

Summary
Homology-based search is commonly used to uncover mobile genetic elements (MGEs) from metagenomes, but it heavily relies on reference genomes in the database. Here we introduce a protocol to extract CRISPR-targeted sequences from the assembled human gut metagenomic sequences without using a reference database. We describe the assembling of metagenome contigs, the extraction of CRISPR direct repeats and spacers, the discovery of protospacers, and the extraction of protospacer-enriched regions using the graph-based approach. This protocol could extract numerous characterized/uncharacterized MGEs.
For complete details on the use and execution of this protocol, please refer to Sugimoto et al. (2021).
Subject areas: Bioinformatics, Sequence analysis, Genomics, CRISPR, Systems biology
Graphical abstract
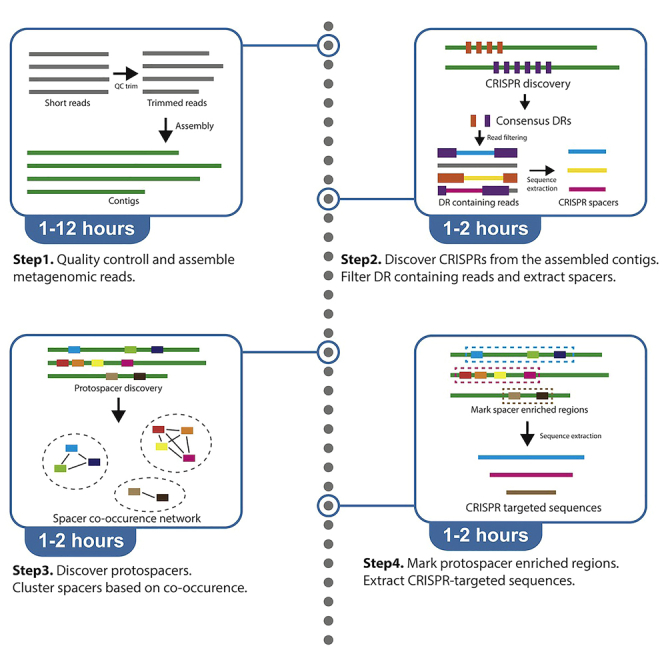
Highlights
-
•
Protocol to extract CRISPR-targeted sequences from the assembled metagenomic sequences
-
•
Extraction of sequences from the human gut metagenome without using a reference database
-
•
Extraction of sequences with various kinds of MGEs including unclassified ones
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Homology-based search is commonly used to uncover mobile genetic elements (MGEs) from metagenomes, but it heavily relies on reference genomes in the database. Here we introduce a protocol to extract CRISPR-targeted sequences from the assembled human gut metagenomic sequences without using a reference database. We describe the assembling of metagenome contigs, the extraction of CRISPR direct repeats and spacers, the discovery of protospacers, and the extraction of protospacer-enriched regions using the graph-based approach. This protocol could extract numerous characterized/uncharacterized MGEs.
Before you begin
Hardware
This section describes the hardware specifications, operating system, and interpreters required to execute the protocol.
-
1.Computer.
-
a.At least 128 GB of memory.
-
b.A hyper-threading CPU with at least 8 threads.
-
a.
-
2.Operating system.
-
a.Linux distribution capable of executing scripts written in the languages of the interpreters specified below.
-
a.
-
3.Interpreters.
-
a.Python3.
-
b.Java.
-
c.Bash.
-
a.
Note: The computer must have sufficient memory for metagenome assembly. A typical human gut metagenome library ranges in size from a few GB to hundreds of GB. Therefore, assembling a large library might require more than 128 GB of memory, which is likely unavailable for a standard desktop machine. In this case, consider using high-capacity servers in a data center.
Software
The software and python packages listed in the key resources table must be downloaded and installed on the system accordingly.
Data collection
Metagenomic raw reads can be downloaded from numerous sources, such as Sequence Read Archive (SRA, https://www.ncbi.nlm.nih.gov/sra), Integrative Human Microbiome Projects (iHMP, https://ibdmdb.org/tunnel/public/summary.html), etc. In this article, we assume that the sequences are stored in paired short-read FASTQ files generated from whole metagenome shotgun sequencing.
