

Abstract
Therapeutic blockade of the programmed cell death protein-1 (PD-1) immune checkpoint pathways has resulted in significant reactivation of T-cell mediated antitumor immunity and is a promising clinical anticancer treatment modality in several tumor types, but the durable response rate remains relatively low (15–20%) in most hepatocellular carcinoma (HCC) patients for unknown reasons. Evidences reveal that the interferon signaling pathway plays a critical role in modulating the efficacy and sensitivity of anti-PD-1 therapy against multiple tumor types, but the mechanisms are unclear. Using Kaplan-Meier survival analysis based on HCC databases, we found that deceased expression of interferon regulatory factor (IRF8) in HCC, among all the nine IRF members which regulate interferon signals, was associated with the poor prognosis of HCC patients. Moreover, gene set enrichment analysis identified the IFN-gama and PD-1 signaling signatures as the top suppressed pathways in IRF8 low HCC patients. Contrarily, overexpression of IRF8 in HCC cells significantly enhanced antitumor effects in immune-competent mice, modulating infiltration of tumor associated macrophages (TAMs) and T cells exhaustion in tumor microenvironment. We further demonstrated that IRF8 regulated recruitment of TAMs by inhibiting the expression of CCL20. Mechanically, IRF8-mediated repression of c-fos transcription resulted in decreased expression of CCL20, rather than directly bound to CCL20 promoter region. Importantly, adeno-associated virus 8-mediated hepatic IRF8 rescue significantly suppressed HCC progression and enhanced the response to anti-PD-1 therapy. In conclusion: This work identified IRF8 as an important prognostic biomarker in HCC patients which predicted the response and sensitivity to anti-PD-1 therapy and uncovered it as a new therapeutic target for enhancing the efficacy of immune therapy.
Keywords: Hepatocellular carcinoma, Interferon signaling, IRF8, Tumor associated macrophage, Immune therapy
Introduction
Hepatocellular carcinoma (HCC) is one of the most fatal human malignancies, accounting for approximately 90% of all primary liver cancers. HCC is often diagnosed at advanced stages of disease, with overall five-year survival rate of only 14% to 18%, for which highly effective therapies are insufficient and has become a serious health concern worldwide(1). Programmed cell death protein-1 (PD-1) and programmed death-ligand 1 (PD-L1) checkpoint blockade is a promising clinical anticancer treatment modality in some patients with metastatic carcinomas of multiple tissue origins, by blocking the interaction between PD-1 and PD-L1 to reactivate T-cell mediated antitumor immunity(2). However, clinical trials show that the durable response rate to anti-PD-1 therapy remains relatively low in patients with HCC, approximately 15%−20%(3–6). Presently, evidence shows that the degree of interferon (IFN) signaling pathway reflects the effectiveness of PD-1 blockade in treatment of several cancers(7–9), but the mechanisms are unclear. The abnormal presence of IFN-inducible genes in HCC lesions is frequent and treatment with IFN is correlated with better outcome of HCC patients(10, 11). Therefore, we suspect that the dysregulated IFN signaling pathway drives HCC progression and disrupts anti-tumor immunity. Consequently, understanding the mechanisms underlying upstream regulation of the IFN signaling pathway will uncover novel targets for HCC immunotherapy.
Interferon regulatory factors (IRFs) are intracellular mediators that mediate transcription of IFN and IFN-inducible genes. In mammals, the IRFs family comprises of nine members (IRF1-IRF9)(12, 13). Previous studies show that IRFs may exert functional roles in regulating progression of several cancers as tumor suppressors or oncogenic factors(9, 14). IRFs are mainly considered transcription factor that regulate IFN signals, however, whether they can modulate the growth of HCC are not clear. We previously analyzed the expression, survival and prognosis of IRFs members in patients with HCC through the The Cancer Genome Atlas (TCGA) database and Kaplan-Meier survival analysis. Importantly, among the members of IRFs, expression of IRF8 was associated with longer overall survival of HCC patients. IRF8, also known as the interferon consensus sequence-binding protein, regulates differentiation of immune cells as well as functioning of the innate immunity(15). Presently, substantial evidence suggests that IRF8 exerts both oncogenic and tumor suppressor activities in hematoma, colon cancer, breast cancer, etc. However, the roles and the underlying mechanisms of hepatic IRF8 in the development and progression of HCC have not been reported up to date.
In this study, we used immunohistochemical (IHC) staining and quantitative real-time PCR (qPCR) to evaluate the expression pattern of IRF8 and its clinical and pathological significance in human HCC tissues. We found that IRF8 is downregulated in HCC and the decreased expression of IRF8 in tumors were positively correlated with suppressed enrichment of genes involved in PD-1 signaling and interferon-gama response. Particularly, hepatic IRF8 regulates anti-HCC immunity by suppressing undesirable infiltration of macrophage to the tumor microenvironment (TME) via modulation of CCL20 secretion. Furthermore, adeno-associated virus 8 (AAV8)-mediated rescue of hepatic IRF8 expression (AAV8-IRF8OE) is a transformational therapy demonstrated to suppress HCC progression in multiple HCC mouse models. Notably, AAV8-IRF8OE based therapy enhances the response to anti-PD-1 blockade. These findings underscore the pivotal role of IRF8 in HCC pathogenesis and immune response. Our findings suggest that hepatic IRF8 is a TME prognostic biomarker that may predict the response to anti-PD-1 therapy and underline the therapeutic potential of IRF8 agonist for immune checkpoint-based treatment of HCC.
Materials and Methods
Animals models and treatment
The C57bl/6 mice, BALB/c, BALB/c-NU NU and BALB /c nude mice were purchased from Charles River Laboratories (Shanghai, China). The NOD-SCID mice were purchased from Shanghai SLAC Laboratory Animal Co., Ltd and NSG mice were purchased from GemPharmatech Co., Ltd (Nanjing, China). All mice used in the experiments were 6–8 week-old male mice (weighing 20–22 g), and housed in specific pathogen free environment with a 12 h/12 h day/night turnover. All animal experiments were approved by the Research Ethics Committee of the Center for New Drug Evaluation and Research (China Pharmaceutical University, China) and in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory animals (NO. B20171127–1).
For the subcutaneous hepatocellular carcinoma mouse model, 2*106 cells suspended in 0.1 mL of Matrigel (Corning) were subcutaneously injected into the flanks of the mice as indicated. Human HCC cells Hep3B, HCCLM3 and PLC/PRF/5 stable cells and control cells were subcutaneously injected into immunodeficient BALB/c nude, BALB/c NU NU and NOD-SCID mice, respectively. Murine Hepa-1-6, Hepa1c1-7 stable cells and control cells were injected subcutaneously into the right posterior flanks of immune-competent C57bl/6 mice or immunodeficient NSG mice.
For the orthotopic hepatocellular carcinoma mouse model, Hepa1-6-Luc and H22-Luc (1–2*105) cells were implanted into the anterior hepatic lobe of BALB/c nude and BALB/c mice, respectively, to mimic the primary HCC. In DEN/CCL4-induced HCC models, fourteen-day-old C57bl/6 mice were administered 25 mg/kg diethylnitrosa-mine (DEN; Sigma Aldrich) via intra-peritoneal (i.p.) injection. Two weeks after DEN treatment, mice were administered 0.5μL/g carbon tetrachloride (CCl4) dissolved in corn oil via i.p. injection once per week for 20 weeks.
Establishment of orthotopic HCC models were evaluated by an in vivo imaging system and magnetic resonance Imaging (MRI). Subcutaneous tumor growth was assessed by caliper measurement once every two days. The tumor volume was calculated using the following formula: Tumor volume (mm3) = (tumor length ×width2)/2.
Human samples
HCC tissue samples used in qPCR for IRF8 and CCL20 gene expression were obtained from The Affiliated Drum Tower Hospital. This study was approved by The Affiliated Drum Tower Hospital Research Ethics Committee, and patients provided their informed consents.
Additional materials and methods are described in the Supporting Materials and Methods.
Results
Decreased expression of IRF8 is associated with poor prognosis of HCC.
To identify the potential IRF family dysregulated between HCC tissues and normal tissues, we first surveyed the expression profile of IRFs from the TCGA data for 374 HCC tumor and 50 adjacent normal tissues. All nine IRFs were submitted to analysis. Of these, Only IRF8 mRNA was remarkably decreased in HCC tissues, whereas IRF3 mRNA tended towards upregulation (Fig.1A and Fig.S1A–B). The expression profile of IRF3 and IRF8 in liver hepatocellular carcinoma (LIHC) was further validated using gene expression omnibus database (GEO) database. Here, IRF8 was downregulated in all the nine GEO data sets. On the other hand, two in nine GEO data sets showed that IRF3 expression in HCC remained largely unaltered (Fig.1B and Fig.S1C). Kaplan-Meier analysis of GEO and TCGA data further revealed that the decreased expression of IRF8 was associated with poor prognosis of HCC (Fig.1C and Fig.S1D). Given the insignificant survival value of IRF3 expression in HCC (Fig.S1E–F), our subsequent analyses only focused on IRF8.
Figure 1.
