

Abstract
Kidney disease is poorly understood due to the organ’s large cellular diversity. We used single cell RNA-sequencing not only to resolve differences in injured kidney tissue cellular composition but also in cell type-specific gene expression in mouse models of kidney disease.
This analysis highlighted major changes in cellular diversity in disease, which markedly impacted whole-kidney transcriptomics outputs. Cell type-specific differential expression analysis identified proximal tubule (PT) cells as the key vulnerable cell type. Through unbiased cell trajectory analyses, we show that PT cell differentiation is altered in kidney disease. Metabolism (fatty acid oxidation and oxidative phosphorylation) in PT cells showed the strongest and most reproducible association with PT cell differentiation and disease. Coupling of cell differentiation and metabolism was established by nuclear receptors (ESRRA and PPARA) that directly control metabolic and PT cell-specific gene expression in mice and patient samples, while protecting from kidney disease in the mouse model.
Keywords: Singe cell RNA-sequencing, single cell ATAC sequencing, kidney, fibrosis, organoids, fatty acid oxidation, PPARA, ESRRA, proximal tubule cells, chronic kidney disease
Introduction
Kidney disease is becoming a major health issue in modern society. Chronic kidney disease (CKD) is the tenth leading cause of death worldwide with a steadily increasing incidence affecting eight hundred million people globally (Levin et al., 2017). The large number of people affected by CKD is of concern because some will progress to end-stage renal disease (ESRD), severely affecting quality of life. In addition, kidney disease is a massive personal and societal economic burden (Breyer and Susztak, 2016; Kovesdy et al., 2013).
Genetic studies examining the heritability of kidney function, such as integration of genome wide association studies (GWASs) (Wuttke et al., 2019) and functional genomic studies, highlighted the role of proximal tubule-specific genes in kidney function (Hellwege et al., 2019; Park et al., 2018; Qiu et al., 2018). PT cells are highly susceptible to toxic and hypoxic injury, representing the primary cause of acute kidney injury (AKI) (Qiu et al., 2018). PT cell-specific injury observed in AKI probably has the most rapid effect on kidney function. CKD, which is defined by more than 40% decline in GFR for more than 3 months, is characterized by PT cell atrophy almost independent of disease etiology. PT cell atrophy strongly correlates with kidney function in CKD (Chang-Panesso and Humphreys, 2017; Reidy et al., 2014) (Liu et al., 2014) (Kang et al., 2015; Li et al., 2012).
Comprehensive genome-wide kidney tissue transcriptomics analysis has been used to define the molecular hallmarks of this complex process, both in patient samples and mouse models (Beckerman et al., 2017; Qiu et al., 2018; Woroniecka et al., 2011). These studies highlighted a correlation between a large number of transcripts and kidney fibrosis. Cellular metabolism, such as genes involved in lipid metabolism, fatty acid oxidation (FAO) and oxidative phosphorylation (OXPHOS) showed strong correlation with disease state, both in patients and mouse CKD models (Chung et al., 2019; Kang et al., 2015). Pharmacological or genetic approaches that enhance FAO and mitochondrial biogenesis improved kidney function; however, the exact mechanism is not fully understood (Gomez et al., 2015; Tran et al., 2011, Tran et al., 2016; Zheng et al., 2019). Further, mitochondrial defects can lead to the leakage of the mitochondrial DNA into the cytoplasm resulting in the activation of the cGAS/STING innate immune system pathway, cytokine release and influx of immune cells and downstream fibrosis development (Chung et al., 2019: Maekawa et al. 2019).
Single-cell RNA-sequencing (scRNA-seq) analysis is transforming our understanding of complex diseases. In our previous study, we identified 21 distinct cell types, including 3 novel cell types in the kidney (Park et al., 2018). At the same time, we defined cell identity genes that can stably and reproducibly classify key kidney cell types in mice and humans (Young et al., 2018).
Here we analyzed the transcriptome of different CKD mouse models, human kidney samples and human organoids. Prior studies mostly relied on single nuclear sequencing and did not properly capture the immune cell diversity (Lake et al., 2019; Wu et al., 2019). We identify several PT cell subgroups, and using cell trajectory analyses we show an alteration in the differentiation state of PT cells in diseased kidneys. Single-cell epigenetics and transcriptomics indicate the critical role of HNF4A, HNF1B (hepatocyte nuclear factor 4A and 1B), PPARA (peroxisomal proliferation-activated receptor alpha) and ESRRA (estrogen related receptor alpha) in defining PT cell identity. Using mouse knock-out and human kidney transcriptomics data we further demonstrate the protective role of ESRRA, which links energy metabolism, proximal tubule differentiation and kidney function by directly binding to PT cell-specific genes and regulating their expression.
Results
The single-cell landscape shows increased cellular heterogeneity in fibrotic kidneys
To unravel cellular changes associated with kidney fibrosis, first we analyzed the transcriptome of 65,091 individual cells from 6 mouse control kidneys and 2 folic acid-induced fibrotic kidneys (folic acid nephropathy: FAN) (Figure 1A). This is a well- established kidney disease model presenting both with structural damage (fibrosis) and kidney function decline indicated by serum BUN level (Figure S1A–S1C). We observed that PT cells represented the majority of cell types in the dataset. To accurately cluster smaller cell populations, we first focused on non-PT cells (Figure 1B). Our unbiased clustering identified 30 cell populations, including kidney epithelial, immune and endothelial cells based on marker gene expression (Figure 1C and Figure S1D, Table S1). The proportion of cells were relatively stable in biological replicates but were substantially different between control and FA samples (Figure S1E). Gene sets previously used to define cell types (cell identity genes) showed conserved expression in disease state (Figure S1F). Immune cell diversity was markedly increased in the FAN mice (Figure S1D and S1E). Such as we identified 14 immune cell clusters in our FAN model while we our previous study characterized 5 immune clusters (Park et al., 2018). Amongst the newly identified, we observed granulocytes, macrophages, dendritic cells (DCs) and basophils. DCs were further subclustered into DC 11b+ (Cd209a and Cd11b), DC 11b- (Cd24a and Clec9a) and plasmacytoid DC clusters (Siglech and Cd300c) (Figure 1D and 1E). A large number of lymphoid cells were also identified, including B cells, T cells, and natural killer cells (Figure 1B and 1C). T lymphocytes were subclustered into CD4+ T, Treg, gamma delta T, NKT, and CD8+ effector cells (Figure 1F and 1G). We made this dataset publicly available on our interactive website (http://susztaklab.com/VisCello/).
Figure 1. The cellular diversity of diseased kidney samples.
