

Summary
Beta-catenin (CTNNB1) is commonly mutated in hepatocellular carcinoma (HCC). CTNNB1-mutated HCC has important clinical correlates, such as being immune cold and less likely to respond to immune checkpoint inhibitor therapies. It remains unclear, however, if they are a morphologically homogenous group of tumors. To better understand the association between the morphology, CTNNB1 mutations, and other molecular features, a detailed study of 338 The Cancer Genome Atlas cases was performed. A characteristic histological morphology was strongly associated with CTNNB1 mutations but was present in only 58% of CTNNB1-mutated HCCs. Tumors with APC mutations tended to have the classic morphology; those with AXIN mutations did not. Pseudoglands are a key feature of the classic morphology, and they were associated with CTNNB1 mutations, male gender, specific CTNNB1 mutation site, and lack of TP53 mutations. Differential gene expression analysis stratified by the presence/absence of pseudoglands identified 60 differentially expressed genes (FDR <5%); clustering according to these differentially expressed genes revealed three groups of tumors, one with pseudoglands and a strong association with genes regulated by Wnt signaling; within this group, TP53 mutations were associated with a loss of the typical morphology of CTNNB1-mutated HCCs. When stratified by gender, further differential gene expression showed Wnt-regulated genes were associated with pseudoglands in men but not women. These findings indicate HCC with CTNNB1 mutations are morphologically heterogeneous, with gene penetrance for morphology dependent in part on gender, specific CTNNB1 mutations, and co-occurring TP53 mutations. This heterogeneity has important implications for the classification of HCC.
Keywords: CTNNB1, APC, AXIN, Survival, Differential gene expression
1. Introduction
Hepatocellular carcinomas (HCCs) have heterogeneous molecular aberrations [1]. Beta-catenin (CTNNB1) mutations are among the most common mutations in HCC, present in up to 35% of cases [2]. Beta-catenin is a key protein of the Wnt signaling pathway, with activating mutations leading to enhanced signaling of the Wnt pathway. Additional key proteins are encoded by AXIN and APC; these proteins normally suppress Wnt pathway activation, and inactivating mutations can lead to increased Wnt pathway signaling. HCCs with CTNNB1 mutations are more likely to be well-differentiated, have a thin trabecular growth pattern (synonym is microtrabecular), have pseudoglands (synonym is microacini), and produce bile [3–5], histological findings that together create a composite morphological pattern for CTNNB1-mutated HCCs. Recent studies have identified important clinical correlates for CTNNB1-mutated tumors, with a subset that has less inflammation by gene expression analysis (“immune cold”) [6] and a subset that appears less likely to respond to checkpoint inhibitor therapies [7]. Clinical outcome studies have been inconsistent, with studies showing less aggressive [4] and more aggressive behavior [2] for CTNNB1-mutated HCCs.
An important question for morphological/molecular tumor classification is whether CTNNB1-mutated HCCs represent a homogenous entity. For example, the reported correlations between CTNNB1 mutations and the classic morphology are statistically strong, but a significant proportion of HCCs with CTNNB1 mutations lack the classic morphology [3–5]. On the other hand, anecdotal experience has shown that HCCs can sometimes have the classic morphology but lack CTNNB1 mutations. To better understand the morphology of CTNNB1-mutated HCCs and to refine the morphological-molecular classification, detailed studies were undertaken of the clinical-histological-molecular correlations for CTNNB1-mutated HCCs.
2. Materials and methods
2.1. Case selection
The study was approved by the Mayo Clinic Rochester Institutional Review Board. HCC images were reviewed, and histological data were collected from The Cancer Genome Atlas (TCGA) data set by a liver pathologist (M.S.T.) using the https://portal.gdc.cancer.gov/website. The TCGA collection consists of a single scanned slide for each case; for those cases with CTNNB1, APC, or AXIN mutations that originated from Mayo Clinic, the full set of slides from the original resection specimen was also reviewed to confirm the quality and representativeness of the TCGA image. Morphological findings were then correlated with clinical and sequencing data from TCGA, followed by gene expression analysis of specific subgroups, focusing on key morphological features such as pseudogland formation (Fig. 1).
Fig. 1. Study design.

(Panel A) The histology was reviewed and classified without regard to clinical or sequencing results. Next, the histology was correlated with clinical and sequencing results, identifying a strong but inconsistent relationship. Factor analysis then focused on cases with CTNNB1 or APC mutations to identify correlates of the classic morphology. This was followed up by subset analysis of cases with pseudoglands. (Panel 2B) Approach for analysis of cases with pseudoglands. In the first step, HCCs were divided by morphology into those with and without pseudoglands. DGE identified a specific set of gene expression associated with pseudoglands. This set of DGE was then used to classify HCC without regards to morphology or mutation status and identified three distinct clusters.
2.2. Exclusions
After review, 41 TCGA cases were excluded for these reasons: no hematoxylin and eosin (H&E)-stained slide/poor quality H&E-stained slide/necrotic tumor/biopsy with insufficient material (n = 14); no sequencing information (n = 13); a diagnosis of HCC could not be confirmed on the scanned H&E (would need further evaluation to rule out hepatic adenoma or cholangiocarcinoma, n = 7); the H&E showed tumors that were not HCC (n = 5); and the same histological image was posted for different TCGA case numbers (n = 2).
2.3. Definition for beta-catenin-mutated morphology
In concordance with the literature, the classic morphology for beta-catenin-mutated HCCs was defined as the following: well-differentiated tumors with tumor cells having abundant eosinophilic cytoplasm, a thin trabeculae growth pattern (1—2 cells), and pseudoglands. Tumors with somewhat thicker trabeculae (5 cells or less in thickness), but otherwise classic morphology, were considered acceptable. To accommodate differences in H&E staining between cases, also acceptable were tumors with all of the preceding features but more amphophilic or basophilic cells. Cases were classified as having possibly compatible morphologies if (1) the tumor was well differentiated and had pseudoglands but had a predominately solid growth pattern; (2) the classic morphology was present, except for pseudoglands; or (3) the classic morphology was present except the tumor was not well differentiated.
