

Abstract
A method was developed to screen bacteria for synthesis of mutant proteins with altered assembly and solubility properties using bacteriophage MS2 coat protein as a model self-associating protein. Colonies expressing coat protein from a plasmid were covered with an agarose overlay under conditions that caused the lysis of some of the cells in each colony. The proteins thus liberated diffused through the overlay at rates depending on their molecular sizes. After transfer of the proteins to a nitrocellulose membrane, probing with coat protein-specific antiserum revealed spots whose sizes and intensities were related to the aggregation state of coat protein. The method was employed in the isolation of assembly defective mutants and to find soluble variants of an aggregation-prone coat protein mutant.
INTRODUCTION
The aggregation state of a protein is generally important to its function. The assembly of viruses, for example, relies on the specific aggregation of subunits into helical or icosahedral structures which enclose the viral nucleic acid. On the other hand, inappropriate protein aggregation is frequently associated with loss of function. For example, the over-expression of proteins in Escherichia coli sometimes results in the accumulation of insoluble protein in inclusion bodies from which it may be difficult or impossible to recover active protein in good yield.
The propensity of a protein to aggregate, either appropriately or inappropriately, is a property encoded in its amino acid sequence and is therefore subject to genetic manipulation. We describe here a method to screen bacterial colonies for the production of mutant proteins with altered aggregation properties. It is based on the knowledge that proteins in solution diffuse at rates that vary with their molecular sizes. Bacterial colonies expressing a protein of interest are covered with an agarose gel overlay under conditions that cause the lysis of some cells in each colony. Proteins thus released diffuse into the agarose at rates that depend on their molecular weights. A replica of the released proteins is created by blotting to a nitrocellulose membrane that is then probed with antiserum specific for the protein of interest. Treatment of the membrane with a labeled second antibody results in the appearance of spots whose sizes and intensities are related to the diffusion rates of the proteins that produce them, and whose locations on the filter identify the colonies responsible for their synthesis. In this manner one can screen for mutations that either cause aggregation or disrupt it.
Here we demonstrate the utility of this agarose overlay method for the isolation of mutants of MS2 coat protein with the following properties. (i) Some mutations result in failure to form virus-like particles without disrupting the ability to synthesize large quantities of soluble coat protein. These mutants are apparently defective in specific inter-subunit interactions required for assembly into capsids of otherwise properly folded subunits and identify amino acids that play roles in virus assembly and stability. (ii) Some mutants predominantly form insoluble aggregates or are proteolytically degraded. These represent severe protein folding defects and identify amino acids implicated in protein folding/stability. (iii) Soluble second-site revertants of an insoluble coat mutant were isolated, illustrating the potential of the method for improving the solubility of proteins which otherwise aggregate when expressed in E.coli.
MATERIALS AND METHODS
Mutagenic PCR and cloning
Coat sequences were randomly mutagenized using error-prone polymerase chain reaction as described by Cadwell and Joyce (1). Under these conditions ∼0.7 nt substitutions are introduced per 100 nt. Primers were derived from sequences flanking the MS2 coat protein sequence. Their sequences were 5′-GGTCGACTCTAGATAGAGCCCTC-3′ and 5′-CCCGGGGATCCTTAATTAATTAACCCGGCGTCTA-3′. These primers introduce a XbaI site upstream and a BamHI site downstream of the coat gene. The resulting DNA fragments were cloned under control of the T7 promoter between the BamHI and XbaI sites of pET3d (2) and introduced into strain BL21(DE3) (2) by electroporation (3).