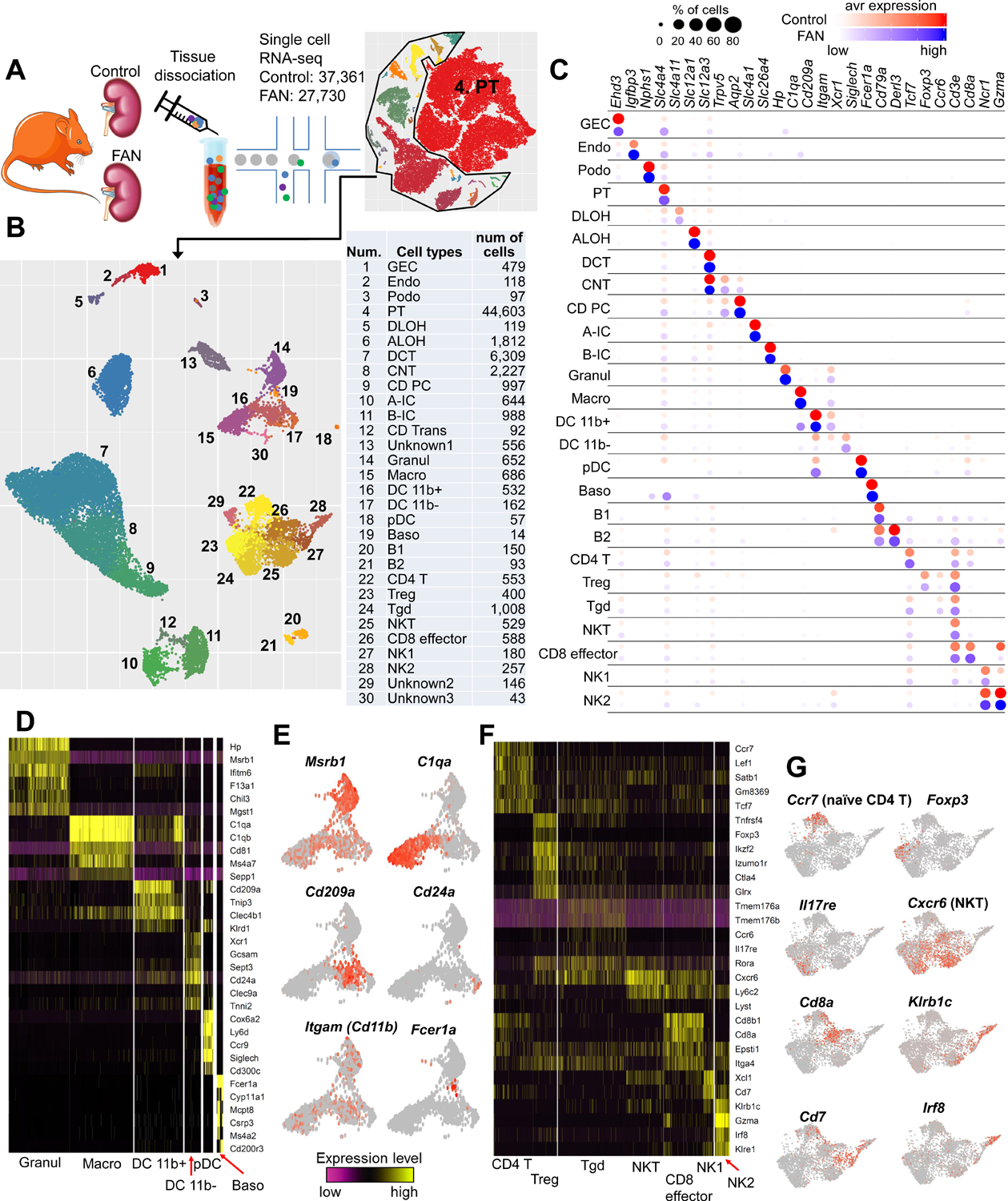
(A) A schematic diagram illustrating the experimental procedure involving the digestion of whole-kidney tissue from 6 control and 2 FAN mice followed by sequencing using a 10x Genomics protocol. and transcriptomic analysis of 65,091 individual cells.
(B) Left, the UMAP of 29 distinct cell types identified by unsupervised clustering after excluding the proximal tubule cells. Right, the tSNE plot for the entire dataset including proximal tubule cells. Assigned cell types are summarized in right panel. GEC: glomerular endothelial cells, Endo: endothelial, Podo: podocyte, PT: proximal tubule, DLOH: descending loop of Henle, ALOH: ascending loop of Henle, DCT: distal convoluted tubule, CNT: connecting tubule, CD-PC: collecting duct principal cell, A-IC: alpha intercalated cell, B-IC: beta intercalated cell, CD-Trans: collecting duct transitional cell, Granul: granulocyte, Macro: macrophage, DC 11b+: CD11b+ dendritic cell, pDC: plasmacytoid DC, Baso: Basophile B: B lymphocyte, Treg: regulatory T cell, Tgd: gamma delta T cell, NK: natural killer cell.
(C) Bubble plots of cell cluster marker genes identified in control and FAN samples (size of the dot indicates the % positive cells, color indicates relative expression).
(D) Heatmap showing expression pattern of myeloid lineage markers.
(E) Gene expression feature plots of myeloid lineage cells projected onto the UMAP. Msrb1 (Methionine Sulfoxide Reductase B1): Granul, C1qa (Complement C1q A Chain): Macro, Cd209a (CD209 antigen-like protein A): DC 11b+, Cd24a (CD24a antigen): DC 11b-, Itgam (Integrin Subunit Alpha M): DC 11b+, and Fcer1a (Fc Fragment Of IgE Receptor Ia): Baso.
(F) Heatmap showing expression pattern of lymphoid lineage markers.
(G) Gene expression feature plots of lymphoid lineage cells projected onto the UMAP. Ccr7 (Chemokine C-C motif receptor 7): CD4 T, Foxp3 (Forkhead box P3): Treg, Il17re (Interleukin 17 receptor E): Tgd, Cxcr6 (Chemokine C-X-C motif receptor 6): NKT, Cd8a (CD8 antigen, alpha chain): CD8 effector, Klrb1c (Killer cell lectin-like receptor subfamily B member 1C): NK1–2, Cd7 (CD7 antigen): NK1 and Irf8 (Interferon regulatory factor 8): NK2
Bulk RNA-sequencing strongly influenced by cell fraction changes
We next performed RNA-sequencing of whole-kidney (i.e. bulk tissue) samples, as single-cell sequencing may suffer from uneven cell drop-out. Differential expression analysis of bulk RNA-seq data indicated changes in expression of more than 4,000 genes (2,776 with higher and 1,361 with lower expression, using FDR of 0.05 and fold change 2) (Figure 2A). Gene ontology analysis highlighted that expression of genes associated with the immune system and inflammation were higher in the FAN model (Figure 2A). Analysis of genes showing the highest increased expression in the bulk dataset indicated that most such genes were exclusively expressed by immune cells (Figure 2B). Genes whose levels were lower in the FAN model were enriched for metabolic processes, such as lipid metabolism, FAO and OXPHOS (Figure 2A). Genes with lower expression in the FAN model showed high enrichment for PT cell expression (Figure 2B), suggesting a strong role for PT cells and immune cells driving transcriptional changes in bulk RNA sequencing data. In addition, we observed that highly expressed and top differentially expressed genes, including Lyz2, Cd52 and Tyrobp, in the bulk RNA-seq data showed similar expression patterns in control and FAN samples at a single-cell level (Figure S2A), suggesting that the majority of genes showing higher expression in disease were related to immune cell proportion changes rather than cell specific changes.
Figure 2. Cell composition and cell type specific changes in kidney fibrosis.
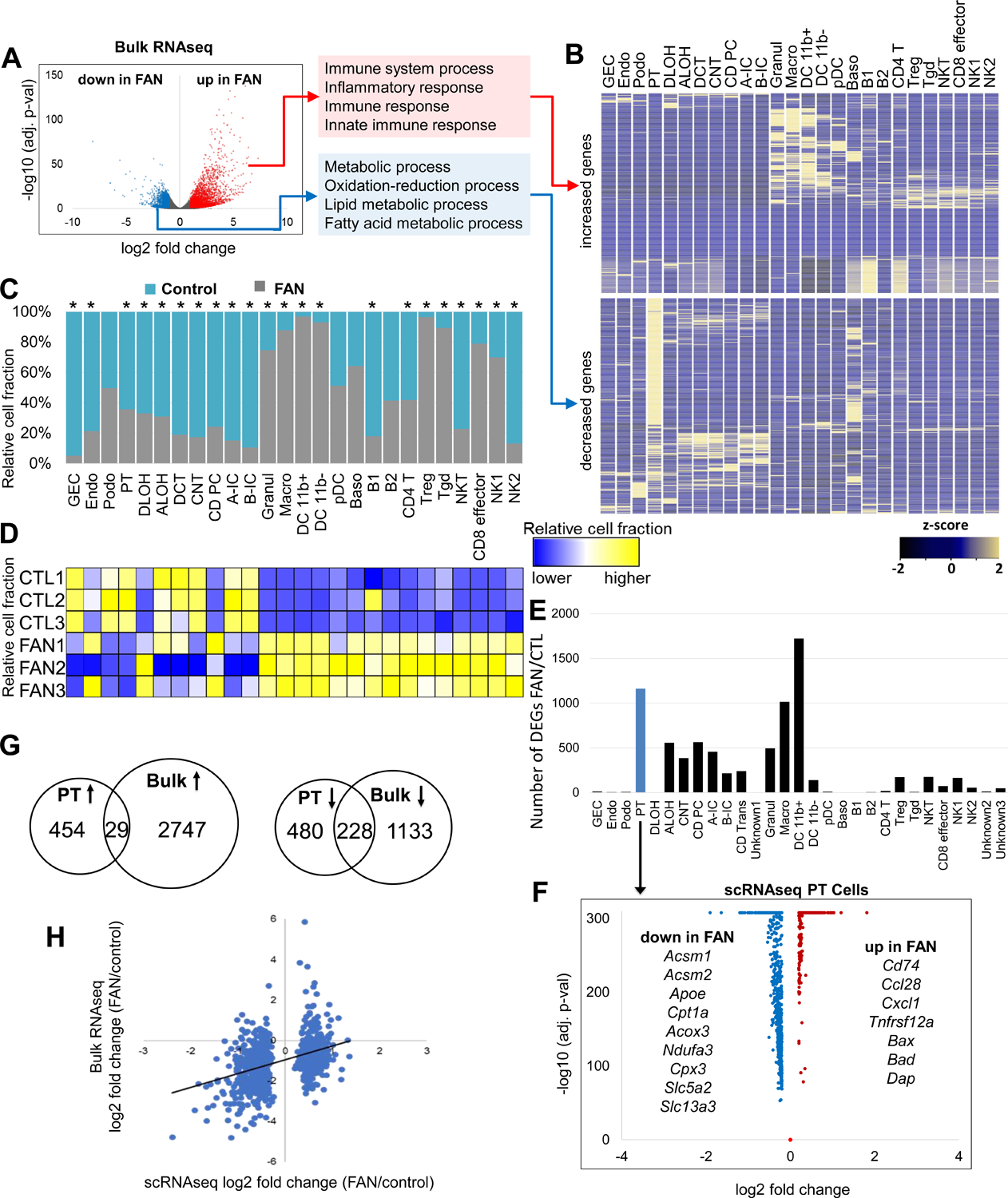
(A) Differentially expressed genes (DEGs) in whole kidneys of control and FAN mice. Volcano plot, the x-axis indicates log2 fold change and Y-axis indicates statistical significance adjusted p −log10. Gene ontology analysis of genes showing higher (red) and lower (blue) in FAN kidneys.
