

Abstract
This study used for the first time LC-MS/MS for the analysis of mitragynine (MIT), a μ-opioid agonist with antinociceptive and antitussive properties, in rat plasma. Mitragynine and the internal standard (amitriptyline) were extracted from plasma with hexane-isoamyl alcohol and resolved on a Lichrospher® RP-SelectB column (9.80 and 12.90 min, respectively). The quantification limit was 0.2 ng/mL within a linear range of 0.2–1000 ng/mL. The method was applied to quantify mitragynine in plasma samples of rats (n = 8 per sampling time) treated with a single oral dose of 20 mg/kg. The following pharmacokinetic parameters were obtained (mean): maximum plasma concentration: 424 ng/mL; time to reach maximum plasma concentration: 1.26 h; elimination half-life: 3.85 h, apparent total clearance: 6.35 L/h/kg, and apparent volume of distribution: 37.90 L/kg.
Keywords: Mitragynine, Mitragyna speciosa, LC–MS/MS, Rats, Pharmacokinetics
1. Introduction
Mitragynine (MIT; Fig. 1) is the main alkaloid extracted from Mitragyna speciosa Korth (Rubiaceae), a species known as “Biak-Biak” in Malaysia and as “Kratom” in Thailand. Kratom leaves are consumed fresh or as dried leaf powder, and are either swallowed or prepared as tea which is strained and then ingested [1]. Kratom is traditionally used in folk medicine to mitigate opioid withdrawal symptoms and is available on the Internet as an alternative to other opioid-replacement medications that can be obtained without a prescription. Unfortunately, there is a growing trend of self-treatment of opioid withdrawal and an increasing number of exposures to Kratom have been reported in the literature [2,3].
Fig. 1.
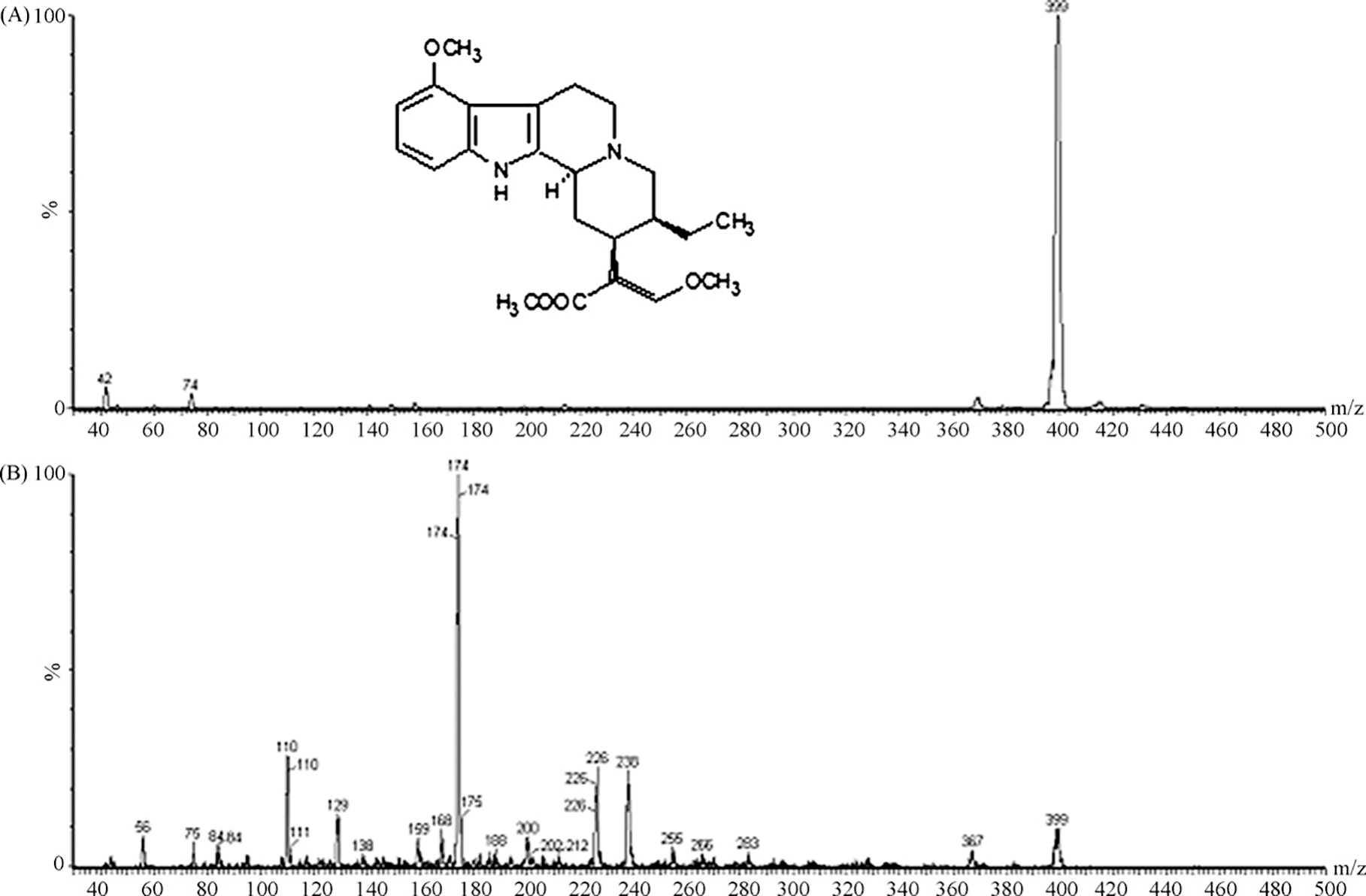
Full-scan mass spectra of the protonated molecular ion (A) and ion product (B) of mitragynine.
MIT has antinociceptive and antitussive properties in animals. In addition, MIT causes much less respiratory depression and does not produce emesis or dyspnea when compared to other opioids such as codeine [4]. MIT has been shown to behave almost as full μ-opioid agonist in guinea-pig ileum [5] and also acts on the serotonin and adrenergic pathways in the spinal cord [6].