Agarose overlay
Transformed bacteria were plated on Luria–Bertani (LB) medium containing 1.5% Bacto agar and ampicillin or carbenicillin at 100 µg/ml and allowed to grow overnight at 37°C in 88 mm Petri dishes. The next morning colonies typically had diameters of ∼2–3 mm. LB medium containing 2% Seaplaque agarose (FMC) was melted, cooled to 45°C and isopropylthio-β-d-galactoside (IPTG, Gold Biotechnologies) and augmentin (SmithKline Beecham) were added, each to concentrations of 2 mg/ml. We used augmentin powder, which is normally administered to patients as an oral suspension. In addition to the antibiotics amoxicillin and clavulanate, this formulation of augmentin contains a number of inactive ingredients. We estimated the concentration of amoxicillin in the overlay at ∼280 µg/ml and of clavulanate at ∼70 µg/ml. Ten milliliters of overlay mixture were added to plates (still warm from the incubator) and allowed to cool at room temperature until the agarose had gelled. The plates were then returned to the 37°C incubator where they were kept for 16–24 h, during which time cell lysis and protein diffusion occurred. Subsequent procedures were performed at room temperature. At the end of the diffusion period a dry nitrocellulose filter (Schleicher and Schuell BA85, 82 mm diameter) was applied to the surface of each plate. Prior to application of membranes to plates, each was given an orientation mark and numbered to aid later in aligning colonies with spots. After 5–10 min filters were removed from the plates, washed briefly in TS (10 mM Tris pH 7.4, 150 mM NaCl), transferred to TS containing 5% bovine serum albumin (BSA) and rabbit polyclonal anti-MS2 serum (a 1:1000 dilution), and gently agitated for 1.5 h. Filters were washed once in TS for 5 min, twice for 5 min each in TS containing 0.5% NP-40, and once again for 5 min in TS. Filters were then transferred to TS-BSA to which had been added alkaline phosphatase-conjugated goat anti-rabbit serum (a 1:3000 dilution, from Bio-Rad). After 30 min of gentle agitation, filters were subjected to the same series of washes described above and placed in 0.1 M Tris pH 9.5 containing a chromogenic alkaline phosphatase substrate (nitroblue tetrazolium and 5-bromo-4-chloro-3-indolyl phosphate, from Bio-Rad) where they were kept until appropriate development of purple color (usually 10–15 min).
Filters and plates were aligned to reveal the correspondence of colonies and spots. Using sterile toothpicks, cells were transferred from colonies to 1 ml LB liquid medium and streaked on an LB plate, both containing carbenicillin.
Protein expression and analysis
One milliliter cultures in LB medium were induced for coat protein expression in mid log phase by the addition of IPTG and grown for an additional 3 h with shaking at 37°C. Cells were harvested by centrifugation, suspended in 0.25 ml 0.1 M NaCl, 0.01 M Tris pH 7.5, 0.001 M MgSO4, 0.0001 M EDTA and sonicated twice for 5 s on ice. Five minutes of centrifugation at 13 000 r.p.m. in a microfuge separated the lysates into soluble (supernatant) and insoluble (pellet) fractions. To determine the distribution of coat protein in the soluble and insoluble fractions, 10 µl of each were subjected to SDS–polyacrylamide gel electrophoresis (4) and the gel was stained with Coomassie Brilliant Blue R250. To determine the presence of virus-like particles, 10 µl of the supernatant fraction were subjected to electrophoresis in a 1% agarose gel in 50 mM potassium phosphate pH 7.0 (5). After completion of electrophoresis the gel was stained with ethidium bromide and photographed under UV illumination. The contents of the gel were then transferred to a Nytran membrane (Schleicher and Schuell) by capillary blotting in 50 mM potassium phosphate pH 7.0. Coat proteins on the membrane were detected with anti-MS2 serum and alkaline phosphatase-conjugated goat anti-rabbit antibody as described above.
RESULTS
The overlay method
The procedure we follow is detailed in Materials and Methods and only briefly described here. Colonies on LB medium in standard 88 mm diameter Petri dishes are overlaid with 10 ml of 2% Seaplaque agarose in LB. The overlay also contains IPTG to induce the expression of MS2 coat protein, and augmentin, a mixture of amoxicillin and clavulanate. Clavulanate is a β-lactamase inhibitor and its presence in the overlay renders cells sensitive to the ampicillin and amoxicillin present in the medium. Actively growing cells are therefore subject to lysis. Clavulanate also has the important effect of preventing growth in the overlay of secondary colonies seeded from the original colonies during introduction of the overlay. After gelling of the overlay the plates are incubated overnight at 37°C, during which time cell lysis and protein diffusion occur. A dry nitrocellulose filter is then placed on the surface of the plate and proteins are allowed to adsorb. The filters are removed and probed with anti-MS2 serum and an alkaline phosphatase-conjugated second antibody to reveal coat protein-specific spots. After identification of colonies producing desired spot phenotypes, live cells are recovered with sterile toothpicks for growth and analysis.
