

Abstract
Although the sonic hedgehog (SHH) signaling pathway has been implicated in promoting malignant phenotypes of prostate cancer, details on how it is activated and exerts its oncogenic role during prostate cancer development and progression is less clear. Here, we show that GLI3, a key SHH pathway effector, is transcriptionally upregulated during androgen deprivation and posttranslationally stabilized in prostate cancer cells by mutation of speckle-type POZ protein (SPOP). GLI3 is a substrate of SPOP-mediated proteasomal degradation in prostate cancer cells and prostate cancer driver mutations in SPOP abrogate GLI3 degradation. Functionally, GLI3 is necessary and sufficient for the growth and migration of AR positive prostate cancer cells, particularly under androgen-depleted conditions. Importantly, we demonstrate that GLI3 physically interacts and functionally cooperates with AR to enrich an AR-dependent gene expression program leading to castration resistant growth of xenografted prostate tumors. Finally, we identify an AR/GLI3 co-regulated gene signature that is highly correlated with castration resistant metastatic prostate cancer and predictive of disease recurrence. Together, these findings reveal that hyperactivated GLI3 promotes castration-resistant growth of prostate cancer and provide a rationale for therapeutic targeting of GLI3 in CRPC patients.
Introduction
Prostate cancer is the most common malignancy and the second leading cause of cancer-related deaths among US men (1). Although advances in early diagnosis and treatment have rendered localized prostate cancer highly curable, more than 32,000 men succumb annually to metastatic disease (2–4). Although first-line androgen deprivation therapy triggers a rapid response in men with metastatic prostate cancer, most patients will progress to lethal castration resistant prostate cancer (CRPC), characterized by androgen receptor (AR)-dependent tumor growth despite castrate levels of circulating androgens (2–6). Mechanisms proposed to underlie CRPC are multifactorial in nature, likely to reflect the unique molecular constitution of individual prostate tumors, and include, but are not limited to, tumor-specific expression of ligand-independent AR isotypes, intratumoral androgen production, and stimulation of AR activity via crosstalk with alternative signaling pathways (7,8). Among the latter, the Sonic hedgehog (SHH) pathway, for which inhibitors are currently approved or in clinical trials for a variety of cancers, is a potential target in CRPC (9).
The SHH pathway is a preeminent developmental signaling circuit essential for developmental morphogenesis and adult tissue homeostasis in a variety of organs and systems, including the prostate (10–12). SHH signaling arises from the specification of unique gene expression programs dependent upon key nuclear effectors of the GLI transcription factor family. In vertebrates, three GLI family members, GLI1–3, subsume the functions of a single ancestral GLI homolog, Cubitus interuptus (Ci), originally identified in invertebrates (13). Whereas GLI1 functions primarily as an activator and downstream signal amplifier, GLI2 and GLI3 more closely resemble Ci as proximal signal effectors and functionally bipartite regulators with the ability to directly repress and activate their target genes, including GLI1, in the absence and presence of SHH, respectively (14–16). In the absence of SHH, its transmembrane receptor Patched (PTCH) inhibits the SHH transducer Smoothened (SMO), a transmembrane G-protein-coupled receptor (17). This inhibition promotes phosphorylation-dependent proteolytic processing of GLI2/GLI3 to N-terminal repressors (18). Binding of SHH to PTCH liberates SMO from inhibition, triggering the accumulation and translocation of full-length GLI2/3 into the nucleus whereupon their site-specific phosphorylation converts them into labile transcriptional activators (17,18). SHH thus dictates the transcriptional output of a responsive cell by altering the ratio of GLI2/3 repressor to activator species.
Notably, SHH signaling has been implicated in castration resistant prostate cancer through a functional interplay with AR (19–21). In this regard, we and others have previously shown that androgen deprivation induces SHH signaling, which reciprocally activates AR-dependent gene expression and prostate cancer cell growth in the absence of androgens (22–24). Mechanistically, this crosstalk occurs through autocrine and/or paracrine SHH signaling in the tumor microenvironment and appears to involve a direct interaction between AR and GLI proteins leading to the stabilization of full-length GLI proteins (19,21,25,26). Together, these findings suggest that activated SHH signaling, in response to androgen deprivation, could support reactivation of AR signaling and progression to CRPC. Herein, we show that this scenario is particularly germane to a prostate cancer subtype supporting hyperactivated GLI3-dependent SHH signaling through mutations in SPOP (Speckle-type POZ protein), a suppressor of the SHH signaling axis (27–29).
Recurrent somatic mutations in SPOP are the most frequent non-synonymous mutations in primary prostate cancer, occurring in up to 15% of cases, and show mutual exclusivity with ETS rearrangements, suggesting that SPOP mutations anchor a distinct genetic subtype (27,28,30). SPOP is a substrate binding subunit of a Cullin-based E3 ubiquitin ligase (27,31). Notably, prostate cancer-associated mutations in SPOP cluster in its substrate-binding groove and abolish substrate binding (27,31). Loss of SPOP expression is also frequently observed in prostate cancer, suggesting that SPOP is a prostate tumor suppressor whose inactivation through somatic mutation or diminished expression drives tumorigenesis (31,32). Established SPOP substrates thus far linked with prostate cancer include AR, the steroid receptor co-activator SRC-3, the pro-apoptotic gene DDIT3, the bromodomain and extraterminal repeat (BET) proteins BRD2–4, and the oncogenes DEK, ERG, and TRIM24 (33–44). In addition, SPOP, through regulation of unknown effector substrates, has been implicated in control of homology directed DNA double-strand break repair and maintenance of genomic integrity; accordingly, oncogenic SPOP mutations disrupt error-free homology-directed DNA repair and instead promote repair through error-prone non-homologous pathways, providing a molecular explanation for high-frequency genomic rearrangements observed in primary SPOP-mutant tumors (45). However, the role of SPOP and the identity of its biologically relevant substrates in mediateing androgen-independent growth of prostate cancer cells are comparably poorly understood.
Herein, we identify GLI3-dependent SHH signaling to be a key driver of prostate cancer cell growth and tumor formation and further show that oncogenic SPOP mutations stabilize GLI3 in its full-length activator form. Mechanistically, we show that pathologically stabilized GLI3 physically interacts and functionally cooperates with AR to enrich an AR-dependent gene expression program. Finally, we identify an AR/GLI3 co-regulated gene signature that correlates with castration resistant metastatic prostate cancer and predicts disease recurrence. Altogether, our findings reveal that hyperactivated GLI3 drives castration-resistant growth of prostate tumors, suggesting that GLI3 inhibitors might prove effective to block CRPC.
Materials and Methods
Animal models.
All animal experiments were performed with approval of the Institutional Animal Care and Use Committee at UT Health San Antonio. Male NSG mice (NOD.Cg-PrkdcScidIL2rgtm1Wjl /SzJ, Stock No. 005557, The Jackson Laboratory, Sacramento, CA) were used for subcutaneous inoculation of LNCaP cells with or without GLI3 knockdown. Male nude mice (Hsd:Athymic Nude-Foxn1nu, Envigo, Frederick, MD) were used for orthotopic inoculation of PacMetUT1 cells with or without Gli3 knockdown.
Cell Lines and cell culture.
All cell lines used in these studies were confirmed to be Myocplasma-free using either the MycoAlert™ Mycoplasma Detection Kit (Lonza) or the Venor™ GeM Mycoplasma Detection Kit (MilliporeSigma). Human prostate cancer cell lines LNCaP, PC-3, 22Rv-1, and C4–2 and were obtained from the American Type Culture Collection (ATCC, Manassas, VA), and authenticated with the DNA markers used by ATCC. The PacMetUT1 cell line was previously described (46). LNCaP and 22Rv-1 cell lines were cultured as described previously (23). Androgen-independent LNCaP and 22Rv-1cells, which were called LNCaP AI and 22Rv-1 AI, were also cultured as described (23). PC-3, C4–2, LNCaP SPOP WT, and LNCaP SPOP F102C cells were cultured at 37°C and 5% CO2 in RPMI 1640 media (Corning Cellgro) supplemented with 10% fetal bovine serum (HyClone) and penicillin-streptomycin-L-glutamine (Invitrogen). For androgen-deplete conditions cells were cultured in RPMI 1640 media without phenol red (Corning Cellgro) supplemented with 10% charcoal stripped fetal bovine serum (Fisher Scientific) and penicillin-streptomycin-L-glutamine (Invitrogen).
Plasmids.
Plasmids pact-FLAG-GLI3 for mammalian expression of FLAG epitope-tagged human GLI3 (14) and those for HA-tagged ubiquitin and MYC-tagged SPOP derivatives (34) have been described previously. Full-length GLI3 cDNA was cloned into PCS2+ vector for in vitro translation. GLI3 truncations for in vitro translation were generated by cloning the following GLI3 fragments (nucleoride coordinates) into PCS2+: NTD (1–1437), DBD (1438–1896), CTD (1897–4744). GST-AR plasmids used for GST pulldown experiments were generated by cloning the following AR fragments (nucleoride coordinates) into pGEX4T1 vector: NTD (1–1675,) DBD (1517–1908), LBD (1980–2764). GST-SPOP plasmids used for GST pulldown experiments were subcloned from pCMV plasmids into pGEX6P1 vector. pLKO.1-based shRNA expression vectors for SPOP and GLI3 knockdown experiments were purchased from Sigma. The sequences are listed in Supplementary Table S7. GLI responsive promoter-luciferase reporter (Gli-Luc) and CMV promoter-driven β-galactosidase expression plasmids were kind gifts of Dr. Hollie Swanson. Plasmid pLV411G effLuc-flag (IRES-hrGFP) for stable expression of luciferase and GFP in prostate cancer cells was a kind gift of Dr. Brian Rabinovich.
Transfections and MG132/Cyclopamine/DHT treatment.
