

Abstract
Background
Mucosal melanoma has characteristically distinct genetic features and typically poor prognosis. The lack of representative mucosal melanoma models, especially cell lines, has hindered translational research on this melanoma subtype. In this study, we aimed to establish and provide the biological properties, genomic features and the pharmacological profiles of a mucosal melanoma cell line that would contribute to the understanding and treatment optimization of molecularly‐defined mucosal melanoma subtype.
Methods
The sample was collected from a 67‐year‐old mucosal melanoma patient and processed into pieces for the establishment of cell line and patient‐derived xenograft (PDX) model. The proliferation and tumorigenic property of cancer cells from different passages were evaluated, and whole‐genome sequencing (WGS) was performed on the original tumor, PDX, established cell line, and the matched blood to confirm the establishment and define the genomic features of this cell line. AmpliconArchitect was conducted to depict the architecture of amplified regions detected by WGS. High‐throughput drug screening (HTDS) assay including a total of 103 therapeutic agents was implemented on the established cell line, and selected candidate agents were validated in the corresponding PDX model.
Results
A mucosal melanoma cell line, MM9H‐1, was established which exhibited robust proliferation and tumorigenicity after more than 100 serial passages. Genomic analysis of MM9H‐1, corresponding PDX, and the original tumor showed genetic fidelity across genomes, and MM9H‐1 was defined as a triple wild‐type (TWT) melanoma subtype lacking well‐characterized “driver mutations”. Instead, the amplification of several oncogenes, telomerase reverse transcriptase (TERT), v‐Raf murine sarcoma viral oncogene homolog B1 (BRAF), melanocyte Inducing transcription factor (MITF) and INO80 complex ATPase subunit (INO80), via large‐scale genomic rearrangement potentially contributed to oncogenesis of MM9H‐1. Moreover, HTDS identified proteasome inhibitors, especially bortezomib, as promising therapeutic candidates for MM9H‐1, which was verified in the corresponding PDX model in vivo.
Conclusions
We established and characterized a new mucosal melanoma cell line, MM9H‐1, and defined this cell line as a TWT melanoma subtype lacking well‐characterized “driver mutations”. The MM9H‐1 cell line could be adopted as a unique model for the preclinical investigation of mucosal melanoma.
Keywords: bortezomib, cell line, extrachromosomal DNA, high‐throughput drug screening, mucosal melanoma, patient derived tumor cells, patient‐derived xenograft, whole‐genome sequencing
1. BACKGROUND
Mucosal melanoma, which arises from melanocytes in the mucosal layer of different anatomical parts, accounts for about 0.8%‐3.7% of all melanomas among Caucasian populations [1, 2], while it is the second most common subtype in Asian melanoma populations, constituting approximately 25% of all melanomas in Asian patients [3]. Although targeted agents, such as BRAF inhibitor, mitogen‐activated protein kinase (MEK) inhibitor, and programmed cell death protein‐1/programmed cell death‐ligand 1 (PD‐1/PD‐L1) immune checkpoint inhibitor, have revolutionized clinical management of melanoma, the effectiveness of these treatments was not satisfactory in mucosal melanoma [4]. The 5‐year survival rate of mucosal melanoma is only 16%‐27% [5, 6], much lower than that of cutaneous melanoma (about 90%) [7], possibly because of its high rates of recurrence and metastasis as well as a lack of effective therapies.
Large‐scale cancer genomic studies, such as The Cancer Genome Atlas (TCGA) project in melanoma, led to the genetic classification of melanoma into four major subtypes based on the mutually exclusive significantly mutated genes: BRAF‐mutant subtype, RAS‐mutant subtype, neurofibromin 1 (NF1)‐mutant subtype, and triple wild‐type (TWT) subtype. Interestingly, TWT melanoma subtype account for only about 10%‐15% of all cutaneous melanoma cases [8], while the majority of acral melanoma and mucosal melanoma (50%‐80%), which is characterized by a lack of significantly mutated genes, could be classified as TWT subtype [9]. KIT proto‐oncogene receptor tyrosine kinase (KIT) is the only recurrent significantly mutated oncogene with a targeted treatment option in TWT mucosal melanoma, and imatinib is currently recommended by clinical practice guidelines of the National Comprehensive Cancer Network (NCCN) [10] and Chinese Society of Clinical Oncology [11] for the treatment of KIT‐mutant mucosal melanoma. Unfortunately, the clinical efficacy of imatinib is still limited because only about 15%‐20% of mucosal melanomas harbor KIT mutations, and drug resistance often occur during either the early phase or the late phase of drug treatment [11, 12]. Currently, there are still few targeted treatment options for mucosal melanoma, especially for TWT mucosal melanoma patients without KIT mutations [13].
More recently, we [14] and several studies [9, 13] using whole‐genome sequencing (WGS) have further revealed the distinct genomic pattern in mucosal melanoma when compared with cutaneous melanoma, indicating the biological significance of large‐scale genomic rearrangements, structure variations (SVs) and copy number variations (CNVs), which could be detected in the majority of mucosal melanomas. Specifically, we have found over 50% of the mucosal melanoma patients harbored concurrent focal amplifications of cyclin‐dependent kinase 4 (CDK4) (12q14) and telomerase reverse transcriptase (TERT) (5p15), an event that co‐occurs with inter‐chromosomal translocations that defined a patient subgroup with significantly worse clinical outcomes [14]. Notably, CDK4/6 inhibitor was further evaluated and demonstrated remarkable antitumor effects in CDK4‐amplified mucosal melanoma patient‐derived xenograft (PDX) models [14]. Accordingly, a phase II clinical trial is currently recruiting CDK4‐amplified mucosal melanoma patients to access the therapeutic potential of CDK4/6 inhibitor (ChiCTR2000031608). However, there are some TWT mucosal melanoma patients who are not eligible for treatment targeting CDK4 amplification or KIT mutation, so that further molecular and biological characterization of TWT mucosal melanoma is needed to identify potential therapeutic options for this melanoma subtype. High‐throughput drug screening (HTDS) is a drug discovery process that allows rapid testing of large numbers of agents which is increasingly used in antitumor drug reposition and translation [15]. Multiple studies have reported the identification of novel treatment options or old drugs for new uses by conducting HTDS using genetically characterized cell lines, which successfully led to the clinical investigation of the potential agents in cancer patients [16, 17]. According to Cellosaurus (https://web.expasy.org/cellosaurus/), the largest cell line database, more than 3000 melanoma cell lines have been documented, while only 14 of these were mucosal melanoma cell lines which lacked detailed genetic annotations. To our knowledge, few such studies have conducted drug screening using mucosal melanoma cell lines or patient‐derived tumor cells (PDC). Thus, the scarcity of stable mucosal melanoma models in vitro, which can reflect the genetic and biological features of mucosal melanoma patients, represents a major challenge for understanding mucosal melanoma biology and for drug screening in mucosal melanoma.
In the present study, we set out to evaluate the feasibility of mucosal melanoma cell line establishment and characterization as an experimental tool for mucosal melanoma translational research. In this effort, we also aimed to determine (1) whether the established cell line would recapitulate the biological properties and genomic features of the original tumor; (2) whether the established cell line could be applied for HTDS that would facilitate the identification of novel therapeutic strategies for the treatment of molecularly‐defined mucosal melanoma patients.
2. MATERIALS AND METHODS
2.1. Mucosal melanoma specimens
Mucosal melanoma biopsy and paraffin‐embedded tissue samples were from a patient (MM108) treated at Shanghai Ninth People's Hospital (Shanghai, China). The collection and use of these mucosal melanoma specimens for this experimental study were approved by The Ethics Committee of Shanghai Ninth Peoples Hospital, Shanghai Jiao Tong University School of Medicine (Shanghai, China), and the patient's consent was obtained. We followed the tenets of the Declaration of Helsinki, in accordance with recommendations approved by the Institutional Ethics Committee. Biopsy tissues were immediately immersed in tissue storage solution (MACS, Bergisch Gladbach, Germany) to maximize the viability of cells. Generally, the sample was processed into 1 mm3 pieces in biosafety cabinet for PDX model establishment and primary culture for cell line establishment.
2.2. Primary culture of cell lines
The dissected primary tumor lesion was immediately delivered from the operating room to the lab after tumor biopsy in a 5 mL tube filled with tissue storage solution. Fresh tumor tissue was then washed 3 times in precooling phosphate buffered saline (PBS) containing 2% penicillin‐streptomycin, 1% gentamicin and 1% amphotericin B (Gibco, Thermo Fisher Scientific, Waltham, MA, USA). The tissue was cut into small fragments (about 0.5‐1 mm3 each) and transferred into pre‐warmed RPMI‐1640 medium (Gibco, Carlsbad, CA, USA), with 20% fetal bovine serum (FBS, Gibco) and 1% penicillin‐streptomycin. The tissue fragments were placed in a collagen coated T‐25 flask containing 2 mL culture medium. The culture bottle was inverted overnight and gently turned over the second day, which was placed in a 37°C incubator containing a humidified atmosphere and 5% CO2. After grown for up to 6 days, cells were suspended with 0.25% trypsin and 0.02% EDTA treatment and passaged with 1:2 or 1:3 split ratio depending on their growth rate. A375, a malignant cutaneous melanoma cell line, was kindly provided by Stem Cell Bank, Chinese Academy of Sciences (Shanghai, China) and was cultured in RPMI‐1640 with 10% FBS. Images were captured with cell imaging systems in Zeiss microscope (Zeiss, Jena, Germany).
2.3. Flow cytometry for purity
Cell purity of the established mucosal melanoma cell line MM9H‐1 was determined by flow cytometric staining for melanoma‐associated chondroitin sulfate proteoglycan (MCSP) on a FACS Calibur (Becton Dickinson, San Jose, CA, USA) [18]. In brief, approximately 100,000 cells were transferred to a flow tube. The cell suspension was then centrifuged at 600× g for 3 min, and the supernatant was discarded. The cells were resuspended in 100 μL PBS. Then viability dye or/and MCSP was added into the tube and mixed by gently blowing. Incubation occurred for 30 min at 4°C protected from light. After the incubation was completed, 1 mL PBS was added and mixed gently. The cell suspension was then centrifuged at 600× g for 3 min, and the supernatant was discarded. The procedure was repeated once. The cells were resuspended in 100 μL PBS and assayed with FACS Aria flow cytometer (Becton Dickinson).