The growing sales of Kratom leaves and the medicinal properties of MIT provide a compelling argument for more research into the pharmacology of MIT. The first study investigating the pharmacokinetics of MIT was conducted on rats treated with a single oral dose of 40 mg/kg (gavage). According to Janchawee et al. [7], MIT is rapidly absorbed after oral administration to rats and its elimination is slow, with an elimination half-life of 9.43 h. The only method developed for this analysis employs HPLC with UV detection. Also, the quantification limit of the method is 100 ng/mL and plasma MIT concentrations can only be determined up to 15 h (only 1.59 times its half-life) [7].
Pharmacokinetic studies require a sensitive and selective analytical method to determine the concentration of MIT in plasma samples. HPLC coupled to mass spectrometry (LC–MS) or tandem mass spectrometry (LC–MS/MS) has almost completely replaced other detection systems in the field of bioanalytical research because of its high sensitivity and specificity. This article describes a highly sensitive LC–MS/MS assay for the determination of MIT in rat plasma (quantification limit of 0.2 ng/mL). The validated method was applied to determine plasma concentrations of MIT up to 24 h after administration of a single oral dose of 20 mg MIT/kg to rats.
2. Materials and methods
2.1. Chemicals and reagents
Mitragynine oxalate was purified according to published procedures [8]. H NMR spectra of purified MIT were analyzed according to characterization data published previously [9]. The structure of the compound was also confirmed by X-ray analysis (data not shown).
Acetonitrile and methanol, both HPLC grade, were purchased from Merck (Darmstadt, Germany). Hexane, HPLC grade, was purchased from Acros (New Jersey, USA). The analytical grade reagents used were ammonium acetate (J. T. Baker, Xastoloc, Mexico), formic acid (J. T. Baker, Phillipsburg, USA), isoamyl alcohol (Reagen Quimibras, Rio de Janeiro, Brazil), and sodium hydroxide pellets (Mallinckrodt Baker, Inc., Xastoloc, Mexico). Amitriptyline hydrochloride was purchased from Sigma–Aldrich (St. Louis, MO, USA) and was used as internal standard. Water was purified with the Milli-Q Plus system (Millipore, Bedford, MA, USA).