For all transfections, cells were seeded 24 hours prior to transfection (∼70% cell confluence upon transfection). 293T cells were transfected with XtremeGene9 transfection reagent (Roche) at a 1:3 ratio and cells were harvested 48 hours post-transfection. LNCaP and PC-3 cells were transfected with GenJet Plus transfection reagent (SignaGen Laboratories) at a 1:3 ratio and cells were harvested 24–48 hours post-transfection. MG132 (Enzo Life Sciences) treatment was always performed using 15 or 20 μM as specified in experiments for 18 hours, cyclopamine (Sigma) treatment at 10 μM for 24 hours, and DHT (gift of K. Xu) treatment at 10 nM for 16 hours.
Generation of model cell lines.
Generation of stable GLI3 knockdown cell lines: pLKO.1 vector containing no shRNA sequence (CTL KD) or either GLI3 KD1 or GLI3 KD2 shRNAs were transfected into HEK 293T cells together with two other plasmids, psPAX2 and pMD2.G, that were purchased from Invitrogen (Grand Island, NY) for lentivirus packaging. Viral supernatants were harvested for infection of LNCaP, LNCaP AI, 22Rv.1 AI and PacMetUT1 cell lines. Infected cells were selected with puromycin (2 mg/ml). Generation of GFP- and Luciferase-expressing stable GLI3 knockdown cell lines: Parental stable GLI3 knockdown cells were infected with a lentivirus generated by 293T cell-packaged pLV411G effLuc-flag (IRES-hrGFP) containing the enhanced green fluorescent protein (GFP) and Luciferase (Luc). Generation of SPOP F102C LNCaP cell lines: SPOP mutant LNCaP cell lines were generated through CRISPR/Cas9 mediated genome editing. SPOP cDNA sequence consisting of exon 6 (containing the c.305 T>G mutation) and flanking upstream and downstream sequence (≈1 kb each) and including engineered and XhoI/EcoRI cut sites (to subclone a neomycin resistance gene) was synthesized and cloned into pBSK vector by Biomatik Corp. After insertion of the neomycin cassette (with flanking LoxP sites), plasmid DNA was linearized and transfected along with pX330 (containing SPOP-specific gRNA sequence: GACAAGTTGTGGCTTTGATC) into LNCaP cells. Targeted cells were selected with G418 (800 μg/ml) and seeded at low density to permit single cell colony formation. Positive colonies were confirmed by genomic DNA and cDNA sequencing and serially subcloned to establish monoclonality. To generate HA-GLI3 overexpressing LNCaP and C4–2 cell lines, HA-epitope tagged full-length GLI3 cDNA, generated by PCR using pACT-FLAG-GLI3 as a template, was sublconed into the lentiviral vector pLenti PGK puro GFPw509–5 (Addgene plasmid #19070) prior to transfection of 293T cells with pLenti PGK puro GFPw509–5-HA-GLI3 along with plasmids psPAX and pMD2.G. Viral supernatants were harvested for infection of LNCaP and C4–2 cells, and infected cells were selected with puromycin. GLI3-FLAG overxpressing LNCaP cells were generated by transfection of LNCaP cells with plasmid hGLI3-GLAG3x (Addgene plasmid #84921) followed by selection with G418 (800 μg/ml).
Animal studies.
Orthotopic and subcutaneous injections of PacMetUT1 and LNCaP Gli3 KD and control cells, respectively, as well as monitoring and measurement of tumor growth through bioluminescence imaging and volumetric determination followed our own previously published protocols (23). Detailed descriptions are provided in Supplementary Data.
Molecular and cell biological Analyses.
RNA isolation and RT-qPCR analyses, cell proliferation and migration, and soft agar and colony formation analyses were peformed essentially as described (23). Detailed descriptions are provided in Supplementary Data. Primer sets used for quantitative real-time PCR are listed in Supplementary Table S7.
RNA sequencing and analysis.
RNA was prepared as described for quantitative real-time PCR assays. RNA library prep and sequencing (on an Illumina HiSeq 2000 or 3000 with >32 million reads/sample) was performed at the UTHSCSA Genomics Sequencing Facility. All sequencing was done on duplicate independent experiments. All samples were aligned to the UCSC human genome build hg19 using TopHat2, and bam files from the alignment were processed using HTSeq-count to obtain the read counts per gene in all samples. Analysis of the read count data was performed using R version 3.3.3. Raw count data was first filtered to exclude genes that had only one or zero reads across all samples. The data was then transformed for visual inspection using regularized log2 transformation function of R package DESeq2 (47), version 1.14.1. Visual inspection was done by performing principal component analysis and by generating a Pearson’s correlation heat map using basic R functions, R package pheatmap and custom R scripts. Data normalization and differential expression (DE) were performed sing R package DESeq2, version 1.14.1. P values of the DE analysis were adjusted for multiple testing using Benjamini-Hochberg procedure. DE analysis results were annotated with gene biotypes, gene descriptions, and Ensembl identifiers using R package biomaRt. Statistically and biologically significant DE genes (DEGs) were defined by applying the following stipulations: adjusted p-values <0.05 and absolute log2 fold change ≥1 (corresponding to a minimal two-fold up- or down-regulation). Biological Process Gene Ontology (GO BP) term over-representation analyses were performed using R package clusterProfiler, with p-values calculated using the hypergeometric distribution. The p-values derived from enrichment analysis were corrected for multiple testing using Benjamini-Hochberg multiple testing of adjustment procedure. Enrichment results were filtered to include only terms that had a least three DEGs annotated to them, and redundant GO terms were excluded using the simply function fo the clusterProfiler package.
Gene set enrichment analysis.
Gene set enrichment analysis (GSEA) was performed using expressed genes, with RPKM (averaged over all cells) > 1, ranked based on average fold-change in control (Ctl. shR) compared to GLI3 knockdown (Gli3 shR1) cells. Significantly enriched Hallmark (MSigDB v7.1) gene sets (48) were identified using GSEA pre-ranked software (v. 4.0.3) with default settings (49). The Hallmark gene sets with false discovery rate (FDR) < 0.25 were identified as significantly enriched.
Survival analyses.
Microarray gene expression data reported in Taylor et al. (50) were retrieved from NCBI GEO database (accession number GSE21032) along with clinical annotation (primary or metastatic disease) from the analyzed tumor set. Expression values from 46 transcripts corresponding to each of the androgen and GLI3 co-upregulated genes (31 of the 32 genes in Fig. 6b as LGALS8-AS1 was not found in the dataset) were used for the unsupervised clustering of all 150 patients. Available biochemical recurrence (BCR) data from 140 patients were used to generate Kaplan-Meier (KM) survival plots using survival R package (51). The patients were categorised as high expression and low expression groups based on the hierarchical clustering (Fig. 6c). Log‐rank test was applied to identify significant difference of BCR free survival between patients from the two groups. Nine (Supplementary Table 6) of the 46 transcripts were found significantly (p-value < 0.05) upregulated in 19 metastatic tumors compared to 131 primary tumors. The expression values of the 9 transcripts for each patient were averaged. The patients with expression above and below the overall average expression of all 150 samples were classified as high and low expression groups, respectively, and were used for the BCR free survival analysis The KM survival plots and log‐rank test was performed in the same way described above.
Figure 6. GLI3 and AR co-regulated genes correlate with metastatic CRPC and predict recurrence.

a Gene set enrichment analysis shows significant enrichment of Hallmark Androgen Response genes in control (CTL KD) compared to GLI3 knockdown (GLI3 KD1) LNCaP AI cells. b Venn diagram comparison of genes found significantly upregulated by ≥ 2 fold in androgen replete (+DHT) compared to androgen-depleted (-DHT) LNCaP cells versus genes with significant ≥ 2 fold upregulation in control (CTL KD) compared to GLI3 knockdown (GLI3 KD1) androgen-depleted LNCaP cells. c Heatmap representation of unsupervised hierarchical clustering of 150 tumor samples derived from prostate cancer patients(50) based on log2 (expression) values of 46 transcripts (from 31 androgen and GLI3 co-upregulated genes). One of the 32 genes in Panel b, LGALS8-AS1, was not found in the gene expression data provided by Taylor et al.(50) and hence excluded. The heatmap was plotted using R based ‘gplots’ package with default parameters. Red, green, and pink bars indicate metastatic tumor, primary tumor, and primary tumor samples from patients with observed clinical metastasis, respectively. d Kaplan-Meier plots depicting BCR-free survival of 140 prostate cancer patients(50) stratified by expression clusters from panel (c). e Kaplan-Meier plots depicting BCR-free survival of 140 prostate cancer patients stratified by average expression of 9 transcripts (from 6 genes), which were found significantly (p-value < 0.05) upregulated in 19 metastatic tumors compared to 131 primary tumors. The expression values of the 9 transcripts for each patient were averaged. The patients with expression above and below the overall average expression of all 150 samples were classified as high and low expression groups, respectively.
GLI3 Immunohistochemitry.
Formalin-fixed paraffin-embedded (FFPE) samples from 86 patients with clinically localized prostate cancer were randomly selected from Mayo Clinic Tissue Registry under approval of the Mayo Clinic institutional review board (IRB). FFPE tissue blocks were sectioned at thickness of 20 µm for Sanger sequencing with SPOP-specific primers and 4 µm for IHC. The IHC protocol was followed as previously described (52) using Gli3 primary antibody (Novus, NBP2–29627; 1 mg/ml) at a dilution of 1:100 at 4°C overnight. Staining was developed with SignalStain® DAB Substrate Kit (Cell Signaling Technology, Cat# 8059). Staining intensity and staining percentage for each tissue was graded using set criteria into four categories: 0, 1, 2 and 3. Specifically, 0 = very week or no staining, 1 = staining obvious only at 40X, 2 = staining obvious at 10X but not 4X, and 3 = staining obvious at 4X. Stain percentage was graded 1 for 0%−33% positive cells, 2 for 34%−66%, and 3 for 67%−100%. The final staining index (SI) score for each staining was obtained by multiplying values obtained from staining percentage and intensity and used for correlation analysis. The IHC scoring was performed by two GU pathologists independently and blindly.
Immunoprecipitation and GST pulldown assays.
