

Abstract
Cardiorenal syndromes (CRS) describe concomitant bidirectional dysfunction of the heart and kidneys in which 1 organ initiates, perpetuates, and/or accelerates decline of the other. CRS are common in heart failure and universally portend worsened prognosis. Despite this heavy disease burden, the appropriate diagnosis and classification of CRS remains problematic. In addition to the hemodynamic drivers of decreased renal perfusion and increased renal vein pressure, induction of the renin-angiotensin-aldosterone system, stimulation of the sympathetic nervous system, disruption of balance between nitric oxide and reactive oxygen species, and inflammation are implicated in the pathogenesis of CRS. Medical therapy of heart failure including renin-angiotensin-aldosterone system inhibition and β-adrenergic blockade can blunt these deleterious processes. Renovascular disease can accelerate the progression of CRS. Volume overload and diuretic resistance are common and complicate the management of CRS. In heart failure and CRS being treated with diuretics, worsening creatinine is not associated with worsened outcome if clinical decongestion is achieved. Adjunctive therapy is often required in the management of volume overload in CRS, but evidence for these therapies is limited. Anemia and iron deficiency are importantly associated with CRS and might amplify decline of cardiac and renal function. End-stage cardiac and/or renal disease represents an especially poor prognosis with limited therapeutic options. Overall, worsening renal function is associated with significantly increased mortality. Despite progress in the area of CRS, there are still multiple pathophysiological and clinical aspects of CRS that need further research to eventually develop effective therapeutic options.
RÉSUMÉ
Le syndrome cardiorénal (SCR) est caractérisé par une dysfonction bidirectionnelle et concomitante du cœur et des reins dans laquelle l’un de ces deux organes déclenche, perpétue et/ou accélère le déclin de l’autre. Le SCR est fréquent dans l’insuffisance cardiaque et, dans tous les cas, laisse présager un pronostic plus défavorable. Malgré le lourd fardeau du SCR, son diagnostic et sa classification demeurent difficiles. Outre les causes hémodynamiques comme la diminution de l’irrigation rénale et l’augmentation de la pression dans la veine rénale, d’autres facteurs comme l’induction du système rénine-angiotensine-aldostérone, la stimulation du système nerveux sympathique, la perturbation de l’équilibre entre l’oxyde nitrique et le dérivé réactif de l’oxygène et l’inflammation jouent également un rôle dans la pathogenèse du SCR. Le traitement médical de l’insuffisance cardiaque, y compris par l’inhibition du système rénine-angiotensine-aldostérone et le blocage des récepteurs bêta-adrénergiques, peut atténuer ces processus délétères. La maladie rénovasculaire peut accélérer la progression du SCR. La surcharge volémique et la résistance diurétique sont fréquentes et compliquent la prise en charge du SCR. Lorsque l’insuffisance cardiaque et le SCR sont traités par des diurétiques, l’élévation du taux de créatinine n’est pas associée à une aggravation du problème si une décongestion clinique peut être obtenue. Un traitement d’appoint est souvent nécessaire pour contrer la surcharge volémique dans le SCR, mais les données probantes sur l’efficacité d’un tel traitement sont rares. L’anémie et la carence en fer sont associées de façon importante au SCR et pourraient amplifier le déclin des fonctions cardiaque et rénale. L’insuffisance cardiaque et l’insuffisance rénale terminales ont un pronostic particulièrement défavorable et leurs options thérapeutiques sont très limitées. Dans l’ensemble, la détérioration de la fonction rénale est associée à une augmentation significative de la mortalité. Malgré les progrès réalisés dans le traitement du SCR, il subsiste de multiples facettes de cette affection, tant physiopathologiques que cliniques, qui devraient faire l’objet de recherches dans le but de trouver des options thérapeutiques efficaces.
Cardiorenal syndromes (CRS) is an all-encompassing term that describes the complex interplay between concomitant cardiac and renal dysfunction in which disease of 1 organ initiates, perpetuates, and/or accelerates decline in the other. In the setting of heart failure, CRS comprise one-quarter to one-third of presentations and universally portend worse clinical outcomes.1–7 Accordingly, there is considerable interest in the appropriate classification, diagnosis, and management of CRS in heart failure.7,8 In this review, we describe challenges related to the classification of CRS, pathophysiological mechanisms leading to CRS, and clinical considerations in the management of patients with heart failure and concomitant renal dysfunction.
Diagnosis of CRS
A widely published classification scheme separates CRS into 4 categories on the basis of the primary organ driving bidirectional dysfunction and the acuity of decline, as well as a fifth category describing CRS occurring in the presence of systemic disease (Table 1).9,10 However, this classification scheme has not resulted in a change in clinical practice or research directions11 and there are several noteworthy shortcomings that limit its utility.12 Primarily, heterogenous characterizations of renal and cardiac dysfunction are ubiquitous. Three commonly cited classification systems, the Kidney Disease: Improving Global Outcomes Clinical Practice Guideline for Acute Kidney Injury,13 Acute Dialysis Quality Initiative Risk, Injury, Failure, Loss, and End-stage renal disease criteria,14 and the Acute Kidney Injury Network classification15 have been used to define acute kidney injury (AKI) in the nephrology literature. Further complicating assessment of renal injury, absolute and percentage increases in creatinine have been used to define renal impairment or worsening renal function in numerous heart failure studies.16 Although unifying criteria have been proposed, widespread acceptance remains elusive to date.3 A failure of this classification scheme to highlight the mechanisms of action further restricts its utility. Although the same organ might be affected by an insult, pathophysiological drivers, and thus response to therapy, can widely differ between precipitants of CRS. Moreover, in acute presentations it can be difficult to ascertain temporal relationships between cardiac and renal dysfunction.4 Finally, the classification does not exclude patients with criteria that would obfuscate the clinical picture, such as patients with severe gastrointestinal bleeding.
Table 1.
Classification of CRS
| Classification | Timing | Primary organ affected | Description | Examples of disease states |
|---|---|---|---|---|
| CRS type 1 (acute cardiorenal) | Acute | Heart | Acute heart failure causing acute kidney injury |
|
| CRS type 2 (chronic cardiorenal) | Chronic | Heart | Chronic heart failure causing chronic kidney disease |
|
| CRS type 3 (acute renocardiac) | Acute | Kidneys | Acute kidney injury leading to acute heart failure |
|
| CRS type 4 (chronic renocardiac) | Chronic | Kidneys | Chronic kidney disease leading to chronic heart failure |
|
| CRS type 5 (systemic disease) | Acute or chronic | Heart and kidneys | Systemic disease causing concomitant heart failure and kidney disease (acute or chronic) |
|
CRS, cardiorenal syndrome.
Data from Ronco et al.10
These challenges have led researchers to identify biomarkers that might have utility in the diagnosis of CRS. In the Neutrophil Gelatinase-Associated Lipcalin (NGAL) Evaluation Along With B-type Natriuretic Peptide in Acutely Decompensated Heart Failure (GALLANT; ClinicalTrials.gov identifier: NCT00693745) trial the utility of plasma neutrophil gelatinase-associated lipocalin (NGAL; a measure of AKI), was evaluated in acute heart failure (AHF).17 Plasma NGAL was a strong prognostic biomarker for rehospitalization for heart failure within 30 days.17 On a short-term basis (ie, 48–72 hours after presentation), NGAL was independently associated with worsening renal function in patients with AHF.18 However, the utility of plasma NGAL above creatinine was called into question in a subsequent trial.19 In chronic heart failure, increased levels of urinary kidney injury molecule-1 (KIM-1), N-acetyl-β-D-glucosaminidase (NAG), and NGAL are associated with worsening renal function, which portends poor prognosis.20 Interestingly, KIM-1 and NAG can be elevated in heart failure patients with normal kidney function on the basis of creatinine, suggesting that some patients might be developing renal injury, particularly tubular damage, in the context of heart failure without a decline in glomerular filtration rate (GFR).21 Indeed, elevated excretion of NAG and KIM-1 was associated with progression of chronic kidney disease (CKD) in heart failure patients.22 Further research is needed to establish whether these biomarkers might be useful in establishing the chronicity of declining renal function, differentiating tubular injury from a rise in creatinine without true injury, and identifying patients who have tubular injury in chronic heart failure despite normal kidney function.
Pathophysiology of CRS
The interaction between the heart and kidneys in CRS has been the subject of considerable interest but mechanistic pathways have yet to be fully elucidated.12 Hemodynamic factors, previously thought to be sole contributors to the CRS, cannot fully explain observed clinical consequences.12 Indeed, outpatients with heart failure without significant hemodynamic derangements suffer from a greater rate of estimated GFR (eGFR) decline than would be expected from aging alone.23 The other major pathophysiological mechanisms linking the heart and the kidneys have been denoted as cardiorenal connectors. These include activation of the renin-angiotensin-aldosterone system (RAAS), stimulation of the sympathetic nervous system (SNS), inflammation, and dysregulation of the balance between nitric oxide (NO) and reactive oxygen species (ROS; Fig. 1).8 There is significant interplay between these cardiorenal connectors, allowing them to potentiate each other and further disrupt cardiac and renal function. Fibrosis, a common result of inflammation and oxidative stress, is also a marker of more severe irreversible heart failure and CKD, which has led some authors to label it as a key driver in the pathophysiology of CRS.11
Figure 1.
