

Abstract
Recent increases in human longevity have been accompanied by a rise in the incidence of dementia, raising an important question about how to preserve cognitive functioning in an aging population. Interestingly, a small percentage of individuals with pathological hallmarks of neurodegenerative disease are able to maintain normal cognition. The molecular mechanisms that govern this neuro-protected state remain unknown, but individuals that exhibit cognitive resilience (CgR) represent a unique source of insight into potential therapies that could preserve brain function in the face of aging and neurodegenerative disease. In both humans and animal models, living in an enriched, cognitively stimulating environment is the most effective known inducer of CgR. To gain insight into potential modulators of this phenomenon, we began by studying the molecular changes that arise from environmental enrichment in mice. Global chromatin and transcriptomic profiling of cortical neurons led to the identification of Mef2a and Mef2c (Mef2a/c) transcription factors, known to be induced by neuronal activity, as potential coordinators of the neuronal response to enrichment. Conditional knockdown of Mef2a/c prior to enrichment blocks the cognitive enhancement afforded by an enriched environment. In order to assess the importance of Mef2 activity in regulating CgR in humans, we utilize repositories of clinical and brain transcriptomic data, where we find that levels of MEF2C are positively associated with cognitive ability, and that MEF2 target genes are significantly overrepresented among the genes that are most predictive of cognition. Through single-nucleus RNA-sequencing of cortical tissue from a cohort of resilient and non-resilient individuals, we pinpoint upregulation of MEF2C and its transcriptional network in resilient individuals to a subpopulation of excitatory neurons. Finally, to determine the causal impact of Mef2 on cognition in the context of neurodegeneration, we overexpress Mef2a/c in the PS19 mouse model of tauopathy. Remarkably, Mef2a/c overexpression alone is sufficient to improve cognition and reduce hyperexcitability in PS19 mice. Overall, our findings reveal a novel role for MEF2 TFs in promoting cognition and cognitive resilience, highlighting their potential as novel biomarkers or therapeutic targets for healthy aging and neurodegeneration.
One Sentence Summary:
Mef2 is induced upon chronic enrichment in mice, reduces disease-associated hyperexcitability, and is a feature of cognitive resilience in AD patients.
Introduction:
Human lifespan has increased substantially – nearly a decade in the Unites States in the past fifty years – and great strides are being made to identify modifiable targets that might further enhance longevity(1). However, this extension of lifespan is often associated with a decrease in quality of life(2), highlighting that measures to extend lifespan must also address how to maintain functional capacity as we age. One of the biggest obstacles to healthy aging is the significant burden of age-related neurodegenerative diseases, like Alzheimer’s Disease, which almost always result profound cognitive impairment and decline. Here, we aim to identify molecular features that preserve cognitive function and could serve as therapeutic targets for neurodegenerative diseases and healthy aging.
Clinicians and scientists have long documented a robust epidemiological link between cognitive, social, and physical stimulation throughout life and subsequent reduced manifestation of neurological disease in adulthood and old age(3). As a whole, these healthy lifestyle factors, or forms of enrichment, are the most effective strategies that exist for enhancing cognitive function at baseline and enabling cognitive resilience (CgR) to multiple forms of brain pathology later in life. They represent the only known preventative measure against aging- and disease-related cognitive decline.
Rodent studies have further confirmed the causal role that an enriched lifestyle has in mediating CgR: providing environmental enrichment is sufficient to preserve cognition in the context of genetically-induced neurodegeneration(4, 5). In addition to improved cognitive performance, enrichment in rodents also reverses behavioral sensitization to cocaine(6), and reduces symptoms of anxiety and depression after exposure to chronic stress(7). The extensive positive effects of an enriched environment that manifest throughout life and in response to a variety of stressors underscore the need to understand the mechanisms through which this neuroprotection and resilience are afforded. A limited number of studies have begun to identify targets/pathways that can be harnessed to confer CgR(8), but the molecular drivers endogenously recruited to actualize the phenotypic benefits of cognitive stimulation remain unknown. Elucidation of these mechanisms could lead to novel therapeutic targets for promoting cognitive health across the lifespan. To advance these efforts, we (i) use a mouse model of environmental enrichment to recapitulate the benefits of sustained cognitive stimulation and identify key molecular regulators, and (ii) analyze three cohorts of clinically-curated brain RNA-sequencing data to characterize the transcriptional underpinnings of CgR in humans. These complementary approaches led us to uncover the Mef2 family of transcription factors as candidate targets, and by upregulating Mef2 TFs in a mouse model of neurodegeneration, we confirm MEF2s critical role as central drivers of environment-induced neuroprotection to cognitive decline.
Previous work on the Mef2 family (Mef2a-d) has examined their role in neurodevelopment, implicating various members in maintaining appropriate neural transmission and regulating synaptic density(9, 10). Additionally, Mef2 transcriptional activity is induced by neuronal activity(11), highlighting the importance of this family in regulating molecular responses to neural stimulation. Interestingly, genetic variants within the MEF2C loci have been associated with differences in human intelligence(12), implicating this family in the expression of higher order cognitive ability. Our findings extend the function of MEF2 beyond its well-studied roles in neurodevelopment and activity-dependent gene regulation, by establishing it as a critical and specific mediator of cognitive health across the lifespan and in the context of neurodegeneration.
Results:
Subhead 1: Cognitive stimulation improves cognition and results in global epigenomic changes in cortical neurons of both male and female mice
The strongest epidemiological predictors of CgR are mentally stimulating activities such as high educational attainment and cognitively demanding occupations(3). These forms of enrichment have been shown to afford neurological protection from the harmful pathology of neurodegenerative disease, delaying the onset of dementia by years. We examined clinical data from the Religious Orders Study (ROS)(13) and the Rush Memory and Aging Project (MAP)(14) cohort and found that among individuals spanning a range of dementia diagnoses and pathological burdens, there was a significant positive correlation between frequency of cognitive activity (e.g. reading and writing) and end-stage cognition (Figure 1A; Supplemental Table 1). To study enriching lifestyle factors – including cognitive stimulation – in mice, researchers have employed models of environmental enrichment. As in humans, enrichment in mice has been shown to enhance cognition(15) and delay the onset of neurodegeneration in numerous disease models(5, 16). We hypothesized that the molecular effects of enrichment throughout life might underlie the neural processes that are induced and maintained in individuals who ultimately display CgR later in life.
Figure 1 |. Cognitive stimulation improves cognition and results in global epigenomic changes in cortical neurons of both male and female mice.

A, Lifetime cognitive activity score (frequency of participation in cognitively stimulating activities) plotted against global cognitive function at last visit (an average of 19 tests) for patients in the ROSMAP cohort (n = 676 individuals). Solid red line represents best-fit line; dashed red lines represent 95% confidence interval. Linear regression: Y=0.3874X – 2.1; p-value for slope<0.0001. B, Schematic of experimental paradigm for environmental enrichment. Wildtype Swiss Webster mice were housed under standard conditions prior to weaning. At postnatal day 25 mice were weaned into either enriched environment (EE) or standard housing (SH) conditions. At P60, mice were subjected to molecular or behavioral analysis. C, Left: EE mice display enhanced associative learning in a contextual fear conditioning paradigm (n = 24 mice per group, unpaired t-test, p = 0.0012). Right: When compared to mice housed in standard conditions (SH), EE mice show enhanced rates of fear extinction (n = 10 SH mice vs. 9 EE mice, unpaired t-test comparing % freezing post 2-shocks and % freezing on last day of extinction training, p=0.5340 for SH and p=0.0341 for EE). D, Top: Heatmap of significant (q-value ≦ 0.1) differentially accessible regions (DARs) as identified via ATACseq of neurons isolated from the frontal cortex of EE (red, 450 DARs) and SH (blue, 225 DARs) female mice (n = 3 mice per group). Bottom: Heatmap of significant (q-value ≦ 0.1) differentially accessible regions (DARs) as identified via ATACseq of neurons isolated from the frontal cortex of EE (red, 140 DARs) and SH (blue, 59 DARs) male mice (n = 6 EE vs. 7 SH). E, Pie chart distribution of genomic annotations for each set of ATACseq peaks in females (left) and males (right) for all peaks (top), gained EE peaks (middle) and gained SH peaks (bottom). Annotations are based on overlap with genomic reference annotations computed using the R package ChiPseeker and the UCSC reference of the mouse genome version mm10. DARs that are gained in EE are enriched for intergenic regions. F, Left: Venn diagram of overlap between statistically overrepresented transcription factor (TF) binding motifs among DARs in male and female EE mice. Right: List of all overlapping TFs along with their average rank (between male and female mice). Table indicating occurrence of TF in major neurocognitive-relevant GWAS studies.
We employed a paradigm in which wildtype Swiss Webster mice were weaned into environmental enrichment (EE) or standard housing (SH) cages and maintained in these conditions for one month prior to downstream molecular and behavioral analysis (Figure 1B). EE chambers were large mouse cages containing two nesting/burrowing materials, a running wheel, and toys that were changed every 3–4 days for novelty and exploration. By contrast, SH control mice were housed with an equal number of littermates in smaller, standard cages containing only one nesting material and no toys or running wheel. One month of exposure to EE did not affect general locomotion or anxiety-related behaviors, and resulted in slightly increased body weight (Supplemental Figure 1). To validate the cognitive impact of our enrichment model, we conducted a memory test based on contextual fear conditioning and found, as anticipated, that EE mice display better associative memory compared to SH controls (Figure 1C left). Additionally, EE mice showed significantly more robust fear extinction than SH animals, consistent with previous research (Figure 1C right)(17).
Previous work from our lab demonstrated that enrichment leads to chromatin remodeling and an increase in histone modifications associated with active transcriptional states in neural tissue(18). However, the genomic loci and functional significance associated with this remodeling remain unknown. To unbiasedly assess the changes in global chromatin landscape that occur in the neurons of enriched mice, we performed ATAC-sequencing (ATACseq) to identify regions of the genome that become more accessible after enrichment. Epigenomic accessibility reflects functional relevance, as these loci are more likely to be transcribed or operate as regulatory sites(19). We first used an antibody for Rbfox3 (NeuN) to sort neuronal nuclei from the dissociated frontal cortices of female mice reared in EE (n = 3 mice) or SH (n = 3 mice) conditions. ATACseq of these nuclei revealed 675 differentially accessible regions (DARs), 67% of which were more accessible in EE mice (Figure 1D top), consistent with previous work from our lab showing an increase in active chromatin marks following enrichment(18) (Supplemental Tables 2–5). Principal component analysis showed modest separation of EE and SH samples based on all ATACseq peaks (Supplemental Figure 2A). We repeated this experiment in male mice and similarly found more DARs in EE mice compared to their age-matched SH controls (Figure 1D bottom; Supplemental Figure 2B).
To probe the functional significance of the genomic regions that became more accessible after enrichment, we used multiple, complementary approaches to annotate all accessible regions and DARs identified across the genome (Figure 1E). First, accessible regions in male and female neurons showed a characteristic distribution of openness in introns, intergenic regions, and promoters as is characteristic of published ATACseq datasets(19). Interestingly in both males and females, regions that were more accessible in enriched neurons showed an over-representation of distal intergenic regions, suggesting that distal enhancers and their associated binding proteins could be preferentially recruited in response to cognitive stimulation. Second, utilizing ChromHMM, genome-wide chromatin states were inferred from existing ChIP-sequencing data of histone marks from adult mouse cortical neurons. This analysis revealed that DARs showed a significant enrichment for histone marks associated with enhancer sites (Supplemental Figure 3A). Finally, DARs were assessed for statistical enrichment of transcription factor (TF) binding motifs using the software package Homer, which identified eleven motifs that were significantly overrepresented in the DARs of both enriched female and male mice (Figure 1F; Supplemental Table 6). This list could be broadly separated into the Mef2 family (Supplemental Figure 2C–H) and members of the AP-1 complex, including JunB and Fosl2/Fra2. Interestingly, only one TF from this list, MEF2C, has been causally implicated in both Alzheimer’s disease and general intelligence (as well as neurodevelopmental disorders) via genome-wide association studies (Figure 1F). Consistent with genomic annotation and chromHMM modelling that demonstrated differential openness of putative intergenic, enhancer regions in enriched neurons, Mef2 TFs have been shown to predominantly bind such regions(20).
Subhead 2: Enrichment induces a specific transcriptional signature associated with Mef2 activity
In order to determine the transcriptional consequences of the altered chromatin structure after enrichment, we performed RNA-sequencing from cortical neurons of both female and male mice and examined gene expression after one month of rearing in EE or SH conditions. Neurons were again isolated using an antibody for Rbfox3 (NeuN) to sort nuclei from dissociated frontal cortices. Nuclear RNA expression correlated highly with open regions from our ATAC-sequencing data (Supplemental Figure 3B left). Additionally, promoter regions of the most highly expressed genes were enriched for active histone marks from a dataset of cortical neurons (Supplemental Figure 3B right), indicating successful purification of neurons. Principal component analysis showed modest separation of the EE and SH samples based on all identified genes (Figure 2A; Supplemental Figure 4A). We identified 679 and 657 differentially expressed genes (DEGs) in response to enrichment (FDR<0.1) in female and male mice, respectively (Figure 2B; Supplemental Figures 4B; Supplemental Tables 7–11).
Figure 2 |. Enrichment induces a transcriptional signature associated with Mef2 activity.
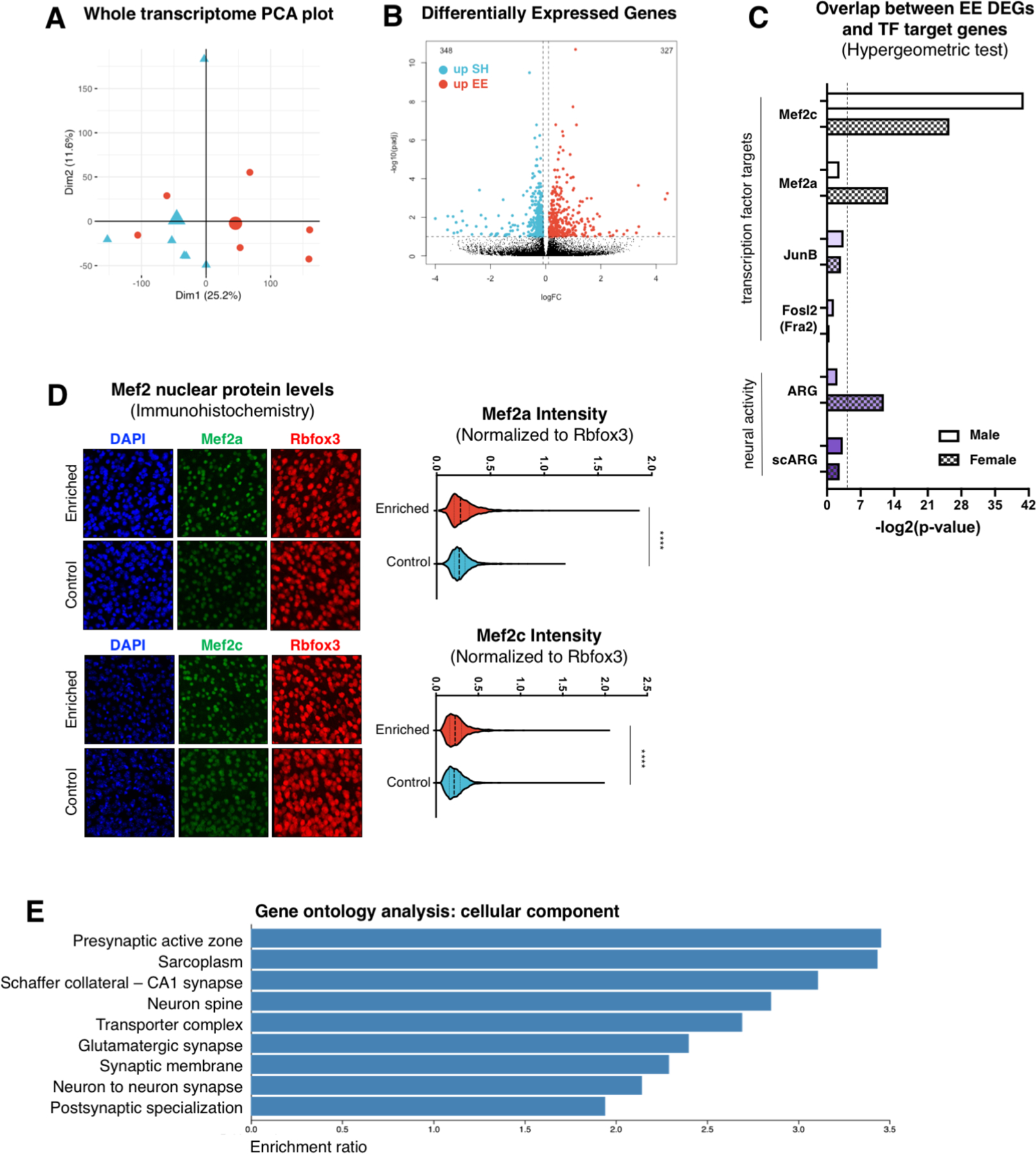
A, PCA plot of nuclear RNAseq expression data from frontal cortical neurons of EE (n=6) and SH (n=6) female mice. B, Volcano plot of DEG analysis of nuclear RNA isolated from the frontal cortex of EE and SH female mice (n = 3 EE vs. 3 SH), showing 327 (red) and 348 (blue) significantly up-regulated in EE (upEE) and SH (upSH) groups, respectively (significance cut-off q-value ≦ 0.1). C, Overlap between EE DEGs (male mice=solid fill; female mice=checkered fill) and lists of TF target genes (Fosl2/Fra2, JunB, Mef2a, Mef2c) or activity-induced genes (ARG, scARG). Dashed line indicates statistical significance threshold via hypergeometric test. D, Top left: Immunohistochemistry (IHC) from the frontal cortex of EE and SH female mice, staining for DAPI (blue), Rbfox3 (red), and Mef2a (green). Top right: Imaris quantification of IHC staining in top left with Mef2a/NeuN intensity plotted for individual nuclei across 3 EE and 3 SH mice. EE mice showed increased Mef2a intensity (unpaired t-test, p-value < 0.0001) compared to SH mice. Bottom left: IHC from the medial prefrontal cortex of EE and SH male mice, staining for DAPI (blue), Rbfox3 (red), and Mef2c (green). Bottom right: Imaris quantification of IHC staining with Mef2c/NeuN intensity plotted for individual nuclei across 3 EE and 3 SH mice. EE mice showed increased Mef2c intensity (unpaired t-test, p-value < 0.0001) compared to SH mice. E. Gene set enrichment analysis (cellular component) for EE vs. SH DEGs in female mice. Enrichment ratio is calculated by dividing the number of observed genes within a gene ontology (GO) category by the number of expected genes. All GO terms listed show a FDR<0.05.