2.4. Tumor grade
For grading tumors, the predominate grade was recorded, with the requirement that at least 5% of the tumor show the given grade when multiple grades were present. Tumor grades were assigned using the same criteria used in prior TCGA studies [1]. This approach is similar to that of the current WHO [8], with the addition of another grade for HCC with minimal cytological atypia: very-well-differentiated HCCs (grade 1) have no more than minimal cytological atypia; well-differentiated tumors (grade 2) show unequivocal hepatocellular differentiation on H&E, along with mild cytological atypia and/or mild architectural atypia; moderately differentiated tumors (grade 3) show hepatocellular differentiation that is clearly present or strongly suspected from H&E, along with moderate cytological and or architectural atypia; poorly differentiated tumors (grade 4) showed marked cytological and or architectural atypia.
2.5. Tumor growth patterns
The predominate growth patterns in every tumor were classified as solid, pseudoglandular, trabecular, or macrotrabecular (trabeculae at least 10 cells in thickness).
2.6. Other histological findings
Macrovesicular steatosis was scored as positive or negative, with a minimal cut-off of 5%. Intratumoral lymphocytic inflammation was scored as none or minimal, mild, moderate, or marked (on average, more inflammatory cells than tumor cells). Bile within the tumor was scored as none, mild (less than 5% of tumor area), moderate (6—50% of tumor area), or marked (greater than 50% of tumor area). Intratumoral fibrosis was scored as none or minimal, mild (intratumoral fibrosis less than 5—25% of surface area), moderate (intratumoral fibrosis 26—50% of surface area), or marked (fibrosis is equal to or greater than the number of tumor cells). The percent surface area of the tumor involved by pseudoglands was scored as none, 1%, 5%, and then in 10% increments. The size of pseudoglands was scored as small (luminal diameter <2 tumor cell diameters), medium (luminal diameter 2—10 tumor cells in diameter), and large (luminal diameter 10 or more tumor cells in diameter). For analysis, cases were classified by the largest size of pseudoglands present in the section.
2.7. Statistical analysis
Categorical variables were compared using chi-square test, and numerical data were compared by Student’s t-test or one-way analysis of variance, with P values <0.05 considered significant. Overall survival (OS) was defined as time from HCC diagnosis to death from any cause. The distribution of OS was estimated using the Kaplan—Meier methods and compared using the log-rank test and Cox proportional hazards model.
2.7.1. RNASeq gene expression analysis
The RNA sequencing (RNASeq) samples analyzed in this study were obtained by TCGA Research Network. TCGA-processed expression data were downloaded from Genomic Data Commons (GDC; https://gdc-hub.s3.us-east1.amazonaws.com/latest/TCGA-LIHC.htseq_counts.tsv.gz). Gencode’s Homo sapiens hg38 assembly, v22, was used for the reference transcriptome, and GDC’s GRCh38.d1.vd1 reference was used to define the human genome. The GDC RNASeq analysis pipeline leverages FASTQC and Picard Tools to assess the quality of the data and uses a combination of STAR and HTSeq to align the RNASeq reads and quantify gene expression. More details can be found at https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/.
Genes with an average of ≥25 reads were kept for differential expression analysis. The R package, edgeR, was used to identify which genes were statistically differentially expressed from defined group comparisons. The categorical clinical metadata on these samples was used to define which samples belong to which groups. For each comparison, unsupervised clustering was implemented with the ClustVis application on genes with a false discovery rate of <0.05 and with an absolute log2 fold change of >1.5 from the differential expression analysis. ClustVis was additionally used to visualize these findings through heat maps.
3. Results
A total of 338 cases had both histologic images of sufficient quality for review and molecular data, comprising seven fibrolamellar carcinomas and 331 conventional HCCs. A total of 128 tumors had mutations in CTNNB1, APC, or AXIN genes. There were 94 cases with CTNNB1 mutations (one synonymous mutation and one intron mutation were classified as no mutation). Tumors showed CTNNB1 mutations alone (N = 88), CTNNB1 and APC mutations (N = 4), CTNNB1 and AXIN mutations (N = 2), AXIN mutations alone (N = 26), APC mutations alone (N = 7), and AXIN and APC mutations (N = 1).
The predominant growth pattern in HCCs with CTNNB1 mutations alone (without concomitant APC or AXIN mutations) were trabecular (N = 39/88, 44%), solid (N = 32/88, 36%), pseudoglandular (N = 12/88, 14%), and macrotrabecular (N = 5/88, 6%). In contrast, the remainder of the HCC were less likely to have pseudoglandular predominant growth patterns and more likely to have macrotrabecular predominant growth patterns (P = 0.007); growth patterns were as follows: trabecular (N = 126/250, 50%), solid (N = 91/250, 36%), pseudoglandular (N = 9/250, 4%), and macrotrabecular (N = 24/250, 10%).
CTNNB1 mutations were more common in men (80/231, 35%) than in women (14/107, 13%; P = 0.000039). In contrast, APC mutations did not differ in frequency between men (10/231, 4%) and women (2/107, 2%; P = 0.26). AXIN mutations also did not differ in frequency between men (19/231, 8%) and women (11/107, 10%; P = 0.54).
Cases with APC mutations, but lacking CTNNB1 or AXIN mutations, were limited (N = 7), with two cases classified as having the classical CTNNB1-mutated morphology, three cases classified as possible CTNNB1-mutated morphologies, and two as showing non-CTNNB1-mutated morphologies. Given these results, tumors with APC mutations were included with CTNNB1-mutated tumors for subsequent analysis.
3.1. Classic CTNNB1-mutated HCC morphology
The classic morphological pattern associated with CTNNB1-mutated HCC was studied (Fig. 2; Table 1), with cases classified as having, possibly having, or not having the classic morphology. As expected, given the criteria used to identify the classic CTNNB1 mutated morphological pattern, the classic pattern was strongly associated with the presence of pseudoglands (P < 0.00001) and bile production (P < 0.00001). The classic pattern was also associated with lower tumor grade: 42 of 63 (67%) of tumors with the classic morphology were very well or well differentiated, compared with 73 of 257 (28%) of tumors without the classic morphology. In addition, tumors with a classic CTNNB1-mutated morphology were less likely to have necrosis (P = 0.004).
