Petrakova K, Blackwell KL, Winer EP, Janni W, Verma S, Conte P, Arteaga CL, Cameron DA, Mondal S, Su F, Miller M, Elmeliegy M, Germa C, & O’Shaughnessy J (2018). Updated results from MONALEESA-2, a phase III trial of first-line ribociclib plus letrozole versus placebo plus letrozole in hormone receptor-positive, HER2-negative advanced breast cancer. Ann Oncol, 29, 1541–1547. [DOI] [PubMed] [Google Scholar]
- Houdaihed L, Evans JC, & Allen C (2019). In Vivo Evaluation of Dual-Targeted Nanoparticles Encapsulating Paclitaxel and Everolimus. Cancers, 11, 752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q, van Rooijen N, & Liu D (1996). Effect of Macrophage Elimination Using Liposome-Encapsulated Dichloromethylene Diphosphonate on Tissue Distribution of Liposomes. Journal of Liposome Research, 6, 681–698. [Google Scholar]
- Huang B, Omoto Y, Iwase H, Yamashita H, Toyama T, Coombes RC, Filipovic A, Warner M, & Gustafsson J (2014). Differential expression of estrogen receptor α, β1, and β2 in lobular and ductal breast cancer. Proc Natl Acad Sci U S A, 111, 1933–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida T, Harada M, Wang XY, Ichihara M, Irimura K, & Kiwada H (2005). Accelerated blood clearance of PEGylated liposomes following preceding liposome injection: effects of lipid dose and PEG surface-density and chain length of the first-dose liposomes. J Control Release, 105, 305–317. [DOI] [PubMed] [Google Scholar]
- Johnston SR, Martin LA, Head J, Smith I, & Dowsett M (2005). Aromatase inhibitors: combinations with fulvestrant or signal transduction inhibitors as a strategy to overcome endocrine resistance. J Steroid Biochem Mol Biol, 95, 173–181. [DOI] [PubMed] [Google Scholar]
- Kariagina A, Aupperlee MD, & Haslam SZ (2008). Progesterone receptor isoform functions in normal breast development and breast cancer. Crit Rev Eukaryot Gene Expr, 18, 11–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersten K, de Visser KE, van Miltenburg MH, & Jonkers J (2017). Genetically engineered mouse models in oncology research and cancer medicine. EMBO Mol Med, 9, 137–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehn J, Iwamoto JF, Kraft JC, McConnachie LA, Collier AC, & Ho RJY (2018). Extended cell and plasma drug levels after one dose of a three-in-one nanosuspension containing lopinavir, efavirenz, and tenofovir in nonhuman primates. AIDS, 32, 2463–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima C (2015). Preclinical studies of dendrimer prodrugs. Expert Opin Drug Metab Toxicol, 11, 1303–1315. [DOI] [PubMed] [Google Scholar]
- Kozma GT, Shimizu T, Ishida T, & Szebeni J (2020). Anti-PEG antibodies: Properties, formation, testing and role in adverse immune reactions to PEGylated nano-biopharmaceuticals. Advanced Drug Delivery Reviews, 154-155, 163–175. [DOI] [PubMed] [Google Scholar]
- Kraft JC, Freeling JP, Wang Z, & Ho RJ (2014). Emerging research and clinical development trends of liposome and lipid nanoparticle drug delivery systems. J Pharm Sci, 103, 29–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft JC, McConnachie LA, Koehn J, Kinman L, Collins C, Shen DD, Collier AC, & Ho RJ (2017). Long-acting combination anti-HIV drug suspension enhances and sustains higher drug levels in lymph node cells than in blood cells and plasma. AIDS, 31, 765–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroep JR, Loves WJ, van der Wilt CL, Alvarez E, Talianidis I, Boven E, Braakhuis BJ, van Groeningen CJ, Pinedo HM, & Peters GJ (2002). Pretreatment deoxycytidine kinase levels predict in vivo gemcitabine sensitivity. Mol Cancer Ther, 1, 371–376. [PubMed] [Google Scholar]