To evaluate whether the differential chromatin accessibility of certain TF binding motifs following enrichment, as identified via ATACseq, could explain the gene expression changes observed between EE and SH neurons, we compiled lists of target genes for the top TFs whose motifs were overrepresented among regions that gained accessibility following enrichment: JunB(21), Fosl2/Fra2(22), Mef2a(23, 24), and Mef2c(24, 25). We observed the greatest degree of overlap between enrichment DEGs and target genes for Mef2c in both sexes (Figure 2C top). Additionally, enrichment DEGs in female mice showed a significant degree of overlap with Mef2a targets. As there can be a discordance between nuclear RNA expression and mature RNA transcript levels, increases in known Mef2-regulated genes (Zmat4, Astn2, Slc16a7, and Daam1) were also confirmed via qPCR of mature transcripts (Supplemental Figure 4C) from an independent cohort of male mice (n=3 per group). Downstream targets of JunB or Fosl2 (Fra2), however, did not show a statistically meaningful overlap with enrichment DEGs in any sex. Taken together, this data suggests that enhanced accessibility and transcriptional activity of the Mef2 family specifically – in particular Mef2c and Mef2a – are important features of the molecular response to environmental enrichment.
Mef2 family members exhibit activity-dependent regulation(9), and it could be hypothesized that the benefits of enrichment are simply due to increased neural activity. To assess whether upregulation of the Mef2 network simply reflects a transcriptional signature associated with generic neuronal activity, or is a specific response to sustained enrichment, we compared enrichment DEGs to two published datasets of transcriptional output following neuronal activity (Figure 2C bottom). These datasets, termed scARG and ARG, were generated from single nucleus RNA-sequencing of dentate gyrus neurons after brief exposure to a novel environment(26), and bulk RNA-sequencing after stimulating neurons in vitro and in vivo with KCl and light stimulation(27), respectively. Interestingly, enrichment DEGs did not show significant overlap with generic neuronal activity DEGs, with the exception of the ARG list in female mice. These results suggest that the type of neural activity arising from sustained enrichment leads to somewhat specific upregulation of the Mef2 transcriptional network, and cannot be explained by known transcriptional networks associated with generic neuronal activity alone.
Nuclear localization of Mef2 TFs is important for the activation of their transcriptional network. We next assessed protein levels of Mef2a and Mef2c within the nuclei of prefrontal cortex neurons following enrichment and found that Mef2a was significantly increased in both male and female mice, and Mef2c was significantly increased in female mice (Figure 2D; Supplemental Figure 4D). This data is consistent with the notion that Mef2 activity is increased upon chronic enrichment. To determine if the resulting transcriptional changes are consistent with known functional roles of Mef2 TFs, we performed gene ontology analysis of enrichment DEGs. Genes that are altered in enriched mice are statistically enriched for cellular components localized to dendritic spines and synapses, including ion channels and receptor complexes (Figure 2E, Supplemental Figure 5). This is consistent with previous work implicating various Mef2 family members in regulating synaptic density(9, 10). Widespread changes in the expression of synaptic and membrane proteins likely result in alterations at the level of neuronal transmission.
Subhead 3: Loss of Mef2a/c in the frontal cortex of enriched WT mice impairs cognitive performance and leads to neuronal hyperexcitability
Epigenomic and transcriptomic profiling of enriched neurons suggests that Mef2 activity might play a role in mediating the phenotypic effects of EE. To causally link Mef2 and determine whether increased Mef2 activity is required for the cognitive benefits of EE, we first used a lentiviral construct to generate short hairpin RNA (shRNA) that would reduce endogenous levels of Mef2a and Mef2c. Both family members were targeted because of our earlier results implicating an increase in nuclear Mef2a and Mef2c activity following enrichment, and previous work that has shown they share a significant proportion of chromatin binding sites, suggesting redundancy in their targets and thus function (28). Control mice received a scrambled (Scrmb) shRNA construct. In vitro cell culture was used to demonstrate that Scrmb virus did not affect levels of any Mef2 members, and that Mef2a and Mef2c shRNA constructs specifically knocked down only their targeted TF (Supplemental Figure 6A). Bilateral lentiviral injections into the medial frontal cortices were performed on postnatal day 30 (P30), after which mice were reared in standard cages for two weeks to allow for viral shRNA expression. All mice were then moved to enrichment cages on P42 and remained in enriched environments for thirty days before undergoing fear extinction training and further experiments.
Injection of Mef2 viruses led to efficient knockdown (Figure 3A), and did not affect weight or locomotion of the animals (Supplemental Figure 6B,C). Both groups (Mef2a/c, Scmrb) displayed equivalent levels of initial memory acquisition, as there was no difference in the proportion of time spent freezing after receiving two foot shocks in the fear conditioning chamber (Figure 3B). However, Mef2a/c-knockdown in both males and females resulted in an impairment in memory extinction, as animals spent significantly more time freezing after multiple exposures to the chamber without foot shock (Figure 3B; Supplemental Figure 6D). These results suggest that the absence of Mef2a/c might impair the molecular processes required to effectively update previously formed associations.
Figure 3 |. Loss of Mef2a/c in the frontal cortex of enriched WT mice impairs cognitive performance and leads to neuronal hyperexcitability.
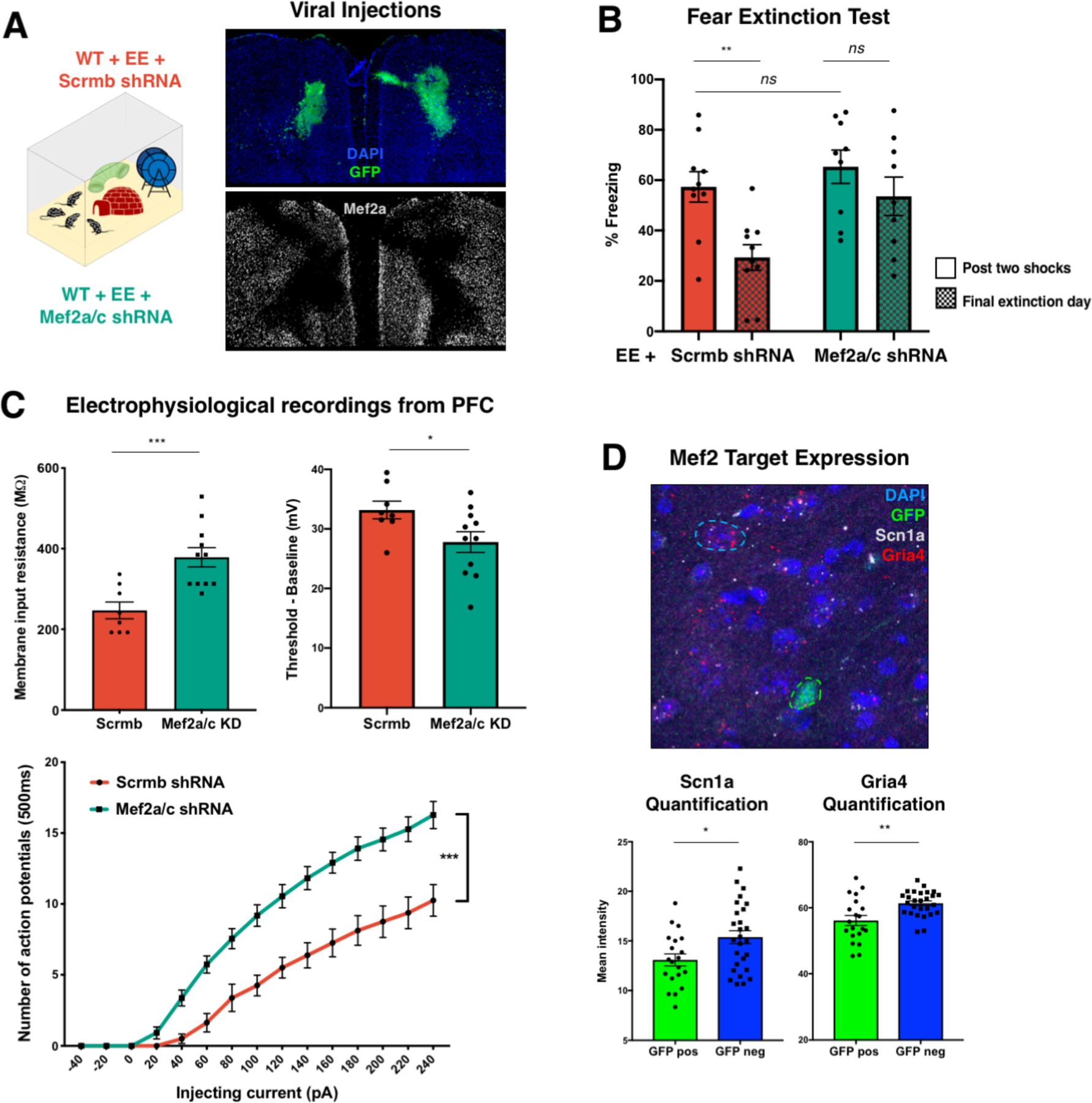
A, Left: Schematic of experimental paradigm for shRNA knockdown in the context of environmental enrichment. On postnatal day 30, wildtype Swiss Webster mice received bilateral frontal cortex injections of lentiviral vectors containing shRNA cassettes against either a scrambled sequence (Scrmb, red) or Mef2a and Mef2c (Mef2a/c, green). shRNA cassettes also constitutively expressed GFP to aid in determining the knockdown region. Right: Immunohistochemistry staining for DAPI (blue, top), GFP (green, top) and Mef2a (white, bottom), to show representative viral expression and Mef2 knockdown. B, Mef2a/c shRNA-treated mice show significantly impaired fear extinction when compared to Scrmb-treated mice (n = 10 mice per group, unpaired t-test comparing % freezing post 2-shocks and % freezing on last day of extinction training, p=0.3047 for Mef2a/c KD and p=0.0101 for Scrmb). C, Voltage-clamp recordings from pyramidal neurons (KD n = 11 cells from four animals; Ctrl n = 8 cells from 4 animals) in shRNA-expressing regions (identified by GFP fluorescence) revealed that neurons with decreased levels of Mef2a and Mef2c (green) showed increased membrane input resistance (left, unpaired t-test, p-value=0.0010) and a reduced difference between baseline voltage and action potential threshold (right, unpaired t-test, p-value=0.0391). Bottom: Mef2a/c knockdown (green line) was associated with increased hyperexcitability, as measured by number of action potentials in 500 ms for a given injecting current (two-way ANOVA, p-value < 0.0001 for group and interaction). D, Top: RNA in situ hybridization for Scn1a (white) and Gria4 (red) in parallel with immunohistochemistry for GFP (green) in the frontal cortex of animals injected with Mef2a/c shRNA showed decreased levels of Scn1a and Gria4 transcripts in cells expressing GFP along with the shRNA construct (top, representative image). Bottom: Quantification of Scn1a (left; t-test p-value = 0.0168) and Gria4 (right; t-test p-value = 0.0015) intensity in either GFP positive (n=20 from four animals) of GFP negative (n=27 from 4 animals) cells.
Previous studies have demonstrated that depletion of Mef2 family members has a significant impact on the electrophysiological properties of cortical excitatory neurons. Embryonic deletion of Mef2c in cortical excitatory neurons led to a significant decrease in cortical activity(25). In contrast, postnatal deletion of Mef2c led to increased spontaneous firing of excitatory neurons in L2/L3 of the neocortex(29). To better understand the functional consequences of loss of cortical Mef2a/c postnatally in the context of an enriched enrichment, we performed electrophysiological recordings from pyramidal neurons harboring either Mef2a/c shRNA or Scrmb shRNA (identified by the presence of GFP) after mice were exposed to enrichment for thirty days. Mef2a/c-knockdown neurons exhibited greater membrane input resistance than control neurons (Figure 3C top left). We also observed non-significant trends toward reduced action potential amplitude and reduced IPSC frequency (Supplemental Figure 6E). The difference between a neuron’s action potential threshold and its baseline resting membrane potential was significantly reduced after Mef2a/c knockdown, indicating a lower threshold to fire (Figure 3C top right). These changes in membrane properties were accompanied by a considerable increase in hyperexcitability after Mef2a/c knockdown, evidenced by significantly more action potentials fired at a given current injection, compared to neurons containing the Scrmb virus (Figure 3C bottom).
Together, these electrophysiological findings highlight Mef2’s critical role in tempering excitatory transmission, and are consistent with previous findings in which Mef2 activity was modulated postnatally(29). They are also reminiscent of early electrophysiological changes that occur in numerous neurodegenerative disorders, including AD, in which neurons tend to show hyperexcitability in early pathological states, even before the onset of cognitive symptoms(30).
Several Mef2 targets are known to regulate the resting membrane potential of neurons(31), so we next examined how Mef2a/c knockdown in enriched animals affected downstream members of the Mef2 regulatory network to better understand the mechanism by which hyperexcitability could be achieved. We focused on two of these genes, Scn1a and Gria4, which were also upregulated in cortical neurons of male, enriched mice. Scn1a encodes part of a sodium channel and is critically involved in maintaining appropriate excitability thresholds, as hundreds of mutations in this gene have been found to cause genetic epilepsy(32, 33). Similarly, Gria4 encodes part of a glutamate-gated ion channel, and loss-of-function mutations in this gene have also been associated with epileptic discharge(34). We used RNA in situ hybridization to measure transcript levels of these targets in GFP-positive neurons that received shRNA against Mef2a/c and confirmed that levels of Scn1a and Gria4 are decreased in neurons lacking Mef2a/c, suggesting that a Mef2-directed transcriptional program might contribute to the altered electrophysiological properties of these neurons (Figure 3D). Collectively, these results confirm that Mef2 activity is required during enrichment to enable cognitive benefits, and that blocking Mef2 activity conditionally leads to a hyperexcitability in cortical neurons.
Subhead 4: The MEF2 transcriptional network is associated with cognition and cognitive resilience in humans
To gain insight into the molecular factors that promote cognitive health and resilience in humans and assess the translational potential of Mef2-mediated cognitive benefits, we used clinical and neurotranscriptomic data from two repositories that contain both healthy control subjects and individuals suffering from AD: (i) Mount Sinai Brain Bank, MSBB, n=301, and (ii) Religious Orders Study/Memory and Aging Project, ROSMAP, n=636. While MSBB and ROSMAP contain transcriptomic data for multiple brain regions, we chose to focus on the anterior prefrontal cortex (aPFC) to parallel our mouse epigenomic and transcriptomic studies. Additionally, the aPFC is critically involved in working memory and other executive functions that deteriorate in AD (35).
