

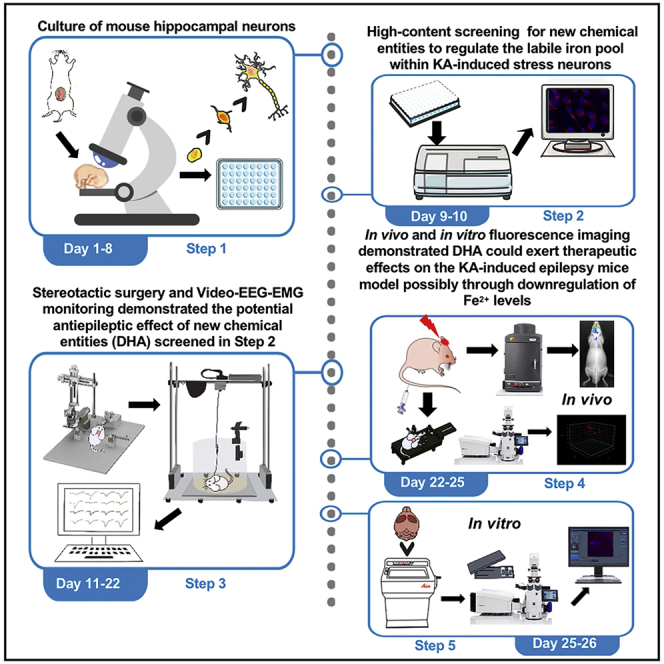
Summary
The development of techniques for tracking active ferrous iron (Fe2+) distribution has greatly promoted the biological studies of iron. Here, we present an innovative application of a 3D two-photon fluorescent probe for Fe2+ tracking in the epileptic mouse brain, which has expanded the toolbox of screening for iron homeostasis regulators and contributed to the discovery of new chemical entities for the treatment of epilepsy.
For complete details on the use and execution of this protocol, please refer to Shao et al. (2022).
Subject areas: Health Sciences, Chemistry
Graphical abstract
Highlights
-
•
A screening protocol for iron homeostasis modulators based on high-throughput imaging
-
•
A protocol to understand FeP in vitro and in vivo applications
-
•
Map ferrous iron in the epileptic mice brain with a 3D two-photon fluorescent probe
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
The development of techniques for tracking active ferrous iron (Fe2+) distribution has greatly promoted the biological studies of iron. Here, we present an innovative application of a 3D two-photon fluorescent probe for Fe2+ tracking in the epileptic mouse brain, which has expanded the toolbox of screening for iron homeostasis regulators and contributed to the discovery of new chemical entities for the treatment of epilepsy.
Before you begin
This protocol requires the use of a high content cell imaging analysis system, an in vivo confocal laser scanning microscope, a fluorescence in vivo imaging system, a suite of stereotactic surgery to implant the electrodes, an electrophysiology suite for recording EEG-EMG signals, and an isoflurane anesthesia system.
Institutional permissions
Prior authorization must be obtained before performing animal experiments. In this study, all mice were purchased from the Model Animal Research Centre of Nanjing University (Nanjing, China) or Shanghai Slake Laboratory Animal Co., LTD. All mice were maintained under specific pathogen-free conditions and used in accordance with the animal experimental guidelines set by the Institute of Animal Care and Use Committee. All animal experiments were approved by the Institutional Animal Care and Use Committee at Nanjing Normal University.
Preparing stock solutions
Preparing stock solutions for high-content screening
Timing: 3 h
The protocol below describes the steps for preparing the stock solution of reagents for the high-content screening.
-
1.Prepare the stocks of kainic acid (KA) and a variety of natural products (see also Table 1).Note: Natural products are small molecular compounds occurring in nature and derived from any living organism, including primary and secondary metabolites. The natural products have potential biological activity and can be used as lead compounds for drug discovery for commercial development. According to different targets, researchers can select different natural product libraries (redox compound library, cytokine inhibitor library, ion channel inhibitor library, ferroptosis compound library, etc.) for high-throughput screening. The natural products in this protocol are made up of natural products that have been reported to have certain oxidation-reducing properties and potential antiepileptic effects, and the quantity is also limited by our laboratory. The specific compound information can be referred to (Shao et al., 2022).
-
a.Dissolve KA in distilled water to 50 mM and natural products in DMSO to 20 mM, respectively.
-
b.Store at ≤−20°C for up to 1 month in dark.
-
a.
-
2.Prepare the stock of the 3D two-photon fluorescent probe (FeP).Note: The chemical structure of FeP can be referred to (Shao et al., 2022).
-
a.Dissolve FeP (< 1 mg) in DMSO to 10 mM.
-
b.Store at ≤−20°C for up to 1 month in dark.
 CRITICAL: The stock solution should be aliquoted to avoid repeated freezing and thawing.
CRITICAL: The stock solution should be aliquoted to avoid repeated freezing and thawing.
-
a.
Table 1.
List of reagents to be prepared
| Reagent | Target of reagent | Stock concentration | Solvent | Storage of stock solutions | Working concentration |
|---|---|---|---|---|---|
| kainic acid (KA) | neuronal stress model inducer | 50 mM | distilled water | ≤−20°C, for up to 1 month with protection from light | 500 μM |
| Natural products | potential Fe2+ scavenge | 20 mM | DMSO | ≤−20°C, for up to 1 month | 20 μM |
Preparing the stock solutions for video-EEG-EMG monitoring
The protocol below describes the steps for preparing the stock solutions of KA (a classic epileptic mouse model inducer) and dihydroartemisinin (DHA, the potential antiepileptic drug screened by high-content) for Video-EEG-EMG monitoring.
-
3.Prepare the stocks of KA and DHA (see also Table 2).
-
a.Dissolve KA in distilled water to 8 mg/mL and DHA in DMSO to 60 mg/mL, respectively.
-
b.Store at ≤−20°C for up to 1 month in a dark place.
-
a.
Table 2.
List of reagents to be prepared
| Reagent | Target of reagent | Stock concentration | Solvent | Storage of stock solutions | Working concentration |
|---|---|---|---|---|---|
| KA | epileptic mouse model inducer | 8 mg/mL | distilled water | ≤−20°C, for up to 1 month with protection from light | 20 mg/kg |
| DHA | potential antiepileptic drug | 60 mg/mL | DMSO | ≤−20°C, for up to 1 month | 60 mg/kg |
