

Abstract
Increased ecological disturbances, species invasions, and climate change are creating severe conservation problems for several plant species that are widespread and foundational. Understanding the genetic diversity of these species and how it relates to adaptation to these stressors are necessary for guiding conservation and restoration efforts. This need is particularly acute for big sagebrush (Artemisia tridentata; Asteraceae), which was once the dominant shrub over 1,000,000 km2 in western North America but has since retracted by half and thus has become the target of one of the largest restoration seeding efforts globally. Here, we present the first reference-quality genome assembly for an ecologically important subspecies of big sagebrush (A. tridentata subsp. tridentata) based on short and long reads, as well as chromatin proximity ligation data analyzed using the HiRise pipeline. The final 4.2-Gb assembly consists of 5,492 scaffolds, with nine pseudo-chromosomal scaffolds (nine scaffolds comprising at least 90% of the assembled genome; n = 9). The assembly contains an estimated 43,377 genes based on ab initio gene discovery and transcriptional data analyzed using the MAKER pipeline, with 91.37% of BUSCOs being completely assembled. The final assembly was highly repetitive, with repeat elements comprising 77.99% of the genome, making the Artemisia tridentata subsp. tridentata genome one of the most highly repetitive plant genomes to be sequenced and assembled. This genome assembly advances studies on plant adaptation to drought and heat stress and provides a valuable tool for future genomic research.
Keywords: Artemisia tridentata, keystone species, genomic resources
Introduction
Sagebrush ecosystems, comprising shrub and steppe dominated communities, are distributed across 14 western US states and two Canadian provinces (Fig. 1), and are dominated by endemic keystone sagebrush species of Artemisia L. subgenus Tridentatae (Rydb.) McArthur (McArthur et al. 1981; Garcia et al. 2011; Remington et al. 2021). These ecosystems are valued for livestock grazing, recreation, and wildlife habitat, but are pressured by altered climate, plant invasions, and wildfire, and thus intensive restoration efforts are underway (Baker 2006; Brabec et al. 2015; Remington et al. 202,1). Sagebrush communities are recognized as some of the most imperiled suites of ecosystems worldwide with >350 species of plants and animals of conservation concern (Remington et al. 2021). Climatic niche models predict a 39% range reduction for the mid- to low-elevation sagebrush populations by mid-century due to rising temperatures (Still and Richardson 2015). This alarming prediction calls for research to prioritize the conservation and restoration of these taxa.
Fig. 1.

Map highlighting the sagebrush ecosystems and the site of collection of IDT3 within the Soda Fire site (burned in 2015) in Idaho, USA. Sagebrush ecosystems (also called the “Sagebrush Biome” per Rigge et al. 2020) currently cover an estimated range of 653,316 km2. The inset shows a landscape photo of the Soda Fire site.
Big sagebrush (Artemisia tridentata Nutt.) shrublands once occupied ∼1,000,000 km2, but have been reduced by half due to the compound effects of climate change (Miller et al. 2012; Pilliod et al. 2017; O’Connor et al. 2020). Because big sagebrush does not re-sprout post-fire, ecosystem recovery only occurs via seedling recruitment (Wijayratne and Pyke 2012; Germino et al. 2018). Novel climatic conditions caused by climate change are creating conditions unsuitable for seedling recruitment therefore threatening the sustainability of sagebrush ecosystems (Pilliod et al. 2017).
Big sagebrush is a polyploid complex including three major subspecies—A. tridentata subsp. tridentata, A. tridentata subsp. vaseyana, and A. tridentata subsp. wyomingensis (hereafter referred to by subspecific epithets)—distributed across an environmental gradient with polyploids dominating the landscape (McArthur and Sanderson 1999). Subspecies tridentata and vaseyana exhibit both diploid (2n = 2× = 18) and tetraploid (2n = 4× = 32) cytotypes, whereas subspecies wyomingensis is only known as a polyploid (2n = 4×, 2n = 6× = 54) (McArthur and Sanderson 1999). Common garden experiments indicated that demographic phenotypes are under gene-by-environment control (Chaney et al. 2017). For example, a common garden experiment focusing on growth and fecundity rates was conducted to compare 2× tridentata and 4× wyomingensis performance across environments (Richardson et al. 2021). This study demonstrated that 2× tridentata outperformed 4× wyomingensis, even in environments dominated by polyploids (Richardson et al. 2021). The higher performance of 2× tridentata raised the question of how polyploids could be more prevalent in the landscape. A reference genome would provide genomic resources for future research aimed at increasing our understanding of observed phenotypes in common gardens, allow researchers to assess how big sagebrush populations have adapted to environmental changes, explain cytotype distributions, and provide a key resource to estimate the effect of climate change on its populations.
Here, we describe the first reference-quality genome assembly for 2× A.tridentata based on a clonally propagated individual line. A combination of short- and long-read and conformation capture sequencing technologies was used to assemble the 4.2 Gb haploid genome.
Materials and methods
Sample collection, in vitro tissue propagation, and biomass production
Previous studies have estimated the genome sizes of 2× tridentata and 4× wyomingensis to be 8.2 Gb/2C and 14.7 Gb/2C, respectively, suggesting an allopolyploid origin of the latter taxon (Garcia et al. 2008). This hypothesis was confirmed by phylogenetic analyses supporting polyphyly of 4× wyomingensis, and monophyly of 2× tridentata (Richardson et al. 2012). Previous research on a 2× tridentata draft genome has also suggested high genome complexity and levels of heterozygosity (Melton et al. 2021). Given the high heterozygosity, difference of genome sizes, and nonmonophyly of 4× wyomingensis, we focused on producing a reference genome for 2× tridentata. Due to the high genome complexity and outbred nature of the genome, an in vitro tissue propagation method was developed to provide sufficient biomass for genome sequencing and to allow for further experiments using plants of a single genotype that is shared with the reference genome (Barron et al. 2020).
