

Abstract
Screening microbial secondary metabolites is an established method to identify novel biologically active molecules. Preparation of biological screening samples from microbial fermentation extracts requires growth conditions that promote synthesis of secondary metabolites and extraction procedures that capture the secondary metabolites produced. High-performance liquid chromatography (HPLC) analysis of fermentation extracts can be used to estimate the number of secondary metabolites produced by microorganisms under various growth conditions but is slow. In this study we report on a rapid (approximately 1 min per assay) surrogate measure of secondary metabolite production based on a metabolite productivity index computed from the electrospray mass spectra of samples injected directly into a spectrometer. This surrogate measure of productivity was shown to correlate with an HPLC measure of productivity with a coefficient of 0.78 for a test set of extracts from 43 actinomycetes. This rapid measure of secondary metabolite productivity may be used to identify improved cultivation and extraction conditions by analyzing and ranking large sets of extracts. The same methods may also be used to survey large collections of extracts to identify subsets of highly productive organisms for biological screening or additional study.
Microbial extracts have been and continue to be a productive source of new biologically active molecules for drug discovery (2, 13). It is estimated that more than 30% of worldwide human pharmaceutical sales have compounds from natural sources as their origin (12). With advances in genomics and high-throughput screening (HTS) technology, many new therapeutic targets are accessible for identifying pharmaceutical agents. In addition to the historical practice of screening microbial fermentation extracts for antibiotic activity, extracts can now routinely be screened with a variety of new functional, receptor binding, enzyme inhibition, and protein-protein interaction assays. HTS formats used at many large pharmaceutical research organizations are, however, generally incompatible with complex fermentation extracts. The reasons for the incompatibility include nonspecific interference with assay systems, cost in dollars and time to identify and dereplicate active components from a complex mixture, and adverse physical properties for automated liquid-handling equipment. Therefore, additional investment in selection and preparation of fermentation extracts is one strategy to align natural product drug discovery with today's automated HTS assay systems. A typical approach to improve the compatibility of fermentation extracts with HTS was recently reported by Schmid et al., who described a multistage automated solid-phase extraction (SPE) system (12).
To make the best use of finite resources in natural product discovery organizations, it is important to identify collections of organisms that produce secondary metabolites, culture conditions that generally support secondary metabolite synthesis, and sample preparation processes that retain secondary metabolites while maintaining compatibility with HTS formats. It is well known that production of secondary metabolites by microorganisms is influenced by fermentation conditions. In a study of 29 Nodulisporium strains, Monaghan et al. found that synthesis of secondary metabolites was directly influenced by fermentation conditions and that there were differences of up to 400-fold in the concentrations of secondary metabolites between conditions (11). Additionally, Monaghan et al. found that rare metabolites were consistently found in extracts containing larger numbers of secondary metabolites. Yarbrough et al. reported that several different growth conditions were required to elicit secondary metabolite production in a set of 760 microorganisms (16).
Given that additional investment is required to make fermentation extracts compatible with modern HTS and that many fermentation extracts may contain insufficient secondary metabolites to warrant such investment, methods to characterize extracts from an industrial, high-throughput drug discovery perspective are needed. There are a number of previously developed methods which use direct chemical measurement to classify microorganisms (5–8, 14). Most previous studies focused on characterizing microorganisms by detecting the presence of known secondary metabolites. Frisvad et al. used high-performance liquid chromatography (HPLC) diode array detection and flow injection analysis together with electrospray ionization mass spectrometry (ES-MS) to detect secondary metabolites characteristic of fungal strains responsible for spoilage of stored cereals (7, 8, 14). Feistner employed HPLC–ES-MS to determine metabolic profiles of strains of the bacterial genus Erwinia (6). In our laboratories, Julian et al. developed an HPLC–ES-MS system with automatic data processing that compares crude fermentation extracts via an overall quantitative mixture similarity measure (10).
The objective of this study was to investigate a rapid ES-MS method to estimate the production of secondary metabolites in fermentation extracts. If throughput were not a consideration, an HPLC-evaporative light scattering detection (ELSD) analysis would provide a quasi-universal estimate of the number of secondary metabolites contained in an extract. ELSD is preferred due to the nonselective nature of the detection of compounds. Our goal was to identify a rapid ES-MS surrogate method for secondary metabolite production whose results correlate with the results of HPLC-ELSD analysis and provide some chemical information concerning the molecular and fragment ions of secondary metabolites. With a rapid estimator of secondary metabolite productivity, large collections of fermentation extracts could be characterized. Such a characterization would be a useful tool for identifying growth conditions that support synthesis of secondary metabolites. A tool for rapid characterization of extracts would also be a useful survey tool for identifying productive organisms for more detailed chemical analysis, purification, and biological screening.