Preparing the stock solutions for fluorescence imaging in vivo
The protocol below describes the steps for preparing the stock solutions for fluorescence imaging in vivo.
-
4.Prepare the stocks of KA and DHA (see also Table 3).Note: FeP is an innovative near-infrared excited two-photon fluorescent probe synthesized by us which can be applied to 3D imaging of the spatiotemporal distribution of Fe2+ in the brain of living epileptic mouse models. FeP is not commercially available currently, but detailed synthesis steps can refer to (Shao et al., 2022) and FeP will also be made available on request upon completion of a Materials Transfer Agreement. In the article (Shao et al., 2022), we have listed eleven fluorescent probes with FeP, although they have been successfully applied in imaging Fe2+ of living cells and fixed tissues, no probes have been reported to monitor the distribution of Fe2+ in the epileptic brain. Other Fe2+ probes may use instead, but the performance needs to be tried and demonstrated.
-
a.Dissolve KA in distilled water to 2.4 mg/mL, DHA in DMSO to 60 mg/mL, and FeP (< 1 mg) in DMSO to 2 mg/mL, respectively.
-
b.Store at ≤−20°C for up to 1 month in a dark place.
-
a.
Table 3.
List of reagents to be prepared
| Reagent | Target of reagent | Stock concentration | Solvent | Storage of stock solutions | Working concentration |
|---|---|---|---|---|---|
| KA | epileptic mouse model inducer | 2.4 mg/mL | distilled water | ≤−20°C, for up to 1 month with protection from light | 6 or 20 mg/kg |
| DHA | potential Fe2+ scavenge | 60 mg/mL | DMSO | ≤−20°C, for up to 1 month | 60 mg/kg |
| FeP | fluorescent Fe2+ marker |
2 mg/mL | DMSO | ≤−20°C, for up to 1 month with protection from light | 0.5 mg/kg |
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| DMSO | Meiluncell | Cat# PWL064 |
| Poly-D-lysine | Beyotime | Cat# ST508 |
| B27 Plus | Gibco | Cat# A3582801 |
| Phosphate-buffered saline (PBS), pH 7.4 | WISENT | Cat# 311-010-CL |
| Hanks’ balanced salt solution without calcium and magnesium (HBSS) | WISENT | Cat# 311-512-CL |
| 0.25% Trypsin including 0.1% EDTA | WISENT | Cat# 325-043-EL |
| Neurobasal Plus Medium | Gibco | Cat# A3582901 |
| L-Glut | Agilent Technologies | Cat# 103579-100 |
| Kainic acid | Sigma-Aldrich | Cat# K0250 |
| Hoechst 33342 | Beyotime | Cat# C1022 |
| Optimal Cutting Temperature | Sakura | Cat# 4583 |
| High glucose DMEM | WISENT | Cat# 319-050-CL |
| Recombinant DNase I (RNase-free) | Takara | Cat# 2270A |
| Penicillin-streptomycin | WISENT | Cat# 450-201-EL |
| DAPI (4′,6-diamidino-2-phenylindole) | Beyotime | Cat# C1002 |
| Other | ||
| 96-well culture plate (flat and clear bottom; black side) | PerkinElmer | CellCarrier-Ultra |
| 100 mm Cell Culture Dishes, TC-treated | BeyoGold | Cat# FCD100 |
| Syringe Filters (0.22 μm/33 mm, PES, Sterile) | BeyoGold | Cat# FF362-100pcs |
| DHA | Yuanye Bio | Cat# S71411 |
| MAP2 | Abcam | Cat# ab32454 |
| Stainless Steel Screws | Pinnacle Technology | Cat# KS66046 |
| Experimental models | ||
| Mouse: ~8 weeks male wild type BALB/c mice at stereotactic EEG-EMG electrodes surgery | Shanghai Slake Laboratory Animal Co., LTD |
https://www.jax.org/strain/000651 Strain #:000651 |
| Mouse: ~6 weeks male wild type BALB/c nude mice (BALB/cJNju-Foxn1nu/Nju) at fluorescence imaging | The Model Animal Research Centre of Nanjing University | http://www.nrcmm.cn/strain/172 |
| Mouse: female wild type BALB/c mice at 18–20 days of pregnancy at culturing mouse hippocampal neurons | Shanghai SLAC Laboratory Animal Co., LTD |
https://www.jax.org/strain/000651 Strain #:000651 |
| Software and algorithms | ||
| Columbus Image Data Storage and Analysis System | Columbus | http://114.212.161.24/file/download/Columbus/MacOSX_x86/Release_32bit/2.4.1/ColumbusClients.zip |
| Living_Image_4.2 | Living Image Software from PerkinElmer | https://www.perkinelmer.com.cn/lab-products-and-services/resources/in-vivo-imaging-software-downloads.html |
| BrainWave software | 3Brain, Wadenswil | N/A |
| Sirenia Seizure Pro | Pinnacle Technology | https://www.pinnaclet.com/index.html |
| Image-Pro Plus 6.0 | Media Cybernetics | N/A |
| ZEN 2.1 (blue edition) | Carl Zeiss Microscopy GmbH | N/A |
Materials and equipment
Inoculation medium for hippocampal neurons
| Reagent | Final concentration | Amount |
|---|---|---|
| Heat Inactivated FBS | 10% | 5 mL |
| Penicillin-streptomycin (100×) | 0.5% | 0.25 mL |
| High glucose DMEM | n/a | 44.75 mL |
| Total | n/a | 50 mL |
Store at 4°C, for up to 1 month.
Growth medium for hippocampal neurons
| Reagent | Final concentration | Amount |
|---|---|---|
| L-Glut (200 mM) | 500 μM or 0 μM | 0.125 mL or 0 mL |
| B27 Plus (50×) | 2% | 1 mL |
| Penicillin-streptomycin (100×) | 0.5% | 0.25 mL |
| Neurobasal Plus Medium | n/a | 48.625 mL or 48.75 mL |
| Total | n/a | 50 mL |
Store at 4°C, for up to 1 week.
Alternatives: The growth medium can replace the inoculation medium when hippocampal neurons are inoculated. It is more expensive but will minimize the division of glial cells as much as possible.
Step-by-step method details
Preparing plates for culturing mouse hippocampal neurons
The protocol below describes the specific steps to prepare plates for mouse hippocampal neurons (see details in troubleshooting 1).