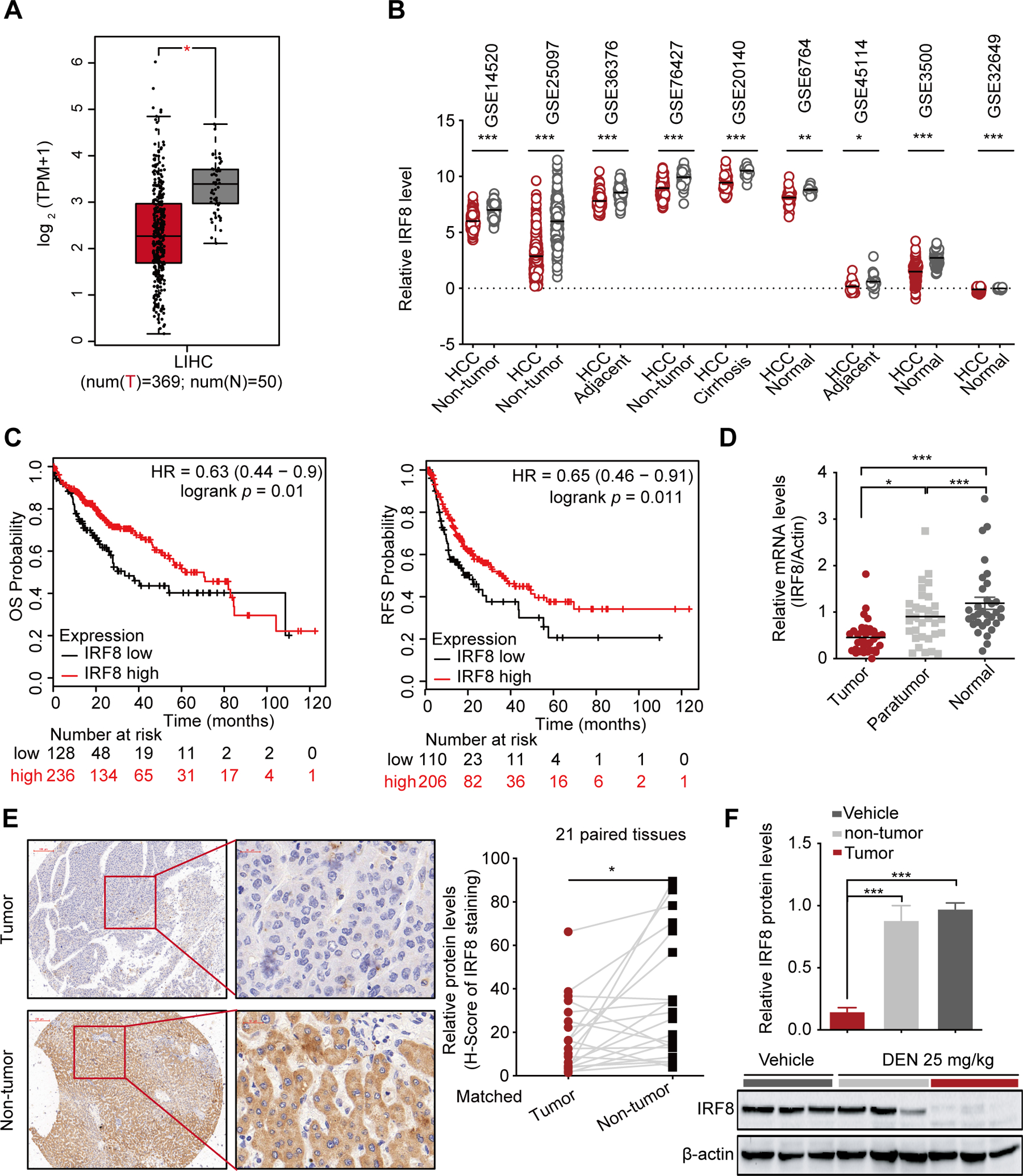
Decreased expression of IRF8 in HCC significantly correlates with the poor prognosis. (A) The comparative expression of IRF8 in 369 HCC tissues and 50 adjacent normal liver tissue samples based on TCGA data using GEPIA website. (B) The comparative expression of IRF8 in HCC and non-tumor tissues in 9 independent GEO data sets. (C) Kaplan-Meier overall survival (OS) and relapse-free survival (RFS) curves for HCC patients based on expression levels of IRF8 using Kaplan-Meier Plotter website. (D) qPCR for the relative expression levels of IRF8 mRNA in 30-pairs of tumor compared with adjacent and normal liver tissues. (E) Representative IHC images for IRF8 expression in 21-pairs of HCC and adjacent normal tissues. The relative protein expression was based on H-score. *p<0.05 (Paired t-test). (F) Western blot analysis for IRF8 protein levels in DEN-induced murine HCC tissues.
Meanwhile, qPCR analyses of 30 paired HCC tissues with adjacent noncancerous liver tissues and 30 normal liver tissues validated the decreased expression of IRF8 mRNA in HCC, relative to the normal tissues (Fig.1D and Fig.S1G). IHC analysis using human HCC tissue microarray of 21 paired tumor and non-tumor specimens further revealed that not only mRNAs, but also the corresponding IRF8 proteins were decreased in human HCC tissues (Fig.1E). The downregulated expression of IRF8 proteins was also observed in N-nitrosodiethylamine (DEN)-induced HCC tissues in mouse model (Fig.1F and Fig.S1H). Overall, these findings have demonstrated that among the IRFs, only IRF8 is downregulated in HCC tissues and lower IRF8 predicts poor clinical outcomes in patients with HCC.
Building the above findings, we investigated mechanisms underlying modulated-expression of IRF8 in HCC cells. Studies have reported that IRF8 expression can be regulated by IFN signaling or epigenetic modification(16–19), but IFN gene levels were not changed between the HCC and normal tissues, so we focused on the epigenetic modulations at IRF8 gene focus. We found that the decreased expression of IRF8 in HCC is not associated with genomic deletion/mutation of IRF8 gene, DNA CpG hypomethylation, histone methylation but with histone deacetylation. Histone deacetylase (HDAC) inhibitor trichostatin A treatment upregulated the expression of IRF8 in 3 HCC cells, in a dosage dependent manner (Fig.S2A). Similarly, drastic elevation of IRF8 expression was also observed in HCC cells treated with another two HDAC inhibitors vorinostat and panobinostat (Fig.S2B). As acetylation of histone H3 at Lys9 (H3K9) is linked to transcriptional activation of IRF8 promoter (Fig.S2C), we performed chromatin immunoprecipitation (ChIP) experiments and found that acetylation of H3K9 at the IRF8 promoter was significantly increased in HCC cells after TSA treatment (Fig.S2D–E). Furthermore, ChIP-PCR analysis verified that the IRF8 gene locus showed reduced levels of H3K9ac in HCC as compared to that in nontumor tissues (Fig.S2F). Taken together, these findings suggest that lower IRF8 expression in HCC is at least in part deregulated by H3K9-acetylation at the IRF8 gene locus.
IRF8 inhibits HCC progression in vivo via the host immune system
We developed HCC cells overexpression (OE) IRF8 or knockdown (KD) cell lines through the stable transduction of IRF8 CDS domain associated plasmids or IRF8-shRNA lentiviral particles in mouse and human HCC cells. Similar models were also developed in mice using doxycycline hydrochloride (DOX) treatment. We found dysregulated IRF8 expression had no effect on cell proliferation and clonogenic ability of HCC cells in vitro (Fig. 2A–B and Fig. S3A–C). However, inoculation of IRF8OE cells into immunocompetent syngeneic C57bl/6 showed significant tumor growth inhibition, improving the overall survival of mice compared to those in control animals (Fig. 2C–D). We found IRF8 overexpression impaired growth of tumor cells with lower PCNA and Ki67 expression in the cancer tissues, relative to controls (Fig. 2E). Collectively, these findings demonstrated that hepatic IRF8 regulates proliferation of HCC in vivo but not in vitro. Interestingly, overexpression or knockdown of IRF8 had no effect on proliferation of HCC cell lines xenografted into immune compromised BALB/c nude, BALB/c-nu/nu or NOD-SCID mice (Fig. S3D–F).
Figure 2.