2.4. Chromosome analysis
The karyotype of MM9H‐1 cells was analyzed by GTG‐banding techniques (G‐bands by trypsin using Giemsa) using standard protocols. In brief, when cells had grown to 70% confluence, the cells were fed with fresh medium containing 0.2 μg/mL colchicine (Biological Industries, Beit HaEmek, Israel), and the culture was continued for an additional 4 h. Then, cells were harvested and treated with hypotonic solution (0.075 mol/L KCl) for 20 min at 37°C. The cells were fixed in freshly prepared methanol/glacial acetic acid (3:1) solution before they were evenly spread on a pre‐cooled slide and stained with Giemsa solution (Gibco). The slides were scanned with an automated slide scanner (GSL120, Leica, Nussloch, Germany) and analyzed with Applied Imaging Software CytoVision (Applied Imaging, Santa Clara, CA, USA).
2.5. Immunofluorescence
For immunofluorescence analysis, staining was performed according to our previous report [19]. In brief, cells were seeded into 12‐well plates containing glass coverslips to create a cell climbing slice. When the cell confluence reached about 50%, cells were fixed with 4% paraformaldehyde for 20 min at 22‐25°C and permeabilized with 0.3% Triton X‐100 for 10 min. After blocking with 5% bovine serum albumin at 22‐25°C for 1 h, the cells were incubated with primary antibodies against S100A6 (1:100, 10245‐1‐AP, Proteintech, Wuhan, Hubei, China), S100 beta (1:100, 15146‐1‐AP, Proteintech), Melan‐A (1:100, 60348‐1‐Ig, Proteintech), human melanoma black 45 (HMB45) (1:1000, ab212829, Abcam, Cambridge, UK), and a triple anti‐melanoma antibody cocktail against HMB45, Melan‐A, and Tyrosinase (1:200, ab733, Abcam) overnight at 4°C. Solutions containing secondary antibodies conjugated to fluorescein isothiocyanate (FITC, Thermo Fisher Scientific) were applied at 22‐25°C for 1 h. Diamidino‐phenyl‐indole (DAPI, 1:1000, Abcam) was used for nuclear counter staining.
2.6. Immunohistochemistry
Immunohistochemistry (IHC) staining was performed according to our previous research [14, 19]. Briefly, 4‐μm‐thick formalin‐fixed, paraffin‐embedded (FFPE) tissue sections of tumor samples were cut and processed for IHC detection using antibodies against Ki67, INO80, protection of telomeres 1 (POT1), protein tyrosine phosphatase receptor type N2 (PTPRN2), pan‐cytokeratin (AE1/AE3), HMB45, Melan‐A, melanocyte‐inducing transcription factor (MITF), S100, sry‐related hmg‐box‐10 protein (SOX‐10), and Tyrosinase. Detailed information about the primary antibodies is showed in Supplementary Table S1. Slides were developed with 3‐amino‐9‐ethylcarbazole (AEC, Dako, Copenhagen, Denmark) for peroxidase reaction and counterstained with Mayer's hematoxylin.
2.7. Real‐time cell analyzer
Cell proliferation was analyzed by the real‐time cell analysis (RTCA) system (xCELLigence, ACEA Biosciences, Inc., San Diego, CA, USA). In total, 3000 A375 cells or 6000 MM9H‐1 cells (100 μL/well) were seeded into an E‐Plate. After seeding, the cells were automatically monitored, and the cell index was recorded every 15 min by the RTCA software (version 2.0).
2.8. Wound healing assay
Cell migration was analyzed using IBIDI culture inserts (IBIDI, Munich, Germany). A375 and MM9H‐1 cells were seeded into culture insert plates at a concentration of 5 × 106 cells/mL, 100 μL per well. When the cells reached 90%‐100% confluence after 24 h of incubation, the culture inserts were removed. Floating cells or debris was washed away gently by PBS and resupplied with fresh serum‐free medium. Photographs of the migration of the cells into the scratch area were taken every 24 h.
2.9. Spheroid formation assays
For spheroid formation assays, single‐cell suspensions of 500 or 4000 cells were seeded in 6‐well or 96‐well plates, ultra‐low attachment surface (Corning, New York, NY, USA). The number of spheroids in 6‐well plates was measured after about one week of incubation in a 5% CO2 incubator at 37°C. Images were captured with Zeiss microscope. The spheroids visualized with the naked eye in 96‐well plates were collected and translated into a 12‐well plate using liquid transfer gun after about one week of incubation. Frozen sections of the MM9H‐1 spheroids were counterstained with hematoxylin and eosin (HE), and the expression of melanoma markers in MM9H‐1 spheroids were detected by immunofluorescence staining using the frozen sections.
2.10. Fluorescence in situ hybridization (FISH)
FISH was performed according to our previous research [14]. CDK4, TERT and BRAF amplification tests were performed on FFPE tissue sections using commercial probes, ZytoLight SPEC CDK4/CEN12 Dual Color Probe, BRAF/CEN 7 Dual Color Probe and TERT/CEN5 Dual Color Probe (Zytovision, Bremerhaven, Germany). The experiment procedures were performed according to the manufacturer's instructions.
2.11. In vivo tumorigenesis and metastasis experiments
Three 5‐week‐old male BALB/c nude mice (Shanghai Laboratory Animal Center, Shanghai, China) were used for growing xenografts. Cells were inoculated subcutaneously into the right flank of mice at a concentration of 1 × 104 cells/μL in 100 μL or orthotopically in vestibular of the oral cavity in 20 μL. Tumor growth was measured twice weekly. The hepatic and lung metastasis models were also established in BALB/c nude mice or NOG mice (5 weeks old, Weitong Lihua Limited Company, Beijing, China) via intrasplenic or tail vein injection of 100 μl PBS containing 1 × 105 MM9H‐1 cells. When the subcutaneous tumor grown to approximately 1000 mm3 or 5 weeks after splenic or tail vein injection, the mice were euthanized by CO2 inhalation and cervical dislocation. Tumor tissues, spleens, livers and lungs were collected and fixed in 4% paraformaldehyde, and paraffin‐embedded sections were stained with HE. The expression of HMB45, Melan‐A, MITF, AE1/AE3 and Ki67 in subcutaneous tumor was examined by IHC. The corresponding antibody information is listed in Supplementary Table S1.
2.12. High‐throughput drug Screening
A library of anticancer agents was screened for their antitumor activities against MM9H‐1. Cells were seeded in 90 μL growth medium using an auto dispenser into 96‐well polystyrene tissue culture‐treated Corning plates at a density of 6000 cells/well to allow compounds to be present during exponential growth phase. After cell adhesin, 103 anticancer agents, including the investigational agents focusing on melanoma and US Food and Drug Administration (FDA)‐approved drugs, were added using a 9‐point concentration gradient ranging from 20 μmol/L to 3 nmol/L. Plates were incubated for 72 h at standard incubator conditions and covered by a stainless steel gasketed lid to prevent evaporation. Finally, 10 μL of CCK‐8 reagent were added to each well according to the manufacturer's protocol (Beyotime, Shanghai, China), and plates were incubated at 37°C for 2‐3 h. The reads were calculated using SynergyTM H1 (BioTek, Winooski, VT, USA), and compound dose‐response curves were normalized to dimethylsulfoxide (DMSO, Sigma‐Aldrich, Rehovot, Israel) and empty well controls on each plate. The list of anticancer agents is provided in Supplementary Table S2.
2.13. PDX establishment and in vivo evaluation of therapeutic agents
PDX model was established as we previously reported [14]. Fresh tissue biopsy of the donor patient was subcutaneously inoculated into BALB/c nude mice. Xenograft from the established PDX model was trimmed into about 20 mm3 fragments for subcutaneous implantation in nude mice. Xenograft‐bearing mice with an average tumor volume about 100‐200 mm3 were randomized into the control arm and the treatment arm on the basis of tumor size, tumor growth rate, and mouse body weight and were treated with vehicle and different types of inhibitors. The specific usages were as follows: vehicle (the drug solvent), palbociclib (Selleck, Shanghai, China; orally, daily) at a dose of 90 mg/kg, dabrafenib (Selleck; orally, daily) at a dose of 30 mg/kg, cisplatin (Selleck; intraperitoneal injection, once a week) at 2 mg/kg, and bortezomib (Selleck; intraperitoneal injection, twice a week) at 0.5 mg/kg. Tumor size (calculation formula: V = 0.5× length × width2) and body weight for mice were measured twice weekly, using a digital caliper and electronic scale. Tumor growth inhibition (TGI) was defined as the formula: % growth inhibition = 100 × [1 − (V t treated − V t0 treated)/(V t placebo − V t0 placebo)], where V t represents the average tumor volume at the end of study and V t0 represents the average tumor volume on the first day of treatment. Animal care and experiments were performed under the approval and supervision of the Institutional Animal Care and Use Committee of the Shanghai Jiao Tong University School of Medicine.
2.14. Whole‐genome sequencing (WGS)
WGS (Mingma Technologies Co., Ltd, Shanghai, China) was performed using genomic DNA extracted from MM9H‐1 passage 10, PDX passage 3, and the primary tumor and matched blood samples collected from patient MM108. We used a paired comparison with blood cells from the tissue donor to distinguish somatic mutations and copy number aberrations in the tumor cells. Paired‐end sequencing is performed using an Illumina NovaSeq6000 system (Illumina, San Diego, CA, USA) following the manufacturer's protocols for 2 × 150 paired‐end sequencing (Mingma Technologies Co., Ltd, Shanghai, China). The average depth was 69× in MM9H‐1, PDX, primary tumor and the blood cells. Sequence data were aligned to the UCSC human reference genome (hg19) using Burrows‐Wheeler Aligner (BWA v0.7.12‐r1044 (http://bio‐bwa.sourceforge.net/bwa.shtml), and binary files (BAM) were generated in a sorted manner. Mark duplication was performed with Sention (version 201611.02) on each BAM file. Then somatic single‐nucleotide variants (SNVs) and insertion‐deletions (InDels) were detected with Sention TNseq. Mutations in low‐complexity regions, such as tandem repeat and highly homologous regions, were filtered out. All high‐confidence mutations were then annotated with ANNOVAR (Version 2016‐02‐01, https://annovar.openbioinformatics.org/en/latest/user‐guide/download).
2.15. Classification and annotation of amplified regions detected by WGS
To quantify and characterize the architecture of amplified regions detected by WGS, AmpliconArchitect, a software which was developed to reconstruct the fine structure of focally amplified regions [20], was used. AmpliconArchitect was run on WGS data with default parameters as described in the documentation (https://github.com/virajbdeshpande/AmpliconArchitect). Only aligned reads in regions with copy number greater than 5 above the sample ploidy were used as seeds. As recently described [21], amplification regions were classified as (1) ‘circular’ or extrachromosomal DNA (ecDNA)‐like, representing extrachromosomal amplicons; (2) ‘BFB’ if they bore a signature16 of having been created by a BFB; (3) ‘rearranged’, for noncircular amplicons containing pieces of DNA (DNA segments) from different chromosomes or regions that were very far apart on chromosomes (>1 Mb); or (4) ‘linear’ for linear amplifications. OncoKB Cancer Gene List (https://www.oncokb.org/cancerGenes), which includes 1064 cancer genes from published research [22], and Sanger Cancer Gene Census was used to annotate the detected genes within the amplification regions.