Note: The steps must be performed in a biosafety cabinet.
-
1.Preparing plates coated with Poly-D-lysine.
-
a.Sterilize distilled water using a syringe filter with a 0.22 μm pore size.
-
b.Dilute Poly-D-lysine in distilled water to 50 mg/mL (molecular weight: 150,000–300,000), and store at 4°C for up to 1 week.
-
c.Add 50 μL of the diluted Poly-D-lysine solution to each well of a 96-well culture plate (flat and clear bottom; black side).
-
d.Incubate at 37°C for at least 4 h.
-
e.Aspirate the Poly-D-lysine solution and rinse the plate at least three times with phosphate-buffered saline (PBS).
-
f.Dry the 96-well plate in the biosafety cabinet for later use in a week.
-
a.
Isolation and culture of hippocampal neurons
The protocol below describes the specific steps to culture mouse hippocampal neurons for screening drugs that can regulate iron homeostasis (see details in troubleshooting 1).
-
2.Isolation of mouse hippocampal neurons.
-
a.Prepare a dissecting microscope, sterilized fine tweezers, scissors, 100-mm culture dishes, and 15 mL centrifuge tubes.
-
b.Prepare female BALB/c mice at 18–20 days of pregnancy.
-
c.Prepare 75% ethanol, pre-cooled Hanks’ Balanced Salt Solution without Ca2+ and Mg2+ (HBSS), high glucose DMEM, 0.25% trypsin (containing 0.1% EDTA), Recombinant DNase I (RNase-free), heat-inactivated fetal bovine serum (FBS, 56°C for 30 min with shaking every 5 min).
-
d.Sacrifice a mouse by cervical dislocation after deep anesthetization with 1.5%–2% isoflurane.
-
e.Immerse the mouse in 75% ethanol for 2–5 s and place on ice to lower body temperature to 0°C as fast as possible.
-
f.While keeping the mouse at low temperature (working on ice or an ice pad), dissect the mouse, find the placenta, and put it into the pre-cooled soaking solution (HBSS without Ca2+ and Mg2+) in a 100-mm culture dish.
-
g.Carefully remove the embryo brains one by one and place them into another 100-mm culture dish with cold HBSS (without Ca2+ and Mg2+).
-
h.Isolate the entire hippocampal region of the mouse under a dissecting microscope as much as possible.
-
i.Carefully remove all blood vessels and membranes on the hippocampal tissue surface using fine tweezers.
-
j.Transfer the hippocampal tissue into another culture dish with pre-cooled sterile serum-free high glucose DMEM.
-
k.Wash three times with serum-free high glucose DMEM.
-
l.Cut the hippocampal tissue into 1–2 mm fragments with sterile scissors.
-
m.Carefully remove the supernatant.
-
n.Add 1.5 mL trypsin and 30 μL Recombinant DNase I (RNase-free) to the hippocampal tissue and incubate at 37°C.
-
o.Shake every 2 min for three times.
-
p.Transfer the digested tissue to a 15 mL centrifuge tube, then add 3 mL of 10% inactivated FBS-containing complete medium to terminate trypsin digestion.
-
q.Centrifuge at 319 × g for 5 min and remove the supernatant.
-
r.Resuspend the pellet in complete medium.
-
s.Centrifuge at 319 × g for 5 min to remove the supernatant again.
-
t.Add 2 mL of complete medium, and slowly pipette 10 times.
-
u.After standing for 5 min, remove 1.5 mL of the supernatant, and place it in another centrifuge tube.
-
v.Add 1.5 mL complete medium and pipette 10 times.
-
w.After standing for 5 min, transfer 1.5 mL of the supernatant to the same centrifuge tube in step u.
-
x.Add 1.5 mL complete medium again, pipette 10 times.
-
y.After standing for 5 min, transfer 1.5 mL of the supernatant to the same centrifuge tube in steps u and w.
-
z.Centrifuge the centrifuge tube containing a total of 4.5 mL cell suspension for 10 min at 319 × g.
-
a.
-
3.Culture of mouse hippocampal neurons.
-
a.Prepare the plating medium: 10% heated-inactivated FBS, 0.5% penicillin-streptomycin, high glucose DMEM.
-
b.Prepare the growth medium: 500 μM L-Glut, 2% B27 Plus, 0.5% penicillin-streptomycin, Neurobasal Plus.
-
c.Remove the supernatant from step 1z, resuspend the pellet of neuron cells in the plating medium, count the cells and adjust to a final concentration of 2×104/mL with the plating medium.
-
d.Plate 100 μL of the neuron cell suspension into each well of a 96-well plate that has been pre-coated with Poly-D-lysine.
-
e.Place the 96-well plate in a 37°C, 5% CO2 incubator for 4 h.
-
f.Replace the plating medium with a growth medium completely.
-
g.Replace half of the growth medium every three days.
-
h.Culture hippocampal neurons for at least one week before using them for subsequent experiments.
-
a.
Alternatives: The steps listed above are methods that we have used to successfully culture mature neurons, but they could be substituted by similar protocols.
-
4.Monitoring the maturation of neurons.
-
a.(Day 1, 10 min) Check the adherent state of hippocampal neurons in the growth medium.
-
b.(Day 4, 30 min) Replace half of the culture medium with fresh medium.Note: Exclude L-Glut when making the fresh culture medium. Check the status of neurite outgrowth (Figure 1).
-
c.(Day 7, 30 min) Replace half of the culture medium with fresh medium.Note: Exclude L-Glut when making the fresh culture medium. Observe the neurite outgrowth (Figure 2).Optional: To ensure the successful culture of mature neurons, immunofluorescence staining of synapsin could be used. An abundance of synapsin-positive puncta per neuron should be observed in mature neuronal cells (Figure 3).
-
a.
Figure 1.