- Lei M, Sha S, Wang X, Wang J, Du X, Miao H, Zhou H, Bai E, Shi J, & Zhu Y (2019). Co-delivery of paclitaxel and gemcitabine via a self-assembling nanoparticle for targeted treatment of breast cancer. RSC Advances, 9, 5512–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Newman RA, Wu Q-P, Ke S, Chen W, Hutto T, Kan Z, Brannan MD, Charnsangavej C, & Wallace S (2000). Biodistribution of paclitaxel and poly(l-glutamic acid)-paclitaxel conjugate in mice with ovarian OCa-1 tumor. Cancer Chemotherapy and Pharmacology, 46, 416–422. [DOI] [PubMed] [Google Scholar]
- Liedtke C, Mazouni C, Hess KR, André F, Tordai A, Mejia JA, Symmans WF, Gonzalez-Angulo AM, Hennessy B, Green M, Cristofanilli M, Hortobagyi GN, & Pusztai L (2008). Response to Neoadjuvant Therapy and Long-Term Survival in Patients With Triple-Negative Breast Cancer. Journal of Clinical Oncology, 26, 1275–1281. [DOI] [PubMed] [Google Scholar]
- Litton JK, Rugo HS, Ettl J, Hurvitz SA, Goncalves A, Lee KH, Fehrenbacher L, Yerushalmi R, Mina LA, Martin M, Roche H, Im YH, Quek RGW, Markova D, Tudor IC, Hannah AL, Eiermann W, & Blum JL (2018). Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N Engl J Med, 379, 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HM, Xie YD, Zhang YF, Cai YF, Li BY, Mao HL, & Yu RT (2016). CA4-loaded doxorubicin prodrug coating Fe3O4 nanoparticles for tumor combination therapy. Rsc Advances, 6, 113933–113939. [Google Scholar]
- Liu YR, Fang JX, Joo KI, Wong MK, & Wang P (2014). Codelivery of Chemotherapeutics via Crosslinked Multilamellar Liposomal Vesicles to Overcome Multidrug Resistance in Tumor. Plos One, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longmire MR, Ogawa M, Choyke PL, & Kobayashi H (2011). Biologically optimized nanosized molecules and particles: more than just size. Bioconjug Chem, 22, 993–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Zi X, Zhao Y, Mascarenhas D, & Pollak M (2001). Insulin-like growth factor-I receptor signaling and resistance to trastuzumab (Herceptin). J Natl Cancer Inst, 93, 1852–1857. [DOI] [PubMed] [Google Scholar]
- Luo K, Li C, Li L, She W, Wang G, & Gu Z (2012). Arginine functionalized peptide dendrimers as potential gene delivery vehicles. Biomaterials, 33, 4917–4927. [DOI] [PubMed] [Google Scholar]
- Mager DE, Mody V, Xu C, Forrest A, Lesniak WG, Nigavekar SS, Kariapper MT, Minc L, Khan MK, & Balogh LP (2012). Physiologically based pharmacokinetic model for composite nanodevices: effect of charge and size on in vivo disposition. Pharm Res, 29, 2534–2542. [DOI] [PubMed] [Google Scholar]
- Marcinkowska M, Sobierajska E, Stanczyk M, Janaszewska A, Chworos A, & Klajnert-Maculewicz B (2018). Conjugate of PAMAM Dendrimer, Doxorubicin and Monoclonal Antibody-Trastuzumab: The New Approach of a Well-Known Strategy. Polymers (Basel), 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markman M (2003). Managing taxane toxicities. Support Care Cancer, 11, 144–147. [DOI] [PubMed] [Google Scholar]
- Maurer-Jones MA, Bantz KC, Love SA, Marquis BJ, & Haynes CL (2009). Toxicity of therapeutic nanoparticles. Nanomedicine, 4, 219–241. [DOI] [PubMed] [Google Scholar]