2.16. RNA‐sequencing and analysis
RNA‐sequencing was performed according to our previous research [19]. RNA was isolated from snap‐frozen tumors from vehicle‐ and bortezomib‐treated mice. Total RNA was extracted using RNeasy Mini Kit (Qiagen, GmBH, Germany) following the manufacturer's instructions and checked for a RIN number to inspect RNA integrity by an Agilent Bioanalyzer 2100 (Agilent technologies, Santa Clara, CA, US). Qualified total RNA was further purified by RNAClean XP Kit (Beckman Coulter, Inc., Brea, CA, USA) and RNase‐Free DNase Set (Qiagen). The Illumina TruSeq RNA Sample Preparation Kit (Illumina) was used for library preparation. RNA‐sequencing was performed on the Illumina Hiseq 2000 platform. Raw reads were trimmed with Skewer (v0.2.2) to remove adapter sequences and then aligned to the reference human genome GRCh37/hg19 with STAR (v2.4.2a). Gene expression abundance was estimated by RSEM (1.2.29) based on the uniquely mapped reads. Differentially expressed genes was determined using negative binomial model via EdgeR by comparing vehicle‐ and bortezomib‐treated tumors, and P value was calculated using Wald test, multiple test correction was performed using B‐H procedure, with a fold change ≥ 2 and an adjusted P value < 0.05 as the cut‐off criteria. Pathway analysis on differentially expressed genes was performed using kyoto encyclopedia of genes and genomes (KEGG) pathway analysis.
2.17. Gene set enrichment analysis (GSEA)
Gene expression levels were quantified by RNA‐sequencing and subjected to GSEA for pathway enrichment analysis. Enrichment score (ES) indicates the degree to which the specified gene sets are overrepresented at the top/bottom of a ranked list of total protein‐coding genes in individual PDX. Normalized enrichment score (NES) computes density of modified genes in the dataset with random expectancies, normalized by gene numbers found in each gene cluster. False discovery rate (FDR) was calculated by comparing actual data with 1000 Monte‐Carlo simulations.
2.18. Statistical analyses
Spearman rank‐based correlation coefficients were separately calculated to demonstrate variant allele frequency (VAF) stability between a PDX and its patient origin. Antitumor activity of bortezomib, cisplatin, palbociclib and dabrafenib in PDX models was analyzed using a generalized linear mixed model of tumor volume fold changes, with random effects and treatment arm and time as covariates. Statistical analyses were done with GraphPad Prism 8 (GraphPad Software, San Diego, CA, USA). Unpaired Student's t‐tests were performed to compare differences between the two groups. P < 0.05 was considered statistically significant.
3. RESULTS
3.1. Patient background
The donor was a 67‐year‐old woman who was admitted to Shanghai Ninth People's Hospital due to black mass in gingiva of the mandibular anterior teeth with cervical lymphadenopathy on the right side in March 2020. Computed tomography (CT) revealed the presence of right mandibular gingival soft tissue thickening on the lingual side, with metastasis to levels IB and II in right cervical lymph nodes (stage T4aN1M0, Figure 1A, Supplementary Figure S1A‐B). Biopsy was performed on frozen tissue for pathological diagnosis, which was confirmed by H&E staining and IHC using melanoma‐specific markers including Melan‐A, HMB45, AE1/AE3, MITF, Tyrosinase, S100, and SOX‐10 (Figure 1B, Supplementary Figure S1C). The patient initially received three cycles of preoperative treatment using recombinant human endostatin (Endostar, Shandong Simcere Medgenn Bio‐Pharmaceutical Co., Ltd, Yantai, Shangdong, China) combined with dacarbazine (Nanjing Pharmaceutical Factory Co., Ltd, Nanjing, Jiangsu, China). The patient had tumor progression within two months after the initiation of preoperative treatment and received palliative surgery. Local recurrence and left lymph node metastasis occurred approximately one month after surgery, and the patient received four cycles of PD‐1 inhibitor immunotherapy. Again, the tumor progressed, and the patient underwent a second palliative surgery. The patient died five months after the surgery. The main course of the disease and the detailed treatment schedule are provided in Supplementary Figure S2.
FIGURE 1.
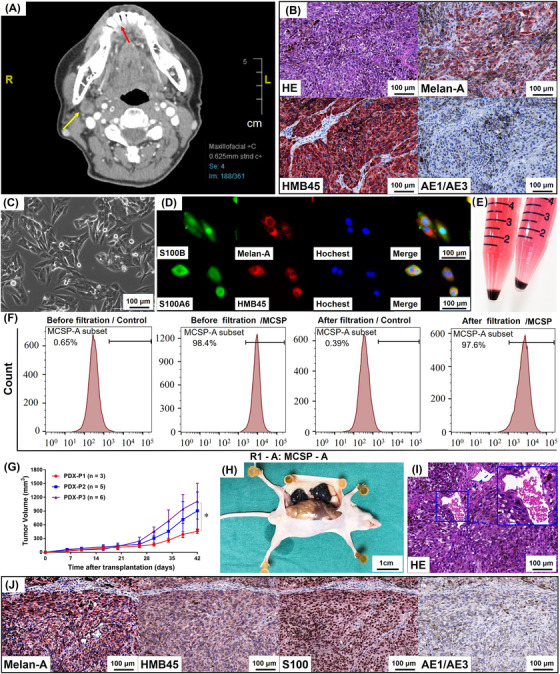
Establishment and characterization of MM9H‐1 cell line. (A) Clinical imaging of the patient. The patient was a 67‐year‐old woman who was diagnosed with mucosal melanoma at T4aN1M0 stage. CT axial view reveals a low‐density mass around the lower anterior teeth, and the boundary display is not clear. The arrows indicate the primary foci (red), the location of the cryobiopsy specimens, and the metastatic lymph node (yellow). (B) Histopathology of the patient. HE staining of the biopsy shows abundance of tumor cells with pigmentation. Immunohistochemistry analysis shows positive staining for HMB45 and Melan‐A and negative for AE1/AE3. (C) Phase contrast image of cultured mucosal melanoma MM9H‐1 cells. The cells exhibit a spindle cell‐type appearance, a typical fusiform morphology (the 5th passage). (D) MM9H‐1 cells are positive for staining of melanoma‐specific biomarkers Melan‐A, S100B HMB45, S100A6. (E) Cell suspension from the 10th‐passage cells formed a black clump at the bottom of the centrifuge tube after centrifugation. (F) Flow cytometry for the purity of MM9H‐1. MM9H‐1 cells were filtered with or without anti‐melanoma (MCSP) microbeads and then stained with or without MCSP antibody. The purity of MCSP‐positive cells is >97%. (G‐J) Establishment of PDX model. (G) Growth curves of different PDX passages, which start to increase in size at around 10 days after transplantation (P1, P2, P3 = PDX tumors in first, second and third passages). Tumor volume was analyzed with Student's t test. (H) Representative image of PDX‐P1 tumors subcutaneously grow in nude mice. (I) H&E staining of the subcutaneous PDX‐P3 tumor, with vessel formation indicated by the yellow box. (J) Immunohistochemistry analysis of PDX‐P3 shows positive staining for Melan‐A, HMB45, S100 and negative for AE1/AE3. * P < 0.05. Abbreviations: CT, computed tomography; HE, hematoxylin and eosin; HMB45, human melanoma black 45; MCSP, melanoma‐associated chondroitin sulfate proteoglycan; PDX, patient‐derived tumor xenografts
3.2. Establishment of a novel mucosal melanoma cell line (MM9H‐1) and the corresponding PDX model
The mucosal melanoma cell line MM9H‐1 was established using the biopsy tumor tissue from the above‐mentioned case (MM018), and it exhibited a spindle cell‐type appearance which was typical for malignant melanoma cell morphology (Figure 1C, Supplementary Figure S3A). Melanoma‐specific biomarkers HMB45, Melan‐A, Tyrosinase, and S100 were found to be highly expressed in MM9H‐1 cells (Figure 1D). Notably, the MM9H‐1 cell line retained its cell morphology and proliferation potential, in addition to stably expressing melanoma‐specific biomarkers, after more than 100 passages (Supplementary Figure S3A‐B). Harvesting of tumor cells from different passages by trypsinization followed by centrifugation produced a black cell pellet at the bottom of centrifuge tube (Figure 1E, Supplementary Figure S3C), indicating that the cells produced significant amounts of melanin. The purity of MM9H‐1 cells was further checked by flow cytometry, and the percentage of MCSP antigen‐positive cells was 98.4% and 97.6% before and after filtration, respectively (Figure 1F), which indicated the high purity of this cell line. We evaluated the chromosomal karyotype of MM9H‐1 cells by GTG‐banding, which revealed an abnormal human female karyotype with chromosome numbers ranging from 68 to 72. Structural aberrations were highly complicated, with frequent observation of deletions, translocations, isochromosomes, and chromosomes of unknown derivation (Supplementary Figure S3D).
In addition to cell lines that grow in vitro under non‐physiological conditions, PDX model established by direct implantation of tumor tissue growing in vivo could mostly retain the genomic and phenotypic profiles of the primary patient tumor. Consequently, PDX models had become the preferred personalized experiment tools for preclinical drug evaluation as well as development of novel treatment methods [23, 24]. To this end, we also used the biopsy tumor tissue to establish a PDX model in immunodeficient mice. Tumor growth curves showed that PDX tumors from the second and third passages (P2 and P3) exhibited relatively faster growth rates after serial transplantation (Figure 1G). The macroscopic appearance and melanoma‐specific histological/molecular features of the xenografts indicated the stable establishment of the PDX model (Figure 1H‐J). Thus, these results supported the successful establishment of a novel mucosal melanoma cell line that could retain its expression of melanoma‐specific markers during transplantation, and PDX models established with MM108 tumor tissue could also preserve the histopathological and basic genetic features of the original tumor.
3.3. Biological features of MM9H‐1 cells
Migration and sustained chronic proliferation are two major hallmarks of cancer cells, which reflect the potential for metastasis and proliferation capacity of tumor cells [25]. To evaluate the biological features of MM9H‐1 cells, we measured the proliferation ability of MM9H‐1 cells in comparison with A375 cells, a widely used cutaneous melanoma cell line in preclinical melanoma studies. The doubling time of MM9H‐1 cells was approximately 36 h, similar to that of A375 cells (Figure 2A). To evaluate the migratory capability of MM9H‐1 cells, we performed wound healing assays and analyzed the relative migration area over a 96‐hour observation period. MM9H‐1 cells exhibited relatively rapid migration compared with A375 cells (P < 0.001) (Supplementary Figure S3E‐F).