Note: We suggest that at least 10 independent samples from an environment are required to collect a sufficient number of spacers and candidate protospacers to build a versatile spacer co-occurrence network.
Key resources table
Note: Some software versions specified in this table might not be available. In such a case, use the latest version of the software and refer to their manuals.
Materials and equipment
Alternatives: Our repository also includes custom Bash scripts used in the previous study to automate the analysis pipelines. With modifications, these scripts could be used as starting points to build the custom pipeline for the reader’s purpose.
| Custom scripts | (Sugimoto et al., 2021) | https://doi.org/10.5281/zenodo.6621424 |
CRITICAL: These automation pipeline scripts are mainly written in Bash language (files ending their names with .sh) and require modifications to specify the file paths in their codes. The parts that require changes are noted with comments in the Bash scripts (see README files for details). Conversely, the python scripts used in this protocol do not require any change.
| Dataset from the previous study | (Sugimoto et al., 2021) | https://doi.org/10.5281/zenodo.6503687 |
Alternatives: We also provide FASTA and BED-formatted files, which contain spacers, protospacers, CRISPR direct repeats, and CRISPR-targeted terminally redundant sequences extracted from approximately ten thousand human gut metagenome libraries in the previous study. This dataset includes approximately two million unique spacers, ten thousand unique CRISPR direct repeats, and approximately ten thousand CRISPR-targeted unique sequences. The protocol described in this article does not require these files. However, for the case of human gut metagenome analysis, this previously extracted dataset could be used to skip the processes and reduce the analysis time.
Step-by-step method details
Preprocess raw paired FASTQ files
Timing: 1–2 h
This process is quality control of the input sequences.
Alternatives: A pipeline script for preprocessing, assembling, and spacer collection is provided in the repository (pipeline_scripts/assembly_pipeline.sh).
-
1.
Trim poor-quality bases, adapters, and PhiX spikes.
$ bbduk.sh -Xmx100g t=8 in1=r1.fastq.gz in2=r2.fastq.gz out=trimmed.fastq.gz ftm=5 qtrim=rl trimq=20 ftl=15 ref=adapters,phix ktrim=r k=23 mink=11 hdist=1 tpe tbo maq=20
-
2.
Remove human-derived reads.
$ bbmap.sh -Xmx100g t=8 ref={path to the human reference FASTA file} in=trimmed.fastq.gz outu=decontaminated.fastq.gz interleaved=t minratio=0.9 maxindel=3 bwr=0.16 bw=12 fast=t minhits=2 qtrim=r trimq=10 untrim=t idtag=t printunmappedcount=t kfilter=25 maxsites=1 k=14
Note: We used the human genome reference FASTA file posted by the author of BBtools.
-
3.
Correct errors in the reads.
$ tadpole.sh threads=8 -Xmx100g interleaved=t in=decontaminated.fastq.gz out=ecc.fastq.gz mode=correct
Metagenome assembly
Assemble metagenome contigs from the preprocessed FASTQ files.
-
4.
Assemble preprocessed FASTQ file using SPAdes.
$ spades.py -t 8 -m 100 --meta --only-assembler --12 ecc.fastq.gz -o {path to spades output directory}
Note: We skipped the error-correction step in the SPAdes program to avoid the occasional endlessly running problem. Please refer to the troubleshooting section for detail.
-
5.
Assign unique identifiers to all assembled contigs.
$ cd {path to spades output directory}
$ id=$(openssl rand -hex 12)
awk -v id=${id} '/ˆ>/ {n++; print ">contig_"id"_"n;} !/ˆ>/{print}' scaffolds.fasta > scaffolds.renamed.fasta
Optional: We recommend removing contigs shorter than 1k bases to reduce the file size.
Note: The pre and assembling processes are independently conducted for each paired FASTQ file.
Optional: We advise that the system has sufficient disk space to store all preprocessed FASTQ and assembled FASTA files. The rest of the files, including intermediate and temporary files, can be deleted to save the disk space.
Extraction of CRISPR direct repeats from the assembled contigs
From the assembled contigs, discover CRISPR consensus direct repeats.
-
6.
Run CRISPRDetect to discover consensus CRISPR direct repeats.
$ CRISPRDetect.pl -q 0 -f scaffolds.renamed.fasta -o crispr_detect_output -array_quality_score_cutoff 2
-
7.
