

Abstract
We introduce a probabilistic model that enables study of myriad, disparate and fundamental problems in genome science and expands the scope of inference currently possible. Our model formulates an unrecognized unifying goal of many biological studies – to discover sample-specific sequence diversification – and subsumes many application-specific models. With it, we develop a novel algorithm, NOMAD, that performs valid statistical inference on raw reads, completely bypassing references and sample metadata. NOMAD’s reference-free approach enables data-scientifically driven discovery with previously unattainable generality, illustrated with de novo prediction of adaptation in SARS-CoV-2, novel single-cell resolved, cell-type-specific isoform expression, including in the major histocompatibility complex, and de novo identification of V(D)J recombination. NOMAD is a unifying, provably valid and highly efficient algorithmic solution that enables expansive discovery.
One-Sentence Summary:
We present a unifying formulation of disparate genomic problems and design an efficient, reference-free solution.
Introduction
Sequence diversification – mutation, reassortment or rearrangement of nucleic acids – is fundamental to evolution and adaptation across the tree of life. Diversification of pathogen genomes enables host range expansion, and as such host-interacting genes are under intense selective pressure (1). Sequence diversity in these genes is sample-dependent in hosts and in time as dominant strains emerge. In jawed vertebrates, V(D)J recombination and somatic hypermutation generate sequence diversity during the adaptive immune response that varies across cells, and is thus sample-dependent. Sequence diversification in the transcriptome takes the form of regulated RNA-isoform expression. This diversification enables varied phenotypes from the same reference genome including expression programs that enable cell specialization: cell-type specific splicing yields sample-dependent sequence diversity, samples being cells or cell-types. These examples are snapshots of sequence diversification which is core to wide-ranging biological functions, including adaptation, with applications in disparate fields ranging from plant biology to ecological metagenomics, among others (Fig. 1A, Supplement).
Figure 1: Overview of sample-dependent sequence diversity and NOMAD model and pipeline.
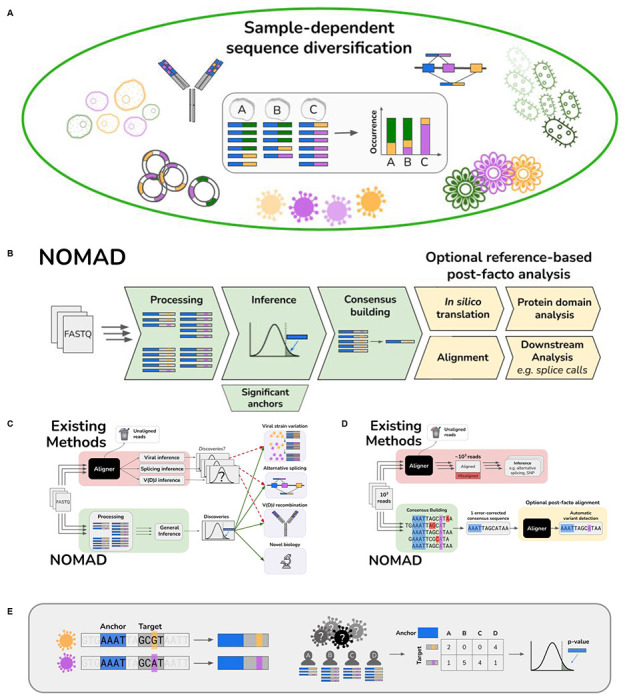
A. Biological generality of sample-dependent sequence diversification. The study of sample-dependent sequence diversification unifies problems in disparate areas of genomics which are currently studied with application-specific models and algorithms. Viral genome mutations, alternative splicing, and V(D)J recombination all fit under this framework, where sequence diversification depends on the sample (through cell-type or infection strain type). Myriad problems in plant genomics, metagenomics and biological adaptation are subsumed by this framework.
B. Overview of NOMAD pipeline. NOMAD takes as input raw FASTQ files for any number of samples >1 and processes them in parallel, counting (anchor, target) pairs per sample. NOMAD performs inference on these aggregated counts, outputting statistically significant anchors. For each significant anchor, a denoised per-sample consensus sequence is built (Fig. 1D). NOMAD also enables optional reference-based post-facto analysis. If a reference genome is available, NOMAD can align the consensus sequences to the reference, enabling denoised downstream analysis (e.g. SNPs, indels, or splice calls). In silico translation of consensuses can optionally be used to study relationships of anchors to protein domains by mapping to databases such as Pfam (Methods).
C. Overview of NOMAD versus existing workflows. Existing workflows (red) discard low-quality reads during FASTQ processing and alignment, only then performing statistical testing after algorithmic bias is introduced; p-values are then not unconditionally valid. Further, for every desired inferential task, a different inference pipeline must be used. NOMAD (green) performs direct statistical inference on raw FASTQ reads, bypassing alignment and enabling data-scientifically driven discovery. Due to its generality, NOMAD can simultaneously detect myriad biological examples of sample-dependent sequence diversification.
D. NOMAD consensus building. NOMAD constructs a per-sample consensus sequence for every significant anchor by taking all reads in which the anchor (blue) appears, and recording plurality votes for each nucleotide, denoising reads while preserving the true variant; sequencing errors in red and biological mutations in purple. Existing approaches require alignment of all reads to a reference prior to error correction, requiring orders of magnitude more computation, discarding reads in both processing and alignment, and potentially making erroneous alignments due to sequencing error. They further require inferential steps, e.g. to detect if there is a SNP or alternatively spliced variant.
E. Example construction of NOMAD anchor, target pairs. A stylized expository example of viral surveillance: 4 individuals A-D are infected with one of two variants (orange and purple), differing by a single basepair (orange and purple). NOMAD anchor k-mers are blue (k=4), followed by a lookahead distance of L=2, and the corresponding k-mer targets. Given sequencing reads from the 4 individuals as shown, NOMAD generates a target by sample contingency table for this blue anchor, and computes a p-value to test if this anchor has sample-dependent sequence diversity.
In this work, we identify the conceptually uniting biological goal in the above problems: to determine sequences with sample-dependent diversity. Biological studies today typically provide deep sampling of the nucleic acid composition, for example through RNA-seq or DNA-seq. In principle, this data provides an opportunity to identify sample-dependent sequence diversity with powerful and efficient statistical models. A timely example is sequencing of SARS-CoV-2: sequences in the spike glycoprotein are diversified as strains compete, for example during the emergence of the omicron variant (2). Patient samples collected in late 2021 include sampling of Delta and Omicron strains, thus containing sample-dependent sequence diversity in regions that differentiate the strains, including but not limited to ones in the spike protein coding region. Statistical approaches should be able to capture this diversity without a reference genome.
Today, genomic algorithms to detect sample-specific diversity are highly specialized to each application and lack conceptual unification, creating several issues. First, biological inference is limited if a workflow is chosen that does not have power to detect significant signals in the data. For example, if the workflow does not map transposable element insertions, none will be found, as these variants are missing from references.
Second, the first step in existing approaches almost always requires the use of references through alignment or their construction de novo. In human genomics, workflows beginning with reference alignment miss important variation absent from assemblies, even with pangenomic approaches. It is well-known that genetic variants associated with ancestry of under-studied populations are poorly represented in databases and result in health disparities (3). In disease genomics (4), sequences of pathogenic cells may be missing from reference genomes, or no reference genome exists, as in genomically unstable tumors (5). Many species do not have reference genomes even when they in principle could be attained due to logistical and computational overheads. In these cases, alignment-based methods we call “reference-first” approaches fail. In viral surveillance, reference-first approaches are even more problematic (6). Viral reference genomes cannot capture the complexity of viral quasispecies (7) or the vast extent of polymorphism (8). New viral assemblies are constantly being added to reference databases (9, 10). In the microbial world, pre-specifying a set of reference genomes is infeasible due to its inherent rapid genomic changes. References also cannot capture insertional diversity of mobile elements, which have significant phenotypic and clinical impact (11) and are only partially cataloged in references (12).