Fig. 2. Morphological findings.
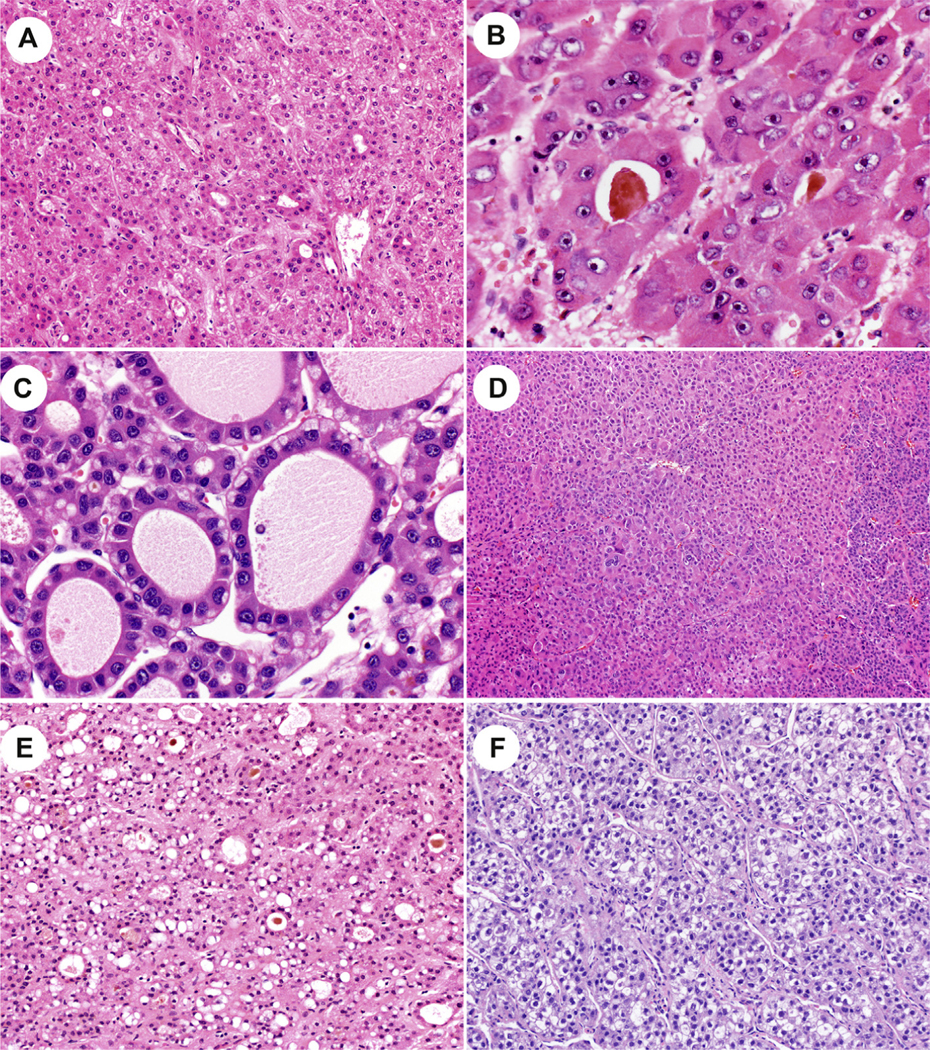
(Panel A) The classic beta-catenin morphology with thin trabeculae and pseudoglands. (Panel B) A higher power image of pseudoglands. (Panel 1C) Bile production is seen. (Panel 1D) This beta-catenin-mutated hepatocellular carcinoma lacks the classic morphology. (Panel 1E) This APC-mutated hepatocellular carcinoma shows the classic morphology. (Panel 1F) This AXIN-mutated hepatocellular carcinoma lacks the classic morphology.
Table 1.
Clinical, morphological, and mutational correlates of hepatocellular carcinoma with the classic beta-catenin mutation morphology of thin trabeculae, pseudoglands, and often bile.
| Classic morphology | Possible classic morphology | Not classic morph | P | |
|---|---|---|---|---|
|
| ||||
| Total | 63 | 18 | 257 | |
| Age | 60 ± 14 | 59 ± 12 | 58 ± 14 | 0.6 |
| Gender | 0.17 | |||
| Male | 49 | 13 | 169 | |
| Female | 14 | 5 | 88 | |
| Ethnicity | 0.462 (grouped together: Hispanic, native American and not stated) | |||
| White | 25 | 5 | 122 | |
| Asian | 32 | 12 | 112 | |
| Black | 4 | 0 | 9 | |
| Hispanic | 1 | 1 | 8 | |
| Native American | 0 | 0 | 1 | |
| Not stated | 1 | 0 | 5 | |
| Tumor subtype | ||||
| NOS | 61 | 17 | 190 | |
| Steatohepatitic | 0 | 1 | 21 | |
| Scirrhous | 0 | 0 | 18 | |
| Clear cell | 2 | 0 | 12 | |
| FLC | 0 | 0 | 7 | |
| Lymphocyte rich | 0 | 0 | 3 | |
| Cirrhotomimmetic | 0 | 0 | 3 | |
| Sarcomatoid | 0 | 0 | 2 | |
| Myxoid | 0 | 0 | 1 | |
| Tumor grade (predominate) | <0.001 | |||
| 1 | 8 | 3 | 18 | |
| 2 | 33 | 3 | 55 | |
| 3 | 19 | 8 | 120 | |
| 4 | 3 | 4 | 64 | |
| NA | ||||
| Tumor necrosis | 0.004 | |||
| Yes | 5 | 1 | 63 | |
| No | 58 | 17 | 194 | |
| Tumor hyaline bodies | 0.46 | |||
| Yes | 9 | 2 | 50 | |
| No | 54 | 16 | 207 | |
| Pseudoglands | <0.001 | |||
| Yes | 63 | 11 | 58 | |
| No | 0 | 7 | 199 | |
| Tumor bile | <0.001 | |||
| Yes | 33 | 4 | 42 | |
| No | 30 | 14 | 215 | |
| Tumor inflammation | 0.80 | |||
| Yes | 15 | 5 | 72 | |
| None or minimal | 48 | 13 | 185 | |
| Tumor steatosis (>5%) | 0.09 | |||
| Yes | 10 | 2 | 68 | |
| No | 53 | 16 | 189 | |
| Tumor fibrosis | 0.004 | |||
| Yes | 7 | 3 | 79 | |
| No | 56 | 15 | 178 | |
| No. somatic mutations | 161 + 100 | 164 + 71 | 170 + 199 | 0.6 |
| Any Wnt mutation (CTNNB1, | <0.001 | |||
| AXIN, APC) | ||||
| Yes | 41 | 9 | 78 | |
| No | 22 | 9 | 179 | |
| CTNNB1 mutation alone | <0.001 | |||
| Yes | 34 | 3 | 51 | |
| No | 29 | 15 | 206 | |
| AXIN mutation alone | 0.54 | |||
| Yes | 4 | 2 | 20 | |
| No | 59 | 16 | 237 | |
| CTNNB1 and/or APC | <0.001 | |||
| Yes | 37 | 7 | 58 | |
| No | 26 | 11 | 199 | |
| TP53 mutation alone (point, indel, or CNV-loss) | <0.001 | |||
| Yes | 10 | 4 | 157 | |
| No | 53 | 14 | 100 | |
| CTNNB1 and TP53 mutation | 0.88 | |||
| Yes | 4 | 1 | 20 | |
| No | 59 | 17 | 237 | |
Excluding clonal progression.