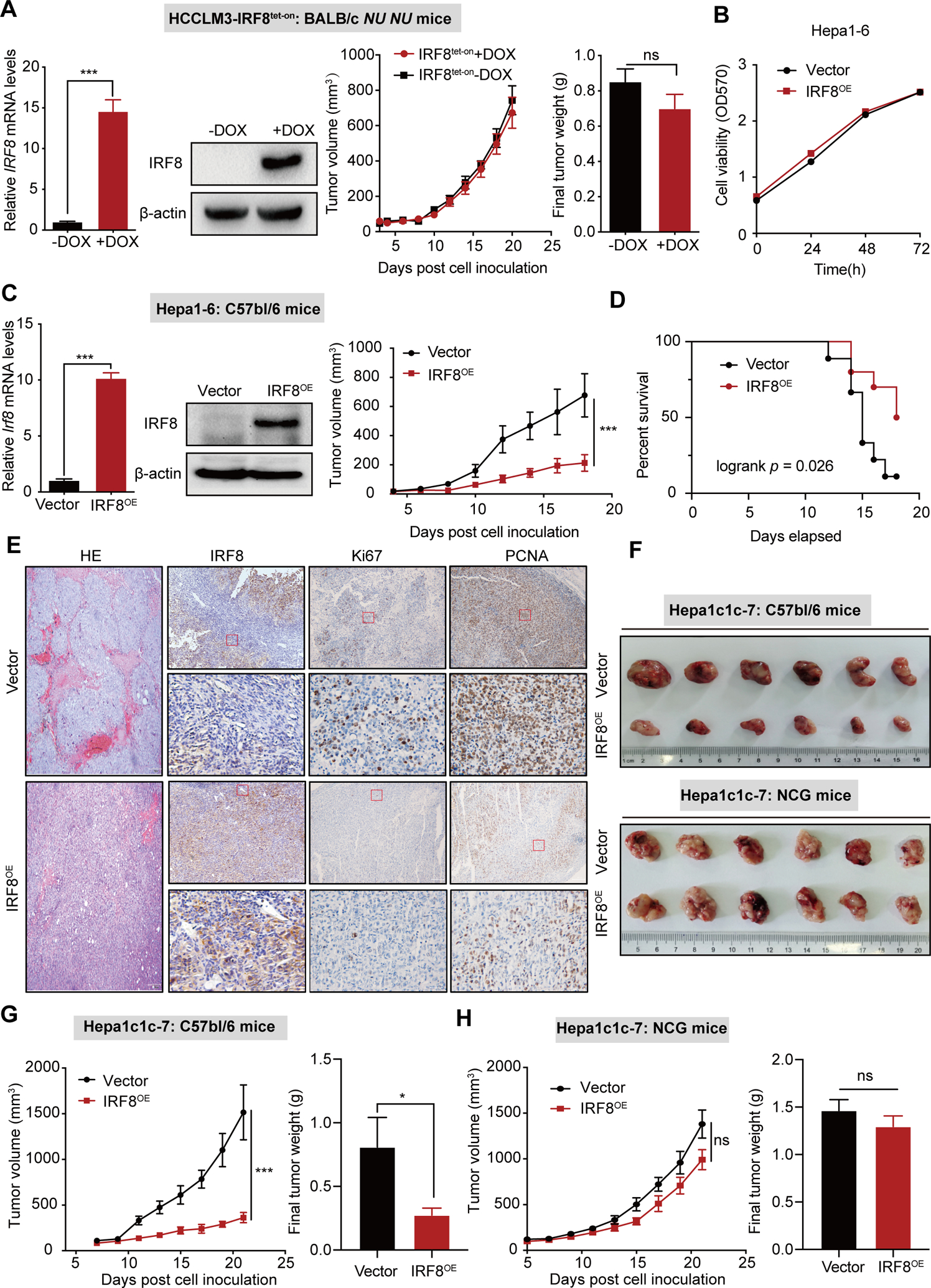
IRF8 inhibits HCC progression in vivo. (A) qPCR, western blot analyses, tumor growth curves and final tumor weight of IRF8 overexpression (IRF8OE) in human HCCLM3 cells xenografted into immune-deficient BALB/c-NU NU mice. (B) Sulforhodamine B based cell viability analysis of IRF8 overexpression (IRF8OE) in murine Hepa1-6 cells. (C) qPCR and western blot analyses and tumor growth curves of for murine Hepa1-6- IRF8OE cells transplanted into immunocompetent syngeneic mice. (D) Kaplan-Meier survival curves for the Hepa1-6-mouse with IRF8OE tumors or corresponding controls. (E) HE and IHC for IRF8, PCNA and Ki67 expression in IRF8OE tumors or vector controls in the Hepa1-6-mouse model. (F-H) Tumors, tumor growth curves and final tumor weight of the Hep1c1c-7-mouse model bearing IRF8OE tumors or vector control in immunocompetent C57bl/6 and immune-deficient NSG mice.
To validate the role of the immune system in hepatic IRF8-mediated regulation of HCC, we injected murine IRF8OE HCC cell lines or their control into syngeneic mouse hosts (C57bl/6) or NCG/NOD-SCID, mice deficient of T cells, B cells and natural killer cell (Fig. 2F–H and Fig. S3G–H). Notably, the anti-HCC functions of IRF8 were substantially impaired in immune-deficient mice (Fig. S3I). These findings suggest that the tumor suppression function of IRF8 in vivo is mediated by the host immune system.
IRF8 overexpression in HCC cells induces anti-tumor immunity by inhibiting infiltration of tumor associated macrophages in HCC
Based on the above findings, we hypothesized that the anti-HCC IRF8 properties are mediated by the host antitumor immune responses. We therefore injected both IRF8OE and corresponding wild type hepa1-6 cells into either side of the same mouse. Strikingly, co-injection of IRF8OE cells significantly suppressed tumor growth of WT Hepa1-6 tumor cells (Fig. 3A). These findings suggested that IRF8 against HCC is associated with immunity of TME.
Figure 3.
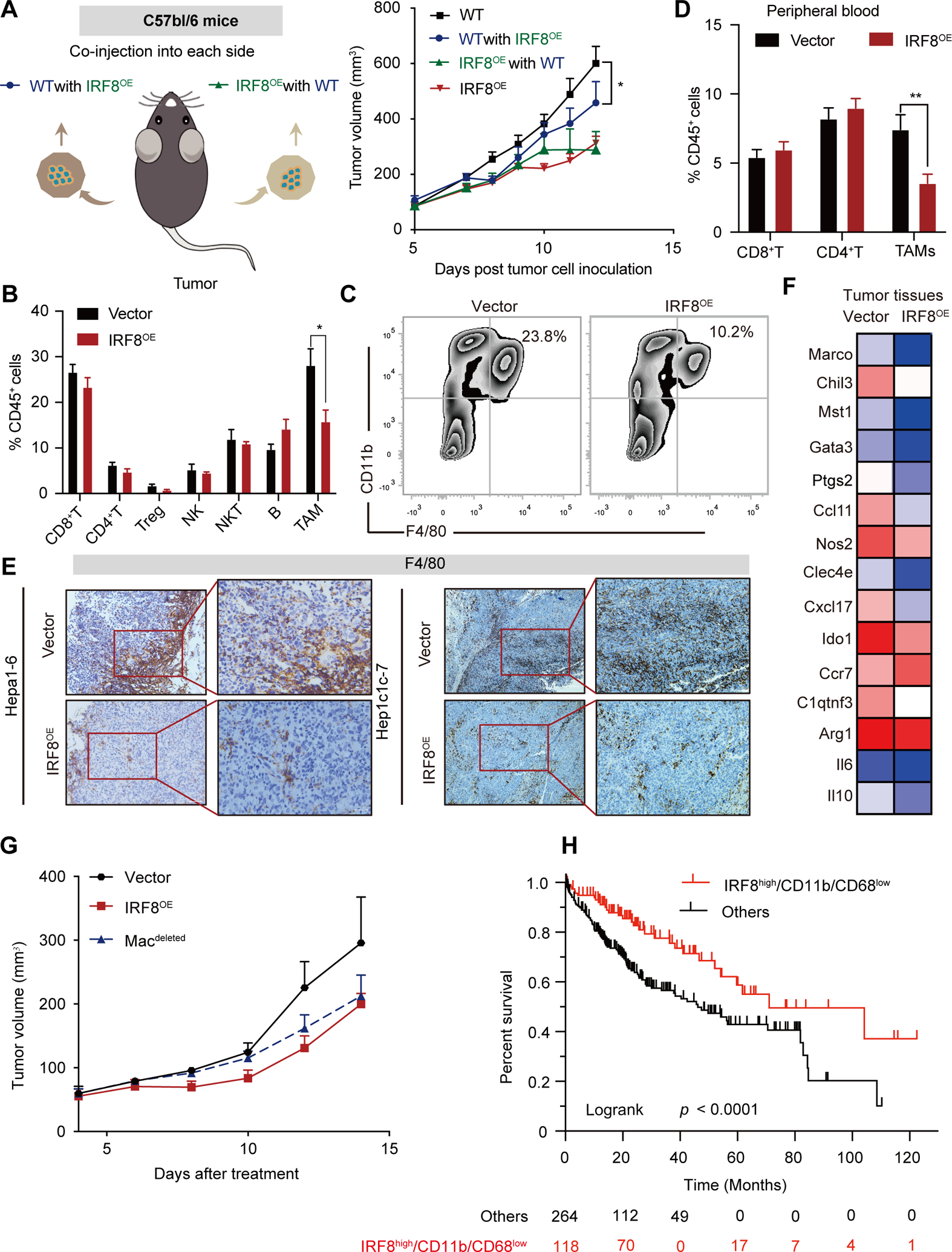
IRF8 inhibits tumor growth in a macrophage-dependent manner. (A) Tumor growth curve for IRF8OE Hepa1-6 cells or vector controls which were co-injected into opposite flanks into C57BL/6 mice (left panel). “Vector with IRF8OE” (blue line) indicates vector tumor growth, and “IRF8OE with vector” (green line) indicates IRF8OE tumor growth, in the co-injected mice. (B) Quantification by flow cytometry of the ratio of CD8+ T cells, CD4+ T cells, CD25+ Treg cells, NK1.1+ NK cells, NK49b+ NKT cells, CD19+ B cells, and CD11b+F4/80+ TAMs within the CD45+ population to total live cells in Hepa1-6 tumors. (C) Representative flow cytometric analysis of TAMs in IRF8OE and vector controls. (D) Quantification by flow cytometry of the ratio of CD8+ T cells, CD4+ T cells, and CD11b+ F4/80+ TAMs within the CD45+ population to total live cells in peripheral blood of mice bearing with Hepa1-6 tumors. (E) Representative IHC analysis of F4/80+ TAMs in Hepa1-6 and Hepa1c1c-7 tissues with IRF8OE and vector control tumors. (F) Heatmap of a selected gene list of TAMs showed by gene FPKM expression based on RNA-seq data of IRF8OE and vector control tumors in Hepa1-6 models. (G) Tumor growth curve for Hepa1-6 models with IRF8OE, vector control or with Macrophage deletion (Macdeleted). Macrophage deletion was induced using clodronate liposomes treatment (intravenous injection, 100μL/time for 2 times). (H) Kaplan-Meier survival analysis for IRF8highCD11b/CD68low in HCC patients.