Comparing wild type and assembly defective coat proteins
To determine whether the overlay method could detect different aggregation states of MS2 coat protein, we compared the spots produced by the wild-type protein, which assembles into a capsid containing 180 subunits, with those produced by two mutant coat proteins (Fig. 1). One of these, dlFG, is assembly defective and accumulates principally as dimers (6). Another, m3, produces a protein that aggregates extensively and is found almost exclusively in the insoluble fraction of the cell (see later). We obtained spot sizes and intensities which were correlated with the different aggregation states of these proteins, the assembly defective mutant producing much larger spots than those derived from the wild-type protein, whereas the m3 mutant was undetectable.
Figure 1.
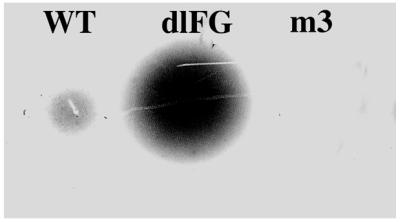
Spots formed by wild type coat protein, which assembles into capsids (i.e. 180mers) and by two mutants. The dlFG mutant makes coat protein dimers predominantly, whereas the m3 coat protein is found almost entirely in the insoluble fraction of cell lysates (see Fig. 4).
Screening a library of coat mutants for different spot phenotypes
The coat sequence was randomly mutagenized by error-prone PCR (1), cloned under T7 promoter control in pET3d (2), introduced into E.coli by electroporation, plated at a density of ∼100 colonies per plate, and subjected to the overlay protocol. A diversity of spot phenotypes was obtained. Some were similar to wild type, others were larger and more intense. Still others produced small, weak spots, or even no spots at all. Several colonies corresponding to each of these spot phenotypes were transferred with sterile toothpicks to 1 ml LB medium and grown to mid log phase when they were induced with IPTG for 3 h. Cells were then harvested for analysis. Cells from the same colonies were also tooth-picked to an LB plate in a grid pattern where they were subjected to the overlay method a second time. The individual clones were labeled m1 through m27 (note there is no m2). We attempted to correlate the spot phenotypes thus produced (Fig. 2) with the amounts of capsid found by agarose gel electrophoresis (Fig. 3) and with the quantities of coat protein found in the soluble and insoluble fractions of cell lysates separated by SDS gel electrophoresis (Fig. 4). The amino acid substitutions of the mutants were also determined by DNA sequence analysis (Table 1).
Figure 2.

The spots produced by a number of coat mutants show that random mutagenesis results in a range of spot phenotypes. The labels at left correspond to spots on the right.
Figure 3.


The capacities of the mutants of Figure 2 to assemble into virus-like particles (capsids) as judged by their behavior upon agarose gel electrophoresis. (A) The ethidium bromide-stained gel. (B) A blot of the same gel probed with anti-MS2 serum.
Figure 4.

SDS gel electrophoresis of the soluble (left member of each pair of lanes) and the insoluble (right member of each pair) fractions of lysates produced from cells containing the indicated mutant coat sequences. The lane labeled M contains protein from purified MS2 virus and an arrow to the left of each panel locates the position at which coat protein migrates. WT means wild type. Numbers correspond to the m1 to m27 mutants shown in Figures 2 and 3.
Table 1. The amino acid substitutions found in the mutant proteins whose spot phenotypes and assembly and solubility properties are shown in Figures 2–4.
| m1 | S51T A96T |
|---|---|
| m3 | N24S C46R A96V N116S |
| m4 | A26T I118F |
| m5 | Q109L |
| m6 | V48A Q109H |
| m7 | no amino acid substitutions |
| m8 | I104V |
| m9 | N27S |
| m10 | A107T |
| m11 | K66R |
| m12 | A53E |
| m13 | S2T N27K |
| m14 | I94V |
| m15 | V79I A107V N116S |
| m16 | Q40L |
| m17 | F4S W32R Q50R |
| m18 | not determined |
| m19 | N12D S34G S52P I92M C101R Q109L S120T |
| m20 | S37P A41T P59A S84P |
| m21 | K43STOP |
| m22 | A21S N24D Q40R C46Y V79A |
| m23 | Q6L N12D I33T R56C F95L |
| m24 | I33V P65T |
| m25 | not determined |
| m26 | T15N N24S V29A W32C T45S I60T N98Y I104N S126P |
| m27 | V61E L103F K106R Y129H |
For purposes of discussion, we arbitrarily identify three classes of spot phenotypes.