Third, current approaches have severe technical limitations. Statistical analysis in reference-first approaches is conditional on the output of the noisy alignment step, making it difficult or impossible to provide valid statistical significance levels in downstream analyses, such as differential testing or expression analysis. When available, computationally intensive resampling is required.
NOMAD is a statistics-first approach to identify sample-dependent sequence diversification
In this work we first show that detecting sample-dependent sequence diversification can be formulated probabilistically. Second, we reduce this to a statistical test on raw sequencing read data (e.g. FASTQ files). Third, we implement the test in a highly efficient algorithmic workflow, providing novel discovery across disparate biological disciplines (Fig. 1B,C).
To begin, we define an “anchor” k-mer in a read, and say the anchor has sample-dependent diversity if the distribution of k-mers starting R basepairs downstream of it (called “targets”) depends on the sample (Fig. 1E) (13). Inference can be performed for much more general constructions of anchors and targets: any tuple of disjoint subsequences from DNA, RNA or protein sequence data can be analyzed in this framework (Supplement).
This formulation unifies many fundamental problems in genome science. It allows us to develop a novel statistics-first approach, NOMAD (Novel multi-Omics Massive-scale Analysis and Discovery), that is reference-free and operates directly on raw sequencing data. It is an extremely computationally efficient algorithm to detect sample-dependent sequence diversification, through the use of a novel statistic of independent interest that provides closed form p-values (Methods). NOMAD makes all predictions blind to references and annotations, though they can be optionally used for post-facto interpretation. This makes NOMAD fundamentally different from existing methods. To illustrate, as a special case NOMAD is detection of differential isoform expression. In this domain, the closest approach to NOMAD is Kallisto (14) which requires a reference transcriptome and statistical resampling for inference, and is further challenged to provide exact quantification for more than a handful of paralogous genes and isoforms. Unlike NOMAD, Kallisto cannot discover spliced isoforms de novo.
NOMAD’s calls are “significant anchors”: sequences a where, given observing a in a read, the conditional distribution of observing a target sequence t a distance R downstream of a is sample-dependent (Fig. 1E). NOMAD is by default an unsupervised algorithm that does not require any sample identity metadata. It finds approximate best splits of data into two groups (Methods), or it can use user-defined groups if desired. Anchors are reported with an effect size in [0,1], a measure of target distribution difference between sample groups: 0 if the groups have no difference in target distributions and increasing to 1 when the target distributions of the two groups are disjoint. NOMAD has multiple major technical innovations: 1) a parallelizable, fully containerized, and computationally efficient approach to parse FASTQ files into contingency tables, 2) novel statistical analysis of the derived tables, using concentration inequalities to derive closed form p-values, 3) a micro-assembly-based consensus sequence representing the dominant error-corrected sequence, similar to (15, 16), downstream of the anchor for post-facto interpretation and identification of SNPs, indels or isoforms, to name a few (Fig. 1D). If post anchor-identification inference is desired, the consensus, rather than raw reads, is aligned. This reduces the number of reads to align by ~1000x in real data.
Together, NOMAD’s theoretical development yields an extremely computationally efficient implementation. We ran NOMAD on a 2015 Intel laptop with an Intel(R) Core(TM) i7-6500U CPU @ 2.50GHz processor, generating significance calls for single cell RNA-seq totaling over 43 million reads in only 1 hour 45 min. When performed on a compute cluster, the same analysis is completed in an average of 2.28 minutes with 750 MB of memory for 4 million reads, a dramatic speed up over existing methods for de novo splicing detection and significance calls.
NOMAD discovers sequence diversification in proteins at the host-viral interface without a genomic reference
We first show that NOMAD automatically detects viral strain evolution without any knowledge of the sample origin. Existing approaches are computationally-intensive, require genome assemblies and rely on heuristics. Yet, emergent viral threats or variants of concern, e.g. within SARS-CoV-2, will necessarily be absent from reference databases. Because virus’ genomes are under selective pressure to diversify when infecting a host, NOMAD should prioritize anchors near genome sequences that are under selection, in theory, based purely on their statistical features: sequences flanking variants that distinguish strains have consistent sample-dependent sequence diversity. When NOMAD is run on patients differentially infected by Omicron and Delta strains of SARS-CoV-2, significant anchors are expected to be called adjacent to strain-specific mutations (Fig. 2A); they should be, and as we show are, discoverable without any knowledge of a reference.
Figure 2: NOMAD analysis of SARS-CoV-2 data.
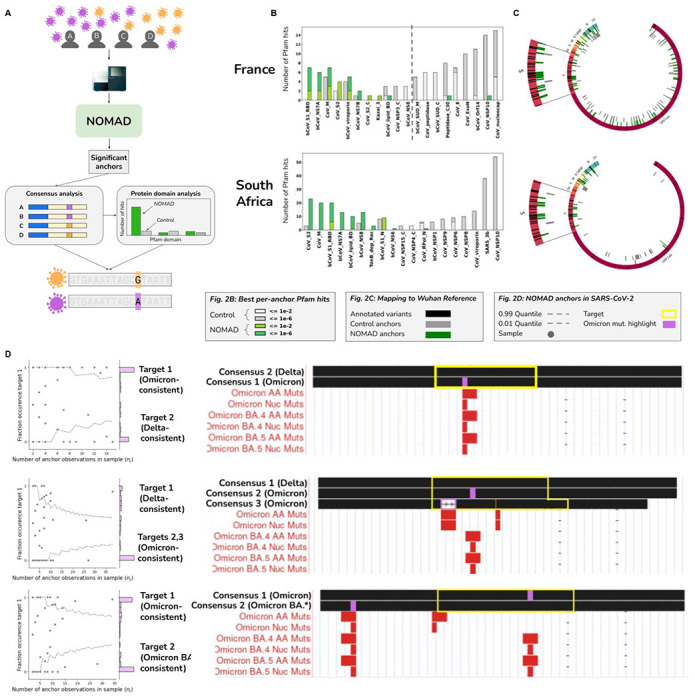
A. Stylized example representing NOMAD workflow for viral data. Patients with varying viral strains are sampled; two representative strains with differentiating mutations are depicted in orange and purple. NOMAD is run on raw FASTQs generated from sequencing patient samples. Significant anchors are called without a reference genome or clinical metadata. Optional post-facto analysis quantifies domain enrichment via in silico translation of consensus sequences derived from NOMAD-called anchors versus controls. Consensuses can also be used to call variants de novo and can be compared to annotated variants e.g. in SARS-CoV-2, Omicron.
B. NOMAD protein profile analysis of SARS-CoV-2. NOMAD SARS-CoV-2 protein profile hits (anchor effect size >.5) to the Pfam database (greens) and control (greys) for France and South Africa datasets; ordered by enrichment in NOMAD hits compared to control showing large distributional differences (chi-squared test p-values France: < 1.1E-12, SA: <2.5E-39). Spike protein domains are highly enriched in the NOMAD versus control. In the France data, the most NOMAD-enriched domain is the betacoronavirus S1 receptor binding domain (hypergeometric p=2.9E-4, corrected) followed by Orf7A (hypergeometric p=1.6E-3, corrected), known to directly interact with the host innate immune defense. In the South Africa data, the most enriched NOMAD profiles are CoV S2 (p=2.9E-6) and the coronavirus membrane protein (p=8.4E-8). Plots were truncated for clarity of presentation as indicated by dashed grey lines (Fig. S2A, B).