The blood samples used for the development and validation of the analytical method were obtained from male Wistar rats provided by the Central Animals House of the University of São Paulo, Ribeirão Preto Campus, Brazil. Heparin (Liquemine®, 5000 IU; Roche, Rio de Janeiro, Brazil) was used as anticoagulant in all samples. Plasma was obtained by centrifugation and stored at −70 °C until the time for use.
2.2. Chromatographic analysis
The HPLC system consisted of a Shimadzu chromatograph (Kyoto, Japan) equipped with an LC-10 AD pump and a CTO-10 AS oven. MIT and the internal standard were separated on a Lichrospher60® RP-SelectB column (particle size 5 μm, 125 × 3 mm; Merck). The mobile phase consisted of 20 mM ammonium acetate:acetonitrile (70:30, v/v) containing 0.5% formic acid and the flow rate was 1.2 mL/min. The column was kept at 23 ± 1 °C.
The Quattro Micro triple quadrupole mass spectrometer (Micromass, Manchester, UK) equipped with an electrospray interface (ESI) was used as the mass spectrometry detection system (MS/MS) for the analysis of MIT. Analysis was carried out in the positive electrospray mode. The capillary voltage of the ESI was 3.0 kV. The temperature of the source and desolvation system was kept at 120 and 200 °C, respectively. Nitrogen was used as the nebulizer gas at a flow rate of 415 L/h. Argon was used as the collision gas at a pressure of ~1.0 × 10−3 mbar. The cone voltage was maintained at 30 V. The collision energy was 20 eV.
The conditions for the optimization of MS/MS were determined by direct infusion of standard solution (10 μg/mL) prepared in the mobile phase with an infusion pump at a flow rate of 20 μL/min. Analysis was carried out in the multiple reaction monitoring mode. Protonated ions [M + H]+ and their respective ion products were monitored at the following transitions: 398 > 174 for MIT and 278.4 > 90.8 for the internal standard. Data acquisition and sample quantification were performed with the MassLynx program, version 3.5 (Micromass).
2.3. Standard and working solutions
The stock solution of MIT was prepared in methanol at a concentration of 1 mg/mL. This solution was then diluted in methanol to concentrations of 4, 20, 100, 400, 5000, 10,000, and 20,000 ng/mL. A solution of amitriptyline (internal standard; Sigma) was prepared at a concentration of 5 μg/mL in methanol.
2.4. Sample preparation
Before analysis, 500 μL plasma samples were spiked with 25 μL amitriptyline solution (internal standard, 5 μg/mL). The samples were added to 200 μL of 1 M sodium hydroxide and 6 mL hexane: isoamyl alcohol (99:1, v/v). Extraction was performed by shaking for 30 min in a horizontal shaker (model MA 139/CTF, Marconi, Piracicaba, Brazil) (220 ± 10 cycles/min) and centrifugation at 670 × g for 10 min in a refrigerated Beckman centrifuge (model TJ-6, 6000 g). The organic phases (5 mL) were transferred to conic tubes and evaporated to dryness in a vacuum evaporation system (RCT90 and RC10.22, Jouan AS, St. Herblain, France) at 25 °C. The residues were reconstituted in 140 μL of the mobile phase. An aliquot of 120 μL was injected into the chromatographic column.
2.5. Experimental study
The study was approved by the Ethics Committee on the Use of Animals, University of São Paulo, Ribeirão Preto Campus. Male Wistar rats weighing 200–250 g were obtained from the Central Animal House, USP, Ribeirão Preto Campus, and were allowed to adapt to the Institutional Animal House in a temperature- (21–23 °C) and humidity (40–60%)-controlled room under a 12 h light/dark cycle for 1 day before the beginning of the experiment. Chow and water were available ad libitum.