As expected in both datasets, end-stage cognitive function was significantly correlated with both beta-amyloid plaque density (MSBB p<2×10−16; ROSMAP p<2×10−16) and tau burden (Braak stage; MSBB p<2×10−16; ROSMAP p<2×10−16), with higher levels of pathology predicting worse cognition (Figure 4A). However, a small subset (<10%) of individuals exhibited cognitive resilience, defined as the absence of dementia despite the presence of extensive amyloid and tau pathology (Supplemental Figure 7A). For each dataset, we generated two lists of genes that were associated with cognitive function. The first list, termed Cog, contained all genes whose expression levels significantly (FDR < 0.05) correlate with end-stage cognition in a simple linear regression. The second list, termed CgR, included all genes whose expression levels correlate (FDR < 0.05) with end-stage cognition in a multiple linear regression controlling for other potential clinical predictors of cognitive decline – age of death, sex, and neuropathological data available for all patients in the dataset. For the MSBB dataset, we controlled for plaque mean, neuropathology score, and Braak stage, while for the ROSMAP dataset, we controlled for severity of global AD pathology, cerebral arteriolosclerosis, cerebral amyloid angiopathy, gross and micro cerebral infarctions, and hippocampal sclerosis. The Cog lists (MSBB = 6544 genes; ROSMAP = 5998 genes) and CgR lists (MSBB = 553 genes; ROSMAP = 745 genes) were significantly correlated across the two human cohorts, increasing confidence in their biological validity (Figure 4B, Supplemental Table 12). Within each human cohort, there was a high degree of correlation between the Cog and CgR lists, indicating that many of the same genes remain significantly correlated with end-stage cognition even when taking other, potentially-confounding clinical variables into consideration (Figure 4B). Both Cog and CgR lists were enriched for molecular functions related to signal transduction, including PDZ domain binding, Rho GTPase binding and ion channel binding, consistent with the types of gene categories that are upregulated following chronic enrichment (Figure 4C, Supplemental 7B).
Figure 4 |. The MEF2 transcriptional network is associated with cognitive resilience in humans.
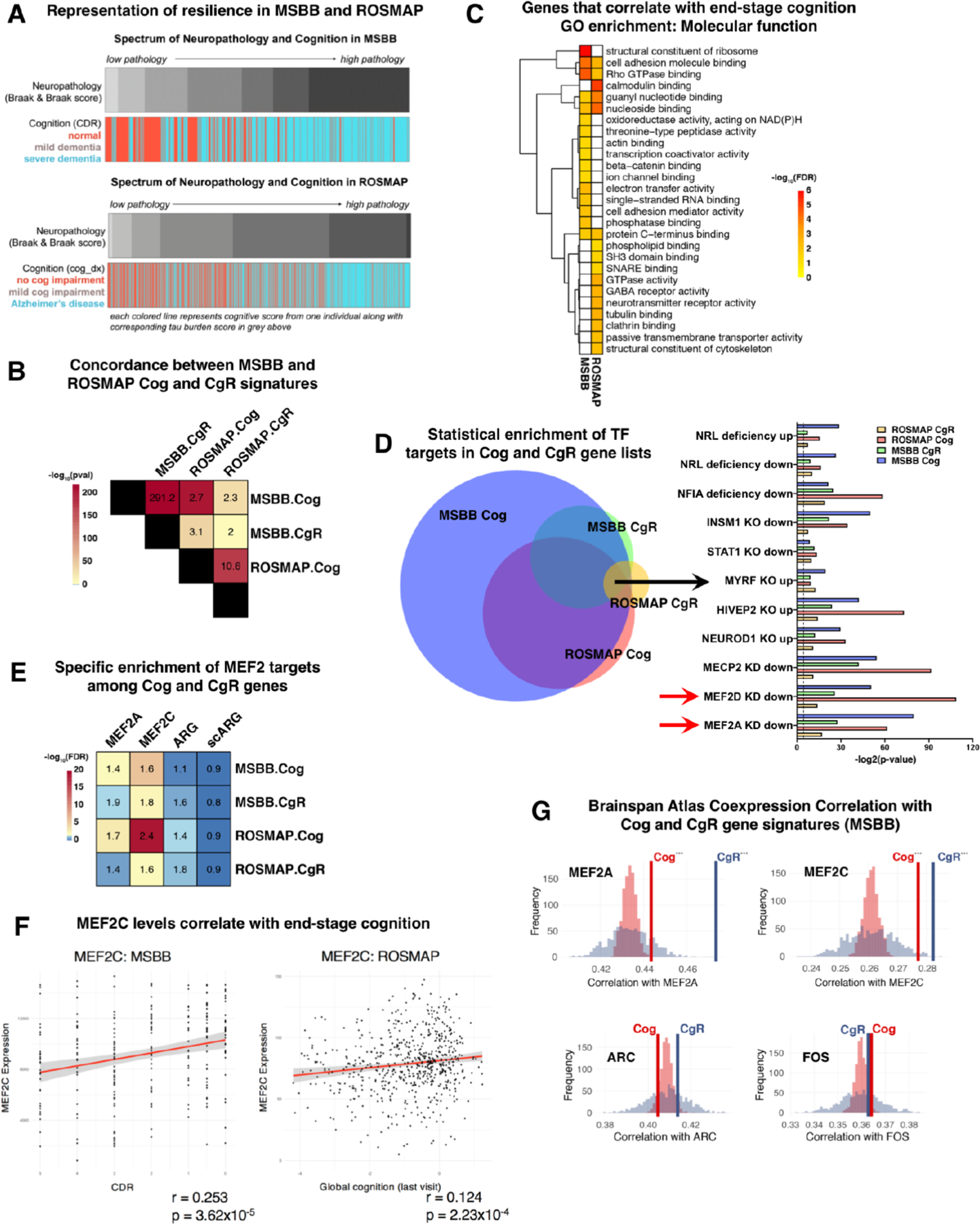
A, Spectrum of neuropathology and cognition in the MSBB (n = 364, top) and ROSMAP (n = 1494, bottom) cohort, with each individual represented as a vertical line across the Neuropathology (Braak and Braak score) and Cognition bars, depicts a clear association between pathology and cognition. B, Overlap analysis of Cognition (Cog) and Cognitive resilience (CgR) associated genes in MSBB (n=260 individuals) and ROSMAP (n=636 individuals) cohorts reveals a significant concordance in genes associated with cognition and cognitive resilience in the two independent datasets. Numbers in the heatmap refer to odds ratios of overlap, and significance (−log10FDR) denoted by color shading. C, Gene ontology enrichment of Cog gene sets in MSBB and ROSMAP demonstrates several conserved molecular functions that are associated with better cognition across the two datasets. Shading denotes the significance (−log10FDR) of GO term enrichment. D, Statistical enrichment of TF targets in Cog and Cgr gene lists, from Enrichr database. Venn diagram reflects all significant (p<0.01) TFs identified in each list, with arrow indicating the overlap of ten distinct TFs in all four lists. Right: Bars indicate significance (−log2[p-value]) for overlap between each list and TF targets in a hypergeometric test. Red arrows indicate highest level of overall enrichment for targets of MEF2. E, Cog and CgR gene sets from the MSBB and ROSMAP cohorts were assayed for enrichment among four different gene sets: MEF2A targets, MEF2C targets, Activity related genes (ARG), and genes associated with neuronal activity determined by single cell sequencing of neurons (scARG). See Text and Methods for more description of gene sets. Overlap analysis revealed specific and significant enrichment for MEF2A and MEF2C targets, but not general markers of activity, within the Cog and CgR gene signatures. Numbers in the heatmap refer to odds ratios of overlap, and significance (−log10FDR) denoted by color shading. F, Gene expression of MEF2C from the anterior prefrontal cortex in the MSBB cohort (left, n = 260, r = 0.253, p-value = 3.62×10−5) and from the dorsolateral prefrontal cortex in the ROSMAP cohort (right, n = 888, r = 0.124, p = 2.23×10−4) plotted against clinical dementia rating (MSBB) and global cognitive function at last visit (ROSMAP). Each individual represented by a dot. Red line denotes regression line with standard error (grey shading). G, Pearson correlation coefficient values measuring co-expression between MEF2A (top left) and either MSBB Cog (bold red line) or CgR (bold blue line) gene sets across developmental time from cortical samples of the BrainSpan Atlas of the Developing Human Brain. Resampling-based statistical test performed contrasting average correlation values observed (red and blue lines) with random expectation estimated by computing average MEF2A co-expression values across BrainSpan Atlas for 10,000 randomly selected, same-sized gene sets (Cog = red histogram, CgR = blue histogram). Asterix denote significant results (p-values for MEF2A Cog = 0.001; MEF2A Cgr < 1×10−4 ). Similar analysis performed for MEF2C (top right, p-values for Cog < 1×10−4; Cgr = 0.005), ARC (bottom left, p-values for Cog = 0.916; MEF2C Cgr = 0.295), and FOS (bottom right, p-values for Cog = 0.04; MEF2C Cgr = 0.362).
Our mouse work demonstrated that Mef2 TFs are drivers of the molecular processes induced upon enrichment, the most robust predictor of cognitive resilience. To unbiasedly assess the robustness of the Mef2 signal in human signatures of cognition, we first probed Cog and CgR lists for statistical enrichment of TF targets. We used the Enrichr database(36) which contains transcriptional changes following many TF perturbations (e.g. knockdown, overexpression). To generate a high confidence list, we overlapped the TFs that were identified from each of the four gene lists (MSBB Cog, MSBB CgR, ROSMAP Cog, ROSMAP CgR), yielding a final set of ten TFs whose targets are enriched among the genes that predict end-stage cognition (Figure 4D). Included in this list were MEF2A and MEF2D, indicating that unbiased interrogation of transcriptional predictors of cognitive function and resilience in humans also points to the MEF2 circuit as a network of genes whose expression levels are associated with the maintenance of cognitive function. Of note, while the Enrichr database contains targets for most TFs, including MEF2A and MEF2D, it lacks information for MEF2C. To validate the enrichment of MEF2 targets in the Cog and CgR lists, we assessed the degree of overlap between each list and the MEF2A and MEF2C targets we previously defined by overlapping ChIP-seq data and conditional mouse knockout data. MEF2C targets were significantly enriched in all lists, while MEF2A targets were significantly enriched in Cog gene lists only (Figure 4E). Interestingly, transcriptional targets of generic neural activity (ARG, scARG) were not enriched in any of the Cog or CgR lists, again indicating specificity of the MEF2 network in the genes associated with cognitive function and resilience. Finally, we found that expression levels of MEF2C itself are positively and significantly correlated with higher cognitive function in the MSBB and ROSMAP cohorts (Figure 4F). This was also true for MEF2A and MEF2D in the ROSMAP cohort (Supplemental Figure 7C–E). Intriguingly, MEF2A appears to be more lowly expressed in the human cortex compared to MEF2C, unlike in the mouse brain, which could provide an explanation of why MEF2C appears more strongly correlated with cognition and cognitive resilience in humans (Supplemental Figure 7F).
Using a separate dataset of RNA-sequencing from human cortical brain tissue across development (BrainSpan Atlas of the Developing Human Brain(37)), we determined the co-expression values, defined as the absolute value of r in a Pearson’s correlational analysis, between MEF2C and all other expressed genes from 8 weeks post-conception to 12 months of age postnatally. Interestingly, the average correlation of all genes in the Cog and CgR lists with MEF2C was significantly higher than that observed for randomly selected, similar-sized groups of genes (10,000 iterations) in a bootstrapping analysis (Figure 4G; Supplemental Figure 7G). This was also true for MEF2A, but not for FOS or ARC, two classic markers of neuronal activity. Our findings suggest that MEF2 TFs are part of a specific regulatory network that is transcriptionally more active in cognitively resilient individuals.
Subhead 5: Single nucleus RNA-sequencing of cortical tissue from a cognitively resilient AD cohort reveals that resilience-promoting MEF2 activity is specific to a subpopulation of excitatory neurons
To further dissect the molecular signature of CgR, we next performed single-nucleus RNA sequencing (snRNAseq) on prefrontal cortex tissue from nine resilient and nine non-resilient individuals identified from the ROSMAP brain bank. All individuals were matched for sex (all female), age, postmortem interval, and pathological variables (Supplemental Figure 8A). Resilient subjects, however, remained cognitively unimpaired prior to death, while non-resilient individuals received a final diagnosis of AD. Using the 10X Chromium single cell platform and applying stringent quality control metrics (see Methods), we sequenced a total of 94,552 cells across the eighteen individuals. Dimensionality reduction and clustering, via Uniform Manifold Approximation and Projection (UMAP) analysis, revealed that all cells fell into six distinct transcriptional clusters (Figure 5A). By interrogating the expression patterns of known gene markers, we determined that the six clusters corresponded to the known major cell-types in the brains: excitatory neurons, oligodendrocytes, inhibitory neurons, astrocytes, oligodendrocyte precursor cells (OPCs), and microglia (Figure 5A, Supplemental Figure 8B). Within each cell-type cluster, we then performed differential gene expression analysis between resilient and non-resilient cells (Supplemental Table 13). Gene ontology (GO) analysis revealed that differentially expressed genes (DEGs) in excitatory and inhibitory neurons shared similar signatures, with enrichment for molecular functions related to protein targeting and localization, regulation of translation, and RNA catabolic processes, suggesting that a signature of resilience may be the presence of neurons that are actively engaged in protein synthesis (Figure 5B). Both astrocytes and microglia showed enrichment for distinct molecular functions, though the terms for both cell types were generally related to paracrine and nonautonomous functioning; astrocytes gene sets suggest the induction of a response to the extracellular environment while microglia displayed enrichment for genes that regulate cell secretion and signal release. No functional enrichment was found for DEGs from oligodendrocytes or OPCs. To determine whether MEF2 activity was driving differential gene expression, we first assessed overall expression levels of MEF2A and MEF2C in each of the cell-types. Interestingly, MEF2A was virtually un-expressed across all the cell-types with only minimal expression in excitatory neurons and microglia. On the other hand, MEF2C was more robustly present, predominantly in excitatory neurons, inhibitory neurons and microglia (Figure 5C). Neither MEF2A nor MEF2C, were differentially expressed between the resilient and non-resilient cells of any major cell-type (Supplemental Figure 8C). Given the heterogeneity known to be present within the major cell-types of the brain, we next performed subclustering analysis, focusing first on the excitatory neurons, which revealed seven distinct subclusters (Figure 5D).
Figure 5 |. Single nucelus RNA-sequencing of cortical tissue from a cognitively resilient AD cohort reveals that resilience-promoting MEF2 activity may be specific to a unique subpopulation of excitatory neurons.