Immunoprecipitiaton of endogenous and ectopically expressed proteins as well as GST pull-down assays were peformed as described (53,54). Detailed descriptions are provided in Supplementary Data.
Quantification and statistical analysis.
All the experiment results are expressed as mean ± SEM. Two-tailed Student’s t-tests were used to compare two groups. One-way ANOVA was used for the comparison among more than two Groups followed by Tukey–Kramer post hoc test. A Mann–Whitney U test is used for comparisons made between any two groups of data within an experiment that are not normally distributed. Only a probability value of ≤0.05 was considered as statistically significant. All statistical analysis was performed with Excel or Prism 6 software of GraphPad (La Jolla, CA).
Data Availability Statement.
RNA Sequencing data is available in the NCBI Gene Expression Omnibus (GEO) repository under accession number GSE134682 (reviewer token: ahcroumuppgfveb).
Results
GLI3-dependent SHH signaling supports androgen-independent prostate cancer cell growth.
Prior studies have revealed that androgen deprivation induces SHH signaling, which in turn, supports prostate cancer cell growth in the absence of androgens (19,23–25). We confirmed induction of SHH signaling upon androgen withdrawal and maintenance of active SHH signaling following long-term androgen deprivation in human prostate cancer cell lines. Thus, short-term androgen deprivation in androgen sensitive LNCaP cells led to upregulated expression of SHH target genes that was maintained in the androgen-independent LNCaP clonal derivative C4–2, obtained following serial passage in castrated mice (Fig. 1a, b). As expected, enhanced expression of SHH target genes following androgen deprivation was reversed by cyclopamine, an inhibitor of the SHH transducer SMO (Fig. 1a, b). Cyclopamine also significantly impaired androgen-independent proliferation of both LNCaP and C4–2 cells, confirming a requirement for active SHH signaling in this process (Fig. 1c, d).
Figure 1. GLI3-dependent SHH signaling supports androgen-independent prostate cancer cell growth.

a, b LNCaP or C4–2 cells were transitioned from androgen-replete to androgen-deplete (-DHT) medium and 10 days thereafter processed by RT-qPCR using primers for the indicated SHH/GLI3-target genes and β-actin (a non-target gene). Where indicated, cells were infected with control (CTL KD) or GLI3-specific (GLI3 KD1) shRNA-expressing lentiviruses 1 day prior to transition or treated with DMSO or Cyclopamine (CYCLO) 1 day prior to harvest. mRNA levels were normalized to GAPDH mRNA and expressed relative to their levels in androgen replete (+DHT) medium. Data are the mean ± SEM of 3 experiments in triplicate. Asterisks denote statistically significant differences vs CTL knockdown or DMSO treated cells (Student’s t-test: ***p < 0.001). n.s., not significant. c, d LNCaP or C4–2 cells were transitioned from androgen-replete to androgen-deplete (-DHT) medium and thereafter monitored for proliferation using an IncuCyte ZOOM® system. Where indicated, cells were infected with control (CTL KD) or GLI3-specific (GLI3 KD1) shRNA-expressing lentiviruses or treated with DMSO or Cyclopamine (CYCLO) 1 day prior to transition. Data are the mean ± SEM of 3 experiments in triplicate. Asterisks denote statistically significant differences vs CTL knockdown or DMSO treated cells (Student’s t-test: *p < 0.05; p** < 0.01; p*** < 0.001). e, f LNCaP androgen-dependent (AD) cells were transfected with a control shRNA (CTL KD) or either of 2 GLI3-specific (GLI3 KD1, GLI3 KD2) shRNAs and monitored for proliferation for 5 days in androgen replete (+DHT) or androgen deplete (-DHT) media by MTT assay (e) or viable cell counting (f). Data are the mean ± SEM of 3 experiments in triplicate. Asterisks denote statistically significant differences (Student’s t-test: p** < 0.01; p*** < 0.001). g-i LNCaP androgen-independent (AI), 22Rv.1, and PacMetUT1 cells were transfected with control (CTL KD) or either of 2 GLI3-specific (GLI3 KD1, GLI3 KD2) shRNAs, and monitored for proliferation for 5 or 6 days as indicated in androgen deplete (-DHT) medium by MTT assay. Data are the mean ±SEM from 4 replicate assays. Asterisks denote statistically significant differences vs CTL knockdown cells (Student’s t-test: *p < 0.05; **p < 0.01; ***p < 0.001). j-n 22Rv.1, LNCaP AI, and PacMetUT1 cells were transfected with control (CTL KD) or either of 2 GLI3-specific (GLI3 KD1, GLI3 KD2) shRNAs, cultured in androgen deplete medium, and monitored for androgen-independent colony formation (j and k) or transwell migration (l-n). Data are the mean ± SEM from 4 replicate assays. Asterisks denote statistically significant differences vs CTL knockdown cells (Student’s t-test: *p < 0.05; **p < 0.01; ***p < 0.001).
Within the SHH pathway, GLI2 and GLI3 function as primary transcriptional effectors, and heretofore, only the former has been implicated in SHH signal-dependent prostate cancer cell growth in the absence of androgens (25,26,55). However, we found GLI3 to be highly expressed among the three GLI isoforms in various human prostate cancer cell lines (Supplementary Fig. 1a), CWR22 xenograft tumors (Supplementary Fig. 1b), and human prostate tumors (Supplementary Fig. 1c). Notably, GLI3 is further upregulated in AR-positive, but not AR-negative, human prostate cancer cell lines cultured in androgen-depleted medium (Supplementary Fig. 2a). Conversely, treatment of AR-positive prostate cancer cell lines with the synthetic androgen R1881 downregulated GLI3 mRNA and protein levels (Supplementary Fig. 2b, c). This negative regulation of GLI3 by androgen was also observed in vivo, as GLI3 mRNA was found to be significantly upregulated in both mouse prostate and human CWR22 prostate xenograft tumors following castration of mice (Supplementary Fig. 2d, e).
To examine the role of GLI3 in androgen-independent prostate cancer cell growth, we monitored the impact of RNAi-mediated GLI3 depletion on SHH-dependent gene expression and proliferation in androgen-deprived LNCaP and C4–2 cells. Notably, GLI3 knockdown (Supplementary Fig. 3a, b) reduced androgen-independent SHH-target gene expression and proliferation in both cell lines comparably to cyclopamine treatment (Fig. 1a-d). Although GLI3 knockdown also reduced androgen-dependent LNCaP cell proliferation, this inhibitory effect was nonetheless much more pronounced for androgen-independent cell growth (Fig. 1e, f; Supplementary Fig. 3c-e). Furthermore, knockdown of GLI3 prevented LNCaP cells from acquiring androgen-independent growth in culture following extended androgen depletion (Supplementary Fig. 4), and also impaired androgen-independent proliferation, colony formation in soft agar, and migration of LNCaP-AI and 22Rv-1 AI cell lines, both derived from long-term androgen deprivation in culture, as well as PacMetUT1 cells, derived from a lymph node metastatic tumor arising in a prostate cancer patient following ADT (Fig. 1g-n; Supplementary Fig. 3c-e). Finally, we observed that ectopic overexpression of GLI3 signficantly enhanced both androgen-dependent and androgen-indpendent proliferation of LNCaP and C4–2 cells, with a more prounounced effect observed under androgen-independent conditions (Supplementary Fig. 5). Altogether, these findings identify GLI3 as a critical effector of SHH-driven androgen-independent prostate cancer cell growth.
GLI3 is targeted for proteasomal degradation by WT but not oncogenic mutant SPOP.
GLI3 is one of three vertebrate GLI family proteins (GLI1–3) that collectively subsume the function of a single ancestral invertebrate GLI homolog, Cubitus interuptus (Ci) (56). Notably, Ci was previously shown to be targeted for proteasomal degradation by HIB (Hh-induced MATH and BTB domain containing protein), the Drospohila homolog of SPOP (57,58). More recently, GLI3 was reported to be a substrate of SPOP-mediated degradation in mammalian embryonic tissue-derived cells, thus revealing an evolutionarily conserved mechanism for maintenance of physiological GLI3 effector levels in the SHH pathway (29,59–62). We confirmed endogenous GLI3 to be a target of SPOP-mediated proteasomal degradation in LNCaP cells; thus, SPOP and GLI3 proteins physically associate (Fig. 2a and Supplementary Fig. 6), and SPOP knockdown enhanced (Fig. 2b), while SPOP overexpression reduced (Fig. 2c), GLI3 protein levels in a manner reversible by the proteasome inhibitor MG132 (Fig. 2C).
Figure 2. GLI3 is targeted for proteasomal degradation by WT but not oncogenic mutant SPOP.

a LNCaP whole cell lysates were subjected to immunoprecipitation (IP) with antibodies specific for GLI3 or isotype matched IgG. IPs were processed by western blot (WB) analysis using GLI3- and SPOP-specific antibodies as indicated. INPUT, 10% of lysate used in IP. b, c LNCaP cells were transfected with control (CNTL) or either of two different SPOP-specific shRNAs (SPOP KD1, SPOP KD2) (b) or increasing amounts of WT SPOP (c). Transfected whole cell lysates were processed by WB using antibodies specific for GLI3, SPOP, or TFIIEβ (loading control). In (c) cells were treated at 24 hrs post-transfection without or with MG132 (20 μM; 18 hrs) as indicated. d-e LNCaP cells transfected without or with Myc-tagged SPOP (M-SPOP; WT or mutant as indicated), were treated at 24 hrs post-transfection with MG132 (20 μM; 18 hrs). In (e), cells were also transfected with HA-tagged ubiquitin (Ub). Transfected whole cell lysates were subjected to IP with GLI3-specific antibodies and processed by WB using antibodies specific for GLI3, the Myc or HA epitopes, or TFIIEβ. INPUT, 10% of lysate used in IP. f LNCaP cells transfected without or with Myc-tagged SPOP (M-SPOP; WT or mutant as indicated) were treated at 24 hrs post-transfection without or with MG132 (20 μM;18 hrs). Transfected whole cell lysates were directly processed by WB using antibodies specific for GLI3, the Myc epitope, or TFIIEβ.