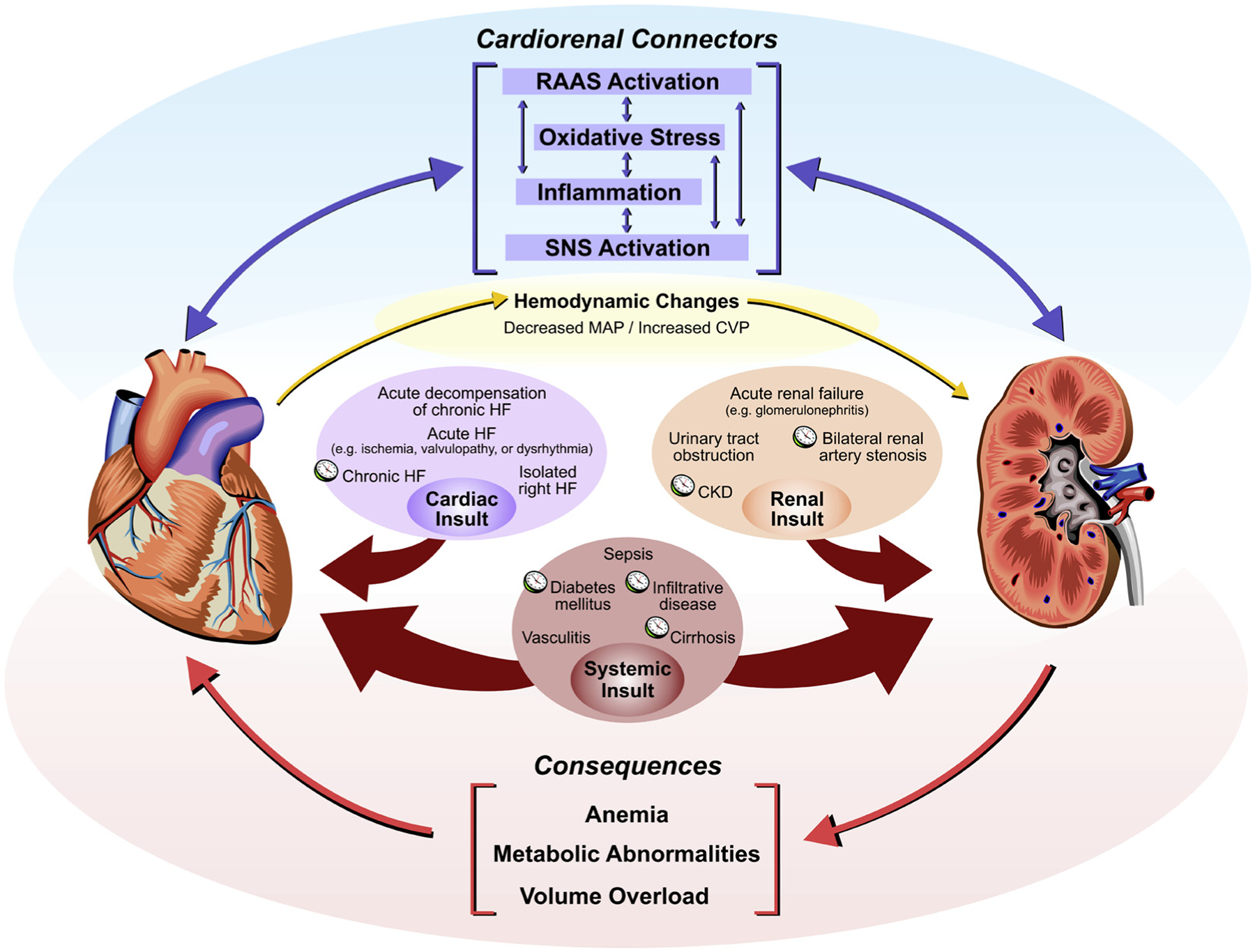
A schematic depiction of the cardiorenal syndrome. Cardiac, renal, or systemic insults result in dysfunction of the heart and/or kidneys. Clocks are used to denote conditions that typically present chronically. The cardiorenal connectors mediate cross-talk between the heart and the kidneys, causing concomitant organ dysfunction. Cardiac dysfunction results in hemodynamic changes, which results in renal dysfunction. The consequences of cardiorenal syndrome include anemia, metabolic abnormalities, and volume overload. CKD, chronic kidney disease; CVP, central venous pressure; HF, heart failure; MAP, mean arterial pressure; RAAS, renin-angiotensin-aldosterone system; SNS, sympathetic nervous system.
Hemodynamic Factors
Classically, inadequate renal perfusion due to decreased cardiac output was thought to be primary precipitant of worsening renal function in CRS.24 However, it is now recognized that decreased cardiac output and venous congestion are hemodynamic contributors to CRS.25 In situations such as severe sepsis, decreased renal perfusion due to excessive vasodilatation is an important precipitant of CRS. In contrast, most patients with heart failure maintain adequate renal perfusion yet are still susceptible to worsening renal function and CRS. On the basis of this observation and a wealth of data, venous congestion is now recognized as a primary hemodynamic precipitant of worsening renal function.26 Increasing right atrial pressure results in altered flow patterns in renal veins.27 Clinically, elevated central venous pressure is associated with decreased renal function and worsening prognosis in heart failure.26,28 Specifically, increased renal vein pressure increases interstitial and tubular hydrostatic pressure and induces strong renal vasoconstriction, leading to decreased renal blood flow and glomerular filtration.29,30 Worsening renal function precipitates further volume overload, which can, in turn, lead to increased central venous pressure and increased renal venous pressure. Importantly, merely the presence of altered intrarenal venous flow patterns, independent of right atrial pressure, also correlate with clinical outcomes in heart failure.31 Altogether the association between systemic congestion with increased renal vein pressure as a consequence and worsening renal function is now well established. It remains to be elucidated, however, how increased renal vein pressure leads to renal vasoconstriction. An extended discussion regarding diuresis to control volume overload is presented later in this review.
RAAS
Activation of the RAAS is a feature of heart failure and CKD.8,32 Decreased renal perfusion is sensed by the juxtaglomerular apparatus, resulting in secretion of renin into the circulation via the juxtaglomerular cells. Subsequently, angiotensin II is produced, which, in turn, stimulates the adrenal cortex to release aldosterone. Activation of the RAAS has numerous physiologic consequences in the human body, including peripheral vasoconstriction, increased sodium and water retention, and activation of the SNS.8 In addition, increased RAAS activity has been observed after experimentally increased renal vein pressure.33 In heart failure, activation of the RAAS can decrease cardiac energy supply and cardiac efficiency.34 Chronically elevated aldosterone levels are implicated in myocardial and renal fibrosis.35
Importantly, the RAAS has a significant effect on other cardiorenal connectors. RAAS activation results in increased activity of the SNS through central stimulation to increase sympathetic outflow, adrenally-mediated effects, and local activation at sympathetic nerve endings.36 Increased levels of inflammatory biomarker tumour necrosis factor in patients with chronic heart failure are associated with activation of the RAAS.37 Furthermore, mineralocorticoid excess is associated with activation of the inflammatory cascade, which ultimately leads to cardiac and renal fibrosis.38 Chronic activation of RAAS impairs mitochondrial function and increases oxidative stress.39 Activation of the RAAS increases myocardial nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity, disrupting the balance between NO and ROS in the myocardium, resulting in heart failure.40,41
RAAS blockade, in the form of angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers, or mineralocorticoid receptor antagonists, is an integral component in the management of heart failure and CKD.24 In heart failure, RAAS blockade prevents excessive sodium reabsorption and adverse cardiac remodelling.42 In patients with renal failure, RAAS blockade reduces sympathetic hyperactivity, further supporting the previously described interplay between cardiorenal connectors.43,44 Although adverse effects of RAAS blockade such as hypotension and hyperkalemia can complicate management of patients, a clear mortality benefit of RAAS blockade in heart failure has been shown in numerous clinical trials.6 In CKD, RAAS inhibition results in lower proteinuria and a decreased rate of GFR decline in a wide range of patients.45 Despite these benefits in isolated heart or renal failure, there is a surprising dearth of studies in well defined cardiorenal patients on the effect of initiation, maintenance, or cessation of RAAS blockade on renal function, diuresis, and other clinical outcomes. The ACE 2/angiotensin 1–7 axis has emerged as the protective arm of the RAAS and is protective in the cardiovascular and renal systems suggesting that clinical trials with these agents are warranted.46,47
SNS
Overactivation of the SNS is a feature common to heart failure and CKD.48,49 In chronic heart failure, the SNS is activated by suppression of inhibitory cardiovascular reflexes including the arterial baroreceptor reflex, augmentation of the excitatory cardiovascular reflexes such as the arterial chemoreceptor reflex, and alterations in the central nervous system response to these aforementioned reflexes; these changes result in the induction of apoptosis, oxidative damage, and chronic adverse cardiac remodelling.48 Heart failure itself is associated with increased renal norepinephrine spillover.50 In the kidney, excess sympathetic drive is also precipitated by renal ischemia, chemoreflex activation, and/or NO imbalance, which results in sodium retention, decreased renal blood flow, and increased plasma renin.49,51 In CKD, the heart becomes desensitized to the action of β-adrenergic stimulation.52 Overall, increased renal sympathetic activity is associated with decreased GFR and left ventricular (LV) ejection fraction.53 SNS hyperactivity can potentiate other cardiorenal connectors by inducing inflammation via increased cytokine production in the liver and heart, increasing ROS production, and directly stimulating the RAAS via renin release.8 Inappropriate activation of the RAAS in CKD is considered a primary driving force for activation of the SNS, resulting in a vicious circle of deleterious RAAS and SNS activation.