The classic morphology was strongly associated with CTNNB1 mutations (P < 0.00001), but not with AXIN mutations (P = 0.5). HCCs with CTNNB1 mutations, however, were histologically heterogeneous, with 58% (37/63) showing the classic morphology. TP53 mutations were rare in tumors with the classic morphology (Table 1).
As tumors with possible and definite beta-catenin mutation morphologies appeared similar in most characteristics, they were combined for further analysis. There were at least five different CTNNB1 specific missense mutations that were each present in at least five cases of HCC. Although the numbers are modest, the frequency of classic morphology varied sufficiently to suggest an association: S33P (4/5 have classic beta-catenin morphology), D32V (3/5), T41A (2/5), D32G (2/7), N387 (1/5), and K335I (0/5). Follow-up analysis of the affected codon, regardless of the specific mutation, further suggests that mutations in codons 335 and 387 are not associated with the development of the classic morphology (Supplemental Table 1).
3.2. Pseudogland formation and bile production
Pseudogland formation is an important component of the classic beta-catenin-mutated HCC morphological pattern, so the association with clinicopathological findings was further investigated as an independent variable (Table 2). As anticipated, there were strong associations with CTNNB1/APC mutations, lower tumor grade, bile production, and less frequent tumor necrosis. When analyzing the entire cohort of cases, male gender was strongly associated with the formation of pseudoglands (P = 0.001). Likewise, when analyzing the subset of HCCs with CTNNB1/APC mutations (N = 102), male gender was associated with the formation of pseudoglands (P = 0.014).
Table 2.
Clinical, morphological, and mutational correlates of pseudoglands.
| Factor | Total | Pseudoglands present | Pseudoglands negative | P (yes versus no pseudoglands) |
|---|---|---|---|---|
|
| ||||
| Total | 338 | 132 (39%) | 206 (61%) | |
| Gender | 0.001 | |||
| Male | 231 | 104 (45% of gender) | 127 (55%) | |
| Female | 107 | 28 (26%) | 79 (74%) | |
| Age (mean ± SD) | ||||
| All | 58.5 ± 13.8 | 59.5 ± 14.2 | 58.0 ± 13.4 | 0.31 |
| Male | 57.8 ± 12.9 | 59.4 ± 12.6 | 56.5 ± 13 | 0.10 |
| Female | 60.2 ± 15.3 | 60.0 ± 18.9 | 60.2 ± 14.0 | 0.96 |
| Ethnicity | 0.32 (grouped together: Hispanic, native American and not stated) | |||
| White | 159 | 61 | 91 | |
| Asian | 157 | 56 | 100 | |
| Black | 14 | 8 | 5 | |
| Hispanic | 10 | 5 | 5 | |
| Native American | 1 | 0 | 1 | |
| Not stated | 6 | 2 | 4 | |
| Tumor subtype | 0.002392 (FLC and below grouped together) | |||
| NOS | 275 | 116 | 152 | |
| Steatohepatitic | 22 | 3 | 19 | |
| Scirrhous | 21 | 2 | 16 | |
| Clear cell | 14 | 3 | 11 | |
| FLC | 6 | 5 | 2 | |
| Lymphocyte rich | 3 | 1 | 2 | |
| Cirrhotomimmetic | 3 | 2 | 1 | |
| Sarcomatoid | 2 | 0 | 2 | |
| Myxoid | 1 | 0 | 1 | |
| Tumor grade predominate | 0.014 | |||
| 1 | 22 | 10 | 19 | |
| 2 | 71 | 48 | 43 | |
| 3 | 148 | 53 | 94 | |
| 4 | 103 | 21 | 50 | |
| Tumor necrosis | 0.002 | |||
| Yes | 69 (20%) | 16 (12%) | 53 (26%) | |
| No | 269 (80%) | 116 (78%) | 153 (74%) | |
| Tumor hyaline bodies | 0.155 | |||
| Yes | 43 (44%) | 15 | 35 | |
| No | 304 | 117 | 171 | |
| Tumor bile | <0.0001 | |||
| Yes | 79 | 64 | 15 | |
| No | 288 | 68 | 191 | |
| Tumor inflammation | 0.33 | |||
| Yes | 93 | 32 | 60 | |
| No | 254 | 100 | 146 | |
| Tumor steatosis (>5%) | 0.057 | |||
| Yes | 80 (24%) | 24 | 56 (x%) | |
| No | 258 | 108 | 150 | |
| Tumor fibrosis | 0.5 | |||
| Yes | 95 | 27 | 62 | |
| No | 252 | 105 | 144 | |
| No. somatic mutations | ||||
| All | 166 ± 177 | 170 ± 172 | 167 ± 184 | 0.89 |
| Male | 179 ± 203 | 181 ± 189 | 177 ± 218 | 0.90 |
| Female | 141 ± 100 | 128 ± 69 | 150 ± 111 | 0.33 |
| Any Wnt mutation (CTNNB1, AXIN, and APC) | <0.001 | |||
| Yes | 128 | 70 | 58 | |
| No | 210 | 62 | 148 | |
| CTNNB1 mutation | <0.001 | |||