Nearly all prostate cancer-associated mutations in SPOP thus far discovered occur within its substrate-binding pocket and disrupt its interaction with established substrates (27,31). To determine if this pathological defect extends to the interaction of SPOP with GLI3, we monitored the impact of prostate cancer-associated mutations in SPOP on its ability to interact with, ubiquitylate, and promote the degradation of GLI3. These experiments were performed in both androgen-dependent (LNCaP) and androgen-independent (PC-3) prostate cancer cells, with identical results. Thus, among four oncogenic SPOP mutant derivatives tested, including Y87C, F102C, W131G, and F133V, all but Y87C were severely compromised, compared to WT SPOP, in their respective abilities to interact with, ubiquitylate, and promote the degradation of GLI3 (Fig. 2d-f; Supplementary Fig. 7a-c). Notably, ectopic expression of SPOP mutant derivatives F102C, W131G, and F133V augmented the level of endogenous GLI3 beyond that observed in the absence of ectopic SPOP (Fig. 2f; Supplementary Fig. 7c), consistent with a dominant negative effect of mutant SPOP that has been observed for other SPOP substrates (34,40,42,63). Furthermore, MG132-mediated proteasomal inhibition did not promote further stabilization of endogenous GLI3 in LNCaP or PC-3 cells transfected with mutant SPOP (Fig. 2f; Supplementary Fig. 7c), confirming that SPOP promotes GLI3 degradation through the ubiquitin proteasome pathway. Importantly, our observation that GLI3 and SPOP physically interact in AR-negative PC-3 cells (Supplementary Fig. 7a) reveals their association to be independent of AR, an established target of SPOP. Finally, the ability of SPOP mutant Y87C to promote the degradation of GLI3, but not other previously reported SPOP substrates, including AR, ERG, DDIT3, TRIM24, and BRD2–4 (33–35,41,43,44), reveals substrate specificity among prostate cancer-associated SPOP mutations, and raises the possibility that tumor-specific differences in the functional integrity of SPOP substrates exist, with biological and possible therapeutic implications for development and progression of SPOP mutant prostate tumors. Nonetheless, these findings reveal for the first time that prostate cancer-associated mutations in SPOP, including hotspot mutations F102C and F133V, disrupt its ability to interact with and promote the degradation of GLI3.
To confirm these findings in a more clinically relevant setting, we monitored GLI3 protein levels in a cohort of 86 primary prostate tumors, including 12 tumors that were determined by Sanger sequencing to be SPOP mutation-positive. The SPOP mutation frequency (∼14%) in this sample set is consistent with the reported frequency of SPOP mutations in other prostate cancer cohorts (27,44,63). Immunohistochemical analysis revealed that among the 12 SPOP mutant tumors, nine expressed elevated levels of GLI3 (i.e., individual IHC scores exceeding the mean IHC score for SPOP WT tumors) (Fig. 3a-c). Notably, among the three SPOP mutant tumors that did not express elevated levels of GLI3, one carried a Y87C mutation that we found comparable to WT SPOP in its ability to bind, ubiquitylate, and promote the degradation of GLI3 (Fig. 2d-f; Fig. 3a, b). Exclusion of this Y87C SPOP-mutant specimen from statistical consideration further enhanced the significant difference in mean GLI3 signal intensity between SPOP WT and mutant tumors, revealing GLI3 protein levels to be elevated in the latter (Fig. 3a). Altogether, our analyses in both prostate cancer cell lines and tumor tissues confirm GLI3 to be a clinically relevant substrate of SPOP-mediated proteasomal degradation and further reveal substrate selectivity among individual prostate cancer-associated SPOP mutations.
Figure 3. GLI3 protein expression is elevated in SPOP mutant prostate cancer specimens.
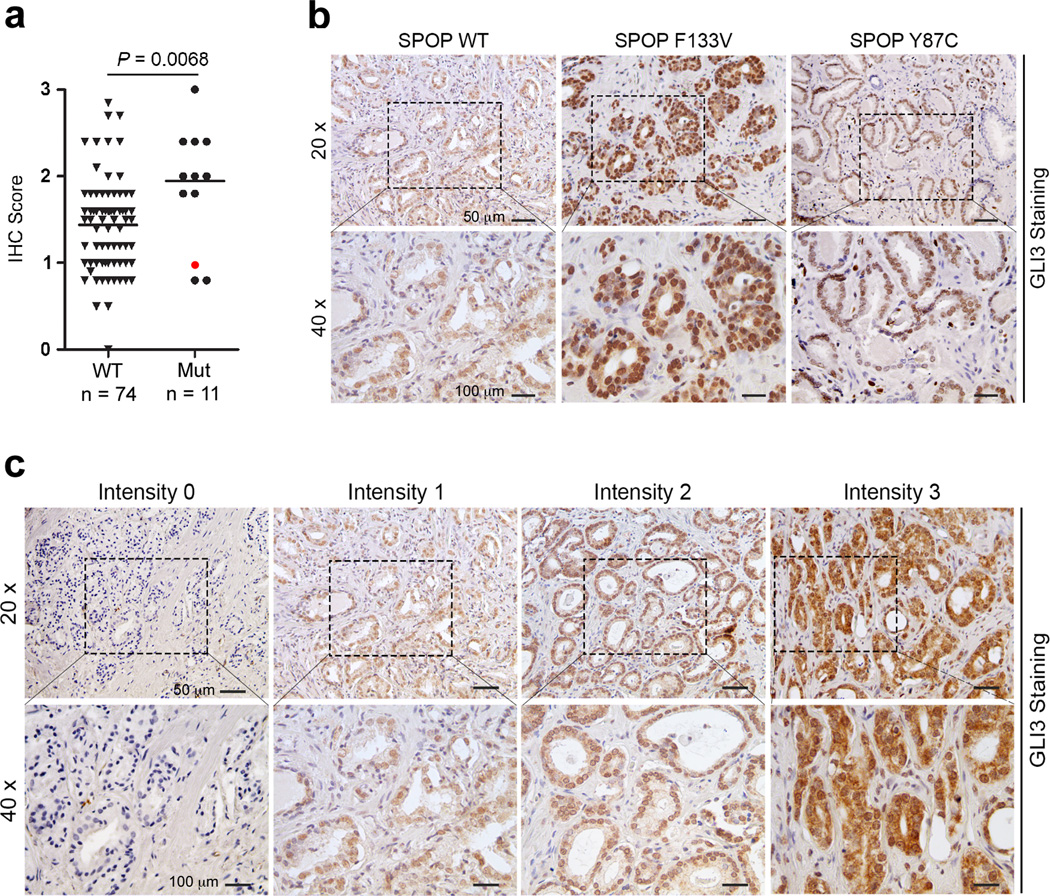
a Quantitative GLI3 IHC analysis from 86 cases of SPOP mutation-negative (n=74) and SPOP mutation-positive (n=12) prostate cancers. A single SPOP mutant Y87C tumor specimen (red dot) was excluded from statistical consideration. Significance was determined by two-tailed t-test. b Representative images of GLI3 IHC performed on one SPOP WT and two SPOP mutant (F133V and Y87C) prostate tumors. Note that GLI3 is not elevated in the SPOP mutant prostate tumor carrying a Y87C mutation that retains the ability to bind, ubiquitylate, and promote the degradation of GLI3. c GLI3 IHC scoring schema.
Pathologically stabilized GLI3 promotes androgen-independent growth of SPOP mutant prostate cancer cells through reactivation of an AR signaling axis.
Our identification of GLI3 as a critical effector of SHH in androgen-independent prostate cancer cell growth led us to investigate the impact of mutant SPOP, and thus pathologically stabilized GLI3, on these processes. To this end, we used CRIPSR/Cas9-mediated genome editing to engineer clonal LNCaP cell lines expressing one of the most frequent SPOP mutations found in prostate cancer (c.305 T>G, p.F102C; (Supplementary Fig. 8a). Two independent sequence-verified clonal lines heterozygous for SPOP mutation c.305 T>G (F102C.1 and F102C.2; (Supplementary Fig. 8b) were used for subsequent molecular phenotypic analyses. We validated defects in the SPOP-binding and ubiquitylation status of endogenous GLI3, leading to enhanced steady state levels of GLI3 protein, but not GLI3 mRNA, in SPOP mutant versus SPOP WT LNCaP cells (Fig. 4a-d). Notably, the steady state level of GLI3 protein in SPOP mutant cells was comparable to that observed in SPOP WT cells following proteasome inhibition, and enhanced GLI3 protein levels in SPOP mutant cells were unchanged by treatment with MG132 (Fig. 4c). Together, these findings confirm pathological stabilization of GLI3 protein in LNCaP cell lines expressing oncogenic SPOP mutant F102C.
Figure 4. Pathologically stabilized GLI3 promotes androgen-independent growth of SPOP mutant prostate cancer cells through reactivation of an AR signaling axis.