MATERIALS AND METHODS
Organisms and cultivation.
Broth cultures of 43 actinomycetes (17) were started from cryogenic stocks in a vegetative medium that contained (per liter) 30 g of tryptic soy broth (Difco Laboratories, Detroit, Mich.), 3 g of yeast extract (Sigma Chemical Co., St. Louis, Mo.), 2 g of MgSO4, 5 g of glucose, and 4 g of maltose. At the mid-log phase, 60 μl (∼0.4% inoculum) of each vegetative culture was transferred into 12 ml of a complex growth medium containing (per liter) 10 g of glucose, 40 g of potato dextrin (Avedex, Keokuk, Iowa), 15 g of cane molasses (Cargill, Minneapolis, Minn.), 10 g of Hy-case amino (Sheffield Products, Norwich, N.Y.), 1 g of MgSO4, and 2 g of CaCO3. The fermentation vessel consisted of a rectangular Axid (Eli Lilly and Co., Indianapolis, Ind.) polypropylene bottle that was approximately 3.5 cm long by 4.25 cm wide by 6 cm high. The closure used for each small shake flask fermentation bottle consisted of a γ-irradiated, vented polypropylene cap lined with a gas-permeable membrane (Performance Systematix, Inc., Caledonia, Mich.). Submerged fermentations were incubated at 30°C for 7 days on an orbital shaker (stroke length, 2 in.) at 110 rpm. Fermentations were prepared for chemical analysis by solublization and extraction of secondary metabolites by the addition of an equal volume of absolute ethanol to the fermentation vessels. Fermentation vessels containing ethanol were agitated for 2 h on an orbital shaker (stroke length, 2 in.) at 200 rpm, and then the contents were allowed to settle for 16 h at 4°C. The ethanol extracts were then filtered through a 100-μm-pore-size polypropylene screen and transferred to 96-well plates for solid-phase extraction.
Extract bioassay.
Gram-positive bactericidal bioassays were performed by applying 6-μl portions of ethanol-solublized fermentation broth to 0.25-in.-diameter sterile paper discs. The dried discs were placed on seeded agar assay plates containing Micrococcus luteus ATCC 9341 (Food and Drug Administration strain PCI1001) and were incubated for 2 days at 37°C.
SPE of actinomycete extracts.
Nonpolar components were enriched from 0.7-ml samples of the actinomycete extracts by SPE on Empore octadecyl SD high-performance extraction disc plates (3M Company, St. Paul, Minn.). The SPE stationary phase was conditioned for use by sequential washing with 2 ml of distilled H2O, with 2 ml of methanol, and finally with 2 ml of 1 mM ammonium acetate (pH 5.5) in distilled H2O. The ethanol was removed from 0.7-ml samples under a vacuum (−80 kPa) at 20°C for 16 h. The residual aqueous material was resuspended to a total volume of 0.5 ml with 1 mM ammonium acetate (pH 5.5) and loaded on the conditioned SPE stationary phase. The SPE stationary phase was washed with 5 ml of 1 mM ammonium acetate (pH 5.5), and the fraction analyzed, enriched for relatively nonpolar metabolites, was eluted with 0.7 ml of a 70% acetonitrile–30% (vol/vol) methanol solution which contained 6.5 mM ammonium acetate (pH 5.5). The secondary metabolite-containing fraction was transferred into microwell plates for chemical analyses.
Chemical analyses.