To further explore the specific immune component critical in anti-tumor IRF8 function, we analyzed cell infiltrates in TME using flow cytometry. We found that the infiltration of CD11b+ and F4/80+ macrophages (F4/80 and CD11b are mouse tumor associated macrophage (TAMs) markers) and the percentage of PD1+CD4+T cells and PD1+CD8+T cells significantly decreased in IRF8OE tissues and in peripheral blood (Fig. 3B–D, Fig. S4A–E). IHC staining for F4/80 in tumor xenografs confirmed the decreased infiltration of F4/80 positive TAMs in the IRF8OE xenografts, both in Hepa1-6 and Hepa1c1c-7 models (Fig. 3E). To further validate the effect of IRF8OE in HCC tissues against TAMs, we performed RNA sequence (RNA-seq) in hepa1-6-IRF8OE and control Hepa1-6 tumor tissues. We found the majority of TAMs signature genes including Marco, Chil3, Mst1 were down-regulated in IRF8OE HCC (Fig. 3F). To analyze the essential relationship between IRF8 overexpression in HCC and modulated infiltration of TAMs, we investigated the effect of inhibiting infiltration of macrophages following treatment with liposomal clodronate or colony-stimulating factor 1 (CSF-1) inhibitors for 2 weeks, after tumor xenograft establishment. We found selective blockade of macrophage in TME with anti-CSF-1 antibody or clodronate liposomes treatment resulted in dramatically decreased tumor weight and the antitumor effects of IRF8 were completely eliminated (Fig. 3G and Fig. S4F–G). This suggests that TAMs substantially influence IRF8 mediated anti-tumor activity in HCC cells. Moreover, EPIC database analysis for the proportion of tumor infiltrating immune cells in TCGA-LIHC samples revealed that macrophages were the most decreased immune cells infiltrated in IRF8high HCC patients with lower immune cell score, consistent with the results that IRF8 inhibits TAMs infiltration (Fig. S4H). We further investigated whether IRF8 and decreased TAM markers were associated with prognosis of HCC patients in TCGA. Kaplan-Meier analysis revealed that high expression of IRF8 but low infiltration of CD11b+/CD68+ TAMs was associated with better prognosis of HCC (Fig. 3H). Taken together, these findings suggest that hepatic IRF8 enhances antitumor immune response in HCC by regulating the infiltration of TAMs in the TME.
CCL20 signaling promotes TAMs recruitment
We first verified in vitro that conditioned media (CM) of IRF8 overexpressing Hepa1-6 cells reduced macrophage chemotaxis (Fig. 4A). To identify potential IRF8 downstream effector(s) mediating decreased-infiltration of TAMs in tumor cells over-expressing IRF8, we analyzed RNA-seq profiles of Hepa1-6-IRF8OE and control cells. We identified 164 and 135 downregulated and upregulated genes, respectively, in Hepa1-6-IRF8OE cells (more than 2-fold change; p < 0.0005; Fig. 4B). GO enrichment analysis revealed that the differentially expressed genes involved in regulation of cytokine production, innate immune response and cytokine metabolic process, were the most significantly enriched pathways (Fig. S5A). Notably, CCL20 was the most down-regulated immune related molecule in cells over-expressing IRF8 (Fig. 4B–C and Fig. S5B). CCL20, also known as liver and activation-regulated chemokine, is one of the most important chemokines that regulates the infiltration of immune cells in liver tissues. Analysis of TCGA and GEO data revealed that CCL20 was substantially upregulated in HCC tissues. Intriguingly, up-regulation of CCL20 was strongly and negatively associated with short overall survival and poor prognosis of HCC (Fig. S5C–D). Further qPCR analysis revealed that overexpression of IRF8 decreased the expression of CCL20 in HCC cells or tumor tissue. ELISA assay further validated the decreased expression of CCL20 in IRF8OE cells and tumor mass (Fig. 4D–E). Meanwhile, we found CCL20 expression was upregulated in IRF8KD cells, relative to the controls (Fig. S5E). Additionally, a strong negative correlation between IRF8 and CCL20 expression were observed in three HCC cell lines (Fig. S5F). Further qPCR analysis validated the increased expression of CCL20 and the negative correlation between IRF8 and CCL20 expression in HCC tissues (Fig. 4F–G, Hepa1-6 tumors: R = −0.64, p = 0.002; HCC patients: R = −0.467, p < 0.001). Further IHC staining analysis of clinical samples using TMA confirmed the up-regulation of CCL20 in HCC patients, relative to the adjacent normal tissues. Consistently, patients of HCC with low expression of IRF8 exhibited higher CCL20 expression (Fig. 4H–I), further confirming the negative correlation in the expression of the two proteins in HCC specimens (Fig. S5G). Finally, we confirmed that IRF8 decreases macrophage recruitment via downregulating CCL20 expression by supplementing CCL20 in Hepa1-6-IRF8OE cells and tumor sites. As shown in figure 4J, the suppressing role of CM from IRF8OE cells on macrophage infiltration was largely compromised by addition of recombinant CCL20, indicating that CCL20 enhances the migratory ability of macrophages. Similarly, supplementing CCL20 in subcutaneous tumor models by intratumoral injection largely weakened the antitumor effect induced by IRF8OE, establishing that CCL20 is critical in IRF8 mediated antitumor effects in tumor-bearing mice (Fig. S5H). Overall, these findings demonstrated that IRF8 modulates infiltration of macrophages in TME by down-regulating the expression of CCL20.
Figure 4.
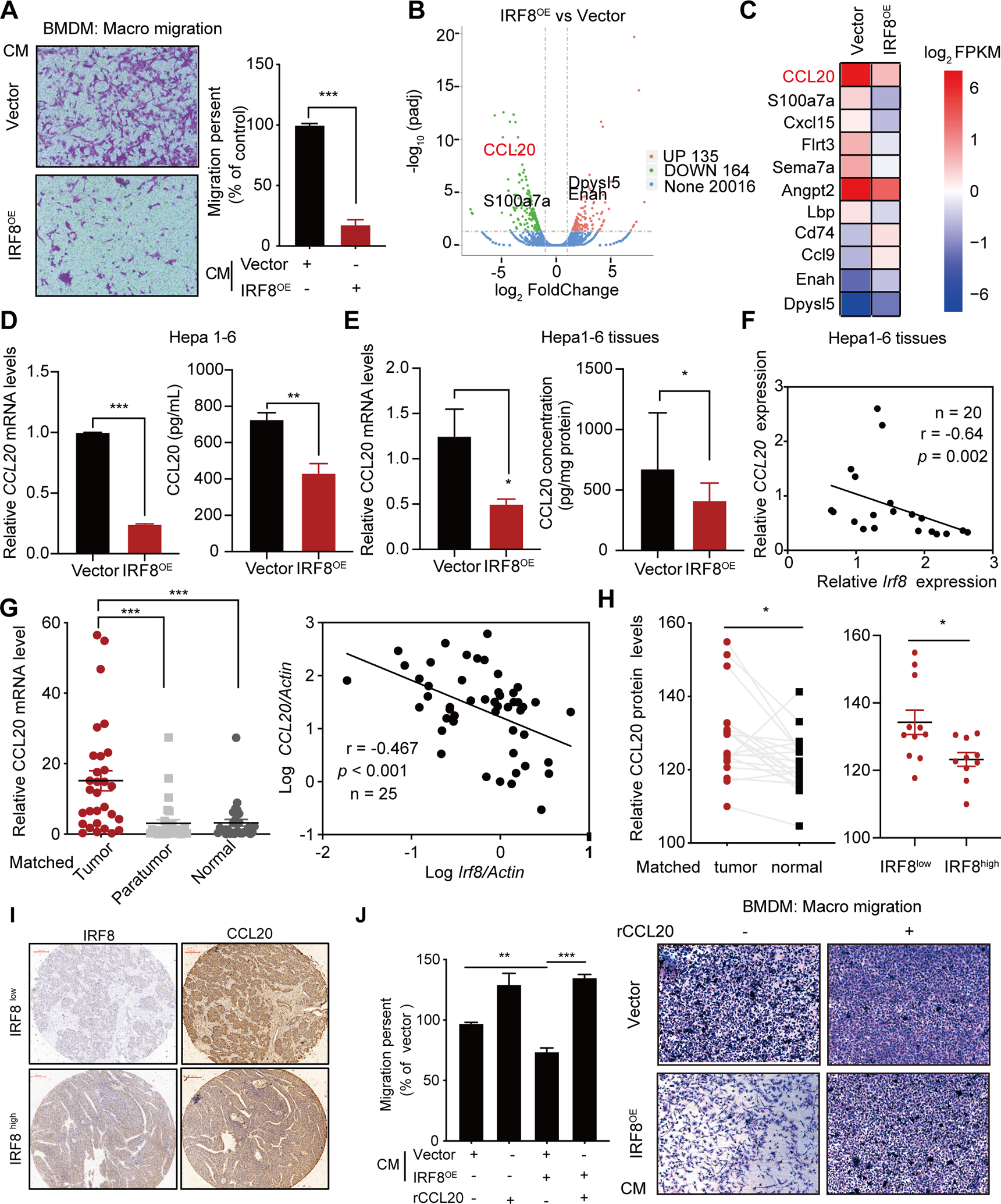
Mechanism underlying suppression of macrophage infiltration by IRF8. (A) Migration assay and quantitative analysis of bone marrow-derived macrophages (BMDM) cultured for 24 hours in conditioned medium (CM) of Hepa1-6-IRF8OE or vector control. (B) Volcano map of RNA-seq profiles of Hepa1-6-IRF8OE compared with vector control cells. (C) Heatmap of representative secreted molecules in Hepa1-6-IRF8OE based on RNA-Seq. (D-E) Relative CCL20 mRNA expression and ELISA assay of supernatant (D) and tumor burdens (E) in Hepa1-6-IRF8OE compared with vector control. (F) The negative correlations of IRF8 and CCL20 mRNA level was measured by qPCR in Hepa1-6 tumor and denoted with Pearson’s correlation coefficients assay. (G) qPCR for the expression of mRNA levels of CCL20 in 30-pairs of tumor and adjacent nontumorous and normal liver tissues (left). The negative correlations between CCL20 and IRF8 expression in HCC tumor were based on Pearson’s correlation coefficients (right). (H) Relative expression of CCL20 proteins in 20 pairs of HCC patients (left), and in IRF8high and IRF8low HCC tissues (right). The classification was based on the median cut-off of IRF8 protein level in HCC tissues. (I) Representative IHC staining images for CCL20 in IRF8high and IRF8low HCC tissues. (J) Quantitative analysis assay for BMDM migration toward basal medium supplemented with recombinant CCL20 (rCCL20, 100pg /mL). The experiment was performed for 72h.