a, b SPOP WT and F102C mutant LNCaP cells (clones F102C.1 and F102C.2) were treated with MG132 (20 μM) for 18 hrs prior to harvest and preparation of whole cell lysates. In (b) cell were additionally transfected for 48 hrs with HA-tagged Ub. Whole cell lysates were subjected to GLI3-specific IP followed by WB analysis using antibodies specific for GLI3, SPOP, the HA epitope, and TFIEβ as indicated. INPUT, 10% of lysate used in IP. c SPOP WT and F102C mutant LNCaP cells were treated without (−) or with (+) MG132 (20 μM) for 18 hrs. Thereafter, whole cell lysates were processed directly by WB analysis using antibodies specific for GLI3, SPOP, and TFIEβ as indicated. d RNA from SPOP WT and F102C mutant LNCaP cells was processed by RT-qPCR. SPOP and GLI3 mRNA levels were normalized to GAPDH and expressed relative to their mRNA levels in WT SPOP cells. Data are the mean ± SEM of 3 experiments performed in triplicate. e-g SPOP WT and F102C mutant LNCaP cells were seeded at low density in androgen-depleted medium. (e) Cells were monitored for proliferation by live cell counting. Data are the mean ± SEM of 3 experiments in triplicate. Asterisks denote statistically significant differences vs SPOP WT cells (Student’s t-test: **p < 0.01; ***p < 0.001). (f, g) Cells were infected with control (CT), GLI3 (GLI3 KD1), or AR (AR KD1) shRNA-expressing lentiviruses 24 hrs prior to seeding or treated with DMSO (DM) or Cyclopamine (CYCLO) at the time of seeding. 30 days later, cell colonies were stained with crystal violet. A representative experiment is shown in (f) and 3 independent experiments were quantified in (g). h-j SPOP WT and F102C LNCaP cells transduced without (−) or with (+) GLI3-specific shRNA-expressing lentivirus (GLI3 KD1) were subjected to RNA-seq. (h) Venn diagram depicts overlap among genes differentially expressed as a function of: (i) androgen (±DHT) in WT SPOP cells (blue); (ii) SPOP mutation status (-DHT; green); (iii) GLI3 (-DHT; orange). (i) Heat map of 184 overlapping genes from (h). Heatmap was generated from duplicate RNA-seq runs (colored number bars) for SPOP WT (aqua and yellow bars) and each of two clonal SPOP F102C mutant LNCaP (purple and orange) cell lines under the condition specified (−/+ DHT; −/+ GLI3 KD). Note that gene expression levels [up (red); down (green)] in SPOP mutant cells (-DHT; purple bar) approach gene expression levels in WT SPOP cells (+DHT; aqua bar), indicating partial restoration of AR signaling in the former. Notably, GLI3 knockdown reverses this effect, diminishing restored androgen signaling in SPOP mutant cells (-DHT; orange bar). (j) Top 5 biological functions for the 184 genes co-regulated by androgen and GLI3 in SPOP mutant cells.
To examine the impact of pathologically stabilized GLI3 on androgen-independent LNCaP cell growth, we seeded SPOP WT and mutant cell lines at low density in the absence of androgen, and thereafter monitored cell proliferation and colony formation for 9 and 30 days, respectively. As expected based on their androgen-sensitive phenotype, parental LNCaP cells initially proliferated poorly, and 5 days after androgen deprivation all but ceased to proliferate with no evidence of colony formation 30 days after androgen withdrawal (Fig. 4e-g). By striking contrast, SPOP mutant cells exhibited robust androgen-independent proliferation, forming substantive lawns of colonies 30 days following androgen deprivation (Fig. 4e-g). Importantly, androgen-independent growth of SPOP mutant LNCaP cells was similarly reversed by cyclopamine or RNAi-mediated knockdown of GLI3 or AR (Fig. 4f, g). These findings reveal that SPOP mutant prostate cancer cells acquire robust androgen-independent growth through enhanced GLI3-dependent SHH signaling.
To explore the basis by which GLI3 promotes androgen-independent growth of SPOP mutant prostate cancer cells, we profiled the transcriptomes of SPOP WT and mutant LNCaP cells before and after androgen deprivation by RNA-sequencing. Using standard criteria (minimum 2-fold change), we identified 973 genes that were differentially expressed in SPOP mutant vs SPOP WT LNCaP cells grown in the absence of androgens (Fig. 4h; Supplementary Table 1). Strikingly, a significant fraction (588 or ∼60%) of these genes overlapped with genes (3436 in number) differentially expressed as a function of androgen in WT LNCaP cells (Fig. 4h; Supplementary Table 2), indicating partial restoration of AR signaling in SPOP mutant cells following androgen withdrawal. Supporting this possibility, RNAi-mediated knockdown of AR completely blocked androgen independent growth of SPOP mutant LNCaP cells (Fig. 4f, g). To determine the contribution of GLI3 to this process, we performed RNA-sequencing following GLI3 knockdown in androgen-deprived SPOP mutant LNCaP cells (Supplementary Table 3). Among the 588 androgen-responsive genes in LNCaP cells whose hormone-independent expression is regulated by SPOP (Fig. 4h; overlap of blue and green), 184 (or ∼31%) are also regulated by GLI3 (Fig. 4h; overlap of blue, green, and orange; Supplementary Table 4), thus revealing a prominent role for GLI3 in restoration of AR signaling in SPOP mutant cells following androgen withdrawal (Fig. 4i; Supplementary Fig. 9). Enrichment analysis using Gene Ontology Biological Processes revealed these 184 GLI3-regulated genes to be prominently linked with “Cell division”, “DNA replication”, “G1/S transition of mitotic cell cycle”, “Regulation of ubiquitin protein ligase activity”, and “chromatin silencing” among others, suggesting possible molecular genetic bases for androgen independent growth of SPOP mutant cells (Fig. 4j). Taken together, these findings indicate that SPOP mutant prostate cancer cells acquire robust androgen-independent growth through reestablishment of an AR signaling axis that involves functional crosstalk with the SHH/GLI3 pathway.
To explore a mechanistic basis for functional crosstalk between AR and GLI3, we investigated their possible physical interaction. Co-immunoprecipitation analyses revealed an association between ectopic GLI3 and AR in HEK293T cells, as well as their endogenous counterparts in LNCaP cells where this interaction was significantly enhanced in the absence of androgens (Supplementary Fig. 10a, b). Importantly, comparative analyses of the GLI3-AR interaction in SPOP WT and mutant LNCaP cells revealed the strongest association to occur in the latter (Supplementary Fig. 10b), likely reflecting enhanced steady state levels of both GLI3 and AR proteins. Immobilized protein affinity chromatography using recombinant GLI3 and AR truncation derivatives revealed their interaction to be direct and mediated by their respective DNA-binding domains (Supplementary Fig. 10c; Supplementary Fig. 11). Thus, GLI3 and AR interact directly in prostate cancer cells, and this association is enhanced significantly by androgen withdrawal and mutation of SPOP.
GLI3 supports castration resistant tumor growth.
The increased GLI3 expression under androgen deprivation (Supplementary Fig. 2) and stabilization of its full-length activator form by SPOP mutations as well as by AR (21) led us to determine next if GLI3 is necessary for the development of CRPC by examining the impact of GLI3 knockdown on the outgrowth of castration resistant LNCaP tumor xenografts in mice. To this end, control and two GLI3 knockdown LNCaP cell lines, each stably expressing firefly luciferase and GFP, were subcutaneously injected into male NOD scid gamma (NSG™) mice, and subsequent tumor burden was quantified by volume measurement as well as bioluminescence imaging. We observed that mice injected with control knockdown cells developed larger tumors than those injected with GLI3 knockdown cells four weeks after inoculation, confirming a requirement for GLI3 in androgen-dependent LNCaP cell growth (Supplementary Fig. 12). At this point (four weeks post-inoculation), mice bearing control knockdown tumors were surgically castrated, while those bearing GLI3 knockdown tumors were castrated once their mean tumor volumes reached statistically comparable levels to four-week post-inoculation control tumor volumes. Following castration, control and GLI3 knockdown tumor xenografts were monitored for an additional eight weeks. As expected, control tumors initially regressed after castration, but three weeks thereafter developed castration resistance and initiated androgen-independent growth (Fig. 5a, b). By contrast, GLI3 knockdown tumors, following regression after castration, did not become castration resistant (Fig. 5a, b). Accordingly, the mean tumor weights of the GLI3 knockdown xenografts were significantly lower than that of the control group at the termination of the experiment (Fig. 5c).
Figure 5. GLI3 signaling supports castration resistant tumor formation.

a-c Subcutaneous tumors were generated by injection of gonadally intact mice with control (CTL KD) or either of 2 GLI3-specific (GLI3 KD1, GLI3 KD2), shRNA-expressing LNCaP cell lines. Once mean volumes in control and GLI3 knockdown tumors reached statistically comparable levels, mice were castrated and castration-resistant tumor growth was measured by tumor volume (a) and total photon flux following bioluminescence imaging (b). Eight weeks post-castration, control and GLI3 knockdown tumors were collected and weighed (c). Each data point in (a and b) represents the mean ± SEM from twelve single tumors. Asterisks denote statistically significant differences vs GLI3 knockdown tumors. In (c) asterisks denote statistically significant differences vs control knockdown tumors (Student’s t-test: *p < 0.05; **p < 0.01; ***p < 0.001). d-i Mice (10 per group) were orthotopically injected with control (CTL KD) or either of 2 GLI3-specific (GLI3 KD1, GLI3 KD2), shRNA-expressing PacMetUT1 cell lines. (d) Tumor growth was monitored for 7 weeks post-inoculation by weekly bioluminescence imaging, and mean total photon flux (± SEM) was plotted for each group of 10 tumors. Asterisks denote statistically significant differences vs control tumors (Mann Whitney test: *p < 0.05; **p < 0.01). (e) Representative bioluminescence image from each group. (f) Comparison of tumor incidence in control versus GLI3 knockdown tumors (Fisher’s exact test, p<0.05). (g) Tumors were collected at 7 weeks post-inoculation and weighed. Data represents the mean ± SEM from single tumors. Asterisks denote statistically significant differences vs control tumors (Student’s t-test: *p < 0.05). (h) Lungs were harvested at 7 weeks post-inoculation and GFP metastatic cancer cell colonies were visualized and counted. (i) Representative fluorescence image of GFP metastatic cancer cell colonies in the lung.
We also examined the impact of GLI3 knockdown on the castration resistant growth of androgen-independent PacMetUT1 cells following orthotopic transplantation into mice. Thus, control and two GLI3 knockdown PacMetUT1 cell lines, each stably expressing luciferase and GFP, were inoculated into the prostate glands of male nude mice one week prior to castration, and tumor growth monitored for seven weeks thereafter. Control tumors grew significantly faster than the two GLI3 knockdown tumors, as reflected by the total photon flux from bioluminescence imaging (Fig. 5d, e). Autopsies revealed that all mice inoculated with control cells developed tumors, while only 50%−60% of mice inoculated with GLI3 knockdown cells did so (Fig. 5f). The mean weights of tumors derived from GLI3 knockdown cells was also significantly lower than the mean weight of tumors derived from control knockdown cells (Fig. 5g). Furthermore, four mice within the control group exhibited lung micrometastases, whereas only one mouse in each of the GLI3 knockdown groups showed lung micrometastases (Fig. 5h, i). Thus, GLI3 supports both the development and the malignant phenotypes of CRPC in multiple human prostate cancer models.