(i) Spots close to wild type in size and intensity. One isolate (m7) appeared wild type with respect to all the parameters we consider here. That is, it gave a wild-type spot and produced normal quantities of coat protein that was found almost entirely in the soluble fraction. Consistent with its wild-type phenotype, sequence analysis showed that although this mutant contained three different nucleotide substitutions, each of these was a silent change. Three other mutants (m4, m9 and m10) gave spots of about the same intensity as wild type, but a little larger and more diffuse. Their capsid yields were reduced and the amount of protein found in the insoluble fraction was increased. Each of these was the result of different amino acid substitutions (Table 1).
(ii) Very weak or undetectable spots. Fourteen of the 26 clones examined here (m3, m5, m6, m8, m15, m17, m18, m19, m21, m22, m23, m25, m26 and m27) produced no detectable spot and little or no detectable capsid. Most of these produced near normal amounts of coat protein, but nearly all of it was confined to the insoluble fraction. These coat proteins apparently formed aggregates of such size that little or none of it could diffuse to the overlay surface. Clones m15 and m18 made significantly reduced amounts of coat protein, while another, m21, turned out to contain a nonsense mutation at codon 43.
(iii) Spots larger and more intense than wild type. Eight clones, m1, m11, m12, m13, m14, m16, m20 and m24, were judged to be in this class. Most produced normal or near normal amounts of soluble coat protein, although m1 and m20 were only ∼50% soluble. All of these were at least partially defective for capsid assembly. In fact m1, m14, m16, m20 and m24 made no detectable capsids.
It should be noted that the frequency of occurrence of clones in the various classes is expected to be a function of the mutation rate. We expect that wild-type spot phenotypes will be favored by a low mutation rate. On the other hand, insoluble-folding defects should predominate at high mutation rates where most clones accumulate multiple substitutions. We have not actually measured the frequency of mutation in our library. However, we used PCR conditions that are reported to result in an error rate of 0.7% (1) and assume therefore that most clones have several nucleotide substitutions. Indeed, most of the clones listed in Table 1 contain several mutations. One (m26) has as many as nine amino acid substitutions. Such a high-mutation frequency is consistent with the relatively small number of spots with a wild-type phenotype (∼5%) and the predominance of clones that make no spot (∼50%).
Using the method to isolate soluble, second-site revertants of an insoluble coat mutant
The existence of coat mutants that produce no spot because they synthesize an insoluble coat protein provided an opportunity to test another potential application of the agarose overlay method, the identification of mutational variants that produce soluble versions of otherwise insoluble proteins. As many proteins form inclusions when expressed in E.coli, such a method could be widely useful.
We arbitrarily selected the m3 mutant for an attempt at reversion to solubility. In retrospect m3 may have been a less than optimal choice as it has four amino acid substitutions and we did not know which is/are responsible for the m3 folding defect. Nevertheless, the m3 sequence was mutagenized by error-prone PCR, cloned in pET3d, and introduced by electroporation into strain BL21(DE3). A few hundred colonies were screened by the agarose overlay method, <1% of which restored the ability to make a detectable spot. We picked five m3 revertants. Three of them (r303, r304 and r305) retained their enhanced spot phenotypes upon re-screening. Analysis showed that each produced about half its coat protein in the soluble fraction (Fig. 5). None produced capsids, but this is probably due to the presence of a substitution at residue 96, a disruption that is probably sufficient by itself to prevent assembly. In this regard, we note the assembly defects of m1, which has a substitution at amino acid 96, and of m14, which changes the nearby amino acid 94. These residues are located in a loop near the end of β-strand G and the beginning of α-helix A in a region that forms H-bonds with the DE-loop of an adjacent dimer at the quasi 3-fold axes (7).
Figure 5.
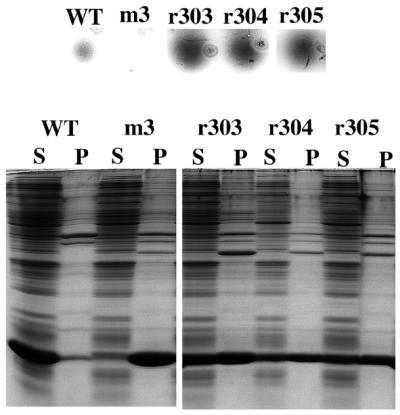
Spot (top) and coat protein solubility (bottom) phenotypes of wild type coat protein, the m3 mutant, and of r303–305, which were isolated as variants of m3 with improved solubility.