C. NOMAD anchors are enriched near annotated variants of concern. NOMAD anchors (effect size >.5) for SARS-CoV2 mapping to the Wuhan reference (NC_045512) show enrichment near variants of concern. SARS-CoV2 genome depicted with annotated ORFs and lines depicting positions of variants of concern (VOC) annotated as Omicron and Delta variants. No control anchor maps to spike or other areas of VOC density except in N (nucleocapsid).
D. NOMAD consensuses identify variants of concern de novo. Examples of NOMAD-detected anchors in SARS-CoV2 (France data). Scatterplots (left) show the fraction of each sample’s observed fraction of target 1 (the most abundant target) for three representative anchors, binomial confidence intervals: (.01,.99), p=empirical fraction occurrence of target 1 (Supplement). y-axis shows histogram of the fraction occurrence of target 1. Mutations (right)found in the targets are highlighted in purple, BLAT shows single nucleotide mutations match known Omicron mutations. Binomial p-values of 6.8E-8, 3.1E-7, and 6.7E-15 respectively (Methods). The anchor in (top) maps to the coronavirus membrane protein; anchors in (middle and bottom) map to the spike protein. One sample (out of 26) depicted in the bottom plot has a consensus mapping perfectly to the Wuhan reference; 3 other consensuses contain annotated Omicron mutations, some designated as VOC in May of 2022, 3 months after these samples were collected.
To test this, we analyzed Oropharyngeal swabs from patients with SARS-CoV-2 from 2021-12-6 to 2022-2-27 in France, a period of known Omicron-Delta coinfection (17). We ran NOMAD and analyzed anchors with effect size >.5, as high effects are predicted if samples can be approximately partitioned by strain, though results are similar without this threshold (fig. S2). For each anchor, we assigned a protein domain label based on in silico translation of its consensus sequence (18). The protein domain with best mapping to the Pfam database is assigned to the anchor, producing a set of “NOMAD protein profiles” (Methods, table S1), and is compared to matched controls (Methods). NOMAD protein profiles are significantly different from controls (p=1.1E-12, chi-squared test, Fig. 2B). The most NOMAD-enriched domains are the receptor binding domain of the betacoronavirus spike glycoprotein (7 NOMAD vs 0 control hits, p=2.9E-4 hypergeometric p-value, corrected,) and Orf7A a transmembrane protein that inhibits host antiviral response (6 NOMAD vs 0 control hits, p=1.6E-3 hypergeometric p-value, corrected). All analysis is blind to the data origin (SARS-CoV-2).
We further analyzed patient samples from the original South African genomic surveillance study that identified the Omicron strain during the period 2021-11-14 to 2021-11-23 (19), again without metadata, reference genomes and directly on input FASTQ files (Methods). NOMAD protein profiles were significantly different from controls (chi-squared test, p=2.5E-39, Fig. 2B). NOMAD-enriched domains in France and South African are highly consistent: the top four domains in both datasets are permutations of each other. The most NOMAD-enriched domains versus controls were the betacoronavirus S2 subunit of the spike protein involved in eliciting the human antibody response (20) (23 NOMAD vs 2 control hits, p=2.9E-6 hypergeometric p-value, corrected), the matrix glycoprotein which interacts with the spike (20 NOMAD vs 0 control hits, p=8.4E-8 hypergeometric p-value, corrected), and the receptor binding domain of the spike protein and to which human antibodies have been detected (21) (20 NOMAD vs 0 control hits, p=8.4E-8 hypergeometric p-value, corrected). All domains are biologically predicted to be under strong selective pressure; NOMAD discovers this de novo.
We further aligned NOMAD anchors with effect size >.5 to the Wuhan reference strain to test if NOMAD anchors rediscovered known strain-defining mutations. We defined a mutation-consistent anchor as one consistent with detecting an Omicron or Delta variant mutation (Supplement). NOMAD anchors are significantly enriched for being mutation-consistent: in the French data, of the uniquely mapped anchors (Wuhan reference), 30.5% (44/144) of NOMAD’s calls were mutation consistent versus 7.6% (6/78) in the control (p=4.0E-5, hypergeometric test). For the South African data, 67.9% (89/131) of NOMAD’s called anchors were mutation consistent vs 20% (12/60) for the control (p=4.4E-10, hypergeometric test).
Examples of mutation-consistent anchors are presented in Fig. 2 including in the membrane (Fig. 2D, top) and spike protein (Fig. 2D, middle and bottom). Differences between consensus sequences and Wuhan reference illustrate NOMAD’s unsupervised rediscovery of annotated strain-specific variants, including co-detection of a deletion and a mutation (Fig. 2D). While the Wuhan reference genome was used for post-facto interpretation, no alignment or sample metadata was used to generate NOMAD’s calls, only to interpret them (Fig. 2). NOMAD consensuses also extend discovery (Fig. 2D), identifying strain-specific variants beyond the target: both are annotated omicron variants (Fig. 2D). NOMAD’s statistical approach automatically links discovered variants within patients de novo: one consensus contains the omicron Variant of Concern (VOC) T22882G; a second consensus has a single VOC T22917G identified in Omicron strains BA.4 and BA.5 in May of 2022, 3 months after the analyzed samples were collected; a third consensus contains the VOC as well as the VOC G22898A; a single further consensus shows no mutations, consistent with Delta infection. Together, this suggests that mutations in BA.4 and BA. 5 were circulating well before the VOC was called in May 2022.
We further analyzed 499 samples collected in California (2020) before viral strain divergence in the spike had been reported (22) as a negative control. No enrichment of NOMAD protein profiles related to the spike or Orf7a domains were observed (fig. S2B), supporting the idea that NOMAD calls are not false positives. To explore the generality of NOMAD for reference-free discovery in other viral infections, we additionally ran NOMAD on a study of influenza-A and of rotavirus breakthrough cases (Fig. S2A,B, Methods). NOMAD Protein profile analysis showed enrichment in domains involved in viral suppression of the host response and regulated alternative splicing (Supplement). Together, this suggests that NOMAD analysis could aid in viral surveillance, including detecting emergence of variants of concern directly from short read sequencing, bypassing a requirement for reference genomes and without manual scrutiny of individual samples or their assembled genomes (23).
NOMAD discovers isoform-specific expression in single cell RNA-seq
NOMAD is a general algorithm to discover sample-dependent sequence diversification in disparate applications including RNA expression and beyond. To illustrate the former, we ran NOMAD without any parameter tuning on single cell RNA-seq datasets, testing if it could perform the fundamental but previously distinct tasks of identifying regulated expression of paralogous genes, alternative splicing and V(D)J recombination (Fig. 3A).
Figure 3: Detection of differentially regulated alternative splicing and isoforms from single cell RNA-seq.