Chemical analysis of SPE eluates was performed by (HPLC–ES-MS–ELSD) as described by Julian et al. and by direct-infusion ES-MS. For the HPLC–ES-MS–ELSD experiments, ethanol-solublized analytes (50 μl) were separated on a Novapak C18 analytical column (3.9 by 150 mm) by using a 30-min linear gradient from 2% methanol–15 mM ammonium acetate to 95% methanol–15 mM ammonium acetate at a flow rate of 710 μl min−1. The column effluent was split between a Finnigan Navigator (160 μl min−1) equipped with a Finnigan electrospray source and a Sedex 55 evaporative light scattering detector (570 μl min−1; Sedere, Alfortville, France) in order to provide qualitative and quantitative data, respectively. The lower limit of detection for the evaporative light scattering detector was estimated to be approximately 2 μg/ml based on dilution of a standard mixture. UV and visible light absorption spectra (220 to 550 nm) were acquired with a Hewlett-Packard series 1100 photodiode array spectrophotometer by using column effluent prior to analysis by evaporative light scattering. The electrospray source was switched between positive ion mode and negative ion mode at 0.4-s intervals to acquire both positive and negative ion spectra. During data acquisition, the mass spectrometer probe voltage was maintained at 3.6 kV, the cone voltage was maintained at 35 V, the source temperature was kept at 180°C, and the drying gas flow rate was 500 liters · h−1. A standard solution consisting of 2-amino-6-chloropurine (50 μg/ml), caffeine (50 μg/ml), m-cresol purple (50 μg/ml), tylosin (25 μg/ml), and spinosyn A (50 μg/ml) was injected at the beginning of the analysis and then after every 10th experimental sample to assess instrument stability and performance.
Direct-infusion ES-MS was performed with SPE-treated samples by using a Finnigan Navigator mass spectrometer equipped with a Finnigan electrospray source. The mass spectrometer parameters were identical to those described above for the HPLC–ES-MS analysis. Exactly 20 μl of a sample was injected into the carrier gas stream of the electrospray source at a flow rate of 160 μl min−1 through a Gilson 215 liquid handler and a Gilson 819 injection valve actuator by using a Shimadzu LC-10ADVP liquid chromatograph pump. The standard solution described above was injected at the beginning of the analysis and after every 10th experimental sample to assess instrument stability and performance. The time between injections was approximately 60 s.
Data collected by the mass spectrometer data system were converted to the netCDF file format (15) and transferred to a Sun compute server for analysis. The C programming language was used to automate execution of the algorithms described in this paper.
RESULTS AND DISCUSSION
Analytical HPLC and collection of fractions of biologically active fermentation extracts are routinely done by natural product drug discovery groups to identify the active component(s) in a complex mixture (1). We coupled these familiar analytical techniques with automated algorithms to identify organisms that produced secondary metabolites under specific growth conditions. In this study we analyzed 43 actinomycete extracts by using a 30-min HPLC-ELSD method to identify the numbers and relative amounts of secondary metabolites present in ethanol extracts of fermentation broth. SPE of ethanol extracts on C18 activated silica was employed to reduce the polar components, presumably including salts, cellular macromolecules, spent medium components, and other cellular debris present in the ethanol extracts. Despite some influence of solvent concentration and the molecular structure of an analyte, ELSD is generally considered a quasi-universal detector of the quantity of an analyte in a sample (3, 9). For the growth conditions considered here, we observed a broad range of apparent secondary metabolite production in organisms. Some extracts contained no detectable metabolites (Fig. 1d), while others contained many (Fig. 1f). In order to monitor detector sensitivity and column performance, a standard mixture consisting of five known compounds at known concentrations was injected between sets of 10 extracts. The mean natural log (loge) of the area under the HPLC-ELSD chromatogram for the control injections was 11.0 with a coefficient of variation (COV) of 1.3%, indicating that sufficient reproducibility could be achieved in automated analyses of relatively complex samples. Because of the exponential nature of ELSD, a log transform was done in order to work in units that are linearly related to the log of the concentration of a solute (3). While the 30-min HPLC-ELSD method is generally effective for identifying the numbers and approximate concentrations of secondary metabolites present in fermentation extracts, the throughput is not sufficient for studies in which assessment of larger numbers of organisms is required. Additionally, the HPLC-ELSD method provides no information regarding the chemical composition of the metabolites present in an extract. For these reasons we investigated a higher-throughput ES-MS method that we believed had the potential to be a surrogate for the HPLC-ELSD method for estimating secondary metabolite production. In addition to serving as a rapid surrogate for the HPLC-ELSD method, the mass fragments from the rapid ES-MS method provided information about the chemical compositions of compounds in an extract.
FIG. 1.