The representative image of primary mouse hippocampal neurons in culture for 4 days
Figure 2.
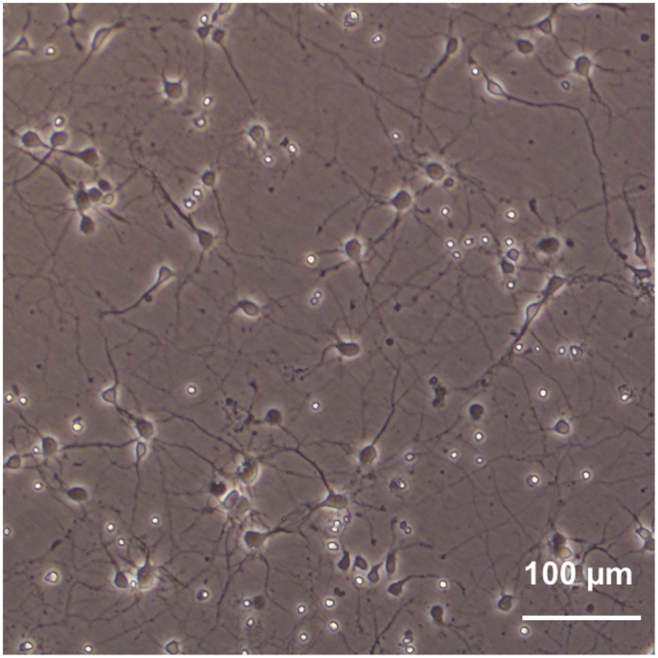
The representative image of primary mouse hippocampal neurons in culture for 8 days
Figure 3.
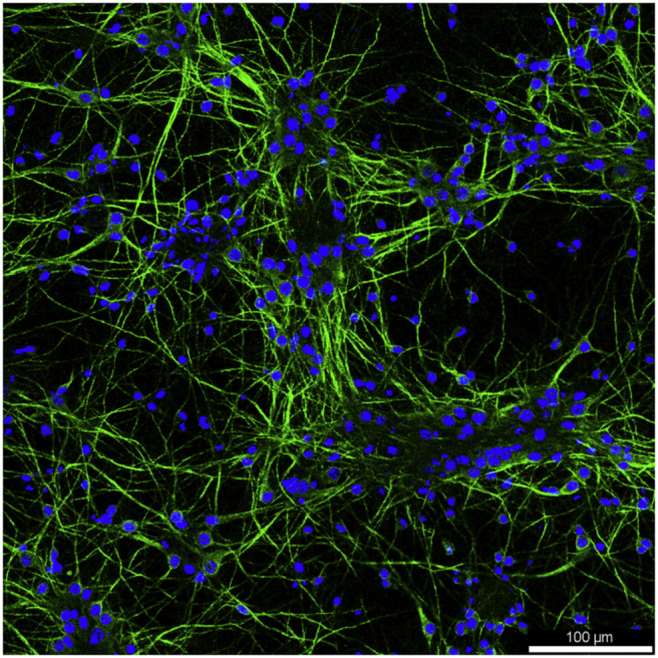
MAP2 (green) and DAPI (blue) staining of primary mouse hippocampal neurons cultured in the B-27 Plus supplement for 10 days
Employing drug treatment to modulate iron homeostasis
Note: In all steps, pre-warm the hippocampal neuron culture medium, PBS, and HBSS at 37°C to avoid cold stimulation of the neurons. Perform steps 5–7 in a biosafety cabinet.
-
5.(Day 9, 30 min) Treatment of hippocampal neurons with kainic acid.
-
a.Make sure the neurons are mature enough before use.
-
b.Discard the medium.
-
c.Add fresh medium with 500 μM kainic acid.
-
d.Incubate at 37°C under 5% CO2 for 12 h.
-
a.
-
6.(Day 9, 30 min) Treatment of hippocampal neurons with different natural products.
-
a.Discard the medium.
-
b.Wash with PBS twice.
-
c.Add fresh medium with 20 μM of different natural products.
-
d.Incubate at 37°C under 5% CO2 for 12 h.
-
a.
-
7.(Day 10, 30 min) Incubation of hippocampal neurons with fluorescence probes of Fe2+ and the nucleus.Note: We would like to promote the use of specific target probes for high-throughput screening of reported active compounds rather than being limited to a few natural products covered in this protocol. If readers have more natural product library resources, they can use the FeP or other Fe2+ probe and methods provided in this protocol to expand screening.
-
a.Discard the medium.
-
b.Wash the cells twice with PBS.
-
c.Add 100 uL fresh medium with 5 μg/mL Hoechst 33342 and 10 μM FeP.
-
d.Incubate at 37°C under 5% CO2 for 30 min.
-
e.Discard the medium.
-
f.Wash with PBS twice.
-
g.Add 50 μL HBSS and proceed to high-content analysis.
-
a.
Setting up the protocol for the high-content analysis
The protocol below describes the method for the measurement of Fe2+ flux in the Columbus Image Data Storage and Analysis System (see details in troubleshooting 2 and 3).
-
8.(Day 10, 2 h) Fluorescence image acquisition.
-
a.Turn on the high-content screening (HCS) machine (PerkinElmer Operetta CLS) 30 min before use.
-
b.Place the 96-well culture plates into the HCS machine.
-
c.Choose a normal-focus 40× magnification lens.
-
d.Set a combination of 380 nm excitation wavelength and emission filter 525/15 in channel 1 for Hoechst 33342.
-
e.Set a combination of 475 nm excitation wavelength and emission filter 615/15 in channel 2 for FeP.
-
f.Set the optimal exposure time for all channels.
-
g.Adjust the best focal length.
-
h.Set 10 fields of view per well.
-
i.Set the parameters of the living cell studio at 37°C and 5% CO2.
-
j.Start scanning.
-
a.
Note: Approximately 400 cells per well are imaged.
Analysis of Fe2+ flux with FeP
This section describes a method for analyzing the fluorescence intensity mediated by Fe2+ flux per body part of neurons area from the images captured by FeP over the number of Hoechst-positive objects.
-
9.(Day 10, ∼1 h) Set up the measurement protocol of Columbus Image Data Storage and Analysis System software as follows (Figures 4, 5, 6, 7, and 8).Note: To avoid interference from the intricate connections between neurons, we only measured fluorescence in the cell body of each neuron.
-
a.Create an analysis protocol (termed “step 9.a”) to detect neurons.Note: Set stack processing as the maximum intensity projection. Set flatfield correction as basic mode. Adjust the contrast of two channels until the contrast between the body of neurons and background is clear and the image is not overexposed, respectively.
-
b.Create an analysis protocol (termed “step 9.b”) to detect neuronal nuclei from images of neurons stained by Hoechst 33342.Note: Set the method as B mode: common threshold as 0.4, area > 30 μm2, output population as nuclei.
-
c.Create an analysis protocol (termed “step 9.c”) to detect neuronal cytoplasm from images of neurons stained by FeP based on identified nuclei.Note: Set the method as A mode: common threshold as 0.15.
-
d.Create an analysis protocol (termed “step 9.d”) to analyze the images of FeP fluorescence staining captured in 9.c.Note: Set channel as FeP, the population as nuclei, the region as whole-cell, method as standard, output properties as “Intensity Cell FeP Mean”.
-
e.Create an analysis protocol (termed “step 9.e”) to define the results analyzed in step 9.d.Note: Choose method as standard out. Export the intensity cell FeP mean value from the analysis results as an Excel file, which represents the content of Fe2+ per cell. If necessary, download the value of the number of objects, which represents the number of neurons analyzed.
-
a.
Figure 4.