(B) Cell type-specific expression of top DEGs identified in bulk RNA-seq analysis in the single-cell dataset. Mean expression values of the genes were calculated in each cluster. The color scheme is based on z-score distribution.
(C) Cell proportion changes in control and FAN kidneys revealed by single cell RNA-sequencing. * indicates significant changes by proportion test.
(D) Cell proportion changes revealed by in silico deconvolution of bulk RNA sequencing data.
(E) The numbers of cell type-specific differentially expressed genes identified in control FAN kidneys in the 30 cell clusters.
(F) Volcano plot for differentially expressed genes (DEGs) between control and FAN proximal tubules identified in the single cell data. X-axis is log2 fold change and Y-axis is statistical significance adjusted p −log10.
(G) Venn diagrams showing the overlaps between the identified differentially expressed genes in PT cell by scRNA-seq data and bulk RNA-seq data in control vs FAN kidneys (Up arrow: upregulated genes and down arrow : downregulated genes).
(H) Scatter plot showing the correlation of DEGs identified in PT cell and bulk data. X-axis shows the fold change expression in PT cells in the single cell data, Y-axis shows the fold change expression in whole kidney (bulk) samples.
Next, we determined cell proportion changes using single-cell and bulk RNA-seq data. We found marked differences in cell proportion, such as a distinct increase in myeloid and lymphoid cell proportion (e.g. macrophages (7.2-fold), granulocytes (2.9-fold), Tregs (27-fold) and CD8 effector cells (3.8-fold)), while the proportion of tubule epithelial cells (e.g., PT (0.55-fold) and distal convoluted tubule (0.23-fold)) was lower in the single-cell data of the FAN model (Figure 2C). On the other hand, consistent with prior observations, proportion of podocytes did not show clear changes in the FAN model (Figure 2C). We also performed in silico deconvolution of bulk RNA-seq data implemented in the CellCODE package. This analysis yielded results broadly consistent with the single-cell RNA sequencing data (Figure 2D), such as higher immune cell fractions and lower epithelial cell fractions. Finally, cell proportion changes in epithelial and immune cells were confirmed by histological analysis (Figure S1A).
To further understand the contribution of cell type-specific and cell fraction changes in bulk RNA-seq results, we directly compared the single-cell with the bulk data. After adjusting the data to the observed cell fraction changes, the number of genes showing differential expression were markedly reduced. Among the 4,137 differentially expressed genes in bulk data, only 14, 2, 902 and 753 genes remained significant after adjustment by proximal convoluted tubule (PCT), proximal straight tubule (PST), myeloid or lymphoid cell fractions, respectively (Figure S2B).
To unravel cell type-specific gene expression changes in the FAN model, we performed differential expression analysis in all identified cell types. Keeping in mind the limitation of this analysis, such as the complete confounding of the disease state and possible batch effect, we found that myeloid cells, such as macrophages, showed a large number of differentially expressed genes (Figure 2E). We found that amongst the epithelial cells, PT cells showed the largest number of differentially expressed genes (Figure 2E, Table S2). Genes that showed lower expression levels in diseased PT cells are solute carrier (cell differentiation-related genes) such as Slc5a2 and Slc13a3, as well as genes involved in FAO and OXPHOS (e.g., Acsm1, Acsm2, Cpt1a, Acox3) (Figure 2F).
Even though PT cells represented a large portion of the bulk dataset, only a small fraction of differentially expressed genes observed in PT cells was shared in the bulk RNA-seq data (Figure 2G) and correlation between PT cell-specific differentially expressed genes in single-cell and bulk data was weak. Less than 10% of PT cell-specific differentially expressed genes showed direction-consistent and observable changes in the bulk gene expression data (Figure 2H).
Altered differentiation drives proximal tubule response during fibrosis
To better understand cell state changes in PT cells, we performed sub-clustering and cell trajectory analysis of healthy and diseased samples from CKD mouse models. In healthy controls, we identified several PT cells subtypes, including PST cells expressing Slc22a30, and several subgroups of PCT expressing Slc5a2 and Slc5a12 (Figure 3A, Table S3). RNA velocity analysis has opened up new ways of studying cellular differentiation (La Manno et al., 2018) by predicting the future state of individual cells. Our analysis indicated that in control kidneys, PT cells differentiated into 2 major cell types: PCT and PST segments (Figure 3B and 3C). Interestingly, the analysis highlighted that PT cells originated from a common precursor-like cell, expressing higher levels of Med28 and Cycs (Figure 3D). Importantly, our analysis also suggested that PT cell differentiation did not necessitate cell proliferation, as we did not observe changes in the expression of proliferation markers (Figure 3D).
Figure 3. Heterogeneous proximal tubule cell populations in fibrotic kidneys.