- Mayer LD, Tardi P, & Louie AC (2019). CPX-351: a nanoscale liposomal co-formulation of daunorubicin and cytarabine with unique biodistribution and tumor cell uptake properties. Int J Nanomedicine, 14, 3819–3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnachie LA, Kinman LM, Koehn J, Kraft JC, Lane S, Lee W, Collier AC, & Ho RJY (2018). Long-Acting Profile of 4 Drugs in 1 Anti-HIV Nanosuspension in Nonhuman Primates for 5 Weeks After a Single Subcutaneous Injection. J Pharm Sci, 107, 1787–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonnell DP, & Wardell SE (2010). The molecular mechanisms underlying the pharmacological actions of ER modulators: implications for new drug discovery in breast cancer. Curr Opin Pharmacol, 10, 620–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medeiros B, & Allan AL (2019). Molecular Mechanisms of Breast Cancer Metastasis to the Lung: Clinical and Experimental Perspectives. Int J Mol Sci, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng H, Wang M, Liu H, Liu X, Situ A, Wu B, Ji Z, Chang CH, & Nel AE (2015). Use of a lipid-coated mesoporous silica nanoparticle platform for synergistic gemcitabine and paclitaxel delivery to human pancreatic cancer in mice. ACS Nano, 9, 3540–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercier C, Raynal C, Dahan L, Ortiz A, Evrard A, Dupuis C, Blesius A, Duluc M, Franceschini F, Giacometti S, Salas S, Milano G, Favre R, Seitz JF, & Ciccolini J (2007). Toxic death case in a patient undergoing gemcitabine-based chemotherapy in relation with cytidine deaminase downregulation. Pharmacogenet Genomics, 17, 841–844. [DOI] [PubMed] [Google Scholar]
- Modi S, Saura C, Yamashita T, Park YH, Kim SB, Tamura K, Andre F, Iwata H, Ito Y, Tsurutani J, Sohn J, Denduluri N, Perrin C, Aogi K, Tokunaga E, Im SA, Lee KS, Hurvitz SA, Cortes J, Lee C, Chen S, Zhang L, Shahidi J, Yver A, Krop I, & Investigators DE-B (2020). Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N Engl J Med, 382, 610–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moo TA, Sanford R, Dang C, & Morrow M (2018). Overview of Breast Cancer Therapy. PET Clin, 13, 339–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss DM, & Siccardi M (2014). Optimizing nanomedicine pharmacokinetics using physiologically based pharmacokinetics modelling. Br J Pharmacol, 171, 3963–3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu Q, Yu J, Griffin JI, Wu Y, Zhu L, McConnachie LA, & Ho RJY (2020). Novel drug combination nanoparticles exhibit enhanced plasma exposure and dose-responsive effects on eliminating breast cancer lung metastasis. PLoS One, 15, e0228557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu Q, Yu J, McConnachie LA, Kraft JC, Gao Y, Gulati GK, & Ho RJY (2018). Translation of combination nanodrugs into nanomedicines: lessons learned and future outlook. J Drug Target, 26, 435–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murayama T, & Gotoh N (2019). Patient-Derived Xenograft Models of Breast Cancer and Their Application. Cells, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy RK, Loi S, Okines A, Paplomata E, Hamilton E, Hurvitz SA, Lin NU, Borges V, Abramson V, Anders C, Bedard PL, Oliveira M, Jakobsen E, Bachelot T, Shachar SS, Muller V, Braga S, Duhoux FP, Greil R, Cameron D, Carey LA, Curigliano G, Gelmon K, Hortobagyi G, Krop I, Loibl S, Pegram M, Slamon D, Palanca-Wessels MC, Walker L, Feng W, & Winer EP (2020). Tucatinib, Trastuzumab, and Capecitabine for HER2-Positive Metastatic Breast Cancer. N Engl J Med, 382, 597–609. [DOI] [PubMed] [Google Scholar]