After a 12 h fast, animals (n = 8 per sampling time) received by gavage a single oral dose of 20 mg MIT/kg dissolved in 1% acetic acid, pH 4.7 (adjusted with 1 M NaOH) [10]. Blood samples were collected by decapitation of the animals at times zero, 0.5, 1, 2, 3, 4, 5, 6, 8, 10, 15 and 24 h after administration of the drug. Plasma was obtained by centrifugation at 670 × g for 10 min and stored at −70 °C until the time of analysis.
The pharmacokinetic parameters were calculated based on the plasma concentration versus time curves using the WinNonlin program, version 4.0 (Pharsight Corp, Mountain View, CA, USA). The calculations were performed using first-order kinetics and a monocompartment model. The experimental data were analyzed statistically using the Graphpad Instat® software for the calculation of mean, median, and 95% confidence interval.
2.6. Matrix effect
The matrix effect was assessed by direct comparison of the peak areas of MIT and the internal standard injected directly into the mobile phase, and spiked into extracts originating from six different sources of rat plasma.
2.7. Method validation
Method validation was performed according to US FDA guidance for industry on bioanalytical method validation [11]. To evaluate the linearity of the method, calibration curves were constructed by spiking 500 μL blank plasma with 25 μL of each standard solution of MIT, in duplicate. The resulting plasma concentrations were 0.2, 1, 5, 20, 250, 500 and 1000 ng MIT/mL. The correlation coefficients and the linear regression equation used for the determination of MIT concentration in the samples were obtained by plotting plasma MIT concentration versus MIT/internal standard peaks area ratios.
Recovery of MIT was evaluated by comparing the peak areas obtained when plasma samples supplemented with the standard solutions (0.5, 500 and 800 ng/mL plasma) were submitted to the extraction procedure with peak areas obtained when MIT standard solutions were added to blank plasma extracts at the same concentrations as reported above.
The quantification limit was assayed by analyzing plasma samples spiked with MIT at concentrations as low as 0.2 ng/mL plasma, in quintuplicate, using a calibration curve. The quantification limit was defined as the lowest plasma MIT concentration determined with an error of 20% or lower.
The precision and accuracy of the method were evaluated at concentrations of 0.4, 400 and 800 ng/mL plasma. For the evaluation of intra-assay precision and accuracy, five aliquots of each sample were analyzed using a single calibration curve. For interassay precision and accuracy, five aliquots of each sample were analyzed in three consecutive assays.
The stability of the drug was guaranteed by three freeze (−20 °C) and thaw (25 °C) cycles lasting 12 h each and by the evaluation of short-term stability (4 h at room temperature, 25 °C) and post-processing stability (12 °C for 16 h). For this purpose, blank plasma samples spiked with MIT concentrations of 0.4 and 800 ng/mL plasma were analyzed in quintuplicate. The results of the stability tests are reported as accuracy in relation to freshly prepared samples.
3. Results and discussion
The present study used for the first time a liquid chromatography system coupled to a mass spectrometry detector (MS/MS) for the analysis of MIT in rat plasma samples.
A typical chromatogram of drug-free rat plasma used for the validation of the analytical method was initially used to determine the absence of interference peaks (Fig. 2A).
Fig. 2.