Seeds used for tissue propagation came from a 2× tridentata mother plant known as IDT3 originating from the Soda Fire site (43.336 N, 116.964 W; Fig. 1) in the Northern Basin and Range ecoregion of Idaho, USA (Richardson et al. 2012). The taxonomy and ploidy level of the mother plant were confirmed using morphological features coupled with phylogenetic analyses and flow cytometry (Richardson et al. 2012; Chaney et al. 2017). An in vitro method of propagation for 2× tridentata developed by Barron et al. (2020) was used to produce biomass for IDT3 “G1_b2” by harvesting leaf tissue (average of 1.7 g per plantlet) from 15-week-old plantlets. The ploidy level and genome size of “G1_b2” were confirmed using flow cytometry (see below). Genome complexity and level of heterozygosity were estimated using a k-mer approach as implemented in GenomeScope (Vurture et al. 2017) using Illumina short-reads (see below). Based on these results, we estimated that 120 g of fresh leaf biomass was required to extract sufficient high quality and high-molecular weight DNA (fragment size greater than 50 kb) using a CTAB DNA extraction protocol for genome sequencing to sequence a genome at 100× coverage, de novo genome assembly, and scaffolding using OmniC proximity-ligation sequencing and the HiRise pipeline. This amount of tissue corresponded to 71 “G1_b2” plantlets. It took seven months to generate the necessary biomass while also maintaining the individual line in culture at Boise State University. Prior to biomass harvesting, plantlets were dark-treated for 48 h. The biomass was then flash frozen using liquid nitrogen and shipped overnight on dry ice to DovetailGenomics (Scotts Valley, California, USA) where DNA and RNA extractions were conducted (see below). For RNA extractions, 1 g of root biomass was also provided to complement the leaf biomass, both of which were used for genome annotation (see below).
Flow cytometry and genome complexity analysis
Flow cytometry was performed using methods outlined in Garcia et al. (2008) and Pellicer and Leitch (2014). Briefly, leaf material of G1_b2 was cochopped with the calibration standard Petunia hybrida Vilm. “PxPc6” (2C = 2.85 pg) in General Purpose Buffer (Loureiro et al. 2007) and stained using the base-independent fluorochrome propidium iodide. The samples were analyzed at Boise State University using a BD Accuri C6 Flow Cytometer with approximately 10,000 events (i.e. DNA fluorescence for approximately 10,000 nuclei) being recorded. Genome size was calculated per equation in (Pellicer and Leitch 2014).
The QIAGEN DNeasy Plant mini kit (Hilden, Germany; catalogue # 69204) was used to extract genomic DNA for short-read sequencing using 20 mg of dried leaf tissue per manufacturer protocol. To assess genome size and complexity, whole-genome sequencing (2 × 150 bp; genome coverage ∼160× read depth) was conducted on five lanes of Illumina HiSeq X (San Diego, CA, USA) by GeneWiz (New Jersey, NJ, USA). Raw read data were cleaned using Trimmomatic v.0.36 (Bolger et al. 2014). A subset of 1.05 × 1011 cleaned reads were then used to generate k-mers (k = 21) with KMCTools V3.1.1 (Kokot et al. 2017) for assessing genome size and complexity with the online GenomeScope portal (Vurture et al. 2017) and the R package “Smudgeplot” V0.2.4 (Ranallo-Benavidez et al. 2020). The lower and upper thresholds for k-mer coverage were 18 and 3,700, respectively, per the cutoff function from the Smudgpelot python script for the Smudgeplot analysis, limiting the inclusion of sequencing error (lower limit) and homozygous duplicate k-mers (upper limit).
PacBio and Omni-C sequence data generation
PacBio long-read and OmniC proximity-ligation sequence data production for the “G1_b2” genome assembly were performed as follows: (1) extract high-molecular weight DNA from 120 g of leaf biomass, (2) conduct whole-genome sequencing using PacBio long-read technology to produce ∼100× raw data coverage, and (3) prepare and sequence Dovetail Omni-C proximity-ligation libraries to further scaffold the de novo genome. These analyses were performed by DovetailGenomics.
High-molecular weight DNA was extracted using the CTAB method (Doyle and Doyle, 1987). DNA samples were quantified using Qubit 2.0 Fluorometer (Life Technologies, Carlsbad, CA, USA). A total of five PacBio SMRTbell libraries (∼20 kb) for PacBio Sequel were constructed using a SMRTbell Express Template Prep Kit 2.0 (PacBio, Menlo Park, CA, USA) following the manufacturer-recommended protocol. Each library was bound to polymerase using the Sequel II Binding Kit 2.0 (PacBio) and loaded onto the PacBio Sequel II instrument. Each library was sequenced individually on PacBio Sequel II 8M SMRT cells for a total of five sequencing runs.
Three Dovetail Omni-C libraries were prepared for proximity-ligation analysis. To prepare these libraries, chromatin was fixed with formaldehyde in the nucleus and then extracted using the QIAGEN blood and cell culture DNA mini kit (Hilden, Germany; catalogue # 13343). Fixed chromatin was digested with DNAse I, chromatin ends were repaired and ligated to a biotinylated bridge adapter followed by proximity ligation of adapter containing ends. After proximity ligation, crosslinks were reversed and the DNA was purified. Purified DNA was treated to remove biotin that was not internal to ligated fragments. Sequencing libraries were generated using NEBNext Ultra enzymes and Illumina-compatible adapters (New England BioLabs, Hitchin, UK). Biotin-containing fragments were isolated using streptavidin beads before PCR enrichment of each library. The libraries were sequenced on an Illumina HiSeq X platform at approximately 30× sequence coverage.
PacBio long-read de novo assembly and validation
A de novo assembly of the resulting PacBio continuous long reads was performed using WTDBG2 v2.5 (Ruan and Li 2020) with the following parameters: genome size 5.0 Gb, minimum read length 20,000, and minimum alignment length of 8,192 bp. Additionally, realignment was enabled with the -R option and read type was set with the option -x sq. To identify potential contaminants, the de novo assembly was assessed using a BLAST (Altschul et al. 1990) search against a database of nucleotide sequences from NCBI. BLAST results of the de novo assembly against the nucleotide database were assessed using blobtools v1.1.1 (Laetsch et al. 2020). Scaffolds identified as possible contamination using BLAST and blobtools were then removed from the assembly. Finally, purge_dups v1.2.3 (Guan et al. 2020) was used to remove haplotigs and highly overlapping contigs.
Pseudomolecule construction with HiRise
The de novo assembly and Dovetail Omni-C library reads were used as input data for HiRise, a software pipeline designed specifically for using proximity ligation data to scaffold genome assemblies (Putnam et al. 2016). Dovetail Omni-C library sequences were aligned to the draft input assembly using bwa (Li and Durbin 2009). The separations of Dovetail Omni-C read pairs mapped within draft scaffolds were analyzed by HiRise to produce a likelihood model for genomic distance between read pairs, and the model was used to identify and break putative misjoins, to score prospective joins, and make joins above a threshold (Fig. 2). The final HiRise assembly was assessed for completeness using the eukaryota_odb10 database in BUSCO V4.0.5 (Benchmarking Universal Single-Copy Orthologs; Simão et al. 2015).