- Murugan C, Rayappan K, Thangam R, Bhanumathi R, Shanthi K, Vivek R, Thirumurugan R, Bhattacharyya A, Sivasubramanian S, Gunasekaran P, & Kannan S (2016). Combinatorial nanocarrier based drug delivery approach for amalgamation of anti-tumor agents in bresat cancer cells: an improved nanomedicine strategies. Scientific Reports, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada T, Sugihara K, Jikoh T, Abe Y, & Agatsuma T (2019). The Latest Research and Development into the Antibody–Drug Conjugate, [fam-] Trastuzumab Deruxtecan (DS-8201a), for HER2 Cancer Therapy. Chemical and Pharmaceutical Bulletin, 67, 173–185. [DOI] [PubMed] [Google Scholar]
- Nanayakkara AK, Follit CA, Chen G, Williams NS, Vogel PD, & Wise JG (2018). Targeted inhibitors of P-glycoprotein increase chemotherapeutic-induced mortality of multidrug resistant tumor cells. Sci Rep, 8, 967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathanson SD, Krag D, Kuerer HM, Newman LA, Brown M, Kerjaschki D, Pereira ER, & Padera TP (2018). Breast cancer metastasis through the lympho-vascular system. Clin Exp Metastasis, 35, 443–454. [DOI] [PubMed] [Google Scholar]
- Ng IO, Lam KY, Ng M, Kwong DL, & Sham JS (1998). Expression of P-glycoprotein, a multidrug-resistance gene product, is induced by radiotherapy in patients with oral squamous cell carcinoma. Cancer, 83, 851–857. [DOI] [PubMed] [Google Scholar]
- Nicholson RI, Gee JMW, Manning DL, Wakeling AE, Montano MM, & Katzenellenbogen BS (1995). Responses to Pure Antiestrogens (Ici-164384, Ici-182780) in Estrogen-Sensitive and Estrogen-Resistant Experimental and Clinical Breast-Cancer. Steroid Receptors and Antihormones, 761, 148–163. [DOI] [PubMed] [Google Scholar]
- Noh I, Kim HO, Choi J, Choi Y, Lee DK, Huh YM, & Haam S (2015). Co-delivery of paclitaxel and gemcitabine via CD44-targeting nanocarriers as a prodrug with synergistic antitumor activity against human biliary cancer. Biomaterials, 53, 763–774. [DOI] [PubMed] [Google Scholar]
- Northfelt DW, Martin FJ, Working P, Volberding PA, Russell J, Newman M, Amantea MA, & Kaplan LD (1996). Doxorubicin encapsulated in liposomes containing surface-bound polyethylene glycol: pharmacokinetics, tumor localization, and safety in patients with AIDS-related Kaposi’s sarcoma. J Clin Pharmacol, 36, 55–63. [DOI] [PubMed] [Google Scholar]
- O’Brien ME, Wigler N, Inbar M, Rosso R, Grischke E, Santoro A, Catane R, Kieback DG, Tomczak P, Ackland SP, Orlandi F, Mellars L, Alland L, Tendler C, & Group CBCS (2004). Reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (CAELYX/Doxil) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Ann Oncol, 15, 440–449. [DOI] [PubMed] [Google Scholar]
- Okuda T, Kawakami S, Akimoto N, Niidome T, Yamashita F, & Hashida M (2006). PEGylated lysine dendrimers for tumor-selective targeting after intravenous injection in tumor-bearing mice. Journal of Controlled Release, 116, 330–336. [DOI] [PubMed] [Google Scholar]
- Okuda T, Kawakami S, Maeie T, Niidome T, Yamashita F, & Hashida M (2006). Biodistribution characteristics of amino acid dendrimers and their PEGylated derivatives after intravenous administration. Journal of Controlled Release, 114, 69–77. [DOI] [PubMed] [Google Scholar]
- Osborne CK, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, Fuqua SA, Wong J, Allred DC, Clark GM, & Schiff R (2003). Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J Natl Cancer Inst, 95, 353–361. [DOI] [PubMed] [Google Scholar]