Positive ion ES-MS spectra and HPLC-ELSD chromatograms for the actinomycete extract with the minimum positive ES-MS productivity index (1,607) (a and d), the actinomycete extract with the positive ES-MS productivity index nearest the median (2,277) (b and e), and the actinomycete extract with the maximum positive ES-MS productivity index (5,852) (c and f).
Analysis of direct-infusion ES-MS data and algorithm description.
We found that simple summation of peak intensities in the ES-MS spectra determined for the extracts was only marginally effective at predicting the productivities of broths as benchmarked against the HPLC-ELSD analyses (data not shown). In response, we examined preprocessing steps to improve the prediction. Mass spectra are commonly binned by partitioning the m/z range into fixed, equal-size partitions and assigning each ion intensity to the appropriate partition or bin. The sum of all ion intensities that fall within each m/z bin is used as the intensity value for the bin. This simple binning scheme works well as long as the m/z values of ions do not lie near a bin boundary. For ions with m/z values near a bin boundary, small differences in the m/z values estimated by mass spectrometry can place the same ion from two samples into different bins, which results in errors when two binned spectra are compared. To address this problem, an overlapping binning scheme was developed in order to assign the intensity of an ion to two bins if the m/z value was within an overlap region centered at the bin boundary. The proportion of intensity assigned to each bin for an ion falling within an overlap region was scaled to be linearly proportional to the difference between the measured m/z value and the bin boundary. Thus, all examples presented in this paper are based on spectra that were preprocessed by binning the measured intensities into 5,800 m/z bins between 150 and 1,600 atomic mass units (amu) with a 0.2-amu bin overlap region forming a vector of fixed length for each extract analyzed. This transformation of raw data to vectors allowed spectral averaging and background subtraction in order to obtain an improved estimate of fermentation productivity.
A background corrected mass spectrum was computed by averaging all of the binned spectra within a selected signal window and subtracting the average of the binned spectra within a defined background window. For the ES-MS method, the signal window of interest included scans detected between 0.15 and 0.60 min after injection. The background region was defined by scans obtained between 0.0 and 0.10 min after injection.
Fermentation growth media often contain numerous complex components to support growth and expression of secondary metabolites. Many times these medium components are not fully consumed and may therefore remain in broth media at harvest time. In order to reduce any systematic bias introduced by medium components in our analysis, the averaged and background subtracted spectrum from an uninoculated medium blank was subtracted from the binned, background subtracted mass spectrum of each extract. Ion counts less than zero following this subtraction were given a value of zero. This medium subtraction strategy works well for unaltered medium components in an extract but would not work well for components transformed by an organism.
Following background and medium subtraction, the productivity of an extract was estimated by examining the number and intensity of ions in the preprocessed spectrum. Extracts containing more secondary metabolites were observed to have more ions present in their preprocessed mass spectra (Fig. 1). Metabolites present at higher concentrations were observed to produce larger ion signals. We believe that any measure of overall productivity should consider both the number of ions and their intensity. A simple ion counting procedure was developed to capture information about the number and intensity of the ions detected. For each extract, the number of ions with intensities above an intensity threshold value was recorded. The threshold was then increased, and the number of ions with intensities above the new threshold value were added to the initial count. This process was repeated until the threshold value reached a specified maximum value. This procedure is represented graphically in Fig. 2. For the productivity indices reported for the extracts described here we used 1,000 intensity threshold values between a minimum intensity threshold value of zero and a maximum intensity threshold value of 106 counts. Analysis after repeated injections of the growth medium indicated that a six- to sevenfold decrease in productivity index for the growth medium was obtained when medium subtraction was included. Productivity indices were computed independently for positive ion mass spectra and negative ion mass spectra. The mathematical manipulations of mass spectra described above were coded into a C language computer program to facilitate automated, high-throughput analysis of spectra.
FIG. 2.

Background- and medium-subtracted positive ion ES mass spectrum of a solid-phase actinomycete extract. The productivity index was computed by adding all of the ions above a series of ion count thresholds. In this example 12 thresholds are shown as horizontal dotted lines, resulting in a productivity index of 57.
System reproducibility.