Fig. 2.

Linkage–density histogram for the HiRise assembly generated by Dovetail Genomics. The axes represent the mapping positions along the genome assembly of the first (x-axis) and second (y-axis) read in the read pair, grouped into bins. The color of each square represents the number of reads within a given bin, with darker colors indicating more reads being mapped within the given bin. Vertical and horizontal lines have been added to delimit the scaffolds (smaller scaffolds are not visible in the plot due to scale and are represented by the large gray lines at the upper limits of the X- and Y-axes). X and Y-axes represent the position within the genome assembly in Gb, with pseudo-chromosomal scaffolds ordered largest to smallest.
Genome annotation
The genome was annotated for both noncoding repetitive DNA and for functional, coding genes. Preliminary functional annotation was performed using ab initio gene discovery and transcriptional data.
RNA sequencing
Illumina short-read RNA-Seq was performed to support annotation of the genome assembly. Total RNA extractions for leaf and root tissues were performed using the QIAGEN RNeasy Plus Kit following manufacturer protocols (Hilden, Germany). Total RNA was quantified using Qubit RNA Assay (Thermo Fisher Scientific, Waltham, MA, USA) and TapeStation 4200 (Agilent, Santa Clara, CA, USA). Prior to library prep, DNase treatment was performed followed by AMPure bead clean up (Beckman Coulter Life Sciences, Indianapolis, IN, USA) and QIAGEN FastSelect HMR rRNA depletion (Hilden, Germany). Library preparation was performed with the NEBNext Ultra II RNA Library Prep Kit following manufacturer protocols. These libraries were then sequenced on an Illumina NovaSeq6000 instrument in the 2 × 150 bp configuration.
Repeat identification
Repeat families found in the genome assemblies of 2× tridentata were identified de novo and classified using the software package RepeatModeler v.2.0.1 (Flynn et al. 2020). RepeatModeler depends on the programs RECON v.1.08 (Bao and Eddy 2002) and RepeatScout v.1.0.6 (Price et al. 2005) for the de novo identification of repeats within the genome. The custom repeat library obtained from RepeatModeler was used to discover, identify, and mask the repeats in the assembly file using RepeatMasker v.4.1.0 (Smit et al. 2013 ).
Functional annotation
Coding sequences from Cynara cardunculus L., Erigeron canadensis L., Helianthus annuus L., Lactuca sativa L., and Mikania micrantha Kunth. were used to train the initial ab initio gene discovery model for 2× tridentata using the AUGUSTUS software v.2.5.5 (Stanke et al. 2004). Six rounds of prediction optimization were done with the software package provided by AUGUSTUS. The same coding sequences were also used to train a separate ab initio gene discovery model for 2× tridentata using SNAP v.2006-07-28 (Korf 2004). RNA-Seq reads were mapped onto the genome using the STAR aligner software v.2.7 (Dobin et al. 2013) and intron hints (i.e. extrinsic evidence about the location and structure of genes) generated with the bam2hints tools within the AUGUSTUS software. MAKER (Cantarel et al. 2008), SNAP (Korf 2004), and AUGUSTUS (Stanke et al. 2004) (with intron–exon boundary hints provided from RNA-Seq) were then used to predict genes in the repeat-masked reference genome. To help guide the prediction process, Swiss-Prot peptide sequences from the UniProt (UniProt Consortium 2019) database were downloaded and used in conjunction with the protein sequences from C. cardunculus, E. canadensis, H. annuus, L. sativa, and M. micrantha to generate peptide evidence in the MAKER pipeline. Only genes that were predicted by both SNAP and AUGUSTUS were retained in the final gene sets. To help assess the quality of the gene prediction, Annotation Edit Distance scores (Eilbeck et al. 2009), a metric to quantify the amount of change between individual annotations, were generated for each of the predicted genes as part of the MAKER pipeline. Genes were further characterized for their putative function by performing a BLAST search of the peptide sequences against the UniProt database. tRNA were predicted using the software tRNAscan-SE v.2.05 (Chan et al. 2021). Finally, to meet NCBI genome submission standards, seven scaffolds of 200 bases or less and one scaffold comprising a mitochondrial genome fragment were removed from the annotated HiRise assembly.
Results and discussion
Validation of genome assembly and annotation
The final processed 2× tridentata genome assembly comprises 5,492 scaffolds, nine of which are pseudo-chromosomes (L90 = 9 = n), and 4,198,553,833 bases (4.20 Gb; Fig. 3a). The pseudo-chromosome scaffolds range from 0.528 to 0.338 Gb in length (Fig. 3a and Table 1). Flow cytometry on the IDT3 “G1_b2” sample estimated the genome size to be 4.19 Gb/1C, which is in line with previous estimates of the 2× tridentata genome sizes (i.e. 4.1 Gb/1C per Garcia et al. 2008). The GenomeScope and Smudgeplot analyses further confirmed the genome to be diploid, with two distinct k-mer peaks in the GenomeScope plot and greatest density of k-mers in the diploid AB “smudge” in the Smudgeplot, and revealed high levels of genome complexity, with evidence of past hybridization, polyploidization-to-diploidization events, and high levels of out-crossing (Fig. 3, b and c). These results are consistent with previous studies that found evidence of past polyploidy and hybridization events within Artemisia (e.g. Garcia et al. 2008; Barron et al. 2020).
Fig. 3.