- Osborne CK, Shou J, Massarweh S, & Schiff R (2005). Crosstalk between estrogen receptor and growth factor receptor pathways as a cause for endocrine therapy resistance in breast cancer. Clin Cancer Res, 11, 865s–870s. [PubMed] [Google Scholar]
- Osborne CK, Wakeling A, & Nicholson RI (2004). Fulvestrant: an oestrogen receptor antagonist with a novel mechanism of action. Br J Cancer, 90 Suppl 1, S2–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padera TP, Kadambi A, di Tomaso E, Carreira CM, Brown EB, Boucher Y, Choi NC, Mathisen D, Wain J, Mark EJ, Munn LL, & Jain RK (2002). Lymphatic Metastasis in the Absence of Functional Intratumor Lymphatics. Science, 296, 1883. [DOI] [PubMed] [Google Scholar]
- Paliwal R, Babu RJ, & Palakurthi S (2014). Nanomedicine Scale-up Technologies: Feasibilities and Challenges. Aaps Pharmscitech, 15, 1527–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan H, Gray R, Braybrooke J, Davies C, Taylor C, McGale P, Peto R, Pritchard KI, Bergh J, Dowsett M, Hayes DF, & Ebctcg. (2017). 20-Year Risks of Breast-Cancer Recurrence after Stopping Endocrine Therapy at 5 Years. N Engl J Med, 377, 1836–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park MK, Lee CH, & Lee H (2018). Mouse models of breast cancer in preclinical research. Lab Anim Res, 34, 160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perazzolo S, Shireman LM, Koehn J, McConnachie LA, Kraft JC, Shen DD, & Ho RJY (2018). Three HIV Drugs, Atazanavir, Ritonavir, and Tenofovir, Coformulated in Drug-Combination Nanoparticles Exhibit Long-Acting and Lymphocyte-Targeting Properties in Nonhuman Primates. J Pharm Sci, 107, 3153–3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira ER, Kedrin D, Seano G, Gautier O, Meijer EFJ, Jones D, Chin SM, Kitahara S, Bouta EM, Chang J, Beech E, Jeong HS, Carroll MC, Taghian AG, & Padera TP (2018). Lymph node metastases can invade local blood vessels, exit the node, and colonize distant organs in mice. Science, 359, 1403–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernas S, & Tolaney SM (2019). HER2-positive breast cancer: new therapeutic frontiers and overcoming resistance. Therapeutic Advances in Medical Oncology, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters GJ, Clavel M, Noordhuis P, Geyssen GJ, Laan AC, Guastalla J, Edzes HT, & Vermorken JB (2007). Clinical Phase I and Pharmacology Study of Gemcitabine (2’, 2’-Difluorodeoxycytidine) Administered in a Two-Weekly Schedule. Journal of Chemotherapy, 19, 212–221. [DOI] [PubMed] [Google Scholar]
- Pistelli M, Mora AD, Ballatore Z, & Berardi R (2018). Aromatase inhibitors in premenopausal women with breast cancer: the state of the art and future prospects. Curr Oncol, 25, e168–e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preusser M, De Mattos-Arruda L, Thill M, Criscitiello C, Bartsch R, Ruhstaller T, de Azambuja E, & Zielinski CC (2018). CDK4/6 inhibitors in the treatment of patients with breast cancer: summary of a multidisciplinary round-table discussion. ESMO Open, 3, e000368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probst CE, Zrazhevskiy P, Bagalkot V, & Gao XH (2013). Quantum dots as a platform for nanoparticle drug delivery vehicle design. Advanced Drug Delivery Reviews, 65, 703–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulaski BA, & Ostrand-Rosenberg S (2001). Mouse 4T1 breast tumor model. Curr Protoc Immunol, Chapter 20, Unit 20.22. [DOI] [PubMed] [Google Scholar]