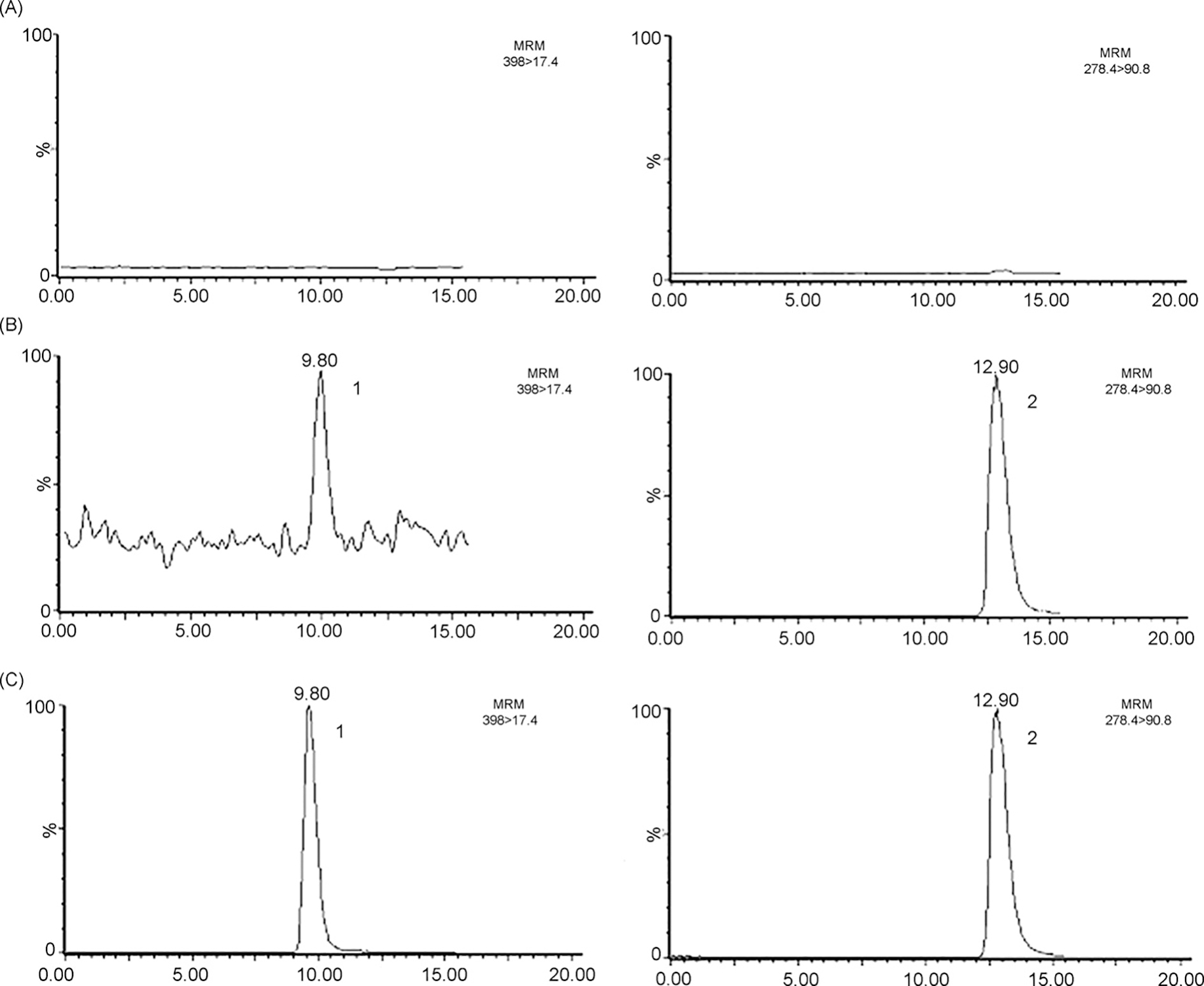
Chromatograms referring to the analysis of MIT in rat plasma. (A) Blank plasma, (B) plasma spiked with 0.2 ng MIT/mL and internal standard, (C) rat plasma sample collected 2 h after the administration of 20 mg/kg of mitragynine oxalate to rats. (1) Mitragynine (9.8 min); (2) internal standard (amitriptyline, 12.9 min).
The matrix effect using rat plasma samples spiked at the concentrations of 0.5, 500 and 800 ng/mL for MIT showed values of 97.99, 86.45 and 100.49, respectively. The matrix effect evaluated for the internal standard amitriptyline was 86.05%. These data indicated that practically no matrix effect was observed for MIT or the internal standard.
The method was validated by evaluating recovery, linearity, precision, accuracy, quantification limit, and stability. Coefficients of variation and relative errors of less than 15% were considered to be acceptable, except for the quantification limit for which a value of 20% was established [11].