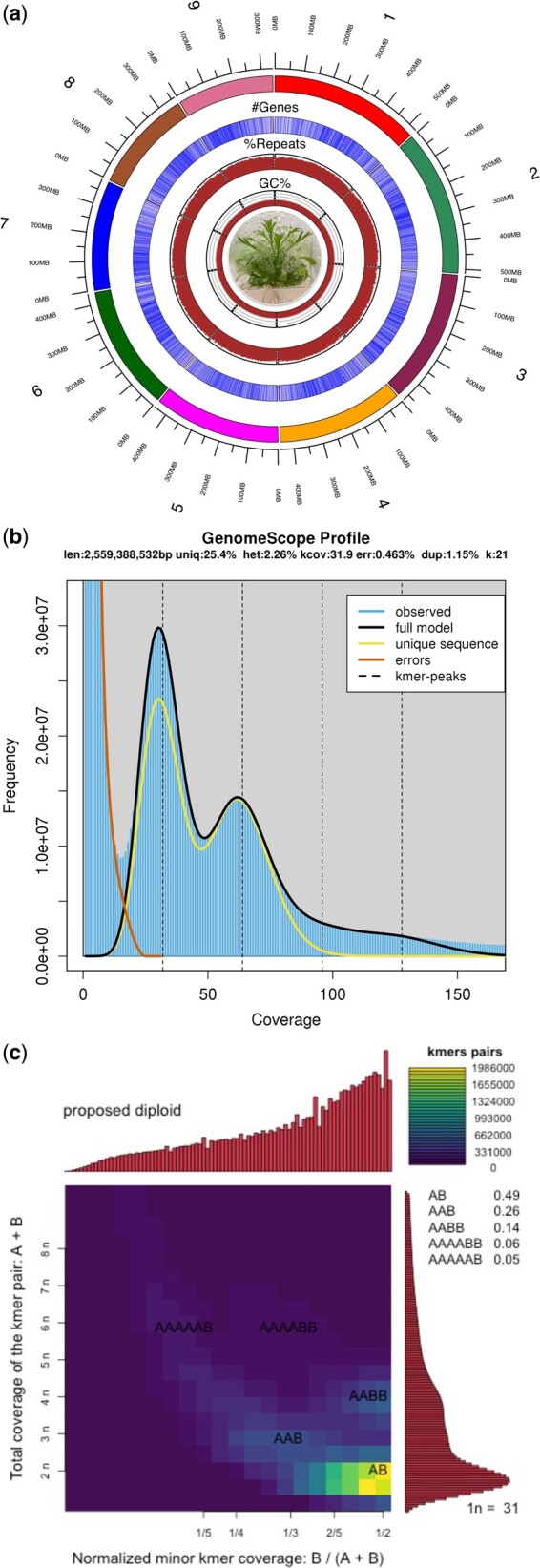
Density plot of k-mer analysis in GenomeScope and genome map showing GC content (%), % repeat per 1 million nucleotides, number of genes per 1 million nucleotides, and the size of the scaffold for the nine pseudo-chromosomal scaffolds. Subset (a) shows the genome feature mapping for the nine pseudo-chromosomal scaffolds, subset (b) shows GenomeScope results, and subset (c) shows the Smudgeplot results. GenomeScope summary statistics, including heterozygosity rate (listed as “het”), are listed at the top of plot (b). Two primary k-mer peaks are present, indicating that the genome is diploid. The Smudgeplot shows the frequency of k-mer pairs within the genome, with darker colors indicating the group is less frequent and bright yellow indicating the group is more frequent. When visualized, the plot shows distinct “smudges” representing each k-mer pair with the greatest of density of k-mers relating to the ploidy level of the genome (e.g. the diploid A. tridentata genome has the brightest “smudge” for the diploid AB k-mer pair).
Table 1.
Summary statistics for the 9 pseudo-chromosomal scaffolds within the IDT3 “G1_b2” genome assembly.
| Scaffold | Length in Gb (% of assembly) | Protein coding genes | Total gene length in Gb (% of assembly) | Repeat occurrences | Repeat length total in Gb (% of assembly) |
|---|---|---|---|---|---|
| 1 | 0.528 (12.58) | 5,869 | 0.018 (3.49) | 709,220 | 0.444 (84.00) |
| 2 | 0.514 (12.23) | 5,153 | 0.015 (2.99) | 682,886 | 0.443 (86.21) |
| 3 | 0.472 (11.24) | 4,781 | 0.015 (3.15) | 624,680 | 0.406 (86.04) |
| 4 | 0.446 (10.62) | 4,707 | 0.015 (3.33) | 591,412 | 0.378 (84.73) |
| 5 | 0.445 (10.59) | 4,951 | 0.017 (3.73) | 591,818 | 0.371 (83.43) |
| 6 | 0.439 (10.46) | 4,358 | 0.013 (3.04) | 580,217 | 0.379 (86.38) |
| 7 | 0.385 (9.18) | 4,096 | 0.013 (3.30) | 513,867 | 0.330 (85.52) |
| 8 | 0.361 (8.61) | 3,520 | 0.011 (3.03) | 480,240 | 0.311 (86.11) |
| 9 | 0.338 (8.06) | 3,430 | 0.011 (3.11) | 446,444 | 0.295 (87.12) |
| Total | 3.929 (93.58) | 40,865 | 0.128 (3.25) | 5,220,784 | 3.356464852 (85.43) |
Repeat identification analysis revealed that the 2× tridentata genome is highly repetitive. A total of 77.99% of the genome consisted of repetitive sequences (Fig. 3a), with the largest class being Class I Transposable Elements (TE; 36.20%), with Class II TEs being the second most common repeat (2.19%) (Tables 1 and 2). Low complexity and simple repeats comprise 0.10% and 0.82% of the genome assembly, respectively. A total of 85.43% of the pseudo-chromosome scaffold sequence was found to be repetitive, with an average of 85.50% for each pseudo-chromosome (Table 1). This level of repetitive DNA sequence is high since the average repetitive DNA content for plant genomes is 57%, with relatively few plant genomes containing >75% repetitive sequence (Michael and Jackson 2013; Michael 2014), making the 2× tridentata genome one of the most highly repetitive plant genomes sequenced.
Table 2.
Summary statistics for the de novo and HiRise genome assembly outputs.
| De novo assembly | HiRise assembly | |
|---|---|---|
| Total length (bp) | 4,197,847,053 | 4,198,560,453 |
| N50 | 965,994 | 444,777,032 |
| L50 | 1,188 | 5 |
| N90 | 246,927 | 338,336,202 |
| L90 | 4,521 | 9 |
| Largest scaffold (bp) | 10,654,198 | 528,210,163 |
| Number of scaffolds | 12,613 | 5,500 |
| Number of scaffolds >1 kb | 12,577 | 5,464 |
| Number of gaps | 1,859 | 8,993 |
| Number of N's/100 kb | 1 | 18 |
| Complete BUSCOs (C) | 232 (90.98%) | 233 (91.37%) |
| Complete and single-copy BUSCOs (S) | 175 (68.63%) | 188 (73.73%) |
| Complete and duplicated BUSCOs (D) | 57 | 45 |
| Fragmented BUSCOs (F) | 2 | 5 |
| Missing BUSCOs (M) | 21 | 17 |
| Total BUSCO groups searched | 255 | 255 |
The final assembly, with scaffolds <200 bases in length and 1 mitochondrial fragment removed, totaled 4,198,553,833 bases and comprised 5,492 scaffolds.