- Rahman M, & Mohammed S (2015). Breast cancer metastasis and the lymphatic system. Oncol Lett, 10, 1233–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid OM, & Takabe K (2015). Animal models for exploring the pharmacokinetics of breast cancer therapies. Expert Opin Drug Metab Toxicol, 11, 221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redig AJ, & McAllister SS (2013). Breast cancer as a systemic disease: a view of metastasis. J Intern Med, 274, 113–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid JM, Qu W, Safgren SL, Ames MM, Krailo MD, Seibel NL, Kuttesch J, & Holcenberg J (2004). Phase I trial and pharmacokinetics of gemcitabine in children with advanced solid tumors. J Clin Oncol, 22, 2445–2451. [DOI] [PubMed] [Google Scholar]
- Rizza P, Barone I, Zito D, Giordano F, Lanzino M, De Amicis F, Mauro L, Sisci D, Catalano S, Dahlman Wright K, Gustafsson JA, & Andò S (2014). Estrogen receptor beta as a novel target of androgen receptor action in breast cancer cell lines. Breast Cancer Res, 16, R21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts JC, Bhalgat MK, & Zera RT (1996). Preliminary biological evaluation of polyamidoamine (PAMAM) StarburstTM dendrimers. Journal of Biomedical Materials Research, 30, 53–65. [DOI] [PubMed] [Google Scholar]
- Rojas K, & Stuckey A (2016). Breast Cancer Epidemiology and Risk Factors. Clin Obstet Gynecol, 59, 651–672. [DOI] [PubMed] [Google Scholar]
- Rowinsky EK, Jiroutek M, Bonomi P, Johnson D, & Baker SD (1999). Paclitaxel steady-state plasma concentration as a determinant of disease outcome and toxicity in lung cancer patients treated with paclitaxel and cisplatin. Clin Cancer Res, 5, 767–774. [PubMed] [Google Scholar]
- Rugo HS, Im SA, Cardoso F, Cortes J, Curigliano G, Musolino A, Pegram MD, Wright GS, Saura C, Escriva-de-Romani S, De Laurentiis M, Levy C, Brown-Glaberman U, Ferrero JM, de Boer M, Kim SB, Petrakova K, Yardley DA, Freedman O, Jakobsen EH, Kaufman B, Yerushalmi R, Fasching PA, Nordstrom JL, Bonvini E, Koenig S, Edlich S, Hong S, Rock EP, Gradishar WJ, & Group SS (2021). Efficacy of Margetuximab vs Trastuzumab in Patients With Pretreated ERBB2-Positive Advanced Breast Cancer: A Phase 3 Randomized Clinical Trial. JAMA Oncol, 7, 573–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraf S, Jain A, Tiwari A, Verma A, Panda PK, & Jain SK (2020). Advances in liposomal drug delivery to cancer: An overview. Journal of Drug Delivery Science and Technology, 56, 101549. [Google Scholar]
- Scaltriti M, Verma C, Guzman M, Jimenez J, Parra JL, Pedersen K, Smith DJ, Landolfi S, Ramon y Cajal S, Arribas J, & Baselga J (2009). Lapatinib, a HER2 tyrosine kinase inhibitor, induces stabilization and accumulation of HER2 and potentiates trastuzumab-dependent cell cytotoxicity. Oncogene, 28, 803–814. [DOI] [PubMed] [Google Scholar]
- Schiff R, Massarweh SA, Shou J, Bharwani L, Mohsin SK, & Osborne CK (2004). Cross-Talk between Estrogen Receptor and Growth Factor Pathways as a Molecular Target for Overcoming Endocrine Resistance. Clinical Cancer Research, 10, 331s–336s. [DOI] [PubMed] [Google Scholar]