Numerous trials in heart failure have shown the significant benefit of blunting SNS overactivation by β-blockade, and, accordingly, β-blockers remain an integral component of the medical management of heart failure.6 With regard to other non-β-blocker agents, the Moxonidine Congestive Heart Failure (MOXCON; unlisted on ClinicalTrials.gov) trial examined the use of the sympatholytic drug moxonidine in heart failure patients.54 The trial was prematurely terminated because of increased morbidity and mortality, suggesting that central sympathetic inhibition in heart failure might be deleterious.54 In contrast, renal denervation might improve left ventricular LV hypertrophy and diastolic dysfunction in hypertensive patients independent of an antihypertensive effect.55 Emerging data suggest that renal denervation improves symptoms and LV function in heart failure with reduced ejection fraction patients, whereas improvements in surrogate end points might be expected in heart failure with preserved ejection fraction patients.56 However, the failure of renal denervation to improve outcomes in resistant hypertension has dampened enthusiasm for this potential therapy.57 Of note, vagal nerve stimulation and baroreceptor activation trials have shown equivocal results thus far.58 Similar benefits of SNS inhibition appear to be present in the kidney, but strong clinical data are lacking. Independent of blood pressure, β-blockade reduced the progression of glomerulosclerosis in rats.59 In humans, studies involving β-blockers and CKD have shown benefit only when used as an antihypertensive agent, and was inferior to RAAS blockade.60 Otherwise, there is weak evidence that renal denervation and the aforementioned sympatholytic drug moxonidine could be renoprotective in patients with CKD.61,62 Unfortunately, there are no trials regarding SNS inhibition in well defined cardiorenal patients; further research is needed to explore the therapeutic potential of interrupting nervous system traffic in the various CRS.
Inflammation
CKD, especially end-stage renal disease, and chronic heart failure are associated with elevation of proinflammatory cytokines, which portends worse outcomes.37,63–66 Indeed, worsening heart failure correlates with increased levels of proinflammatory cytokines.67 Traditional sources of proinflammatory cytokines include production by circulating cells, particularly monocytes, and locally by the heart and kidneys in response.63 More recently, it has been shown that venous congestion and/or intestinal ischemia can lead to endotoxin absorption from the bowel and subsequent systemic inflammation.68 Protein-bound uremic toxins induce inflammation in endothelial cells through multiple mechanisms.69 Regardless of the source, inflammation is at the basis of progression of endothelial dysfunction and vascular disease.70 Inflammation causes local oxidative stress at the endothelium, disrupting the endothelial barrier and causing tissue injury.70 Worsening inflammation is associated with progression of cardiovascular and renal disease.64,68 In heart failure, worsening inflammation causes cardiomyocyte apoptosis.71 Elevated C-reactive protein concentrations are associated with increased all-cause and cardiovascular mortality in uremic patients.72 An evolving paradigm implicates inflammation in the pathogenesis of heart failure with preserved ejection fraction,73 as well as concomitant renal dysfunction.74,75
Inflammation can potentiate the other cardiorenal connectors. Inflammation can cause the release of renin, activating the RAAS.8 Cytokine production activates the SNS by increasing serum norepinephrine concentrations.8 Finally, inflammation causes release of ROS from leukocytes.76 However, directed anti-inflammatory therapies have yet to improve outcomes in cardiovascular and renal disease. The Anti-TNF Therapy Against Congestive Heart Failure (ATTACH; unlisted on ClinicalTrials.gov) trial examined the effect of the anti-tumour necrosis factor drug infliximab in patients with heart failure and showed no benefit and potential harm with higher doses of the drug.77 A similar trial has not been conducted to date in CKD. Because of the heterogeneous effects of cytokines and potential adverse events, it is understandable that anti-inflammatory therapies in CRS are limited; further studies are needed to identify potential therapeutic targets to decrease inflammation in patients with combined heart and kidney failure.
Oxidative Stress, NO, and Antioxidant Capacity
Another crucial mechanism involved in combined heart and kidney failure is the balance between oxidative stress, NO, and antioxidant forces. Increased ROS production results in increased NADPH oxidase and xanthine oxidase activity, as well as NO synthase uncoupling.8 Heightened NADPH oxidase activity is correlated with myocardial oxidative stress.41 Increased endothelium-bound xanthine-oxidase activity results in vascular oxidative stress and endothelial dysfunction in patients with heart failure.78 Diminished antioxidant capacity is also a major component of CKD,79 and, in conjunction with inflammation, can contribute to cardiovascular complications.80 Uremic solutes such as β-2 microglobulin, cysteine, homocysteine, and advanced glycosylated end products cause oxidative injury, whereas malnutrition and loss via dialysis can diminish levels of antioxidants.8 Overall, oxidative stress is implicated in the pathogenesis of heart failure,41 promotes endothelial dysfunction,24 and induces cardiomyocyte apoptosis.71
NO-ROS imbalance also potentiates other cardiorenal connectors. Oxidative stress can activate the SNS, increasing heart rate and blood pressure.8 Importantly, oxidative stress damages proteins, carbohydrates, and lipids and initiates an inflammatory response by increasing production and activation of proinflammatory cytokines, causing endothelial dysfunction.8,81 Finally, oxidative damage to renal tubular or interstitial cells might impair feedback mechanisms, causing increased RAAS activity.82 Animal studies have shown that NO reduction causes permanent cardiac and renal dysfunction in CKD.83 Conversely, an NO donor was shown to ameliorate the cardiac dysfunction in an animal model of CRS.84 Although therapies to diminish oxidative stress and improve cardiac and renal dysfunction have been evaluated in trials, there is no solid evidence in the setting of combined heart and kidney failure. The association of xanthine oxidase activity with endothelial dysfunction has led to the evaluation of this therapeutic option in heart failure patients,85 and might represent a potential target in CRS. As with the other cardiorenal connectors, more research is needed to identify new therapeutic options to limit oxidative stress in patients with CRS.
Renal Artery Stenosis
Atherosclerotic renal artery stenosis is a common cause of CKD, secondary hypertension, and heart failure and represents a prototypical potentiator of CRS.86,87 Situations in which renal artery stenosis should be considered and diagnosis is reviewed elsewhere.88 Bilateral renal artery stenosis can present with flash pulmonary edema, also known as Pickering syndrome, and hypertensive heart failure, and is independently associated with decreased survival.89–91 Bilateral renal artery stenosis leads to decreased renal perfusion and subsequent activation of the RAAS and SNS, which eventually result in sodium retention, resistant hypertension, renal injury, and, ultimately, heart failure.92 Approximately 75% of patients with atherosclerotic renal artery stenosis have diastolic dysfunction and only 5% have normal cardiac structure and function.93 Revascularization procedures in patients with bilateral renal artery stenosis failed to improved outcomes over medical therapy in several trials.94–96 However, these trials were underpowered to discriminate whether there could be benefit in patients with flash pulmonary edema, resistant hypertension, or progressive renal decline. Accordingly, Canadian guidelines reserve revascularization for patients for these specific indications.88
Management of Consequences of CRS: Volume Overload
Volume overload is a consequence of CRS. Because worsening volume overload results in increasing central venous pressure and renal vein pressure, management of volume overload is an important therapeutic goal in the management of heart failure with CRS. Loop diuretics are first-line therapy for relieving symptoms of congestion and improving venous hemodynamics97 but can be complicated by diuretic resistance, especially in the presence of CRS.98 Although new treatment pathways involving relative blood volume measurement, bioimpedance vector analysis, and other monitoring devices are emerging, the optimal diuretic strategy in CRS has yet to be elucidated.7 Specifically, the evidence to guide choice of diuretic, mode of administration, use of adjunctive medications such as ACE inhibitors, and role of device therapy is lacking.
Universal measures for patients with CRS and volume overload include nonpharmacologic therapies and medication review. Dietary sodium and fluid restriction have been considered a cornerstone of therapy for AHF, especially in the setting of chronic renal disease, and has been extrapolated to patients with CRS. Despite a potential physiological basis for this recommendations, the benefit of these interventions is inconclusive99; Canadian heart failure guidelines make weak recommendations regarding dietary sodium and fluid intake.6 Definitive studies are required to guide the management of patients with CRS. In presentations complicated by hypotension, discontinuation of antihypertensive medications is warranted, along with potential initiation of vasoactive medications. Nonsteroidal anti-inflammatory drugs and other medications known to increase the risk of heart failure should be discontinued.100
Although preservation of renal function has been classically described as a target in management of AHF,101 there is increasing evidence that holding or decreasing diuretics in the face of apparent worsening renal function to prevent further renal decline via optimization of the Frank-Starling curve to maintain cardiac output might be deleterious.102 As described previously, it is in fact renal venous congestion that is the primary driver of AKI in heart failure, rather than decreased effective circulating volume.26 Indeed, multiple studies in AHF have shown that rising creatinine level alone is not independently associated with worsening clinical outcomes; it is only deleterious when there is persistent evidence of congestion.103,104 Patients with good diuretic response have improved clinical outcomes, but, interestingly, worsening renal function was noted in patients with the best and worst response to diuretics.105 This paradigm shift has led some authors to suggest new terminology such as pseudo-AKI or pseudo-worsening renal function to denote situations in which creatinine level rises and GFR declines without true renal injury (Table 2).3 In patients with CRS, clinical signs of congestion should supersede rising creatinine level.97
Table 2.