Mass spectrometer sensitivity was monitored throughout the analysis of experimental samples due to the potential for variable sensitivity caused by the plugging and coating of surfaces in the instruments. Detector sensitivity was monitored by bracketing sets of 10 extract injections with injections of the standard mixture. The background-corrected positive ion mass spectrum of the standard mixture (Fig. 3) had a mean positive ion productivity index of 3,490 with a COV of 7.5% (n = 12). The mean negative ion productivity index for the control injections was 59.6 with a COV of 19.7% (n = 12). The higher variability observed in the negative ion mode was attributed to the overall lower sensitivity of the mass spectrometer in the negative ion detection mode. For large studies conducted with multiple instruments or over long periods of time, the productivity indices for the bracketing controls may be used to scale the productivity indices of extracts.
FIG. 3.

Direct-infusion positive ES-MS of control mixture containing 2-amino-6-chloropurine (50 μg/ml), caffeine (50 μg/ml), m-cresol purple (50 μg/ml), tylosin (25 μg/ml), and spinosyn A (50 μg/ml). [M+H]+ ions and adduct ions are shown for each analyte in the control mixture.
Comparison of the rapid ES-MS and HPLC-ELSD methods.
Examination of the positive ion mass spectrum and its associated HPLC-ELSD chromatogram for the extracts with minimum, median, and maximum positive ion productivity illustrated the visual correlation between the positive ion productivity and the HPLC-ELSD chromatogram (Fig. 1). To quantitatively assess this correlation, the natural log of the area under the HPLC-ELSD chromatogram was computed for each solid-phase extract and compared to positive and negative ion productivity indices via Pearson's correlation coefficient. Additionally, the positive and negative ion productivity indices were combined into a single composite productivity index by first linearly scaling the positive and negative ion productivity indices to be on the [0, 1] scale and then adding the two scaled indices. Scaling was required due to the large differences in productivity index values between the positive and negative modes. Pearson correlation coefficients for comparisons between the positive ion, negative ion, and combined productivity indices and the loge HPLC-ELSD areas under the curve are shown in Table 1. A 90% confidence interval for each correlation coefficient was estimated by using the bias-corrected and accelerated (BCa) method (10,000 bootstrap samples) described by Efron and Tibshirani (4). The bootstrap method is used as an alternative to Fisher's transformation of the correlation coefficient for estimating confidence intervals (mean ± 1.96 × standard error of the transformed data). Bootstrap confidence intervals are estimated by drawing many random samples (with replacement) from the original data, computing the correlation coefficient for each random sample, and then examining the distribution (histogram) of the correlation coefficients. Confidence intervals estimated with the bootstrap method are essentially identical to those estimated by using Fisher's transformation. The combined productivity index correlated best with the HPLC-ELSD measure of secondary metabolite productivity with a correlation coefficient (rc) of 0.78 (Fig. 4). While the positive and negative ion productivity indices had correlation coefficients that were approximately equal (r+ = 0.72 and r− = 0.70, respectively), the length of the 90% confidence interval for the correlation coefficient was much narrower for positive ion data (0.24 for the positive ion data and 0.51 for the negative ion data). A bootstrap hypothesis testing procedure (4) using 10,000 bootstrap samples was used to test the one-sided hypotheses:
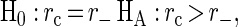 |
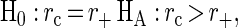 |
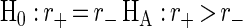 |
Based on these hypothesis tests, the combined productivity index appears to be better than the negative and positive ion productivity indices at the approximate achieved significance levels of 0.13 and 0.21, respectively (Table 2). The approximate significance levels were estimated by dividing the number of bootstrap samples for which the alternative hypothesis was false by the total number of bootstrap samples. There appeared to be no significant difference between the correlations of the positive and negative ion productivity indices with the HPLC-ELSD benchmark (approximate achieved significance level, 0.40). A direct-infusion ES-MS analysis of the crude extracts, bypassing the SPE step, was done in order to assess the correlation of a simplified sample preparation method with an HPLC-ELSD benchmark. The correlation coefficient for comparisons between the combined positive and negative ion productivity indices for the non-SPE approach and the HPLC-ELSD benchmark (rc,nospe) was 0.68, a value somewhat lower than the correlation coefficient obtained when the SPE step preceded the ES-MS analysis. A one-sided test of the hypothesis H0: rc = rc,nospe, HA: rc > rc,nospe was rejected at the approximate achieved significance level of 0.08 with 10,000 bootstrap samples, indicating only marginal improvement with SPE.
TABLE 1.