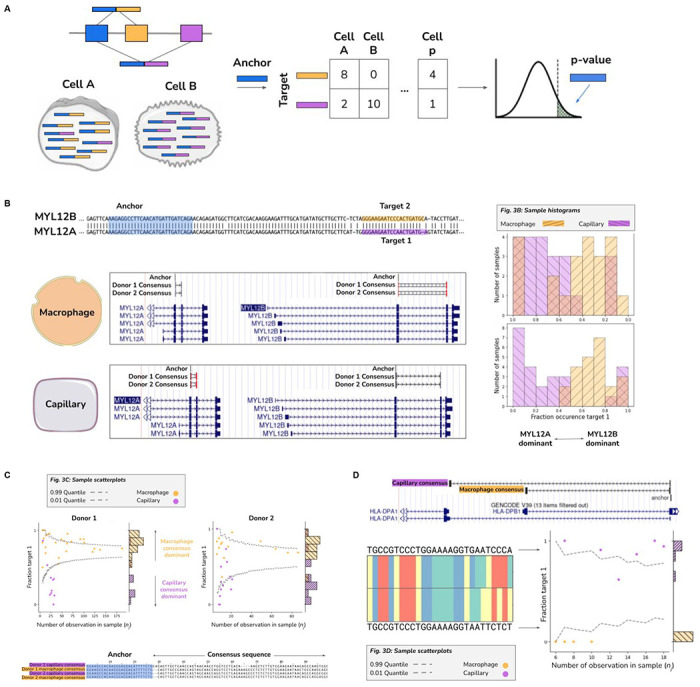
A. Stylized diagram depicting differentially regulated alternative splicing detection with 3 exons and 2 isoforms with NOMAD. Isoform 1 consists of exon 1 (blue) and exon 2 (orange), and is predominantly expressed in cell A. Isoform 2 consists of exon 1 (blue) and exon 3 (purple) and is primarily expressed in cell B.. An anchor sequence in exon 1 (blue), then generates target sequences in exon 2 (orange) or exon 3 (purple). Counts are used to generate a contingency table, and NOMAD’s statistical inference detects this differentially regulated alternative splicing.
B. Detection of differential regulation of MYL12A/B isoforms. (top-left) Shared anchor (q-value 2.5E-8, donor 1, 2.3E-42 for donor 2) highlighted in yellow, maps post fact to both MYL12 isoforms, highlighting the power of NOMAD inference: MYL12A and MYL12B isoforms share >95% nucleotide identity in coding regions. (bottom-left) NOMAD’s approach automatically detects target and consensus sequences that unambiguously distinguish the two isoforms. (right) In both donors, NOMAD reveals differential regulation of MYL12A and MYL12B in capillary cells (MYL12A dominant) and macrophages (MYL12B dominant).
C. NOMAD identifies single-cell-type regulated expression of HLA-DRB1 alleles. NOMAD shared anchor, q-value of 4.0E-10 for donor 1, 1.2E-4 for donor 2. Scatter plots show cell-type regulation of different HLA-DRB1 alleles not explained by a null binomial sampling model p<2E-16 for donor 1, 5.6E-8 for donor 2, finite sample confidence intervals depicted in gray (Methods). Each (donor, cell-type) pair has a dominant target, per-cell fractions represented as “fraction target 1” in scatterplots, and a dominant consensus mapping to the HLA-DRB1 3’ UTR (multiway alignment); donor 1 capillary consensus contains an insertion and deletion.
D. Cell-type specific splicing of HLA-DPA1 in capillary versus macrophage cells. Anchor q-value: 7.9E-22. Detected targets are consistent with macrophages exclusively expressing the short splice isoform which excises a portion of the ORF and changes the 3’ UTR compared to the dominant splice isoform in capillary cells; splice variants found de novo by NOMAD consensuses. Binomial hypothesis test as in D for cell-type target expression depicted in scatter plots (binomial p<2.8E-14).
First, we tested if NOMAD discovers alternatively spliced genes in single cell RNA-seq (Smart-seq 2) of human macrophage versus capillary lung cells, chosen because they have a recently established positive control of alternative splicing, MYL6, a subunit of the myosin light chain (24). NOMAD rediscovered MYL6 and made new discoveries not reported in the literature. For example, we discovered reproducible cell-type specific regulation of MYL12 isoforms, MYL12A and MYL12B. Like MYL6, MYL12 is a subunit of the myosin light chain. In humans (as in many species) two paralogous genes, MYL12A and MYL12B, sharing >95% nucleotide identity in the coding region, are located in tandem on chromosome 18 (Fig. 3B). Reference-first algorithms fail to quantify differential expression of MYL12A and MYL12B due to mapping ambiguity. NOMAD automatically detects targets that unambiguously distinguish the two paralogous isoforms, and demonstrates their clear differential regulation in capillary cells and macrophages (Fig. 3B). We confirmed MYL12 isoform specificity in pairwise comparisons of the same cell types in two further independent single cell sequencing studies of primary cells from the same cell-types (Supplement). MYL12 was recently discovered to mediate allergic inflammation by interacting with CD69 (25); while today little is known about differential functions of the two MYL12 paralogs, the distinct roles of highly similar actin paralogs provides a precedent (26, 27).
NOMAD also called reproducible cell-type specific allelic expression and splicing in the major histocompatibility (MHC) locus (Fig. 3C), the most polymorphic region of the human genome which carries many significant disease risk associations (28). Despite its central importance in human immunity and complex disease, allotypes are difficult to quantify, and statistical methods to reliably distinguish them do not exist. NOMAD finds (i) allele-specific expression of HLA-DRB within cell types and (ii) cell-type specific splicing, predicted to change the amino acid and 3’ UTR sequence of HLA-DPA1 (Methods, Fig. 3D). These empirical results bear out a snapshot of the theoretical prediction that NOMAD’s design gives it high statistical power to simultaneously identify isoform expression variation and allelic expression, including that missed by existing algorithms (table S3).
Unsupervised discovery of B, T cell receptor diversity from single-cell RNA-seq
We next tested if NOMAD could identify T cell receptor (TCR) and B cell receptor (BCR) variants. B and T cell receptors are generated through V(D)J recombination and somatic hypermutation, yielding sequences that are absent from any reference genome and cannot be cataloged comprehensively due to their diversity (>1012) (29). Existing methods to identify V(D)J rearrangement require specialized workflows that depend on receptor annotations and alignment and thus fail when the loci are unannotated (30, 31). In many organisms, including Microcebus murinus, the mouse lemur, T cell receptor loci are incompletely unannotated as they must be manually curated (32)).
We tested if NOMAD could identify TCR and BCR rearrangements in the absence of annotations on 111 natural killer T and 289 B cells isolated from the spleen of two mouse lemurs (Microcebus murinus) profiled by Smart-Seq2 (SS2) (32), and performed the same analysis on a random choice of 50 naive B cells from the peripheral blood and 128 CD4+ T cells from two human donors profiled with SS2 (33) for comparison (Fig. 4A, Methods).
Figure 4: Unsupervised identification of V(D)J recombination in human and lemur immune cells.
