

Abbreviations
- AA
arachidonic acid
- ACSL4
acyl coenzyme A synthetase long chain family member 4
- AdA
adrenic acid
- CoA
coenzyme A
- FIN
ferroptosis inducer
- ICI
immune checkpoint inhibitor
- IFNγ
interferon gamma
- IRF1
interferon regulatory factor 1
- KEAP1
kelch‐like ECH‐associated protein 1
- LPCAT3
lysophosphatidylcholine acyltransferase 3
- PCBP1
poly(RC) binding protein 1
- PKC
protein kinase C
- PUFA‐PL
polyunsaturated‐fatty‐acid‐containing phospholipid
- ROS
reactive oxygen species
- STAT1
signal transducer and activator of transcription 1
- VB
glutathione peroxidase 4
Ferroptosis is an iron‐dependent form of regulated cell death that results from oxidative damages of membrane phospholipids, and is mechanistically and morphologically unique compared to other cell death modalities, such as apoptosis and necroptosis [1]. Excessive ferroptosis is indicative of many pathological conditions, including cardiovascular diseases, neurodegenerative diseases, and acute organ injury, whereas ferroptosis impairment has been shown to fuel tumor progression and metastasis; therefore, targeting ferroptosis represents a promising strategy for treating these diseases [2, 3, 4, 5, 6, 7, 8, 9, 10]. Ferroptosis is triggered by a lethal accumulation of lipid peroxides on the cell membrane. Cells have evolved effective antioxidant defense systems to counteract membrane damages caused by lipid peroxidation and escape from their ferroptotic fate. The most potent defense system is powered by glutathione peroxidase 4 (GPX4), a peroxidase that requires glutathione as its cofactor to quench the otherwise toxic lipid peroxides [11]. This ferroptosis defense system can be overridden by ferroptosis inducers (FINs) that inactivate GPX4 or deplete glutathione pools in cells; consequently, ferroptosis defense systems collapse and ferroptosis ensues [12].
Polyunsaturated‐fatty‐acid‐containing phospholipids (PUFA‐PLs), particularly arachidonic acid (AA)‐ and adrenic acid (AdA)‐containing PLs, are critical for triggering lipid peroxidation [12]. To be incorporated into PLs, AAs and AdAs are first catalyzed by acyl‐coenzyme A synthetase long‐chain family member 4 (ACSL4) to generate their acyl‐coenzyme A (CoA) derivatives (AA‐CoA and AdA‐CoA) [13]. These PUFA‐CoAs are further integrated into PLs to form PUFA‐PLs by lysophosphatidylcholine acyltransferase 3 (LPCAT3) and other enzymes [14]. Due to their unstable double bonds, PUFAs are especially vulnerable to peroxidation within reactive oxygen species (ROS)‐ and iron‐rich cellular environments, driving lipid peroxidation chain reactions in PUFA‐PLs in the cellular membrane through the Fenton reaction and other enzymes, and ultimately resulting in membrane rupture and ferroptotic cell death if left unchecked by ferroptosis defense systems [12]. However, the sensing and amplification mechanisms of lipid peroxidation remain poorly understood, and whether or how ACSL4 is activated in ferroptosis remains elusive. In fact, ACSL4‐mediated PUFA‐PL synthesis is generally believed to operate in a passive way to constitutively provide substrates for lipid peroxidation.
Protein kinase C (PKC) constitutes a family of protein kinases, including eight PKC isozymes (namely PKCα, PKCβΙ, PKCβΙΙ, PKCγ, PKCδ, PKCɛ, PKCθ and PKCη), which control the activity of downstream proteins by phosphorylating their serine/threonine residues [15]. PKCs play important roles in a wide variety of cellular processes, such as cell proliferation, cell survival/death, and oxidative stress responses [15]. Notably, PKCs have been shown to promote ROS generation [15, 16]. The increased levels of ROS can further amplify PKC signaling, thus forming a positive feedback loop [17]. Given that ferroptosis is a form of oxidative stress‐induced cell death, one interesting question is whether PKCs are also engaged in lipid peroxidation and ferroptosis. A recent study by Zhang et al. [18] showed that lipid peroxidation activates PKCβII; subsequently, the activated PKCβII further amplifies lipid peroxidation by phosphorylating and activating ACSL4, constituting a lipid peroxidation‐PKCβII‐ACSL4 positive feedback loop for ferroptosis execution.