GLI3 and AR co-regulated genes correlate with metastatic CRPC and predict recurrence.
The striking difference in xenograft growth between the control and GLI3 knockdown prostate cancer cells after castration suggests that GLI3 may reactivate an AR transactivation program under androgen depleted conditions. To address this question, we profiled gene expression by RNA sequencing after knocking down GLI3 in androgen-independent LNCaP cells, which were adapted to grow in androgen-depleted culture medium. Gene set enrichment analysis (GSEA) showed significant enrichment of the Hallmark Androgen Resonse gene set in the control cells in comparison to the GLI3 knockdown cells (Fig. 6a). Because the genes in this gene set are androgen-stimulated genes, their reduced enrichment after GLI3 knockdown suggests that part of the GLI3 transactivation program overlaps with that of AR. Indeed, among the 392 differentially expressed genes (2-fold or greater changes with adjusted P-value < 0.05) due to GLI3 knockdown (Supplementary Table 5), one-hundred fifty-nine genes are among the 3436 genes differentially regulated by DHT shown in Supplementary Table 2. Among the 159 genes, there are 32 commonly upregulated genes by both DHT and GLI3 (Fig. 6b). We used 31 genes (46 transcripts) of the 32 AR/GLI3 co-stimulated genes that can be identified in the expression data derived from 150 prostate cancer patients (50) to perform unsupervised hierarchical clustering as shown in Fig. 6c. Interestingly, the majority of metastatic castration resistant tumors show high expression of the 46 annotated transcripts and are clustered together. The 50 patients clustered in the high expression group showed significantly worse biochemical recurrence (BCR)-free survival than the low expression group (Fig. 6d). To explore whether a smaller set of transcripts is equally effective in predicting the survival, we found that 9 of the 46 transcripts are significantly overexpressed in the 19 metastatic tumors versus the 131 primary tumors in the cohort (Supplementary Table 6) and their average expression can also predict BCR-free survival (Fig. 6e).
Discussion
Our findings provide new insight concerning the role, regulation, and possible therapeutic implications of GLI3 in SHH-driven development of CRPC. While prior studies have implicated SHH signaling in prostate cancer progression and the development of hormone therapy resistance, little is currently known regarding the molecular underpinnings of this process, including whether and how the downstream SHH effector GLI3 contributes to this process. Within the SHH pathway, GLI2 and GLI3 are primary transcriptional effectors and functionally bipartite regulators that directly repress and activate their respective target genes, including the pathway amplifier and dedicated activator GLI1, in the absence and presence of SHH, respectively (10,56). Prior studies have implicated GLI2 in the development of CRPC through its ability to function as a direct binding coactivator of AR. Thus, GLI2 is upregulated during clinical progression to CRPC, promotes androgen independent prostate cancer cell growth, and functionally stimulates and physically co-occupies AR-target genes (19,25,26,55,64,65). More recently, GLI3 was shown capable of interacting with transcriptionally active AR, which, in turn, could stimulate GLI3-dependent transcription (21). However, the role and regulation of GLI3 in prostate carcinogenesis have remained obscure. Herein, we bridge this considerable knowledge gap and identify a prominent role for GLI3-dependent SHH signaling in the development and progression of prostate cancer.
First, we observed that among GLI family members, GLI3 is expressed at much higher levels than either GLI1 or GLI2 in prostate cancer cell lines, tumor xenografts, and human prostate tumor tissues. Second, we found that GLI3 mRNA and protein levels are negatively regulated by androgens, leading to upregulation of GLI3 upon androgen withdrawal in both cultured prostate cancer cells and human prostate tumor xenografts. Third, we showed that targeted depletion of GLI3 in cultured prostate cancer cells not only prevented the acquisition of androgen-independent growth when implemented prior to long-term androgen deprivation, but also impaired cell growth and migration when implemented following acquisition of the androgen-independent phenotype. Finally, GLI3 knockdown blocked castration-resistant formation and metastatic spread of human prostate tumor xenografts in mice. Collectively, these findings reveal for the first time a prominent role for GLI3-dependent SHH signaling in the development and malignant phenotypes of CRPC in a variety of human prostate cancer models. Accordingly, factors regulating GLI3 expression and/or activity could significantly impact the trajectory of prostate cancer development and progression.
In this regard, we identified GLI3 to be targeted for ubiquitin-mediated proteasomal degradation in prostate cancer cells by the E3 ubiquitin ligase adaptor protein SPOP, whose corresponding gene is the most frequently mutated in primary prostate cancer. Importantly, we found that oncogenic SPOP mutations occurring within its substrate binding cleft, including mutants F102C, W131G, and F133V, lead to pathological stabilization of GLI3, and we further confirmed these findings in patient-derived prostate tumor tissues, wherein GLI3 protein levels were found to be significantly higher in SPOP-mutation positive compared to SPOP mutation-negative tumors. Functionally, we showed that SPOP-mutant prostate cancer cells support androgen-independent growth and effectively recapitulate an androgen-dependent gene expression profile, consistent with cell cycle progression, characteristic of androgen-stimulated SPOP WT cells, and furthermore, that GLI3 contributes prominently to this process. As a result, individual depletion of GLI3 or AR similarly inhibited androgen-independent growth of SPOP-mutant cells, suggesting possible coordinate transcription control.
Notably, we observed that oncogenic SPOP mutant Y87C nonetheless retained wild-type-like ability to bind to and promote the ubiquitylation and degradation of GLI3, thus indicating substrate selectivity among SPOP mutations initially suggested in earlier studies (43,63). Our findings further complement and extend those from recent work providing evidence for unique substrate selectivity among cancer type-specific SPOP mutations (41). In this regard, comprehensive genome sequencing studies have identified recurrent SPOP mutations in endometrial cancer at frequencies similar those found in prostate cancer (4–14%) (66–68). Although the overwhelming majority of oncogenic SPOP mutations found in both endometrial and prostate cancer lie within the substrate recognition (MATH) domain of SPOP, their distribution within this domain is nonetheless divergent. Whereas prostate cancer-specific SPOP mutations cluster predominantly in the substrate binding cleft, endometrial-specific SPOP mutations instead tend to accumulate in an uncharacterized region of the MATH domain (27,66–69). Notably, recent work has revealed opposing effects of these cancer type-specific mutations in SPOP on its biological function in protein turnover. Thus, SPOP mutations specific to endometrial or prostate cancer were shown, respectively, to promote or prevent SPOP-mediated degradation of BRD2–4 and, accordingly, elicit opposing BET inhibitor sensitivities in mutant-expressing cells (41). Furthermore, based on their correlative relationship with ubiquitylation and abundance levels of established SPOP substrates, endometrial cancer-specific SPOP mutations themselves could be segregated based on their apparent propensity to promote or impair SPOP-mediated degradation of established substrates. Our findings herein extend these earlier observations in endometrial cancer and offer evidence for substrate selective activity among prostate cancer-specific SPOP mutations.
Given that SPOP mutation is an early event during prostate tumorigenesis (70,71) and leads to stabilization of AR (34,39), our results suggest that SPOP mutation-mediated stabilization and activation of an AR/GLI3 axis may be a main mechanism driving prostate cancer development. Prostate tumors with SPOP mutation are likely addicted to an AR/GLI3 axis for cell cycle progression, which promotes androgen-independent growth of LNCaP cells in vitro and may render hormone therapy naïve tumors highly sensitive to androgen deprivation. This may explain why prostate cancers with SPOP mutation were more sensitive to androgen deprivation therapy than prostate tumors without SPOP mutation (72,73). The fact that SPOP mutations are less common in CRPC (74–76) and that GLI3 is shown to promote CRPC in our study suggests contextual models of AR/GLI3 axis-mediated prostate tumorigenesis and progression. In the context of SPOP mutation, the stabilization of AR and GLI3 proteins along with other oncogenic SPOP substrates likely contributes to prostate cancer development. On the other hand, in the context of hormone deprivation without SPOP mutation, transcription-mediated upregulation of AR (23,77) and the SHH pathway, including GLI3, as shown in this study likely cooperate with other oncogenic pathways to promote prostate cancer progression and castration resistance.
The molecular basis by which enhanced GLI3 drives castration-resistant growth of prostate tumors appears to involve, at least in part, reactivation of an AR signaling axis. Thus, we showed that GLI3 signaling significantly enriches the Hallmark Androgen Response genes under androgen depletion. Furthermore, genes significantly stimulated by both GLI3 and AR appears to contribute to metastasis of CRPC and are associated with poor BCR-free survival. Further studies are need to validate the prognostic value of the 9 transcripts of the 6 genes (Table S6) that are co-stimulated by GLI3 and AR. The underlying molecular basis for the crosstalk between GLI3 and AR remains to be definitively established. One possibility is that AR and GLI3 interact directly on chromatin, whereupon they function cooperatively to regulate AR-target genes and thus sustain AR signaling in the absence of androgens. This model is consistent with prior studies showing that AR and GLI proteins can function as reciprocal co-activators of one another (21,25,26). Our current study also confirmed a physical interaction between GLI3 and AR. Future studies should establish whether and how AR and GLI3 co-occupy specific genomic loci, including co-regulated genes revealed through the transcriptomic analysis.