From the output gff file, extract direct repeats, and convert them into a FASTA file.
$ grep 'repeat_region' crispr_detect_output.gff | cut -f 9 | tr ';' '∖n' | egrep 'ˆNote=' | cut -f 2 -d '=' | awk '{n++; printf(">DR_%i∖n%s∖n", n, $0)}' > crispr_dr.fasta
Note: Again, these direct repeat extractions are independently conducted for each assembled FASTA file.
-
8.
After discovering all direct repeats from each assembled FASTA file, we merge them into a single file, assign unique identifiers, and remove redundancy.
$ cat {all crispr dr fasta files} > merged_crispr_dr.fasta
$ awk '/ˆ>/ {n++; print ">dr_"n; } !/ˆ>/ {print}' merged_crispr_dr.fasta > merged_crispr_dr.renamed.fasta #giving new unique identifiers
$ cd-hit-est -S 1 -c 1 -i merged_crispr_dr.renamed.fasta -o merged_crispr_dr.repr.fasta
Optional: We provided the direct repeat sequences extracted from the human gut metagenomes in the previous study. The sequences are stored in a FASTA formatted file located in the supplementary folder (supplementary_data/crispr/drs/all_dr.clustered.fasta).
Extraction of CRISPR spacers from the preprocessed reads
Alternatives: A pipeline script for the spacer collection is provided in the repository (pipeline_scripts/collect_spacers.sh).
Here, we return to the preprocessed FASTQ files. From them, we extract CRISPR spacers from reads containing CRISPR direct repeats.
-
9.
Extract direct repeat-containing reads. This initial filtering step significantly reduces the number of reads, effectively speeding up the following spacer extraction processes.
$ bbduk.sh in=ecc.fastq.gz ref=merged_crispr_dr.repr.fasta outm=dr_containing_reads.fastq.gz interleaved=t k=21 hdist=1 rename=t
-
10.
Second filter to mask the direct repeats in the reads.
$ bbduk.sh in=dr_containing_reads.fastq.gz ref=merged_crispr_dr.repr.fasta kmask=R k=19 hdist=1 out=dr_masked.fastq mink=15
-
11.
Extract spacers from the masked reads using our python script.
$ {path to the virome_scripts directory}/pipeline_scripts/extract_spacers.py -s {sample name} dr_masked.fastq > spacers.fasta
Note: The python script used above is available from our repository (pipeline_scripts/extract_spacers.py).
Optional: The python program used above does not output short (<20 bp) and long (>50 bp) sequences by default. These parameters can be changed by options (-s and -l).
Note: Spacer extraction is conducted for each preprocessed FASTQ file.
-
12.
Merge all discovered spacers, assign unique identifiers, and remove redundancy.
$ cat {all spacer fasta files} > all_spacers.fasta
$ awk '/ˆ>/ {sub(/ˆ[ˆ[:space:]]+[[:space:]]/, ""); n++; printf(“>spacer_"n"∖t"$0);} !/ˆ>/ {print}' all_spacers.fasta > all_spacers.renamed.fasta #giving new unique identifiers
$ cd-hit-est -T 8 -M 10000 -d 0 -sf 1 -s 0.9 -c 0.98 -i all_spacers.renamed.fasta -o all_spacers.repr.fasta
Optional: It is highly advisable to make each spacer and direct repeat trackable to the original samples, contigs, and/or associated direct repeats. This can be achieved using FASTA descriptions in each record.
Optional: We provided the spacer sequences extracted from the human gut metagenomes in the previous study. The sequences are stored in a FASTA formatted file located in the supplementary folder (supplementary_data/crispr/spacers/all_spacers.clustered.removed_short.fasta).
Protospacer discovery
Here, we discover protospacers by aligning the spacer sequences to CRISPR-masked contigs.
-
13.
Search CRISPR direct repeats from the contigs.
$ makeblastdb -dbtype nucl -in scaffolds.renamed.fasta
$ blastn -evalue 1e-5 -task ‘blastn-short’ -outfmt 6 -num_threads 8 -query merged_crispr_dr.repr.fasta -db scaffolds.renamed.fasta > dr.blastn
-
14.
Mask sequences around direct repeat aligned regions.