The authors set out to identify novel ferroptosis regulatory genes by using CRISPR‐ Cas9 screening, through which they uncovered several known ferroptosis regulators, such as ACSL4, GPX4, Kelch‐like ECH‐associated protein 1 (KEAP1), and Poly(RC) Binding Protein 1 (PCBP1), thereby validating the robustness of their CRISPR screens [18]. Of note, PKCβ was identified as a top suppressor hit (whose deficiency suppresses ferroptosis and whose guide RNAs were enriched upon FIN treatment) with previously unknown function in ferroptosis. The authors also showed that PKC inhibitors exerted the strongest inhibitory effect on ferroptosis among 644 compounds from a kinase inhibitor library [18]. Furthermore, they excluded the potential involvement of other PKC isoforms, such as PKCα, PKCγ, PKCδ and PKCζ, in regulating ferroptosis, and found that only PKCβII isoform contributed directly to the susceptibility of cancer cells to ferroptosis: cancer cell lines with high levels of PKCβII were generally more sensitive to ferroptosis than those with low levels of PKCβII, and PKCβII deficiency resulted in resistance to FIN‐induced lipid peroxidation and ferroptosis [18]. Interestingly, Zhang et al. [18] showed that FINs could induce PKCβII activation by promoting its phosphorylation and membrane localization, and lipophilic radical‐trapping antioxidants completely inhibited PKCβII activation, indicating that lipid peroxidation is vital for PKCβII activation.
These findings raised an intriguing possibility that PKCβII might act as a lipid peroxidation sensor and prompted the question of how PKCβII is engaged in lipid peroxidation and ferroptosis. As noted above, the ferrous‐catalyzed Fenton reaction is required for lipid peroxidation and ferroptosis [12]. In addition, inactivation of GPX4‐mediated ferroptosis defense promotes an overload of lipid peroxides and ferroptosis [11, 12]. Zhang et al. [18] therefore tested the potential connection of PKCβII to iron and GPX4, but found that PKCβII inactivation did not affect GPX4 levels or cellular ferrous levels. Instead, they showed that PKCβII was capable of interacting with ACSL4 and directly phosphorylating ACSL4 at Thr328. ACSL4 Thr328 phosphorylation was increased during ferroptotic stress, which could be inhibited by PKCβII deletion or inactivation; likewise, blocking PKCβII activation by scavenging lipid peroxides restrained FIN‐induced phosphorylation of ACSL4 at Thr328 [18]. Mechanistically, the authors revealed that PKCβII‐mediated ACSL4 phosphorylation at Thr328 is imperative for the dimerization of ACSL4 (which represents the activation form of ACSL4) [18]. Therefore, they speculated that PKCβII‐mediated ACSL4 phosphorylation activates ACSL4, thereby promoting PUFA‐PL generation and ferroptosis. In support of this hypothesis, non‐targeted lipidomic analyses established that FINs increased PUFA‐PL generation in ACSL4 wild‐type cells, but not in ACSL4‐Thr328Ala mutant‐expressing cells, and that PKCβII inhibition impaired FIN‐induced increases of PUFA‐PL levels in ACSL4 wild‐type cells. Furthermore, phosphorylation of ACSL4 at Thr328 was found to be important for ACSL4‐mediated AA/AdA‐CoA biosynthesis. Consistently, FINs induced less potent lipid peroxidation and ferroptosis in ACSL4‐Thr328Ala mutant‐expressing cells than in ACSL4 wild‐type counterparts [18]. Together, these analyses provide compelling evidence that PKCβII‐mediated phosphorylation of ACSL4 at Thr328 is critical for ACSL4 activation and ferroptosis execution.
On the basis of these findings, the authors proposed a positive feedback loop model for lipid peroxidation and ferroptosis execution, wherein PKCβII senses lipid peroxidation and can be activated by lipid peroxides, and its activation further promotes ACSL4 dimerization and activity by phosphorylating the Thr328 site in ACSL4 to enhance PUFA‐PL synthesis and amplify the accumulation of lipid peroxides to a lethal level, resulting in ferroptosis (Figure 1). To place their findings in the context of ferroptosis‐associated diseases, Zhang et al. [18] investigated the potential role of ACSL4 Thr328 phosphorylation in ferroptosis‐related ischemia‐reperfusion. ACSL4 Thr328 phosphorylation was found to increase progressively during ischemia‐reperfusion, suggesting that this phosphorylation may serve as a ferroptosis biomarker in this pathological model and that targeting the PKCβII‐ACSL4 axis is likely to be a potential strategy for treating patients with ischemia‐reperfusion injury.
FIGURE 1.