| CTNNB1 mutation alone | 88 | 55 | 33 | |
| CTNNB1 + AXIN/APC | 6 | 3 | 3 | |
| No CTNNB1 mutation | 244 | 74 | 170 | |
| AXIN mutation | 0.34 | |||
| AXIN mutation alone | 26 | 7 | 19 | |
| AXIN + CTNNB1/APC | 3 | 0 | 3 | |
| No AXIN mutation | 309 | 125 | 184 | |
| CTNNB1 and or APC | <0.001 | |||
| Yes | 104 | 63 | 39 | |
| No | 243 | 69 | 167 | |
Once a tumor had pseudoglands present, neither the size of the pseudoglands nor the percent of the HCC with pseudoglands differed between men and women (Supplemental Table 2); moderate bile, however, was more common in HCC arising in men. In addition, once pseudoglands were present in a tumor, neither the percent of the tumor with pseudoglands nor the size of the pseudoglands correlated with the presence or absence of CTNNB1/APC mutations (Supplemental Table 3). Finally, the polyphen score for CTNNB1 mutations did not correlate with classic morphology nor with the presence or absence of pseudoglands (Supplemental Table 4).
For tumors with pseudoglands and bile production, male gender was associated with moderate (versus mild) cholestasis (Supplemental Table 5). For carcinomas with pseudoglands in men, pseudoglands were associated with younger age (Supplemental Table 5). Younger aged men with tumors also showed more bile production (Supplemental Table 5). Together, these findings suggest that pseudogland formation and bile production are gender and age related, implying a role for androgen. Multivariate analysis to predicate pseudogland was then performed using age, gender, CTNNB1 mutation, and TP53 mutation. CTNNB1 mutations were the most significant variable positively associated with pseudoglands (odds ratio [OR] = 3.2, P = 2.5e-5), followed by male gender (OR = 1.8), whereas TP53 mutations predicated the absence of pseudoglands (OR = 0.5, P = 0.01). Age at diagnosis was not significant.
3.3. Gene expression analysis
To better understand if pseudogland formation is a homogenous process at the gene expression level, differential gene expression analysis was performed for the entire cohort of HCC, comparing all tumors with pseudoglands to all tumors without pseudoglands: 60 differentially expressed genes were identified. This set of 60 genes was then used to classify the entire cohort of all HCC and demonstrated three distinct clusters of tumors (Fig. 3A). Analysis of these three clusters showed that cluster 1 had a low frequency of pseudoglands, was enriched for younger age, female gender, and had a low frequency of CTNNB1 mutations (Table 3). Cluster 2 was enriched for pseudoglands, male gender, and CTNNB1 mutations. Cluster 3 was enriched for pseudoglands but had a low frequency of CTNNB1 mutations. These same three clusters were then mapped to 21 known positive targets and two known negative targets of Wnt signaling (Supplement Table 6) [9] and identified a very strong correlation with cluster 2 (Fig. 3B). In addition, of the 23 genes in this panel, seven were statistically significant in their association with the formation of pseudoglands: NKD1, NOTUM, REG3A, ODAM, RHBG, SLC1A2, and CYP2E1. Together, these findings indicate that tumors with pseudoglands have a distinctive gene expression profile and that a subset of cases (cluster 2) is strongly associated with Wnt signaling. Unexpectedly, cluster 2 was also enriched for the cooccurrence of CTNNB1 and TP53 mutations (Table 2), and with clonal progression, defined as the same tumor showing multiple distinct morphologies [10]. This suggests that morphological features of CTNNB1 mutations may be lost with tumor progression. Next, the analysis showed the set of 60 differentially expressed genes was significantly associated with the Wnt signaling pathway using GO Terms and Metascape, but not with IPA-canonical pathways (data not shown). Analysis of IPA upstream regulators, however, found CTNNB1 to be the most important regulator.
Fig. 3. Pseudoglands, differential gene expression analysis.