FIGURE 2.
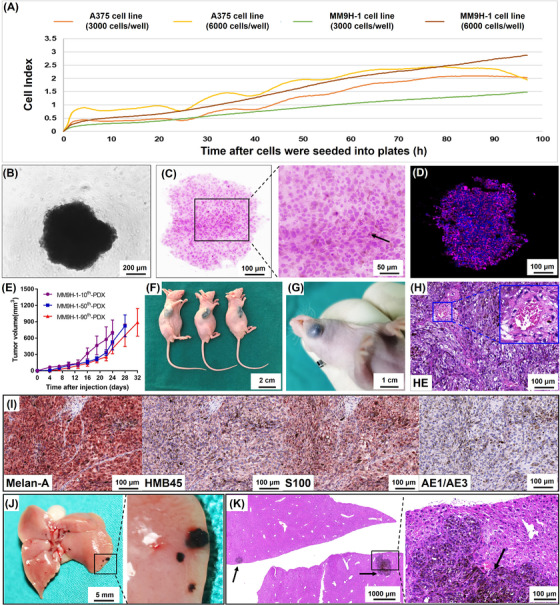
Spheroid formation and tumorigenicity of MM9H‐1 cells and establishment of mucosal melanoma PDX model. (A) Proliferation of MM9H‐1 and A375 cells was monitored by xCELLigence RTCA system. (B‐D) Sphere‐forming assay. (B) Phase contrast image of a sphere from MM9H‐1 cells in 96‐well plate. (C) HE staining shows that MM9H‐1 cells aggregated into regular spheres after 4 days of suspension culture, and the arrow indicates melanin pigmentation in cells. (D) Immunohistochemistry analysis of the sphere shows positive staining for a triple‐antibody cocktail against cytoplasmic HMB45, Melan‐A, and Tyrosinase (red). Hochest was used to stain nucleus (blue). (E‐H) Tumor formation in immunodeficient mice. (E) Tumor growth curves of the subcutaneous tumors formed by the 10th, 50th and 90th passage MM9H‐1 cells (n = 3). Tumor volume was analyzed with Student's t test. Image of tumor formation of MM9H‐1 cells in the subcutaneous (F, 10th passage cells) and orthotopic regions (G, 90th passage cells). (H) H&E staining of the MM9H‐1‐xenografted tumor in subcutaneous regions, with vessel formation indicated by the yellow box. (I) Immunohistochemistry analysis of MM9H‐1‐xenografted tumor shows positive staining for Melan‐A, HMB45, S100 and negative for AE1/AE3. (J) Typical gross appearance of liver metastasis in nude mice injected intrasplenically with MM9H‐1 cells. (K) HE staining for the liver metastasis. The black arrows indicate the metastatic foci with extensive melanin deposition. Abbreviations: HE, hematoxylin and eosin; HMB45, human melanoma black 45
The ability to form spheres was related to the capacity for self‐renewal and cancer stem cell activity of single cells, and patient‐derived spheroids had also been utilized for cancer biology investigation and drug screening [26, 27]. We thus measured the potential of this tumor cell line for spheroid formation by culturing MM9H‐1 cells from different passages in ultra‐low attachment plates. Rapid generation of homogeneous tumor spheres was observed within 2 days in 96‐well spheroid microplates (Figure 2B) and within 4 days in 6‐well plates (Supplementary Figure S4A). These spheroids were visible to the naked eye in about 4 days (Supplementary Figure S4B‐C). Histological analysis using H&E staining of the spheroids revealed a close spatial association between cells, and melanin deposition was observed in the cytoplasm of some cancer cells (Figure 2C, Supplementary Figure S4D).
Immunofluorescence showed that the cells were positive for a triple‐antibody cocktail against HMB45, Melan‐A, and Tyrosinase (Figure 2D). To confirm the tumorigenic ability of MM9H‐1 in vivo, cells from different passages were subcutaneously injected into nude mice. MM9H‐1 cells could induce tumor formation within 2 weeks in the subcutaneous environment, and the tumors featured a black and solid appearance (Figure 2E‐F, Supplementary Figure S4E‐F). Additionally, orthotopic injection of MM9H‐1 cells into the oral cavity also led to the development of orthotopic tumor in the mouse (Figure 2G). The macroscopic appearance and histological features of the xenografts (Figure 2H‐I, Supplementary Figure S4G) indicated that MM9H‐1 cells could be used to establish stable xenograft tumor models. To assess metastatic capability of MM9H‐1 cells in vivo, we performed tail vein and splenic injection experimental metastasis assay. Representative liver and lung metastatic tumors were observed in immunodeficient mice 4 weeks after splenic injection and 5 weeks after tail vein injection, and HE staining showed tumor metastatic foci in the liver and lungs with extensive melanin deposition (Figure 2J‐K, Supplementary Figure S4H‐I).
Taken together, these findings revealed that different passages of the MM9H‐1 cell line demonstrated potent proliferation and migratory ability, as well as robust tumorigenic activities both in vitro and in vivo.
3.4. MM9H‐1 was a TWT subtype melanoma cell line lacking well‐known significantly mutated genes
To further characterize the genomic basis underlying the newly established MM9H‐1 cell line and to assess the genetic fidelity of MM9H‐1 cells and the PDX model, we performed WGS on the MM108 primary tumor, PDX, MM9H‐1 cells, and the matched blood sample. We detected a total of 1139, 1295, and 1208 somatic SVs in the MM108 primary tumor, PDX, and MM9H‐1 cells, respectively (Supplementary Tables S3‐5). Circos plots for MM9H‐1 cells, PDX, and the MM108 primary tumor genomes showed closely matching patterns of somatic SVs in which clustered translocations were detected between chr5 and chr7 as well as between chr14 and chr9/chr22 in all samples, indicating the genomic fidelity of the established PDX model and MM9H‐1 cells (Figure 3A).
FIGURE 3.
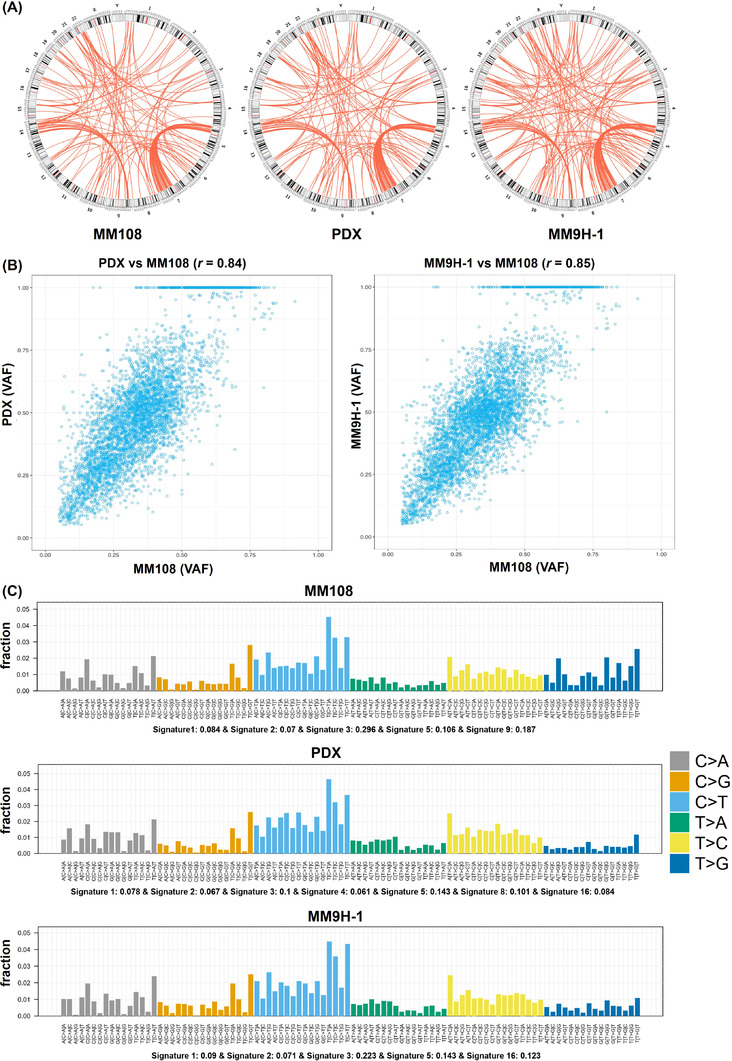
Structure variation feature and the mutation signature of the MM108 primary tumor, PDX, and MM9H‐1 cells. (A) Overview of circos plots depicts the structure variations detected in the MM108 primary tumor, PDX, and MM9H‐1 cells. (B) Scatterplot of the variant allele frequency (VAF). The read counts for each mutant and wild‐type allele are expressed as a percentage of all reads at that position and analyzed by scatterplot and correlation coefficient. Circles close to the mathematical median line represent mutations with preserved allelic frequencies in PDX or MM9H‐1 tumor pairs. (C) The contribution of the mutation signature to MM108, PDX and MM9H‐1 cells, represented by the proportion of somatic substitutions (C > A, C > G, C > T, T > A, T > C and T > G) detected in these samples. Abbreviations: PDX, patient‐derived tumor xenografts
Consistent with our previously reported data [14], analysis of SNVs across all three samples revealed a low overall mutation burden with 2.45, 2.35 and 1.95 mutations per megabase in the MM108 primary tumor tissue, PDX, and MM9H‐1 cells, respectively. The stability of the VAF indicating the frequencies of the detected mutant alleles was maintained between MM9H‐1 cells and MM108 primary tumor as well as between PDX and MM108 primary tumor, with high correlation coefficients of 0.85 and 0.84, respectively (Figure 3B). Additionally, similar patterns were observed in the mutation signature of the MM108 primary tumor, PDX, and MM9H‐1 cells, in which Mutation Signature 3 (associated with failure of DNA double‐strand break‐repair) was one of the most prevalent signatures that could contribute to the development of mucosal melanoma (Figure 3C).