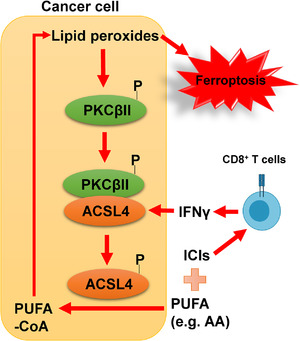
PKCβII‐ACSL4‐mediated positive feedback loop for sensing and amplifying lipid peroxidation and its relevance to ferroptosis execution and anti‐cancer immunity. PKCβII senses lipid peroxidation. Even a moderate accumulation of lipid peroxides can induce the activation of PKCβII (by promoting its phosphorylation and membrane localization). Subsequently, PKCβII activates ACSL4 by phosphorylating the Thr328 site of ACSL4, promoting PUFA‐CoA formation and therefore amplifying the accumulation of lipid peroxides to a lethal level, ultimately triggering ferroptosis. Interferon gamma (IFNγ) secreted by CD8+ T cells stimulates ACSL4 expression and increases ACSL4‐mediated biosynthesis of PUFA‐CoA, thereby promoting tumoral ferroptosis. Accordingly, a promising strategy to induce tumoral ferroptosis in cancer therapy is to combine immune checkpoint inhibitors (ICIs) with PUFA (such as AA) supplementation or drugs that activate the PKCβII‐ACSL4 pathway
Ferroptosis has been shown to partly mediate CD8+ T cell‐mediated antitumor immunity [6]. Specifically, interferon‐gamma (IFNγ) secreted by CD8+ T cells can downregulate the Solute Carrier Family 7 Member 11(SLC7A11)‐mediated cystine uptake and glutathione synthesis in cancer cells, thereby promoting lipid peroxidation and ferroptosis [6]. A recent study by Liao et al. [19] demonstrated that IFNγ could also stimulate ACSL4 expression through signal transducer and activator of transcription 1 (STAT1)‐interferon regulatory factor 1 (IRF1) signaling, thereby promoting ACSL4‐mediated synthesis of PUFA‐PLs (particularly AA‐containing PLs) to boost ferroptosis. Therefore, although AA or IFNγ alone does not trigger strong ferroptosis, AA in combination with IFNγ can synergistically induce potent tumoral ferroptosis. Furthermore, ACSL4 deficiency impaired T cell‐mediated anti‐tumor responses, thereby hastening tumor progression, suggesting that ACSL4 can control anti‐tumor immunity by modulating tumoral ferroptosis [19]. Importantly, low doses of AA could enhance anti‐tumor immunity by promoting ferroptosis and increase the sensitivity of tumors to immune checkpoint inhibitors (ICIs). Furthermore, the analysis of tumor samples from patients showed that higher ACSL4 expression was associated with higher scores of T cell‐mediated anti‐tumor immunity and may be correlated with better clinical responses of these patients to immunotherapy [19]. These results were confirmed by Zhang et al. [18] who demonstrated a positive correlation between ACSL4 expression in tumors and the sensitivity of corresponding tumors to ICIs in a cohort of melanoma patients. Moreover, preclinical models showed that Thr328 phosphorylation‐inactivating mutation in ACSL4 or PKCβII deletion compromised the efficacy of ICIs by suppressing ferroptosis. These results indicate that PKCβII‐ACSL4 pathway‐mediated PUFA‐PL production and ferroptosis execution are critical for T cell‐mediated anti‐tumor immunity (Figure 1).
Collectively, the study by Zhang et al. [18] proposed the importance of a positive feedback loop by the lipid peroxidation‐PKCβII‐ACSL4 pathway in sensing and amplifying lipid peroxidation, and revealed that PKCβII activation is required for ACSL4 activation and ferroptosis execution. Together, Zhang et al. [18] and Liao et al. [19] suggested that ACSL4 expression may serve as a predictor for immunotherapy responses in cancer patients and that targeting the PKCβII‐ACSL4 pathway in combination with ICIs might be a promising strategy for cancer therapy (Figure 1). These studies also raised several outstanding questions. In addition to PUFA‐PLs, PUFA‐containing ether phospholipids (PUFA‐ePLs) are also important substrates for lipid peroxidation [20]. Whether PKCβII can sense and amplify the accumulation of PUFA‐ePL peroxides and therefore regulate ferroptosis remains a fascinating area for future studies. It will be also interesting to study exactly how lipid peroxidation promotes PKCβII activation during ferroptotic stress. Importantly, this study challenges the conventional view that ACSL4‐mediated PUFA‐PL synthesis promotes ferroptosis in a passive and incidental manner. Whether there exist other regulatory mechanisms that actively participate in ferroptosis execution awaits further investigations. Finally, from a therapeutic perspective, it remains to be established whether there exist appropriate therapeutic windows for PKCβII activators (or AA) in combination with different ICIs to selectively kill tumors without inducing obvious toxicities in normal tissues. Whether targeting the PKCβII‐ACSL4 pathway can be effective in treating other ferroptosis‐related diseases, such as acute kidney injury or degenerative diseases, also deserves further exploration.