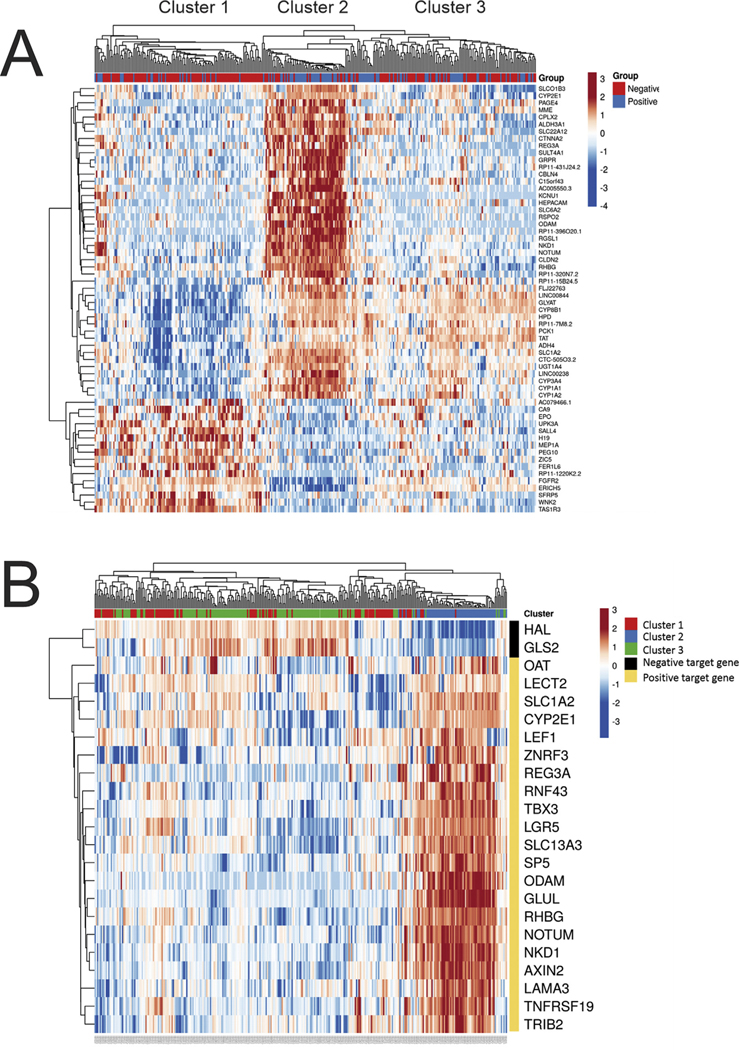
The differentially expressed (DE) genes in HCC with pseudoglands versus without pseudoglands was studied in the entire set of study cases, as well several subsets, which controlled for factors that likely influence pseudogland formation, namely, gender and CTNNB1 mutations. The DE genes identified in each study were then used to classify HCC within the cohort to see if the selected genes accurately predicted pseudogland formation, and the results are shown as heat maps. In the top bar, blue indicates pseudoglands were present, and red indicates their absence. (Panel A) A total of 60 DE genes were identified when examining all cases with sufficient histology and gene expression data (N = 333). When the 60 DE genes were used to classify the HCC, three distinct groups/clusters emerged. (Panel B) These three clusters were then mapped to expression of genes known to be regulated by CTNNB1. Cluster 2, but not clusters 1 and 3, is strongly associated with genes regulated by CTNNB1. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Table 3.
Three case clusters identified by differential gene expression analysis.
| Cluster 1 | Cluster 2 | Cluster 3 | P | |
|---|---|---|---|---|
|
| ||||
| N | 110 | 66 | 142 | |
| Gender M/F (ratio) | 58/52 | 56/10 | 104/38 | 0.00001 |
| Age (average ± SD) | 55.5 ± 12 | 61.6 ± 13 | 60 ± 14 | 0.005 |
| Pseudoglands | 22 (20%) | 43 (65%) | 57 (40%) | <0.00001 |
| CTNNB1/APC mutations | 14 (13%) | 58 (88%) | 25 (18%) | <0.00001 |
| AXIN mutations | 20 (18%) | 0 | 7 (5%) | <0.00001 |
| Classic beta-catenin morphology | 15 (14%) | 33 (50%) | 28 (20%) | <0.00001 |
| CTNNB1 mutations and classic beta-catenin morphology | 2 (2%) | 28 (42%) | 10 (7%) | <0.00001 |
| Intratumoral inflammation | 37 (33%) | 15 (23%) | 37 (45%) | 0.23 |
| Ploidy (average ± SD) | 2.9 ± 1 | 2.5 ± 0.8 | 2.4 ± 0.7 | 0.00013 |
| TP53 mutations | 44 (40%) | 18 (26%) | 44 (31%) | 0.22 |
| Combined CTNNB1 and TP53 mutations | 6 (5%) | 17 (26%) | 2 (1%) | |
| 0 with clonal progression | 8 with clonal progression | |||
| HBV infection | 25 (23%) | 21 (32%) | 27 (19%) | 0.12 |
Because of evidence that both CTNNB1/APC mutations and male gender are associated with pseudoglands, additional differential gene expression studies were performed controlling for these variables. First, the subset of tumors in women with CTNNB1/APC mutations was studied to see if gene expression could identify a signature associated with pseudogland formation. A set of 128 differentially expressed genes were identified and, when applied back to this same group, were able to completely separate tumors with pseudoglands (Fig. 4A). The same analysis in men identified 81 differentially expressed genes (Fig. 4B). Interestingly, there was little overlap in the differentially expressed genes associated with pseudoglands in tumors with CTNNB1/APC mutations for women versus men (Fig. 4C). These results indicate that pseudogland formation in HCC is associated with different gene expression pathways in men versus women, although both groups have CTNNB1/APC mutations.
Fig. 4. Pseudoglands, gender, survival.

(Panel A) DE gene expression for HCC in women with CTNNB1 mutations identified 128 DE genes. This set of 128 DE genes could accurately classify all cases in this cohort as having or not having pseudoglands. (Panel B) DE gene expression for HCC in men with CTNNB1 mutations identified 81 DE genes. This set of 81 DE genes could classify cases as having or not having pseudoglands with moderate accuracy. (Panel C) DE genes that predicated pseudogland formation showed relatively little overlap between the three groups. (Panel D) No survival differences are seen in patients whose HCC have the classic, possible classic, or nonclassic morphology for CTNNB1 mutations. (Panel E) No survival differences are seen in patients whose HCC have pseudoglands versus HCC without pseudoglands. (Panel F) Patients with HCC that had CTNNB1 mutations and pseudogland formation had better survival, but the results are not statistically significant.
3.4. Survival analysis
There was no difference in OS for patients who had HCCs with classic morphology, probable classic morphology, or nonclassic morphology (Fig. 4D); likewise, no survival differences were discernible for CTNNB1 mutations versus no mutations (data not shown) or for pseudoglands versus no pseudoglands (Fig. 4E) in the overall cohort (5-year OS: 59.8% versus 58.6%). For HCCs with CTNNB1 mutations, there appeared to be better survival in tumors that showed pseudoglands (5-year OS: 54.5% versus 37.4%), but the results were not statistically significant, likely because of the small sample size, with survival data available for only 40 patients with HCC that had CTNNB1 mutations (Fig. 4F).