The image shows neurons stained with FeP (red) and Hoechst33342 (blue)
Figure 5.

Display of operation interface to detect the cell nucleus stained with Hoechst 33342 in the Columbus Image Data Storage and Analysis System
The resulting interface is shown in the following figure.
Figure 6.

Display of operation interface to detect the cell cytoplasm stained with FeP in the Columbus Image Data Storage and Analysis System
The resulting interface is shown in the following figure.
Figure 7.

Display of operation interface to calculate the intensity properties for the channel of FeP in the Columbus Image Data Storage and Analysis System
The resulting interface is shown in the following figure.
Figure 8.

Output viewer of fluorescence intensity in the Columbus Image Data Storage and Analysis System
Stereotactic surgery
The protocol below describes how to perform stereotactic surgery to implant EEG-EMG electrodes in mice and investigate the potential antiepileptic effects of the DHA.
Note: Since the wound is hermetically sealed by dental cement, post-operational analgesics and antibiotics were not used. These mice are allowed to recover for one week after surgery.
-
10.(Day 11, 20 h) Prepare mice for stereotactic EEG-EMG electrodes surgery.
-
a.Anesthetize mice by using isoflurane. The induction and maintenance doses of isoflurane are 3% and 1.5%–2%.
-
b.Disinfect stainless steel screws (used as electrodes) with 75% alcohol, wash with PBS, and finally blot with absorbent paper.
-
c.Place the stainless steel screws on top of the dura mater through small holes drilled in the skull with a high-speed drill.
-
d.Implant a total of five electrodes. Place the front two electrodes over the motor cortex (approximately 1 mm anterior to bregma and 1.5 mm lateral to the midline).
-
e.Place the other two electrodes approximately 1.5 mm anterior to lambda and 1.5 mm lateral to the midline.
-
f.Place the ground electrode in the midline approximately 1.5 mm posterior of lambda (Figure 9).
-
g.In the process, hold the electrode still with a tweezer using the left hand, and turn the screw with a screwdriver with the right hand slowly until it passes through the skull (Figure 10).
-
h.Fix the stainless steel screws to the skull by using dental cement.
-
i.Wire EEG-EMG electrodes to the head mount.
-
j.Fix the head mount to the skull by using dental cement.
-
a.
Optional: Although EEG-EMG electrodes are used in this protocol, only the EEG signal is relevant and it also works if only EEG electrodes are implanted for recording. The EMG signal is just a reference to determine whether mice are asleep or awake.
Figure 9.

Surgical procedures for the implantation of recording electrodes
(A) The in-kind real shoot of the electrode adaptor.
(B) Small openings marked on the skull that allow electrode insertion.
Figure 10.

Surgical procedures for the implantation of recording electrodes and dental cement fixation
Video-EEG-EMG monitoring
This section describes the steps to prepare the mice for video-EEG-EMG recording.
-
11.
(Day 11–18, 7 days) Recovery from electrode implantation surgery.
Place mice into the stereotactic apparatus for surgical implantation of electrodes. After surgery, return mice into the home cage and allow them to recover for one week (see section Stereotaxic surgery).
-
12.
(Day 19–21, 2 h) Establishment of an epileptic mouse model and DHA treatment.
Treat mice with DHA to establish epileptic mouse models as detailed in Figure 11:-
a.Dilute the stock solution of DHA to 6 mg/mL with PBS to prepare the working solution. The final injection solvent of DHA is DMSO-PBS buffer (10 mM, pH 7.4, 1:9, v/v).
-
b.Inject mice with DHA (60 mg/kg) via intraperitoneal (i.p.) injection.
-
c.Dilute the stock solution of kainic acid to 4 mg/mL with PBS to prepare the working solution.
-
d.Inject mice with KA (20 mg/kg) via intraperitoneal (i.p.) injection.
-
a.
-
13.(Day 21–22, 10 h) Video-EEG-EMG recording (see details in troubleshooting 4).
-
a.Place each mouse into a 21 cm × 19 cm transparent cage individually.
-
b.Insert a pre-amplifier into the 8-pin head cap and connect to a multichannel commutator (Bio-Signal Technologies).
-
c.Adjust EEG-EMG signals at 500 Hz and bandpass filtered at 1–100 Hz.
-
d.Adjust recording of videos as 10 frames/s.
-
e.Record EEG-EMG signals and synchronized videos by using the Athena system (Bio-Signal Technologies) for 2 h.
-
f.Use Sirenia Seizure Pro (Pinnacle Technology) to analyze the whole course EEG-EMG recordings and representative traces.
-
g.Analyze the EEG signals for electrographic seizure events.
-
a.
Note: Seizure events were defined as high amplitudes at two-fold of the baseline and durations lasting at least 10 s, using the Sirenia Seizure Pro software (Pinnacle Technology Inc, Lawrence, KS).
Figure 11.