$ samtools faidx scaffolds.renamed.fasta
$ awk ‘BEGIN{FS="∖t"; OFS="∖t"} $10<$9{t=$9; $9=$10; $10=t} {print $2,$9–1,$10}' dr.blastn | sort -k1,1 -k2,2n | bedtools merge -i /dev/stdin -d 60 | bedtools slop -b 60 -i /dev/stdin -g scaffolds.renamed.fasta.fai | bedtools maskfasta -bed /dev/stdin -fi scaffolds.renamed.fasta -fo crispr_masked.fasta
-
15.
Align spacers to the masked contigs.
$ makeblastdb -dbtype nucl -in crispr_masked.fasta
$ blastn -evalue 1e-5 -perc_identity 93 -task ‘blastn-short’ -outfmt 6 -num_threads 8 -query all_spacers.repr.fasta -db crispr_masked.fasta > spacers.blastn
Note: Protospacer search is conducted for each assembled FASTA file.
-
16.
Merge all discovered protospacers into a single BED-formatted file.
$ cat {all spacer blastn output files} | awk ‘BEGIN{FS="∖t"; OFS="∖t"} $10<$9{t=$9; $9=$10; $10=t} {print $2,$9–1,$10,$1}' | sort -k1,1 -k2,2n > all_protospacers.bed
Optional: We provided the protospacers discovered from the assembled human gut metagenome contigs in the previous study. The protospacers are stored in a BED-formatted file located in the supplementary folder (supplementary_data/targeted_sequences/protospacers/all_protospacers.bed).
Spacer clustering based on co-occurrence
Here, we cluster spacers using a co-occurrence network calculated from the spacer alignment result. From this network, we discover the graph communities, representing subsets of spacers that corresponding protospacers co-occur together across the assembled contigs. We extract the protospacer-enriched regions using these graph communities, i.e., spacer clusters. This process effectively reduces the false-positive ratio by excluding lone or randomly scattered protospacers.
-
17.
Initial clustering is based on distance.
$ bedtools cluster -d 50000 -i all_protospacers.bed | awk ‘BEGIN{FS="∖t"; OFS="∖t"} {print $1"_cluster_"$5,$2,$3,$4}' > initial_cluster.bed
-
18.
Calculate a weighted graph from the spacer co-occurrence and generate an abc formatted file.
$ {path to the virome_scripts directory}/graph_clustering/generate_abc_edgefile.py initial_cluster.bed > protospacers.abc
Note: The python script used above is available in our repository (graph_clustering/generate_abc_edgefile.py).
-
19.
Convert the abc file into an mci format.
$ mcxload -abc protospacers.abc --stream-mirror -write-tab data.tab -o protospacers.mci
-
20.
Run an mcl program to discover graph communities.
$ mcl protospacers.mci -I 4 -pi 0.4 -te 8
-
21.
Convert the mcl output to a tabular format.
$ mcxdump -icl out.protospacers.mci.I40pi04 -tabr data.tab > protospacers_cluster.tab
-
22.
Mark CRISPR-targeted regions using the clustering result and the merged protospacer bed file.
$ {path to the virome_scripts directory}/graph_clustering/mark_crispr_targeted.py all_protospacers.bed protospacers_cluster.tab > crispr_targeted.bed
Note: The python script used above is available in our repository (graph_clustering/mark_crispr_targeted.py).
Note: The fourth column in the output file is formatted as {cluster id}:{protospacer count in the region}.
-
23.
Merge the regions to rescue the fragmented clusters.
$ sort -k1,1 -k2,2n crispr_targeted.bed > crispr_targeted.sorted.bed
$ bedtools merge -d 1000 -i crispr_targeted.sorted.bed -o collapse -c 4 > crispr_targeted.merged.bed
-
24.
Extract CRISPR-targeted regions from the assembled contigs.
$ samtools faidx scaffolds.renamed.fasta
$ cut -f 1,2 scaffolds.renamed.fasta.fai | sort -k 1,1 | join -t $'∖t' - <(sort -k 1,1 crispr_targeted.merged.bed) | awk 'BEGIN{OFS="∖t"; FS="∖t"} $4>$2{$4=$2} {print $1,$3,$4,$5}' | bedtools getfasta -fi scaffolds.renamed.fasta -bed /dev/stdin -fo crispr_targeted.fasta
Note: The extraction of CRISPR-targeted sequences is conducted for each assembled FASTA file.