In summary, we describe two clinically relevant mechanisms that promote an oncogenic function of GLI3. In prostate cancer cells with SPOP mutation, GLI3 is stabilized to promote their proliferation and androgen-independent growth. GLI3 is also transcriptionally upregulated during androgen deprivation and acts as a key mediator of hormone-refractory prostate cancer cell growth and CRPC. Mechanistically, this occurs through a functional interaction between GLI3 and AR that restores AR signaling under conditions of low hormone availability. Thus, various mechanisms can hyperactivate GLI3 signaling and enhance AR/GLI3 crosstalk, suggesting that GLI3-specific inhibitors might prove effective to block prostate cancer development or delay CRPC.
Supplementary Material
Implications:
We describe two clinically relevant mechanisms leading to hyperactivated GLI3 signaling and enhanced AR/GLI3 crosstalk, suggesting that GLI3-specific inhibitors might prove effective to block prostate cancer development or delay CRPC.
Acknowledgements
This work was supported by a pilot grant from the Institute for Integration of Medicine & Science/Clinical Translational Science Award (L. Sun), Cancer Prevention and Research Institute of Texas (CPRIT) awards RP120290-IIRA (L. Sun) and RP140435-HIHRRA (T. Boyer), and National Cancer Institute Cancer Center Support Grant P30 CA054174 (Optical Imaging, Next Generation Sequencing, and Biostatistics & Bioinformatics Shared Resources). M. Burleson and A. Easley were supported by NIH training grant T32CA148724. M. Burelson., T.M. Duong., and X. Gu. were supported by CPRIT Research Training Program grants RP140105 and RP170345 T. Qin was supported by a fellowship from Xiangya School of Medicine, Central South University, Hunan, China.
Footnotes
The authors declare no potential conflicts of interest
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020;70(1):7–30 doi 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 2.Attard G, Parker C, Eeles RA, Schroder F, Tomlins SA, Tannock I, et al. Prostate cancer. Lancet 2016;387(10013):70–82 doi 10.1016/S0140-6736(14)61947-4. [DOI] [PubMed] [Google Scholar]
- 3.Cookson MS, Lowrance WT, Murad MH, Kibel AS. Castration-resistant prostate cancer: AUA guideline amendment. J Urol 2015;193(2):491–9 doi 10.1016/j.juro.2014.10.104. [DOI] [PubMed] [Google Scholar]
- 4.Crawford ED, Higano CS, Shore ND, Hussain M, Petrylak DP. Treating Patients with Metastatic Castration Resistant Prostate Cancer: A Comprehensive Review of Available Therapies. J Urol 2015;194(6):1537–47 doi 10.1016/j.juro.2015.06.106. [DOI] [PubMed] [Google Scholar]
- 5.Heidegger I, Massoner P, Eder IE, Pircher A, Pichler R, Aigner F, et al. Novel therapeutic approaches for the treatment of castration-resistant prostate cancer. J Steroid Biochem Mol Biol 2013;138:248–56 doi 10.1016/j.jsbmb.2013.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer 2015;15(12):701–11 doi 10.1038/nrc4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karantanos T, Corn PG, Thompson TC. Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches. Oncogene 2013;32(49):5501–11 doi 10.1038/onc.2013.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yuan X, Cai C, Chen S, Yu Z, Balk SP. Androgen receptor functions in castration-resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis. Oncogene 2013. doi 10.1038/onc.2013.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin TL, Matsui W. Hedgehog pathway as a drug target: Smoothened inhibitors in development. Onco Targets Ther 2012;5:47–58 doi 10.2147/OTT.S21957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lum L, Beachy PA. The Hedgehog response network: sensors, switches, and routers. Science 2004;304(5678):1755–9 doi 10.1126/science.1098020. [DOI] [PubMed] [Google Scholar]
- 11.Peng YC, Joyner AL. Hedgehog signaling in prostate epithelial-mesenchymal growth regulation. Dev Biol 2015;400(1):94–104 doi 10.1016/j.ydbio.2015.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shaw A, Bushman W. Hedgehog signaling in the prostate. J Urol 2007;177(3):832–8 doi 10.1016/j.juro.2006.10.061. [DOI] [PubMed] [Google Scholar]
- 13.Hui CC, Angers S. Gli proteins in development and disease. Annu Rev Cell Dev Biol 2011;27:513–37 doi 10.1146/annurev-cellbio-092910-154048. [DOI] [PubMed] [Google Scholar]
- 14.Dai P, Akimaru H, Tanaka Y, Maekawa T, Nakafuku M, Ishii S. Sonic Hedgehog-induced activation of the Gli1 promoter is mediated by GLI3. J Biol Chem 1999;274(12):8143–52. [DOI] [PubMed] [Google Scholar]
- 15.Ikram MS, Neill GW, Regl G, Eichberger T, Frischauf AM, Aberger F, et al. GLI2 is expressed in normal human epidermis and BCC and induces GLI1 expression by binding to its promoter. J Invest Dermatol 2004;122(6):1503–9 doi 10.1111/j.0022-202X.2004.22612.x. [DOI] [PubMed] [Google Scholar]
- 16.Ruiz i Altaba A, Mas C, Stecca B. The Gli code: an information nexus regulating cell fate, stemness and cancer. Trends Cell Biol 2007;17(9):438–47 doi 10.1016/j.tcb.2007.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Briscoe J, Therond PP. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol 2013;14(7):416–29 doi 10.1038/nrm3598. [DOI] [PubMed] [Google Scholar]
- 18.Robbins DJ, Fei DL, Riobo NA. The Hedgehog signal transduction network. Sci Signal 2012;5(246):re6 doi 10.1126/scisignal.2002906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen M, Carkner R, Buttyan R. The hedgehog/Gli signaling paradigm in prostate cancer. Expert Rev Endocrinol Metab 2011;6(3):453–67 doi 10.1586/EEM.11.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ibuki N, Ghaffari M, Pandey M, Iu I, Fazli L, Kashiwagi M, et al. TAK-441, a novel investigational smoothened antagonist, delays castration-resistant progression in prostate cancer by disrupting paracrine hedgehog signaling. Int J Cancer 2013;133(8):1955–66 doi 10.1002/ijc.28193. [DOI] [PubMed] [Google Scholar]
- 21.Li N, Truong S, Nouri M, Moore J, Al Nakouzi N, Lubik AA, et al. Non-canonical activation of hedgehog in prostate cancer cells mediated by the interaction of transcriptionally active androgen receptor proteins with Gli3. Oncogene 2018;37(17):2313–25 doi 10.1038/s41388-017-0098-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen M, Tanner M, Levine AC, Levina E, Ohouo P, Buttyan R. Androgenic regulation of hedgehog signaling pathway components in prostate cancer cells. Cell Cycle 2009;8(1):149–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gowda PS, Deng JD, Mishra S, Bandyopadhyay A, Liang S, Lin S, et al. Inhibition of hedgehog and androgen receptor signaling pathways produced synergistic suppression of castration-resistant prostate cancer progression. Mol Cancer Res 2013;11(11):1448–61 doi 10.1158/1541-7786.MCR-13-0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shaw G, Price AM, Ktori E, Bisson I, Purkis PE, McFaul S, et al. Hedgehog signalling in androgen independent prostate cancer. Eur Urol 2008;54(6):1333–43 doi 10.1016/j.eururo.2008.01.070. [DOI] [PubMed] [Google Scholar]
- 25.Chen M, Feuerstein MA, Levina E, Baghel PS, Carkner RD, Tanner MJ, et al. Hedgehog/Gli supports androgen signaling in androgen deprived and androgen independent prostate cancer cells. Mol Cancer 2010;9:89 doi 10.1186/1476-4598-9-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li N, Chen M, Truong S, Yan C, Buttyan R. Determinants of Gli2 co-activation of wildtype and naturally truncated androgen receptors. Prostate 2014;74(14):1400–10 doi 10.1002/pros.22855. [DOI] [PubMed] [Google Scholar]
- 27.Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet 2012;44(6):685–9 doi 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hieronymus H, Sawyers CL. Traversing the genomic landscape of prostate cancer from diagnosis to death. Nat Genet 2012;44(6):613–4 doi 10.1038/ng.2301. [DOI] [PubMed] [Google Scholar]
- 29.Wang C, Pan Y, Wang B. Suppressor of fused and Spop regulate the stability, processing and function of Gli2 and Gli3 full-length activators but not their repressors. Development 2010;137(12):2001–9 doi 10.1242/dev.052126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012;487(7406):239–43 doi 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mani RS. The emerging role of speckle-type POZ protein (SPOP) in cancer development. Drug Discov Today 2014;19(9):1498–502 doi 10.1016/j.drudis.2014.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garcia-Flores M, Casanova-Salas I, Rubio-Briones J, Calatrava A, Dominguez-Escrig J, Rubio L, et al. Clinico-pathological significance of the molecular alterations of the SPOP gene in prostate cancer. Eur J Cancer 2014;50(17):2994–3002 doi 10.1016/j.ejca.2014.08.009. [DOI] [PubMed] [Google Scholar]
- 33.An J, Ren S, Murphy SJ, Dalangood S, Chang C, Pang X, et al. Truncated ERG Oncoproteins from TMPRSS2-ERG Fusions Are Resistant to SPOP-Mediated Proteasome Degradation. Mol Cell 2015;59(6):904–16 doi 10.1016/j.molcel.2015.07.025. [DOI] [PubMed] [Google Scholar]