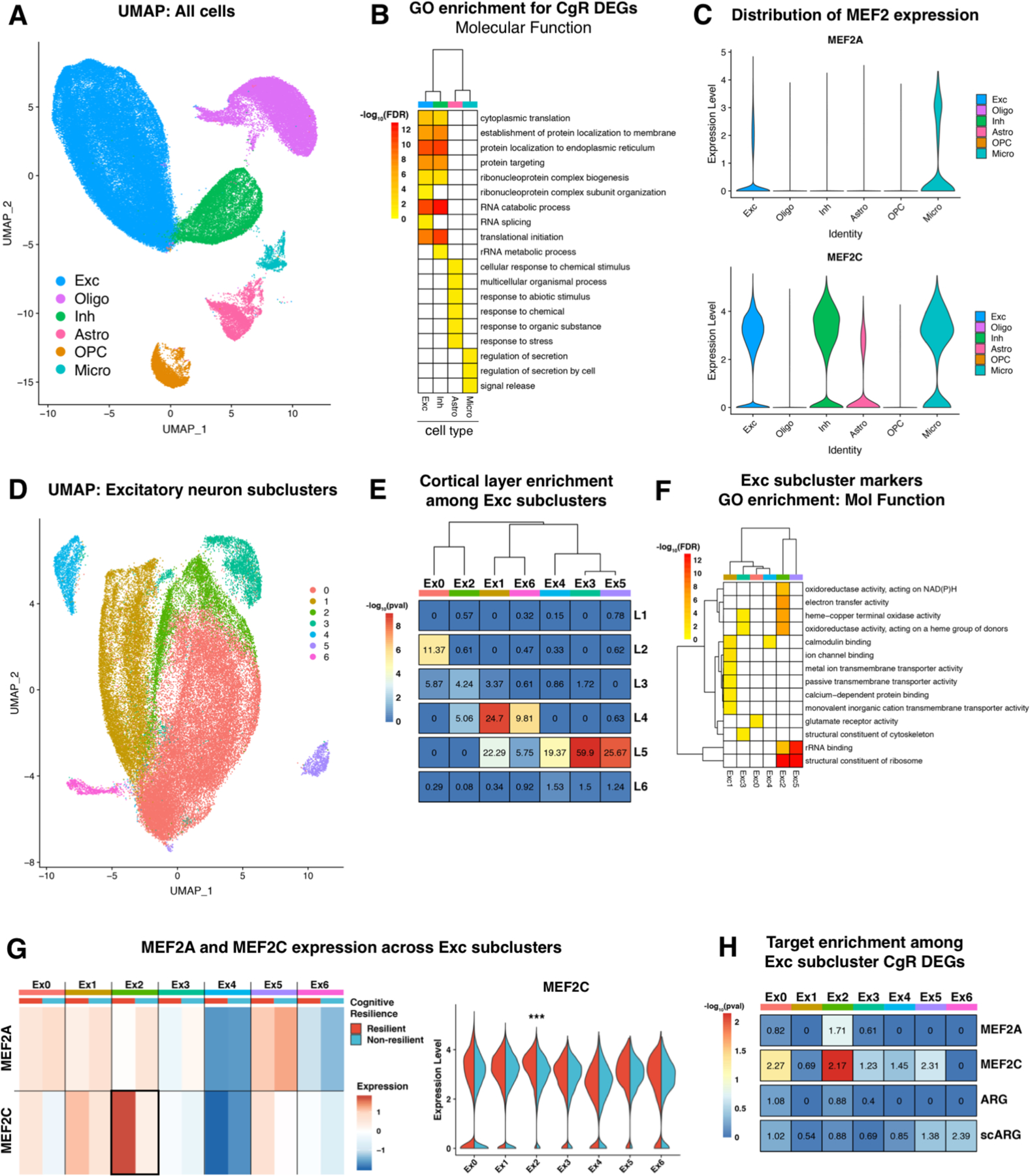
A, Uniform manifold approximation and projection (UMAP) of all annotated cells (n = 94,552 cells from 9 resilient and 9 non-resilient individuals) demonstrates separation of classically-described cell types found in the brain. B, Gene ontology enrichment for molecular function terms of differentially expressed genes (DEGs) between Resilient and Non-resilient cells, for all cell types. Cell types not shown did not have significant GO enrichment for their DEGs. Cognitive resilience was associated with a conserved gene response in excitatory and inhibitory neurons. Shading denotes the significance (−log10FDR) of the GO term enrichment. C, Violin plots displaying the expression level of MEF2A (top) and MEF2C (bottom) for each identified cell-type cluster. MEF2C is more abundant globally than MEF2A and both are only expressed in excitatory neurons, inhibitory neurons, and microglia. D, UMAP plot of Exc cluster from UMAP in (A) reveals seven distinct subclusters of excitatory neurons. E, Enrichment of Exc subcluster markers (Ex0-Ex6) for cortical layer markers (L1-L6) as identified previously (He et al, 2017), demonstrates that excitatory subclusters have specific enrichment for particular cortical layers. Numbers in the heatmap are odds ratios of overlap, and significance (−log10FDR) denoted by color shading. F, Gene ontology enrichment for molecular function terms of Exc subcluster markers. Shading denotes the significance (−log10FDR) of the GO term enrichment. G, Heatmap (left) of MEF2A and MEF2C expression in each excitatory neuron subcluster, differentiated by cells belonging to Resilient (red) and Non-resilient (blue) individuals. The only significant difference observed was for MEF2C in the Ex2 subcluster. Shading denotes expression level, normalized across the row to show gene’s relative expression across all subclusters. Violin plot (right) of MEF2C expression level in each subcluster, differentiating contribution of resilient and non-resilient cells. Ex2 was the only cluster for which MEF2C was differentially expressed between resilient and non-resilient cells (FDR = 2.22×10−57). H, DEGs between Resilient and non-resilient cells in each of the excitatory cell subclusters were assayed for enrichment among four different gene sets: MEF2A targets, MEF2C targets, Activity related genes (ARG), and genes associated with neuronal activity determined by single cell sequencing of neurons (scARG). See Text and Methods for more description of gene sets. Overlap analysis revealed specific and significant enrichment for MEF2C targets, but not general markers of activity, in Ex0 and Ex2 DEGs. Numbers in the heatmap are odds ratios of overlap, and significance (−log10FDR) denoted by color shading.
Comparing the markers of each subcluster with genes enriched in unique cortical layers as defined previously(38), it emerged the excitatory neuron subclusters are defined to a certain extent by the cortical layer from which they originate (Figure 5E). GO enrichment analysis of the subcluster markers also showed functional heterogeneity in the different subclusters (Figure 5F). Differential expression analysis between resilient and non-resilient cells in each of the subclusters demonstrated that MEF2C was specifically and uniquely upregulated in the resilient neurons from subcluster Ex2 (Figure 5G). Ex2 showed weak enrichment for markers of L3/L4 cortical neurons, and strong enrichment ribosomal and rRNA binding genes, suggesting that Ex2 may be a translationally active subset of neurons. Resilient DEGs in Ex2 were also enriched for known MEF2C targets (Figure 5H). No enrichment was observed for MEF2A targets or general markers of neuronal activity (ARG and scARG lists), suggesting that the differential activity within Ex2 neurons is specific to MEF2C. Functionally, the resilient DEGs across the individual subclusters showed some commonality particularly in terms related to the structure constituent of the ribosome, electron transfer activity, and oxidoreductase activity (Supplemental Figure 8D). We also performed a similar subclustering analysis on inhibitory neurons and microglia, the other MEF2C-expressing cell-types, and found no differential abundance of the MEF2 TFs or enrichment of MEF2 targets in the DEGs between resilient and non-resilient cells in any of the inhibitory neuron or microglial subclusters (Supplemental Figure 8E–G). This suggests that the upregulation of MEF2C observed in resilient cells and enrichment of known targets in the DEGs is specific to subpopulation of excitatory neurons in the prefrontal cortex.
Subhead 6: Mef2a/c overexpression improves cognition and rescues tauopathy-induced hyperexcitability in a mouse model of neurodegeneration
With human bulk and single-cell transcriptomic analysis pointing to a role for Mef2 transcriptional activity in excitatory neurons promoting cognitive resilience (CgR), we next asked if enrichment and Mef2 activity could actively rescue and promote resilience in the context of neurodegeneration. To do this we turned to the PS19 transgenic mouse model of tauopathy and neurodegeneration, which expresses the human tau protein harboring the P301S disease-causing mutation that leads to frontal temporal dementia (39). First, we examined RNA sequencing from neurons purified from 2-month-old PS19 mice (Supplemental Table 14) and found that genes that are down-regulated in PS19 mice are enriched for components of the GABA receptor complex and inhibitory ligand-gated ion channels (Supplemental Figure 9A top). PS19 down-regulated genes are also significantly enriched for targets of the Mef2 family (Supplemental Figure 9A bottom). This data suggests that the transcriptional network preserved in CgR might share salient features with the one that is suppressed in P301S-induced tauopathy.
Mice harboring mutant human tau display neuronal hyperexcitability in early stages of the disease(40). We recorded from the PFC in 4-month-old male PS19 mice exposed to either EE or SH conditions. As anticipated, cortical neurons from PS19 mice were more hyperexcitable, with significantly more action potentials fired at a constant injecting current, when compared to wildtype neurons (Figure 6A top). This was accompanied by increased resting membrane potential and membrane input resistance (Figure 6A bottom). Exposure to EE completely rescued this hyperexcitability phenotype. As expected, in a PS19 background, exposure to EE increased nuclear Mef2a intensity compared to mice reared in SH conditions (Figure 6B).
Figure 6 |. Mef2a/c overexpression improves cognition and rescues tauopathy-induced hyperexcitability in a mouse model of neurodegeneration.

A, Voltage-clamp recordings from pyramidal neurons in the medial prefrontal cortex (PS19+SH n = 20 cells from four animals; PS19+EE n = 15 cells from three animals; WT n = 9 cells from two animals) showed a rescue of hyperexcitability phenotypes after housing PS19 mice in EE conditions. Top: EE decreased hyperexcitability, as measured by number of action potentials in 500 ms for a given injecting current, (bottom, 2-way ANOVA for each pairwise comparison: WT vs. PS19 p-value = 0.0045, PS19 vs. PS19+EE p-value < 0.0001, WT vs. PS19+EE p-value = 0.0220). Bottom left: EE decreased membrane resistance of PS19 mice (independent t-test for each pairwise comparison: WT vs. PS19 p-value = 0.0050, PS19 vs. PS19+EE p-value = 0.0036, WT vs. PS19+EE p-value = 0.8173). Bottom right: EE reduced resting membrane potential of PS19 mice (independent t-test for each pairwise comparison: WT vs. PS19 p-value = 0.0163, PS19 vs. PS19+EE p-value < 0.0001, WT vs. PS19+EE p-value = 0.7077). B, Top: IHC from the medial prefrontal cortex of PS19 mice exposed to EE and SH conditions, staining for DAPI (blue), Rbfox3 (red), and Mef2a (green). Bottom: Imaris quantification of IHC with Mef2a/NeuN intensity plotted for individual nuclei across 3 EE and 3 SH mice. EE mice showed increased Mef2a intensity (unpaired t-test, p-value < 0.0001) compared to SH mice. C, Mef2a/c overexpression in 6-month-old PS19 mice enhances rates of fear extinction (n = 9 mice per group, unpaired t-test comparing % freezing post 2-shocks and % freezing on last day of extinction training, adj p=0.0057). D, Top: Voltage-clamp recordings from GFP-positive pyramidal neurons (OE n = 17 cells from 3 animals; Ctrl n = 10 cells from 3 animals) revealed that neurons overexpressing Mef2a and Mef2c (red) showed no difference in membrane input resistance (left) but an increased difference between baseline voltage and action potential threshold (right, unpaired t-test, p-value=0.0143), Bottom: Pyramidal neurons overexpressing Mef2a/c (identified by GFP fluorescence) exhibit reduced excitability as measured by voltage clamp recordings of number of action potentials fired in 500 ms after current injection (two-way ANOVA, p-value < 0.01 for group).
If the pro-cognitive benefits of enrichment are mediated by the transcriptional activity of Mef2, then Mef2 overexpression in the absence of enrichment might be sufficient to enhance cognitive function and rescue the hyperexcitability in P301S-induced tauopathy. Postnatal modulation of Mef2 levels has been shown to have a differential to no effect on behavior(41–43). To test whether Mef2 activity could enhance a cognitive function in a disease context, we generated an AAV-PHP.eB virus and a lentivirus to overexpress Mef2a and Mef2c in addition to GFP in wildtype and PS19 mice. Control animals were injected with viral constructs that expressed GFP alone. With the AAV-PHP.eB construct injected retro-orbitally, we achieved broad expression throughout the cortex with mostly neuronal colocalization. RNA in situ hybridization revealed viral GFP expression along with modest but significant upregulation of Mef2c and two target genes, Scn1a and Gria4 (Supplemental Figure 9B). Young (3 month old) wildtype mice overexpressing Mef2a/c displayed enhanced rates of extinction after fear conditioning compared to control animals (Supplemental Figure 9C) without displaying differences in baseline freezing or locomotion (Supplemental Figure 9C,D), suggesting a general role for the Mef2 family in promoting cognitive flexibility. At 6 months of age, we performed fear extinction training on PS19 mice to determine if Mef2a/c overexpression is sufficient to enhance cognitive function in the absence of enrichment. While there was no difference in body weight or the initial memory acquisition following 2-shock fear conditioning, PS19 mice overexpressing Mef2a/c showed faster extinction of the fear memory compared to their control littermates (Figure 6C; Supplemental Figure 9E). Finally, using the lentiviral construct, which enabled easier ex-vivo identification of transfected neurons, we performed electrophysiological recordings from the PFC of PS19 mice overexpressing Mef2a/c. While we did not observe a change in membrane resistance, we found a significant increase in the voltage required to achieve action potential firing threshold from baseline, consistent with a reduced propensity for neuronal hyperexcitability (Figure 6D top). We also observed a modest but significant reduction in action potential frequency upon current injection (Figure 6D bottom). Collectively, these results highlight that promoting activity of the Mef2 transcriptional network, in the absence of enrichment, is sufficient to induce cognitive resilience even in the presence of neurodegenerative pathology. Furthermore, a potential mechanism of the benefits afforded by Mef2-mediated CgR could be the buffering of the hyperexcitability that is commonly associated with neurodegenerative disease progression and pathology.
Discussion:
The molecular mechanisms that endogenously prevent against cognitive decline are largely unknown; if identified and harnessed, they have the potential to improve a central facet of human health in our aging population. Toward this end, our work uncovers a link between Mef2-dependent transcriptional programs and cognitive health throughout life, in which Mef2 activity contributes to enhanced cognitive function at baseline and cognitive resilience in the face of neuropathology. We began by studying the molecular changes that arise from the most potent known inducer of cognitive resilience, environmental enrichment. Analysis of global chromatin and transcriptional changes in cortical neurons led to the identification of Mef2 TFs as key modulators of neural function following enrichment. Transcriptional activity of the Mef2 regulatory network is increased upon chronic enrichment, and by knocking down Mef2a and Mef2c, we show that these TFs are required to actualize the cognitive benefits of an enriched environment. Importantly, this work was conducted in both male and female mice to ensure equal representation of the sexes in translational research.
The molecular mechanisms mediating cognitive health and resilience in humans have been difficult to define due to a lack of integration between clinical measures and unbiased, comprehensive molecular data. We interrogated phenotypic and bulk brain RNA-sequencing data from two large independent databases (ROSMAP and MSBB) and found that increased levels of MEF2C and downstream targets were significantly associated overall cognition and CgR. Additionally, we used single nucleus RNA-sequencing and showed that the increase in MEF2C expression in cognitively resilient cells occurs in a subset of excitatory neurons that appear to be translationally active. This was particularly intriguing as it suggests that cortical neurons that are actively responding to and encoding environmental stimuli require upregulation of the MEF2 network in order to maintain cognitive function.
Returning to mice, where we can powerfully manipulate gene targets and assess for a causative link between Mef2 TF levels and CgR, we show that Mef2a/c overexpression can rescue tauopathy-induced hyperexcitability and is sufficient to enhance fear extinction learning, in a mouse model of neurodegeneration. The observation that Mef2 overexpression alone phenocopies environmental enrichment suggests that Mef2 may be a master regulator of environmentally-mediated cognitive enhancement and resilience. This direct evidence for a mechanistic link between the Mef2 regulatory network and a clinical demonstration of cognitive resilience suggests that activation of Mef2 transcriptional activity could be a potential therapeutic avenue for preventing cognitive decline and ensuring healthy aging in humans.
An intuitive explanation for the link between sustained stimulation, Mef2 network activity, and cognitive enhancement is that the benefits of enrichment result from the transcriptional output of increased neuronal activity, and that Mef2 TFs are part of this activity-induced network, as they have long been implicated in activity-dependent regulation of gene expression in neurons(9). To test this hypothesis, we compared both enrichment DEGs in mice and cognitive resilience-associated genes in humans to two datasets capturing transcriptional changes following generic neuronal activity (ARG, scARG), but did not observe significant overlap. This finding emphasizes the specificity of Mef2’s involvement in enrichment-induced cognitive resilience, potentially pointing to a unique type of activity or a specific subset of molecular drivers that are enabled in the context of positive and stimulating experiences to afford a phenotypic benefit. In fact, genetic variants within the Mef2c loci have been associated with differences in human intelligence(12), and it could be that these variants may also impart an effect in an environmentally-dependent manner. We examined one aspect of cognitive function often linked to intelligence, the ability to quickly and effectively integrate new information into existing schemas. Manipulating levels of Mef2 had profound effects on this ability, thereby providing insight into the potential neural mechanisms of cognitive flexibility that extend beyond implications for neurodegenerative disease.
How does Mef2-dependent activity in the prefrontal cortex link environmental enrichment to cognitive resilience? Undoubtedly, this most likely occurs in a temporally-and spatially-specific manner as previous work has shown that increasing expression of MEF2 in the dentate gyrus blocks spatial memory formation, while overexpression in the cortex can facilitate learning. In fact, our single-cell results point to a particular subset of neurons within the cortex being important mediators of Mef2-dependent cognitive resilience. The increases in Mef2 TF activity that we observed here must have a myriad of effects that will need to be further elucidated in subsequent studies, but one critical aspect uncovered here and consistent with the literature is their ability to modulate excitatory:inhibitory (E/I) balance in neural circuits(25, 44). Interestingly, an increase in E/I ratio in the prefrontal cortex has already been linked to a profound impairment in cellular information processing and impairments in conditioned learning(45). As hyperexcitability has a toxic effect in multiple models of neurodegenerative and neuropsychiatric diseases, a Mef2-induced dampening of the E/I balance could decrease hyperexcitability in a diseased state thereby explaining how enrichment and Mef2-activity buffer cognitive capacity in the context of neuropathology. Mef2 might also act in a mechanism similar to Arc, helping to establish homeostasis in a highly stimulated brain.
While in humans MEF2C appears to be more strongly correlated with cognition than MEF2A, our mouse work points to a potential role for both Mef2a and Mef2c in coordinating cognitive function and resilience after enrichment. This discrepancy could have multiple origins, and does remain a limitation of the study. First, we could actually be elucidating slightly different processes in mice and humans. Unfortunately, due to the limitations of human research and the MSBB/ROSMAP databases, we cannot ascertain definitively what led to the induction of increased cognitive resilience or the reason why individuals have differing levels of cognition. This could be due to multiple factors, only one of which is differential cognitive stimulation. In our mice work, however, we specifically control the exposure (environmental enrichment) that induces cognitive enhancement and resilience. As a result, the Mef2 family may govern different facets of ensuring optimal cognitive function and neuronal resilience. Taken in the context of previous work that showed no impact on learning memory with postnatal knockdown of Mef2c, it may be that Mef2a and Mef2c activity are conditionally required for actualizing cognitive benefits in the presence of a positive stimulus, such as mental stimulation or environmental enrichment. Second, there appears to be a divergence in the expression levels of Mef2a between mice and humans. Both our single cell experiments and analysis of bulk transcriptomic data show a much more modest expression of MEF2A compared to MEF2C in the human cortex. In mice, however, neuronal expression of Mef2a and Mef2c appear more equal, suggesting that Mef2a may have differential roles in mice and human cortical neurons. To this end, the unique presence of MEF2 response elements in higher-order mammals, and their corresponding absence in lower mammals like mice, has led to the identification of novel roles for Mef2 enhancer activity specifically in non-human primates(46). The clear involvement of Mef2 in regulating cognition in mice points to a conservation of this particular function in rodents, but further work will be required to further elucidate the individual contribution of Mef2a and Mef2c to cognitive health, and what aspects of this function may be unique to humans versus conserved across a broader subset of mammalian species.