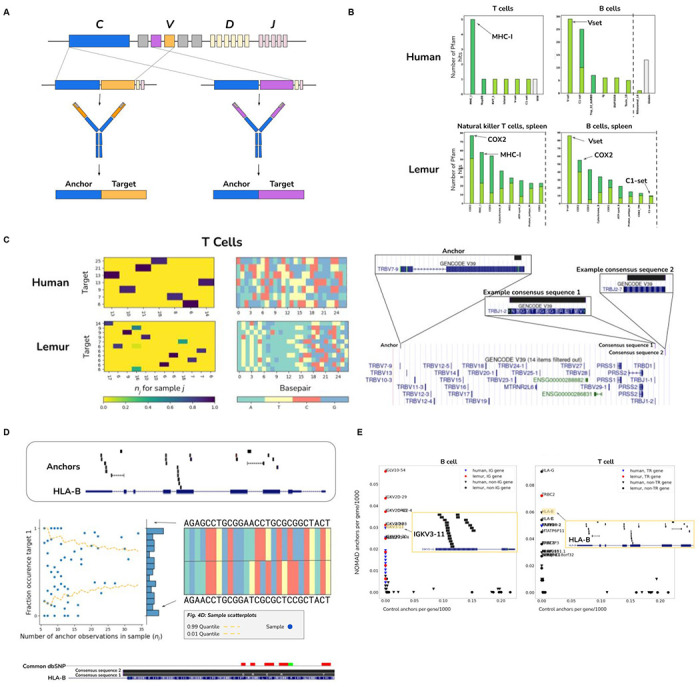
A. Stylized diagram depicting NOMAD detection of V(D)J recombination, with example variable regions in the heavy chain. An anchor sequence in the constant region (blue), generates target sequences (orange and purple) during V(D)J recombination, in which immunoglobulins may receive different gene segments during rearrangement. NOMAD is able to rediscover and detect these recombination events by prioritizing sample-specific TCR and BCR variants.
B. Unsupervised NOMAD protein profile analysis shows MHC and immunoglobulin variable regions are enriched in B and T cells. NOMAD recovers domains known to be diversified in adaptive immune cells, bypassing any genome reference or alignment; control hits computed from the most abundant anchors have no such enrichment. In B cells, hits in the V set, IG like domains resembling the antibody variable region, are at a relatively high E-value, as predicted by protein diversification generated during V(D)J, making matching to reference domains imperfect. The third most hit domain is Tnp_22_dsRBD, a double stranded RNA binding domain, suggesting potential activation of LINE elements in B cells. COX2, known to be involved in immune response, is highly ranked in both lemur T and B cells. Plots were truncated for clarity of presentation as indicated by dashed grey line (Fig. S2F–H).
C. NOMAD detects combinatorial expression of T cell receptors in immune cells de novo. In human T cells (right), we show a NOMAD anchor in the TRVB7-9 gene, and two example consensuses which map to disjoint J segments, TRBJ1-2 and TRBJ2-7. Histograms of this anchor depict combinatorial single-cell (columns) by target (row) expression of targets detected by NOMAD. Histogram for lemur T cells depicted similarly; lemur T cell anchor maps to the human gene TBC1D14.
D. NOMAD detects cell-type and allele-specific expression of HLA-B and HLA-B alleles de novo. NOMAD-annotated anchors are enriched in HLA-B (top Fig. 4.D.1). Sample scatterplot (middle)Fig. 4.D.2 shows that T cells have allelic-specific expression of HLA-B, not explicable by low sampling depth (binomial test as in Fig. 3d,e described in Methods, p< 4.6E-24). Fig. 4.D.3: HLA-B sequence variants are identified de novo by the consensus approach (bottom), including allele-specific expression of two HLA-B variants, one annotated in the genome reference, the other with 5 SNPs coinciding with annotated SNPs.
E. NOMAD analysis of lemur and human B (left) and T (right) cells recovers B, T cell receptors and HLA loci as most densely hit loci. Human genes are depicted as triangles; lemur as circles. Post facto alignments show variable regions in the kappa light chain in human B cells are most densely hit by NOMAD anchors and absent from controls; in T cells, the HLA loci and TRB including its constant and variable region are most densely hit, which are absent from controls. x-axis indicates the fraction of the 1000 control anchors (most abundant anchors) that map to the named transcript, y-axis indicates the fraction of NOMAD’s 1000 most significant anchors that map to the named transcript. Each inset depicts anchor density alignment in the IGKV region (left) and HLA-B in CD4+ T cells (top right) and TRBC-2 (bottom right), showing these regions are densely hit.
NOMAD protein profiles (Fig. 4B), which are blind to the sample’s biological origin and do not require any reference genome, revealed that NOMAD’s most frequent hits in lemur B cell were IG-like domains resembling the antibody variable domain (86 hits), and COX2 (55 hits) a subunit of cytochrome c oxidase, known to be activated in the inflammatory response (34). NOMAD’s top hits for Lemur T cell were COX2 and MHC_I (77 and 58 hits, respectively). Similar results were obtained for the human samples (Fig. 4B). They include novel predictions of cell-type specific allelic expression of HLA-B in T cells (Fig. 4D, Supplement) where NOMAD found statistical evidence that cells preferentially express a single allele (p< 4,6E-24); consensus analysis shows SNPs in HLA-B are concordant with known positions of polymorphism.
We further predicted that BCR and TCR rearrangements would also be discovered by investigating the transcripts most hit by NOMAD anchors. We mapped NOMAD-called lemur B and T cell anchors to an approximation of its transcriptome: that from humans which diverged from lemur ~60-75 million years ago (35). Lemur B cell anchors most frequently hit the immunoglobulin light and kappa variable regions; lemur T cell anchors most frequently hit the HLA and T cell receptor family genes (Methods, Fig. 4C). Similar results are found in human B and T cells (Fig. 4E, Supplement). Transcripts with the most hits in the control were unrelated to immune function. To further illustrate NOMAD’s power, consider its comparison to existing pipelines. They cannot be run without the annotation for the lemur TCR locus; for assembling BCR sequences, pipelines e.g. BASIC (31) cannot always identify V(D)J rearrangement, including in some cells profiled in the lemur dataset (32). We selected the 35 B/plasma cells where BASIC could not programmatically identify variable gene families on the light chain variable region. NOMAD automatically identified anchors mapping to the IGLV locus, with consensus sequences that BLAST to the light chain variable region (Supplement). Together, this shows that NOMAD identifies sequences with adaptive immune function including V(D)J in both B and T cells de novo, using either no reference genome (protein profile analysis) or only an annotation guidepost from a related organism (human). In addition to being simple and unifying, NOMAD can extend discovery compared to custom pipelines.
Conclusion
We have shown that problems from disparate subfields of genomic data-science are unified in their goal to discover sample-dependent sequence diversification; NOMAD is a statistics-first algorithm that formulates and efficiently solves this task, with great generality.
We provided a snapshot of NOMAD’s discoveries in disparate areas in genome science. In SARS-CoV-2 patient samples during the emergence of the omicron variant, it finds the spike protein is highly enriched for sequence diversification, bypassing genome alignment completely. NOMAD provides evidence that Variants of Concern can be detected well before they are flagged as such or added to curated databases. This points to a broader impact for NOMAD in viral and other genomic surveillance, since emerging pathogens will likely have sequence diversification missing from any reference.
NOMAD finds novel cell-type specific isoform expression in homologous genes missed by reference-guided approaches, such as in MYL12A/B and in the MHC (HLA) locus, even in a small sample of single cell data. Highly polymorphic and multicopy human genes have been recalcitrant to current genomic analyses and are critical to susceptibility to infectious and complex diseases, e.g. the MHC. NOMAD could shed new light on other polymorphic loci including non-coding RNAs, e.g. spliceosomal variants (36, 37). In addition, NOMAD unifies detection of many other examples of transcriptional regulation: intron retention, alternative linear splicing, allele-specific splicing, gene fusions, and circular RNA. Further, NOMAD prioritizes V(D)J recombination as the most sample-specific sequencing diversifying process in B and T cells of both human and mouse lemur, where inference in lemur is made using only an approximate genomic reference (human) which diverged from lemur ~60 million years ago.