- Schneeweiss A, Ruckhäberle E, & Huober J (2015). Chemotherapy for Metastatic Breast Cancer - An Anachronism in the Era of Personalised and Targeted Oncological Therapy? Geburtshilfe Frauenheilkd, 75, 574–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya M (2011). Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes Cancer, 2, 1097–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson ER, & Davis SR (2001). Minireview: aromatase and the regulation of estrogen biosynthesis--some new perspectives. Endocrinology, 142, 4589–4594. [DOI] [PubMed] [Google Scholar]
- Singer JW, Shaffer S, Baker B, Bernareggi A, Stromatt S, Nienstedt D, & Besman M (2005). Paclitaxel poliglumex (XYOTAX; CT-2103): an intracellularly targeted taxane. Anticancer Drugs, 16, 243–254. [DOI] [PubMed] [Google Scholar]
- Sliwkowski MX, Lofgren JA, Lewis GD, Hotaling TE, Fendly BM, & Fox JA (1999). Nonclinical studies addressing the mechanism of action of trastuzumab (Herceptin). Semin Oncol, 26, 60–70. [PubMed] [Google Scholar]
- Smith CL, Nawaz Z, & O’Malley BW (1997). Coactivator and corepressor regulation of the agonist/antagonist activity of the mixed antiestrogen, 4-hydroxytamoxifen. Mol Endocrinol, 11, 657–666. [DOI] [PubMed] [Google Scholar]
- Sowinska M, & Urbanczyk-Lipkowska Z (2014). Advances in the chemistry of dendrimers. New Journal of Chemistry, 38, 2168–2203. [Google Scholar]
- Sun CY, Qin C, Wang XL, & Su ZM (2013). Metal-organic frameworks as potential drug delivery systems. Expert Opinion on Drug Delivery, 10, 89–101. [DOI] [PubMed] [Google Scholar]
- Sun JJ, Liu YH, Chen YC, Zhao WC, Zhai QY, Rathod S, Huang YX, Tang SQ, Kwon YT, Fernandez C, Venkataramanan R, & Li S (2017). Doxorubicin delivered by a redox-responsive dasatinib-containing polymeric prodrug carrier for combination therapy. Journal of Controlled Release, 258, 43–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan B, Piwnica-Worms D, & Ratner L (2000). Multidrug resistance transporters and modulation. Curr Opin Oncol, 12, 450–458. [DOI] [PubMed] [Google Scholar]
- Tan M, & Yu D (2007). Molecular mechanisms of ErbB2-Mediated breast cancer chemoresistance. Breast Cancer Chemosensitivity, 608, 119–129. [DOI] [PubMed] [Google Scholar]
- Tardi P, Johnstone S, Harasym N, Xie S, Harasym T, Zisman N, Harvie P, Bermudes D, & Mayer L (2009). In vivo maintenance of synergistic cytarabine:daunorubicin ratios greatly enhances therapeutic efficacy. Leukemia Research, 33, 129–139. [DOI] [PubMed] [Google Scholar]
- Thakkar JP, & Mehta DG (2011). A review of an unfavorable subset of breast cancer: estrogen receptor positive progesterone receptor negative. Oncologist, 16, 276–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolaney S (2014). New HER2-Positive Targeting Agents in Clinical Practice. Current Oncology Reports, 16. [DOI] [PubMed] [Google Scholar]
- Trock BJ, Leonessa F, & Clarke R (1997). Multidrug resistance in breast cancer: a meta-analysis of MDR1/gp170 expression and its possible functional significance. J Natl Cancer Inst, 89, 917–931. [DOI] [PubMed] [Google Scholar]
- van der Wall E, Beijnen JH, & Rodenhuis S (1995). High-dose chemotherapy regimens for solid tumors. Cancer Treatment Reviews, 21, 105–132. [DOI] [PubMed] [Google Scholar]
- Vangijzegem T, Stanicki D, & Laurent S (2019). Magnetic iron oxide nanoparticles for drug delivery: applications and characteristics. Expert Opinion on Drug Delivery, 16, 69–78. [DOI] [PubMed] [Google Scholar]
- Waks AG, & Winer EP (2019). Breast Cancer Treatment: A Review. JAMA, 321, 288–300. [DOI] [PubMed] [Google Scholar]