Benchmarking Universal Single-Copy Orthologs (BUSCO) analysis recovered 91.37% (233 of 255) of single-copy BUSCOs from the HiRise assembly. A total of 1.2% of BUSCOs were found to be duplicated. Only 3.1% of BUSCOs were fragmented and 9.0% were missing (Table 2). This result indicated a high level of completeness in the genome assembly and that the genome was sufficiently assembled for annotation. Using ab initio gene discovery and transcriptomic evidence, a total of 43,377 genes were identified, with coding regions comprising 0.59 Gb. Of the 43,477 genes identified, 40,865 were located on the pseudo-chromosome scaffolds, with each scaffold containing an average of 4,541 genes (Table 1). The average length of these genes was 1,358 bp. A total of 8,759 genes were found to comprise a single exon.
Genome complexity and evidence of past polyploidization
The GenomeScope analysis showed that the 2× tridentata genome is a highly heterozygous genome, with an estimated level of heterozygosity of 2.26% (listed as het: 2.26% in Fig. 3b). This is relatively high when compared to other plants, such as Arabidopsis thaliana (1.04%), and slightly less than the highly heterozygous Vitis vinifera genome (2.6%; Jaillon etal. 2007). The Smudgeplot analysis (Fig. 3c) revealed that while diploid (as shown by the highest k-mer coverage being that of 2n k-mers), there are varying levels of coverage depth for the different k-mer pairs, indicating a complex evolutionary history including prior hybridization and polyploidization events. The diploid AB k-mer pairs were most prominent (49% of k-mers), the AAB and AABB k-mer pairs were the next most common at 26% and 14% of k-mers, respectively (Fig. 3c). Greater AABB k-mer pairs than AAAB k-mer pairs would be indicative of past allopolyploidization via hybridization and genome doubling, with equivalent contributions of the A and B parental genomes (Ranallo-Benavidez et al. 2020). The higher proportion of AAB would suggest backcrossing with the diploid parental A genome after the allopolyploidization event. While our results indicated “G1_b2” is a diploid, the 2× tridentata genome demonstrated evidence of past polyploidization followed by chromosomal rearrangements leading to diploidy (i.e. diploidization; Dodsworth et al. 2016). Such a process has been advocated to be one of the main drivers of the evolutionary success of flowering plants and further studying it in sagebrush could shed light into the mechanisms of adaptations leading to the diversification of this lineage in the sagebrush steppes (Dodsworth et al. 2016).
Comparing the A. tridentata and A. annua genome assemblies
Artemisia annua L., commonly known as sweet wormwood, is the only other species of Artemisia to have its genome sequenced (Shen et al. 2018). The A. annua genome assembly represents a fairly high-quality draft assembly, containing 39,579 scaffolds (Shen et al. 2018). While the divergence of the clades containing A. annua and A. tridentata occurred ∼10.8 MYA (Sanz et al. 2011), these species maintain a conserved ploidy level, with the base karyotype number for each species comprising nine chromosomes (2n = 2× = 18; McArthur et al. 1981). While these species contain the same number of chromosomes, there are distinct differences in their genomes. The genome size for A. tridentata, and other members of the North American Tridentatae subgenus (Garcia et al. 2008; Pellicer et al. 2010), is nearly 2.5 times the size of the A. annua genome (4.20 Gb/1C vs. 1.74 Gb/1C). The current genome assembly of A. annua has been found to contain more genes (63,226 genes; Shen et al. 2018) than identified here in the genome assembly and annotation for A. tridentata (43,377 genes). This difference in gene content may be partially explained by incomplete annotation of paralogues, particularly tandem paralogues whose annotations can be merged into one (Campbell et al. 2014). Tandem paralogues have been previously identified in a draft assembly of the A. tridentata genome, in which two tandem Aquaporin paralogues were found on one scaffold (Melton et al. 2021). Future comparative genomic and transcriptomic analyses will need to be performed to ascertain whether gene content is higher in the A. annua genome than in the A. tridentata genome or if incorrect annotation of tandem paralogs in the A. tridentata genome has led to an underestimation of gene content.
The genome of A. tridentata is far more heterozygous (2.26% vs. 1.0–1.5%) and repetitive (77.99% vs. 61.57%) than the A. annua genome. These aspects of the A. tridentata genome are likely the result of a polyploidization, likely due to an allopolyploidization event, early within the divergence of subgenus Tridentatae followed by diploidization (Garcia et al. 2008; Pellicer et al. 2010), also supported by high proportion of AB k-mer pair, with lower proportions of AAB and AABB k-mer pairs, and greatest density of k-mers in the diploid AB “smudge” presented in the Smudgeplot results here (Fig. 3c). Differences in the assembly levels may also contribute to the perceived differences in repetitiveness, as repetitive genome sequences are difficult to quantify in more fragmented genomes.
Applications of the sagebrush reference genome
The 2× tridentata genome sequence data will serve as a valuable resource for a broad range of researchers. This species has been used to study abiotic stress responses using physiological and eco-physiological methods for decades (DePuit and Caldwell 2017; Richards and Caldwell 1987; Kolb and Sperry 1999; Ryel et al. 2004; Germino 2012; Copeland et al. 2022). This genome will allow for greater connectivity between field-based and ecophysiological research and genomic research, which aims to elucidate genome-to-phenome and stress-response pathways. Artemisia tridentata also belongs to the ecologically and economically important Asteraceae family comprising 10% of angiosperm diversity (Mandel et al. 2019), allowing this genome to serve as an important contribution to our understanding of Asteraceae evolution. Currently, 24 Asteraceae genomes are available through NCBI and this genome fills a taxonomic and phylogenetic gap in Asteroideae (Supplementary Table 1). For these genome assemblies, the average size is 1.59 Gb (standard deviation = ± 1.06 Gb), much smaller than the 4.20 Gb assembly for Artemisia tridentata. This new Asteraceae genome assembly and the variation in genome size within the family allow for further research into the processes that shape genome size. Artemisia is also amongst the largest genera of Asteraceae with species of agricultural, horticultural, medicinal, and pharmaceutical importance (Bora and Sharma 2011; Pellicer et al. 2011, 2018). The antimalarial agent artemisinin was detected in multiple species of Artemisia, including Artemisia tridentata, demonstrating the potential usage of genomic data for studying the evolution of biochemical pathways relevant to potential drug discovery (Pellicer et al. 2018).
