

Abstract
The PRPS1 gene encodes phosphoribosyl pyrophosphate synthetase 1 (PRS-1). The phenotypes associated with PRPS1 mutations include DFN2 (mild PRS-1 deficiency), X-linked Charcot-Marie-Tooth disease type 5 (CMTX5) (moderate PRS-1 deficiency), Arts syndrome (severe PRS-1 deficiency), and PRS-1 superactivity1. CMTX5 is a very rare hereditary neuropathy characterized by deafness, optic atrophy, and polyneuropathy. We herein report a Japanese patient with CMTX5 who had a novel hemizygous mutation c.82 G>C in PRPS1. Despite showing a typical clinical picture, the decrease in enzyme activity measured in the patient's erythrocytes was milder than in previously reported cases.
Keywords: CMTX5, hereditary neuropathy, DFN2, PRPS1, PRS-1
Introduction
X-linked Charcot-Marie-Tooth disease type 5 (CMTX5) is a very rare hereditary neuropathy characterized by deafness, optic atrophy, and polyneuropathy. It is caused by missense mutations in the PRPS1 gene encoding phosphoribosyl pyrophosphate synthetase 1 (PRS-1) (1). PRS1 catalyzes the first step of nucleotide synthesis. The phenotypes associated with PRPS1 mutations include X-linked nonsyndromic sensorineural deafness (DFN2) (mild PRS-1 deficiency), CMTX5 (moderate PRS-1 deficiency), Arts syndrome (severe PRS-1 deficiency), and PRS-1 superactivity (1).
We herein report a Japanese man with CMTX5 caused by a novel mutation in PRPS1.
Case Report
The patient was a 33-year-old Japanese man. He had congenital bilateral hearing loss. A delay in walking had been observed until he was three years old. Reduced visual acuity was recognized at 11 years old. He showed normal intellectuality at school but had difficulty grasping chopsticks and writing. He was able to walk with an ankle foot orthosis until high school but subsequently became wheelchair-bound. He was able to use a personal computer and mobile phone until 30 years old. His visual acuity slowly decreased, and bilateral optic atrophy was recognized by an ophthalmologist. He was thus admitted to our hospital for a precise diagnosis.
The patient's family tree is shown in the Figure. No other family members, including his mother, had polyneuropathy or optic atrophy.
Figure.
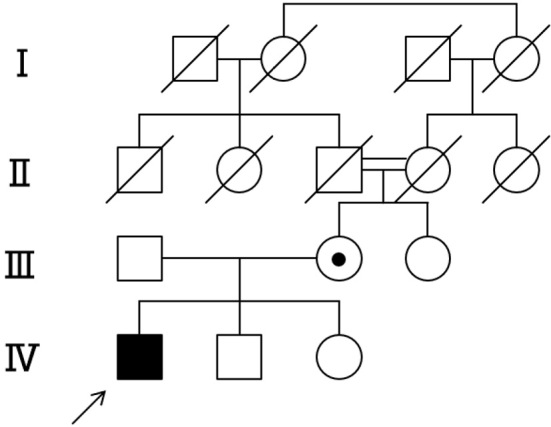
Pedigree of the family with CMTX5. The index patient (marked by an arrow) has optic atrophy, deafness and polyneuropathy and is shown in all black. His mother is an obligate carrier.
Neurological examinations showed bilateral optic atrophy and hearing loss. His visual acuity was limited to light sense (S.I.). He had both ape and claw hands as well as pes cavus deformity. Distal muscle atrophy was recognized both in the upper and lower extremities. Manual muscle testing revealed distal muscle weakness, as follows: deltoid (5/5), biceps (5/5), triceps (5/5), wrist extensors (0/0), wrist flexors (1/1), finger extensors (0/0), finger flexors (1/1), iliopsoas (4/4), quadriceps (5/5), hamstrings (4/4), tibialis anterior (0/0), and gastrocnemius (0/0) (MRC rating scale). Hypesthesia was observed distally in the lower extremities. His vibration sense was absent in the ankles, and his position sense in the toes was disturbed. His tendon reflexes were reduced in both the upper and lower extremities. Routine hematological testing revealed normal findings. The serum level of creatine kinase was elevated at 1,118 U/L (normal range, 54-324 U/L). The serum uric acid level was within normal limits, as were the cell count, protein and sugar levels, and IgG index in the cerebrospinal fluid.
Nerve conduction studies revealed severe motor sensory neuropathy (Table 1). Brainstem auditory-evoked potentials were not evoked bilaterally. He had normal brain magnetic resonance imaging findings.
Table 1.
Nerve Conduction Studies.
| Side | (Normal value) | Left side | Right side | |||
|---|---|---|---|---|---|---|
| Median nerve | ||||||
| DL (ms) | <4.5 | 5.2 | 5.2 | |||
| CMAP (mV) | >8.6 | 0.3 | 0.3 | |||
| MCV (m/s) | >45.6 | 31.7 | 37.2 | |||
| SNAP (µV) | >7.0 | NE | NE | |||
| SCV (m/s) | >43.9 | - | - | |||
| Ulnar nerve | ||||||
| DL (ms) | <3.6 | 3.7 | 3.9 | |||
| CMAP (mV) | >8.8 | 0.9 | 1.0 | |||
| MCV (m/s) | >51.2 | 25.9 | 28.3 | |||
| SNAP (µV) | >7.0 | NE | NE | |||
| SCV (m/s) | >44.4 | - | - | |||
| Tibial nerve | ||||||
| DL (ms) | <5.1 | - | 10 | |||
| CMAP (mV) | >12.0 | NE | 0.04 | |||
| MCV (m/s) | >32.1 | - | 38.3 | |||
| Sural nerve | ||||||
| SNAP (µV) | >7.0 | NE | NE | |||
| SCV (m/s) | >36.4 | - | - |
DL: distal latency, CMAP: compound muscle action potential, MCV: motor nerve conduction velocity, SNAP: sensory nerve action potential, SCV: sensory nerve conduction velocity
With informed consent, systematic DNA tests for CMT were performed, identifying a novel hemizygous mutation c82 G>C (p.G28R) in the PRPS1 gene. His mother was heterozygous for this mutation, and his aunt had only a normal allele.