IRF8 regulates CCL20 expression by repressing c-fos
Research shows that IRF8 represses transcription of interferon consensus sequence (ICS) motif A/GNGAAANNGAAACT or positive regulatory domain I (PRDI, TCACTT or AAGTGA)(12, 13). However, ChIP-qPCR revealed that IRF8 does not bind on CCL20 promoter (Fig. S6A), suggesting that IRF8 does not directly repress CCL20 transcription, consistent with the public ChIP-sequence analysis of IRF8 on CCL20 proomoter. Based on the above findings, we speculated that IRF8 regulates CCL20 expression via a separate pathway.
We therefore analyzed the RNA-seq data of Hepa1-6-IRF8OE cells in the transcriptional regulatory networks database to identify probable transcription factor/pathways (including Pias1, Foxa2, Foxo1, Stat3, AP1 and NF-κB pathway) that regulates CCL20 expression. We found that only the AP1 pathways (c-fos, c-Jun and Junb) were suppressed in IRF8OE HCC cells (Fig. 5A). Further qPCR analysis revealed that IRF8 overexpression only suppressed the expression of c-fos mRNA. Conversely, IRF8 knockdown induced up-regulated expression of c-fos mRNA (Fig. 5B–C and Fig. S6B). These findings were confirmed by western blots analysis. The results revealed that the c-fos protein levels were greatly disrupted in IRF8OE or IRF8KD cells, particularly in the nucleus (Fig. 5D and Fig. S6C). Unchanged phosphorylation levels of NF-κB and STAT3 measured by western blots analysis confirmed that neither the NF-κB nor the STAT3 pathway were involved in IRF8-mediated CCL20 suppression (Fig.5D). Consistent with the other cancers(20), ChIP-qPCR assay confirmed that c-fos transactivates CCL20 gene levels by binding directly to the 5´-TGA(C/G) TCA-3´ sequence on the CCL20 promoter in HCC cells (Fig. S6D). To confirm whether IRF8 regulates the expression of CCL20 via the c-fos signaling pathway, we assessed the expression of CCL20 in IRF8OE HCC cells transfected with a c-fos-expressing plasmids. As expected, the IRF8 mediated CCL20 decrease is greatly reversed by forcing c-fos expression in HCC HCCLM3 and Hep3B IRF8OE cellls (Fig. 5E, Fig. S6E). Moreover, luciferase reporter analysis revealed that c-fos-mediated activation of CCL20 reporter ware significantly abolished by over-expression of IRF8 in HEK293T and Hepa1-6 cells (Fig. 5F). We therefore performed several experiments to assess whether IRF8 directly regulates c-fos expression. Analysis of JASPAR database and public ChIP- sequence data of IRF8 in the Cistrome database revealed that c-fos is a putative IRF8 target in immune cells (Fig. S6F). Moreover, we found several ICS and PRDI motifs both at the human c-fos gene locus. ChIP-qPCR assays and a luciferase reporter assay carrying the c-fos promoter in HCCLM3 cells revealed that IRF8 directly interacts with the c-fos promoter region, and thus repress transcription of c-fos in HCC cells (Fig. 5G–I and Fig. S6G). Overall, the overwhelming evidence suggests that IRF8 regulates CCL20 expression by repressing the transcription of c-fos.
Figure 5.
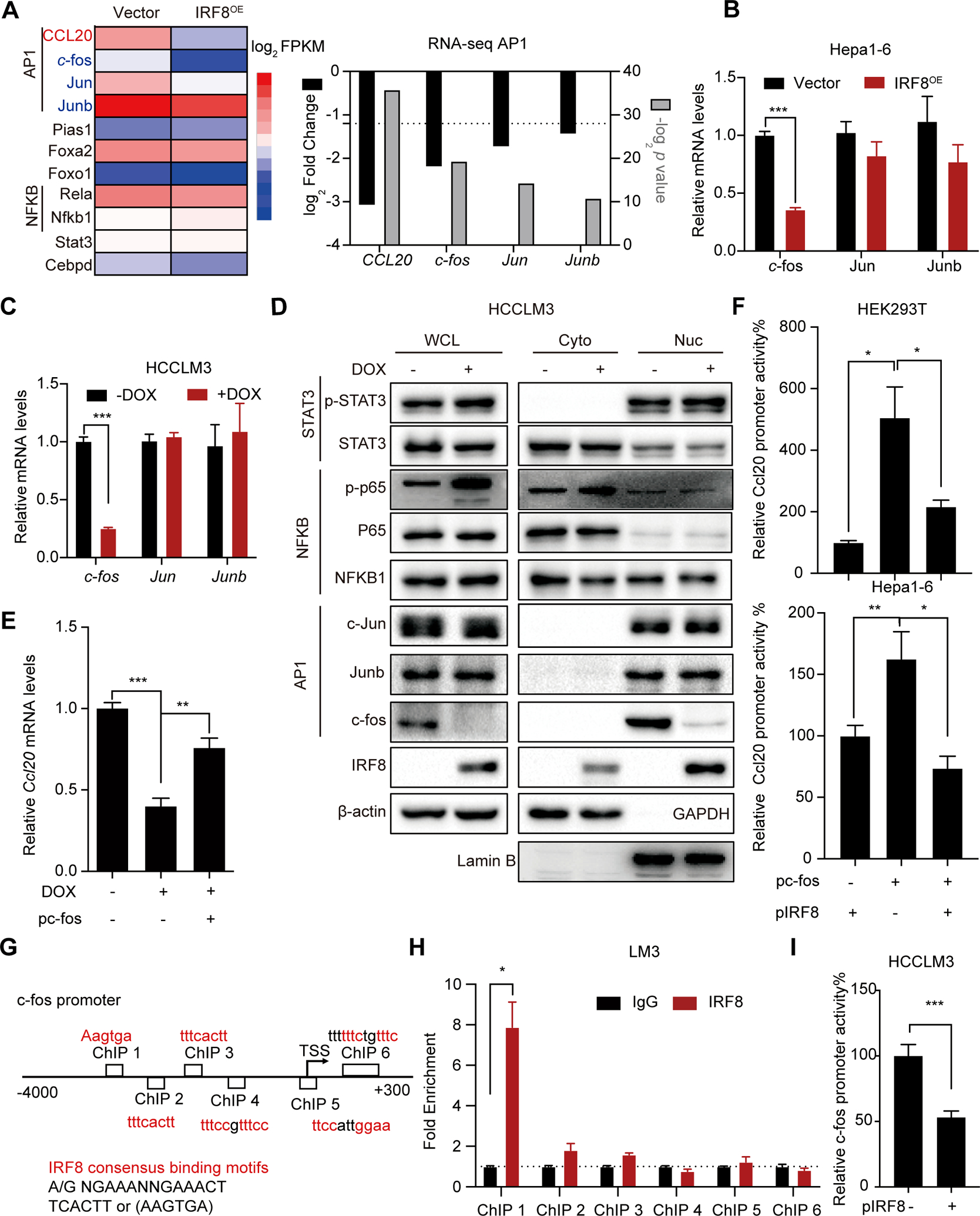
Mechanism underlying IRF8 repression of c-fos transcription. (A) RNA seq data for the expression of differential transcription factor regulating transcription of CCL20 gene in Hepa1-6-IRF8OE cells. (B-C) qPCR analysis for the expression of the AP1 pathway (c-fos, Jun and Junb) expression in 2 HCC cell lines (Hepa1-6, B; HCCLM3,C) with IRF8OE or IRF8KD verus control cells. (D) Western blots assay for the protein expression of the AP1 pathway (c-fos, Jun and Junb), STAT3 (total STAT3 and p-STAT3) and NFκB pathway (NFκB1, P65 and p-P65) in the whole cell lysates (WCL), nucleus(Nuc) and cytoplasm(Cyto) with IRF8 induction. (E) The fold changes in the expression of CCL20 gene by qPCR analysis in HCCLM3 cells transfected with c-fos plasmid (pc-fos) at 24 h after IRF8 expression induction. (F) The effect of IRF8 expression on c-fos-mediated activation of CCL20 transcription. HEK293T or Hepa1-6 cells were co-transfected with IRF8 plasmid (pIRF8) and c-fos expressing plasmid, and thereafter assessed for the relative luciferase activity. (G) The probable IRF8 binding sites identified in the c-fos promoter region. (H) ChIP assays were performed with anti-IRF8 antibodies in HCCLM3 cells. (I) Analysis of the repression of IRF8 in the proximal promoter of c-fos using luciferase reporters in HCCLM3 cells.