Suggested definitions of worsening renal function and acute kidney injury
| Term and setting | Measure | Temporality | sCr or eGFR | Other criteria |
|---|---|---|---|---|
| AKI (acute HF) | Relative sCr | Within 1–7 days before or during hospitalization |
|
Deterioration in heart failure status or failure to improve or requirement of inotropic support, UF, or RRT |
| Absolute sCr | Within 48 hours |
|
Deterioration in heart failure status or failure to improve or requirement of inotropic support, UF, or RRT | |
| Urine output | Over 6–12 hours |
|
Deterioration in heart failure status or failure to improve or requirement of inotropic support, UF, or RRT | |
| Pseudo-AKI (acute HF) | Multiple | Within 1–7 days before or during hospitalization | Not meeting | AKI criteria* |
| WRF (Chronic HF) | Absolute and relative sCr | Over 1–26 weeks |
|
Deterioration in HF status but not leading to hospitalization |
| eGFR | Over 1–26 weeks |
|
Deterioration in HF status but not leading to hospitalization | |
| Pseudo-WRF | Multiple | Over 1–26 weeks | Not meeting | WRF criteria* |
AKI definitions do not include eGFR because eGFR calculations assume steady-state renal function.
AKI, acute kidney injury; eGFR, estimated glomerular filtration rate; HF, heart failure; RRT, renal replacement therapy; sCr, serum creatinine; UF, ultrafiltration; WRF, worsening renal function.
Damman et al.3 suggest that doubling or a > 88.4 μmol/L rise in serum creatinine should trigger further investigation even if falling into the pseudo-AKI or pseudo-WRF categories.
Modified from Damman et al.3 with permission from Oxford University Press.
In AHF with CRS, there are currently no trials available that have examined the effect of loop diuretics on the cardiorenal interaction. Therefore, diuresis in heart failure with CRS has defaulted to extrapolating data from heart failure with or without concomitant renal dysfunction. In the Diuretic Optimization Strategies Evaluation (DOSE; ClinicalTrials.gov identifier: NCT00577135) trial continuous infusion of loop diuretics vs intermittent boluses were compared; at 72 hours, continuous infusion made no significant difference in global assessment of symptoms or mean change in creatinine level, relative to intermittent boluses.106 Further studies specifically in patients with CRS are needed to examine if loop diuretics and the achievement of euvolemia can modify the cardiorenal interaction.
Diuretic resistance in patients with AHF is associated with hypotension, worsening renal function, decreased urine output, increased risk of rehospitalization, and increased risk of death.97,105,107,108 Major mechanisms of diuretic resistance include compensatory distal reabsorption of sodium109 and activation of the RAAS and the SNS.110,111 Although diuretic resistance remains a major problem, there are limited clinical trial data on the efficacy of adjuncts to loop diuretic therapy. Classically, thiazide-like diuretics (eg, metolazone), which act at the distal convoluted tubule, were used in addition in cases of resistant heart failure.112 However, some data suggest that uptitration of loop diuretics is initially preferred over the additional use of thiazide-like diuretics because of an increased prevalence of hypokalemia, hyponatremia, worsening renal function, and increased mortality after propensity adjustment with the latter strategy.113 Several classes of medications that precipitate diuresis have been trialed in the management of diuretic-resistant AHF. Mineralocorticoid receptor antagonists, vasopressin antagonists, dopamine, and natriuretic peptides have all been evaluated in heart failure with poor results, but not specifically in CRS.114–116 Vasopressin antagonists in particular, such as tolvaptan, have been used in patients with severe hyponatremia,97 but otherwise do not improve morbidity or mortality in AHF.115 Although poorly studied in patients with heart failure and CRS, increased intraabdominal pressure might contribute to diuretic resistance and is correlated with renal dysfunction in patients with heart failure, presumably via diminished capacitance of splanchnic vasculature.117 Clearly, further study is needed to determine whether increased abdominal pressure is an important consideration in CRS and whether compartment volume removal is beneficial in these patients.
Extracorporeal therapy is a potentially useful adjunct to diuretic therapy in patients with heart failure, particularly in patients with advanced renal dysfunction with significant resistance to diuretic therapy. Various trials of ultrafiltration in heart failure alone have shown limited benefit.118–120 Nevertheless, some authors have argued that the failure of ultrafiltration to clearly show superiority in these previous trials is attributable to infrequent hemodynamic measurement and suboptimal tailoring of ultrafiltration therapy.121 European guidelines suggest that a high score (≥ 12) during objective assessment of congestion during diuretic therapy and decreased urine output (< 1000 mL in 24 hours) is indicative of diuretic resistance and an indication for ultrafiltration.122 Further research regarding ultrafiltration in the management of heart failure and CRS, especially in context of diuretic resistance, is warranted.
Essentially, there is distinct lack of evidence to help guide the management of volume overload in CRS. Further trials on choice of diuretic, diuretic regimen, adjunct medications (eg, RAAS inhibition), and the use of ultrafiltration in well defined cardiorenal patients would perhaps shed light on this clinically challenging area.
Anemia, Erythropoietin, and Iron Deficiency
Anemia, along with erythropoietin deficiency and resistance, in CKD represents a potential cardiorenal connector.123 Indeed, some authors have proposed the existence of a cardiorenal anemia syndrome on the basis of worsening mortality in heart failure patients with coexisting renal dysfunction and anemia.124 Erythropoietin receptors are present in the heart, kidney, and vascular system, and, accordingly, erythropoietin has notable effects outside of its role in erythropoiesis. Erythropoietin has several effects on the aforementioned cardiorenal connecters: it can modulate NO release, diminish ROS, dampen inflammation, and might diminish RAAS activation.123
The prevalence of anemia in the setting of chronic heart failure is difficult to estimate because of varying definitions but is likely close to 50%,125–127 and is even more common in patients with acute decompensated heart failure.128 Anemia is associated with increased mortality in chronic heart and renal failure patients.126,129,130 There are many mechanisms implicated in the anemia observed in heart failure and its associated deleterious outcomes but little is known about iron handling in these patients. Conflicting evidence shows the potential benefit for parenteral iron therapy for patients with heart failure.131–134 With regard to erythropoietin, although hematocrit levels are associated with improved cardiovascular and renal outcomes, targeting higher levels using erythropoietin does not appear to confer any benefit and is associated with potential harm, likely related to increased blood viscosity.135 In patients with systolic heart failure, 2 randomized controlled trials showed no significant clinical benefits to erythropoietin supplementation.136,137 Because there are no specific trials regarding anemia and erythropoietin deficiency in cardiorenal patients, more studies are warranted.
End-Stage Disease
In patients with end-stage renal disease and secondary heart failure, renal transplantation is a viable therapeutic option. After renal transplantation, LV hypertrophy is reversed and improved LV function, via speckle-tracking echocardiography, is observed.138 Unsurprisingly, renal transplantation in patients with end-stage renal disease and heart failure with reduced ejection fraction results in an increase in LV ejection fraction, New York Heart Association functional class, and improves survival.139 Current Canadian guidelines endorse the use of renal transplantation for this purpose.140 Of note, persistent CRS despite renal transplantation has been attributed to persistence of a high-flow arteriovenous fistula, leading to high-output heart failure.141,142
There is debate regarding cardiac transplantation in patients with end-stage heart failure and concomitant renal dysfunction.143 Current international guidelines suggest that irreversible renal dysfunction with an eGFR < 30 mL/min/1.73 m2 is a relative contraindication for heart transplantation.143 However, it can be difficult to ascertain the proportion of renal dysfunction attributable to underlying heart disease that would be expected to reverse after transplantation.143 Early guideline-directed management of heart failure aimed at preservation of renal function is important to prevent irreversible progression of renal dysfunction.144 Renal dysfunction secondary to calcineurin inhibitor use is an important post-transplantation consideration. In patients with a combined irreversible CRS, simultaneous heart and kidney transplantation is a rare but possible option.145 Similar outcomes data for LV assist devices (LVADs) are lacking, but these devices are being implanted more in patients with CRS.146 Registry data suggest that preimplantation renal dysfunction predicts higher mortality in patients who receive an LVAD, and therefore early LVAD implantation should be considered before the CRS becomes severe.147
Prognosis
Although renal dysfunction is recognized as a marker of worsened clinical outcomes, data regarding prognosis of patients with CRS are lacking, in large part because of the aforementioned problems related to diagnosis and classification of CRS. Nevertheless, data regarding concomitant heart failure and renal dysfunction might be cautiously extrapolated to patients with CRS. A meta-analysis of heart failure patients with renal impairment showed increased mortality with any renal impairment (defined as a creatinine of > 88.4 μmol/L and/or an eGFR of < 90 mL/min/1.73 m2; hazard ratio = 1.56) and moderate-to-severe impairment (defined as a creatinine of > 132.6 μmol/L and/or an eGFR of < 53 mL/min/1.73 m2; hazard ratio, 2.31), corresponding to 7% increased risk for every 10 mL/min/1.73 m2 decrease in eGFR.148 A prospective cohort study reported a 1% increase in mortality for each 1 mL/min decrease in creatinine clearance after adjustment for all other prognostic factors, which was significantly attenuated by ACE inhibition (odds ratio, 0.46) and β-blockade (odds ratio, 0.40) in those with creatinine clearance < 60 mL/min.149 Studies pertaining to prognosis in patients with well defined CRS are needed.