Disparate data – DNA and protein sequence, or any “-omics” experiment, from Hi-C to spatial transcriptomics – can be analyzed in the NOMAD framework. NOMAD may also be impactful in analysis of plants, microbes, and mobile elements, including transposable elements and retrotransposons, which are far less well annotated, and are so diverse that references may never capture them.
NOMAD illustrates the power of statistics-first genomic analysis with optional use of references for post-facto interpretation. It translates the field’s “reference-first” approach to “statistics-first”, performing direct statistical hypothesis tests on raw sequencing data, enabled by its probabilistic modeling of raw reads rather than of alignment outputs. By design, NOMAD is highly efficient: it will enable direct, large-scale study of sample-dependent sequence diversification, completely bypassing the need for references or assemblies. NOMAD promises data-driven biological study previously impossible.
Limitations of the study
Naturally, some problems cannot be formulated in the manner posed, such as cases where the estimand is RNA or DNA abundance. However, the problems that can be addressed using this formulation span diverse fields which are of great current importance (Supplement), including those previously discussed. Further, NOMAD’s statistical test can be applied to tables of gene expression (including as measured by k-mers) by samples.
Supplementary Material
Acknowledgements:
We thank Elisabeth Meyer for extensive edits, figure help, and assistance with exposition; Arjun Rustagi for extensive discussion and assistance in interpretation of viral biology; Roozbeh Dehghannasiri for selecting and collating data from HLCA/TSP/Tabula Microcebus analysis, running the SpliZ, and feedback on presentation; the Tabula Microcebus Consortium (Camille Ezran and Hannah Frank) for data sharing and discussion of B and T cell receptor detection algorithms; Jessica Klein for extensive figure assistance; Michael Swift for discussion of B and T cell receptor detection algorithms; Julia Olivieri for feedback on presentation and detailed comments on the manuscript; Andy Fire for useful discussions and feedback on the manuscript; Robert Bierman for figure assistance and laptop timing; and Aaron Straight, Catherine Blish, Peter Kim, and all members of the Salzman lab for feedback and comments.
Funding:
Stanford University Discovery Innovation Award (J.S.)
National Institute of General Medical Sciences grant R35 GM139517 (J.S.)
National Science Foundation Faculty Early Career Development Program Award MCB1552196 (J.S.)
National Science Foundation Graduate Research Fellowship Program (T.Z.B.)
Stanford Graduate Fellowship (T.Z.B.)
Footnotes
Competing interests
K.C., T.Z.B., and J.S. are inventors on provisional patents related to this work.
The authors declare no other competing interests.
Data and materials availability
The code used in this work is available as a fully-containerized Nextflow pipeline (38) at https://github.com/kaitlinchaung/nomad.
The human lung scRNA-seq data used here is accessible through the European Genome-phenome Archive (accession number: EGAS00001004344); FASTQ files from donor 1 and donor 2 were used. The FASTQ files for the Tabula Sapiens data (both 10X Chromium and Smart-seq2) were downloaded from https://tabula-sapiens-portal.ds.czbiohub.org/: B cells were used from donor 1 and T cells from donor 2. The mouse lemur single-cell RNA-seq data used in this study was generated as part of the Tabula Microcebus consortium; the FASTQ files were downloaded from https://tabula-microcebus.ds.czbiohub.org. Viral data was downloaded from the NCBI: SARS-CoV-2 from France (SRP365166), SARS-CoV-2 from South Africa (SRP348159), 2020 SARS-CoV-2 from California (SRR15881549), influenza (SRP294571), and rotavirus (SRP328899).
The sample sheets used as pipeline input, including individual sample SRA accession numbers, for all analyses are uploaded to pipeline GitHub repository. Similarly, scripts to perform supplemental analysis can be found on the pipeline repository.
References and Notes
- 1.Yang L., Emerman M., Malik H. S., McLaughlin R. N., Retrocopying expands the functional repertoire of APOBEC3 antiviral proteins in primates. eLife. 9 (2020), doi: 10.7554/eLife.58436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang L., Cheng G., Sequence analysis of the emerging SARS-CoV-2 variant Omicron in South Africa. J. Med. Virol. 94, 1728–1733 (2022). [DOI] [PubMed] [Google Scholar]
- 3.West K. M., Blacksher E., Burke W., Genomics, health disparities, and missed opportunities for the nation’s research agenda. JAMA. 317, 1831–1832 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang T., Antonacci-Fulton L., Howe K., Lawson H. A., Lucas J. K., Phillippy A. M., Popejoy A. B., Asri M., Carson C., Chaisson M. J. P., Chang X., Cook-Deegan R., Felsenfeld A. L., Fulton R. S., Garrison E. P., Garrison N. A., Graves-Lindsay T. A., Ji H., Kenny E. E., Koenig B. A., Human Pangenome Reference Consortium, The Human Pangenome Project: a global resource to map genomic diversity. Nature. 604, 437–446 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang C.-Z., Spektor A., Cornils H., Francis J. M., Jackson E. K., Liu S., Meyerson M., Pellman D., Chromothripsis from DNA damage in micronuclei. Nature. 522, 179–184 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.The Nucleic Acid Observatory Consortium, A Global Nucleic Acid Observatory for Biodefense and Planetary Health. arXiv (2021), doi: 10.48550/arxiv.2108.02678. [DOI] [Google Scholar]
- 7.Kirkegaard K., van Buuren N. J., Mateo R., My Cousin, My Enemy: quasispecies suppression of drug resistance. Curr. Opin. Virol. 20, 106–111 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim D., Lee J.-Y., Yang J.-S., Kim J. W., Kim V. N., Chang H., The Architecture of SARS-CoV-2 Transcriptome. Cell. 181, 914–921.e10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Edgar R. C., Taylor J., Lin V., Altman T., Barbera P., Meleshko D., Lohr D., Novakovsky G., Buchfink B., Al-Shayeb B., Banfield J. F., de la Peña M., Korobeynikov A., Chikhi R., Babaian A., Petabase-scale sequence alignment catalyses viral discovery. Nature. 602, 142–147 (2022). [DOI] [PubMed] [Google Scholar]
- 10.Zayed A. A., Wainaina J. M., Dominguez-Huerta G., Pelletier E., Guo J., Mohssen M., Tian F., Pratama A. A., Bolduc B., Zablocki O., Cronin D., Solden L., Delage E., Alberti A., Aury J.-M., Carradec Q., da Silva C., Labadie K., Poulain J., Ruscheweyh H.-J., Wincker P., Cryptic and abundant marine viruses at the evolutionary origins of Earth’s RNA virome. Science. 376, 156–162 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Evans D. R., Griffith Μ. P., Sundermann A. J., Shutt K. A., Saul Μ. I., Mustapha Μ. M., Marsh J. W., Cooper V. S., Harrison L. H., Van Tyne D., Systematic detection of horizontal gene transfer across genera among multidrug-resistant bacteria in a single hospital. eLife. 9 (2020), doi: 10.7554/eLife.53886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wright R. J., Comeau A. M., Langille M. G. I., From defaults to databases: parameter and database choice dramatically impact the performance of metagenomic taxonomic classification tools. BioRxiv (2022), doi: 10.1101/2022.04.27.489753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abante J., Wang P. L., Salzman J., DIVE: a reference-free statistical approach to diversity-generating and mobile genetic element discovery. BioRxiv (2022), doi: 10.1101/2022.06.13.495703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bray N. L., Pimentel H., Melsted P., Pachter L., Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Motahari A., Ramchandran K., Tse D., Ma N., in 2013 IEEE International Symposium on Information Theory (IEEE, 2013), pp. 1640–1644.