Of particular interest, we did not find any well‐characterized significantly mutated genes for melanoma reported by the TCGA project (n = 42), for example, mutations of BRAF, neuroblastoma RAS viral oncogene homolog (NRAS), NF‐1, tumor protein p53 (TP53), AT‐rich interaction domain 2 (ARID2), and cyclin dependent kinase inhibitor 2A (CDKN2A), in all three samples. More recently, a harmonized molecular and clinical analysis on 1048 melanomas defined the mutational landscape of TWT melanomas and further identified 19 TWT‐specific significantly mutated genes, for example, mutations in KIT, G protein subunit alpha 11 (GNA11), G protein subunit alpha q (GNAQ), splicing factor 3b subunit 1 (SF3B1), and DEAD‐box helicase 3 X‐linked (DDX3X) [28]. Similarly, none of these 19 TWT‐specific significantly mutated genes were detected in MM108 primary tumor, PDX, and MM9H‐1 cells. The gene list of evaluated significantly mutated genes and all of the coding SNVs and InDels of these three samples are provided in Supplementary Tables S6‐7. Thus, MM9H‐1 could be defined as a TWT subtype melanoma cell line lacking well‐known significantly mutated genes in melanoma.
3.5. Amplified regions were associated highly rearranged SVs
Previously, we reported that over 50% of the mucosal melanoma patients harbored recurrent focal amplification of oncogenes CDK4, mouse double minute 2 (MDM2), and ArfGAP with GTPase domain, ankyrin repeat and PH domain 2 (AGAP2), located at 12q13‐15, which co‐occurred significantly with amplification of TERT at 5p15 [14]. In this study, significant amplified regions containing well‐characterized oncogenes were found to be located in 5p15 (TERT), 7q34 (BRAF), and 3p12 (MITF), while CDK4 amplification was not detected in the MM108 primary tumor, PDX, or MM9H‐1 cells (Figure 4A). Loss of well‐characterized tumor suppressors was found in 17q15 (TP53) and 17q11 (NF1) (Figure 4A). TERT and BRAF amplifications were validated by FISH analysis, while CDK4 amplification was not observed (Figure 4B). All CNV data of these three samples are provided in Supplementary Table S8.
FIGURE 4.
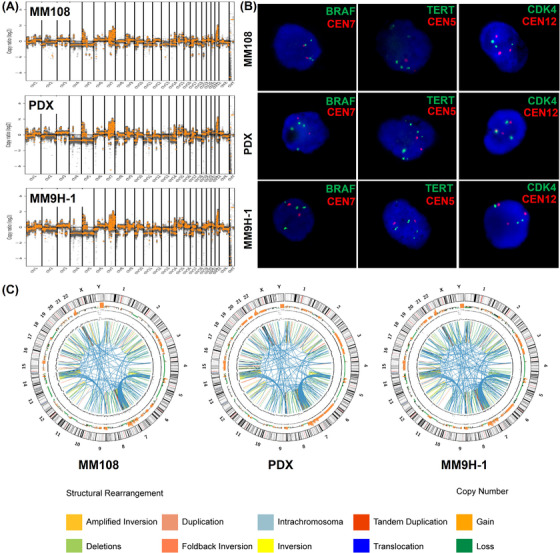
The distribution of the amplified regions and the highly rearranged regions in MM108 primary tumor, PDX and MM9H‐1 cells. (A‐B) The feature of copy number variations and FISH analysis of the amplified regions including BRAF, TERT and CDK4 amplification in MM108 primary tumor, PDX and MM9H‐1 cells. (C) Circos plot depicts the genomic location in the outer ring and chromosomal copy number in the inner ring (orange, copy gain; green, copy loss). Different structural arrangements are shown in different colors, respectively. Abbreviations: PDX, patient‐derived tumor xenografts; FISH, fluorescence in situ hybridization; BRAF, v‐raf murine sarcoma viral oncogene homolog b1; TERT, telomerase reverse transcriptase; CDK4, Cyclin‐dependent kinase 4.
Notably, we also found clusters of breakpoints, which indicate complex genomic rearrangements as defined in our previous study [14], in amplified regions harboring well‐characterized oncogenes on chromosomes 5, 7, 9 and 22 across the MM108 primary tumor, PDX and MM9H‐1 genomes (Figure 4C). Detailed illustrations of the SV and CNV chromosomal distribution patterns revealed a high number of interchromosomal translocations between chr5 and chr7 in the amplified 7q34 and 5p15 regions (Supplementary Figure S5).
3.6. Several oncogenes induced by genomic rearrangement potentially contribute to MM9H‐1 oncogenesis
Recently, the occurrence of breakage‐fusion‐bridge (BFB) mechanism and chromothripsis was detected in several types of malignancies, which appeared to be associated with large‐scale genomic expansion and rearrangement, as well as aggressive clinicopathological behaviors of cancer [29]. In agreement with this report, we found that the MM9H‐1 cell genome exhibited substantial rearrangements, which suggested the involvement of these genetic events. Therefore, we examined the chromosomal distribution of BFB and chromothripsis in MM9H‐1 cells and corresponding MM108 primary tumor and PDX. These two genomic events were found to distribute across several chromosomes, with BFB detected on chromosomes 4, 7, 9, 20, and 22, and chromothripsis detected on chromosomes 5, 7, 9, 15, and 22 (Supplementary Table S9). Although we found similarities in the distribution pattern of these events among the genomes of MM108 primary tumor, PDX, MM9H‐1 cells, with obvious enrichment on chromosomes 7 and 22, we also identified BFB and chromothripsis that were exclusive to each.
We next applied AmpliconArchitect, a tool to reconstruct the fine structure of focally amplified regions using WGS, to characterize the amplified regions in MM108 primary tumor, PDX, MM9H‐1 cells for subsequent classification as “circular” or ecDNA‐like, “BFB”, “rearranged” or “linear”. Additionally, the OncoKB Cancer Gene List was used to identify all classified amplicons that overlapped with these reported cancer genes (Supplementary Tables S10‐11).
Most of the identified amplicons were classified as rearranged (45.5%) or linear (53.0%) with significant enrichment in chromosomes 22 (62.0%) and 7 (31.6%). Specifically, although only a few amplicons were ecDNA‐like (0.6%) or BFB (0.9%), these categories contained several well‐recognized and potential oncogenes (Supplementary Table S12), including INO80, POT1, serine protease 1 (PRSS1), and PTPRZ2 in ecDNA‐like amplicons (Figure 5A) as well as BRAF and ring finger protein 185 (RNF185) in BFB amplicons (Figure 5B). In particular, well‐recognized oncogenes, including BRAF and TERT, and potential oncogenes, including PRSS1, POT1, smoothened, frizzled class receptor (SMO), thyroid stimulating hormone receptor (TSHR), BCR activator of RhoGEF and GTPase (BCR), mitogen‐activated protein kinase 1 (MAPK1), platelet derived growth factor subunit B (PDGFB) and Rac family small GTPase 2 (RAC2), were also detected in the rearranged and linear amplicons, respectively (Figure 5C‐D).
FIGURE 5.
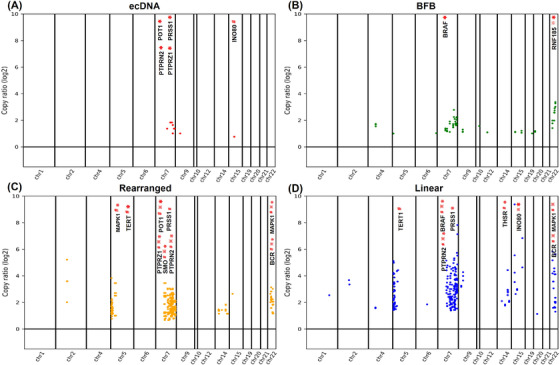
Classification and distribution of the detected amplifications across the chromosomes in MM108 primary tumor, PDX and MM9H‐1 cells. Distribution of well‐recognized or potential oncogenes classified as (A) ecDNA‐like, (B) BFB, (C) rearranged and (D) linear by AmpliconArchitect and annotated by the OncoKB Cancer Gene List in these samples. “#”, “※”, and “*” indicate the amplifications identified in the MM108 primary tumor, PDX, and MM9H‐1 cells, respectively. Abbreviations: BFB, Breakage‐Fusion‐Bridge; PDX, patient‐derived tumor xenografts
INO80, a chromatin remodeling complex, had been demonstrated to function as an oncogene that could promote DNA replication and maintain cancer cell proliferation and viability [30]. INO80 was found to govern super‐enhancer‐mediated oncogenic transcription and tumor growth in melanoma [31]. IHC analysis using serial sections of the MM108 primary tumor further confirmed that INO80, POT1 and PTPRN2 were highly expressed in MM9H‐1 cells and the corresponding patient (Supplementary Figure S6). This analysis thus revealed strong evidence of multiple BFB and chromothripsis events in MM108 tumor genome that could cause massive chromosomal rearrangements and the formation of oncogenes that could potentially drive the oncogenic processes of mucosal melanoma development in MM9H‐1 cells and the MM108‐derived PDX.
3.7. HTDS revealed the pharmacological landscape of MM9H‐1
HTDS could rapidly profile the phenotypic responses of hundreds of agents in numerous cancer cell lines, thereby providing highly informative reference for anticancer drug assessment and accelerating clinical drug translation [15]. To determine the response of MM9H‐1 cells, we assessed the effects of 103 anticancer drugs on proliferation using a 9‐point concentration gradient assay. The drug panel included many US FDA‐approved agents (i.e., most of conventional chemotherapeutics, anti‐angiogenic agents, and targeted agents that are currently used in the treatment of melanoma; https://www.cancer.gov/about‐cancer/treatment/drugs/melanoma). The half maximal inhibitory concentration (IC50) values of 103 anticancer drugs are listed in Supplementary Table S2 with a heatmap of cell viability responses presented in Figure 6A.
FIGURE 6.