Data availability
Supplementary Table 2 lists all sequence data generated in this project. All sequence data for this project are available from the National Center for Biotechnology Information (NCBI) under BioProject accession number PRJNA795150 and BioSample accession number SAMN24662005. The Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession JAKJXK000000000. All raw sequence files are available from the NCBI SRA database (PacBio long read data SRR17863255 Omni-C proximity-ligation data SRR17863200, SRR17870744 and SRR17870745; Illumina HiSeq short read data SRR17870775 and SRR17863368; RNASeq paired end reads from leaf tissue SRR17779362; RNASeq paired end reads from root tissue SRR17779361). Genome annotation results and supporting data files are openly available via the G3 figshare repository at https://doi.org/10.25387/g3.19651260.
All software used in this work is in the public domain, with parameters being clearly described in Materials and methods. If parameters were not detailed for a software, default parameters were used as suggested by the developer.
Supplemental material is available at G3 online.
Supplementary Material
Acknowledgments
The authors are grateful to Denise Pfeifer (Boise State University) and Rick Schumaker (EPSCoR Idaho) for their support with the project management and Jasmine Haimovitz (Dovetail Genomics) for overseeing the data production and genome assembly. We are thankful to the GEM3 Sagebrush Mechanism team as well as the GEM3 leadership team and colleagues for their invaluable support with this project. The authors would also like to thank an anonymous reviewer and Associate Editor Stuart Macdonald for their thoughtful comments that improved the manuscript. Any use of trade, firm, or product names is for descriptive purposes only and does not imply endorsement by the U.S. Government.
SB, PM, SJN, and JSF conceived the study. JSF, AK, and SB secured funding. BAR provided the seeds. PM, MS, CDD, AEM, and SB conducted in vitro tissue propagation and collected biomass for sequencing. IJL, JP, DS, and RB conducted flow cytometry. AEM and CDD conducted the genomic DNA extraction for Illumina short-read sequencing. AC, AEM, and SB conducted the data management and submission to NCBI. AEM and SB performed bioinformatics analyses. AEM and SB wrote the manuscript. All authors read, contributed to, and approved the final manuscript.
Funding
This research was made possible by 2 NSF Idaho EPSCoR grants (award numbers OIA-1757324 and OIA-1826801), as well as a Dovetail Genomics Tree of Life Award.
Conflicts of interest
None declared.
Contributor Information
Anthony E Melton, Department of Biological Sciences, Boise State University, Boise, ID 83725, USA.
Andrew W Child, University of Idaho, Moscow, ID 83844, USA.
Richard S Beard, Jr, Department of Biological Sciences, Boise State University, Boise, ID 83725, USA.
Carlos Dave C Dumaguit, Department of Biological Sciences, Boise State University, Boise, ID 83725, USA.
Jennifer S Forbey, Department of Biological Sciences, Boise State University, Boise, ID 83725, USA.
Matthew Germino, Forest and Rangeland Ecosystem Science Center, United States Geological Survey, Boise, ID 83706, USA.
Marie-Anne de Graaff, Department of Biological Sciences, Boise State University, Boise, ID 83725, USA.
Andrew Kliskey, University of Idaho, Moscow, ID 83844, USA.
Ilia J Leitch, Royal Botanic Gardens, Richmond TW9 3AE, UK.
Peggy Martinez, Department of Biological Sciences, Boise State University, Boise, ID 83725, USA.
Stephen J Novak, Department of Biological Sciences, Boise State University, Boise, ID 83725, USA.
Jaume Pellicer, Royal Botanic Gardens, Richmond TW9 3AE, UK; Institut Botànic de Barcelona (IBB, CSIC-Ajuntament de Barcelona), Barcelona 08038, Spain.
Bryce A Richardson, Rocky Mountain Research Station, United States Forest Service, Moscow, ID 83843, USA.
Desiree Self, Department of Biological Sciences, Boise State University, Boise, ID 83725, USA.
Marcelo Serpe, Department of Biological Sciences, Boise State University, Boise, ID 83725, USA.
Sven Buerki, Department of Biological Sciences, Boise State University, Boise, ID 83725, USA.
Literature cited
- Baker WL. Fire and restoration of sagebrush ecosystems. Wildl Soc Bull. 2006;34(1):177–185. [Google Scholar]
- Bao Z, Eddy SR.. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 2002;12(8):1269–1276. doi: 10.1101/gr.88502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barron R, Martinez P, Serpe M, Buerki S.. Development of an in vitro method of propagation for Artemisia tridentata subsp. tridentata to support genome sequencing and genotype-by-environment research. Plants. 2020;9(12):1717. doi: 10.3390/plants9121717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B.. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bora KS, Sharma A.. The genus Artemisia: a comprehensive review. Pharm Biol. 2011;49(1):101–109. doi:10.3109/13880209.2010.497815. [DOI] [PubMed] [Google Scholar]
- Brabec MM, Germino MJ, Shinneman DJ, Pilliod DS, McIlroy SK, Arkle RS.. Challenges of establishing big sagebrush (Artemisia tridentata) in rangeland restoration: effects of herbicide, mowing, whole-community seeding, and sagebrush seed sources. rangel. Ecol Manag. 2015;68(5):432–435. doi: 10.1016/j.rama.2015.07.001. [DOI] [Google Scholar]
- Cantarel BL, Korf I, Robb SMC, Parra G, Ross E, Moore B, Holt C, Alvarado AS, Yandell M.. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008;18(1):188–196. doi: 10.1101/gr.6743907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MS, Holt C, Moore B, Yandell M.. Genome annotation and curation using MAKER and MAKER]P. Current protocols in bioinformatics. 2014;48(1):4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PP, Lin BY, Mak AJ, Lowe TM.. TRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021;49(16):9077–9096. doi: 10.1093/nar/gkab688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaney L, Richardson BA, Germino MJ.. Climate drives adaptive genetic responses associated with survival in big sagebrush (Artemisia tridentata). Evol Appl. 2017;10(4):313–322. doi:10.1111/eva.12440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland SM, Hamerlynck EP, Holfus CM, Campbell EE, Boyd CS.. Stomatal conductance relates to sagebrush transplant survival across planting season and size-class. Rangel Ecol Manag. 2022;80:26–30. doi: 10.1016/j.rama.2021.09.008. [DOI] [Google Scholar]