To reveal the significance of this mutation, the PRS-I enzymatic activity in the patient's erythrocytes was measured, and a marked reduction to 7.4 nmol/h per mg hemoglobin was found [normal values: 39±7 nmol/h per mg hemoglobin (2)]. The PRS-I enzymatic activities in his mother and aunt were 34.5 and 46.1 nmol/h per mg hemoglobin, respectively.
Discussion
A Japanese man with CMTX5 characterized by congenital hearing loss, optic atrophy, and peripheral neuropathy was reported. A genetic analysis revealed c.82 G>C (p.G28R) in the PRPS1 gene, a new mutation that has not yet been reported. Since glycine at position 28 is strictly conserved from birds to humans, a substitution in this amino acid can have a significant impact on the protein function. In fact, the enzyme activity of this patient was reduced to about 20% of the level in healthy individuals. In addition, the clinical phenotype was segregated with the mutation among family members. Given these findings, we concluded that the mutation causes CMTX5.
CMTX5 is a very rare disease, and to our knowledge, only eight families have been reported thus far (Table 2) (1,3-7). E43D and M115T mutations in PRPS1 were first identified in Korean families with typical manifestations (1). A121G, V309F, M115V, and I109V were associated with the phenotype without optic atrophy (3,6). A heterozygous female patient with Q277P mutation showed only hearing loss (DFN2) in a family where a hemizygous brother exhibited a more severe phenotype of CMTX5/Arts syndrome. Although reports of cases in which PRS-1 enzyme activity was accurately measured are limited, variable clinical phenotypes associated with PRPS1 mutations were thought to be determined by the residual enzyme activity (5). Our patient has a milder reduction in PRS-I activity (7.4 nmol/mg/h) than other reported cases with PRPS1 mutations, even though he showed the typical CMTX5 phenotype. This indicates that the residual enzyme activity does not necessarily determine the clinical phenotype of PRPS1 mutations.
Table 2.
PRPS1 Mutations Identified in CMTX5 Patients.
| Disorder | Gene mutation | Amino acid change | Neuropathy | Hearing loss | Optic atrophy | PRS-I activity (±SD) | Reference |
|---|---|---|---|---|---|---|---|
| CMTX5 | c. 129 A>C | p. E43D | + | + | + | Decreased | 1 |
| CMTX5 | c. 344 T>C | p. M115T | + | + | + | Decreased | 1 |
| CMTX5 | c. 362 C>G | p. A121G | + | + | - | Not described | 3 |
| CMTX5 | c. 46 T>C | p. S16P | + | + | + | Decreased | 4 |
| CMTX5/Arts syndrome | c. 830 A>C | p. Q277P | + | + | + | <0.06 | 5 |
| DFN2* | - | + | - | 6.00 | |||
| DFN2 and peripheral neuropathy | c. 925 G>T | p. V309F | + | + | - | 2.11 (±1.32) | |
| 1.06 (±0.19) | 6 | ||||||
| c. 343 A>G | p. M115V | + | + | - | 0.84 (±0.1) | ||
| CMTX5 | c. 319 A>G | p. I109V | + | + | - | Not described | 7 |
| CMTX5 | c. 82G>C | p. G28R | + | + | + | 7.40 | Our case |
*DFN2: deafness, X-linked 2
In the future, gathering more cases and examining the relationship between residual enzyme activity and phenotype will be necessary.
The authors state that they have no Conflict of Interest (COI).
References
- 1. Kim HJ, Sohn KM, Shy ME, et al. Mutations in PRPS1, which encodes the phosphoribosyl pyrophosphate synthetase enzyme critical for nucleotide biosynthesis, cause hereditary peripheral neuropathy with hearing loss and optic neuropathy (CMTX5). Am J Hum Genet 81: 552-558, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Micheli V, Rocchigiani M, Pompucci G. An HPLC-linked assay of phosphoribosylpyrophosphate synthetase activity in the erythrocytes of adults and children with neurological disorders. Clin Chim Acta 227: 79-86, 1994. [DOI] [PubMed] [Google Scholar]
- 3. Park J, Hyun YS, Kim YJ, et al. Exome sequencing reveals a novel PRPS1 mutation in a family with CMTX5 without optic atrophy. J Clin Neurol 9: 283-288, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Almoguera B, He S, Corton M, et al. Expanding the phenotype of PRPS1 syndromes in females: neuropathy, hearing loss and retinopathy. Orphanet J Rare Dis 9: 190, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Matthis S, Jennifer MH, Tobias BH, et al. X-linked Charcot-Marie-Tooth disease, Arts syndrome, and prelingual non-syndromic deafness form a disease continuum: evidence from a family with a novel PRPS1 mutation. Orphanet J Rare Dis 9: 24, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Robusto M, Fang M, Asselta R, et al. The expanding spectrum of PRPS1-associated phenotypes: three novel mutations segregating with X-linked hearing loss and mild peripheral neuropathy. Eur J Hum Genet 23: 766-773, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nishikura N, Yamagata T, Morimune T, et al. X-linked Charcot-Marie-Tooth disease type 5 with recurrent weakness after febrile illness. Brain Dev 41: 201-204, 2019. [DOI] [PubMed] [Google Scholar]