- 16.Magoč T., Salzberg S. L., FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 27, 2957–2963 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bal A., Simon B., Destras G., Chalvignac R., Semanas Q., Oblette A., Queromes G., Fanget R., Regue H., Morfin F., Valette M., Lina B., Josset L., Detection and prevalence of SARS-CoV-2 co-infections during the Omicron variant circulation, France, December 2021 - February 2022. medRxiv (2022), doi: 10.1101/2022.03.24.22272871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mistry J., Chuguransky S., Williams L., Qureshi M., Salazar G. A., Sonnhammer E. L. L., Tosatto S. C. E., Paladin L., Raj S., Richardson L. J., Finn R. D., Bateman A., Pfam: The protein families database in 2021. Nucleic Acids Res. 49, D412–D419 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Viana R., Moyo S., Amoako D. G., Tegally H., Scheepers C., Althaus C. L., Anyaneji U. J., Bester P. A., Boni M. F., Chand M., Choga W. T., Colquhoun R., Davids M., Deforche K., Doolabh D., du Plessis L., Engelbrecht S., Everatt J., Giandhari J., Giovanetti M., de Oliveira T., Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature. 603, 679–686 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poh C. M., Carissimo G., Wang B., Amrun S. N., Lee C. Y.-P., Chee R. S.-L., Fong S.-W., Yeo N. K.-W., Lee W.-H., Torres-Ruesta A., Leo Y.-S., Chen M. I.-C., Tan S.-Y., Chai L. Y. A., Kalimuddin S., Kheng S. S. G., Thien S.-Y., Young B. E., Lye D. C., Hanson B. J., Ng L. F. P., Two linear epitopes on the SARS-CoV-2 spike protein that elicit neutralising antibodies in COVID-19 patients. Nat. Commun. 11, 2806 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jörrißen P., Schütz P., Weiand M., Vollenberg R., Schrempf I. M., Ochs K., Frömmel C., Tepasse P.-R., Schmidt H., Zibert A., Antibody Response to SARS-CoV-2 Membrane Protein in Patients of the Acute and Convalescent Phase of COVID-19. Front. Immunol. 12, 679841 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gorzynski J. E., De Jong Η. N., Amar D., Hughes C. R., Ioannidis A., Bierman R., Liu D., Tanigawa Y., Kistler A., Kamm J., Kim J., Cappello L., Neff N. F., Rubinacci S., Delaneau O., Shoura M. J., Seo K., Kirillova A., Raja A., Sutton S., Parikh V. N., High-throughput SARS-CoV-2 and host genome sequencing from single nasopharyngeal swabs. medRxiv (2020), doi: 10.1101/2020.07.27.20163147. [DOI] [Google Scholar]
- 23.Jacot D., Pillonel T., Greub G., Bertelli C., Assessment of SARS-CoV-2 Genome Sequencing: Quality Criteria and Low-Frequency Variants. J. Clin. Microbiol. 59, e0094421 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olivieri J. E., Dehghannasiri R., Wang P. L., Jang S., de Morree A., Tan S. Y., Ming J., Ruohao Wu A., Quake S. R., Krasnow M. A., Salzman J., RNA splicing programs define tissue compartments and cell types at single-cell resolution. eLife. 10 (2021), doi: 10.7554/eLife.70692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hayashizaki K., Kimura M. Y., Tokoyoda K., Hosokawa H., Shinoda K., Hirahara K., Ichikawa T., Onodera A., Hanazawa A., Iwamura C., Kakuta J., Muramoto K., Motohashi S., Tumes D. J., Iinuma T., Yamamoto H., Ikehara Y., Okamoto Y., Nakayama T., Myosin light chains 9 and 12 are functional ligands for CD69 that regulate airway inflammation. Sci. Immunol. 1, eaaf9154 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Vedula P., Kurosaka S., Leu N. A., Wolf Y. I., Shabalina S. A., Wang J., Sterling S., Dong D. W., Kashina A., Diverse functions of homologous actin isoforms are defined by their nucleotide, rather than their amino acid sequence. eLife. 6 (2017), doi: 10.7554/eLife.31661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perrin B. J., Ervasti J. M., The actin gene family: function follows isoform. Cytoskeleton (Hoboken). 67, 630–634 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matzaraki V., Kumar V., Wijmenga C., Zhemakova A., The MHC locus and genetic susceptibility to autoimmune and infectious diseases. Genome Biol. 18, 76 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Briney B., Inderbitzin A., Joyce C., Burton D. R., Commonality despite exceptional diversity in the baseline human antibody repertoire. Nature. 566, 393–397 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lindeman I., Emerton G., Mamanova L., Snir O., Polanski K., Qiao S.-W., Sollid L. M., Teichmann S. A., Stubbington M. J. T., BraCeR: B-cell-receptor reconstruction and clonality inference from single-cell RNA-seq. Nat. Methods. 15, 563–565 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Canzar S., Neu K. E., Tang Q., Wilson P. C., Khan A. A., BASIC: BCR assembly from single cells. Bioinformatics. 33, 425–427 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.The Tabula Microcebus Consortium, Ezran C., Liu S., Chang S., Ming J., Botvinnik O., Penland L., Tarashansky A., de Morree A., Travaglini K. J., Hasegawa K., Sin H., Sit R., Okamoto J., Sinha R., Zhang Y., Karanewsky C. J., Pendleton J. L., Morn M., Perret M., Krasnow M. A., Tabula Microcebus: A transcriptomic cell atlas of mouse lemur, an emerging primate model organism. BioRxiv (2021), doi: 10.1101/2021.12.12.469460. [DOI] [Google Scholar]
- 33.Tabula Sapiens Consortium, Jones R. C., Karkanias J., Krasnow M. A., Pisco A. O., Quake S. R., Salzman J., Yosef N., Bulthaup B., Brown P., Harper W., Hemenez M., Ponnusamy R., Salehi A., Sanagavarapu B. A., Spallino E., Aaron K. A., Concepcion W., Gardner J. M., Kelly B., et al. , The Tabula Sapiens: A multiple-organ, single-cell transcriptomic atlas of humans. Science. 376, eabl4896 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Groeger A. L., Cipollina C., Cole Μ. P., Woodcock S. R., Bonacci G., Rudolph T. K., Rudolph V., Freeman B. A., Schopfer F. J., Cyclooxygenase-2 generates anti-inflammatory mediators from omega-3 fatty acids. Nat. Chem. Biol. 6, 433–441 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ezran C., Karanewsky C. J., Pendleton J. L., Sholtz A., Krasnow M. R., Willick J., Razafindrakoto A., Zohdy S., Albertelli M. A., Krasnow M. A., The mouse lemur, a genetic model organism for primate biology, behavior, and health. Genetics. 206, 651–664 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buen Abad Najar C. F., Yosef N., Lareau L. F., Coverage-dependent bias creates the appearance of binary splicing in single cells. BioRxiv (2019), doi: 10.1101/2019.12.19.883256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuo S.-M., Chen C.-J., Chang S.-C., Liu T.-J., Chen Y.-H., Huang S.-Y., Shih S.-R., Inhibition of Avian Influenza A Virus Replication in Human Cells by Host Restriction Factor TUFM Is Correlated with Autophagy. MBio. 8 (2017), doi: 10.1128/mBio.00481-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Di Tommaso P., Chatzou M., Floden E. W., Barja P. P., Palumbo E., Notredame C., Nextflow enables reproducible computational workflows. Nat. Biotechnol. 35, 316–319 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Salzman J., Jiang H., Wong W. H., Statistical Modeling of RNA-Seq Data. Slat. Sci. 26 (2011), doi: 10.1214/10-STS343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Agresti A., A Survey of Exact Inference for Contingency Tables. Stat. Sci. 7, 131–153 (1992). [Google Scholar]