DNA sequence analysis revealed that r303, r304 and r305 retain all the m3 substitutions, but that each has also acquired the additional substitution of proline for serine at position 37.
Factors influencing the performance of the overlay method
Choice of expression vector. The results we report here were obtained expressing the coat protein from the T7 promoter on the pET3d vector in BL21(DE3) cells (2). SDS gel electrophoresis of cell lysates shows that coat protein is expressed as a large fraction of total cell protein. However, in some experiments we have expressed coat protein at much lower levels (a few percent of total protein) from the lac promoter in XL1-blue or CSH41 cells using pCT119 (5) and find that spots are easily detected even at these lower expression levels (D.S.Peabody, unpublished results). The vectors we used all confer resistance to ampicillin and this is the reason for augmentin in the overlay.
Colony density. We typically plate cells at no more than 200–300 per 10 cm plate and overlay the colonies when they attain a size of 2–3 mm. At least in cases where most of the colonies produce coat protein, densities approaching 1000 per plate generally give indistinct spots. Higher densities may be possible when looking for the appearance of a spot against a spot-less background.
Agarose concentration and volume of the overlay. For coat protein in the several aggregation states we describe here, the effects of agarose concentration on spot size and intensity over the range of 1–4% are not very pronounced, and our decision to use 2% is somewhat arbitrary. However, the effects of agarose concentration might vary depending on the diffusion coefficients of the species being detected and may require optimization in some applications.
We generally use an overlay volume of 8–10 ml on an 88 mm Petri dish. Much smaller volumes might not reliably cover the colonies. However, volumes larger or smaller than those we used here might help discriminate some diffusing species.
Diffusion time. We have varied the interval between application of the overlay and the introduction of the membrane from 1 to 24 h. Naturally, spots increase in size as time passes, and the discrimination between capsids and coat protein dimers is enhanced by longer diffusion times. In the work described here we routinely allowed 16–24 h for diffusion, but this is another parameter that may require optimization for a specific application. Ultimately, the allowable diffusion time may be limited by the viability of cells in the overlay. With prolonged incubation (i.e. >2 or 3 days) it is sometimes difficult to recover viable cells from these colonies.
Blotting time and number. We typically apply nitrocellulose membranes to the overlay for 5–10 min. Duplicate membranes can be produced by the application of a second filter after removal of the first. However, spot intensity on the second filter is reduced and spots near the limit of detection on the first membrane may become undetectable on the second. Reducing the blotting time of the first membrane relative to the blotting time for the second would presumably equalize the signals.
So far we have restricted ourselves to the use of nitrocellulose membranes, but other media able to bind proteins with a similar capacity should be suitable.
Detection method. We usually employ an alkaline phosphatase-conjugated second antibody to detect anti-MS2 bound to coat protein in spots. We have used [125I]protein A detected by exposure to X-ray film or a phosphorimager screen with similar results.
DISCUSSION
Correlation of spot size and intensity with coat protein assembly and solubility
At the outset of this work we expected that a complex relationship between spot phenotype and the aggregation properties of coat protein mutants was possible. We assumed that spot size and intensity would be functions not only of a protein’s diffusion coefficient, but also of the concentration of the most diffusible species present in what could be a mixture of species in different aggregation states. Indeed, users of this method should bear in mind that spot size and intensity may not always exhibit a strict correspondence with properties like assembly and overall solubility. For example, even a highly diffusible protein could produce a small weak spot if it were present only in small amounts. However, in our collection of more than two dozen coat mutants, the correspondence of spot phenotype with assembly and solubility properties was quite consistent. Proteins with wild-type assembly properties were found in the soluble fraction as 180mer capsids and produced spots of modest size and intensity. Mutants that made little coat protein, or produced it in an insoluble form, generally made spots that were small and weak or even below the threshold of detection, whereas coat proteins that were abundantly synthesized in an assembly-incompetent but soluble form made large, intense spots. Thus, the method provides an efficient means of enriching for mutants with different aggregation properties.