Conclusions
CRS describes concomitant bidirectional dysfunction of the heart and kidneys in which 1 organ initiates, perpetuates, and/or accelerates decline of the other, resulting in worsened prognosis. Despite its high prevalence and effect on clinical outcomes, the diagnosis and management of CRS is fraught with difficulty. Further research is needed to better understand the pathogenesis of CRS, enable appropriate diagnosis and classification, optimize existing therapies, and discover potential new avenues of treatment in CRS.
Funding Sources
Gavin Y. Oudit is supported by operating grants from the Canadian Institute of Health Research and the Heart and Stroke Foundation of Canada. Branko Braam holds the Translational Kidney Health Translational Research Chair from the Faculty of Medicine and Dentistry, Department of Medicine, Division of Nephrology at the University of Alberta. Mark Chappell is funded by the National Institutes of Health and the American Heart Association.
Disclosures
Gavin Y. Oudit has received research funding from Sanofi-Genzyme. The remaining authors have no conflicts of interest to disclose.
References
- 1.Ronco C, Cicoira M, McCullough PA. Cardiorenal syndrome type 1: pathophysiological crosstalk leading to combined heart and kidney dysfunction in the setting of acutely decompensated heart failure. J Am Coll Cardiol 2012;60:1031–42. [DOI] [PubMed] [Google Scholar]
- 2.Damman K, Testani JM. The kidney in heart failure: an update. Eur Heart J 2015;36:1437–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Damman K, Tang WH, Testani JM, McMurray JJ. Terminology and definition of changes renal function in heart failure. Eur Heart J 2014;35:3413–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bagshaw SM, Cruz DN, Aspromonte N, et al. Epidemiology of cardio-renal syndromes: workgroup statements from the 7th ADQI Consensus Conference. Nephrol Dial Transplant 2010;25:1406–16. [DOI] [PubMed] [Google Scholar]
- 5.Campbell RC, Sui X, Filippatos G, et al. Association of chronic kidney disease with outcomes in chronic heart failure: a propensity-matched study. Nephrol Dial Transplant 2009;24:186–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ezekowitz JA, O’Meara E, McDonald MA, et al. 2017 Comprehensive update of the Canadian Cardiovascular Society Guidelines for the management of heart failure. Can J Cardiol 2017;33:1342–433. [DOI] [PubMed] [Google Scholar]
- 7.Rangaswami J, Bhalla V, Blair John EA, et al. Cardiorenal syndrome: classification, pathophysiology, diagnosis, and treatment strategies: a scientific statement from the American Heart Association. Circulation 2019;139:e840–78. [DOI] [PubMed] [Google Scholar]
- 8.Bongartz LG, Cramer MJ, Doevendans PA, Joles JA, Braam B. The severe cardiorenal syndrome: “Guyton revisited.” Eur Heart J 2005;26: 11–7. [DOI] [PubMed] [Google Scholar]
- 9.McCullough PA, Kellum JA, Haase M, et al. Pathophysiology of the cardiorenal syndromes: executive summary from the eleventh consensus conference of the Acute Dialysis Quality Initiative (ADQI). Contrib Nephrol 2013;182:82–98. [DOI] [PubMed] [Google Scholar]
- 10.Ronco C, Haapio M, House AA, Anavekar N, Bellomo R. Cardiorenal syndrome. J Am Coll Cardiol 2008;52:1527–39. [DOI] [PubMed] [Google Scholar]
- 11.Zannad F, Rossignol P. Cardiorenal syndrome revisited. Circulation 2018;138:929–44. [DOI] [PubMed] [Google Scholar]
- 12.Braam B, Joles JA, Danishwar AH, Gaillard CA. Cardiorenal syndrome–current understanding and future perspectives. Nat Rev Nephrol 2014;10:48–55. [DOI] [PubMed] [Google Scholar]
- 13.Khwaja A KDIGO clinical practice guidelines for acute kidney injury. Nephron Clin Pract 2012;120:c179–84. [DOI] [PubMed] [Google Scholar]
- 14.Bellomo R, Ronco C, Kellum JA, Mehta RL, Palevsky P. Acute renal failure - definition, outcome measures, animal models, fluid therapy and information technology needs: the Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit Care 2004;8:R204–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mehta RL, Kellum JA, Shah SV, et al. Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care 2007;11:R31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Damman K, Valente MA, Voors AA, et al. Renal impairment, worsening renal function, and outcome in patients with heart failure: an updated meta-analysis. Eur Heart J 2014;35:455–69. [DOI] [PubMed] [Google Scholar]
- 17.Maisel AS, Mueller C, Fitzgerald R, et al. Prognostic utility of plasma neutrophil gelatinase-associated lipocalin in patients with acute heart failure: the NGAL EvaLuation Along with B-type NaTriuretic Peptide in acutely decompensated heart failure (GALLANT) trial. Eur J Heart Fail 2011;13:846–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alvelos M, Pimentel R, Pinho E, et al. Neutrophil gelatinase-associated lipocalin in the diagnosis of type 1 cardio-renal syndrome in the general ward. Clin J Am Soc Nephrol 2011;6:476–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maisel AS, Wettersten N, van Veldhuisen DJ, et al. Neutrophil gelatinase-associated lipocalin for acute kidney injury during acute heart failure hospitalizations: the AKINESIS study. J Am Coll Cardiol 2016;68:1420–31. [DOI] [PubMed] [Google Scholar]
- 20.Damman K, Masson S, Hillege HL, et al. Tubular damage and worsening renal function in chronic heart failure. JACC Heart Fail 2013;1: 417–24. [DOI] [PubMed] [Google Scholar]
- 21.Jungbauer CG, Birner C, Jung B, et al. Kidney injury molecule-1 and N-acetyl-beta-D-glucosaminidase in chronic heart failure: possible biomarkers of cardiorenal syndrome. Eur J Heart Fail 2011;13:1104–10. [DOI] [PubMed] [Google Scholar]
- 22.Jungbauer CG, Uecer E, Stadler S, et al. N-acteyl-ss-D-glucosaminidase and kidney injury molecule-1: new predictors for long-term progression of chronic kidney disease in patients with heart failure. Nephrology (Carlton) 2016;21:490–8. [DOI] [PubMed] [Google Scholar]
- 23.Mascarenhas J, Laszczynska O, Severo M, et al. Prognostic effect of renal function in ambulatory patients with heart failure and reduced ejection fraction: the kidney is a marker of cardiac function. Can J Cardiol 2018;34:1325–32. [DOI] [PubMed] [Google Scholar]
- 24.Hatamizadeh P, Fonarow GC, Budoff MJ, et al. Cardiorenal syndrome: pathophysiology and potential targets for clinical management. Nat Rev Nephrol 2013;9:99–111. [DOI] [PubMed] [Google Scholar]
- 25.Damman K, Navis G, Smilde TD, et al. Decreased cardiac output, venous congestion and the association with renal impairment in patients with cardiac dysfunction. Eur J Heart Fail 2007;9:872–8. [DOI] [PubMed] [Google Scholar]
- 26.Mullens W, Abrahams Z, Francis GS, et al. Importance of venous congestion for worsening of renal function in advanced decompensated heart failure. J Am Coll Cardiol 2009;53:589–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang WW, Kitai T. Intrarenal venous flow: a window into the congestive kidney failure phenotype of heart failure? JACC Heart Fail 2016;4:683–6. [DOI] [PubMed] [Google Scholar]
- 28.Damman K, van Deursen VM, Navis G, et al. Increased central venous pressure is associated with impaired renal function and mortality in a broad spectrum of patients with cardiovascular disease. J Am Coll Cardiol 2009;53:582–8. [DOI] [PubMed] [Google Scholar]
- 29.Gottschalk CW, Mylle M. Micropuncture study of pressures in proximal tubules and peritubular capillaries of the rat kidney and their relation to ureteral and renal venous pressures. Am J Physiol 1956;185: 430–9. [DOI] [PubMed] [Google Scholar]
- 30.Braam B, Cupples WA, Joles JA, Gaillard C. Systemic arterial and venous determinants of renal hemodynamics in congestive heart failure. Heart Fail Rev 2012;17:161–75. [DOI] [PubMed] [Google Scholar]
- 31.Iida N, Seo Y, Sai S, et al. Clinical implications of intrarenal hemodynamic evaluation by Doppler ultrasonography in heart failure. JACC Heart Fail 2016;4:674–82. [DOI] [PubMed] [Google Scholar]
- 32.Hene RJ, Boer P, Koomans HA, Mees EJ. Plasma aldosterone concentrations in chronic renal disease. Kidney Int 1982;21:98–101. [DOI] [PubMed] [Google Scholar]
- 33.Kishimoto T, Maekawa M, Abe Y, Yamamoto K. Intrarenal distribution of blood flow and renin release during renal venous pressure elevation. Kidney Int 1973;4:259–66. [DOI] [PubMed] [Google Scholar]