The liquid–liquid extraction procedure yielding the best results involved a fast alkalinization step, followed by extraction with a mixture of hexane-isoamyl alcohol (99:1, v/v). The extraction of MIT from plasma supplemented with concentrations of 0.5, 500 and 800 ng/mL resulted in a recovery rate of 95.7%, 90.0% and 99.1%, respectively (Table 1). The MIT recovery rates are similarly to those reported by Janchawee et al. [7] who used diethyl ether under alkaline conditions.
Table 1.
Confidence limits obtained for the analysis of MIT in rat plasma.
| Recovery (%) (n = 6) | |||
| 0.5 ng/mL | 95.7 | Precision (% RSD, n = 5) | |
| 500 ng/mL | 90.0 | Intra-assay precision | |
| 800 ng/mL | 99.1 | 0.4 ng/ml | 11.7 |
| Linearity (ng/mL) | 400 ng/ml | 5.6 | |
| Linear equation | y = 0.0063x–0.0156 | 800 ng/ml | 5.1 |
| Range (ng/mL) | 0.2–1000 | Intra-assay precision | |
| Correlation coefficient (r2) | 0.999 | 0.4ng/ml | 13.0 |
| LOQ (n = 5) | 400 ng/ml | 4.54 | |
| Concentration (ng, ml) | 0.2 | 800 ng/ml | 5.5 |
| Precision (%RSD) | 13.4 | ||
| Accuracy (%DEV) | 2.5 | Accuracy (%DEV, n = 5) | |
| Stability (%DEV, n = 5) | Intra-assay accuracy | ||
| Freeze-thaw cycles (−20 to 25 °C) | 0.4 ng/ml | −3.3 | |
| 0.4 ng/ml | 2.9 | 400 ng/ml | −5.7 |
| 800 ng/ml | −9.8 | 800 ng/ml | −5.9 |
| Room temperature for 4th | Intra-assay accuracy | ||
| 0.4 ng/ml | 6.6 | 0.4 ng/ml | −3.2 |
| 800 ng/ml | −4.2 | 400 ng/ml | −3.9 |
| Post-processing | 800 ng/ml | −2.7 | |
| 0.4 ng/ml | 0.3 | ||
| 800 ng/ml | −5.7 |
The MIT/internal standard peak area ratios in rat plasma samples varied in a linear manner within a concentration range of 0.2–1000 ng/mL using linear least-square regression. The quantification limit was established at 0.2 ng/mL (Table 1). The present method was found to be more sensitive than the recently described HPLC-UV method whose quantification limit was 100 ng/mL [7].
The coefficients of variation and relative errors obtained for the three concentrations in the intra- and interassays are shown in Table 1. The results show that the analytical method is accurate (± 15% different from the nominal concentration) and precision, expressed as coefficient of variation, is within the accepted limits of 15% or less for the concentrations studied.
The tests of short-term (4 h at room temperature, 25 °C), freeze-thaw (3 cycles) and post-processing (16 h at 12 °C) stability demonstrated the good condition of the samples during freezing and from collection to analysis, with no variation of more than 15% at any of the concentrations tested (Table 1).
Fig. 3 shows the plasma concentration–time profiles for MIT in Wistar rats after a single oral dose (gavage) of 20 mg MIT/kg. A monocompartment model was used to calculate the pharmacokinetic parameters with the WinNonlin software. The results of the pharmacokinetic parameters are shown in Table 2. The maximum plasma concentration was detected at 1.26 h, with a mean value of 423.68 ng/mL. The tmax value of 1.83 h reported by Janchawee et al. [7] is with in the 95% confidence interval shown in Table 2. MIT could still be quantified in rat plasma after 24 h.
Fig. 3.