- Wang LR, Liu J, Huang MZ, & Xu N (2007). Comparison of pharmacokinetics, efficacy and toxicity profile of gemcitabine using two different administration regimens in Chinese patients with non-small-cell lung cancer. J Zhejiang Univ Sci B, 8, 307–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss RB, Donehower RC, Wiernik PH, Ohnuma T, Gralla RJ, Trump DL, Baker JR Jr., Van Echo DA, Von Hoff DD, & Leyland-Jones B (1990). Hypersensitivity reactions from taxol. J Clin Oncol, 8, 1263–1268. [DOI] [PubMed] [Google Scholar]
- Whittle JR, Lewis MT, Lindeman GJ, & Visvader JE (2015). Patient-derived xenograft models of breast cancer and their predictive power. Breast Cancer Research, 17, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicki A, Witzigmann D, Balasubramanian V, & Huwyler J (2015a). Nanomedicine in cancer therapy: Challenges, opportunities, and clinical applications. Journal of Controlled Release, 200, 138–157. [DOI] [PubMed] [Google Scholar]
- Wicki A, Witzigmann D, Balasubramanian V, & Huwyler J (2015b). Nanomedicine in cancer therapy: challenges, opportunities, and clinical applications. J Control Release, 200, 138–157. [DOI] [PubMed] [Google Scholar]
- Widdison WC, Wilhelm SD, Cavanagh EE, Whiteman KR, Leece BA, Kovtun Y, Goldmacher VS, Xie H, Steeves RM, Lutz RJ, Zhao R, Wang L, Blattler WA, & Chari RV (2006). Semisynthetic maytansine analogues for the targeted treatment of cancer. J Med Chem, 49, 4392–4408. [DOI] [PubMed] [Google Scholar]
- Williams RM, Shah J, Ng BD, Minton DR, Gudas LJ, Park CY, & Heller DA (2015). Mesoscale nanoparticles selectively target the renal proximal tubule epithelium. Nano Lett, 15, 2358–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C-X, Xing L, Chang X, Zhou T-J, Bi Y-Y, Yu Z-Q, Zhang Z-Q, & Jiang H-L (2020). Synergistic Platinum(II) Prodrug Nanoparticles for Enhanced Breast Cancer Therapy. Molecular Pharmaceutics, 17, 1300–1309. [DOI] [PubMed] [Google Scholar]
- Yu J (2020). Enhancement of Gemcitabine and Paclitaxel Uptake and Retention in Metastatic Cancer Cells: A Novel Targeted Combination Delivery Approach to Eliminate Breast Cancer in Blood and Lymph. Unpublished Ph.D., University of Washington, Seattle. [Google Scholar]
- Yu J, Mu Q, Perazzolo S, Griffin JI, Zhu L, McConnachie LA, Shen DD, & Ho RJ (2020). Novel Long-Acting Drug Combination Nanoparticles Composed of Gemcitabine and Paclitaxel Enhance Localization of Both Drugs in Metastatic Breast Cancer Nodules. Pharm Res, 37, 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Yu D, Lane S, McConnachie L, & Ho RJY (2020). Controlled Solvent Removal from Antiviral Drugs and Excipients in Solution Enables the Formation of Novel Combination Multi-Drug-Motifs in Pharmaceutical Powders Composed of Lopinavir, Ritonavir and Tenofovir. J Pharm Sci, 109, 3480–3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang ST, Chang SJ, Shahzad MB, Sun XT, Jiang XR, & Yang HZ (2019). Ceramics-based Drug Delivery System: A Review and Outlook. REVIEWS ON ADVANCED MATERIALS SCIENCE, 58, 82–97. [Google Scholar]
- Zhang J, Zhang P, Zou Q, Li X, Fu J, Luo Y, Liang X, & Jin Y (2018). Co-Delivery of Gemcitabine and Paclitaxel in cRGD-Modified Long Circulating Nanoparticles with Asymmetric Lipid Layers for Breast Cancer Treatment. Molecules, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Sun F, Liu S, & Jiang S (2016). Anti-PEG antibodies in the clinic: Current issues and beyond PEGylation. Journal of Controlled Release, 244, 184–193. [DOI] [PMC free article] [PubMed] [Google Scholar]