Overall, the long-standing clinical and epidemiological observation of cognitive resilience affords us the potential to better understand how the brain naturally adapts and responds to the presence of pathology. Taking advantage of this powerful phenomenon, this work highlights an important step toward elucidating a set of molecular targets that can guide the development of therapeutics to promote general cognitive health and protect the brain from various forms of neural insult.
Materials and Methods:
Animals.
All animal work was approved by the Committee for Animal Care of the Division of Comparative Medicine at the Massachusetts Institute of Technology. Swiss Webster pregnant female mice were purchased from Charles River Laboratories (Wilmington, MA, USA) for enrichment experiments. PS19 Tg mice(39) were obtained from the The Jackson Laboratory. Mice were weaned on postnatal day 25 into single-sex groups of four to five mice per cage. Standard cages consisted of a 7 1/2” x 11 1/2” x 5” plastic chamber containing a layer of wood chip bedding and a single pulped cotton fiber nestlet. Enrichment cages were larger (10 1/2” x 19” x 8”) and contained a layer of wood chip bedding, 2–3 nestlets, Crink-l’Nest, a running wheel, a plastic tube, and three additional rodent toys replaced every 3–4 days. Food and water were provided ad libitum. Mice were housed on a standard 12 h light/12 h dark cycle, and all experiments were performed during the light cycle. One animal was excluded from analysis (Figure 2C) because of extreme aggression during the behavior task, and one was excluded (Figure 6C) because of hindlimb paralysis and subsequent death.
Mouse behavior testing.
Behavior testing consisted of the open field test on day 1, followed by fear conditioning (FC) training on day 3, and re-introduction to the FC chamber with no shock on days 4–7/8 using behavioral apparatus from TSE Systems (Chesterfield, MO USA). Mice were brought to the site of behavioral testing 30 minutes prior to testing for habituation each day. The open field chamber consisted of a well lit 40 cm L x 40 cm W x 35 cm H chamber with a solid floor, while the fear conditioning chamber was 20 cm L x 20 cm W x 50 cm H and had a metallic grid floor. Chamber floors were sprayed with 70% ethanol and all waste was removed before each animal entered. For the open field test, mice were given ten minutes to explore freely. For fear conditioning, mice were given 3 minutes to explore the chamber, followed by a foot shock (2 seconds, 0.8 mA). Recall sessions consisted of 3 minutes of exploration in FC chamber. Movement patterns and freezing behavior were recorded and automatically calculated by TSE software to avoid introducing experimenter bias.
Nuclear preparation and isolation via flow cytometry.
Tissue for analysis was obtained from 2 month old Swiss Webster mice by dissecting fresh cortical tissue enriched for frontal cortices (Bregma coordinates 0.5 to 3.0). Subcortical structures were dissected away prior to further processing. Mortar and pestle were used to break up tissue in Phosphate Buffered Saline (ph 7.4, PBS) with cOmplete protease inhibitor cocktail (Roche) and RNaseOUT Recombinant Ribonuclease Inhibitor (ThermoFisher). Protease and ribonuclease inhibitors were present in all downstream steps. Tissue was then fixed using 1% paraformaldehyde (Electron Microscopy Sciences) in PBS for 20 minutes. Fixation was quenched with 1M Tris base (pH 8.0) to a final concentration of 250 mM of Tris. Tissue was washed twice in NF1 buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA, 5 mM MgCl2, 0.1M sucrose, 0.5% Trixon X-100) to remove residual PFA and lyse cells for nuclear extraction. After washing, pellets were then resuspended in NF1 buffer for 30 rounds of dounce homogenization (Wheaton). After douncing, nuclear pellets were obtained by filtering sample through 70 micron mesh, and one more round of NF1 washing. Nuclear pellets were then resuspended in PBS with 1% Bovine Serum Albumin (BSA, ThermoFisher), and stained with Anti-NeuN antibody conjugated to Alexa Fluor 488 (clone A60, EMD Millipore) at a dilution of 1:2000 for 30 minutes. DAPI stain was also added at a concentration of 1:10,000 to aid in identification of nuclei when cell sorting. After washing, nuclear suspension was subjected to fluorescence-activated cell sorting (FACS) on a BD FACS Aria II with 70 micron nozzle set to 70 PSI. Nuclei were identified via DAPI and neuronal nuclei were selected using the 488 channel. Sorted cells were immediately used for RNA-seq or ATAC-seq library generation.
ATAC-seq library generation.
ATAC-seq libraries were generated from sorted neuronal nuclei using the Nextera DNA Library Preparation Kit (Illumina), as described previously(19). Briefly, nuclear pellet was resuspended in transposase reaction mix buffer, and transposase. Transposition reaction was performed for 30 minutes at 37 °C. As described previously(47), ATAC-seq libraries were generated from fixed nuclei. After transposase reaction, defixation buffer (50 mM Tris-HCl ph8.0, 1 mM EDTA, 0.2 M NaCl, 1% SDS, 5 ng/mL proteinase K) was added to the mixture, which was then incubated at 65°C overnight with agitation at 1000 rpm on a benchtop shaker/heater block. The next morning, the reaction was purified using a Qiagen MinElute kit. Following purification, library fragments were amplified using Nextera PCR primers with cycling and cycling number determined as described in the original ATAC-seq paper(19). All libraries were amplified for a total of 10–12 cycles. Libraries were analyzed using an Agilent 2100 BioAnalzyer (sample statistics provided in Supplemental Table 9).
RNA-seq library generation.
Because tissues had been fixed prior to nuclear isolation, RNA was extracted from nuclei using the RecoverAll Total Nucleic Acid Isolation Kit for FFPE Tissue (ThermoFisher). Nuclear RNA was run on an Agilent 2100 BioAnalyzer for quality control (sample statistics provided in Supplemental Table 9). RNA libraries were generated using the SMARTer Stranded Total RNA-seq for mammalian samples (Takara). Of note, libraries were amplified using random hexamer primers given that the RNA isolated was nuclear and had undergone fixation.
ATAC-seq sequencing, data processing and analysis.
ATAC-seq libraries were sequenced using a NextSeq platform with 40 bp paired-end reads. Sequencing was performed at the MIT BioMicroCenter (https://openwetware.org/wiki/BioMicroCenter). ATAC-seq reads were aligned using Bowtie 2 (version 2.2.9). The mouse genome version mm10 was used as reference. Aligned reads (bam format) were further processed using samtools (version 1.3.1) and bedtools (version 2.26.0). First, mitochondrial and duplicate reads were removed. Next, all reads were shifted, per the original ATAC-seq protocol(19) to more accurately reflect the center of the transposase binding event. Basic statistics for each sample (number of fragments sequenced, alignment rate, duplication frequency) can be found in Supplemental Table 9. Significant ATAC-seq enriched genomic regions (i.e., peaks) were called per sample using MACS2 (version 2.1.1). Peak calling parameters were as follows (-f BAMPE -g mm -q 0.01 -- nomodel). Enriched regions across all samples per dataset were merged using Diffbind (version 2.10.0), keeping only reproducible regions defined as enriched regions present in at least 2 biological replicates. This analysis defined a space of 165,184 potentially enriched regions in females (n=6 samples) and 192,919 in males (n=13 samples). Read coverage within this peak universe was calculated, and this matrix was used for subsequent Differential Accessible Region (DAR) analysis that was performed using the edgeR package through DiffBind (dba.analyze, method = DBA_EDGER). Default FDR values for DAR analysis were used (FDR < 0.1 as significant). Differentially accessible regions in each condition were then assayed for enriched motifs using the Homer (version 4.9) “findMotifsGenome.pl” command.
Annotation of ATAC-seq enriched regions.
ATAC-enriched regions were annotated as Promoter, 5’ UTR, 3’ UTR, Exon, Intron, Downstream, Distal Intergenic based on their overlap with reference genome-wide annotations from the UCSC transcripts mm10 database using the R package ChiPseeker (version 1.24.0). In addition, ATAC-enriched regions were classified as putative enhancers or promoters based the distance to mouse UCSC known gene annotations. Enriched regions that overlapped within 1,000 bp from known transcriptional start sites (TSSs) were annotated as promoters, and enriched regions that were located more than 1,000 bp away from TSSs and did not overlap any genic annotations (5’UTR, 3’UTR, Exon, Intron) were considered putative enhancers. Read coverage for a region of 2 kb (50 bp bins) around TSSs was aggregated, smoothed and plotted using the R package EnrichedHeatmap (version 1.18.2).
RNA-seq sequencing, data processing and analysis.
Mouse RNA-seq libraries were sequenced using a HiSeq platform with 40 bp single-end reads. Sequencing was performed at the MIT BioMicroCenter (https://openwetware.org/wiki/BioMicroCenter). RNA-seq reads were aligned using STAR (version 2.5.4a). The mouse genome version mm10 was used as reference. After alignment, reads were then summarized at the gene level using “featureCounts” within the SubRead package (version 1.6.0), for all coding genes annotated in the Ensembl version GRCm38. Next, Differential Expression Gene (DEG) analyses was conducted using the R package DESeq2 (version 1.16.1), after filtering genes out that were not detected in any samples. Histogram of DEG analysis p-values revealed a conservative distribution, with a maxima close to 1.0, indicating an overestimation of the variance in the null distribution. To correct for this, the R package fdrtool (version 1.2.15) was employed to perform empirical null modeling, using the Walt statistic as input to recompute p-values and FDR values. Unless stated otherwise, genes meeting the criteria FDR<0.1 were considered differentially expressed.
Published mouse cortical Chip-seq and RNA-seq data.
Chip-seq data for the chromatin marks H3K27ac, H3Kme3, and H3K27me3 reported in (48) were used as reference epigenomic signals of the mouse cortex. ChromHMM (version 1.21) was used to build chromatin states based on the above datasets using default commands. JunB targets and Fosl2 targets were derived as reported previously in (21) and (22), respectively. To generate high confidence target lists for Mef2c, published transcriptomic analysis performed on the cortex after conditional knockdown of Mef2c in excitatory mouse neurons(25) was intersected with genes containing Mef2c peaks identified through a Mef2c-specific ChIP-seq dataset(24). For Mef2a, the target list was derived by taking the overlap of genes containing Mef2a ChIP-seq peaks(24) and genes identified via RNA-seq as differentially expressed in neurons after Mef2a/d(23) knockdown. Bulk activity regulated genes were obtained as from (27), and activity regulated genes identified in neurons by single-cell RNA-sequencing were obtained from (26).
Published human RNA-seq data.
Transcriptomic and phenotypic data from the MSBB (Mount Sinai Brain Bank, n = 301 individuals, region = Brodmann area 10) Alzheimer’s Disease (AD) study(49) and the Religious Orders Study and Memory and Aging Project (ROSMAP) study(13, 14) were used to identify human cortical gene expression patterns that correlated with overall cognition or cognitive resilience. For an expanded set of ROSMAP patients (n = 888), we were able to obtain normalized expression values of MEF2 TFs, and so simple linear regressions of MEF2 proteins and cognition in the ROSMAP dataset and their corresponding p-values were calculated on this expanded set. For the rest of the analysis, the publicly available anterior prefrontal cortex (aPFC) transcriptomic dataset containing a normalized count matrix for all genes across individuals (n = 636) was downloaded from the Synapse portal (syn3219045). Normalized RNA-seq data from MSBB was also made available through the Synapse portal (syn3159438). For each dataset, genes that correlated with overall cognition were determined by running a simple linear regression between cognition and gene expression. For MSBB, Clinical Dementia Rating (CDR) score was used as a marker of cognition, while in ROSMAP, global cognitive score was utilized. To determine genes that correlated with cognitive resilience, multiple linear regression analyses were conducted to assess for a relationship with cognitive variables (as above) and gene expression values, while controlling for other potential clinical predictors of cognitive decline – age of death, sex, and neuropathological data available for all patients in the dataset. For the MSBB dataset, we controlled for plaque mean, neuropathology score, and Braak stage, while for the ROSMAP dataset, we controlled for severity of global AD pathology, cerebral arteriolosclerosis, cerebral amyloid angiopathy, gross and micro cerebral infarctions, and hippocampal sclerosis. For each comparison, p-values were adjusted using the Benjamini-Hochberg procedure, and genes were deemed to significantly correlate with cognition if the FDR was less than 0.05.
The BrainSpan Atlas of the Developing Human Brain was used as reference to analyze the co-expression of genes that correlated with end-stage cognition, and MEF2 TFs, or ARC and FOS, as controls. All analyses were conducted with a subset of data including only cortical regions of prenatal and early postnatal stages, normalized and curated previously as described(50). Gene co-expression was quantified using the pairwise Pearson correlation coefficient between the target gene (MEF2A, MEF2C, ARC, or FOS) and each gene within a given gene set. The observed value was taken as an average of the absolute value of all coefficients. Significance of co-expression patterns was assessed by calculating the probability of observing as extreme as the empirically observed under a random null model estimated through resampling of similar-sized groups (n=10,000).Cortical layer markers were defined as identified previously(38), with a particular gene being included as a layer marker if it was more than 2-fold enriched in that cluster compared to all others.
snRNA-seq preparation.
Isolation of nuclei from frozen post-mortem brain tissue was carried out as previously described(51). Nuclei were counted manually and diluted to a concentration of 1,000 nuclei per microliter in PBS containing 0.04% BSA. Libraries were prepared using the Chromium Single Cell 3’ Reagent Kits v.2 according to the manufacturer’s protocol (10x Genomics). snRNA-seq libraries were sequenced using NextSeq 500.
Analysis of droplet-based snRNA-seq data.
Gene counts were obtained by aligning reads to the hg38 genome (GRCh38.p5) using CellRanger software. Seurat (version 3) was used to filter, normalize, and cluster cells. Cells expressing fewer than 200 unique genes were excluded on the basis of quality control. K-means clustering (n=2) was used to determine the threshold of percent mitochondrial reads, and cells in the higher cluster were excluded. Gene counts were log normalized. Following normalization, principle component analysis (PCA) was performed. The first 12 principle components were used to form clusters and determine Uniform Manifold Approximation and Projection (UMAP) embeddings. Clustering was based on a K-nearest neighbors graph, derived from the Euclidean distance in PCA space, followed by the Louvain algorithm for modularity optimization, as implemented by Seurat. Cluster identity was assigned by examining expression of cell-type specific markers. After clustering, the DoubletFinder package was used to remove doublets, using the default settings. Differential genes between cells from cognitively resilient and non-resilient individuals were identified separately for each cluster using the Wilcoxon Rank Sum test within Seurat. For subclustering of the cell-type clusters, raw matrices for each subcluster were extracted and the process above was repeated with the exception of doublet removal.
Gene set enrichment analysis.
All gene ontology (GO) enrichment analysis was conducted using the Webgestalt web interface (www.webgestalt.org). For all GO enrichment analysis, the background was selected as all genes detected in a given experiment. Transcription factor perturbation enrichment analysis was performed using the web-based platform Enrichr (http://amp.pharm.mssm.edu/Enrichr/). No background was used for Enrichr analysis. To determine the significance of overlap between two gene lists, the R package GeneOverlap (version 1.12.0) was used, and each experiment the population size of the background was selected by determining the number of genes detected from our neuronal RNA-seq experiments (size = 16000 genes).
Cell culture for viral production.
Human embryonic kidney 293T cells were cultured in Dulbecco’s Modified Eagle Medium with GlutaMAX (Life Technologies) containing 10% FBS and 1% penicillin/streptomycin. At 85% confluence, lentiviral plasmids (RRE, REV, PMDG) were transduced using Lipofectamine 2000 and Opti-MEM. Eighteen hours later, media was replaced with fresh media. 72 hours later, media was collected and viral particles were concentrated by ultracentrifugation (20,000 RPM for 2.5 hours at 4°C in a SW-32 rotor) and stored at −80°C. Dissociated cortical neurons from E16 Swiss-Webster mice were plated at a density of 3×10^5 per well of a 24-well plate. The plates were coated beforehand by incubation with poly-D-lysine (0.5 mg/ml) and laminin (0.005 mg/ml) for 1 hr at 37°C, followed by washing twice with dH2O. Neurons were maintained in neurobasal media and supplemented with L-glutamine, penicillin/streptomycin, and B27. On DIV5, concentrated lentivirus was added to cultured neurons to test for viability. Adeno-Associated viruses (AAV) were obtained from the Viral Tools facility at Janelia, and Mef2a and Mef2c overexpression plasmids were packaged into AAV php.EB virus for efficient non-invasive gene delivery to the central nervous system, as developed and characterized previously(52).