- 34.An J, Wang C, Deng Y, Yu L, Huang H. Destruction of full-length androgen receptor by wild-type SPOP, but not prostate-cancer-associated mutants. Cell Rep 2014;6(4):657–69 doi 10.1016/j.celrep.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dai X, Gan W, Li X, Wang S, Zhang W, Huang L, et al. Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nat Med 2017;23(9):1063–71 doi 10.1038/nm.4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duan S, Pagano M. SPOP Mutations or ERG Rearrangements Result in Enhanced Levels of ERG to Promote Cell Invasion in Prostate Cancer. Mol Cell 2015;59(6):883–4 doi 10.1016/j.molcel.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gan W, Dai X, Lunardi A, Li Z, Inuzuka H, Liu P, et al. SPOP Promotes Ubiquitination and Degradation of the ERG Oncoprotein to Suppress Prostate Cancer Progression. Mol Cell 2015;59(6):917–30 doi 10.1016/j.molcel.2015.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Geng C, He B, Xu L, Barbieri CE, Eedunuri VK, Chew SA, et al. Prostate cancer-associated mutations in speckle-type POZ protein (SPOP) regulate steroid receptor coactivator 3 protein turnover. Proc Natl Acad Sci U S A 2013;110(17):6997–7002 doi 10.1073/pnas.1304502110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Geng C, Rajapakshe K, Shah SS, Shou J, Eedunuri VK, Foley C, et al. Androgen receptor is the key transcriptional mediator of the tumor suppressor SPOP in prostate cancer. Caner Res 2014;74(19):5631–43 doi 10.1158/0008-5472.CAN-14-0476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Groner AC, Cato L, de Tribolet-Hardy J, Bernasocchi T, Janouskova H, Melchers D, et al. TRIM24 Is an Oncogenic Transcriptional Activator in Prostate Cancer. Cancer Cell 2016;29(6):846–58 doi 10.1016/j.ccell.2016.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Janouskova H, El Tekle G, Bellini E, Udeshi ND, Rinaldi A, Ulbricht A, et al. Opposing effects of cancer-type-specific SPOP mutants on BET protein degradation and sensitivity to BET inhibitors. Nat Med 2017;23(9):1046–54 doi 10.1038/nm.4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Theurillat JP, Udeshi ND, Errington WJ, Svinkina T, Baca SC, Pop M, et al. Prostate cancer. Ubiquitylome analysis identifies dysregulation of effector substrates in SPOP-mutant prostate cancer. Science 2014;346(6205):85–9 doi 10.1126/science.1250255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang P, Gao K, Tang Y, Jin X, An J, Yu H, et al. Destruction of DDIT3/CHOP protein by wild-type SPOP but not prostate cancer-associated mutants. Hum Mutat 2014;35(9):1142–51 doi 10.1002/humu.22614. [DOI] [PubMed] [Google Scholar]
- 44.Zhang P, Wang D, Zhao Y, Ren S, Gao K, Ye Z, et al. Intrinsic BET inhibitor resistance in SPOP-mutated prostate cancer is mediated by BET protein stabilization and AKT-mTORC1 activation. Nat Med 2017;23(9):1055–62 doi 10.1038/nm.4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boysen G, Barbieri CE, Prandi D, Blattner M, Chae SS, Dahija A, et al. SPOP mutation leads to genomic instability in prostate cancer. Elife 2015;4 doi 10.7554/eLife.09207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Troyer DA, Tang Y, Bedolla R, Adhvaryu SG, Thompson IM, Abboud-Werner S, et al. Characterization of PacMetUT1, a recently isolated human prostate cancer cell line. Prostate 2008;68(8):883–92 doi 10.1002/pros.20758. [DOI] [PubMed] [Google Scholar]
- 47.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15(12):550 doi 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 2015;1(6):417–25 doi 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102(43):15545–50 doi 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010;18(1):11–22 doi 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Therneau TM, Grambsch PM The Cox Model. In: Modeling Survival Data: Extending the Cox Model. Modeling Survival Data: Extending the Cox Model. Volume 10.1007/978-1-4757-3294-8_3. New York, NY: Springer; 2000. [DOI] [Google Scholar]
- 52.Yan Y, An J, Yang Y, Wu D, Bai Y, Cao W, et al. Dual inhibition of AKT-mTOR and AR signaling by targeting HDAC3 in PTEN- or SPOP-mutated prostate cancer. EMBO Mol Med 2018;10(4) doi 10.15252/emmm.201708478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou H, Kim S, Ishii S, Boyer TG. Mediator modulates Gli3-dependent Sonic hedgehog signaling. Mol Cell Biol 2006;26(23):8667–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou H, Spaeth JM, Kim NH, Xu X, Friez MJ, Schwartz CE, et al. MED12 mutations link intellectual disability syndromes with dysregulated GLI3-dependent Sonic Hedgehog signaling. Proc Natl Acad Sci U S A 2012;109(48):19763–8 doi 10.1073/pnas.1121120109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xia L, Bouamar H, Gu X, Zeballos C, Qin T, Wang B, et al. Gli2 mediates the development of castrationresistant prostate cancer. Int J Oncol 2020;57(1):100–12 doi 10.3892/ijo.2020.5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev 2001;15(23):3059–87 doi 10.1101/gad.938601. [DOI] [PubMed] [Google Scholar]
- 57.Kent D, Bush EW, Hooper JE. Roadkill attenuates Hedgehog responses through degradation of Cubitus interruptus. Development 2006;133(10):2001–10 doi 10.1242/dev.02370. [DOI] [PubMed] [Google Scholar]
- 58.Zhang Q, Zhang L, Wang B, Ou CY, Chien CT, Jiang J. A hedgehog-induced BTB protein modulates hedgehog signaling by degrading Ci/Gli transcription factor. Dev Cell 2006;10(6):719–29 doi 10.1016/j.devcel.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 59.Cai H, Liu A. Spop promotes skeletal development and homeostasis by positively regulating Ihh signaling. Proc Natl Acad Sci U S A 2016;113(51):14751–6 doi 10.1073/pnas.1612520114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen MH, Wilson CW, Li YJ, Law KK, Lu CS, Gacayan R, et al. Cilium-independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev 2009;23(16):1910–28 doi 10.1101/gad.1794109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wen X, Lai CK, Evangelista M, Hongo JA, de Sauvage FJ, Scales SJ. Kinetics of hedgehog-dependent full-length Gli3 accumulation in primary cilia and subsequent degradation. Mol Cell Biol 2010;30(8):1910–22 doi 10.1128/MCB.01089-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Q, Shi Q, Chen Y, Yue T, Li S, Wang B, et al. Multiple Ser/Thr-rich degrons mediate the degradation of Ci/Gli by the Cul3-HIB/SPOP E3 ubiquitin ligase. Proc Natl Acad Sci U S A 2009;106(50):21191–6 doi 10.1073/pnas.0912008106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ma J, Chang K, Peng J, Shi Q, Gan H, Gao K, et al. SPOP promotes ATF2 ubiquitination and degradation to suppress prostate cancer progression. J Exp Clin Cancer Res 2018;37(1):145 doi 10.1186/s13046-018-0809-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Narita S, So A, Ettinger S, Hayashi N, Muramaki M, Fazli L, et al. GLI2 knockdown using an antisense oligonucleotide induces apoptosis and chemosensitizes cells to paclitaxel in androgen-independent prostate cancer. Clin Cancer Res 2008;14(18):5769–77 doi 10.1158/1078-0432.CCR-07-4282. [DOI] [PubMed] [Google Scholar]
- 65.Sheng T, Li C, Zhang X, Chi S, He N, Chen K, et al. Activation of the hedgehog pathway in advanced prostate cancer. Mol Cancer 2004;3:29 doi 10.1186/1476-4598-3-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cancer Genome Atlas Research N, Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013;497(7447):67–73 doi 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jones S, Stransky N, McCord CL, Cerami E, Lagowski J, Kelly D, et al. Genomic analyses of gynaecologic carcinosarcomas reveal frequent mutations in chromatin remodelling genes. Nat Commun 2014;5:5006 doi 10.1038/ncomms6006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Le Gallo M, O’Hara AJ, Rudd ML, Urick ME, Hansen NF, O’Neil NJ, et al. Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nat Genet 2012;44(12):1310–5 doi 10.1038/ng.2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Blattner M, Lee DJ, O’Reilly C, Park K, MacDonald TY, Khani F, et al. SPOP mutations in prostate cancer across demographically diverse patient cohorts. Neoplasia 2014;16(1):14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rubin MA, Demichelis F. The Genomics of Prostate Cancer: emerging understanding with technologic advances. Mod Pathol 2018;31(S1):S1–11 doi 10.1038/modpathol.2017.166. [DOI] [PubMed] [Google Scholar]
- 71.Testa U, Castelli G, Pelosi E. Cellular and Molecular Mechanisms Underlying Prostate Cancer Development: Therapeutic Implications. Medicines (Basel) 2019;6(3) doi 10.3390/medicines6030082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Boysen G, Rodrigues DN, Rescigno P, Seed G, Dolling D, Riisnaes R, et al. SPOP-Mutated/CHD1-Deleted Lethal Prostate Cancer and Abiraterone Sensitivity. Clin Cancer Res 2018;24(22):5585–93 doi 10.1158/1078-0432.CCR-18-0937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Swami U, Isaacsson Velho P, Nussenzveig R, Chipman J, Sacristan Santos V, Erickson S, et al. Association of SPOP Mutations with Outcomes in Men with De Novo Metastatic Castration-sensitive Prostate Cancer. Eur Urol 2020;78(5):652–6 doi 10.1016/j.eururo.2020.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Abida W, Armenia J, Gopalan A, Brennan R, Walsh M, Barron D, et al. Prospective Genomic Profiling of Prostate Cancer Across Disease States Reveals Germline and Somatic Alterations That May Affect Clinical Decision Making. JCO Precis Oncol 2017;2017 doi 10.1200/PO.17.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I, et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci U S A 2019;116(23):11428–36 doi 10.1073/pnas.1902651116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bernasocchi T, El Tekle G, Bolis M, Mutti A, Vallerga A, Brandt LP, et al. Dual functions of SPOP and ERG dictate androgen therapy responses in prostate cancer. Nat Commun 2021;12(1):734 doi 10.1038/s41467-020-20820-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Olson BM, Gamat M, Seliski J, Sawicki T, Jeffery J, Ellis L, et al. Prostate Cancer Cells Express More Androgen Receptor (AR) Following Androgen Deprivation, Improving Recognition by AR-Specific T Cells. Cancer Immunol Res 2017;5(12):1074–85 doi 10.1158/2326-6066.CIR-16-0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA Sequencing data is available in the NCBI Gene Expression Omnibus (GEO) repository under accession number GSE134682 (reviewer token: ahcroumuppgfveb).