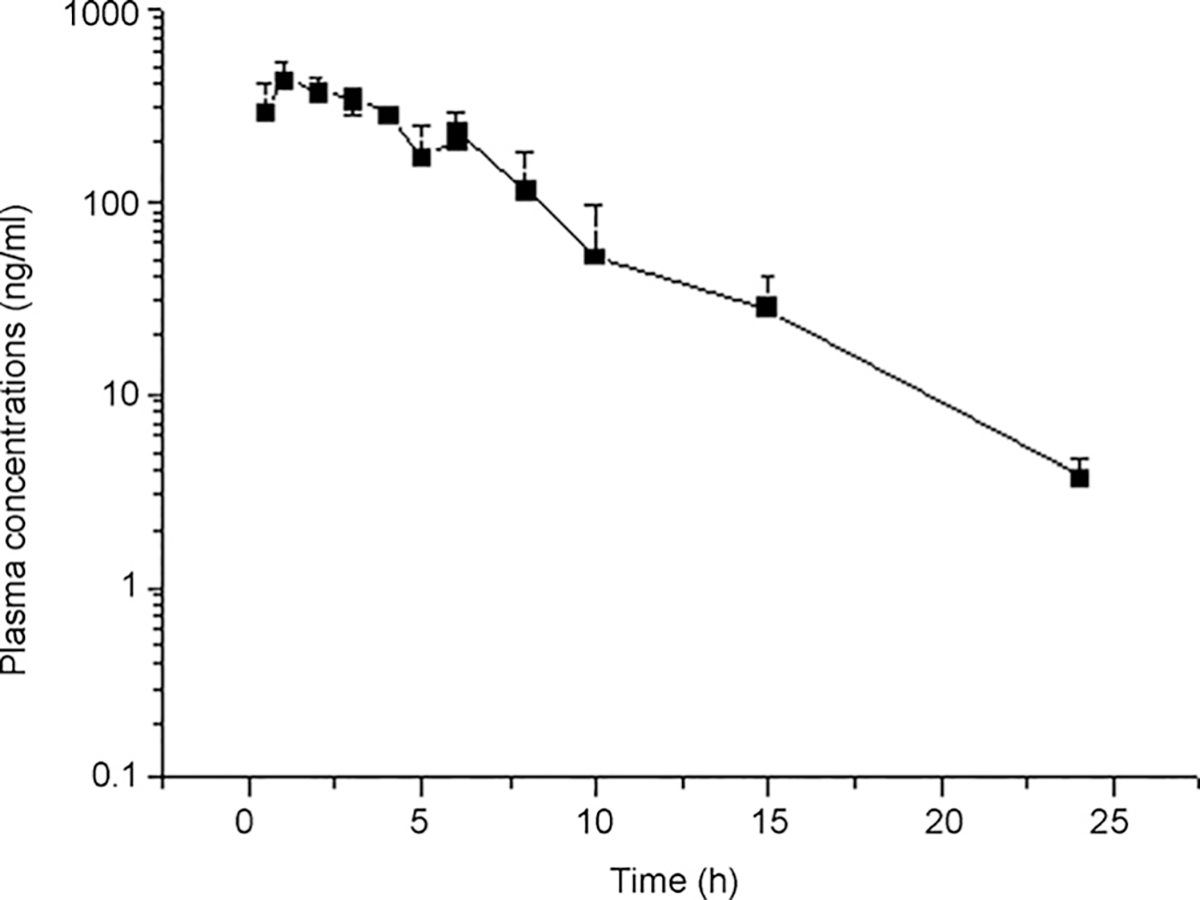
Plasma concentration–time curves for mitragynine after the administration of a single oral dose of 20 mg/kg to Wistar rats (n = 8 per sampling time).
Table 2.
Estimated pharmacokinetic parameters after oral administration of mitragynine (20 mg/kg) to Wistar rats.
| Parameter | Unit | Mean | S.E. |
|---|---|---|---|
|
| |||
| C max | (ng/mL) | 423.68 | 61.79 |
| t max | (h) | 1.26 | 0.20 |
| t 1/2a | (min) | 16.97 | 5.70 |
| K a | (1/min) | 0.04 | 0.01 |
| t 1/2 | (h) | 3.85 | 0.51 |
| K el | (1/h) | 0.18 | 0.02 |
| AUC0–∞ | (μg min/mL) | 188.98 | 1940 |
| V d/f | (L/kg) | 37.90 | 5.41 |
| C1/f | (L/h kg) | 6.35 | 0.43 |
Cmax: peak plasma concentration; tmax: time to reach Cmax: t1/2a, absorption half-life; Ka: absorption rate constant; t1/2: elimination half-life; Kel: elimination rate constant; AUC0–∞: area under the plasma concentration-time curve; Vd/f: apparent volume of distribution; Cl/f: apparent total clearance: S.E.: standard error.