Optional: We provided the CRISPR-targeted terminally redundant sequences extracted from the assembled human gut metagenome contigs in the previous study. The sequences are stored in a FASTA formatted file located in the supplementary folder (supplementary_data/targeted_sequences/tr/tr_sequences.fasta).
Expected outcomes
The final output FASTA files contained CRISPR-targeted sequences. These sequences typically range in size between a few hundred and several hundred thousand bases. The output sequences likely contain multiple kinds of MGEs (Figure 1).
Figure 1.

Classification results of CRISPR-targeted terminally redundant sequences from the human gut metagenome
The most popular elements among the large (>20 kb) CRISPR-targeted sequences in the human gut metagenome were tailed phages. Conversely, ssDNA viruses, plasmid-like elements, and many unclassified sequences were common among the small (<20 kb) sequences. This result was obtained from our previous study (Sugimoto et al., 2021).
Quantification and statistical analysis
-
•
Sequence clustering should be conducted to remove redundancy and fragmentations from the final output sequences.
-
•
Genomic completeness could be investigated by checking terminal redundancy or inverted terminal repeats. In the repository, we provided python scripts that extract terminally redundant or inverted repeat sequences from a FASTA file (utils/circular_contigs.py and utils/inverted_repeat_contigs.py).
-
•
CRISPR-Cas systems target various elements, including viruses, plasmids, transposons, and chromosomes. A protein homology search is a common method to classify the novel genomes. We suggest using a high-sensitivity homology search method based on hidden Markov models.
Limitations
It is possible to predict the CRISPR-targeting hosts by searching protospacer-associated direct repeats in the reference genome database. However, CRISPR undergoes intense horizontal gene transfer. Therefore, combining it with other methods, such as tRNA alignment, is highly advisable to predict the infecting host.
Troubleshooting
Problem 1
Genome assembly takes time (step 4).
Potential solution
The time metagenome assembly takes strongly varies between libraries. If an assembly takes longer than a day, and the library size is very large (>100 Gb), one can downsample the FASTQ file by randomly selecting pairs from the original files or normalizing the depth in silico. Both approaches can reduce the file size and potentially the assembling time.
Problem 2
Why did you skip the error-correction step in the SPAdes program (step 4)?
Potential solution
We found that the SPAdes program is sometimes trapped in the error-correction step and seems to endlessly run for more than four or five days. This phenomenon is sporadic, hard to replicate, and might depend on the system specifications. Such a problem was highly problematic when we were analyzing more than a thousand samples. To avoid this issue, we skipped the error-correction step in the SPAdes program and instead used the tadpole program for the preassemble error correction. We compared the statistics between the assembled contigs, using SPAdes or tadpole, and found no significant difference between them.
Problem 3
Assembled contigs are short (step 4).
Potential solution
The less populated organisms in the sample are likely to be shallowly sequenced, leading to highly fragmented and partial contigs. Therefore, they might require deeper sequencing to assemble the complete genome. Conversely, the metadata of SRA recorded libraries could be wrong sometimes. For example, we encountered several libraries likely produced from 16S amplicon sequencing but recorded as a whole genomic sequencing. It is advisable to avoid such libraries and/or refer to the experimental method described in the original article if necessary.
Problem 4
The constructed spacer co-occurrence network is small and fragmented (step 21).
Potential solution
In the original study, we used millions of spacers extracted from thousands of human gut metagenome datasets to construct a network. A significant number of spacers and spacer-aligned contigs are required to build a versatile network and predict graph communities. If you are attempting to analyze other than the human gut metagenome using a few materials, it might be advisable to skip the network building process entirely and manually check each protospacer containing contigs.
Problem 5
Protein homology searches do not hit the database (quantification and statistical analysis).