AAV8-mediated hepatic IRF8 rescue inhibits HCC progression and enhances the anti-PD1 response
For transformation study, we firstly established a DEN/carbon tetrachloride (CCl4) -induced HCC model in C57bl/6 mice (Fig. 6A). We undertook AAV8 mediated rescue of hepatic IRF8 expression for anti-HCC therapy. After DEN/CCL4 treatment, the mice were injected with AAV8-IRF8OE or AAV8-vector control. We found AAV8 rescue displayed high levels of IRF8 protein, lower HCC incidence, less tumor nodules and lower tumor weight than controls (Fig. 6A–B). Next, we induced the orthotopic tumor model by injecting H22-luc cells under the hepatic membrane of BALB/c mice. Based on tumor fluorescence signals and tumor weight at day 16, AAV8-mediated IRF8 rescue markedly inhibited growth of the tumor (Fig. 6C–D and Fig. S7A), phenotypes not seen in the BALB/c nude mice (Fig. S7B–C). This finding further strengthened the hypothesis that IRF8 exerts its anti-HCC effect via a functional immune system. Notably, the AAV8-mediated rescue of hepatic IRF8 suppressed infiltrations of CD11b+F4/80+ macrophage, PD1+CD4+ T cells, PD1+CD8+ T cells in the TME, accompanied with decreased expression of CCL20 and c-fos in AAV8-treated HCC models (Fig. S7D–H).
Figure 6.
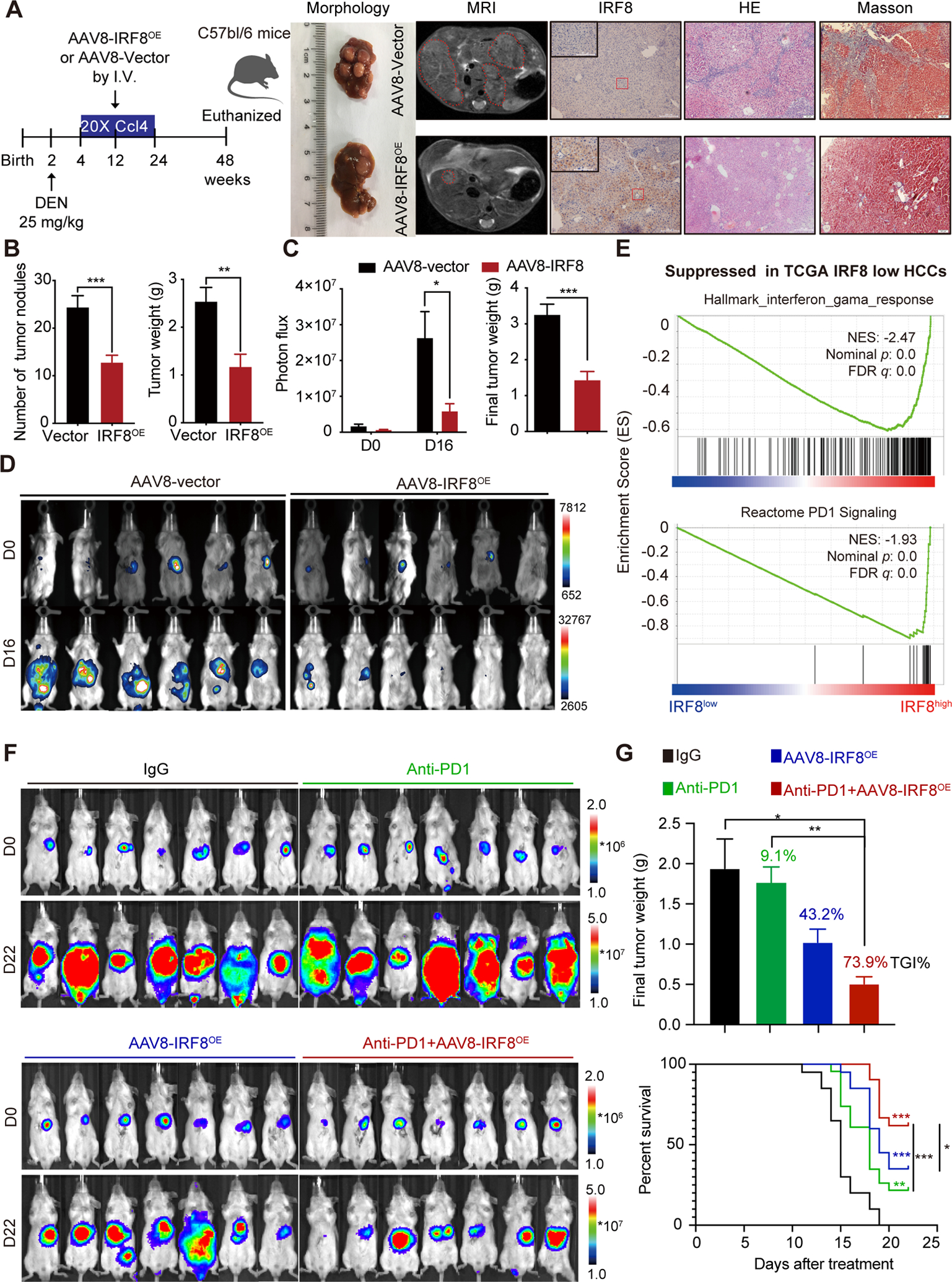
AAV8 mediated rescue of hepatic IRF8 expression suppresses HCC progression and enhances anti-PD-1 response. (A) Scheme of AAV8 mediated hepatic IRF8 rescue (AAV8-IRF8OE), morphology and magnetic resonance imaging (MRI) of liver, IHC images of IRF8, HE and Masson staining of liver tissues in DEN/CCL4- HCC model in C57bl/6 mice. (B) Tumor nodules and tumor weight in AAV8-IRF8OE treated and vector control of DEN/CCL4 HCC models. (C-D) in vivo imaging analysis of AAV8-IRF8OE in BALB/c mice bearing H22-luc cells. H22-luc cells (2*105) were injected into the liver of six-week-old male BALB/C mice. 4 days later, mice were in vivo imaged and randomly divided into two groups according to the total flux of tumor. 100 μl AAV8-IRF8OE or AAV8-vector (1*1012 vg/ml) were injected (I.V.) for once. On day 16, mice were imaged and sacrificed. Results are shown as mean ± SEM. (E) GSEA analysis identified the Hallmark-interferon-gama-response and Reactome PD-1 signaling as the top suppressed pathways in IRF8low HCC patients based on TCGA -LIHC data. GSEA plot based on the gene expression profiles of IRF8high group and IRF8low group according to RNA sequencing data in TCGA-LIHC dataset. NES, normalised enrichment score; FDR, False discovery rate. (F-G) In vivo bioluminescence imaging (F), final tumor weight and overall survival time (G) in murine H22 orthotopic models following IgG control, anti-PD-1, AAV8-IRF8OE, or AAV8-IRF8OE and anti-PD-1 antibody treatment. H22-luc cells (1*105) were injected into the anterior hepatic lobe of six-week-old male BALB/c mice. 4 days later, mice were in vivo imaged and randomly divided into 4 groups and treated with AAV8-IRF8OE, alone or combined with PD-1 (50 μg, q3dx7, I.P.) for 22 days. Tumor growth inhibition (TGI) was used to calculate the antitumor effects with regard to the final tumor weight. Log rank p: ***: p<0.001, **: p<0.01, *: p<0.05.