- DePuit EJ, Caldwell MM.. Seasonal pattern of net photosynthesis of Artemisia tridentata. Am J Bot. 2017;60(5):426–435. http://www.jstor.org/stable/2441499. [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodsworth S, Chase MW, Leitch AR.. Is post-polyploidization diploidization the key to the evolutionary success of angiosperms? Bot J Linn Soc. 2016;180(1):1–5. doi: 10.1111/boj.12357. [DOI] [Google Scholar]
- Doyle J, Doyle J.. A rapid DNA isolation procedure for small quantities of fresh leaf tiss. Phytochem. Bull. 1987;19:11–15. [Google Scholar]
- Eilbeck K, Moore B, Holt C, Yandell M.. Quantitative measures for the management and comparison of annotated genomes. BMC Bioinform. 2009;10:67. doi: 10.1186/1471-2105-10-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn JM, Hubley R, Goubert C, Rosen J, Clark AG, Feschotte C, Smit AF.. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci U S A. 2020;117(17):9451–9457. doi: 10.1073/pnas.1921046117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia S, Canela MÁ, Garnatje T, Mcarthur ED, Pellicer J, Sanderson SC, Vallès J.. Evolutionary and ecological implications of genome size in the North American endemic sagebrushes and allies (Artemisia, Asteraceae). Biol J Linn Soc. 2008;94(3):631–649. doi: 10.1111/j.1095-8312.2008.01001.x. [DOI] [Google Scholar]
- Garcia S, Durant McArthur E, Pellicer J, Sanderson SC, Vallès J, Garnatje T.. A molecular phylogenetic approach to western North America endemic Artemisia and allies (Asteraceae): untangling the sagebrushes. Am J Bot. 2011;98(4):638–653. doi:10.3732/ajb.1000386. [DOI] [PubMed] [Google Scholar]
- Germino M. Selecting sagebrush seed sources for restoration in a variable climate: ecophysiological variation among genotypes, USDA Forest Service, Rocky Mountain Research Station and USDA Bureau of Land Management, 2012. [Google Scholar]
- Germino MJ, Barnard DM, Davidson BE, Arkle RS, Pilliod DS, Fisk MR, Applestein C.. Thresholds and hotspots for shrub restoration following a heterogeneous megafire. Landscape Ecol. 2018;33(7):1177–1194. doi: 10.1007/s10980-018-0662-8. [DOI] [Google Scholar]
- Guan D, Guan D, McCarthy SA, Wood J, Howe K, Wang Y, Durbin R, Durbin R.. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics. 2020;36(9):2896–2898. doi: 10.1093/bioinformatics/btaa025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaillon O, , AuryJ-M, , NoelB, , PolicritiA, , ClepetC, , CasagrandeA, , ChoisneN, , AubourgS, , VituloN, , Jubin C,. et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007;449(7161):463–467. 10.1038/nature06148 17721507 [DOI] [PubMed] [Google Scholar]
- Kokot M, Dlugosz M, Deorowicz S.. KMC 3: counting and manipulating k-mer statistics. Bioinformatics. 2017;33(17):2759–2761. doi: 10.1093/bioinformatics/btx304. [DOI] [PubMed] [Google Scholar]
- Kolb KJ, Sperry JS.. Differences in drought adaptation between subspecies of sagebrush (Artemisia tridentata). Ecology. 1999;80(7):2373–2384. doi: 10.2307/176917. [DOI] [Google Scholar]
- Korf I. Gene finding in novel genomes. BMC Bioinform. 2004;5(1):59–59. doi: 10.1186/1471-2105-5-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laetsch DR, Blaxter ML, Leggett RM.. BlobTools : interrogation of genome assemblies [version 1 ; peer review : 2 approved with reservations.] F1000Research. 2020;1287:1–16. [Mismatch [Google Scholar]
- Li H, Durbin R.. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loureiro J, Rodriguez E, Doležel J, Santos C.. Two new nuclear isolation buffers for plant DNA flow cytometry: a test with 37 species. Ann Bot. 2007;100(4):875–888. doi: 10.1093/aob/mcm152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel JR, Dikow RB, Siniscalchi CM, Thapa R, Watson LE, Funk VA.. A fully resolved backbone phylogeny reveals numerous dispersals and explosive diversifications throughout the history of Asteraceae. Proc Natl Acad Sci U S A. 2019;116(28):14083–14088. doi: 10.1073/pnas.1903871116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur ED, Pope CL, Freeman DC.. Chromosomal studies of subgenus Tridentatae of Artemisia : evidence for autopolyploidy. Am J Bot. 1981;68(5):589–605. [Google Scholar]
- McArthur ED, Sanderson SC.. Cytogeography and chromosome evolution of subgenus Tridentatae of Artemisia (Asteraceae). Am J Bot. 1999;86(12):1754–1775. doi: 10.2307/2656673. [DOI] [PubMed] [Google Scholar]
- Melton AE, Beck J, Galla SJ, Jenkins J, Handley L, Kim M, Grimwood J, Schmutz J, Richardson BA, Serpe M, et al. A draft genome provides hypotheses on drought tolerance in a keystone plant species in Western North America threatened by climate change. Ecol Evol. 2021;11(21):15417–15429. doi: 10.1002/ece3.8245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael TP. Plant genome size variation: bloating and purging DNA. Brief Funct Genomics. 2014;13(4):308–317. doi:10.1093/bfgp/elu005. [DOI] [PubMed] [Google Scholar]
- Michael TP, Jackson S.. The first 50 plant genomes. Plant Genome. 2013;6(2):doi:10.3835/plantgenome2013.03.0001in. [Google Scholar]
- Miller RF, Knick ST, Pyke DA, Meinke CW, Hanser SE, Wisdom MJ, Hild AL.. Characteristics of sagebrush habitats and limitations to long-term conservation. greater sage-grouse: ecology and conservation of a landscape species and its habitats. Stud Avian Biol. 2012;38:144–184. doi: 10.1525/california/9780520267114.003.0011. [DOI] [Google Scholar]
- O’Connor RC, Germino MJ, Barnard DM, Andrews CM, Bradford JB, Pilliod DS, Arkle RS, Shriver RK.. Small-scale water deficits after wildfires create long-lasting ecological impacts. Environ Res Lett. 2020;15(4):044001. doi: 10.1088/1748-9326/ab79e4. [DOI] [Google Scholar]
- Pellicer J, , Leitch IJ.. The application of flow cytometry for estimating genome size and ploidy level in plants. Methods Mol Biol. 2014;1115:279–307. doi: 10.1007/978-1-62703-767-9_14. [DOI] [PubMed] [Google Scholar]