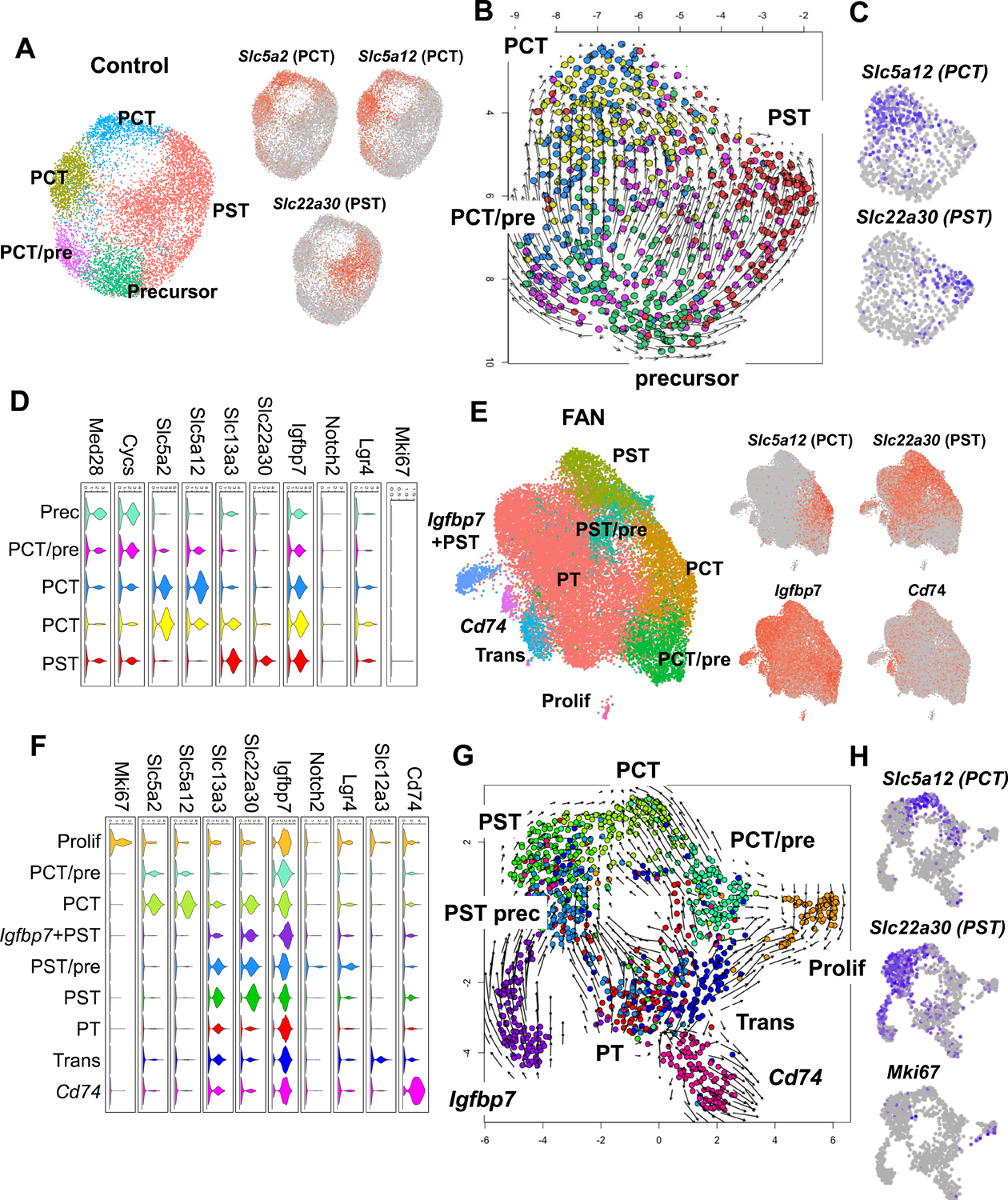
(A) Sub-clustering of PT cells into 5 sub-populations in control kidneys. Feature plots showing expression of key PCT (Slc5a2 and Slc5a12) and PST (Slc22a30) segment markers.
(B) RNA velocity analysis of control PT cells. Each dot is one cell and each arrow represent the time derivative of the gene expression state.
(C) Feature plots showing expression of key PCT (Slc5a12) and PST (Slc22a30) segment markers in control.
(D) Violin plots showing the expression patterns of markers across the PT cell sub-clusters in control. The y-axis shows the log-scale normalized read count.
(E) Sub-clustering of PT cells into 9 sub-populations in FAN kidneys. Feature plots showing expression of key PCT (Slc5a12) and PST (Slc22a30), Igfbp7 (precursor) and Cd74 (immune) PT cell state markers.
(F) Violin plots showing the expression patterns of markers across the PT cell sub-clusters in FAN. The y axis shows the log-scale normalized read count.
(G) RNA velocity analysis of FAN PT cells. Each dot is one cell and each arrow represent the time derivative of the gene expression state.
(H) Feature plots showing expression of key PCT (Slc5a12) and PST (Slc22a30) and proliferating (MKi67) PT cell markers.
PT cells from FAN samples subclustered into 9 groups. Using anchor genes to identify key cell types such as PCT and PST segments, we were able to recognize more heterogeneous cell populations, including proliferating cells, immune marker (Cd74)-expressing cells, transitional cells and precursor cells expressing higher Igfbp7 (Figure 3E, Table S3). In diseased samples, we also identified a prominent proliferating (i.e., Ki67-positive) cell population and it appeared that cells entered and exited this Ki67-positive state. These data are consistent with a facultative progenitor model in kidney tubule cells (Angelotti et al., 2012; Kang et al., 2016) (Figure 3F). Notably, we identified a cell population expressing Notch2 and Lgr4, previously identified as progenitor and transit amplifying cells in the kidney and other organs (de Lau et al., 2011; Zhang et al., 2019). We observed that PCT cells co-expressed PST markers, suggesting that under disease conditions PCT cells may endure transcriptomic changes impacting on their phenotypic signature. Similar to our observation in control samples (Figure 3B), FAN samples showed a differentiation trajectory toward PCT and PST segments (Figure 3G and 3H) but followed a less organized differentiation path than healthy PT cells (Figure 3G). On the other hand, we failed to observe a clear reversal of differentiation of cells already expressing terminal differentiation markers such as Slc5a2 or Slc22a30, indicating that a failure of differentiation rather than dedifferentiation is the reason for the identified cell-state changes.
Differentiation defects in fibrotic proximal tubules track with changes in lipid metabolism
Next, we opted to take advantage of the continuous cell trajectory analysis using the Monocle package by combining all samples under healthy and disease states (Trapnell et al., 2014). Initial exploration showed a clear branching of PT cells into PCT and PST segments (Figure 4A, 4B and Figure S3A), which was mostly consistent with the RNA velocity analysis. To identify genes whose expression changed along the trajectory, we first performed trajectory analysis for the PST segment, as this segment is highly susceptible to injury (Figure 4C, 4D and Figure S3B). Cells from control and diseased kidneys seemed to follow a similar linear trajectory towards PST segment differentiation (i.e., no major branching) but FAN samples were significantly depleted from terminally differentiated PT cells (two sample proportion test, between cells in red and blue area, p-value < 2.2e−16) (Figure 4C and 4E). Trajectory analysis of PCT segment cells showed similar pattern (Figure S3C and S3D). These data are consistent with prior observations indicating lower levels of terminally differentiated markers in FAN samples (Figure 3G).
Figure 4. Cell trajectory analysis identifies differentiation defect in proximal tubule in fibrosis.
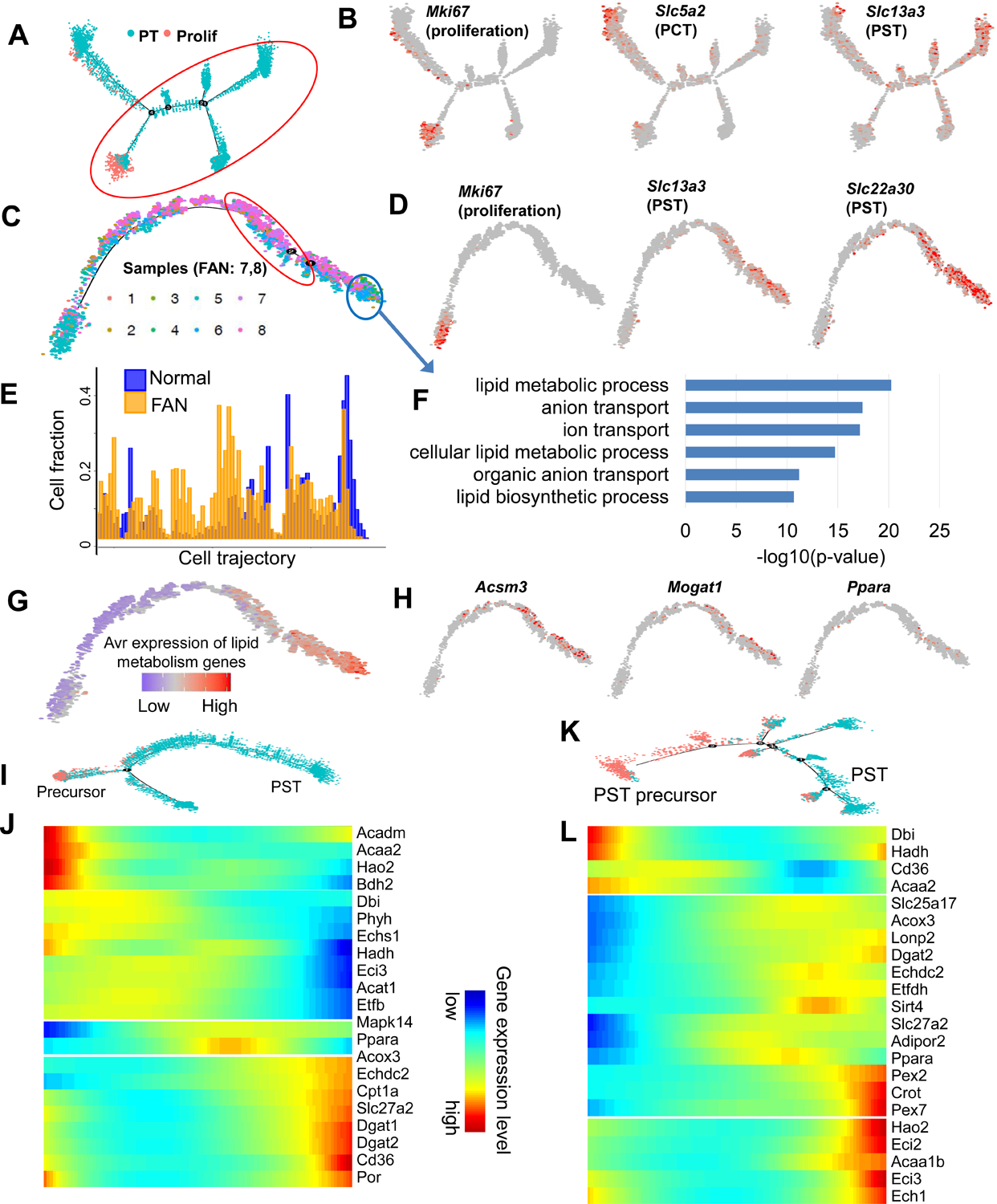
(A) Trajectory analysis of PT cells (including proliferating cells) using Monocle, including all control and FAN samples.