Studies show that dysregulated expression of the IFN signaling is associated with impaired PD-1 blockade in tumors(21). The expression profile of IFN-γ-related mRNA (IFN-gama signature) may predict clinical response to PD-1 blockade in breast cancer. Go analysis of RNA-seq data revealed that the cellular response to IFN-gama was the most enriched pathways in IRF8OE HCC cells (Fig. S5A). Given the decreased expression of IRF8 in HCC, we reasoned that the low response to anti-PD-1 therapy in HCC patients may be related at least in part to the suppression of IRF8 and IFN signaling pathways. Gene set enrichment analysis (GSEA) revealed that IFN-downregulated gene signature was among the top pathways that were activated in IRF8-low HCC. In contrast, the IFN-upregulated gene signature was among the top activated pathways in IRF8-high HCC (Chiang-liver-cancer-subclass-interferon-DN and Chiang-liver-cancer-subclass-interferon-UP (22), Fig. S8A). Further, TCGA-LIHC analysis revealed that the expression of IRF8 positively correlated with expression of IFN-gama signature that may predict response to PD-1 therapy (Fig. S8B). Consistent with this, GSEA of the TCGA LIHC datasets further revealed that two established gene signature Hallmark-interferon-gama-response and Reactome PD-1 signaling were skewed toward high expression of IRF8 (Fig. 6E). These findings suggested that the dysregulated IFN signaling pathway and PD-1 signaling pathway were mediated by decreased expression of IRF8. Importantly, AAV8-mediated rescue of hepatic IRF8 expression activated T cell function at different levels through regulating TAMs-mediated cross-talk between the innate and adaptive immune in the TME, by restoring IFN signaling pathway and IFN signature genes such as MHC-I expression and increasing intratumoural density of GzmB+CD8+ T cells that can affect response to anti-PD1 therapy in solid tumors(23)(Fig. S8C–E). Overall, given that overexpression of IRF8 in HCC cells can restore normal IFN response and activate T cell function, resulting in a remarkable immune remodeling in HCC models, we therefore assessed whether the AAV8-mediated rescue of hepatic IRF8 expression would enhance anti-PD-1 therapy response in the liver. To achieve this, we evaluated H22-luc orthotopic xenograft characteristics using lowest effective dose of AAV8-IRF8OE treatment (50 μl), either alone or in combination with anti-PD-1 antibodies in immune-competent BALB/c mice. In vivo imaging results and final tumor weight of the tumors revealed that most mice treated with anti-PD-1 antibody alone showed no response to anti-PD-1 therapy (9.1% tumor growth inhibition, TGI), but the combination of AAV8-IRF8OE and anti-PD-1 antibody caused a more pronounced suppressive effect on tumor burden (73.9% TGI value, Fig. 6F–G and Fig. S8F). Importantly, combination of AAV-IRF8OE with anti-PD-1 Ab demonstrated a significantly prolonged survival effect in H22 models compared to monotherapy. This suggest that rescue of hepatic IRF8 could enhance response to anti-PD-1 treatment.
Discussion
Immune checkpoint blockade therapy has provided clinical benefit to some patients with advanced cancer. In these malignancies, several biomarkers including PD-1/PD-L1 expression, a high tumor mutational burden, tumor T cell infiltration and high microsatellite instability or mismatch repair-deficient (24–27) have been proposed to predict response. However, the incidence of these defects in HCC is estimated to be low (~3%)(27) and no correlation between PD- L1 expression and clinical benefit for HCC patients were observed in clinic trials (3, 5). The challenges to the clinical use of immune checkpoint inhibitors in patients with HCC is the identification of predictive biomarkers of response(28). In addition, liver immune tolerance is a complex dynamic between the parenchymal cells and immune subsets. Mountainous researchers have focused on the intratumoral immune effector cells such as reduced activation of NK, Th1, and Tfh cells but increased suppressor functions involving regulatory T cells, myeloid cells, and B cells which are thought to significantly influence tumor immunosuppressive microenvironment in HCC (29–31). Here, we focus on the tumor cell-intrinsic change that may limit the therapeutic effects of anti-PD-1 therapy. Unraveling this puzzle may uncover novel genetic and biological biomarkers that would aid in better patient selection and development of personalized immunotherapy. Our findings have identified hepatic IRF8 as a potential biomarker for immune response and prognosis of HCC. AAV8-mediated IRF8 rescue therapy against HCC in animal models has revealed promising prospect. Our findings corroborate with previous hypothesis that the expression profile of IRF8 gene reflects response to PD-1 antibody therapy. A combination of PD-1 antibody and IRF8 gene therapy has achieved remarkable results in the treatment of HCC.
Unlike other IRFs family members, no prior work has shown that IRF8 is restrictively expressed, constitutively in liver tissues. IHC analysis of 21 HCC patients revealed that the expression of IRF8 proteins is substantially downregulated in HCC relative to adjacent noncancerous tissues. qPCR analysis validated the under-expression of IRF8 in clinical and DEN induced HCC tissues. Further analysis suggested that higher IRF8 was associated with better prognosis of HCC (Fig. 1C). These findings underscored the critical role IRF8 play in modulating growth of HCC cells. A key consideration is the mechanism for the downregulation of IRF8 in HCC tissues. Hypoxia and inflammation are the most important hallmarks of TME. However, we found that hypoxia, or LPS treatment had no effect on IRF8 expression (data not shown). In addition to environmental influence, epigenetic factors has a key impact on the regulation of gene expression or transcriptional regulation(32). Existing findings suggest that expression of IRF8 gene in disease conditions is related to epigenetic modification (19, 33–35). Analysis of TCGA-LIHC database on correlation between methylation of IRF8 promoter region and IRF8 expression revealed that IRF8 gene was not very likely to be deleted, mutated or methylated in the HCC, consistent with results of DNA methylation inhibitor treatment in HCC cells (Fig.S9A–F). Trichostatin A up-regulates the expression of IRF8 in HCC cells treated with all HDAC inhibitors treatments (Fig. S2). Related studies show that recruitment of HDAC3 to the ISRE motifs results in transcriptional repression of IFN stimulated genes (36), we hypothesized that deacetylation of H3K9 represses expression of IRF8 by recruiting HDAC3. Overall, these findings suggest that under expression of IRF8 in HCC is, at least in part, mediated by histone deacetylation.
Harnessing and enhancing the adaptive and innate immune system against cancer is becoming an increasingly promising strategy for sustainable anti-tumor responses(37). Surprisingly, the HCC subcutaneous xenograft tumor model demonstrated that IRF8 only inhibits tumor progression in immunocompetent mice. This suggests that the tumor-inhibiting mechanisms of IRF8 are largely related to tumor immunity. Therefore, the present study explored the immunological effect of IRF8 on TME in HCC. IHC and flow cytometry analysis revealed that over-expression of IRF8 modulated infiltration of macrophages and exhaustion of T cells (CD4+PD1+ and CD8+PD1+) in HCC peritumor tissues. Research shows that high infiltration of TAMs after surgery is associated with poor prognosis of HCC(37, 38), consistent with our findings. We found selective blockade of macrophage in TME with anti-CSF-1 monoclonal antibody or chlorophosphate liposomes significantly modulated tumor growth. IRF8 exerted its anti-tumor functions by inhibiting macrophage infiltration to TME, and affected T cell function by remodeling the TAMs-mediated cross-talk between the innate and adaptive immune in the TME. Therefore, inhibiting the infiltration of TAMs to TME is a potential therapeutic module against HCC and response to PD-L1/PD-1 therapy, particularly in IRF8 deficient patients. To some extent, our findings implicate macrophages in tumor progression and impairment of T cells function by inducing tumor immunosuppressive microenvironment(39).
IFN plays a pivotal role in antitumor immunity by promoting secretion of other pro-inflammatory cytokines and chemokines, which enhance anti-tumor immunity by increasing antigen presentation and immune cell infiltration(40). Impaired IFN signaling is a common immune defect in human cancer and may cause immune deficiency in tumors. In fact, as one pleiotropic cytokine that serve as central coordinators of tumor-immune system interaction, current supplementary IFN therapy in the clinic indeed restores anticancer immunity, but achieved limited success due to its dose-limiting toxicity, and simultaneously induces systemic immunotoxicity. Moreover, IFN could induce tumor immune evasion by upregulating multiple immune checkpoints. We found that upregulating downstream interferon-stimulated gene IRF8 could mimic the antitumor effects of IFN and partially rescued the deleterious outcomes of the IFN-induced immune evasion. In our study, we demonstrated a AAV8-mediated gene therapy strategy to simultaneously restore the local function of IFN signaling but decrease CCL20 expression, a chemo-attractant for TAMs, then activate innate and adaptive immune through inhibiting the recruitment of macrophages, especially, without upregulating downstream PD1/PDL1 expression to overcome IFN-regulated multigenic resistance to ICB (Fig.S9G). Further experiments revealed that down-regulating the expression of IRF8 activates the AP1 pathway (c-fos), which up-regulates the expression of CCL20 gene (Fig. 5). This suggests that the early innate immune responses are, at least in part mediated by IRF8 expression, orchestrating the generation of adaptive immune responses through regulated expression of chemo-attractants.
In conclusion, the current findings establish an unequivocal role for HCC-specific decreased expression of IRF8 in regulating hepatic CCL20 secretion and TAMs recruitment to immunosuppressive microenvironment. We further show that IRF8 rescue exhibits inhibited progression of HCC and enhanced tumor response to anti-PD-1 therapy. These findings deepen our understanding on how IRF8 regulates HCC progression and enhances the effectiveness of immune therapy in HCC.
Supplementary Material
Financial Support:
These studies were supported by the National Natural Science Foundation of China (Nos. 82003788, 81903656, 81673468), Natural Science Foundation of Jiangsu Province (No. BK20180560, BK20190801), “Double First-Class” University Project (CPU2018GF10, CPU2018GY46), China Postdoctoral Science Foundation (No. 2018M632430), the Fundamental Research Funds for the Central Universities (2632022ZD13), Youth Project of Natural Science Foundation of Nanjing University of Chinese Medicine (NZY82003788), the Scientific Startup Foundation for High level Scientists of China Pharmaceutical University (No. 3154070026) and the National Institutes of Health Intramural Research Program (Project Z01-ES-101643 to LB).