Correlation coefficients and their 90% BCa bootstrap confidence intervals for comparisons between r+, r−, and rc and loge HPLC-ELSD area under the curve
| Method | Correlation coefficient | 90% BCa confidence interval |
|---|---|---|
| r− | 0.70 | [0.40, 0.91] |
| r+ | 0.72 | [0.57, 0.81] |
| rc | 0.78 | [0.63, 0.91] |
FIG. 4.

Area under the HPLC-ELSD chromatogram versus the combined positive and negative ion productivity index for 43 actinomycete SPE extracts. The correlation coefficient is 0.78 with a 90% BCa bootstrap confidence interval of [0.63, 0.91].
TABLE 2.
Summary of bootstrap hypothesis testing procedure comparing the surrogate secondary metabolite indices to the HPLC-ELSD measure of productivitya
| Hypothesis | Approximate achieved significance level (P value) |
|---|---|
| H0: rc = r− | 0.13 |
| HA: rc > r− } | |
| H0: rc = r+ | 0.21 |
| HA: rc > r+} | |
| H0: r+ = r− | 0.40 |
| HA: r+ > r−} |
A total of 10,000 bootstrap samplings were performed.
A bioassay approach to estimating the production of secondary metabolites is one alternative to the chemical screening methods considered in this study. For example, an antimicrobial bioassay or a battery of antimicrobial assays could be used to identify organisms producing antibacterial secondary metabolites. A major disadvantage of a bioassay profiling approach is that estimation of secondary metabolites is restricted to organisms producing compounds active in the bioassay used and may be dominated by common antibacterial metabolites. The rapid ES-MS chemically based profiling approach is more general in that its only restriction is that compounds must be detectable by either positive or negative ion electrospray ionization. To compare the effectiveness of an antimicrobial measure of secondary metabolite production, gram-positive antibiotic bioassays were performed for each of the actinomycete extracts. Eleven of the 43 actinomycete extracts exhibited non-zero zones of inhibition (data not shown). The correlation coefficient for a comparison between zone of inhibition and the HPLC-ELSD benchmark was 0.25, suggesting that several extracts contained secondary metabolites that exhibited no detectable antimicrobial activity in a single plate assay. It should be emphasized that only one bioassay was conducted for illustration purposes. A higher correlation coefficient might be obtained with a battery of antimicrobial assays, although in practice, this technique would be limited to detecting only metabolites with antimicrobial activity.
Collection of microbial cultures by pharmaceutical research organizations generally results in large numbers of uncharacterized organisms. While some biological classification is useful, direct assessment of secondary metabolites provides highly relevant information for drug discovery applications. Here we report on a rapid ES-MS method (∼50 samples/h) to estimate the presence of secondary metabolites produced by microorganisms. The effectiveness of this rapid method was assessed relative to the effectiveness of HPLC-ELSD quantitation of secondary metabolites contained in ethanol extracts from 43 different actinomycetes. We observed generally good agreement between an HPLC-ELSD method for quantifying secondary metabolite production and the approximately 25-fold-more-rapid ES-MS method (correlation coefficient for combined positive and negative ion ES-MS data, 0.78). Resampling-based hypothesis testing procedures indicated that the combined ES-MS productivity index is better than the individual positive or negative ionization productivity indices with apparent significance levels of 0.13 and 0.21, respectively. Monitoring periodic injections of a control mixture revealed that extracts from microbial fermentation can be analyzed reliably by a high-throughput mass spectrometry system.
The potential applications of a rapid estimator of secondary metabolite production in modern natural product drug discovery are severalfold. Researchers interested in identifying secondary metabolite-eliciting growth conditions for a large, uncharacterized collection of microorganisms may find this rapid tool useful for evaluating multiple growth conditions with statistically large numbers of microorganisms (17). Given a defined set of growth conditions, this tool may also prove to be useful for identifying particularly productive microorganisms for the construction of a screening library. Organisms classified as unproductive by this method could be recycled for additional growth condition development to fully realize the potential of a culture collection for drug discovery applications.
ACKNOWLEDGMENTS
We thank Mike Goodwin, John Scheuring, Dale Duckworth, and Matt Clemens for insightful discussions and for analysis of actinomycete extracts by ES-MS and HPLC–ES-MS–ELSD. We are also grateful to Steve Larsen for assistance with SPE of the actinomycete fermentations.