- 41.Diaconis P., Sturmfels B., Algebraic algorithms for sampling from conditional distributions. Ann. Statist. 26 (1998), doi: 10.1214/aos/1030563990. [DOI] [Google Scholar]
- 42.Chen Y., Diaconis P., Holmes S. P., Liu J. S., Sequential monte carlo methods for statistical analysis of tables. J. Am. Stat. Assoc. 100, 109–120 (2005). [Google Scholar]
- 43.Benjamini Y., Yekutieli D., The control of the false discovery rate in multiple testing under dependency. Arm. Statist. 29, 1165–1188 (2001). [Google Scholar]
- 44.Stahlberg A., Krzyzanowski P. M., Jackson J. B., Egyud M., Stein L., Godfrey T. E., Simple, multiplexed, PCR-based barcoding of DNA enables sensitive mutation detection in liquid biopsies using sequencing. Nucleic Acids Res. 44, el05 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kalvari I., Nawrocki E. P., Ontiveros-Palacios N., Argasinska J., Lamkiewicz K., Marz M., Griffiths-Jones S., Toffano-Nioche C., Gautheret D., Weinberg Z., Rivas E., Eddy S. R., Finn R. D., Bateman A., Petrov A. I., Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 49, D192–D200 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Storer J., Hubley R., Rosen J., Wheeler T. J., Smit A. F., The Dfam community resource of transposable element families, sequence models, and genome annotations. Mob. DNA. 12, 2 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ross K., Varani A. M., Snesrud E., Huang H., Alvarenga D. O., Zhang J., Wu C., McGann P., Chandler M., Tncentral: a prokaryotic transposable element database and web portal for transposon analysis. MBio. 12, e0206021 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leplae R., Hebrant A., Wodak S. J., Toussaint A., ACLAME: a CLAssification of Mobile genetic Elements. Nucleic Acids Res. 32, D45–9 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bi D., Xu Z., Harrison E. M., Tai C., Wei Y., He X., Jia S., Deng Z., Rajakumar K., Ou H.-Y., ICEberg: a web-based resource for integrative and conjugative elements found in Bacteria. Nucleic Acids Res. 40, D621–6 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Couvin D., Bernheim A., Toffano-Nioche C., Touchon M., Michalik J., Néron B., Rocha E. P. C., Vergnaud G., Gautheret D., Poured C., CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 46, W246–W251 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Santamaria M., Fosso B., Licciulli F., Balech B., Larini I., Grillo G., De Caro G., Liuni S., Pesole G., ITSoneDB: a comprehensive collection of eukaryotic ribosomal RNA Internal Transcribed Spacer 1 (ITS1) sequences. Nucleic Acids Res. 46, D127–D132 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Selig C., Wolf M., Muller T., Dandekar T., Schultz J., The ITS2 Database II: homology modelling RNA structure for molecular systematics. Nucleic Acids Res. 36, D377–80 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shen W., Le S., Li Y., Hu F., SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE. 11, e0163962 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Johnson L. S., Eddy S. R., Portugaly E., Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinformatics. 11, 431 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thompson M. G., Dittmar M., Mallory M. J., Bhat P., Ferretti Μ. B., Fontoura B. M., Cherry S., Lynch K. W., Viral-induced alternative splicing of host genes promotes influenza replication. eLife. 9 (2020), doi: 10.7554/eLife.55500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun X., Whittaker G. R., Role of the actin cytoskeleton during influenza virus internalization into polarized epithelial cells. Cell. Microbiol. 9, 1672–1682 (2007). [DOI] [PubMed] [Google Scholar]
- 57.Song Y., Feng N., Sanchez-Tacuba L., Yasukawa L. L., Ren L., Silverman R. H., Ding S., Greenberg Η. B., Reverse genetics reveals a role of rotavirus VP3 phosphodiesterase activity in inhibiting mase L signaling and contributing to intestinal viral replication in vivo. J. Virol. 94 (2020), doi: 10.1128/JVI.01952-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gratia M., Sarot E., Vende R., Charpilienne A., Baron C. H., Duarte M., Pyronnet S., Poncet D., Rotavirus NSP3 Is a Translational Surrogate of the Poly(A) Binding Protein-Poly(A) Complex. J. Virol. 89, 8773–8782 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R., STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kiepiela P., Leslie A. J., Honeyborne I., Ramduth D., Thobakgale C., Chetty S., Rathnavalu P., Moore C., Pfafferott K. J., Hilton L., Zimbwa P., Moore S., Allen T., Brander C., Addo Μ. M., Altfeld M., James I., Mallal S., Bunce M., Barber L. D., Goulder P. J. R., Dominant influence of HLA-B in mediating the potential co-evolution of HIV and HLA. Nature. 432, 769–775 (2004). [DOI] [PubMed] [Google Scholar]
- 61.Elahi S., Dinges W. L., Lejarcegui N., Laing K. J., Collier A. C., Koelle D. M., McElrath M. J., Horton H., Protective HIV-specific CD8+ T cells evade Treg cell suppression. Nat. Med. 17, 989–995 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Francis J. M., Leistritz-Edwards D., Dunn A., Tarr C., Lehman J., Dempsey C., Hamel A., Rayon V., Liu G., Wang Y., Wille M., Durkin M., Hadley K., Sheena A., Roscoe B., Ng M., Rockwell G., Manto M., Gienger E., Nickerson J., Pregibon D. C., Allelic variation in class I HLA determines CD8+ T cell repertoire shape and cross-reactive memory responses to SARS-CoV-2. Sci. Immunol. 7, eabk3070 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Medhekar B., Miller J. F., Diversity-generating retroelements. Carr. Opin. Microbiol. 10, 388–395 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fisher R. A., On the Interpretation of X2 from Contingency Tables, and the Calculation of P. Journal of the Royal Statistical Society. 85, 87 (1922). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The code used in this work is available as a fully-containerized Nextflow pipeline (38) at https://github.com/kaitlinchaung/nomad.
The human lung scRNA-seq data used here is accessible through the European Genome-phenome Archive (accession number: EGAS00001004344); FASTQ files from donor 1 and donor 2 were used. The FASTQ files for the Tabula Sapiens data (both 10X Chromium and Smart-seq2) were downloaded from https://tabula-sapiens-portal.ds.czbiohub.org/: B cells were used from donor 1 and T cells from donor 2. The mouse lemur single-cell RNA-seq data used in this study was generated as part of the Tabula Microcebus consortium; the FASTQ files were downloaded from https://tabula-microcebus.ds.czbiohub.org. Viral data was downloaded from the NCBI: SARS-CoV-2 from France (SRP365166), SARS-CoV-2 from South Africa (SRP348159), 2020 SARS-CoV-2 from California (SRR15881549), influenza (SRP294571), and rotavirus (SRP328899).
The sample sheets used as pipeline input, including individual sample SRA accession numbers, for all analyses are uploaded to pipeline GitHub repository. Similarly, scripts to perform supplemental analysis can be found on the pipeline repository.