- 34.Mori J, Zhang L, Oudit GY, Lopaschuk GD. Impact of the renin-angiotensin system on cardiac energy metabolism in heart failure. J Mol Cell Cardiol 2013;63:98–106. [DOI] [PubMed] [Google Scholar]
- 35.Hostetter TH, Ibrahim HN. Aldosterone in chronic kidney and cardiac disease. J Am Soc Nephrol 2003;14:2395–401. [DOI] [PubMed] [Google Scholar]
- 36.Reid IA. Interactions between ANG II, sympathetic nervous system, and baroreceptor reflexes in regulation of blood pressure. Am J Physiol 1992;262:E763–78. [DOI] [PubMed] [Google Scholar]
- 37.Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med 1990;323:236–41. [DOI] [PubMed] [Google Scholar]
- 38.Chu PY, Zatta A, Kiriazis H, et al. CXCR4 antagonism attenuates the cardiorenal consequences of mineralocorticoid excess. Circ Heart Fail 2011;4:651–8. [DOI] [PubMed] [Google Scholar]
- 39.Giam B, Kaye DM, Rajapakse NW. Role of renal oxidative stress in the pathogenesis of the cardiorenal syndrome. Heart Lung Circ 2016;25: 874–80. [DOI] [PubMed] [Google Scholar]
- 40.Nakagami H, Takemoto M, Liao JK. NADPH oxidase-derived superoxide anion mediates angiotensin II-induced cardiac hypertrophy. J Mol Cell Cardiol 2003;35:851–9. [DOI] [PubMed] [Google Scholar]
- 41.Heymes C, Bendall JK, Ratajczak P, et al. Increased myocardial NADPH oxidase activity in human heart failure. J Am Coll Cardiol 2003;41:2164–71. [DOI] [PubMed] [Google Scholar]
- 42.Weber KT. Aldosterone in congestive heart failure. N Engl J Med 2001;345:1689–97. [DOI] [PubMed] [Google Scholar]
- 43.Ligtenberg G, Blankestijn PJ, Oey PL, et al. Reduction of sympathetic hyperactivity by enalapril in patients with chronic renal failure. N Engl J Med 1999;340:1321–8. [DOI] [PubMed] [Google Scholar]
- 44.Klein IH, Ligtenberg G, Oey PL, Koomans HA, Blankestijn PJ. Enalapril and losartan reduce sympathetic hyperactivity in patients with chronic renal failure. J Am Soc Nephrol 2003;14:425–30. [DOI] [PubMed] [Google Scholar]
- 45.Remuzzi G, Perico N, Macia M, Ruggenenti P. The role of renin-angiotensin-aldosterone system in the progression of chronic kidney disease. Kidney Int Suppl 2005:S57–65. [DOI] [PubMed] [Google Scholar]
- 46.Mori J, Patel VB, Ramprasath T, et al. Angiotensin 1–7 mediates renoprotection against diabetic nephropathy by reducing oxidative stress, inflammation, and lipotoxicity. Am J Physiol Renal Physiol 2014;306:F812–21. [DOI] [PubMed] [Google Scholar]
- 47.Patel VB, Zhong JC, Grant MB, Oudit GY. Role of the ACE2/angiotensin 1–7 axis of the renin-angiotensin system in heart failure. Circ Res 2016;118:1313–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Triposkiadis F, Karayannis G, Giamouzis G, et al. The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J Am Coll Cardiol 2009;54:1747–62. [DOI] [PubMed] [Google Scholar]
- 49.Schlaich MP, Socratous F, Hennebry S, et al. Sympathetic activation in chronic renal failure. J Am Soc Nephrol 2009;20:933–9. [DOI] [PubMed] [Google Scholar]
- 50.Hasking GJ, Esler MD, Jennings GL, et al. Norepinephrine spillover to plasma in patients with congestive heart failure: evidence of increased overall and cardiorenal sympathetic nervous activity. Circulation 1986;73:615–21. [DOI] [PubMed] [Google Scholar]
- 51.Goldsmith SR, Sobotka PA, Bart BA. The sympathorenal axis in hypertension and heart failure. J Card Fail 2010;16:369–73. [DOI] [PubMed] [Google Scholar]
- 52.Leineweber K, Heinroth-Hoffmann I, Ponicke K, et al. Cardiac beta-adrenoceptor desensitization due to increased beta-adrenoceptor kinase activity in chronic uremia. J Am Soc Nephrol 2002;13:117–24. [DOI] [PubMed] [Google Scholar]
- 53.Petersson M, Friberg P, Eisenhofer G, Lambert G, Rundqvist B. Long-term outcome in relation to renal sympathetic activity in patients with chronic heart failure. Eur Heart J 2005;26:906–13. [DOI] [PubMed] [Google Scholar]
- 54.Cohn JN, Pfeffer MA, Rouleau J, et al. Adverse mortality effect of central sympathetic inhibition with sustained-release moxonidine in patients with heart failure (MOXCON). Eur J Heart Fail 2003;5: 659–67. [DOI] [PubMed] [Google Scholar]
- 55.Schirmer SH, Sayed MM, Reil JC, et al. Improvements in left ventricular hypertrophy and diastolic function following renal denervation: effects beyond blood pressure and heart rate reduction. J Am Coll Cardiol 2014;63:1916–23. [DOI] [PubMed] [Google Scholar]
- 56.Nammas W, Koistinen J, Paana T, Karjalainen PP. Renal sympathetic denervation for treatment of patients with heart failure: summary of the available evidence. Ann Med 2017;49:384–95. [DOI] [PubMed] [Google Scholar]
- 57.Bhatt DL, Kandzari DE, O’Neill WW, et al. A controlled trial of renal denervation for resistant hypertension. N Engl J Med 2014;370: 1393–401. [DOI] [PubMed] [Google Scholar]
- 58.van Bilsen M, Patel HC, Bauersachs J, et al. The autonomic nervous system as a therapeutic target in heart failure: a scientific position statement from the Translational Research Committee of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail 2017;19:1361–78. [DOI] [PubMed] [Google Scholar]
- 59.Amann K, Koch A, Hofstetter J, et al. Glomerulosclerosis and progression: effect of subantihypertensive doses of alpha and beta blockers. Kidney Int 2001;60:1309–23. [DOI] [PubMed] [Google Scholar]
- 60.Bakris GL, Hart P, Ritz E. Beta blockers in the management of chronic kidney disease. Kidney Int 2006;70:1905–13. [DOI] [PubMed] [Google Scholar]
- 61.Hering D, Marusic P, Duval J, et al. Effect of renal denervation on kidney function in patients with chronic kidney disease. Int J Cardiol 2017;232:93–7. [DOI] [PubMed] [Google Scholar]
- 62.Vonend O, Marsalek P, Russ H, et al. Moxonidine treatment of hypertensive patients with advanced renal failure. J Hypertens 2003;21: 1709–17. [DOI] [PubMed] [Google Scholar]
- 63.Rastmanesh MM, Braam B, Joles JA, Boer P, Bluyssen HA. Increased SOCS expression in peripheral blood mononuclear cells of end stage renal disease patients is related to inflammation and dialysis modality. Eur J Pharmacol 2009;602:163–7. [DOI] [PubMed] [Google Scholar]
- 64.Rastmanesh MM, Bluyssen HAR, Joles JA, et al. Increased expression of SOCS3 in monocytes and SOCS1 in lymphocytes correlates with progressive loss of renal function and cardiovascular risk factors in chronic kidney disease. Eur J Pharmacol 2008;593:99–104. [DOI] [PubMed] [Google Scholar]
- 65.Zimmermann J, Herrlinger S, Pruy A, Metzger T, Wanner C. Inflammation enhances cardiovascular risk and mortality in hemodialysis patients. Kidney Int 1999;55:648–58. [DOI] [PubMed] [Google Scholar]
- 66.Yeun JY, Levine RA, Mantadilok V, Kaysen GA. C-reactive protein predicts all-cause and cardiovascular mortality in hemodialysis patients. Am J Kidney Dis 2000;35:469–76. [DOI] [PubMed] [Google Scholar]
- 67.Torre-Amione G, Kapadia S, Benedict C, et al. Proinflammatory cytokine levels in patients with depressed left ventricular ejection fraction: a report from the Studies of Left Ventricular Dysfunction (SOLVD). J Am Coll Cardiol 1996;27:1201–6. [DOI] [PubMed] [Google Scholar]
- 68.Colombo PC, Ganda A, Lin J, et al. Inflammatory activation: cardiac, renal, and cardio-renal interactions in patients with the cardiorenal syndrome. Heart Fail Rev 2012;17:177–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Guo J, Lu L, Hua Y, et al. Vasculopathy in the setting of cardiorenal syndrome: roles of protein-bound uremic toxins. Am J Physiol Heart Circ Physiol 2017;313:H1–13. [DOI] [PubMed] [Google Scholar]
- 70.Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal 2014;20:1126–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Virzi GM, Clementi A, Ronco C. Cellular apoptosis in the cardiorenal axis. Heart Fail Rev 2016;21:177–89. [DOI] [PubMed] [Google Scholar]
- 72.Arici M, Walls J. End-stage renal disease, atherosclerosis, and cardiovascular mortality: is C-reactive protein the missing link? Kidney Int 2001;59:407–14. [DOI] [PubMed] [Google Scholar]