3.5. HCC with CTNNB1 and or APC mutations and inflammation
Neither the classic morphology (Table 1) nor the presence of pseudoglands (Table 2) was associated with inflammation. A more detailed analysis of the subset of 102 cases with CTNNB1/APC mutations also found no clear association between the classic beta-catenin morphology and less inflammation (P = 0.2). Cases with classic morphology had no inflammation (N = 36), mild inflammation (N = 7), and moderate inflammation (N = 1); in contrast, HCC without the classic morphology had no inflammation (N = 39), mild inflammation (N = 15), and moderate inflammation (N = 4).
3.6. HCC with CTNNB1 and or APC mutations but without the classic morphology
A total of 58 cases had a CTNNB1 or APC mutation but lacked the classic morphology and could be classified as having a not-otherwise-specified (NOS) morphology (N = 47) or could be classified into a recognized morphological subtype, including steatohepatitic (N = 5), scirrhous (N = 2), clear cell (N = 1), fibrolamellar carcinoma (N = 1), and lymphocyte rich (N = 1).
3.7. AXIN-mutated HCC
Twenty-six HCCs had AXIN mutations, without APC or CTNNB1 mutations, including 17 men and nine women. This cohort had a younger average age of 54 ± 14 years, compared with patients whose HCCs had CTNNB1 mutations alone (P = 0.003).
For the 26 cases with AXIN mutations, the predominate growth patterns were trabecular (n = 14), solid (n = 8), or macrotrabecular (n = 4). Seven cases (27%) had pseudoglands, and six cases (23%) had the classic morphology associated with beta-catenin mutations (four definite and two possible). The 20 cases without classic beta-catenin morphology showed these morphological patterns: NOS (n = 18), steatohepatitic (n = 1), and clear cell (n = 1).
4. Discussion
These findings affirm a strong association between CTNNB1-mutated HCCs and the morphological pattern of well-to-moderately differentiated tumors with thin trabeculae, pseudoglands, and bile production [3–5]. Tumors with the classic morphology were less likely to have TP53 mutations, consistent with prior studies showing that CTNNB1 and TP53 mutations tend to be mutually exclusive, whether defined at the genetic [11] or protein expression levels [12]. Based on our findings, APC mutations in many cases lead to tumor morphology similar to that seen with CTTNB1 mutations, whereas AXIN mutations typically do not. Prior studies examining gene expression/function in human HCCs, cell culture experiments, and animal models have shown that AXIN mutations can promote HCC without activation of the Wnt signaling pathway [13,9], providing a potential explanation for our observation of the lack of a beta-catenin morphological signature in AXIN-mutated tumors. In this study, AXIN-mutated HCCs presented at a younger age compared with CTNNB1-mutated tumors and generally had a NOS morphology.
In this study, CTNNB1-mutated HCCs were enriched for male gender. Likewise, within the subgroup of all HCCs with CTNNB1/APC mutations, the classic morphology was linked to male gender. A detailed analysis of morphology further showed that two key components of the classic beta-catenin-mutated HCC morphology are linked to male gender: pseudoglands and bile production. This association appears to be lost in older men, suggesting androgen may drive the development of pseudoglands and the production of bile. Prior studies provided potential mechanistic insights, showing that androgen receptor (AR) upregulates cell cycle—dependent kinase, driving tumorigenesis in a beta-catenin-dependent fashion [14]. Studies have also identified a positive feedback loop, wherein beta-catenin activation in turn upregulates AR. AR signaling can also lead to repression of Wnt pathway inhibitors via upregulation of EZH2 [15]. At the morphological level, these findings are further paralleled by observations in androgen-driven hepatic adenomas, which characteristically have pseudoglands and bile production [16]. These observations collectively indicate that male gender is not only associated with CTNNB1 mutations, but that male gender in turn affects the gene penetrance of CTNNB1 mutations in terms of distinct histological morphology. It remains unclear if CTNNB1 mutations in the setting of higher levels of androgen expression also lead to differences in clinical behavior, such as prognosis or treatment response, but if so, future classifications may benefit by looking for a combined AR and Wnt signal within tumors. Given published results on the AR and Wnt pathway feedback loop [14], this group of patients may benefit from therapy that interrupts this feedback loop. This may have added clinical relevance, given that previous studies have suggested CTNNB1 mutations promote resistance to anti-PD-1 therapy [7,6], which currently is a key component of the standard of care.
Specific genetic mutations are also relevant to the formation of pseudoglands, presumably engendering stronger Wnt signaling. In this study, neither K335 nor N387 CTNNB1 mutations were consistently associated with the classic beta-catenin-mutated morphology. In keeping with these results, prior studies have shown that these two mutations only weakly activate the Wnt signaling pathway in cell culture [17]. Together, these findings suggest that future molecular-based classification systems will be improved as they incorporate the specific CTNNB1 mutation.
In this study, 58% of HCCs with CTNNB1 mutations had the classic morphology. One of the limitations of this study is that most cases had a single scanned slide for histological review, and HCC can be morphologically heterogenous. Nonetheless, the results in this study are very similar to the results of prior studies [3–5], indicating that 40% of CTNNB1-mutated HCCs do not have the classic morphology. In this study, those cases without the classic morphology could be classified into a specific subtype (10%), such as steatohepatitic or scirrhous HCC, or had a NOS morphology (30%).
Differential gene expression analysis examined the gene expression profile of tumors with and without pseudoglands and identified two distinct clusters of HCC with pseudoglands: clusters 2 and 3. Interestingly, only cluster 2 was associated with the expression of downstream targets of the Wnt signaling pathway and with CTNNB1 mutations, suggesting other signaling pathways can also drive pseudogland formation, as shown by cluster 3. Also of note, although cluster 2 was strongly linked to Wnt pathway activation, only 50% of the tumors had the classic morphology associated with beta-catenin mutations. These latter results suggest that morphology is insufficient to reliably identify subsets of HCC with either beta-catenin mutations or with gene expression indicative of Wnt signaling activation. Part of the explanation for the discrepancy between mutation status and morphology may be that in some cases, the CTNNB1 mutations are late events and not true drivers of the tumor growth. An additional possibility is suggested by further analysis of cluster 2, which showed this cluster was enriched for the presence of shared CTNNB1 and TP53 mutations and was more likely to show clonal progression; these areas of clonal progression may show a different and less well-differentiated morphology. CTNNB1/APC mutations in men versus women also led to distinct gene expression patterns with relatively little overlap. The explanation for this is unclear, but possibilities include the effects of androgens as well the possibility of different timing of mutations in the process of carcinogenesis.