Surgical procedures.
Thirty-day-old mice were anaesthetized with an intraperitoneal (i.p.) injection of a mixture of ketamine (1.1 mg/kg) and xylazine (0.16 mg/kg). Two small craniotomies were made on both sides of the skull at the following coordinates relative to bregma: A/P= +2.2, M/L=+/−0.64. Virus was delivered through a small durotomy by a 33g beveled NanoFil needle (World Precision Instruments) attached to a stereotaxic injector. The micropipette was lowered to 1.62 mm below the brain surface. A bolus of 0.75 nL of virus was injected into the frontal cortex at 0.2 μL min−1. The pipette remained in place for 5 min following the injection before being retracted from the brain. After suturing, animals were placed on a 37° heating pad to recover for 0.5–1 hour.
Animals were monitored for three days following surgery, and moved to enrichment cages two weeks later. AAV php.EB viruses were injected retro-orbitally into thirty-day old mice after brief exposure to inhaled isoflurane for sedation.
Perfusion.
Mice were transcardially perfused with chilled phosphate buffered saline (PBS) followed by 4% paraformaldehyde in PBS. Brains were then removed and post-fixed in 4% PFA overnight at 4°C and subsequently washed with PBS before sectioning.
Immunohistochemistry.
Brains were sliced into 40-micron sections using a vibratome (Leica). Slices were stored in PBS at 4°C. Sections were permeabilized in blocking buffer, consisting of 0.3% Triton and 10% donkey serum in PBS, via gently rocking for 2 hours at room temperature. Sections were incubated overnight at 4 °C in primary antibody diluted to the appropriate concentration in blocking buffer. Primary antibodies used for this study were: Rabbit anti-Mef2a (1/1000, Abcam: ab76063), Guinea Pig anti-NeuN (1/2000, Synpatic Systems: 266004), Mouse anti-Mef2c (1/1000, R&D Systems: MAB6786), Chicken anti-GFP (1/500, Abcam: ab13970), and Mouse anti-phospho-Tau (1/1000, Cell Signaling Technology: 9632S). Primary antibodies were visualized with Alexa-Fluor 488, 594, and 647 secondary antibodies (Molecular Probes), and cell nuclei visualized with Hoechst 33342 (Sigma-Aldrich; 94403). Secondary antibodies and Hoechst were diluted to 1:1000 and 1:5000, respectively, and sections were incubated for one hour at room temperature. Finally, sections were washed and mounted on glass Superfrost Plus Slides (ThermoFisher Scientific). Images were acquired using a confocal microscope (LSM 710; Zeiss) with a 40× objective at equivalent settings for all conditions. Images were quantified using ImageJ or Imaris by an experimenter blind to treatment groups. For each experimental condition, two coronal sections from at least three animals were used for quantification.
RNA In Situ Hybridization.
RNA In Situ Hybridization was performed using the RNAscope Multiplex Fluorescent Reagent Kit v2 Assay (Advanced Cell Diagnostics, Inc). Forty-micron slices were obtained as described above. Slices were mounted on Superfrost Plus Slides (ThermoFisher Scientific), and incubated in 50% EtOH overnight to dehydrate slices and allow optimal slide adherence. As per the v2 Assay protocol for fixed tissues, slices were then subjected to Hydrogen Peroxide treatment, target retrieval, protease treatment, probe hybridization, multiplex amplification, flurophore addition (Alexa-Fluor 488, 594, or 647 depending on the target and experiment), quenching, and finally DAPI staining and slide mounting. Probes used in this study were purchased from Advanced Cell Diagnostics and included probes for the following genes: Mef2a, Mef2c, GFP, Gria4, and Scn1a. Images were acquired using a confocal microscope (LSM 710; Zeiss) with a 40× objective at equivalent settings for all conditions. Images were quantified using ImageJ or Imaris by an experimenter blind to treatment groups. For each experimental condition, two coronal sections from at least three animals were used for quantification.
Imaris Quantification Methods.
Following immunohistochemistry and imaging of 40uM brain slices, 3D reconstruction and analysis was completed using Bitplane Imarisx64 (version 8.3.1). The surfaces module was used to render and count nuclei that stained positive for DAPI, NueN, and PV. Average Mef2 intensity in non-neuronal cells was measuring by first masking the NueN+ nuclei from the DAPI+ channel. A surface was then created around these DAPI+/NueN- nuclei, and the intensity of Mef2 was determined within this non-neuronal nuclei surface. Similarly, Mef2 and NueN signal located within the DAPI+/NueN+ surfaces was measured for neuronal nuclei. Finally, Mef2 signal for PV+ interneurons was measured by creating a surface around and DAPI+/NueN+/PV+ nuclei and quantifying the average intensity within these rendered nuclei. For all other epitopes, mean intensity was calculated over the image area automatically using Imaris. For RNAscope transcript quantification, the Imaris spots tool was used to determine mRNA expression levels. A small radius (~15 microns) was set to analyze all spots within each cell separately. The intensity threshold for inclusion was set such that only cells clearly distinguishable from background were included. Background brightness and contrast were not altered between images, and all quantification was conducted in a blinded fashion. Mean intensity for each spot was calculated by the Imaris software.
Quantitative RT–PCR.
The mPFC was isolated from 2-month-old male Swiss Webster mice. Tissue was rapidly frozen on dry ice and stored at −80 °C. RNA was extracted using the RNeasy kit according to the manufacturer’s protocol (Qiagen). For each sample, 1 μg of RNA was reverse transcribed in a 20 μL reaction volume containing EcoDry Premix (TaKaRa) diluted in water at 42°C for 1 h. cDNA was further diluted 1:10 and 2 μL were used for each RT-qPCR amplification reaction (SsoFast EvaGreen Supermix, Bio-Rad). Relative changes in gene expression were assessed using the 2−ΔΔCt method.
Slice electrophysiology.
Acute brain slices were prepared from 3 month old wildtype Swiss Webster mice who were enriched for 1 month after cortical injections with either Scrmb or Mef2a/c shRNA lentiviruses. In a separate experiment, slices were also prepared from 4–4.5 month old male P301S mice raised in standard conditions, WT controls raised in standard conditions, and P301S mice raised in an enriched environment. For all experiments, mice were anesthetized with isoflurane and transcardially perfused with oxygenated ice-cold dissection buffer containing (in mM) 211 sucrose, 3.3 KCl, 1.3 NaH2PO4, 0.5 CaCl2, 10 MgCl2, 26 NaHCO3 and 11 D-Glucose. Each mouse was then decapitated and the brain was rapidly removed. Coronal slices (250 μm thick) were prepared in ice-cold dissection buffer using a Leica VT1000S vibratome (Leica). Slices were recovered in a holding chamber with 95% O2/5% CO2-saturated artificial cerebrospinal fluid (ACSF) consisting of (in mM) 124 NaCl, 3.3 KCl, 1.3 NaH2PO4, 2.5 CaCl2, 1.5 MgCl2, 26 NaHCO3 and 11 D-Glucose for 1 h at 32 °C and then stored at room temperature. Individual slices for recording were then transferred to a submerged recording chamber and perfused with ACSF at a constant rate of 2–2.5 ml/min at room temperature. Cells were visualized using infrared differential interference contrast (IR-DIC) imaging on an Olympus BX-50WI microscope. Action potentials (APs) in whole-cell current patch clamp from prefrontal cortex were acquired on an EPC10 amplifier (HEKA Elektronik) with Patchmaster software. The pyramidal neuron in layer IV-VI of prefrontal cortex was patched and APs was elicited by current clamp of current steps from −40 pA to +340 pA at 20 pA increments for 500ms. Signals were filtered at 2KHz and stored on a personal computer. A borosilicate glass electrode (resistance of 6–7MΩ) with pipette solution containing (in mM) 130 K gluconate; 20 KCl; 10 HEPES; 0.2 EGTA, 4 MgATP, 0.3 Na2GTP, 10 disodium phosphocreatine was used. APs were analyzed using Patchmaster software (HEKA Electronik) and Prism7 (GraphPad Software, CA) programs.
Statistics.
Molecular results are presented as mean + s.e.m, assuming a normal distribution. All statistical analyses used Prism GraphPad software to compute significance. Data limited to two experimental groups were analyzed by two-tailed unpaired t-tests. Data consisting of three or more groups were analyzed by one- or two-way ANOVAs followed by pairwise multiple comparisons tests. The statistical test, P-values, and sample size (n) for each experiment are specified in the figure legends. No statistical method was used to estimate sample size, but they are consistent with previous publications while attempting to eliminate waste and animal mortality. Molecular analyses used a minimum of three biological replicates per condition, and validation experiments used a distinct set of animals. Behavioral tests were run on at least two independent cohorts of animals, with one reported in the main figures.
Supplementary Material
Fig. S1. Environmental enrichment paradigm does not affect general locomotion in mice.
Fig. S2. Increased accessibility of the Mef2 binding motif after enrichment in both male and female mice.
A, PCA of ATACseq peak counts in EE and SH females (n = 3 EE vs. 3 SH mice). B, PCA of ATACseq peak counts in EE and SH males (n = 7 EE vs. 6 SH mice). C, D, Mef2 motifs are enriched in the DARs that are gained in EE female (C) and male (D) mice. E, Mef2a motif frequency (motif/bp/peak) plotted across the relative length of significant DARs gained in EE (red) and SH female mice (blue). F, Mef2a motif frequency (motif/bp/peak) plotted across the relative length of significant DARs gained in EE (red) and SH male mice (blue). G, Mef2c motif frequency (motif/bp/peak) plotted across the relative length of significant DARs gained in EE (red) and SH female mice (blue). H, Mef2c motif frequency (motif/bp/peak) plotted across the relative length of significant DARs gained in EE (red) and SH male mice (blue).
Fig. S3. Overlap of DARs with histone modifications, and correlation between neuronal ATAC-and RNA-sequencing.
Fig. S4. Enrichment induces a transcriptional signature associated with Mef2 activity in cortical neurons of male mice.
A, PCA plot of nuclear RNAseq expression data from frontal cortical neurons of EE (n=6) and SH (n=6) male mice. B, Volcano plot of DEG analysis of nuclear RNA isolated from the frontal cortex of EE and SH male mice (n = 6 EE vs. 6 SH), showing 280 (red) and 109 (blue) significantly up-regulated in EE (upEE) and SH (upSH) groups, respectively (significance cut-off q-value ≦ 0.1). C, Quantitative PCR (qPCR) on RNA isolated from whole mPFC for a different cohort of animals to validate increases in known downstream targets of Mef2a (n = 4 male animals per group, 2-way ANOVA, p-value for group = 0.0002). D, IHC from the medial prefrontal cortex of wildtype male mice exposed to EE and SH conditions, staining for DAPI (blue), Rbfox3 (red), and Mef2a (green). Top right: Imaris quantification of staining with Mef2a/NeuN intensity plotted for individual nuclei across 3 EE and 3 SH wildtype mice (t-test p-value <0.0001). Bottom left: IHC from the medial prefrontal cortex of EE and SH male mice, staining for DAPI (blue), Rbfox3 (red), and Mef2c (green). Bottom right: Imaris quantification of IHC staining with Mef2c/NeuN intensity plotted for individual nuclei across 3 EE and 3 SH mice (t-test p-value = 0.5700).
Fig. S5. Gene ontology over-representation analysis following environmental enrichment.
A, Gene set enrichment analysis (biological process and molecular function) for EE vs. SH DEGs (q<0.1) in female mice. Enrichment ratio is calculated by dividing the number of observed genes within a gene ontology (GO) category by the number of expected genes. All GO terms listed show a FDR<0.05. B, Gene set enrichment analysis (biological process, molecular function, and cellular component) for EE vs. SH DEGs (q<0.1) in male mice. Enrichment ratio is calculated by dividing the number of observed genes within a gene ontology (GO) category by the number of expected genes. All GO terms listed show a FDR<0.05.
Fig. S6. Effects of Mef2a/c knockdown in cortical neurons and mice.
A, Quantitative PCR (qPCR) was used to determine efficiency and specificity of Mef2a-d knockdown in the presence of a Scrmb (red), Mef2a (solid green), or Mef2c (dotted green) shRNA cassette. Mef2a shRNA led to selective knockdown of Mef2a (unpaired t-test, p-value=0.0004). Mef2c shRNA led to robust knockdown of Mef2c (unpaired t-test, p-value=0.0135) and a modest knockdown of Mef2a (unpaired t-test, p-value=0.0068) and Mef2d (unpaired t-test, p-value=0.0332). B, Mef2a/c and Scrmb shRNA-treated mice have similar body weights (male=left; female=right). C, Mef2a/c and Scrmb shRNA-treated mice show similar distance travelled in a 10-minute open field testing paradigm (male=left; female=right). D, Mef2a/c shRNA-treated female mice show significantly impaired fear extinction (n=12 Scrmb vs. 14 Mef2a/c KD; Scrmb t-test p-value = 0.0450 vs. Mef2a/c KD t-test p-value = 0.9635). E, Voltage-clamp recordings from shRNA-expressing regions (identified by GFP fluorescence and morphology) revealed that excitatory pyramidal neurons with decreased Mef2a/c levels (green) showed no significant difference in action potential amplitude, sIPSC frequency, or cumulative sIPSC probability distribution, relative to Scrmb shRNA-expressing neurons.
Fig. S7. Transcriptional signatures of cognitive resilience in bulk RNA-sequencing of human cortical tissue.
A, Table indicating cognitive distribution of patients across Braak scores (with corresponding average amyloid score) in both MSBB and ROSMAP cohorts. The majority of low-pathology patients (Braak 0–2) are cognitively normal prior to death, and only 20% exhibit dementia. The proportion of mid-pathology patients (Braak 3–4) that exhibit cognitive impairment is substantially higher in both cohorts. ~Three-quarters of all high-pathology patients (Braak 5–6) exhibit dementia prior to death, while fewer than 10% remain cognitively unimpaired. B, Gene ontology enrichment of CgR gene sets in MSBB and ROSMAP demonstrates that distinct molecular functions are associated with resilience in the two datasets. Shading denotes the significance (−log10FDR) of GO term enrichment. C, Gene expression of MEF2A from the anterior prefrontal cortex in the MSBB cohort (left, n = 260, r = −0.108, p = 0.081) and from the dorsolateral prefrontal cortex in the ROSMAP cohort (right, n = 888, r = 0.066, p = 0.049) plotted against clinical dementia rating (MSBB) and global cognitive function at last visit (ROSMAP). Each individual represented by a dot, and red line denotes regression line with standard error (grey shading). D, Same as (C), but for MEF2D (MSBB: r = −0.034, p = 0.5813; ROSMAP: r = 0.140, p = 2.92X10−5). E, Significance of association between MEF2A, MEF2C, and MEF2D expression with cognition in a simple regression (Cog) or multiple regression correcting for age of death, sex, and available pathological variables (CgR) in the MSBB and ROSMAP cohort. Number refers to p-value of association and shading denotes −log10(p-value). F, Histogram plotting all protein-coding genes identified in mouse RNA-seq experiments (top) and the ROSMAP cohort (bottom) by their mean expression revealing a considerable difference in the abundance of MEF2A in the mouse brain (94th percentile), as compared to the human brain (71st percentile). MEF2C appears to be similarly highly expressed in both species. G, Pearson correlation coefficient values measuring co-expression between MEF2A (top left) and either ROSMAP Cog (bold red line) or CgR (bold blue line) gene sets across developmental time from cortical samples of the BrainSpan Atlas of the Developing Human Brain. Resampling-based statistical test performed contrasting average correlation values observed (red and blue lines) with random expectation estimated by computing average MEF2A co-expression values across BrainSpan Atlas for 10,000 randomly selected, same-sized gene sets (Cog = red histogram, CgR = blue histogram). Asterix denote significant results (p-values for MEF2A Cog < 1×10−4; MEF2A Cgr = 0.01). Similar analysis performed for MEF2C (top right, p-values for Cog < 1×10−4; Cgr < 1×10−4), ARC (bottom left, p-values for Cog = 0.787; Cgr = 0.247), and FOS (bottom right, p-values for Cog = 0.49; Cgr < 1×10−4).
Fig. S8. Single nucleus RNA-sequencing of cortical tissue reveals cell type-dependent transcriptomic signatures of cognitive resilience.