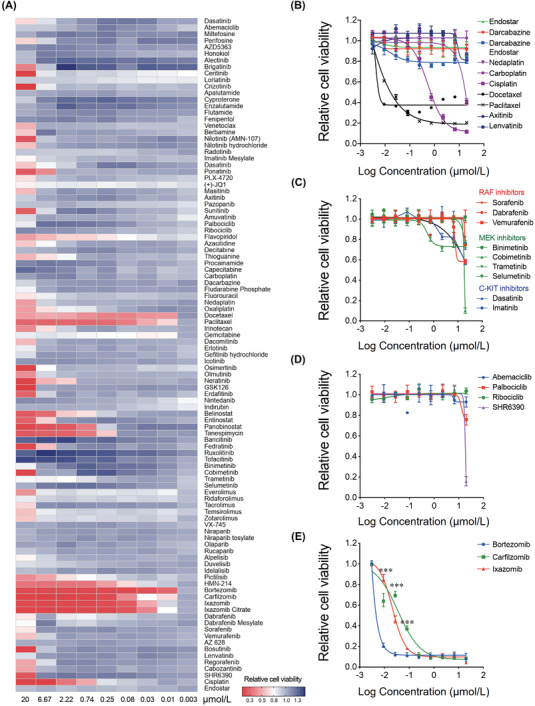
Drug candidates identified by high‐throughput drug screening in MM9H‐1 cells. (A) Heat map representation of the activities of 103 evaluated drugs in MM9H‐1 cells. Activity scores are based on cell viability. (B) Dose‐response curve of Endostar, dacarbazine, nedaplatin, carboplatin, cisplatin, docetaxel, paclitaxel, axitinib and lenvatinib, as well as the simulation results of the clinical medication, the combination of dacarbazine and Endostar in MM9H‐1 cells. (C) Dose‐response curve of the BRAF inhibitors dabrafenib, sorafenib and vemurafenib; MEK inhibitors binimetinib, cobimetinib, trametinib and selumetinib; and C‐KIT inhibitors imatinib and dasatinib in MM9H‐1 cells. (D) Dose‐response curve of CDK4/6 inhibitors palbociclib, ribociclib, abemaciclib and SHR6390 in MM9H‐1 cells. (E) Dose‐response curve of proteasome inhibitors bortezomib, carfilzomib and ixazomib in MM9H‐1 cells. The inhibition efficiency of bortezomib at different concentrations was compared with those of carfilzomib and ixazomib. Statistical analyses were performed by two‐way ANOVA. ***P < 0.001. Abbreviations: CDK4/6, Cyclin‐dependent kinase 4/6; C‐KIT, v‐kit Hardy‐Zuckerman 4 feline sarcoma viral oncogene homolog; MEK, mitogen‐activated protein kinase.
Dacarbazine is a DNA‐alkylating agent which interferes with cancer cell growth. Currently, dacarbazine is the only US FDA‐approved chemotherapeutic drug for metastatic melanoma [32]. The screening results showed that MM9H‐1 cells were insensitive to dacarbazine (IC50 > 20 μmol/L) and other cytotoxic agents such as carboplatin (IC50 > 20 μmol/L), nedaplatin (IC50 = 18.631 μmol/L), and cisplatin (IC50 = 0.6501 μmol/L). Relative sensitivity to microtubule‐damaging agents such as paclitaxel (IC50 = 0.004 μmol/L) and docetaxel (IC50 = 0.001 μmol/L) was detected in MM9H‐1 cells (Figure 6B).
Anti‐angiogenic therapy is a common therapeutic approach for advanced‐stage melanoma [33]. However, anti‐angiogenic small‐molecule kinase inhibitors such as lenvatinib (IC50 > 20 μmol/L), axitinib (IC50 > 20 μmol/L) and Endostar (IC50 > 20 μmol/L), which were potential anti‐melanoma drugs [34, 35], also failed to inhibit MM9H‐1 cell growth (Figure 6B). These results were thus consistent with the poor response to dacarbazine and Endostar in the patient MM108 who experienced tumor progression following treatment (Supplementary Figure S2). The unresponsiveness of MM9H‐1 cells to these agents further illustrated the unique biological and drug responsive features retained by this cell line.
Patients with advanced melanomas harboring BRAF (V600) mutations and C‐KIT mutations/amplification could benefit from BRAF inhibitors (e.g., vemurafenib and dabrafenib) and MEK inhibitors (e.g., trametinib), or C‐KIT inhibitors (e.g., imatinib) [36, 37, 38, 39]. The drug screening results showed that MM9H‐1 cells were insensitive to BRAF inhibitors dabrafenib (IC50 > 20 μmol/L), sorafenib (IC50 > 20 μmol/L) and vemurafenib (IC50 > 20 μmol/L), C‐KIT inhibitors imatinib (IC50 > 20 μmol/L) and dasatinib (IC50 > 20 μmol/L), as well as MEK inhibitors binimetinib (IC50 > 20 μmol/L), cobimetinib (IC50 = 12.48 μmol/L), trametinib (IC50 > 20 μmol/L) and selumetinib (IC50 > 20 μmol/L, Figure 6C). Additionally, US FDA‐approved CDK4/6 inhibitors palbociclib (IC50 > 20 μmol/L), ribociclib (IC50 > 20 μmol/L), abemaciclib (IC50 > 20 μmol/L) and the clinical investigational drug SHR6390 (IC50 = 17.9 μmol/L) showed limited drug efficiency (Figure 6D). The lack of response to these targeted inhibitors could be attributed to the genetic features of MM9H‐1 cells (lacking mutations of BRAF and C‐KIT, as well as CDK4 amplification). Notably, BRAF amplification, a reported potential mechanism of resistance to both MEK and BRAF inhibitors [40], was the only targetable driver gene found in MM9H‐1 cells.
Except for the agents mentioned above, MM9H‐1 cells were less sensitive or insensitive to most of the agents evaluated in HTDS. Strikingly, we found that proteasomal pathway inhibitors, which had been approved for the treatment of hematologic malignancies including multiple myeloma and mantle cell lymphoma [40, 41], demonstrated the highest anti‐proliferation activity across all the evaluated agents. The IC50 values of three proteasome inhibitors bortezomib, carfilzomib and ixazomib was 2 nmol/L, 13 nmol/L and 41 nmol/L (Figure 6E). Several clinical and preclinical studies [42, 43, 44] had been conducted to evaluate the therapeutic effect of bortezomib on melanoma, which suggested that proteasome inhibitors, as potential therapeutic agents for advanced melanoma, were worth investigating. Taken together, the drug efficacy observed was consistent with the theoretically expected results according to the sequencing results of MM9H‐1 cells and HTDS revealed that proteasome inhibitors, especially bortezomib, might become a potential treatment candidate in this TWT mucosal melanoma.
3.8. PDX model recapitulated MM9H‐1 drug responses in HTDS
To further verify the results of the HTDS assays conducted in MM9H‐1 cells, we then evaluated the therapeutic efficacy of selected agents by using the corresponding genetically characterized MM108‐PDX model. Besides bortezomib, which demonstrated the most significant anticancer effect in MM9H‐1 cells, several representative drugs with varied sensitivities in HTDS were also chosen for further evaluation, including dabrafenib and palbociclib, as well as cisplatin, a commonly used chemotherapeutic drug for advanced mucosal melanoma according to guidelines for mucosal melanoma. Additionally, to avoid potential bias caused by different routes of drug administration, we conducted the drug evaluation experiment using two PDX groups: one group received intraperitoneal injection of bortezomib, cisplatin, and vehicle; the other group received oral gavage of dabrafenib, palbociclib and vehicle.
Consistent with the HTDS results in vitro, bortezomib profoundly suppressed the growth of MM108‐PDX tumors when compared with cisplatin and vehicle (TGI = 79.3%, P < 0.001), and cisplatin showed a modest anti‐tumor effect (TGI = 43.8%, P = 0.001) (Figure 7A‐B). As expected, both dabrafenib and palbociclib exerted no appreciable therapeutic effect when compared with vehicle (Figure 7C‐D). Additionally, intraperitoneal injection of bortezomib considerably reduced the tumor weight as compared with vehicle (P < 0.001, Figure 7E), while no significant difference was observed when targeted drugs were administered through oral gavage (Figure 7E). Drug toxicities of all the evaluated agents were all well tolerated (Supplementary Figure S7A‐B). We then used Ki‐67 staining to assess the proliferative status of the PDX tumors after the treatment of bortezomib. Bortezomib resulted in a remarkable reduction in tumor proliferation rate than vehicle, while no significant difference was observed between palbociclib, dabrafenib, and vehicle groups (Figure 7F, Supplementary Figure S7C‐E). To explore the potential mechanisms of the potent inhibitory effects of bortezomib, we also performed RNA‐sequencing in PDXs treated with bortezomib or vehicle. KEGG enrichment analysis was then performed on these differentially expressed genes, and the bubble plot of the top enriched KEGG terms demonstrated the downregulation of a series of cancer‐related pathways (e.g., the Hippo and MAPK signaling pathways) in bortezomib‐treated tumors, which were further validated by GSEA analysis and IHC (Supplementary Figures S8‐S9). Thus, the in vivo studies using the corresponding PDX models further validated the HTDS results conducted in vitro and indicated the promising therapeutic effect of bortezomib in MM9H‐1 cells.
FIGURE 7.
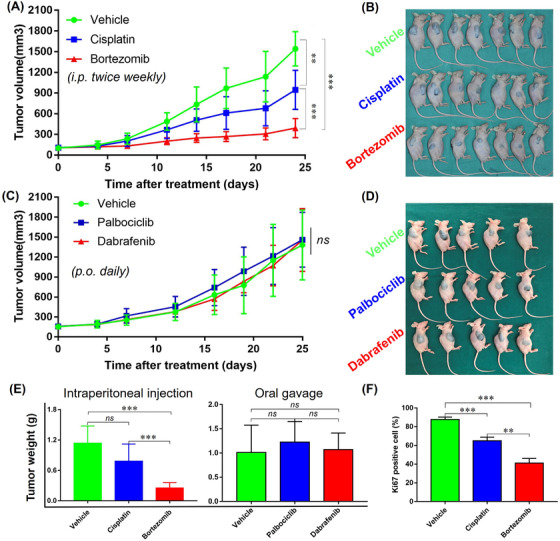
in vivo validations of the most effective agents identified using HTDS in PDX models. (A‐B) Therapeutic efficacy of cisplatin and bortezomib in MM108‐PDX (n = 7). (C‐D) Therapeutic efficacy of palbociclib and dabrafenib in MM108‐PDX (n = 5). (E) The weight of the tumors from different groups. (F) Immunoblot analysis of Ki67 expression in tumors after the last administration with vehicle, cisplatin and bortezomib. Ki‐67 staining scores are presented as the mean ± SEM. Tumor volume and tumor weight were analyzed with Student's t test. * P < 0.05, ** P < 0.01, *** P < 0.001; ns, not significant. Abbreviations: PDX, patient‐derived tumor xenografts; HTDS, high‐throughput drug screening; SEM, structural equation modeling
4. DISCUSSION
Mucosal melanoma is one of the most serious challenges for new melanoma therapies, and limited availability of representative preclinical models has hampered both basic and translational research of this subtype of melanoma [45]. In this study, we established and characterized a TWT mucosal melanoma patient‐derived cell line and PDX. This cell line was identified as a melanoma cell line illustrating (i) stable and consistent expression of melanoma‐specific biomarkers like HMB45, Melan‐A and S100 after over 100 passages both in vitro and in vivo; (ii) stable and robust proliferative property/ability, evidenced by cell proliferation assay using RTCA and Ki67 staining in the xenograft established from MM9H‐1 cells; (iii) tumorigenic property, evidenced by sphere formation capacity and orthodontic or subcutaneous xenograft formation capacity using cancer cells from different passages. Whole‐genome analysis defined MM9H‐1 as a TWT melanoma cell line lacking well‐recognized “driver mutations”, and large‐scale genomic rearrangement events like BFB and chromothripsis leading to the activation of various oncogenes might be responsible for the selective advantage of MM9H‐1 cells. Additionally, MM9H‐1 cells remained the ability of melanin production after 50 passages, which could be associated with the amplification and the expression of MITF, a gene stimulating the melanin synthesis [46].