REFERENCES
- 1.Cordell G A, Shin Y G. Finding the needle in the haystack. The dereplication of natural product extracts. Pure Appl Chem. 1999;71:1089–1094. [Google Scholar]
- 2.Cragg G M, Newman D J, Snader K M. Natural products in drug discovery and development. J Nat Prod (Lloydia) 1997;60:52–60. doi: 10.1021/np9604893. [DOI] [PubMed] [Google Scholar]
- 3.Dreux M, Lafosse M, Morin-Allory L. The evaporative light scattering detector: a universal instrument for non-volatile solutes in LC and SFC. LC-GC Int. 1996;9:148–153. [Google Scholar]
- 4.Efron B, Tibshirani R J. An introduction to the bootstrap. New York, N.Y: Chapman and Hall; 1993. [Google Scholar]
- 5.Erhard M, von Dohren H, Jungblut P. Rapid typing and elucidation of new secondary metabolites of intact cyanobacteria using MALDI-TOF mass spectrometry. Nat Biotechnol. 1997;15:906–909. doi: 10.1038/nbt0997-906. [DOI] [PubMed] [Google Scholar]
- 6.Feistner, G. J. 1994. Profiling of bacterial metabolites by liquid chromatography-electrospray mass spectrometry: a perspective. Am. Lab. (Fairfield, Conn.) 1994(September):32L–32Q.
- 7.Filtenbort O, Frisvad J C, Svendsen J A. Simple screening method for molds producing intracellular mycotoxins in pure cultures. Appl Environ Microbiol. 1983;45:581–585. doi: 10.1128/aem.45.2.581-585.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frisvad J C, Filtenborg O, Thrane U. Analysis and screening for mycotoxins and other secondary metabolites in fungal cultures by thin-layer chromatography and high-performance liquid chromatography. Arch Environ Contam Toxicol. 1989;18:331–335. doi: 10.1007/BF01062357. [DOI] [PubMed] [Google Scholar]
- 9.Guichon G, Moysan A, Holley C. Influence of various parameters on the response factors of the evaporative light scattering detector for a number of non-volatile compounds. J Liq Chromatogr. 1988;11:2547–2570. [Google Scholar]
- 10.Julian R K, Higgs R E, Gygi J D, Hilton M D. A method for quantitatively differentiating crude natural extracts using high-performance liquid chromatography-electrospray mass spectrometry. Anal Chem. 1998;70:3249–3254. doi: 10.1021/ac971055v. [DOI] [PubMed] [Google Scholar]
- 11.Monaghan R L, Polishook J D, Pecore V J, Bills G F, Nallin-Omstead M, Streicher S L. Discovery of novel secondary metabolites from fungi — is it really a random walk through a random forest? Can J Bot. 1995;73(Suppl. 1):S925–S931. [Google Scholar]
- 12.Schmid I, Sattler I, Grabley S, Thiericke R. Natural products in high throughput screening: automated high-quality sample preparation. J Biomol Screening. 1999;4:15–25. doi: 10.1177/108705719900400104. [DOI] [PubMed] [Google Scholar]
- 13.Shu Y. Recent natural products based drug development: a pharmaceutical industry perspective. J Nat Prod (Lloydia) 1998;61:1053–1071. doi: 10.1021/np9800102. [DOI] [PubMed] [Google Scholar]
- 14.Smedsgard J, Frisvad J C. Using direct electrospray mass spectrometry in taxonomy and secondary metabolite profiling of crude fungal extracts. J Microbiol Methods. 1996;25:5–17. [Google Scholar]
- 15.Stranz D, Campbell S, Christopher R, Watt J, Zakett D. Data interchange for mass spectrometry, version 1.0.1. 1993. Analytical Instrument Association. [Google Scholar]
- 16.Yarbrough G G, Taylor D P, Rowlands R T, Crawford M S, Lasure L L. Screening microbial metabolites for new drugs: theoretical and practical issues. J Antibiot. 1993;46:535–544. doi: 10.7164/antibiotics.46.535. [DOI] [PubMed] [Google Scholar]
- 17.Zahn J A, Higgs R E, Hilton M D. Use of direct-infusion electrospray mass spectrometry to guide empirical development of improved conditions for expression of secondary metabolites from actinomycetes. Appl Environ Microbiol. 2001;67:377–386. doi: 10.1128/AEM.67.1.377-386.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]