Schematic of experiment design showing the timeline of the steps involved in preparing epileptic mice for video-EEG-EMG recording
Source: Reproduced with permission from Shao et al. (2022). Copyright 2022, Cell Chem. Biol. (Shao et al., 2022).
Analysis of mouse brain EEG signals using Sirenia Seizure Pro software
This section describes a method to analyze the EEG-based data offline by using Sirenia Seizure Pro software. The advanced analysis generated by Sirenia Seizure Pro software provides user-configured parameters for conveniently analyzing seizure events over a given time period.
-
14.(Day 22 or later, ∼6 h) Set up the measurement protocol of Sirenia Seizure Pro software as follows (Figure 12, Figure 13, Figure 14, Figure 15, Figure 16, and Figure 17).
-
a.Create an analysis protocol (termed “step 14.a”) to read EEG-EMG recordings in EDF format.Note: The BIO file of the whole course EEG-EMG recordings is converted to a standard format of EDF and imported into the Sirenia Seizure Pro software for analysis.
-
b.Create an analysis protocol (termed “step 14.b”) to generate the custom plot.Note: Set the "freq" fields from 0.1 to 100 Hz at the high pass type and click on the" Apply Filter" button to filter the setting, and choose a suitable period. We get used to analyzing 1 h of raw EEG data as a suitable period. The analyzed period choosing based on your collected data generally.
-
c.Create an analysis protocol (termed “step 14.c”) to consolidate and extract data from all channels.
-
d.Create an analysis protocol (termed “step 14.d”) to select seizures.Note: Extract the features from an obvious seizure (Seizures were defined by a rapid and rhythmic spiking activity (>3 Hz) that persisted for >5 s with high amplitudes (>2× baseline level). Here, we didn't distinguish different types of seizure.) as a pattern-matching template in this experiment by the seizure selection tool. Then, click on the" Add type" button to place the event and feature set into a pattern-matching database.
-
e.Create an analysis protocol (termed “step 14.e”) to find all seizures.Note: Adjust the search setting to match patterns with features extracted in step d and identify similar seizures in the whole EEG. Pattern matching includes, but is not limited to, RMS power, frequency band, amplitude, and line length.
-
f.Create an analysis protocol (termed “step 14.f”) to mark all seizures.Note: Please have three experienced researchers confirm the EEG seizure events after sorting by the software. All the matched seizures can be rejected, accepted, or merged with nearby events to better classify seizures.
-
g.Create an analysis protocol (termed “step 14.g”) to export the data for human review and analysis.Note: Analyze data on amplitude during seizures, seizure duration, line length, and total EEG power during seizures based on the entire EEG.
-
a.
Figure 12.

Display window of the EEG signal based on the user requirements
(A and B) (A) User-required type. (B) User-required time period.
Figure 13.

Display window of consolidating and extracting EEG-based data in the Sirenia Seizure Pro software
Figure 14.

Display window of selecting seizure in the Sirenia Seizure Pro software
An obvious seizure is selected in blue and frequency selection is shown in red.
Figure 15.

Display window of finding all seizures in the Sirenia Seizure Pro software
Figure 16.

Display window of pattern matching all seizure events in the Sirenia Seizure Pro software
All similar seizures were selected in red and the features were in yellow.
Figure 17.

The features of all seizure waveforms are exported by Sirenia Seizure Pro software
In vivo fluorescence imaging
The protocol below describes the steps for in vivo fluorescent imaging of drug-treated epileptic mice.
-
15.(Day 22–24, 2 h) Treat mice with DHA to establish epileptic models as described in step 12. Then, perform global imaging of the whole body by using the IVIS Spectrum imaging system.
-
a.Dilute the stock solution of DHA to 6 mg/mL with PBS (10% DMSO) to prepare the working solution.
-
b.Inject DHA (60 mg/kg) into mice via intraperitoneal (i.p.) injection.
-
c.Dilute the stock solution of kainic acid to 1.2 mg/mL with PBS to prepare the working solution.
-
d.Inject mice with kainic acid (6 mg/kg) via intraperitoneal (i.p.) injection.
-
a.
-
16.(Day 23–25, 2 h) Treat the new mice with DHA to establish epileptic models as in step 12. Then, perform local imaging in the primary somatomotor cortex by using a confocal fluorescent microscope (ZEISS LSM 980 with Airyscan).
-
a.Dilute the stock solution of DHA to 6 mg/mL with PBS (10% DMSO) to prepare the working solution.
-
b.Inject DHA (60 mg/kg) into mice via intraperitoneal (i.p.) injection.
-
c.Dilute the stock solution of kainic acid to 4 mg/mL with PBS to prepare the working solution.
-
d.Inject kainic acid (20 mg/kg) into mice via intraperitoneal (i.p.) injection.
-
a.
-
17.(Day 24, 6 h) The whole body imaging using the IVIS Spectrum imaging system (Figure 18).Note: The average radiant efficiency of a good picture is at least on the order of 107 p/sec/cm2/sr/(μW/cm2). The duration to detect the fluorescent depends on the metabolic process of the probe and there will be differences between different probes. The peak of dots per inch fluorescent intensity FeP in mice was 30 min, so we performed fluorescence tracking at different time points within 1 h.
-
a.Dilute the stock solution of FeP to 0.1 mg/mL with PBS (5% DMSO) to prepare the working solution.
-
b.Inject FeP (0.5 mg/kg) into mice via intravenous (i.v.) injection (see details in troubleshooting 5).
-
c.Anesthetize mice with 1.5%–2% isoflurane.
-
d.Set the excitation filter and collection wavelength range of IVIS Lumina XR in vivo imaging system as 465 nm and 575–650 nm, and choose “Auto Exposure”.
-
e.Capture fluorescence images at 5, 15, 30, 45, and 60 min post-injection of FeP.
-
a.
-
18.(Day 25, 8 h) Local imaging using a confocal fluorescent microscope (ZEISS LSM 980 with Airyscan).Note: The body temperature of mice is maintained with an electric heating blanket during the entire surgery-imaging procedure.
-
a.Sterilize all surgical materials (including forceps, scissors, drills, and glass coverslips).
-
b.Warm up the confocal fluorescent microscope (ZEISS LSM 980 with Airyscan) for 30 min.
-
c.Set the excitation filter and collection wavelength range as two-photon 880 nm and 600–680 nm (Figure 19A).Note: the power of imaging light is less than 15%, and the master gain is less than 800.
-
d.Anesthetize mice with 1.5%–2% isoflurane or 2% pentobarbital sodium.
-
e.Place the mouse in a stereotaxic frame.
-
f.Disinfect the scalp with 75% ethanol.
-
g.Cut the scalp with a scalpel and scissors and avoid damaging blood vessels, carefully remove the muscles and periosteum to guarantee access to the cranium.Note: If occasional minor bleeding occurs, stop the bleeding by applying pressure with medical cotton tips not touching the brain surface immediately, and then dip a small amount of warm PBS to gently wipe away the blood.
-
h.By careful drilling, thin the skull in a circular pattern until it is almost detached.
-
i.Remove the skull with fine forceps.
-
j.Dilute the FeP stock solution with PBS (5% DMSO) to 0.1 mg/mL to prepare the working solution. Inject FeP (0.5 mg/kg) into mice by intravenous (i.v.) injection.
-
k.Move the 20× hydroscope lens to the cerebral cortex of the left lateral ventricle, corresponding to the primary somatomotor cortex (Economo et al., 2016) (Wang et al., 2020) (Figure 19B).
-
l.Capture fluorescence images with laser-laminar scanning every 5 min for 1 h (Figure 20).
-
a.
Figure 18.