Previously, we demonstrated that ponatinib was more potent than imatinib against cells bearing KIT mutations using the second or third passages of PDCs from mucosal melanoma PDX models harboring KIT mutations [19]. It is noteworthy that PDCs isolated from the tumor tissues or PDX does not indicate the successful establishment of a mucosal melanoma cell line. Most early passages of PDCs (within 3‐5 generations) could meet experimental needs in drug evaluation, which does not require a large amount of relatively homogeneous tumor cells. However, most PDCs showed growth arrest or were gradually replaced by fibroblasts during serial passage. Our group have been tried to establish an immortalized mucosal melanoma cell line from dozens of mucosal melanoma tumor samples and/or established PDXs using different interventions, such as adjusting the components of the culture medium, using flow cytometry and cell sorting, translating hTERT, the commonly used gene for immortalization, into cancer cells. However, the results were not ideal, and MM9H‐1 cells were the only PDCs presented stable proliferation ability in standard growth media with FBS, such as DMEM, RPMI‐1640 and DMEM/F12, and even without epidermal growth factor (EGF), insulin and B27 after over 100 passages.
Cutaneous melanoma is currently one of the best genetically characterized tumors with well‐characterized molecular subtypes, and the TWT subtype was defined as a subgroup of melanoma characterized by a lack of mutually exclusive mutations of BRAF, N/H/K‐RAS, or NF1 [28]. Recently, our research defined the genomic landscape of a comparatively large group of mucosal melanomas, mostly originated from the head and neck region, and revealed the recurrent CDK4 amplification and oncogenic signature resembling BFB or chromothripsis in over 50% mucosal melanoma samples [14]. Of particular interest, we did not find any well‐recognized significantly mutated genes of melanoma or CDK4 amplification that occurs in over 50% mucosal melanoma, as reported in the present study in MM9H‐1 cells and the corresponding PDX and MM108 primary tumor. Additionally, other significantly mutated genes recurrently observed in mucosal melanoma, such as KIT, GNAQ, SF3B1 and TERT, were also not detected in these models. This result suggested that MM108 is a distinct subtype of TWT melanoma that lacks KIT mutation and CDK4 amplification, while genetic events like BFB and chromothripsis, which cause large‐scale rearrangement and amplification of potential oncogenes, contribute to the development of MM9H‐1 cells.
An inverse relationship in oncogenic signature between the number of refined CNVs and the number of somatic mutations was recently identified, which subdivides cancer into two major subtypes: mutations (M class) or copy number changes (C class) [47]. Furthermore, the distinction is the clearest at the extremes of genomic instability. According to the genomic data we revealed, the MM9H‐1 cell line established in the present study can be readily assigned to C class cancer, characterized by TP53 inactivation (caused by TP53 loss) and multiple recurrent chromosomal alterations involving BRAF, TRET and MITF. Thus, by using the AmpliconArchitect model, gains detected in MM9H‐1 cells were further classified, and we screened a panel of potential oncogenes including INO80, POT1 and PTPRN2. This result strongly suggested that MM9H‐1 represents a distinct subtype of melanoma and MM9H‐1 cells harboring chromothripsis might progress through different genetic evolutions.
The HTDS results showed that MM9H‐1 cells were insensitive to multiple antitumor compounds, including traditional chemotherapeutic drugs and some targeted agents recommended by the NCCN guidelines or currently being investigated in clinical trials for the treatment of melanoma [48]. Specifically, cytotoxic agents, such as dacarbazine, carboplatin and nedaplatin, and anti‐angiogenic agents, such as lenvatinib, albocicli, axitinib and Endostar, failed to inhibit the growth of MM9H‐1 cells [34, 35]. This was generally in line with the patient MM108 who had tumor progression when treated with dacarbazine and Endostar (Supplementary Figure S2). Unsurprisingly, MM9H‐1 cells were also insensitive to BRAF inhibitors (e.g., vemurafenib and dabrafenib), C‐KIT inhibitors (e.g., imatinib), MEK inhibitors (e.g., binimetinib and trametinib), as well as CDK4 inhibitors (e.g., palbociclib, ribociclib, abemaciclib and the clinical investigational drug SHR6390), which could be attributed to the genomic feature of MM9H‐1, a TWT melanoma cell line lacking active mutations of BRAF and C‐KIT or amplification of CDK4.
Bortezomib is a proteasome inhibitor whose anticancer action has been reported to be mediated mainly through the inhibition of the NF‐κB pathway [49]. Bortezomib demonstrated antitumor effects on cutaneous melanoma in vitro and in vivo, as well as in clinical trials [42, 43, 44]. However, to our knowledge, no such study was reported in mucosal melanoma. In our present study, bortezomib treatment conferred remarkable tumor‐growth inhibitory effects both in HTDS assay and the MM108‐PDX model in vivo. Considering that the ubiquitin‐proteasome pathway plays an important role in various physiological and pathophysiological processes [41, 49], our results also indicated that bortezomib could have several putative mechanisms of action by affecting more than one pathway or targets in mucosal melanoma. Further functional studies and mucosal melanoma samples are needed to dissect the mechanisms of action of this compound and develop biomarkers that can help mucosal melanoma classification.
However, the present study also contained some unavoidable limitations that can impede on the generalization of the results. First, only one patient‐derived mucosal melanoma cell line was established and defined as an immortalized mucosal melanoma cell line (MM9H‐1) after systematic characterization. Thus, unlike the mucosal melanoma PDX cohort (n = 24) we previously used for drug evaluation and predictive biomarker discovery [14], MM9H‐1, annotated as a TWT melanoma molecular subtype, cannot capture the complex genetic landscape of mucosal melanoma. Second, although large‐scale genomic rearrangement events like BFB and chromothripsis have been identified in MM9H‐1 cells using WGS, the specific master regulators of the immortalization of MM9H‐1 cells needs to be further explored, for example, the oncogenic functions of the amplified genes such as INO80, POT1 and PTPRN2. Finally, we illustrated the outstanding antitumor ability of bortezomib in both MM9H‐1 cells and matched PDX models. Further studies are needed to clarify the molecular basis of the responsiveness of this agent, and a population‐based “1 × 1 × 1 PDX trial” [24] may further imply the precision application of proteasome inhibitors in specific subgroups of mucosal melanoma patients.
5. CONCLUSIONS
In this study, we established and characterized a new mucosal melanoma cell line, MM9H‐1, and the genomic analysis defined this cell line as a TWT melanoma subtype lacking well‐characterized “driver mutations” with a unique oncogenic feature. Moreover, preclinical drug screening analysis suggested that MM9H‐1 cells could represent a valuable experimental model to investigate the oncogenesis of mucosal melanoma and to discover novel antitumor treatment options.
DECLARATIONS
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
CONSENT FOR PUBLICATION
Not applicable.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Animal care and experiments were performed under the approval and supervision of the Institutional Animal Care and Use Committee of the Shanghai Jiao Tong University School of Medicine. The collection and use of these mucosal melanoma specimens for this experimental study were approved by The Ethics Committee of Shanghai Ninth Peoples Hospital, Shanghai Jiao Tong University School of Medicine (Shanghai, China) and the patients’ consent were obtained. We followed the tenets of the Declaration of Helsinki, in accordance with recommendations approved by the Institutional Ethics Committee.
FUNDING
This study was supported by the National Natural Science Foundation of China (grant No. 82002862), Young Elite Scientist Sponsorship Program by the China Association for Science and Technology (Grant No. 2019QNRC001), Shanghai Clinical Research Center for Oral Diseases (19MC1910600), CAMS Innovation Fund for Medical Sciences (2019‐I2M‐5‐037), Shanghai Municipal Key Clinical Specialty (shslczdzk01601) and Emerging Frontier Technology Joint Research Project (SHDC12018104).
AUTHOR CONTRIBUTIONS
CJS and ZYG performed cell line establishment and characterization of their growth properties. CJS, SMX, YH, CWL contributed to establishment of PDXs and carried out animal model experiments. RZ, JYL, and CJS performed WGS analysis RZ, YTW and JW performed AmpliconArchitect analysis. RZ and CJS performed transcriptome analysis. CJS, ZYG, GXR, ZYZ and RZ contributed to the study concepts and design, and critically revised the manuscript RZ, CJS wrote the paper HYJ, GYZ, and GXR provided samples and performed the clinical information collection. LZW and JL performed pathological examination of specimens. All these works were planned and supervised by ZYZ, RZ and GXR. All authors have read and approved the final manuscript.
Supporting information
Supporting information
ACKNOWLEDGEMENTS
The authors greatly thank Jiawei Wang and Dongjie Deng (from Mingma Technologies Co., Ltd, Shanghai, China) for assistance with bioinformatics analyses. The authors thank Junjie Lu (from the animal lab of Shanghai Ninth People's Hospital of Shanghai Jiao Tong University School of Medicine) for animal studies. The authors would like to thank Cheng Huang (from Shanghai Ninth People's Hospital of Shanghai Jiao Tong University School of Medicine) for statistical analyses.
Shi C, Gu Z, Xu S, Ju H, Wu Y, Han Y, et al. Candidate therapeutic agents in a newly established triple wild‐type mucosal melanoma cell line. Cancer Commun. 2022;42:627–647. 10.1002/cac2.12315
Contributor Information
Guoxin Ren, Email: renguoxincn@sina.com.
Zhiyuan Zhang, Email: Zhiyuan.Zhang@sh9hospital.org.cn.