The results from this study identify key factors that influence heterogeneity in the morphology of CTNNB1-mutated tumors: (1) specific CTNNB1 mutation and (2) gender, but other possibilities include (3) tumor dedifferentiation as well as (4) tumors where CTNNB1 is potentially not the main driver mutation, possibly representing passenger or late-driver mutations [18]. This heterogeneity represents a challenge for sequencing-based classification schemas. Our findings indicate that those HCC with CTNNB1 mutations that occur in men are most likely to be associated with the classic morphology and to be enriched for differentially expressed genes within the Wnt signaling pathway; it is this group (cluster 2) that appears to form the most cohesive morphomolecular group. The observation that some cases have the classic morphology associated with CTNNB1 mutations and/or pseudoglands, but lack CTNNB1 mutations, suggests there are additional pathways that converge on these morphological patterns.
In conclusion, CTNNB1-mutated HCCs are histologically heterogeneous. Factors that influence the morphological findings include gender and the specific mutation site in the gene. These morphological observations can be used to improve the morphomolecular classification of CTNNB1-mutated HCCs.
Supplementary Material
Acknowledgments
Grant support: P50 CA210964 (M.S.T., J.Y., R.R.L., and C.W.).
Abbreviations:
- HCC
hepatocellular carcinoma
Footnotes
Competing interests: None.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.humpath.2021.09.009.
References
- [1].Cancer Genome Atlas Research Network. Electronic address wbe, Cancer Genome Atlas Research N. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 2017; 169:1327e41. e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Nhieu JT, Renard CA, Wei Y, Cherqui D, et al. Nuclear accumulation of mutated beta-catenin in hepatocellular carcinoma is associated with increased cell proliferation. Am J Pathol 1999;155:703–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Audard V, Grimber G, Elie C, Radenen B, et al. Cholestasis is a marker for hepatocellular carcinomas displaying beta-catenin mutations. J Pathol 2007;212:345–52. [DOI] [PubMed] [Google Scholar]
- [4].Dal Bello B, Rosa L, Campanini N, Tinelli C, et al. Glutamine synthetase immunostaining correlates with pathologic features of hepatocellular carcinoma and better survival after radiofrequency thermal ablation. Clin Canc Res 2010;16:2157–66. [DOI] [PubMed] [Google Scholar]
- [5].Kitao A, Matsui O, Yoneda N, Kozaka K, et al. Hepatocellular carcinoma with beta-catenin mutation: imaging and pathologic characteristics. Radiology 2015;275:708–17. [DOI] [PubMed] [Google Scholar]
- [6].Ruiz de Galarreta M, Bresnahan E, Molina-Sanchez P, Lindblad KE, et al. Beta-catenin activation promotes immune escape and resistance to anti-PD-1 therapy in hepatocellular carcinoma. Canc Discov 2019; 9:1124–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Harding JJ, Nandakumar S, Armenia J, Khalil DN, et al. Prospective genotyping of hepatocellular carcinoma: clinical implications of next-generation sequencing for matching patients to targeted and immune therapies. Clin Canc Res 2019;25:2116–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Digestive systems tumors. 5th ed. Lyon, France: International Agency fo Research on Cancer; 2019. [Google Scholar]
- [9].Abitbol S, Dahmani R, Coulouarn C, Ragazzon B, et al. AXIN deficiency in human and mouse hepatocytes induces hepatocellular carcinoma in the absence of beta-catenin activation. J Hepatol 2018; 68:1203–13. [DOI] [PubMed] [Google Scholar]
- [10].Torbenson MS. Hepatocellular carcinoma: making sense of morphological heterogeneity, growth patterns, and subtypes. Hum Pathol 2020. [DOI] [PMC free article] [PubMed]
- [11].Laurent-Puig P, Legoix P, Bluteau O, Belghiti J, et al. Genetic alterations associated with hepatocellular carcinomas define distinct pathways of hepatocarcinogenesis. Gastroenterology 2001;120: 1763–73. [DOI] [PubMed] [Google Scholar]
- [12].Torbenson M, Kannangai R, Abraham S, Sahin F, et al. Concurrent evaluation of p53, beta-catenin, and alpha-fetoprotein expression in human hepatocellular carcinoma. Am J Clin Pathol 2004;122: 377–82. [DOI] [PubMed] [Google Scholar]
- [13].Zucman-Rossi J, Benhamouche S, Godard C, Boyault S, et al. Differential effects of inactivated AXIN1 and activated beta-catenin mutations in human hepatocellular carcinomas. Oncogene 2007;26: 774–80. [DOI] [PubMed] [Google Scholar]
- [14].Feng H, Cheng AS, Tsang DP, Li MS, et al. Cell cycle-related kinase is a direct androgen receptor-regulated gene that drives betacatenin/T cell factor-dependent hepatocarcinogenesis. J Clin Invest 2011;121:3159–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Song H, Yu Z, Sun X, Feng J, et al. Androgen receptor drives hepatocellular carcinogenesis by activating enhancer of zeste homolog 2-mediated Wnt/beta-catenin signaling. EBioMedicine 2018;35: 155–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gupta S, Naini BV, Munoz R, Graham RP, et al. Hepatocellular neoplasms arising in association with androgen use. Am J Surg Pathol 2016;40:454–61. [DOI] [PubMed] [Google Scholar]
- [17].Rebouissou S, Franconi A, Calderaro J, Letouze E, et al. Genotype-phenotype correlation of CTNNB1 mutations reveals different sscatenin activity associated with liver tumor progression. Hepatology 2016;64:2047–61. [DOI] [PubMed] [Google Scholar]
- [18].Friemel J, Rechsteiner M, Frick L, Bohm F, et al. Intratumor heterogeneity in hepatocellular carcinoma. Clin Canc Res 2015;21:1951–61. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.