Potential solution
Many MGE encoded protein sequences such as capsids and polymerases are extremely diversified, therefore it is often difficult to detect their homology to known sequences using the pairwise alignment-based method which is adopted by such as the BLAST program. In order to overcome this issue, hidden Markov models (HMMs) based programs such as HMMER (Eddy, 2011) or HH-suite (Steinegger et al., 2019) could be used for the higher sensitivity homology search. Using the predicted protein sequences from the discovered CRISPR-targeted sequences as seeds, one could iteratively search the bulk of metagenome-derived protein sequence databases such as Metaclust (Steinegger and Söding, 2018). The aligned sequences were used to build an HMM, which is used as a query to search a curated protein database such as Protein Data Bank.
Resource availability
Lead contact
Ituro Inoue: itinoue@nig.ac.jp.
Materials availability
CRISPR spacers and direct repeats extracted in the previous study are available from our repository.
Acknowledgments
R.S. is funded by the Research Organization of Information and Science (https://www.rois.ac.jp). L.N. is funded by JSPS KAKENHI grant number JP21J22509 (https://kaken.nii.ac.jp/en/grant/KAKENHI-PROJECT-21J22509/). P.T.N. is funded by MEXT Doctoral Scholarship (https://www.mext.go.jp). I.I. is funded by JSPS KAKENHI grant number JP20K21405 (https://kaken.nii.ac.jp/grant/KAKENHI-PROJECT-20K21405/). The majority of analysis has been done on the supercomputer systems at the National Institute of Genetics and Okinawa Institute of Science and Technology Graduate University.
Author contributions
R.S. designed this protocol, coded scripts, and wrote the manuscript. L.N. curated the results. P.T.N. executed the protocol in the supercomputer systems. I.I. supervised the study.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Ryota Sugimoto, Email: rsugimot@nig.ac.jp.
Ituro Inoue, Email: itinoue@nig.ac.jp.
Data and code availability
The source codes used in this protocol are available from our repository.
References
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Bankevich A., Nurk S., Antipov D., Gurevich A.A., Dvorkin M., Kulikov A.S., Lesin V.M., Nikolenko S.I., Pham S., Prjibelski A.D., et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas A., Staals R.H., Morales S.E., Fineran P.C., Brown C.M. CRISPRDetect: a flexible algorithm to define CRISPR arrays. BMC Genom. 2016;17:356. doi: 10.1186/s12864-016-2627-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushnell B. BBTools software package. 2014. http://sourceforge. net/projects/bbmap
- Cock P.J.A., Antao T., Chang J.T., Chapman B.A., Cox C.J., Dalke A., Friedberg I., Hamelryck T., Kauff F., Wilczynski B., de Hoon M.J.L. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25:1422–1423. doi: 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P., Bonfield J.K., Liddle J., Marshall J., Ohan V., Pollard M.O., Whitwham A., Keane T., McCarthy S.A., Davies R.M., Li H. Twelve years of SAMtools and BCFtools. GigaScience. 2021;10:giab008. doi: 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy S.R. Accelerated Profile HMM Searches. PLoS Comput Biol. 2011;7 doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright A.J., Van Dongen S., Ouzounis C.A. An efficient algorithm for large-scale detection of protein families. Nucleic acids research. 2002;30:1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris C.R., Millman K.J., van der Walt S.J., Gommers R., Virtanen P., Cournapeau D., Wieser E., Taylor J., Berg S., Smith N.J., et al. Array programming with NumPy. Nature. 2020;585:357–362. doi: 10.1038/s41586-020-2649-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinegger M., Meier M., Mirdita M., Vöhringer H., Haunsberger S.J., Söding J. HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinformatics. 2019;20:473. doi: 10.1186/s12859-019-3019-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinegger M., Söding J. Clustering huge protein sequence sets in linear time. Nat Commun. 2018;9:2542. doi: 10.1038/s41467-018-04964-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto R., Nishimura L., Nguyen P.T., Ito J., Parrish N.F., Mori H., Kurokawa K., Nakaoka H., Inoue I. Comprehensive discovery of CRISPR-targeted terminally redundant sequences in the human gut metagenome: viruses, plasmids, and more. PLoS Comp. Biol. 2021;17:e1009428. doi: 10.1371/journal.pcbi.1009428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virtanen P., Gommers R., Oliphant T.E., Haberland M., Reddy T., Cournapeau D., Burovski E., Peterson P., Weckesser W., Bright J., et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods. 2020;17:261–272. doi: 10.1038/s41592-019-0686-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The source codes used in this protocol are available from our repository.