- 73.Putko BN, Wang Z, Lo J, et al. Circulating levels of tumor necrosis factor-alpha receptor 2 are increased in heart failure with preserved ejection fraction relative to heart failure with reduced ejection fraction: evidence for a divergence in pathophysiology. PLoS One 2014;9: e99495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ter Maaten JM, Damman K, Verhaar MC, et al. Connecting heart failure with preserved ejection fraction and renal dysfunction: the role of endothelial dysfunction and inflammation. Eur J Heart Fail 2016;18: 588–98. [DOI] [PubMed] [Google Scholar]
- 75.Yogasundaram H, Nikhanj A, Putko Brendan N, et al. Elevated inflammatory plasma biomarkers in patients with Fabry disease: a critical link to heart failure with preserved ejection fraction. J Am Heart Assoc 2018;7:e009098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ward RA, McLeish KR. Polymorphonuclear leukocyte oxidative burst is enhanced in patients with chronic renal insufficiency. J Am Soc Nephrol 1995;5:1697–702. [DOI] [PubMed] [Google Scholar]
- 77.Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation 2003;107:3133–40. [DOI] [PubMed] [Google Scholar]
- 78.Landmesser U, Spiekermann S, Dikalov S, et al. Vascular oxidative stress and endothelial dysfunction in patients with chronic heart failure: role of xanthine-oxidase and extracellular superoxide dismutase. Circulation 2002;106:3073–8. [DOI] [PubMed] [Google Scholar]
- 79.Ruiz S, Pergola PE, Zager RA, Vaziri ND. Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Int 2013;83:1029–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Himmelfarb J, Stenvinkel P, Ikizler TA, Hakim RM. The elephant in uremia: oxidant stress as a unifying concept of cardiovascular disease in uremia. Kidney Int 2002;62:1524–38. [DOI] [PubMed] [Google Scholar]
- 81.Witko-Sarsat V, Friedlander M, Capeillere-Blandin C, et al. Advanced oxidation protein products as a novel marker of oxidative stress in uremia. Kidney Int 1996;49:1304–13. [DOI] [PubMed] [Google Scholar]
- 82.Katoh M, Egashira K, Usui M, et al. Cardiac angiotensin II receptors are upregulated by long-term inhibition of nitric oxide synthesis in rats. Circ Res 1998;83:743–51. [DOI] [PubMed] [Google Scholar]
- 83.Bongartz LG, Braam B, Verhaar MC, et al. Transient nitric oxide reduction induces permanent cardiac systolic dysfunction and worsens kidney damage in rats with chronic kidney disease. Am J Physiol Regul Integr Comp Physiol 2010;298:R815–23. [DOI] [PubMed] [Google Scholar]
- 84.Bongartz LG, Braam B, Verhaar MC, et al. The nitric oxide donor molsidomine rescues cardiac function in rats with chronic kidney disease and cardiac dysfunction. Am J Physiol Heart Circ Physiol 2010;299: H2037–45. [DOI] [PubMed] [Google Scholar]
- 85.Hare JM, Mangal B, Brown J, et al. Impact of oxypurinol in patients with symptomatic heart failure. Results of the OPT-CHF study. J Am Coll Cardiol 2008;51:2301–9. [DOI] [PubMed] [Google Scholar]
- 86.Kane GC, Xu N, Mistrik E, et al. Renal artery revascularization improves heart failure control in patients with atherosclerotic renal artery stenosis. Nephrol Dial Transplant 2010;25:813–20. [DOI] [PubMed] [Google Scholar]
- 87.Watson PS, Hadjipetrou P, Cox SV, Piemonte TC, Eisenhauer AC. Effect of renal artery stenting on renal function and size in patients with atherosclerotic renovascular disease. Circulation 2000;102:1671–7. [DOI] [PubMed] [Google Scholar]
- 88.Nerenberg KA, Zarnke KB, Leung AA, et al. Hypertension Canada’s 2018 guidelines for diagnosis, risk assessment, prevention, and treatment of hypertension in adults and children. Can J Cardiol 2018;34: 506–25. [DOI] [PubMed] [Google Scholar]
- 89.Messerli FH, Bangalore S, Makani H, et al. Flash pulmonary oedema and bilateral renal artery stenosis: the Pickering syndrome. Eur Heart J 2011;32:2231–5. [DOI] [PubMed] [Google Scholar]
- 90.Conlon PJ, Little MA, Pieper K, Mark DB. Severity of renal vascular disease predicts mortality in patients undergoing coronary angiography. Kidney Int 2001;60:1490–7. [DOI] [PubMed] [Google Scholar]
- 91.Alyamani M, Thomas J, Shanks M, Oudit GY. Resistant hypertension from renal artery stenosis leading to heart failure with preserved ejection fraction. J Investig Med High Impact Case Rep 2018;6. 2324709618816501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rundback JH, Murphy TP, Cooper C, Weintraub JL. Chronic renal ischemia: pathophysiologic mechanisms of cardiovascular and renal disease. J Vasc Interv Radiol 2002;13:1085–92. [DOI] [PubMed] [Google Scholar]
- 93.Wright JR, Shurrab AE, Cooper A, et al. Left ventricular morphology and function in patients with atherosclerotic renovascular disease. J Am Soc Nephrol 2005;16:2746–53. [DOI] [PubMed] [Google Scholar]
- 94.Cooper CJ, Murphy TP, Cutlip DE, et al. Stenting and medical therapy for atherosclerotic renal-artery stenosis. N Engl J Med 2014;370:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bax L, Woittiez AJ, Kouwenberg HJ, et al. Stent placement in patients with atherosclerotic renal artery stenosis and impaired renal function: a randomized trial. Ann Intern Med 2009;150(840–8), w150–1. [DOI] [PubMed] [Google Scholar]
- 96.Wheatley K, Ives N, Gray R, et al. Revascularization vs medical therapy for renal-artery stenosis. N Engl J Med 2009;361:1953–62. [DOI] [PubMed] [Google Scholar]
- 97.Mullens W, Damman K, Harjola VP, et al. The use of diuretics in heart failure with congestion - a position statement from the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail 2019;21:137–55. [DOI] [PubMed] [Google Scholar]
- 98.Ellison DH, Felker GM. Diuretic treatment in heart failure. N Engl J Med 2017;377:1964–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mahtani KR, Heneghan C, Onakpoya I, et al. Reduced salt intake for heart failure: a systematic review. JAMA Intern Med 2018;178: 1693–700. [DOI] [PubMed] [Google Scholar]
- 100.Heerdink ER, Leufkens HG, Herings RM, et al. NSAIDs associated with increased risk of congestive heart failure in elderly patients taking diuretics. Arch Intern Med 1998;158:1108–12. [DOI] [PubMed] [Google Scholar]
- 101.Gheorghiade M, De Luca L, Fonarow GC, et al. Pathophysiologic targets in the early phase of acute heart failure syndromes. Am J Cardiol 2005;96:11–7. [DOI] [PubMed] [Google Scholar]
- 102.Schrier RW. Role of diminished renal function in cardiovascular mortality: marker or pathogenetic factor? J Am Coll Cardiol 2006;47:1–8. [DOI] [PubMed] [Google Scholar]
- 103.Metra M, Davison B, Bettari L, et al. Is worsening renal function an ominous prognostic sign in patients with acute heart failure? The role of congestion and its interaction with renal function. Circ Heart Fail 2012;5:54–62. [DOI] [PubMed] [Google Scholar]
- 104.van der Meer P, Postmus D, Ponikowski P, et al. The predictive value of short-term changes in hemoglobin concentration in patients presenting with acute decompensated heart failure. J Am Coll Cardiol 2013;61: 1973–81. [DOI] [PubMed] [Google Scholar]
- 105.Valente MA, Voors AA, Damman K, et al. Diuretic response in acute heart failure: clinical characteristics and prognostic significance. Eur Heart J 2014;35:1284–93. [DOI] [PubMed] [Google Scholar]
- 106.Felker GM, Lee KL, Bull DA, et al. Diuretic strategies in patients with acute decompensated heart failure. N Engl J Med 2011;364:797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.ter Maaten JM, Dunning AM, Valente MA, et al. Diuretic response in acute heart failure-an analysis from ASCEND-HF. Am Heart J 2015;170:313–21. [DOI] [PubMed] [Google Scholar]
- 108.Hasselblad V, Gattis Stough W, Shah MR, et al. Relation between dose of loop diuretics and outcomes in a heart failure population: results of the ESCAPE trial. Eur J Heart Fail 2007;9:1064–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rao VS, Planavsky N, Hanberg JS, et al. Compensatory distal reabsorption drives diuretic resistance in human heart failure. J Am Soc Nephrol 2017;28:3414–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Martin PY, Schrier RW. Sodium and water retention in heart failure: pathogenesis and treatment. Kidney Int Suppl 1997;59:S57–61. [PubMed] [Google Scholar]
- 111.Sobotka PA, Mahfoud F, Schlaich MP, et al. Sympatho-renal axis in chronic disease. Clin Res Cardiol 2011;100:1049–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kiyingi A, Field MJ, Pawsey CC, et al. Metolazone in treatment of severe refractory congestive cardiac failure. Lancet 1990;335:29–31. [DOI] [PubMed] [Google Scholar]