(B) Feature plots showing expression levels of key cell state markers (Mki67: proliferating cell, Slc5a2:PCT, Slc13a3:PST) on the cell trajectory.
(C) Cell trajectory analysis narrowed for PST cluster (cells under red circle in panel A). Batches 1–6 represent healthy kidneys, while batches 7–8 were obtained from FAN samples.
(D) Feature plots showing expression levels of key cell state markers (Mki67: proliferating cell, Slc22a30:PST, Slc13a3:PST) on the cell trajectory.
(E) Distributions of cells along the pseudo-time trajectory. Note the shift of Normal (blue) and FAN samples (yellow).
(F) Functional annotation (gene ontology) analysis of genes showing changes along the trajectory (cells highlighted by red and blue circles on panel C).
(G) Average expression levels of the highly variable genes that are involved in lipid metabolism along the cell trajectory.
(H) Feature plots showing expression levels of the lipid metabolism genes (Acsm3, Mogat1, Ppara) along the cell trajectory.
(I) Cell trajectory analysis for PST and precursor clusters identified in control kidneys (Figure 3A).
(J) Heatmap showing the expression changes of highly variable FAO genes along the cell trajectory in control kidneys.
(K) Cell trajectory analysis for PST and precursor clusters identified in FAN samples (Figure 3E).
(L) Heatmap showing the expression changes of highly variable FAO genes along the cell trajectory in FAN samples.
When we interrogated genes and pathways that underlie the PT cell differentiation state, we found that the expression genes associated with terminal differentiation, such as those with ion transport function (i.e., SLC solute careers) increased along the differentiation trajectory (Figure 4F). In addition to ion transport, lipid metabolism showed a positive correlation with cellular differentiation (Figure 4F–4H and Figure S3E). Moreover, we observed changes in FAO genes along the differentiation path from precursor to PT cells in both healthy controls and FAN samples (Figure 4I–4L).
We also generated scRNA-seq data from the unilateral ureteral obstruction (UUO) model of kidney fibrosis and compared cell trajectories in the UUO and the FAN models. Continuous cell trajectory analysis showed selective lack of terminally differentiated PT cells in UUO kidneys, recapitulating the results obtained from the FAN model (Figure S3F–S3H). In addition, when examining pathways associated with differentiation of PT cells along the cell trajectory, we found enrichment for FAO, OXPHOS and ion transport (Figure S3I and S3J). There was a strong (>50%) overlap of gene expression changes along the respective differentiation trajectories in the UUO and FAN models (Figure S3K).
Partial epithelial-mesenchymal transition (EMT) has been used to describe the aberrantly differentiated PT cells (Grande et al., 2015; Lovisa et al., 2015; Zeisberg and Duffield, 2010). We found that EMT markers tended to be lower upon PT cell differentiation (Figure S3L), we failed to observe significant expression of Zeb, Twist and Snai in the different PT cell subclusters in fibrotic samples (Table S4). We also failed to observe cells exhibiting classic senescence markers (SASP; senescence associated secretory phenotype) (Table S4).
Proximal tubule differentiation in kidney organoids correlates with metabolic changes
To distinguish whether lipid metabolism and OXPHOS only enhance PT cell-specific genes expression or they are true drivers of PT cell maturation, we tested the role of FAO and OXPHOS in tubule cell differentiation of developing kidney organoids. Previously, we showed three-dimensional (3D) culture systems that recapitulate architectural and functional features of the human developing kidney; so-called kidney organoids generated from human pluripotent stem cells (hPSCs) (Garreta et al., 2019). We generated hPSCs-kidney organoids in free-floating conditions by assembling nephron progenitor cells (NPCs) derived from hPSCs (Figure 5A). Bulk gene expression analysis of differentiating organoids indicated an increase in expression of PPARGC1A on days 16 and 21. The increase in PPARGC1A expression in organoids correlated with the expression of PT cell markers, such as SLC27A2, SLC3A1, SLC5A12 (Figure 5B). As bulk RNA expression data cannot provide a faithful read-out for proximal tubule differentiation, we performed unbiased scRNA-seq analysis (Figure 5C). Clustering analysis based on cell type-specific marker gene expression indicated, that in addition to mesenchymal clusters we could also identify a variety of kidney cell types resembling those of collecting duct, actively cycling cells, endothelial cells, podocytes, loop of Henle and PT cells (Figure 5C and 5D). Next, we specifically examined the differentiation trajectory of organoid PT cells. Cells differentiated from a SIX1 positive progenitor and gained PT cell marker SLC3A1 expression (Figure 5E). Next, we analyzed genes whose expression changed along this trajectory and found that the expression of differentiation markers such as solute carriers increased along the trajectory and their expression strongly correlated with genes in FAO, including PPARA (Figure 5F). Finally, to confirm that FAO is a driver of cellular differentiation, we cultured kidney organoids in glycolytic (EGM) or OXPHOS promoting (REGM) media. We found higher expression of PPARGC1A mRNA and lipid metabolic genes such as ACOX2, ACOT12, and CPT1A, when organoids were cultured in REGM media for 4 days versus EGM media (Figure 5G). We further assessed the protein levels of mitochondrial OXPHOS proteins in organoids exposed to both REGM and EGM culture media (Figure 5H and Figure S4A, S4B). Concomitantly to these metabolic changes, we observed that an REGM lead to an increase in the expression of PT cell markers such as SLC34A1, SLC27A2, SLC5A12, SLC6A19 and SLC3A1 compared to EGM media (Figure 5G). These results run in parallel with our in vitro observations of cultured PT cells from mice (Figure S4C and S4D). In addition, organoids exhibited a visibly higher number of proximal tubules as observed by immunofluorescence (IF) analysis for LTL labeling (Figure 5I and 5J).
Figure 5. FAO and OXPHOS drives proximal tubule differentiation in human kidney organoid.

(A) Experimental scheme for the generation of human kidney organoid. Briefly, hPSCs were first differentiated into posterior primitive streak (PPS) fate then to intermediate mesoderm (IM). Cells were aggregated (day 0, D0) and further differentiated in 3D culture into renal vesicle (RV) and nephron stage. At D20 of differentiated kidney organoids were stained for podocalyxin (PODXL: podocyte marker, yellow), Wilm’s tumor 1 (WT1, red), and Lotus tetragonolobus Lectin (LTL: PT marker, green). Scale Bar=200μM.