DECLARATIONS
COMPETING INTERESTS
Boyi Gan is an inventor of patent applications involving targeting ferroptosis in cancer therapy. Other authors have no conflicts of interest to declare.
AUTHORS' CONTRIBUTIONS
Guang Lei and Amber Horbath drafted the manuscript. Boyi Gan provided critical revision of the manuscript with additional manuscript editing support from Zhuang Li. All authors read and approved the final manuscript.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
Not applicable.
ACKNOWLEDGMENTS
Research in the authors’ lab has been supported by The University of Texas MD Anderson Cancer Center, National Institutes of Health grants R01CA181196, R01CA244144, and R01CA247992, and Cancer Prevention & Research Institute of Texas grant RP220258 (to Boyi Gan), and by Cancer Center Support (Core) Grant P30 CA016672 from the National Cancer Institute (to The University of Texas MD Anderson Cancer Center).
Guang Lei and Amber Horbath contributed equally.
Contributor Information
Guang Lei, Email: glei@mdanderson.org.
Amber Horbath, Email: adhorbath@mdanderson.org.
Zhuang Li, Email: lzhuang@mdanderson.org.
Boyi Gan, Email: bgan@mdanderson.org.
REFERENCES
- 1. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron‐dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stockwell BR, Jiang X, Gu W. Emerging Mechanisms and Disease Relevance of Ferroptosis. Trends Cell Biol. 2020;30:478–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lei G, Zhuang L, Gan B. Targeting ferroptosis as a vulnerability in cancer. Nat Rev Cancer. 2022. 10.1038/s41568-022-00459-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lei G, Zhang Y, Koppula P, Liu X, Zhang J, Lin SH, et al. The role of ferroptosis in ionizing radiation‐induced cell death and tumor suppression. Cell Res. 2020;30(2):146–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lei G, Zhang Y, Hong T, Zhang X, Liu X, Mao C, et al. Ferroptosis as a mechanism to mediate p53 function in tumor radiosensitivity. Oncogene. 2021;40(20):3533–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569(7755):270–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lei G, Mao C, Yan Y, Zhuang L, Gan B. Ferroptosis, radiotherapy, and combination therapeutic strategies. Protein & Cell. 2021;12(11):836–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. DHODH‐mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593(7860):586–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang Y, Swanda RV, Nie L, Liu X, Wang C, Lee H, et al. mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat Commun. 2021;12(1):1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lei G, Zhuang L, Gan B. mTORC1 and ferroptosis: Regulatory mechanisms and therapeutic potential. Bioessays. 2021;43:e2100093. [DOI] [PubMed] [Google Scholar]
- 11. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1‐2):317–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jiang X, Stockwell BR, Conrad M. Ferroptosis: Mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22:266–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kagan VE, Mao G, Qu F, Angeli JPF, Doll S, St Croix C, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13(1):81‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mochly‐Rosen D, Das K, Grimes KV. Protein kinase C, an elusive therapeutic target? Nat Rev Drug Discovery. 2012;11(12):937–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Inoguchi T, Sonta T, Tsubouchi H, Etoh T, Kakimoto M, Sonoda N, et al. Protein kinase C–dependent increase in reactive oxygen species (ROS) production in vascular tissues of diabetes: role of vascular NAD (P) H oxidase. J Am Soc Nephrol. 2003;14(suppl 3):S227–S32. [DOI] [PubMed] [Google Scholar]
- 17. Lee HB, Yu MR, Song JS, Ha H. Reactive oxygen species amplify protein kinase C signaling in high glucose‐induced fibronectin expression by human peritoneal mesothelial cells. Kidney Int. 2004;65(4):1170–1179. [DOI] [PubMed] [Google Scholar]
- 18. Zhang H‐L, Hu B‐X, Li Z‐L, Du T, Shan J‐L, Ye Z‐P, et al. PKCβII phosphorylates ACSL4 to amplify lipid peroxidation to induce ferroptosis. Nat Cell Biol. 2022:88–9. [DOI] [PubMed] [Google Scholar]
- 19. Liao P, Wang W, Wang W, Kryczek I, Li X, Bian Y, et al. CD8+ T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell. 2022;40(4):365–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zou Y, Henry WS, Ricq EL, Graham ET, Phadnis VV, Maretich P, et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature. 2020;585(7826):603–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.