Effects of the various mutations on coat protein structure
Mutations that result in the production of insoluble or proteolytically degraded (i.e. absent) coat protein must affect amino acids whose identities are important for proper folding and/or stability. Most of the mutants we found in this class contain multiple amino acid substitutions. In fact, the mutant we call m26 has nine. This is presumably due to the high level of mutagenesis we used, as it is clearly the case that single substitutions (e.g. m5) can also produce proteins that are insoluble. Nearly all the insoluble or degraded mutants we isolated have at least one substitution in a hydrophobic core residue that might reasonably be expected to compromise protein stability.
Mutations that result in the production of soluble, assembly defective proteins seem to have subtler defects. These coat proteins probably fold normally into dimers that mutation has rendered defective in an inter-molecular contact required for the assembly of stable capsids. The locations of the amino acid substitutions we found in these mutants are consistent with this view; all of them contain at least one substitution at or near a site of inter-dimer contact where it could interfere with capsid formation. Figure 6 shows the locations of amino acids whose substitution results in the production of a big spot together with a significant capsid assembly defect and retention of the ability to synthesize normal amounts of soluble coat protein. Note that the affected residues reside on the edges of the dimer, each of them in a location where it could directly or indirectly affect an inter-dimer contact. Subsequent isolation of about two dozen additional mutants of this type confirms this pattern (D.S.Peabody and L.Al-Bitar, unpublished results). It is not our purpose in this report to describe the structural consequences of all the mutations that conferred assembly defects. However, as an example we describe briefly below the expected effects of the m16 mutation, a substitution of glutamine40 by leucine (Q40L).
Figure 6.

The locations of amino acids (in green space fill) substituted in mutants m11, m12, m13, m14, m16 and m24. These mutants were selected for this illustration because each exhibits a significant assembly defect while producing a normally soluble coat protein and a larger spot than wild type. The two polypeptide chains of the dimer are shown as blue and red ribbons. Amino acids identified previously in a genetic screen for RNA binding defects are shown as yellow spacefill and illustrate the contrasting locations of amino acids implicated in protein–RNA and protein–protein contacts.
The X-ray structure of MS2 shows that important inter-dimer interactions occur between FG loops at the 5- and 3-fold (quasi-6-fold) symmetry axes of the icosahedral capsid (7), and deletions and point mutations in the FG loop often result in failure of coat protein to assemble into capsids (6,8–10). The conformations of the FG loops and the interactions they form between dimers differ depending on their locations in the virus structure. Surrounding the 3-fold (quasi-6-fold) axes the FG loops of six coat proteins adopt extended β-hairpin conformations, while at the 5-fold axes the FG loops are bent. These conformations, especially the bent one, seem to be stabilized by the interaction of each FG loop with Gln40 in the nearby DE loop of the same polypeptide chain. In its extended conformation the FG loop interacts through the main chain carbonyl of Val79 with an H-bond to the side chain amide of Gln40. Meanwhile, in the bent conformation Gln40 forms several H-bonds from its side chain to side chain and main chain atoms of Thr71, Gly73 and Glu76 in the FG loop. Substitution of Gln40 with leucine in the m16 mutant disrupts these interactions and results in failure to produce capsids. Although Gln 40 does not directly form an inter-dimer contact, its stabilization of at least one of the alternative FG-loop conformations, most likely the bent one, is important to the formation of capsid-stabilizing interactions between FG-loops. For additional structural details and illustrations of the interactions between Gln40 and the FG loops the reader is referred to Golmohammadi et al. (7).
We do not yet know the actual aggregation states of the soluble assembly-defective mutants. Inspection of the structure of MS2 shows that the two monomers of the coat protein dimer are intimately intertwined so that each depends on the other for acquisition of its native fold. This suggests that the dimer is the fundamental unit of protein folding and capsid assembly. When inter-dimer interactions are perturbed, as in the case of the dlFG mutant for example, the dimer seems to be the predominant stable species (6). This may also be the case with the soluble assembly-defective mutants we describe here, but additional work will be needed if their actual aggregation properties are to be known. When inter-dimer contacts are only partially disrupted, a range of oligomeric states may be possible through the utilization of residual capsid-like interactions. Some of these aggregates could resemble capsid assembly intermediates. At the same time, however, we suspect that mutations resulting in soluble monomers are rare. We suppose that monomeric species are, by their nature, folding intermediates and probably are prone to aggregation. Therefore, mutation may be disinclined to produce a soluble monomeric species.