A, Clinical, demographic, and pathological data for resilient and non-resilient individuals selected for scRNA-sequencing CgR cohort B, Heatmap of canonical cell marker expression across the different clusters identified in Figure 4a illustrates that clusters identified correspond to known cell types found in the brain. Shading denotes expression level. C, Heatmap of MEF2A and MEF2C expression in each cell type cluster, differentiated by cells belonging to Resilient (red) and Non-resilient (blue) individuals. No significant differences were found for any MEF2 genes in any cell type. Shading denotes expression level, normalized across the row to show genes relative expression across all cell types. D, Gene ontology enrichment for molecular function terms of Exc subcluster DEGs between resilient and non-resilient patients. Shading denotes the significance (−log10FDR) of the GO term enrichment. E, UMAP plot of subclustering performed on Inhibitory neuron (Inh, top) and Microglia (Micro, bottom) cell clusters. F, Heatmap of MEF2A and MEF2C expression in each of the Inh (left) and Micro (right) subclusters, differentiated by cells belonging to Resilient (red) and Non-resilient (blue) individuals. No significant differences were found for any MEF2 genes in any of the subclusters. Shading denotes expression level, normalized across the row to show genes relative expression across all cell types. G, DEGs between resilient and non-resilient cells in each of the inhibitory neuron and microglia cell subclusters (for which DEGs were identified) were assayed for enrichment among known MEF2A targets and MEF2C targets. No significant enrichment for MEF2A or MEF2C targets was identified in any of the subclusters. Numbers in the heatmap refer to odds ratios of overlap, and significance (−log10FDR) denoted by color shading.
Fig. S9. Tauopathy impacts Mef2 transcriptional network, and Mef2a/c overexpression alters levels of target genes involved in neural transmission without affecting movement or weight.
A, Gene set enrichment analysis for molecular function (top) and transcription factor perturbations (bottom) were performed on the list of genes identified as down-regulated in the neurons of 2-month-old male, C57/BL6 PS19 mice, when compared to age- and sex-matched, wildtype C57/BL6 mice. B, Top left: RNA in situ hybridization from the medial prefrontal cortex of SH PS19 mice after retro-orbital injections of AAV PHP.eB viral vectors containing an overexpression plasmid encoding GFP (GFP Control) or Mef2a-GFP/Mef2c-GFP (Mef2a/c OE), staining for DAPI (blue), Scn1a (red), GFP (green). Top right: quantification of RNA in situ hybridization staining with total Scn1a signal quantified per GFP+ cell (n = 4 animals per group; t-test p-value = 0.0012). Bottom left: RNA in situ hybridization from the medial prefrontal cortex of SH PS19 mice after retro-orbital injections of AAV PHP.eB viral vectors, staining for Mef2c (green) and Gria4 (red). Bottom right: quantification of Mef2c and Gria4 transcript intensity (n = 4 animals per group; t-test p-value < 0.0001 for both Mef2c and Gria4 spots). C, Mef2a/c overexpression via bilateral lentiviral injection in 3 month old wild type C57/BL6 mice enhances rates of fear extinction (n = 9 mice per group, unpaired t-test comparing % freezing post 2-shocks and % freezing on last day of extinction training; t-test p-value for GFP control group = 0.0709; t-test p-value for Mef2a/c OE group = 0.0047). D, Mef2a/c OE- and GFP-injected PS19 mice travel similar distances in a 10-minute open field testing paradigm. E, Injections did not affect body weight of PS19 mice.
Table S1. Clinical variables of ROSMAP and MSBB subjects
Table S2. Male Environmental Enrichment (EE) vs Standard Housing (SH) ATAC-Seq raw count matrix
Table S3. Male EE vs SH ATAC-Seq Differential Accessible Region (DAR) analysis
Table S4. Female EE vs SH ATAC-Seq raw count matrix
Table S5. Female EE vs SH ATAC-Seq Differential Accessible Region (DAR) analysis
Table S6. Transcription factor motif enrichment of ATAC-Seq EE vs SH DARs
Table S7. Male EE vs SH RNA-Seq raw count matrix
Table S8. Female EE vs SH RNA-Seq raw count matrix
Table S9. ATAC-seq and RNA-seq Sequencing Statistics
Table S10. Male EE vs SH RNA-Seq Differentially Expressed Gene (DEG) analysis
Table S11. Female EE vs SH RNA-Seq Differentially Expressed Gene (DEG) analysis
Table S13. snRNAseq Results
Table S12. ROSMAP and MSBB Cog and CgR gene lists
Acknowledgements:
We would like to thank members of the Tsai Lab who helped to edit this work, including Elana Lockshin, Djuna von Maydell, Dr. Priyanka Narayan, Dr. Jay Penney, Dr. Wen-Chin Huang, Dr. Joel Blancahrd, Dr. Hiruy Meharena, Dr. Asaf Marco, and Dr. Mat Victor.
Funding:
We would finally like to acknowledge the following funding sources for financially supporting this work: Glenn for Biology of Aging Research #6935778, NIH 1-R01-AG046174-03, NIH RF1-AG054321, 1-RF1-AG054012 to L-H T; NIH 7-R01-AG058002 to M.K, Cure Alzheimer’s Fund to L-H T and M.K; P30AG10161, R01AG15819, R01AG17917, U01AG61356 to D.A.B; NIH T32 5T32HD098061 to R.M.R.
Footnotes
Table S14. Downregulated genes in PS19 NeuN-positive nuclei from 2 month old males
Competing Interests: The authors have no competing financial interests that could be perceived as being a conflict of interest.
Supplemental Tables: Please see Auxiliary Supplementary Materials and Other Supporting Files as the tables were too large to insert here.
Data availability:
The MSBB clinical and bulk sequencing data can be accessed via the Synapse portal (Synapse ID: syn3159438). The ROSMAP metadata can be accessed via the Synapse portal (Synapse ID: syn3219045). The data are available under controlled use conditions set by human privacy regulations. To access the data, a data use agreement is needed. This registration is in place solely to ensure anonymity of the ROSMAP study participants. A data use agreement can be agreed with either Rush University Medical Center (RUMC) or with SAGE, who maintains Synapse, and can be downloaded from their websites. The snRNA-seq and bulk sequencing data will be available on The Rush Alzheimer’s Disease Center (RADC) Research Resource Sharing Hub at https://www.radc.rush.edu/docs/omics.htm or at Synapse (__) under the doi ___. The mouse RNA- and ATAC-sequencing data will be available after publication under the doi ___. [These will be finalized and inserted once the manuscript is accepted for publication.] Code used throughout this study is available upon request, from the corresponding authors.
References:
- 1.Woolf SH, Schoomaker H, Life Expectancy and Mortality Rates in the United States, 1959–2017, JAMA 322, 1996–21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brown GC, Living too long, EMBO Rep 16, 137–141 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stern Y, Gurland B, Tatemichi TK, Tang MX, Wilder D, Mayeux R, Influence of education and occupation on the incidence of Alzheimer’s disease, JAMA 271, 1004–1010 (1994). [PubMed] [Google Scholar]
- 4.van Dellen A, Blakemore C, Deacon R, York D, Hannan AJ, Delaying the onset of Huntington’s in mice, Nature 404, 721–722 (2000). [DOI] [PubMed] [Google Scholar]
- 5.Jankowsky JL, Environmental Enrichment Mitigates Cognitive Deficits in a Mouse Model of Alzheimer’s Disease, Journal of Neuroscience 25, 5217–5224 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Solinas M, Chauvet C, Thiriet N, El Rawas R, Jaber M, Reversal of cocaine addiction by environmental enrichment, Proc. Natl. Acad. Sci. U.S.A 105, 17145–17150 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lehmann ML, Herkenham M, Environmental Enrichment Confers Stress Resiliency to Social Defeat through an Infralimbic Cortex-Dependent Neuroanatomical Pathway, Journal of Neuroscience 31, 6159–6173 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leon J, Moreno AJ, Garay BI, Chalkley RJ, Burlingame AL, Wang D, Dubal DB, Peripheral Elevation of a Klotho Fragment Enhances Brain Function and Resilience in Young, Aging, and α-Synuclein Transgenic Mice, CellReports 20, 1360–1371 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flavell SW, Cowan CW, Kim T-K, Greer PL, Lin Y, Paradis S, Griffith EC, Hu LS, Chen C, Greenberg ME, Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number, Science 311, 1008–1012 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Pulipparacharuvil S, Renthal W, Hale CF, Taniguchi M, Xiao G, Kumar A, Russo SJ, Sikder D, Dewey CM, Davis MM, Greengard P, Nairn AC, Nestler EJ, Cowan CW, Cocaine Regulates MEF2 to Control Synaptic and Behavioral Plasticity, Neuron 59, 621–633 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaudilliere B, Shi Y, Bonni A, RNA interference reveals a requirement for myocyte enhancer factor 2A in activity-dependent neuronal survival, J. Biol. Chem 277, 46442–46446 (2002). [DOI] [PubMed] [Google Scholar]
- 12.Sniekers S, Stringer S, Watanabe K, Jansen PR, Coleman JRI, Krapohl E, Taskesen E, Hammerschlag AR, Okbay A, Zabaneh D, Amin N, Breen G, Cesarini D, Chabris CF, Iacono WG, Ikram MA, Johannesson M, Koellinger P, Lee JJ, Magnusson PKE, McGue M, Miller MB, Ollier WER, Payton A, Pendleton N, Plomin R, Rietveld CA, Tiemeier H, van Duijn CM, Posthuma D, Genome-wide association meta-analysis of 78,308 individuals identifies new loci and genes influencing human intelligence, Nat Genet 49, 1107–1112 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bennett DA, Schneider JA, Arvanitakis Z, Wilson RS, Overview and findings from the religious orders study, Curr Alzheimer Res 9, 628–645 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bennett DA, Schneider JA, Buchman AS, Barnes LL, Boyle PA, Wilson RS, Overview and findings from the rush Memory and Aging Project, Curr Alzheimer Res 9, 646–663 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zeleznikow-Johnston A, Burrows EL, Renoir T, Hannan AJ, Environmental enrichment enhances cognitive flexibility in C57BL/6 mice on a touchscreen reversal learning task, Neuropharmacology 117, 219–226 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Lahiani-Cohen I, Lourbopoulos A, Haber E, Rozenstein-Tsalkovich L, Abramsky O, Grigoriadis N, Rosenmann H, Moderate environmental enrichment mitigates tauopathy in a neurofibrillary tangle mouse model, J. Neuropathol. Exp. Neurol 70, 610–621 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Lach G, Bicca MA, Hoeller AA, Santos ECDS, Costa APR, de Lima TCM, Short-term enriched environment exposure facilitates fear extinction in adult rats: The NPY-Y1 receptor modulation, Neuropeptides 55, 73–78 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai L-H, Recovery of learning and memory is associated with chromatin remodelling, Nature 447, 178–182 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ, Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position, Nat. Methods 10, 1213–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wales S, Hashemi S, Blais A, McDermott JC, Global MEF2 target gene analysis in cardiac and skeletal muscle reveals novel regulation of DUSP6 by p38MAPK-MEF2 signaling, Nucleic Acids Research 42, 11349–11362 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang X, Jin JY, Wu J, Qin X, Streilein R, Hall RP, Zhang JY, RNA-Seq and ChIP-Seq Reveal SQSTM1/p62 as a Key Mediator of JunB Suppression of NF-κB-Dependent Inflammation, Journal of Investigative Dermatology 135, 1016–1024 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davies JS, Klein DC, Carter DA, Selective genomic targeting by FRA-2/FOSL2 transcription factor: regulation of the Rgs4 gene is mediated by a variant activator protein 1 (AP-1) promoter sequence/CREB-binding protein (CBP) mechanism, J. Biol. Chem 286, 15227–15239 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andzelm MM, Vanness D, Greenberg ME, Linden DJ, A Late Phase of Long-Term Synaptic Depression in Cerebellar Purkinje Cells Requires Activation of MEF2, Cell Reports 26, 1089–1097.e3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Telese F, Ma Q, Perez PM, Notani D, Oh S, Li W, Comoletti D, Ohgi KA, Taylor H, Rosenfeld MG, LRP8-Reelin-Regulated Neuronal Enhancer Signature Underlying Learning and Memory Formation, Neuron 86, 696–710 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harrington AJ, Raissi A, Rajkovich K, Berto S, Kumar J, Molinaro G, Raduazzo J, Guo Y, Loerwald K, Konopka G, Huber KM, Cowan CW, MEF2C regulates cortical inhibitory and excitatory synapses and behaviors relevant to neurodevelopmental disorders, Elife 5, 140 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lacar B, Linker SB, Jaeger BN, Krishnaswami S, Barron J, Kelder M, Parylak S, Paquola AA, Venepally P, Novotny M, Connor COA, Fitzpatrick C, Erwin J, Hsu JY, Husband D, McConnell MJ, Lasken R, Gage FH, Nuclear RNA-seq of single neurons reveals molecular signatures of activation, Nature Communications, 1–13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tyssowski KM, DeStefino NR, Cho J-H, Dunn CJ, Poston RG, Carty CE, Jones RD, Chang SM, Romeo P, Wurzelmann MK, Ward JM, Andermann ML, Saha RN, Dudek SM, Gray JM, Different Neuronal Activity Patterns Induce Different Gene Expression Programs, Neuron 98, 530–546.e11 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma Q, Telese F, Genome-wide epigenetic analysis of MEF2A and MEF2C transcription factors in mouse cortical neurons, Communicative & Integrative Biology 8, e1087624–5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rajkovich KE, Loerwald KW, Hale CF, Hess CT, Gibson JR, Huber KM, Experience-Dependent and Differential Regulation of Local and Long-Range Excitatory Neocortical Circuits by Postsynaptic Mef2c, Neuron 93, 48–56 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Selkoe DJ, Early network dysfunction in Alzheimer’s disease, Science 365, 540–541 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Wang J, Ou S-W, Wang Y-J, Distribution and function of voltage-gated sodium channels in the nervous system, Channels 11, 534–554 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Escayg A, MacDonald BT, Meisler MH, Baulac S, Huberfeld G, An-Gourfinkel I, Brice A, LeGuern E, Moulard B, Chaigne D, Buresi C, Malafosse A, Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2, Nat Genet 24, 343–345 (2000). [DOI] [PubMed] [Google Scholar]
- 33.Ohno Y, Ishihara S, Mashimo T, Sofue N, Shimizu S, Imaoku T, Tsurumi T, Sasa M, Serikawa T, Scn1a missense mutation causes limbic hyperexcitability and vulnerability to experimental febrile seizures, Neurobiology of Disease 41, 261–269 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Beyer B, Deleuze C, Letts VA, Mahaffey CL, Boumil RM, Lew TA, Huguenard JR, Frankel WN, Absence seizures in C3H/HeJ and knockout mice caused by mutation of the AMPA receptor subunit Gria4, Human Molecular Genetics 17, 1738–1749 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stopford CL, Thompson JC, Neary D, Richardson AMT, Snowden JS, Working memory, attention, and executive function in Alzheimer’s disease and frontotemporal dementia, Cortex 48, 429–446 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma’ayan A, Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool, BMC Bioinformatics 14, 128 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miller JA, Ding S-L, Sunkin SM, Smith KA, Ng L, Szafer A, Ebbert A, Riley ZL, Royall JJ, Aiona K, Arnold JM, Bennet C, Bertagnolli D, Brouner K, Butler S, Caldejon S, Carey A, Cuhaciyan C, Dalley RA, Dee N, Dolbeare TA, Facer BAC, Feng D, Fliss TP, Gee G, Goldy J, Gourley L, Gregor BW, Gu G, Howard RE, Jochim JM, Kuan CL, Lau C, Lee C-K, Lee F, Lemon TA, Lesnar P, McMurray B, Mastan N, Mosqueda N, Naluai-Cecchini T, Ngo N-K, Nyhus J, Oldre A, Olson E, Parente J, Parker PD, Parry SE, Stevens A, Pletikos M, Reding M, Roll K, Sandman D, Sarreal M, Shapouri S, Shapovalova NV, Shen EH, Sjoquist N, Slaughterbeck CR, Smith M, Sodt AJ, Williams D, Zöllei L, Fischl B, Gerstein MB, Geschwind DH, Glass IA, Hawrylycz MJ, Hevner RF, Huang H, Jones AR, Knowles JA, Levitt P, Phillips JW, Šestan N, Wohnoutka P, Dang C, Bernard A, Hohmann JG, Lein ES, Transcriptional landscape of the prenatal human brain, Nature 508, 199–206 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.He Z, Han D, Efimova O, Guijarro P, Yu Q, Oleksiak A, Jiang S, Anokhin K, Velichkovsky B, Grünewald S, Khaitovich P, Comprehensive transcriptome analysis of neocortical layers in humans, chimpanzees and macaques, Nat. Neurosci 20, 886–895 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Yoshiyama Y, Higuchi M, Zhang B, Huang S-M, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VMY, Synapse Loss and Microglial Activation Precede Tangles in a P301S Tauopathy Mouse Model, Neuron 53, 337–351 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Maeda S, Djukic B, Taneja P, Yu G-Q, Lo I, Davis A, Craft R, Guo W, Wang X, Kim D, Ponnusamy R, Gill TM, Masliah E, Mucke L, Expression of A152T human tau causes age-dependent neuronal dysfunction and loss in transgenic mice, EMBO Rep 17, 530–551 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mitchell AC, Javidfar B, Pothula V, Ibi D, Shen EY, Peter CJ, Bicks LK, Fehr T, Jiang Y, Brennand KJ, Neve RL, Gonzalez-Maeso J, Akbarian S, MEF2C transcription factor is associated with the genetic and epigenetic risk architecture of schizophrenia and improves cognition in mice, Molecular Psychiatry 23, 123–132 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cole CJ, Mercaldo V, Restivo L, Yiu AP, Sekeres MJ, Han J-H, Vetere G, Pekar T, Ross PJ, Neve RL, Frankland PW, Josselyn SA, MEF2 negatively regulates learning-induced structural plasticity and memory formation, Nat. Neurosci, 1–12 (2012). [DOI] [PubMed] [Google Scholar]
- 43.Adachi M, Lin P-Y, Pranav H, Monteggia LM, Postnatal Loss of Mef2c Results in Dissociation of Effects on Synapse Number and Learning and Memory, Biol. Psychiatry 80, 140–148 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tu S, Akhtar MW, Escorihuela RM, Amador-Arjona A, Swarup V, Parker J, Zaremba JD, Holland T, Bansal N, Holohan DR, Lopez K, Ryan SD, Chan SF, Yan L, Zhang X, Huang X, Sultan A, McKercher SR, Ambasudhan R, Xu H, Wang Y, Geschwind DH, Roberts AJ, Terskikh AV, Rissman RA, Masliah E, Lipton SA, Nakanishi N, NitroSynapsin therapy for a mouse MEF2C haploinsufficiency model of human autism, Nature Communications, 1–12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O’Shea DJ, Sohal VS, Goshen I, Finkelstein J, Paz JT, Stehfest K, Fudim R, Ramakrishnan C, Huguenard JR, Hegemann P, Deisseroth K, Neocortical excitation/inhibition balance in information processing and social dysfunction, Nature, 1–8 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ataman B, Boulting GL, Harmin DA, Yang MG, Baker-Salisbury M, Yap E-L, Malik AN, Mei K, Rubin AA, Spiegel I, Durresi E, Sharma N, Hu LS, Pletikos M, Griffith EC, Partlow JN, Stevens CR, Adli M, Chahrour M, Šestan N, Walsh CA, Berezovskii VK, Livingstone MS, Greenberg ME, Evolution of Osteocrin as an activity-regulated factor in the primate brain, Nature 539, 242–247 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen X, Shen Y, Draper W, Buenrostro JD, Litzenburger U, Cho SW, Satpathy AT, Carter AC, Ghosh RP, East-Seletsky A, Doudna JA, Greenleaf WJ, Liphardt JT, Chang HY, ATAC-see reveals the accessible genome by transposase-mediated imaging and sequencing, Nat. Methods 13, 1013–1020 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Halder R, Hennion M, Vidal RO, Shomroni O, Rahman R-U, Rajput A, Centeno TP, van Bebber F, Capece V, Vizcaino JCG, Schuetz A-L, Burkhardt S, Benito E, Sala MN, Javan SB, Haass C, Schmid B, Fischer A, Bonn S, DNA methylation changes in plasticity genes accompany the formation and maintenance of memory, Nat. Neurosci 19, 102–110 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Wang M, Beckmann ND, Roussos P, Wang E, Zhou X, Wang Q, Ming C, Neff R, Ma W, Fullard JF, Hauberg ME, Bendl J, Peters MA, Logsdon B, Wang P, Mahajan M, Mangravite LM, Dammer EB, Duong DM, Lah JJ, Seyfried NT, Levey AI, Buxbaum JD, Ehrlich M, Gandy S, Katsel P, Haroutunian V, Schadt E, Zhang B, The Mount Sinai cohort of large-scale genomic, transcriptomic and proteomic data in Alzheimer’s disease, Sci Data 5, 180185–16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, Horvath S, Geschwind DH, Integrative Functional Genomic Analyses Implicate Specific Molecular Pathways and Circuits in Autism, Cell 155, 1008–1021 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, Martorell AJ, Ransohoff RM, Hafler BP, Bennett DA, Kellis M, Tsai L-H, Single-cell transcriptomic analysis of Alzheimer’s disease, Nature, 1–24 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chan KY, Jang MJ, Yoo BB, Greenbaum A, Ravi N, Wu W-L, Sánchez-Guardado L, Lois C, Mazmanian SK, Deverman BE, Gradinaru V, Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems, Nat. Neurosci 20, 1172–1179 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Environmental enrichment paradigm does not affect general locomotion in mice.