Abbreviations:
- HCC
Hepatocellular Carcinoma
- LIHC
Liver hepatocellular carcinoma
- IRFs
Interferon regulatory factors
- IRF8
Interferon regulatory factor 8
- IFN
Interferon
- PD-1
Programmed cell death protein-1
- PD-L1
Programmed death-ligand 1
- TAMs
Tumor Associated Macrophages
- TCGA
The Cancer Genome Atlas
- LIHC
Liver hepatocellular carcinoma
- GEO
Gene Expression Omnibus database
- GSEA
Gene set enrichment analysis
- ChIP
Chromatin Immunoprecipitation
- qPCR
Quantitative real-time PCR
- DEN
N-nitrosodiethylamine
- HDAC
Histone deacetylase
- CM
Conditioned media
- ISRE
Interferon-stimulated response element
- AAV8
Adeno-Associated Virus 8
- DOX
Doxycycline Hydrochloride
- OE
Overexpression
- KD
Knockdown
- Luc
Luciferase
- IHC
immunohistochemistry
- TME
Tumor microenvironment
- TGI
Tumor growth inhibition
Footnotes
Conflict of interest
Authors declare no conflict of interest.
References
- 1.Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol 2019;16:589–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 2015;27:450–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, Kim TY, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017;389:2492–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sangro B, Melero I, Wadhawan S, Finn RS, Abou-Alfa GK, Cheng AL, Yau T, et al. Association of inflammatory biomarkers with clinical outcomes in nivolumab-treated patients with advanced hepatocellular carcinoma. J Hepatol 2020;73:1460–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yau T, Park JW, Finn RS, Cheng AL, Mathurin P, Edeline J, Kudo M, et al. LBA38_PR - CheckMate 459: A randomized, multi-center phase III study of nivolumab (NIVO) vs sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC). Annals of Oncology 2019;30:v874–v875. [Google Scholar]
- 6.Finn RS, Ryoo BY, Merle P, Kudo M, Bouattour M, Lim HY, Breder V, et al. Pembrolizumab As Second-Line Therapy in Patients With Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J Clin Oncol 2020;38:193–202. [DOI] [PubMed] [Google Scholar]
- 7.Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, Albright A, et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest 2017;127:2930–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, Cucolo L, et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016;167:1540–1554 e1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liao W, Overman MJ, Boutin AT, Shang X, Zhao D, Dey P, Li J, et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell 2019;35:559–572.e557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang Y, Zhou Y, Hou J, Bai C, Li Z, Fan J, Ng IOL, et al. Hepatic IFIT3 predicts interferon-α therapeutic response in patients of hepatocellular carcinoma. Hepatology 2017;66:152–166. [DOI] [PubMed] [Google Scholar]
- 11.Hou J, Zhou Y, Zheng Y, Fan J, Zhou W, Ng IO, Sun H, et al. Hepatic RIG-I predicts survival and interferon-alpha therapeutic response in hepatocellular carcinoma. Cancer Cell 2014;25:49–63. [DOI] [PubMed] [Google Scholar]
- 12.Nelson N, Marks MS, Driggers PH, Ozato K. Interferon consensus sequence-binding protein, a member of the interferon regulatory factor family, suppresses interferon-induced gene transcription. Mol Cell Biol 1993;13:588–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weisz A, Marx P, Sharf R, Appella E, Driggers PH, Ozato K, Levi BZ. Human interferon consensus sequence binding protein is a negative regulator of enhancer elements common to interferon-inducible genes. J Biol Chem 1992;267:25589–25596. [PubMed] [Google Scholar]
- 14.Klement JD, Paschall AV, Redd PS, Ibrahim ML, Lu C, Yang D, Celis E, et al. An osteopontin/CD44 immune checkpoint controls CD8+ T cell activation and tumor immune evasion. J Clin Invest 2018;128:5549–5560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tamura T, Ozato K. ICSBP/IRF-8: its regulatory roles in the development of myeloid cells. J Interferon Cytokine Res 2002;22:145–152. [DOI] [PubMed] [Google Scholar]
- 16.Hambleton S, Salem S, Bustamante J, Bigley V, Boisson-Dupuis S, Azevedo J, Fortin A, et al. IRF8 Mutations and Human Dendritic-Cell Immunodeficiency. New England Journal of Medicine 2011;365:127–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee K, Geng H, Ng KM, Yu J, Van Hasselt A, Cao Y, Zeng Y, et al. Epigenetic disruption of interferon-γ response through silencing the tumor suppressor interferon regulatory factor 8 in nasopharyngeal, esophageal and multiple other carcinomas. Oncogene 2008;27:5267–5276. [DOI] [PubMed] [Google Scholar]
- 18.Ibrahim ML, Klement JD, Lu C, Redd PS, Xiao W, Yang D, Browning DD, et al. Myeloid-Derived Suppressor Cells Produce IL-10 to Elicit DNMT3b-Dependent IRF8 Silencing to Promote Colitis-Associated Colon Tumorigenesis. Cell Rep 2018;25:3036–3046 e3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fang C, Qiao Y, Mun SH, Lee MJ, Murata K, Bae S, Zhao B, et al. Cutting Edge: EZH2 Promotes Osteoclastogenesis by Epigenetic Silencing of the Negative Regulator IRF8. J Immunol 2016;196:4452–4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ji Z, He L, Regev A, Struhl K. Inflammatory regulatory network mediated by the joint action of NF-kB, STAT3, and AP-1 factors is involved in many human cancers. Proceedings of the National Academy of Sciences 2019;116:9453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng AL, Hsu C, Chan SL, Choo SP, Kudo M. Challenges of combination therapy with immune checkpoint inhibitors for hepatocellular carcinoma. J Hepatol 2020;72:307–319. [DOI] [PubMed] [Google Scholar]
- 22.Chiang DY, Villanueva A, Hoshida Y, Peix J, Newell P, Minguez B, LeBlanc AC, et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res 2008;68:6779–6788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lei Q, Wang D, Sun K, Wang L, Zhang Y. Resistance Mechanisms of Anti-PD1/PDL1 Therapy in Solid Tumors. Front Cell Dev Biol 2020;8:672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patel SP, Kurzrock R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol Cancer Ther 2015;14:847–856. [DOI] [PubMed] [Google Scholar]
- 25.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015;348:124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;357:409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salmon H, Remark R, Gnjatic S, Merad M. Host tissue determinants of tumour immunity. Nat Rev Cancer 2019;19:215–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Q, He Y, Luo N, Patel SJ, Han Y, Gao R, Modak M, et al. Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma. Cell 2019;179:829–845.e820. [DOI] [PubMed] [Google Scholar]
- 30.Liu Z, Lin Y, Zhang J, Zhang Y, Li Y, Liu Z, Li Q, et al. Molecular targeted and immune checkpoint therapy for advanced hepatocellular carcinoma. J Exp Clin Cancer Res 2019;38:447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Llovet JM, Montal R, Sia D, Finn RS. Molecular therapies and precision medicine for hepatocellular carcinoma. Nature Reviews Clinical Oncology 2018;15:599–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marquardt JU, Factor VM, Thorgeirsson SS. Epigenetic regulation of cancer stem cells in liver cancer: Current concepts and clinical implications. Journal of Hepatology 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Q, Zhang L, Li L, Wang Z, Ying J, Fan Y, Xu B, et al. Interferon regulatory factor 8 functions as a tumor suppressor in renal cell carcinoma and its promoter methylation is associated with patient poor prognosis. Cancer Lett 2014;354:227–234. [DOI] [PubMed] [Google Scholar]
- 34.Guo C, Pei L, Xiao X, Wei Q, Chen JK, Ding HF, Huang S, et al. DNA methylation protects against cisplatin-induced kidney injury by regulating specific genes, including interferon regulatory factor 8. Kidney Int 2017;92:1194–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thumbigere-Math V, Foster BL, Bachu M, Yoshii H, Brooks SR, Coulter A, Chavez MB, et al. Inactivating Mutation in IRF8 Promotes Osteoclast Transcriptional Programs and Increases Susceptibility to Tooth Root Resorption. J Bone Miner Res 2019;34:1155–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuwata T, Gongora C, Kanno Y, Sakaguchi K, Tamura T, Kanno T, Basrur V, et al. Gamma interferon triggers interaction between ICSBP (IRF-8) and TEL, recruiting the histone deacetylase HDAC3 to the interferon-responsive element. Molecular & Cellular Biology 2002;22:7439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu Y, Yang J, Xu D, Gao XM, Zhang Z, Hsu JL, Li CW, et al. Disruption of tumour-associated macrophage trafficking by the osteopontin-induced colony-stimulating factor-1 signalling sensitises hepatocellular carcinoma to anti-PD-L1 blockade. Gut 2019;68:1653–1666. [DOI] [PubMed] [Google Scholar]
- 38.Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol 2017;14:399–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li X, Yao W, Yuan Y, Chen P, Li B, Li J, Chu R, et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut 2017;66:157–167. [DOI] [PubMed] [Google Scholar]
- 40.Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, Conway JR, Li Y, et al. LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell 2018;174:549–563 e519. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.