- Pellicer J, Garcia S, Canela MÁ, Garnatje T, Korobkov AA, Twibell JD, Vallès J.. Genome size dynamics in Artemisia L. (Asteraceae): following the track of polyploidy. Plant Biol. 2010;12(5):820–830. doi: 10.1111/j.1438-8677.2009.00268.x. [DOI] [PubMed] [Google Scholar]
- Pellicer J, Garnatje T, Vallès J.. Artemisia (Asteraceae): understanding its evolution using cytogenetic and molecular systematic tools, with emphasis on subgenus Dracunculus. Recent Adv Pharm Sci. 2011;661(2):199–222. [Google Scholar]
- Pellicer J, Saslis-Lagoudakis CH, Carrió E, Ernst M, Garnatje T, Grace OM, Gras A, Mumbrú M, Vallès J, Vitales D, et al. A phylogenetic road map to antimalarial Artemisia species. J Ethnopharmacol. 2018;225(June):1–9. doi: 10.1016/j.jep.2018.06.030. [DOI] [PubMed] [Google Scholar]
- Pilliod DS, Welty JL, Toevs GR.. Seventy-five years of vegetation treatments on public rangelands in the great basin of North America. Rangelands. 2017;39(1):1–9. doi: 10.1016/j.rala.2016.12.001. [DOI] [Google Scholar]
- Price AL, Jones NC, Pevzner PA.. De novo identification of repeat families in large genomes. Bioinformatics. 2005;21(Suppl 1):i351–i358. doi: 10.1093/bioinformatics/bti1018. [DOI] [PubMed] [Google Scholar]
- Putnam NH, O'Connell BL, Stites JC, Rice BJ, Blanchette M, Calef R, Troll CJ, Fields A, Hartley PD, Sugnet CW, et al. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 2016;26(3):342–350. doi:10.1101/gr.193474.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranallo-Benavidez TR, Jaron KS, Schatz MC.. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun. 2020;11(1):1432. doi: 10.1038/s41467-020-14998-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remington TE, Deibert PA, Hanser SE, Davis DM, Robb LA, L WJ.. 2021. Sagebrush conservation strategy—challenges to sagebrush conservation: U.S. Geological Survey Open-File Report 2020–1125, p. 327. https://doi.org/10.3133/ofr20201125.
- Richards JH, Caldwell MM.. Hydraulic lift: substantial nocturnal water transport between soil layers by Artemisia tridentata roots. Oecologia. 1987;73(4):486–489. doi: 10.1007/BF00379405. [DOI] [PubMed] [Google Scholar]
- Richardson BA, Germino MJ, Warwell MV, Buerki S.. The role of genome duplication in big sagebrush growth and fecundity. Am J Bot. 2021;108(8):1405–1416. doi: 10.1002/ajb2.1714. [DOI] [PubMed] [Google Scholar]
- Richardson BA, Page JT, Bajgain P, Sanderson SC, Udall JA.. Deep sequencing of amplicons reveals widespread intraspecific hybridization and multiple origins of polyploidy in big sagebrush (Artemisia tridentata; Asteraceae). Am J Bot. 2012;99(12):1962–1975. [DOI] [PubMed] [Google Scholar]
- Rigge M, Homer C, Cleeves L, Meyer DK, Bunde B, Shi H, Xian G, Schell S, Bobo M.. 2020. Quantifying western U.S. rangelands as fractional components with multi-resolution remote sensing and in situ data. Remote Sensing. 12(3):1–26. doi: 10.3390/rs12030412. [DOI] [Google Scholar]
- Ruan J, Li H.. Fast and accurate long-read assembly with wtdbg2. Nat Methods. 2020;17(2):155–158. doi: 10.1038/s41592-019-0669-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryel RJ, Leffler AJ, Peek MS, Ivans CY, Caldwell MM.. Water conservation in Artemisia tridentata through redistribution of precipitation. Oecologia. 2004;141(2):335–345. doi: 10.1007/s00442-003-1421-2. [DOI] [PubMed] [Google Scholar]
- Sanz M, Schneeweiss GM, Vilatersana R, Vallès J.. Temporal origins and diversification of Artemisia and allies (Anthemideae, Asteraceae). Collect Bot. 2011;30(0):7–15. doi: 10.3989/collectbot.2011.v30.001. [DOI] [Google Scholar]
- Shen Q, Zhang L, Liao Z, Wang S, Yan T, Shi P, Liu M, Fu X, Pan Q, Wang Y, et al. The genome of Artemisia annua provides insight into the evolution of asteraceae family and artemisinin biosynthesis. Mol Plant. 2018;11(6):776–788. doi: 10.1016/j.molp.2018.03.015. [DOI] [PubMed] [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM.. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31(19):3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green P.. RepeatMasker Open-4.0. 2013. http://www.repeatmasker.org [Google Scholar]
- Stanke M, Steinkamp R, Waack S, Morgenstern B.. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 2004;32(WEB server iss.):309–312. doi: 10.1093/nar/gkh379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Still SM, Richardson BA.. Projections of contemporary and future climate niche for wyoming big sagebrush (Artemisia tridentata subsp. wyomingensis): a guide for restoration. Nat Areas J. 2015;35(1):30–43. doi: 10.3375/043.035.0106. [DOI] [Google Scholar]
- The UniProt Consortium. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47(D1):D506–D515. doi: 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vurture GW, Sedlazeck FJ, Nattestad M, Underwood CJ, Fang H, Gurtowski J, Schatz MC.. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics. 2017;33(14):2202–2204. doi: 10.1093/bioinformatics/btx153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijayratne UC, Pyke DA.. Burial increases seed longevity of two Artemisia tridentata (Asteraceae) subspecies. Am J Bot. 2012;99(3):438–447. doi: 10.3732/ajb.1000477. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Supplementary Table 2 lists all sequence data generated in this project. All sequence data for this project are available from the National Center for Biotechnology Information (NCBI) under BioProject accession number PRJNA795150 and BioSample accession number SAMN24662005. The Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession JAKJXK000000000. All raw sequence files are available from the NCBI SRA database (PacBio long read data SRR17863255 Omni-C proximity-ligation data SRR17863200, SRR17870744 and SRR17870745; Illumina HiSeq short read data SRR17870775 and SRR17863368; RNASeq paired end reads from leaf tissue SRR17779362; RNASeq paired end reads from root tissue SRR17779361). Genome annotation results and supporting data files are openly available via the G3 figshare repository at https://doi.org/10.25387/g3.19651260.
All software used in this work is in the public domain, with parameters being clearly described in Materials and methods. If parameters were not detailed for a software, default parameters were used as suggested by the developer.
Supplemental material is available at G3 online.