(B) Transcript expression levels (in bulk organoids) of PPARGC1A, SLC3A1, SLC5A12 and SLC27A2 on day 4, 8, 12, 16 and 20 of organoid differentiation. Data are represented as mean ± SEM. n = 2 independent experimental replicates analyzed from a pool of 12 organoids/group.
(C) Single-cell RNA-seq analysis of human kidney organoid. UMAP showing 9 distinct cell types identified by unsupervised clustering. Mesench: mesenchymal cells, CD: collecting duct, Endo: endothelial cells, cycling: cell cycling cells, Podo: podocytes, LOH: loop of Henle and PT: proximal tubule.
(D) Feature plots of key cell type markers (DES, COL21A1, GATA3; mesenchyme, NPHS1; podocytes, SLC3A1;PT cell, SLC12A1;LOH, PCNA, CCNA2;proliferating cells, PECAM11;endothelial cells).
(E) Expression SIX1 (nephron progenitor marker), MKI67 (proliferation maker) and SLC3A1 (PT cell marker) along the differentiation trajectory.
(F) Heatmap showing the expression changes of highly variable genes involved in FAO identified (Figure 4J, 4L) along the organoid cell differentiation trajectory.
(G) Expression level of genes associated with FAO (PPARGC1A, ACOX2, and CPT1A), and PT cell markers (ATP11A, ACOX12, SLC27A2, SLC34A1, SLC3A1, SLC5A2, and SLC6A19) in kidney organoids cultured in EGM and REGM media. The data are represented as mean ± SEM. n ≥ 2 independent experimental replicates from a pool of 12 organoids/group; *P < 0.05, **P < 0.01 ***P < 0.001 and ****P < 0.0001 paired Student’s t-test.
(H) Quantification of changes in the protein expression OXPHOS proteins in organoids cultured in EGM or REGM. Tubulin is used as loading control. The data are represented as mean ± SEM. n = 3 independent experimental replicates from a pool of 16 organoids/group; *P < 0.05, **P < 0.01 ***P < 0.001 and ****P < 0.0001 two-way ANOVA, followed by Bonferroni post-test.
(I) Representative immunofluorescence staining of LTL (green) and PODXL (red) in kidney organoids cultured in EGM and REGM. Scale Bar=400μM (EGM) and 500μM (REGM).
(J) Quantification of LTL positive cells in kidney organoids cultured in EGM or REGM. Y-axis represent relative fluorescence. The data are presented as mean ± SEM. n = 3 organoids/group.
ESRRA drives proximal tubule differentiation in mouse models and couples metabolism with differentiation
In order to define the key transcriptional regulatory organization of PT cells, we analyzed mouse kidney single cell open chromatin data (scATACseq) (Cao et al., 2018). Using a computation motif search algorithm, we found that the most enriched open binding motifs were HNF4A, HNF1B, PPARA and ESRRA in PCT and PST cells (Figure 6A). Our single cell gene expression analysis confirmed transcript enrichment for these 4 transcription factors in PT cells (Figure S5A). Next, we defined putative PPARA and ESRRA target genes by intersecting PCT- or PST-specific open chromatin region at promoters (transcription start site ± 5kb) or gene body regions chromatin regions that contained PPARA- or ESRRA-binding motifs. Gene set enrichment analysis showed enrichment for PPARA- and ESRRA-target genes (Table S5) in differentiated PST and PCT cells (Figure 6B and Figure S5B). PT-specific PPARA and ESRRA target genes were enriched for kidney development, lipid metabolism and epithelial transport functions (Figure 6C and Figure S5C).
Figure 6. ESRRA drives the PT differentiation state and protects from kidney disease.
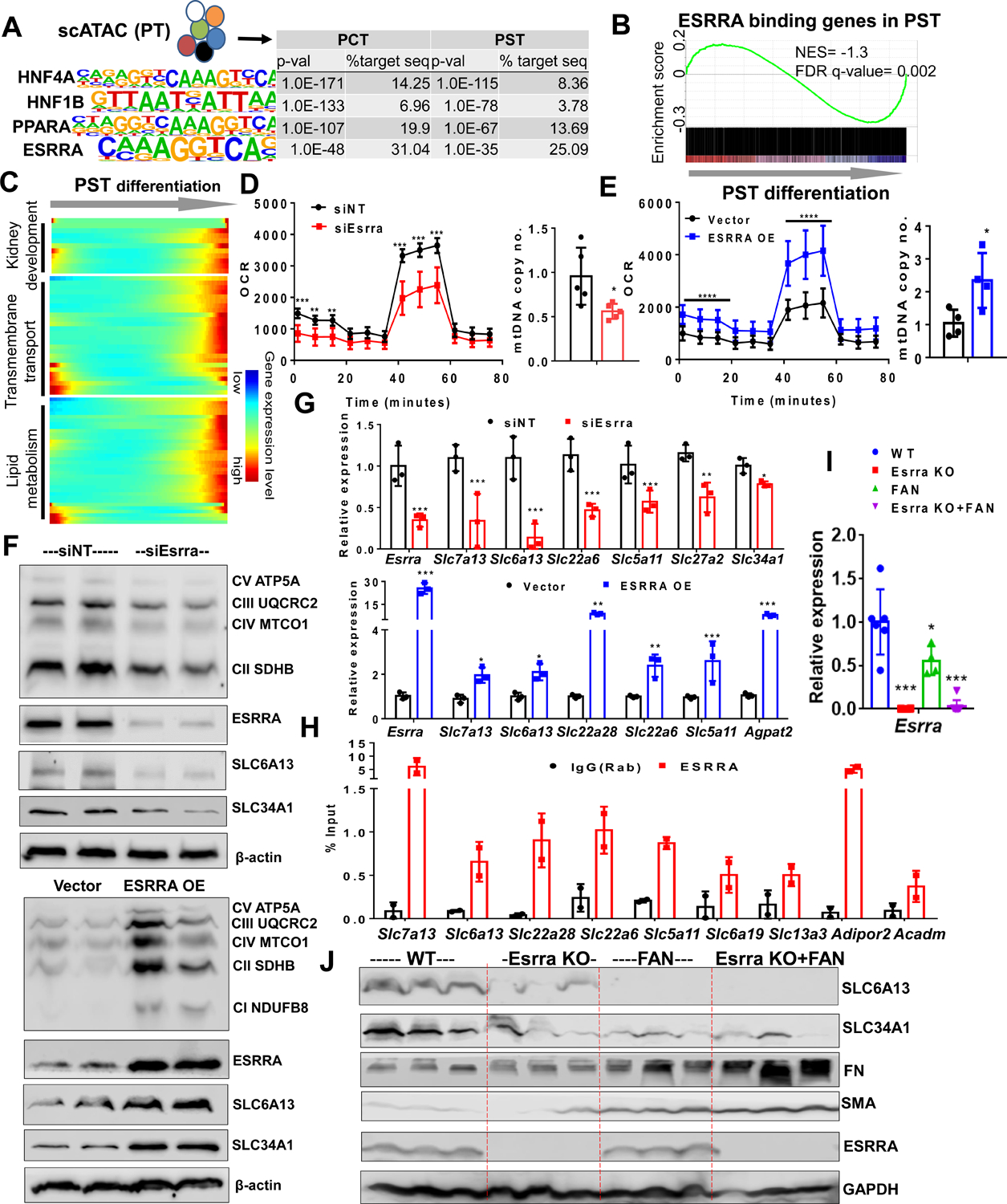
(A) Top transcription factor-binding motifs significantly enriched in PT cell-specific open chromatin regions that are identified from mouse single cell ATAC-sequencing. P-values and percent of target sequences among all open chromatin regions are shown in the table on right.
(B) Gene Set Enrichment Analysis (GSEA) enrichment plot of ESRRA target genes along PST cell differentiation.
(C) Heatmap showing the expression changes of ESRRA target genes along the PST differentiation trajectory (ordered from Figure 4C) grouped by functional annotation (kidney development, transmembrane transport and lipid metabolism).