Schematic representation of IVIS Spectrum imaging system
Figure 19.

Schematic representation of in vivo imaging of Fe2+ in the primary somatomotor cortex by a confocal fluorescent microscope
(A) The basic setup includes a head plate holder, microscope objective (20× hydroscope), excitation light source, and the detector.
(B) The reference position of the primary somatomotor cortex.
Figure 20.

The representative 3D image of the primary somatomotor cortex in mice treated with KA
Source: Reproduced with permission from Shao et al. (2022). Copyright 2022, Cell Chem. Biol. (Shao et al., 2022).
Preparing brain slices for fluorescence imaging
The protocol below describes the steps for preparing brain slices.
-
19.(Day 26, 3.5 h) Prepare the brain slices.
-
a.Pre-cool the freezing microtome to −20°C.
-
b.After injecting inject mice with FeP through the tail vein for 1 h; sacrifice mice by cervical dislocation after deep anesthetization with 1.5%–2% isoflurane.
-
c.Dissect out the brain and flash freeze in liquid nitrogen (see details in troubleshooting 6).
-
d.Embed the frozen brain at the Optimal Cutting Temperature (O.C.T.).
-
e.Place the embedded brain tissue in the freezing microtome for sectioning.
-
f.Dry the slices and store them at −80°C for use.
-
a.
Brain slices fluorescence imaging
This protocol below describes the steps for preparing the brain slices of mice for fluorescence imaging.
-
20.(Day 26, 8 h) Staining and fluorescence imaging.
-
a.Take out the brain slices from the ultra-low temperature freezer.
-
b.Return slices to 25°C.
-
c.Dry for 30 min before staining.
-
d.Dilute DAPI stock solution to 10 μg/mL with PBS to prepare the working solution.
-
e.Use liquid blocker super PAP Pen to form a stable hydrophobic isolation zone surrounding tissue samples.
-
f.Stain with DAPI for 30 min at 37°C.
-
g.Warm up the two-photon laser of the confocal fluorescent microscope (Leica TCS SP8 MP) for 1 h.
-
h.Set the fluorescence excitation filter and emission window as MP 880 nm and 600–680 nm for FeP. Set the fluorescence excitation filter and emission window as 405 nm and 420–480 nm for DAPI.
-
i.Choose the scan selected area in the XY stage to determine the boundaries of the hippocampus.
-
j.Capture individual MP fluorescence images in sequence.
-
k.Join all images of the multi-photon together into the maximum hippocampal surface in brain tissues by Leica TCS SP8 MP (Figure 21).
-
a.
Figure 21.