The apparent total clearance of MIT obtained in this study (6.35 L/h/kg) was higher than the 1.60 L/h/kg reported by Janchawee et al. [7]. However, these authors administered a two times higher dose (40 mg/kg) than that used in the present study (20 mg/kg), a fact suggesting metabolism saturation. In addition, the lack of specificity associated with UV detection may have resulted in higher plasma concentrations and an underestimation of apparent clearance. The volume of distribution reported by Janchawee et al. [7] is about two times higher than that shown in Table 2 (89.5 versus 37.9 L/kg, respectively). This finding might be explained by plasma protein binding saturation or by the overestimation of plasma concentrations due to the lack of specificity of UV detection when compared to LC–MS/MS.
Janchawee et al. [7] also reported a higher elimination half-life for MIT than that obtained in the present study (9.43 versus 3.85 h). Pharmacokinetic parameters were evaluated by these authors using plasma concentrations obtained up to 15 h after drug administration due to the low sensitivity of the analytical method used.
4. Conclusion
A precise, accurate and highly sensitive LC–MS/MS method for the determination of mitragynine in rat plasma was developed and validated. Sample cleanup involves a liquid–liquid extraction step and shows good reproducibility. The method was applied to the pharmacokinetic study of mitragynine in rats and may also be applied, with minor changes, to other biological samples.
Acknowledgments
We gratefully acknowledge CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico) for financial support and for granting research fellowships. Part of this work (CRM) was supported by the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH) (grant number P20 RR021929).
References
- [1].Chittrakarn S, Sawangjaroen K, Prasettho S, Janchawee B, Keawpradub N, J. Ethnopharmacol. 116 (2008) 173. [DOI] [PubMed] [Google Scholar]
- [2].Babu KM, McCurdy CR, Boyer EW, Clin. Toxicol. (Phila) 46 (2008) 146. [DOI] [PubMed] [Google Scholar]
- [3].Boyer EW, Babu KM, Adkins JE, McCurdy CR, Halpern JH, Addiction 103 (2008) 1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Macko E, Weisbach JA, Douglas B, Arch. Int. Pharmacodyn. Ther. 198 (1972) 145. [PubMed] [Google Scholar]
- [5].Watanabe K, Yano S, Syunji H, Yamamoto LT, Life Sci. 60 (1997) 933. [DOI] [PubMed] [Google Scholar]
- [6].Matsumoto K, Mizowaki M, Suchitra T, Takayama H, Sakai SI, Aimi N, Watanabe H, Life Sci. 59 (1996) 1149. [DOI] [PubMed] [Google Scholar]
- [7].Janchawee B, Keawpradub N, Chittrakarn S, Prasettho S, Wararatananurak P, Sawangjareon K, Biomed. Chromatogr. 21 (2007) 176. [DOI] [PubMed] [Google Scholar]
- [8].Ponglux D, Wongseripipatana S, Takayama H, Kikuchi M, Kurihara M, Kitajima M, Aimi N, Sakai S, Planta Med. 60 (1994) 580. [DOI] [PubMed] [Google Scholar]
- [9].Takayama H, Chem. Pharm. Bull. (Tokyo) 52 (2004) 916. [DOI] [PubMed] [Google Scholar]
- [10].Thongpradichote S, Matsumoto K, Tohda M, Takayama H, Aimi N, Sakai S, Watanabe H, Life Sci. 62 (1998) 1371. [DOI] [PubMed] [Google Scholar]
- [11].Food and Drug Administration, Center for Drug Evaluation and Research (May, 2001), Available from: http://www.fda.gov/cder/guidance.