(D) Oxygen consumption rate (OCR) (pmol/min/μg of protein) and mtDNA copy number (ratio of mtDNA to nuclear DNA) in LTL+ PT cells transfected with non-target siRNA (siNT: black) and ESRRA siRNA (siEsrra: Red) for 2 days. * P < 0.05, ** P < 0.01, *** P < 0.001 vs. siNT.
(E) OCR and mtDNA copy no. in LTL+ PT cells transfected with vector alone (black) and ESRRA expressing vector (ESSRA OE: Blue) for 48 hours. * P < 0.05, ** P < 0.01, *** P < 0.001 vs. vector.
(F) Protein levels of OXPHOS, ESRRA, SLC6A13, and SLC34A1 in LTL+ PT cells transfected with siEsrra (upper panel) or ESRRA OE (lower panel) shown by Western Blot. β-actin was used as loading control.
(G) Relative mRNA levels of Esrra and variety of SLCs markers (Slc7a13, Slc6a13, Slc22a6, Slc5a11, Slc27a2, and Slc34a1) in LTL+ PT cells transfected with siNT, siEsrra (red) and ESRRA OE plasmid (blue). * P < 0.05, ** P < 0.01, *** P < 0.001 vs. siNT/vector.
(H) ChIP-qPCR of ESRRA showed enrichment in genes including SLCs markers (Slc7a13, Slc6a13, Slc22a28, Slc5a11, Slc6a19, and Slc13a3) and metabolic genes (Adipor2 and Acadm) in LTL+ PT cells compared to IgG control.
(I) Relative gene expression of Esrra measured by qRT-PCR in kidneys of wild type, Esrra knock-out mice, sham or FAN treated mice. * P < 0.05, ** P < 0.01, *** P < 0.001 vs. WT.
(J) Protein levels of SLC6A13, SLC34A1, FN, SMA, and ESRRA in kidneys of wild type, Esrra knock-out mice, sham or FAN treated mice were analyzed by Western Blot. GAPDH was used as loading control.
To functionally confirm the role of ESRRA in cellular differentiation, we treated LTL+ PT cells with Esrra siRNA or XCT790, a pharmacological inhibitor (inverse agonist) of ESRRA. We observed that reduced Esrra activity led to compromised mitochondrial function, as assessed by oxygen consumption rate (OCR) and OXPHOS proteins levels as well as decreased mitochondrial DNA content. In addition, reduced Esrra led to lower expression of PT differentiation genes (Figure 6D–6G and Figure S6A–S6D). We next transfected LTL+ PT cells with ESRRA, PPARA or all four TFs (ESRRA, PPARA, HNF4A and HNF1B) plasmids. ESRRA overexpression in PT cells not only improved mitochondrial function and mtDNA copy number but also led to an increase in expression SLC genes (Figure 6D–6G and Figure S6D–S6F). We found that overexpression of PPARA, HNF1B and HNF4A had a synergistic effect as evident by the strong additive effect on cellular differentiation, such as SLCs expression (Figure S6E and S6F). To establish the role of ESRRA in human proximal tubule cell differentiation, we treated human primary renal proximal tubule epithelial cells (HPTCs) and kidney organoids cultured in REGM media with XCT790 for 48 hours. Kidney organoids and HPTCs treated with XCT790 showed impaired FAO (Figure S6G and S6I) and reduced expression of SLCs genes such as SLC3A1, SLC27A2, SLC34A1, SLC6A19, and ATP11A (Figure S6G and S6I). Overall, we found that ESRRA inhibition negatively affected PT differentiation as shown by decreased LTL fluorescence intensity (Figure S6H).
Finally, to distinguish whether ESRRA directly (via binding of the promoter) or indirectly (via improving metabolism) regulates PT cell differentiation, we performed chromatin immunoprecipitation (ChIP) coupled to detection by quantitative real-time PCR (ChIP-qPCR) to study ESRRA transcription factor binding to DNA in PT cells. We found enrichment for multiple PT-specific genes, such as Slc5a11, Slc6a13, Slc6a19, Slc13a3, Slc7a13, Slc22a6 and Slc22a28, and metabolic genes, such as Adipor2 and Acadm (Figure 6H).
To define the role of Esrra in kidney disease, we challenged Esrra knock-out mice with FA (Figure 6I). We found that the expression of Esrra was lower in FAN and UUO model of fibrosis when compared to controls (Figure 6I and Figure S6J). We observed that Esrra knock-out mice showed increased susceptibility to FA-induced kidney injury compared to wild-type littermates as detected by histological analysis (Figure S6K). Levels of pro-fibrotic markers such as Col1a1 and Col3a1 were higher in FA-treated Esrra knock-out mice (Figure S6L) compared to wild-type counterparts. Animals showed increased collagen accumulation on Sirius red stain (Figure S6M) and increased cell proliferation by Ki67 staining (Figure S6N). We further confirmed the decrease in SLCs proteins (SLC6A13 and SLC34A1) and increase in pro-fibrotic proteins (SMA and FN) in Esrra KO mice upon FAN injury (Figure 6J and Figure S6O). Further, we found that genetic deletion of ESRRA in PT cells was associated with impaired mitochondrial function despite the compensatory increase in other nuclear receptors, such as Esrrg and Ppara, (Figure S6P–S6Q) that improved by re-expression of ESRRA. Similar to prior results, we found that PPARA also regulated PT metabolism and cellular differentiation both in vitro and in vivo (Figure S4C and S5D) (Kang et al., 2015) indicating a likely complex interaction between the different nuclear receptors in PT cells.
ESRRA driven metabolic changes correlates with kidney disease severity in patient samples
Finally, we wanted to ascertain whether ESRRA-driven metabolism and PT cell differentiation that appears to drive disease development in mouse kidney disease models can also be recapitulated in patients with CKD. We analyzed 91 microdissected human kidney tubule samples obtained from healthy subjects and from patients with diabetic and hypertensive kidney disease (Table S7). First, we examined the expression of genes involved in FAO and found a group of genes, which strongly correlated with kidney fibrosis (Figure 7A). These genes included ADIPOR2, PPARA, ACSM2A, ACSM3, and APOE, for which we had previously demonstrated an increase along the proximal tubule differentiation trajectory (Figure 4J). Correlation analysis revealed that the expression of lipid metabolism genes showed positive correlation with the expression of PT cell differentiation and negative correlation with fibrosis (Figure 7B). In silico deconvolution of bulk transcriptome data from 91 human samples showed that PCT and PST cell proportions were decreased in fibrotic tissues (Figure 7C). Next, we assessed the effect of cell proportion changes on gene expression changes observed in bulk gene profiling data (Figure 7C). Expression of a total of 1,980 genes significantly correlated with fibrosis scores in 91 human kidney samples analyzed by linear regression using age, gender, race and diabetes and hypertension status as covariates (FDR < 0.05). Next, we performed in silico deconvolution analysis of the data using CellCODE. Adjusting the model to the 4 cell lineages (PCT, PST, myeloid and lymphoid cells) reduced the number of differentially expressed genes (DEGs) from 1,980 to 22 genes, indicating the key role of cell heterogeneity driving bulk gene expression changes (Figure S7A).
Figure 7. ESRRA-driven metabolic changes correlate with kidney disease severity in patient samples.

(A) Relative expression levels of the highly variable lipid metabolism genes that were identified along the mouse PT cell differentiation trajectory (Figure 4) in 91 microdissected human tubules. The human kidney samples were ordered based on the degree of fibrosis.
(B) Heatmap showing Pearson’s correlation coefficient between lipid metabolism genes, PT cell markers and fibrosis markers in the human samples (yellow positive correlation, purple negative correlation, intensity indicates the strength of correlation).
(C) Heatmap showing the relative cell fraction changes, calculated by in silico deconvolution (CellCODE) of the 91 human kidney RNA profiling data. The human kidney samples were ordered based on their fibrosis scores.
(D) Heatmap showing correlation coefficients between lipid metabolism genes, PT cell markers and transmembrane transport genes that contain ESRRA binding motifs in their promoter or gene body and fibrosis markers in the human samples (yellow positive correlation, purple negative correlation, intensity indicates the strength of correlation).