Rong Zhou, Email: rongzhou@shsmu.edu.cn.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. McLaughlin CC, Wu XC, Jemal A, Martin HJ, Roche LM, Chen VW. Incidence of noncutaneous melanomas in the U.S. Cancer. 2005;103(5):1000‐7. [DOI] [PubMed] [Google Scholar]
- 2. Yde SS, Sjoegren P, Heje M, Stolle LB. Mucosal Melanoma: a Literature Review. Curr Oncol Rep. 2018;20(3):28. [DOI] [PubMed] [Google Scholar]
- 3. Chi Z, Li S, Sheng X, Si L, Cui C, Han M, et al. Clinical presentation, histology, and prognoses of malignant melanoma in ethnic Chinese: a study of 522 consecutive cases. BMC Cancer. 2011;11:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Klemen ND, Wang M, Rubinstein JC, Olino K, Clune J, Ariyan S, et al. Survival after checkpoint inhibitors for metastatic acral, mucosal and uveal melanoma. J Immunother Cancer. 2020;8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lian B, Cui CL, Zhou L, Song X, Zhang XS, Wu D, et al. The natural history and patterns of metastases from mucosal melanoma: an analysis of 706 prospectively‐followed patients. Ann Oncol. 2017;28(4):868‐73. [DOI] [PubMed] [Google Scholar]
- 6. Marangoni E, Poupon MF. Patient‐derived tumour xenografts as models for breast cancer drug development. Curr Opin Oncol. 2014;26(6):556‐61. [DOI] [PubMed] [Google Scholar]
- 7. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7‐30. [DOI] [PubMed] [Google Scholar]
- 8. Genomic Classification of Cutaneous Melanoma. Cell. 2015;161(7):1681‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hayward NK, Wilmott JS, Waddell N, Johansson PA, Field MA, Nones K, et al. Whole‐genome landscapes of major melanoma subtypes. Nature. 2017;545(7653):175‐80. [DOI] [PubMed] [Google Scholar]
- 10. Meng D, Carvajal RD. KIT as an Oncogenic Driver in Melanoma: An Update on Clinical Development. Am J Clin Dermatol. 2019;20(3):315‐23. [DOI] [PubMed] [Google Scholar]
- 11. Guo J, Si L, Kong Y, Flaherty KT, Xu X, Zhu Y, et al. Phase II, open‐label, single‐arm trial of imatinib mesylate in patients with metastatic melanoma harboring c‐Kit mutation or amplification. J Clin Oncol. 2011;29(21):2904‐9. [DOI] [PubMed] [Google Scholar]
- 12. Beadling C, Jacobson‐Dunlop E, Hodi FS, Le C, Warrick A, Patterson J, et al. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res. 2008;14(21):6821‐8. [DOI] [PubMed] [Google Scholar]
- 13. Newell F, Kong Y, Wilmott JS, Johansson PA, Ferguson PM, Cui C, et al. Whole‐genome landscape of mucosal melanoma reveals diverse drivers and therapeutic targets. Nat Commun. 2019;10(1):3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhou R, Shi C, Tao W, Li J, Wu J, Han Y, et al. Analysis of Mucosal Melanoma Whole‐Genome Landscapes Reveals Clinically Relevant Genomic Aberrations. Clin Cancer Res. 2019;25(12):3548‐60. [DOI] [PubMed] [Google Scholar]
- 15. Held MA, Langdon CG, Platt JT, Graham‐Steed T, Liu Z, Chakraborty A, et al. Genotype‐selective combination therapies for melanoma identified by high‐throughput drug screening. Cancer Discov. 2013;3(1):52‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang C, Cao Y, Yang C, Bernards R, Qin W. Exploring liver cancer biology through functional genetic screens. Nat Rev Gastroenterol Hepatol. 2021;18(10):690‐704. [DOI] [PubMed] [Google Scholar]
- 17. Williams SP, McDermott U. The Pursuit of Therapeutic Biomarkers with High‐Throughput Cancer Cell Drug Screens. Cell Chem Biol. 2017;24(9):1066‐74. [DOI] [PubMed] [Google Scholar]
- 18. Griss J, Bauer W, Wagner C, Simon M, Chen M, Grabmeier‐Pfistershammer K, et al. B cells sustain inflammation and predict response to immune checkpoint blockade in human melanoma. Nat Commun. 2019;10(1):4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Han Y, Gu Z, Wu J, Huang X, Zhou R, Shi C, et al. Repurposing Ponatinib as a Potent Agent against KIT Mutant Melanomas. Theranostics. 2019;9(7):1952‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Deshpande V, Luebeck J, Nguyen ND, Bakhtiari M, Turner KM, Schwab R, et al. Exploring the landscape of focal amplifications in cancer using AmpliconArchitect. Nat Commun. 2019;10(1):392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim H, Nguyen NP, Turner K, Wu S, Gujar AD, Luebeck J, et al. Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers. Nat Genet. 2020;52(9):891‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chakravarty D, Gao J, Phillips SM, Kundra R, Zhang H, Wang J, et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis Oncol. 2017;2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Siolas D, Hannon GJ. Patient‐derived tumor xenografts: transforming clinical samples into mouse models. Cancer Res. 2013;73(17):5315‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, et al. High‐throughput screening using patient‐derived tumor xenografts to predict clinical trial drug response. Nat Med. 2015;21(11):1318‐25. [DOI] [PubMed] [Google Scholar]
- 25. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646‐74. [DOI] [PubMed] [Google Scholar]
- 26. Shiroki T, Yokoyama M, Tanuma N, Maejima R, Tamai K, Yamaguchi K, et al. Enhanced expression of the M2 isoform of pyruvate kinase is involved in gastric cancer development by regulating cancer‐specific metabolism. Cancer Sci. 2017;108(5):931‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gabriel J, Brennan D, Elisseeff JH, Beachley V. Microarray Embedding/Sectioning for Parallel Analysis of 3D Cell Spheroids. Sci Rep. 2019;9(1):16287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Conway JR, Dietlein F, Taylor‐Weiner A, AlDubayan S, Vokes N, Keenan T, et al. Integrated molecular drivers coordinate biological and clinical states in melanoma. Nat Genet. 2020;52(12):1373‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cheng C, Zhou Y, Li H, Xiong T, Li S, Bi Y, et al. Whole‐Genome Sequencing Reveals Diverse Models of Structural Variations in Esophageal Squamous Cell Carcinoma. Am J Hum Genet. 2016;98(2):256‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Morrison AJ, Shen X. Chromatin remodelling beyond transcription: the INO80 and SWR1 complexes. Nat Rev Mol Cell Biol. 2009;10(6):373‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhou B, Wang L, Zhang S, Bennett BD, He F, Zhang Y, et al. INO80 governs superenhancer‐mediated oncogenic transcription and tumor growth in melanoma. Genes Dev. 2016;30(12):1440‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yi JH, Yi SY, Lee HR, Lee SI, Lim DH, Kim JH, et al. Dacarbazine‐based chemotherapy as first‐line treatment in noncutaneous metastatic melanoma: multicenter, retrospective analysis in Asia. Melanoma Res. 2011;21(3):223‐7. [DOI] [PubMed] [Google Scholar]
- 33. Cho WC, Jour G, Aung PP. Role of angiogenesis in melanoma progression: Update on key angiogenic mechanisms and other associated components. Semin Cancer Biol. 2019;59:175‐86. [DOI] [PubMed] [Google Scholar]
- 34. Suyama K, Iwase H. Lenvatinib: A Promising Molecular Targeted Agent for Multiple Cancers. Cancer Control. 2018;25(1):1073274818789361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fruehauf J, Lutzky J, McDermott D, Brown CK, Meric JB, Rosbrook B, et al. Multicenter, phase II study of axitinib, a selective second‐generation inhibitor of vascular endothelial growth factor receptors 1, 2, and 3, in patients with metastatic melanoma. Clin Cancer Res. 2011;17(23):7462‐9. [DOI] [PubMed] [Google Scholar]
- 36. Maio M, Lewis K, Demidov L, Mandalà M, Bondarenko I, Ascierto PA, et al. Adjuvant vemurafenib in resected, BRAF(V600) mutation‐positive melanoma (BRIM8): a randomised, double‐blind, placebo‐controlled, multicentre, phase 3 trial. Lancet Oncol. 2018;19(4):510‐20. [DOI] [PubMed] [Google Scholar]
- 37. Long GV, Hauschild A, Santinami M, Atkinson V, Mandalà M, Chiarion‐Sileni V, et al. Adjuvant Dabrafenib plus Trametinib in Stage III BRAF‐Mutated Melanoma. N Engl J Med. 2017;377(19):1813‐23. [DOI] [PubMed] [Google Scholar]
- 38. Hodi FS, Corless CL, Giobbie‐Hurder A, Fletcher JA, Zhu M, Marino‐Enriquez A, et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun‐damaged skin. J Clin Oncol. 2013;31(26):3182‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Amaria RN, Menzies AM, Burton EM, Scolyer RA, Tetzlaff MT, Antdbacka R, et al. Neoadjuvant systemic therapy in melanoma: recommendations of the International Neoadjuvant Melanoma Consortium. Lancet Oncol. 2019;20(7):e378‐e89. [DOI] [PubMed] [Google Scholar]
- 40. Corcoran RB, Dias‐Santagata D, Bergethon K, Iafrate AJ, Settleman J, Engelman JA. BRAF gene amplification can promote acquired resistance to MEK inhibitors in cancer cells harboring the BRAF V600E mutation. Sci Signal. 2010;3(149):ra84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Palumbo A, Chanan‐Khan A, Weisel K, Nooka AK, Masszi T, Beksac M, et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. N Engl J Med. 2016;375(8):754‐66. [DOI] [PubMed] [Google Scholar]
- 42. Lee MS, Lim SH, Yu AR, Hwang CY, Kang I, Yeo EJ. Carfilzomib in Combination with Bortezomib Enhances Apoptotic Cell Death in B16‐F1 Melanoma Cells. Biology (Basel). 2021;10(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Amiri KI, Horton LW, LaFleur BJ, Sosman JA, Richmond A. Augmenting chemosensitivity of malignant melanoma tumors via proteasome inhibition: implication for bortezomib (VELCADE, PS‐341) as a therapeutic agent for malignant melanoma. Cancer Res. 2004;64(14):4912‐8. [DOI] [PubMed] [Google Scholar]
- 44. Markowitz J, Luedke EA, Grignol VP, Hade EM, Paul BK, Mundy‐Bosse BL, et al. A phase I trial of bortezomib and interferon‐α‐2b in metastatic melanoma. J Immunother. 2014;37(1):55‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Patton EE, Mueller KL, Adams DJ, Anandasabapathy N, Aplin AE, Bertolotto C, et al. Melanoma models for the next generation of therapies. Cancer Cell. 2021;39(5):610‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hwang YS, Oh SW, Park SH, Lee J, Yoo JA, Kwon K, et al. Melanogenic Effects of Maclurin Are Mediated through the Activation of cAMP/PKA/CREB and p38 MAPK/CREB Signaling Pathways. Oxid Med Cell Longev. 2019;2019:9827519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013;45(10):1127‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Seth R, Messersmith H, Kaur V, Kirkwood JM, Kudchadkar R, McQuade JL, et al. Systemic Therapy for Melanoma: ASCO Guideline. J Clin Oncol. 2020:Jco2000198. [Google Scholar]
- 49. Narayanan S, Cai CY, Assaraf YG, Guo HQ, Cui Q, Wei L, et al. Targeting the ubiquitin‐proteasome pathway to overcome anti‐cancer drug resistance. Drug Resist Updat. 2020;48:100663. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.