Fig. S2. Increased accessibility of the Mef2 binding motif after enrichment in both male and female mice.
A, PCA of ATACseq peak counts in EE and SH females (n = 3 EE vs. 3 SH mice). B, PCA of ATACseq peak counts in EE and SH males (n = 7 EE vs. 6 SH mice). C, D, Mef2 motifs are enriched in the DARs that are gained in EE female (C) and male (D) mice. E, Mef2a motif frequency (motif/bp/peak) plotted across the relative length of significant DARs gained in EE (red) and SH female mice (blue). F, Mef2a motif frequency (motif/bp/peak) plotted across the relative length of significant DARs gained in EE (red) and SH male mice (blue). G, Mef2c motif frequency (motif/bp/peak) plotted across the relative length of significant DARs gained in EE (red) and SH female mice (blue). H, Mef2c motif frequency (motif/bp/peak) plotted across the relative length of significant DARs gained in EE (red) and SH male mice (blue).
Fig. S3. Overlap of DARs with histone modifications, and correlation between neuronal ATAC-and RNA-sequencing.
Fig. S4. Enrichment induces a transcriptional signature associated with Mef2 activity in cortical neurons of male mice.
A, PCA plot of nuclear RNAseq expression data from frontal cortical neurons of EE (n=6) and SH (n=6) male mice. B, Volcano plot of DEG analysis of nuclear RNA isolated from the frontal cortex of EE and SH male mice (n = 6 EE vs. 6 SH), showing 280 (red) and 109 (blue) significantly up-regulated in EE (upEE) and SH (upSH) groups, respectively (significance cut-off q-value ≦ 0.1). C, Quantitative PCR (qPCR) on RNA isolated from whole mPFC for a different cohort of animals to validate increases in known downstream targets of Mef2a (n = 4 male animals per group, 2-way ANOVA, p-value for group = 0.0002). D, IHC from the medial prefrontal cortex of wildtype male mice exposed to EE and SH conditions, staining for DAPI (blue), Rbfox3 (red), and Mef2a (green). Top right: Imaris quantification of staining with Mef2a/NeuN intensity plotted for individual nuclei across 3 EE and 3 SH wildtype mice (t-test p-value <0.0001). Bottom left: IHC from the medial prefrontal cortex of EE and SH male mice, staining for DAPI (blue), Rbfox3 (red), and Mef2c (green). Bottom right: Imaris quantification of IHC staining with Mef2c/NeuN intensity plotted for individual nuclei across 3 EE and 3 SH mice (t-test p-value = 0.5700).
Fig. S5. Gene ontology over-representation analysis following environmental enrichment.
A, Gene set enrichment analysis (biological process and molecular function) for EE vs. SH DEGs (q<0.1) in female mice. Enrichment ratio is calculated by dividing the number of observed genes within a gene ontology (GO) category by the number of expected genes. All GO terms listed show a FDR<0.05. B, Gene set enrichment analysis (biological process, molecular function, and cellular component) for EE vs. SH DEGs (q<0.1) in male mice. Enrichment ratio is calculated by dividing the number of observed genes within a gene ontology (GO) category by the number of expected genes. All GO terms listed show a FDR<0.05.
Fig. S6. Effects of Mef2a/c knockdown in cortical neurons and mice.
A, Quantitative PCR (qPCR) was used to determine efficiency and specificity of Mef2a-d knockdown in the presence of a Scrmb (red), Mef2a (solid green), or Mef2c (dotted green) shRNA cassette. Mef2a shRNA led to selective knockdown of Mef2a (unpaired t-test, p-value=0.0004). Mef2c shRNA led to robust knockdown of Mef2c (unpaired t-test, p-value=0.0135) and a modest knockdown of Mef2a (unpaired t-test, p-value=0.0068) and Mef2d (unpaired t-test, p-value=0.0332). B, Mef2a/c and Scrmb shRNA-treated mice have similar body weights (male=left; female=right). C, Mef2a/c and Scrmb shRNA-treated mice show similar distance travelled in a 10-minute open field testing paradigm (male=left; female=right). D, Mef2a/c shRNA-treated female mice show significantly impaired fear extinction (n=12 Scrmb vs. 14 Mef2a/c KD; Scrmb t-test p-value = 0.0450 vs. Mef2a/c KD t-test p-value = 0.9635). E, Voltage-clamp recordings from shRNA-expressing regions (identified by GFP fluorescence and morphology) revealed that excitatory pyramidal neurons with decreased Mef2a/c levels (green) showed no significant difference in action potential amplitude, sIPSC frequency, or cumulative sIPSC probability distribution, relative to Scrmb shRNA-expressing neurons.
Fig. S7. Transcriptional signatures of cognitive resilience in bulk RNA-sequencing of human cortical tissue.
A, Table indicating cognitive distribution of patients across Braak scores (with corresponding average amyloid score) in both MSBB and ROSMAP cohorts. The majority of low-pathology patients (Braak 0–2) are cognitively normal prior to death, and only 20% exhibit dementia. The proportion of mid-pathology patients (Braak 3–4) that exhibit cognitive impairment is substantially higher in both cohorts. ~Three-quarters of all high-pathology patients (Braak 5–6) exhibit dementia prior to death, while fewer than 10% remain cognitively unimpaired. B, Gene ontology enrichment of CgR gene sets in MSBB and ROSMAP demonstrates that distinct molecular functions are associated with resilience in the two datasets. Shading denotes the significance (−log10FDR) of GO term enrichment. C, Gene expression of MEF2A from the anterior prefrontal cortex in the MSBB cohort (left, n = 260, r = −0.108, p = 0.081) and from the dorsolateral prefrontal cortex in the ROSMAP cohort (right, n = 888, r = 0.066, p = 0.049) plotted against clinical dementia rating (MSBB) and global cognitive function at last visit (ROSMAP). Each individual represented by a dot, and red line denotes regression line with standard error (grey shading). D, Same as (C), but for MEF2D (MSBB: r = −0.034, p = 0.5813; ROSMAP: r = 0.140, p = 2.92X10−5). E, Significance of association between MEF2A, MEF2C, and MEF2D expression with cognition in a simple regression (Cog) or multiple regression correcting for age of death, sex, and available pathological variables (CgR) in the MSBB and ROSMAP cohort. Number refers to p-value of association and shading denotes −log10(p-value). F, Histogram plotting all protein-coding genes identified in mouse RNA-seq experiments (top) and the ROSMAP cohort (bottom) by their mean expression revealing a considerable difference in the abundance of MEF2A in the mouse brain (94th percentile), as compared to the human brain (71st percentile). MEF2C appears to be similarly highly expressed in both species. G, Pearson correlation coefficient values measuring co-expression between MEF2A (top left) and either ROSMAP Cog (bold red line) or CgR (bold blue line) gene sets across developmental time from cortical samples of the BrainSpan Atlas of the Developing Human Brain. Resampling-based statistical test performed contrasting average correlation values observed (red and blue lines) with random expectation estimated by computing average MEF2A co-expression values across BrainSpan Atlas for 10,000 randomly selected, same-sized gene sets (Cog = red histogram, CgR = blue histogram). Asterix denote significant results (p-values for MEF2A Cog < 1×10−4; MEF2A Cgr = 0.01). Similar analysis performed for MEF2C (top right, p-values for Cog < 1×10−4; Cgr < 1×10−4), ARC (bottom left, p-values for Cog = 0.787; Cgr = 0.247), and FOS (bottom right, p-values for Cog = 0.49; Cgr < 1×10−4).
Fig. S8. Single nucleus RNA-sequencing of cortical tissue reveals cell type-dependent transcriptomic signatures of cognitive resilience.
A, Clinical, demographic, and pathological data for resilient and non-resilient individuals selected for scRNA-sequencing CgR cohort B, Heatmap of canonical cell marker expression across the different clusters identified in Figure 4a illustrates that clusters identified correspond to known cell types found in the brain. Shading denotes expression level. C, Heatmap of MEF2A and MEF2C expression in each cell type cluster, differentiated by cells belonging to Resilient (red) and Non-resilient (blue) individuals. No significant differences were found for any MEF2 genes in any cell type. Shading denotes expression level, normalized across the row to show genes relative expression across all cell types. D, Gene ontology enrichment for molecular function terms of Exc subcluster DEGs between resilient and non-resilient patients. Shading denotes the significance (−log10FDR) of the GO term enrichment. E, UMAP plot of subclustering performed on Inhibitory neuron (Inh, top) and Microglia (Micro, bottom) cell clusters. F, Heatmap of MEF2A and MEF2C expression in each of the Inh (left) and Micro (right) subclusters, differentiated by cells belonging to Resilient (red) and Non-resilient (blue) individuals. No significant differences were found for any MEF2 genes in any of the subclusters. Shading denotes expression level, normalized across the row to show genes relative expression across all cell types. G, DEGs between resilient and non-resilient cells in each of the inhibitory neuron and microglia cell subclusters (for which DEGs were identified) were assayed for enrichment among known MEF2A targets and MEF2C targets. No significant enrichment for MEF2A or MEF2C targets was identified in any of the subclusters. Numbers in the heatmap refer to odds ratios of overlap, and significance (−log10FDR) denoted by color shading.
Fig. S9. Tauopathy impacts Mef2 transcriptional network, and Mef2a/c overexpression alters levels of target genes involved in neural transmission without affecting movement or weight.
A, Gene set enrichment analysis for molecular function (top) and transcription factor perturbations (bottom) were performed on the list of genes identified as down-regulated in the neurons of 2-month-old male, C57/BL6 PS19 mice, when compared to age- and sex-matched, wildtype C57/BL6 mice. B, Top left: RNA in situ hybridization from the medial prefrontal cortex of SH PS19 mice after retro-orbital injections of AAV PHP.eB viral vectors containing an overexpression plasmid encoding GFP (GFP Control) or Mef2a-GFP/Mef2c-GFP (Mef2a/c OE), staining for DAPI (blue), Scn1a (red), GFP (green). Top right: quantification of RNA in situ hybridization staining with total Scn1a signal quantified per GFP+ cell (n = 4 animals per group; t-test p-value = 0.0012). Bottom left: RNA in situ hybridization from the medial prefrontal cortex of SH PS19 mice after retro-orbital injections of AAV PHP.eB viral vectors, staining for Mef2c (green) and Gria4 (red). Bottom right: quantification of Mef2c and Gria4 transcript intensity (n = 4 animals per group; t-test p-value < 0.0001 for both Mef2c and Gria4 spots). C, Mef2a/c overexpression via bilateral lentiviral injection in 3 month old wild type C57/BL6 mice enhances rates of fear extinction (n = 9 mice per group, unpaired t-test comparing % freezing post 2-shocks and % freezing on last day of extinction training; t-test p-value for GFP control group = 0.0709; t-test p-value for Mef2a/c OE group = 0.0047). D, Mef2a/c OE- and GFP-injected PS19 mice travel similar distances in a 10-minute open field testing paradigm. E, Injections did not affect body weight of PS19 mice.
Table S1. Clinical variables of ROSMAP and MSBB subjects
Table S2. Male Environmental Enrichment (EE) vs Standard Housing (SH) ATAC-Seq raw count matrix
Table S3. Male EE vs SH ATAC-Seq Differential Accessible Region (DAR) analysis
Table S4. Female EE vs SH ATAC-Seq raw count matrix
Table S5. Female EE vs SH ATAC-Seq Differential Accessible Region (DAR) analysis
Table S6. Transcription factor motif enrichment of ATAC-Seq EE vs SH DARs
Table S7. Male EE vs SH RNA-Seq raw count matrix
Table S8. Female EE vs SH RNA-Seq raw count matrix
Table S9. ATAC-seq and RNA-seq Sequencing Statistics
Table S10. Male EE vs SH RNA-Seq Differentially Expressed Gene (DEG) analysis
Table S11. Female EE vs SH RNA-Seq Differentially Expressed Gene (DEG) analysis
Table S13. snRNAseq Results
Table S12. ROSMAP and MSBB Cog and CgR gene lists
Data Availability Statement
The MSBB clinical and bulk sequencing data can be accessed via the Synapse portal (Synapse ID: syn3159438). The ROSMAP metadata can be accessed via the Synapse portal (Synapse ID: syn3219045). The data are available under controlled use conditions set by human privacy regulations. To access the data, a data use agreement is needed. This registration is in place solely to ensure anonymity of the ROSMAP study participants. A data use agreement can be agreed with either Rush University Medical Center (RUMC) or with SAGE, who maintains Synapse, and can be downloaded from their websites. The snRNA-seq and bulk sequencing data will be available on The Rush Alzheimer’s Disease Center (RADC) Research Resource Sharing Hub at https://www.radc.rush.edu/docs/omics.htm or at Synapse (__) under the doi ___. The mouse RNA- and ATAC-sequencing data will be available after publication under the doi ___. [These will be finalized and inserted once the manuscript is accepted for publication.] Code used throughout this study is available upon request, from the corresponding authors.