The method we have described here is able to distinguish the large molecular size differences represented by MS2 capsids, dimers and insoluble aggregates. Thus, it is useful for isolation of assembly-defective virus mutants. Our ability to isolate soluble variants of the insoluble m3 mutant shows that the overlay method can also be used to improve the solubility of a protein that folds poorly in E.coli, although other recently described methods may be better suited to this specific purpose (11,12). The overlay technique is probably applicable to a wide variety of viruses and other large protein aggregates, assuming they can be expressed and assembled in E.coli or other suitable host. Indeed, the method has already been used to isolate assembly-defective mutants of the core protein of Duck Hepatitis B Virus (J.Summers, personal communication). The extent to which it may be usefully applied to other self-assembling systems remains to be determined.
Finally, we point out that other applications of this method can be imagined. For instance, we think it could be adapted to find inhibitors of virus assembly. One approach to achieving this goal would be to screen a peptide library expressed from a plasmid for sequences that inhibit assembly of a coat protein expressed from a second, compatible plasmid.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Jesse Summers for discussions and for first working out some of the overlay conditions. This work was supported by a grant from the National Institutes of Health.
REFERENCES
- 1.Cadwell R.C. and Joyce,G.F. (1992) Randomization of genes by PCR mutagenesis. PCR Methods Appl., 2, 28–33. [DOI] [PubMed]
- 2.Studier F.W., Rosenberg,A.H., Dunn,J.J and Dubendorff,J.W. (1990) Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol., 185, 60–89. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D. Jessee,J. and Bloom,F.R. (1991) Plasmid transformation of Escherichia coli and other bacteria. Methods Enzymol., 204, 63–113. [DOI] [PubMed] [Google Scholar]
- 4.Laemmli U.K. (1970) Cleavage of structural proteins during assembly of the head of bacteriophage T4. Nature, 227, 680–685. [DOI] [PubMed] [Google Scholar]
- 5.Peabody D.S. (1993) The RNA-binding site of MS2 coat protein. EMBO J., 12, 595–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peabody D.S. and Ely,K.R. (1992) Control of translational repression by protein–protein interactions. Nucleic Acids Res., 20, 1649–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Golmohammadi R., Valegard,K., Fridborg,K. and Liljas,L. (1993) The refined structure of bacteriophage MS2 at 2.8Å resolution. J. Mol. Biol., 234, 620–639. [DOI] [PubMed] [Google Scholar]
- 8.Pushko P., Kozlovskaya,T., Sominskaya,I., Brede,A., Stankevica,E., Ose,V., Pumpens,P. and Grens,E. (1993) Analysis of RNA phage fr coat protein assembly by insertion, deletion and substitution mutatgenesis. Protein Eng., 6, 883–891. [DOI] [PubMed] [Google Scholar]
- 9.Stockley P.G., Stonehouse,N.J., Walton,C., Walters,D.A., Medina,G., Macedo,M.B., Hill,H.R., Goodman,S.T.S., Talbot,S.J., Tewary,H.K., Golmohammadi,R., Liluas,L. and Valegard,K. (1993) Molecular mechanisms of RNA-phage morphogenesis. Biochem. Soc. Trans., 21, 627–633. [DOI] [PubMed] [Google Scholar]
- 10.Stonehouse N.J. and Stockley,P.G. (1993) Effects of amino acid substitutions on the thermal stability of MS2 capsids lacking genomic RNA. FEBS Lett., 334, 355–359. [DOI] [PubMed] [Google Scholar]
- 11.Waldo G.S., Standish,B.M., Berendzen,J. and Terwilliger,T.C. (1999) Rapid protein-folding assay using green fluorescent protein. Nature Biotechnol., 17, 691–695. [DOI] [PubMed] [Google Scholar]
- 12.Wigley W.C., Stidham,R.D., Smith,N.M., Hunt,J.F. and Thomas,P.J. (2001) Protein solubility and folding monitored in vivo by structural complementation of a genetic marker protein. Nature Biotechnol., 19, 131–136. [DOI] [PubMed] [Google Scholar]