- 113.Brisco-Bacik MA, Ter Maaten JM, Houser SR, et al. Outcomes associated with a strategy of adjuvant metolazone or high-dose loop diuretics in acute decompensated heart failure: a propensity analysis. J Am Heart Assoc 2018;7:e009149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Butler J, Anstrom KJ, Felker GM, et al. Efficacy and safety of spironolactone in acute heart failure: the ATHENA-HF randomized clinical trial. JAMA Cardiol 2017;2:950–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Konstam MA, Gheorghiade M, Burnett JC Jr, et al. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST outcome trial. JAMA 2007;297:1319–31. [DOI] [PubMed] [Google Scholar]
- 116.Chen HH, Anstrom KJ, Givertz MM, et al. Low-dose dopamine or low-dose nesiritide in acute heart failure with renal dysfunction: the ROSE acute heart failure randomized trial. JAMA 2013;310:2533–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Verbrugge FH, Dupont M, Steels P, et al. Abdominal contributions to cardiorenal dysfunction in congestive heart failure. J Am Coll Cardiol 2013;62:485–95. [DOI] [PubMed] [Google Scholar]
- 118.Kazory A, Ejaz AA, Ross EA. The UNLOAD trial: a “nephrologic” standpoint. J Am Coll Cardiol 2007;50:820 [author reply 820–1]. [DOI] [PubMed] [Google Scholar]
- 119.Bart BA, Goldsmith SR, Lee KL, et al. Ultrafiltration in decompensated heart failure with cardiorenal syndrome. N Engl J Med 2012;367: 2296–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Costanzo MR, Negoianu D, Jaski BE, et al. Aquapheresis vs intravenous diuretics and hospitalizations for heart failure. JACC Heart Fail 2016;4: 95–105. [DOI] [PubMed] [Google Scholar]
- 121.Costanzo MR, Ronco C, Abraham WT, et al. Extracorporeal ultrafiltration for fluid overload in heart failure: current status and prospects for further research. J Am Coll Cardiol 2017;69:2428–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gheorghiade M, Follath F, Ponikowski P, et al. Assessing and grading congestion in acute heart failure: a scientific statement from the Acute Heart Failure Committee of the Heart Failure Association of the European Society of Cardiology and endorsed by the European Society of Intensive Care Medicine. Eur J Heart Fail 2010;12:423–33. [DOI] [PubMed] [Google Scholar]
- 123.Jie KE, Verhaar MC, Cramer MJ, et al. Erythropoietin and the cardiorenal syndrome: cellular mechanisms on the cardiorenal connectors. Am J Physiol Renal Physiol 2006;291:F932–44. [DOI] [PubMed] [Google Scholar]
- 124.Scrutinio D, Passantino A, Santoro D, Catanzaro R. The cardiorenal anaemia syndrome in systolic heart failure: prevalence, clinical correlates, and long-term survival. Eur J Heart Fail 2011;13:61–7. [DOI] [PubMed] [Google Scholar]
- 125.Tang YD, Katz SD. The prevalence of anemia in chronic heart failure and its impact on the clinical outcomes. Heart Fail Rev 2008;13: 387–92. [DOI] [PubMed] [Google Scholar]
- 126.Cleland JG, Zhang J, Pellicori P, et al. Prevalence and outcomes of anemia and hematinic deficiencies in patients with chronic heart failure. JAMA Cardiol 2016;1:539–47. [DOI] [PubMed] [Google Scholar]
- 127.Young JB, Abraham WT, Albert NM, et al. Relation of low hemoglobin and anemia to morbidity and mortality in patients hospitalized with heart failure (insight from the OPTIMIZE-HF registry). Am J Cardiol 2008;101:223–30. [DOI] [PubMed] [Google Scholar]
- 128.Cohen-Solal A, Damy T, Terbah M, et al. High prevalence of iron deficiency in patients with acute decompensated heart failure. Eur J Heart Fail 2014;16:984–91. [DOI] [PubMed] [Google Scholar]
- 129.McClellan WM, Flanders WD, Langston RD, Jurkovitz C, Presley R. Anemia and renal insufficiency are independent risk factors for death among patients with congestive heart failure admitted to community hospitals: a population-based study. J Am Soc Nephrol 2002;13: 1928–36. [DOI] [PubMed] [Google Scholar]
- 130.Jankowska EA, Rozentryt P, Witkowska A, et al. Iron deficiency: an ominous sign in patients with systolic chronic heart failure. Eur Heart J 2010;31:1872–80. [DOI] [PubMed] [Google Scholar]
- 131.van Veldhuisen DJ, Ponikowski P, van der Meer P, et al. Effect of ferric carboxymaltose on exercise capacity in patients with chronic heart failure and iron deficiency. Circulation 2017;136:1374–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Anker SD, Comin Colet J, Filippatos G, et al. Ferric carboxymaltose in patients with heart failure and iron deficiency. N Engl J Med 2009;361: 2436–48. [DOI] [PubMed] [Google Scholar]
- 133.Avni T, Leibovici L, Gafter-Gvili A. Iron supplementation for the treatment of chronic heart failure and iron deficiency: systematic review and meta-analysis. Eur J Heart Fail 2012;14:423–9. [DOI] [PubMed] [Google Scholar]
- 134.Lewis GD, Malhotra R, Hernandez AF, et al. Effect of oral iron repletion on exercise capacity in patients with heart failure with reduced ejection fraction and iron deficiency: the IRONOUT HF randomized clinical trial. JAMA 2017;317:1958–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kazory A, Ross EA. Anemia: the point of convergence or divergence for kidney disease and heart failure? J Am Coll Cardiol 2009;53:639–47. [DOI] [PubMed] [Google Scholar]
- 136.Swedberg K, Young JB, Anand IS, et al. Treatment of anemia with darbepoetin alfa in systolic heart failure. N Engl J Med 2013;368: 1210–9. [DOI] [PubMed] [Google Scholar]
- 137.Ghali JK, Anand IS, Abraham WT, et al. Randomized double-blind trial of darbepoetin alfa in patients with symptomatic heart failure and anemia. Circulation 2008;117:526–35. [DOI] [PubMed] [Google Scholar]
- 138.Hewing B, Dehn AM, Staeck O, et al. Improved left ventricular structure and function after successful kidney transplantation. Kidney Blood Press Res 2016;41:701–9. [DOI] [PubMed] [Google Scholar]
- 139.Wali RK, Wang GS, Gottlieb SS, et al. Effect of kidney transplantation on left ventricular systolic dysfunction and congestive heart failure in patients with end-stage renal disease. J Am Coll Cardiol 2005;45: 1051–60. [DOI] [PubMed] [Google Scholar]
- 140.Knoll G, Cockfield S, Blydt-Hansen T, et al. Canadian Society of Transplantation consensus guidelines on eligibility for kidney transplantation. CMAJ 2005;173:1181–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Samarendra P, Ramkumar M, Sharma V, Kumari S. Cardiorenal syndrome in renal transplant recipientsd—it’s the fistula at fault: a case series. Clin Transplant 2018;32:e13417. [DOI] [PubMed] [Google Scholar]
- 142.Nickel P, Gul S, Puhl G, Poellinger A, Schindler R. Acute cardiorenal syndrome by high flow arteriovenous fistula after kidney transplantation. J Vasc Access 2013;14:394–6. [DOI] [PubMed] [Google Scholar]
- 143.Mehra MR, Canter CE, Hannan MM, et al. The 2016 International Society for Heart Lung Transplantation listing criteria for heart transplantation: a 10-year update. J Heart Lung Transplant 2016;35:1–23. [DOI] [PubMed] [Google Scholar]
- 144.Cowger JA, Radjef R. Advanced heart failure therapies and cardiorenal syndrome. Adv Chronic Kidney Dis 2018;25:443–53. [DOI] [PubMed] [Google Scholar]
- 145.Schaffer JM, Chiu P, Singh SK, et al. Heart and combined heart-kidney transplantation in patients with concomitant renal insufficiency and end-stage heart failure. Am J Transplant 2014;14:384–96. [DOI] [PubMed] [Google Scholar]
- 146.Walther Carl P, Niu J, Winkelmayer Wolfgang C, et al. Implantable ventricular assist device use and outcomes in people with end-stage renal disease. J Am Heart Assoc 2018;7:e008664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kirklin JK, Naftel DC, Kormos RL, et al. Quantifying the effect of cardiorenal syndrome on mortality after left ventricular assist device implant. J Heart Lung Transplant 2013;32:1205–13. [DOI] [PubMed] [Google Scholar]
- 148.Smith GL, Lichtman JH, Bracken MB, et al. Renal impairment and outcomes in heart failure: systematic review and meta-analysis. J Am Coll Cardiol 2006;47:1987–96. [DOI] [PubMed] [Google Scholar]
- 149.McAlister FA, Ezekowitz J, Tonelli M, Armstrong PW. Renal insufficiency and heart failure: prognostic and therapeutic implications from a prospective cohort study. Circulation 2004;109:1004–9. [DOI] [PubMed] [Google Scholar]