Perspective for 3D two-photon fluorescence images at different depths in epileptic mice brain sections
Source: Reproduced with permission from Shao et al. (2022). Copyright 2022, Cell Chem. Biol. (Shao et al., 2022).
Expected outcomes
Using the protocols provided here, one can quickly screen a given compound library for its ability to regulate iron homeostasis. In this protocol, all steps from cell fluorescence imaging to quantification are automated by a high-throughput screening strategy provided by PerkinElmer Operetta CLS and Columbus Image Data Storage and Analysis System, which avoids the observer bias in visual field selection and analysis. The use of this protocol can greatly improve the speed of imaging analysis of primary hippocampal neurons, thus reducing the waste of human and material resources. Meanwhile, the feasibility of high-throughput screening to discover lead compounds for modulating iron homeostasis was verified by the analysis of EEG-EMG signals (a hallmark of epilepsy). The automated seizure detection provided by the Sirenia Seizure Pro software can be used for the rapid identification of seizure events within large-scale datasets. We believe that this protocol, with some modifications and using other probes, would also be applicable to screen drugs that can regulate other diseases.
Limitations
Experimenters may experience technical challenges in the culture of mouse hippocampal neurons and stereotactic surgery. The duration of drug administration and concentration of drugs and probes are both exploratory. Be patient; developing skills and acquiring satisfactory results in these procedures require practice. In this protocol, the instrument platform is an essential factor. From in vitro to in vivo, different instruments have their roles in research and cannot be easily replaced.
Troubleshooting
Problem 1
The survival rate and maturity of hippocampal neurons are low.
Potential solution
Many factors can affect the state of neurons. The culture plates coated with Poly-D-lysine should be kept out of light. Although dilutions of Poly-D-lysine can be reused, adding more than half of the freshly prepared Poly-D-lysine each time is highly recommended. After removing the Poly-D-lysine coating solution, the plates should be washed at least three times with PBS rather than dried directly due to the toxicity of PDL. Brain dissection must always be performed on ice to inhibit neuronal apoptosis and improve neuronal survival. Be sure to remove blood vessels and vascular membranes as much as possible. If the blood vessels and vascular membranes are not fully removed, hippocampal tissue will be difficult to disperse into individual neurons, thus requiring transitional digestion or multiple blowing of discrete neurons, resulting in massive neuronal death. In addition, the cells from the blood vessels and the vascular membrane will mix into the primary neurons, resulting in a lower purity and quality of neurons. Among the more than three culture conditions of neurons (B-27 Plus supplement culture medium: 2% Gibco B-27 Plus, 0.5% Penicillin-streptomycin (100×), Neurobasal Plus Medium; B-27 supplement culture medium: 2% Gibco B-27, 0.5% Penicillin-streptomycin (100×), Neurobasal Medium; W-21 supplement culture medium: 2% WISENT W-21, 0.5% Penicillin-streptomycin (100×), WISENT NEUROCELL Medium) that we have tried, the Gibco B-27 Plus neuronal culture medium is recommended. Compared to a basal medium consisting of B-27 and Neurobasal medium, B-27 Plus can promote a higher survival rate of neurons. L-Glut is unstable in solution, thus add it fresh if possible. After the fourth day of neuronal culture, remove L-Glut from the fresh culture medium. FBS should be inactivated at a high temperature (56°C for 30 min with shaking every 5 min) in the step of neuron isolation. FBS should also be avoided during neuronal culture to inhibit the proliferation of non-neuronal cells.
Problem 2
Does long-term high-content screening affect cell viability?
Potential solution
A live cell imaging system is recommended to maintain cell viability at 37°C at 5% CO2. In our experience, temperature affects the state of neurons more than the concentration of CO2. If your high-content imaging system is not equipped with a live cell platform, please ensure that the temperature of the instrument storage room is 37°C. It is also suggested to slightly increase the laser intensity to reduce the exposure time and to replace the high magnification objective lens with a low magnification objective lens (like 20×) to reduce the total screening time in 96-well plates.
Problem 3
Are there other instruments available to replace the high-content screening and analysis capabilities provided by PerkinElmer Operetta CLS?
Potential solution
Automatic high-speed microscopic imaging, automatic image analysis, and data management are irreplaceable advantages of high-content cell imaging analysis technology. Therefore, other high-content analysis devices (such as High-content analysis CQ1(Sasahara and Hoshi, 2022) that can measure 96-well (or 384-well) culture plates within a short time and in an automated way may be a suitable substitute for the PerkinElmer Operetta CLS.
Flow cytometry and confocal fluorescence microscopy can also be used for quantitative analysis of fluorescence signals, but we discourage their usage for FeP-based high-content screening. Flow cytometry requires a large number of cells, and since primary neurons cannot proliferate, it results in a large number of mice pups that need to be sacrificed and in a huge amount of work. Moreover, flow cytometry cannot be used to acquire fluorescence images. On the other hand, when using confocal microscopes each field of view in the imaged channels needs manual adjustment. The acquisition speed of multicolor fluorescence images in different drug-treated groups is very low. Also, confocal microscopy software generally does not provide fluorescence analysis tools for individual cells, and other image analysis software, such as image pro plus and ImageJ, are needed.
Problem 4
How to assure the accuracy of the electrographic seizure sorted by the software?
Potential solution
The Sirenia Seizure Pro software (Pinnacle Technology Inc, Lawrence, KS) has long been accepted for the detection of electrographic seizure events, and the related references can be found on the official website of this software (Glushakov et al., 2016), (Arain et al., 2015). To ensure accuracy, three experienced researchers confirmed the electrographic seizure events after sorting by the software, and we encourage other groups to follow this practice.
Problem 5
Why is the working concentration of the FeP set at 0.5 mg/kg for in vivo fluorescence imaging?
Potential solution
The selection of FeP concentration is determined from our preliminary experiments. The optimal working concentration of the probe is based on the performance of the probe itself, including detection range, the substrate expression level in vivo, ability to penetrate the blood-brain barrier, spectral performance, etc. We recommend using a low concentration of the probe to minimize false-positive results. The recommended concentration of probe is 0.5–1.5 mg/kg.
Problem 6
Histological morphology of mouse brain tissue frozen sections is incomplete.
Potential solution
The mouse brain tissue needs to be dissected out on ice within 5 min after a cervical dislocation to prevent autolysis and corruption of the tissue. In addition, the brain tissue directly frozen in liquid nitrogen may be ruptured due to uneven freezing. It is recommended that embed the separated mouse brain tissue with the Optimal Cutting Temperature (O.C.T.) and then freeze it in liquid nitrogen. Take out the brain tissue when the center of the embedding medium is white and store at −80°C for subsequent slicing.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Yong Qian (yongqian@njnu.edu.cn).
Materials availability
This study did not generate new unique reagents.
Acknowledgments
This work is financially supported by the National Natural Science Foundation of China (22074065, 21778033, 21877060), the program of Jiangsu Specially-Appointed Professor for Yong Qian, and the Open Project of State Key Laboratory of Pharmaceutical Biotechnology (KF-GN-202002).
Author contributions
Y.Q., C.W.S., and Z.P.C. designed and optimized the protocol; C.W.S. and Z.P.C. wrote the original draft, and C.Y. and Y.Q. reviewed and edited the manuscript.
Declaration of interests
Nanjing Normal University Office of Technology and Intellectual Property is in the process of filing a patent protection of applications of the reported probe; Y.Q. and C.W.S. are named as co-inventors.
Contributor Information
Chao Yan, Email: yanchao@nju.edu.cn.
Yong Qian, Email: yongqian@njnu.edu.cn.
Data and code availability
This study did not generate datasets or codes.
References
- Arain F., Zhou C., Ding L., Zaidi S., Gallagher M.J. The developmental evolution of the seizure phenotype and cortical inhibition in mouse models of juvenile myoclonic epilepsy. Neurobiol. Dis. 2015;82:164–175. doi: 10.1016/j.nbd.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Economo M.N., Clack N.G., Lavis L.D., Gerfen C.R., Svoboda K., Myers E.W., Chandrashekar J. A platform for brain-wide imaging and reconstruction of individual neurons. Elife. 2016;5:e10566. doi: 10.7554/elife.10566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glushakov A.V., Glushakova O.Y., Doré S., Carney P.R., Hayes R.L. Animal models of posttraumatic seizures and epilepsy. Methods Mol. Biol. 2016;1462:481–519. doi: 10.1007/978-1-4939-3816-2_27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasahara T., Hoshi M. High-throughput screening for agonists of ROS production in live human vascular endothelial cells. STAR Protoc. 2022;3:101053. doi: 10.1016/j.xpro.2021.101053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao C., Liu Y., Chen Z., Qin Y., Wang X., Wang X., Yan C., Zhu H.L., Zhao J., Qian Y. 3D two-photon brain imaging reveals dihydroartemisinin exerts antiepileptic effects by modulating iron homeostasis. Cell Chem. Biol. 2022;29:43–56.e12. doi: 10.1016/j.chembiol.2021.12.006. [DOI] [PubMed] [Google Scholar]
- Wang Q., Ding S.L., Li Y., Royall J., Feng D., Lesnar P., Graddis N., Naeemi M., Facer B., Ho A., et al. The allen mouse brain common coordinate framework: a 3D reference atlas. Cell. 2020;181:936–953.e20. doi: 10.1016/j.cell.2020.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate datasets or codes.