Lastly, we examined the expression of ESRRA and its target genes in 431 microdissected human kidneys (Table S7). Expression of ESRRA was lower in disease samples and strongly correlated both with eGFR and kidney fibrosis (Figure S7B). Protein expression of ESRRA was mostly localized to the nuclei of proximal tubules and it was markedly lower in human CKD samples (Figure S7C). Expression of ESRRA in human kidney tubule samples correlated with lipid metabolism and proximal tubule markers as well as with membrane transporter genes that are ESRRA targets (Figure 7D). These results confirm the relationship between proximal tubule (differentiation) state and metabolism via ESRRA and PPARA.
Discussion
Here we present a comprehensive analysis using mouse single-cell RNA and epigenome analysis, cultured cells, mouse models, patient samples and kidney organoids to demonstrate that PT cells exist in different differentiation states in health and disease conditions. Our results highlight PT cellular metabolism as one of the main drivers of PT cell differentiation identifying ESRRA as a central key player in coupling metabolism and differentiation by directly binding and regulating expression of PT genes. Furthermore, our observations describe how ESRRA together with HNF1B, HNF4A, PPARA likely form a complex network regulating PT metabolism and differentiation. ESRRA-driven PT metabolism and differentiation plays a critical role protecting kidney from injury and correlates with kidney disease severity in patient samples.
We show that gene expression changes observed in bulk RNA-seq analysis mostly reflected cell heterogeneity of diseased mouse and human kidney samples. For example, proximal tubule-specific genes had lower expression levels in bulk RNA-seq analysis; however, many of these genes showed no clear change at a single-cell level. Genes that showed higher expression in disease samples were mostly genes exclusively expressed in immune cells, however, they didn’t show marked changes in the single-cell data when control and disease samples were compared. There was a marked increase in cell diversity of healthy and diseased samples, mostly related to the increase in the diversity of immune cells.
We provide a high-resolution comprehensive analysis of cell type-specific changes in two different mouse kidney fibrosis models. We identified different PT cell subtypes in healthy and disease states. In addition, to the known PCT and PST segments, we also identified precursor-like cells. Cell heterogeneity was significantly higher in diseased PT cells as we identified proliferating cells, immune marker expressing cells and transitional cells (such as PCT and PST intermediate cells), and PT cell and LOH intermediate cells. Future studies will determine the role of these cells in disease development. Important to note that these cell populations represented a continuum between the established PCT and PST cells rather than true discrete groups. Using trajectory and clustering methods we identified cell state differences amongst PT cells. We identified precursor cells that expressed high levels of Igfbp7, but lower levels of differentiated PT cell markers. IGFBP7 is one of the best-known biomarkers of AKI (Meersch et al., 2014; Vijayan et al., 2016). Further studies shall examine the connection between kidney and urinary IGFBP7 expression levels and renal injury as well as outcome.
We found that in diseased kidneys fewer cells were in the terminal differentiation state. Increased death of differentiated cells could have contributed to this finding, however, only minimal changes in cell death is observed at the stages examined (Bielesz et al., 2010). Consistent with prior reports, Wnt expression correlated with cell differentiation in one but not in the second kidney fibrosis model (Edeling et al., 2016; He et al., 2009; Kato et al., 2011; Rinkevich et al., 2014). Changes in lipid metabolism, FAO and OXPHOS were consistent in both models. This might be consistent with earlier reports that such developmental pathways regulate metabolic changes in diseased kidneys (Huang et al., 2018). Overall, our results indicate that kidney PT cells exist in different states where higher expression of cell function genes (e.g., SLCs) strongly correlates with higher expression of FAO and OXPHOS genes.
Biologically, coupling of metabolism and cell state makes perfect sense as it harmoniously couples energy production and utilization with cellular function. Indeed, coupling of cell state and metabolism have been best demonstrated in the field of immunometabolism. For example, effector T cells exhibit high glycolysis, whereas regulatory cells have higher FAO and mTORC1 activation, which in turn drives effector differentiation while suppressing regulatory generation (Angelin et al., 2017; Delgoffe et al., 2009; Michalek et al., 2011). Dysregulated metabolism contributes to disease development, as T cells from systemic lupus erythematosus patients exhibit increased glycolysis and OXPHOS, whereas increased fatty acid biosynthesis and reduced ROS levels are associated with rheumatoid arthritis (Shen et al., 2017; Yang et al., 2013; Yin et al., 2015). Recently, sodium glucose cotransporter 2 inhibitors have shown remarkable success in improving kidney function decline (Barnett et al., 2014), it is possible that reducing the cellular energy requirement is part of their mechanism of action.
Our results for the first time define the key role of several nuclear receptors, such as PPARA, ESRRA in driving PT cell differentiation. ESRRA is a critical transcription factor that regulates mitochondrial biogenesis and FAO (Singh et al., 2018, Soriano et al., 2006). ESRRA remains an underappreciated nuclear receptor and metabolic target due to its diverse role in multiple cellular signaling pathways and its function as a co-regulator of metabolism. Here, we show that ESRRA not only transcriptionally regulates mitochondrial and metabolic genes but also directly binds to genes associated with PT differentiation such as a variety of SLCs. ESRRA target gene expression shows consistent changes in PT cell differentiation in vivo in mice and in patients supporting the key role of ESRRA in driving cell state. ESRRA overexpression not only improved metabolism and mitochondrial function but also PT differentiation and its inhibition resulted in impaired FAO and OXPHOS with altered differentiation state. We found that other transcription factors such as ESRRG, PPARA levels increased in the absence of ESRRA which might be responsible for the lack of phenotypic changes at baseline, however this compensation was insufficient to protect Esrra KO mice during kidney injury (Zhao et al., 2018, Marable et al., 2020). Previous studies showed the role of Esrrg in kidney tubules (Zhao et al., 2018).
It has been difficult to induce PT cell differentiation in cultured organoids (Combes et al., 2019; Wu et al., 2018). Using human kidney organoids, we show that FAO and OXPHOS directly drive differentiation of PT cells. Our data indicates that increasing the activity of Esrra (and Ppara) could be beneficial for PT cell differentiation.
In summary, we show the continuum of PT cell states in health and disease and the key role of metabolism in driving PT cell state. ESRRA couples cell differentiation state and metabolism by not only regulating the expression of cellular metabolism but also the expression of key cell type–specific genes. The work provides new opportunities to manipulate cell fate, PT cell differentiation and metabolism based on their reliance on nuclear receptors such as ESRRA.
Limitations of Study
There are several limitations of our study and future studies shall carefully examine changes observed in non-PT cells in the context of kidney fibrosis and in patients with kidney disease. Follow-up studies should examine the large number of genes and cell type changes identified by single-cell analysis. Future studies are needed to study the detailed ESRRA regulatory network in the kidney and its interaction with other key PT transcription factors, such as PPARA, HNF1B and HNF4A.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact: Katalin Susztak. ksusztak@pennmedicine.upenn.edu
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Processed and raw data can be downloaded from NCBI GEO (GSE156686 and GSE152765). Furthermore, the data is available via an interactive web browser at http://susztaklab.com/VisCello/.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse Models
Animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Pennsylvania. Mice were housed in the Institute pathogen free animal house (12 h dark/light cycle) in a temperature- and humidity-controlled environment (23 ± 1°C) and fed with standard mouse diet and water ad libitum. 5- to 8-week-old male C57BL/6 wild type mice were used in the study. Esrra KO mice were kindly provided by Dr. Liming Pei (University of Pennsylvania) and littermates were used from in-house matings. All animals were pathogen free and healthy prior to the beginning of experiments. For all the mice experiments, mice were randomly assigned to experimental groups, unless stated otherwise.
For fenofibrate experiment, the PPARA agonist fenofibrate (50 mg/kg for 3 days and 100 mg/kg for 5 days) was administered by oral gavage starting one day before the folic acid (FA) injection. Mice were injected with FA (250 mg/kg once, dissolved in 300 mM NaHCO3) intraperitoneally and sacrificed on day 7. For the unilateral ureteral obstruction (UUO) model, mice underwent ligation of the left ureter and were sacrificed on day 7.
Isolation and culture of LTL+ PT cells
Primary mouse proximal tubule epithelial cells were isolated from kidneys of 4 weeks old wild type mice and LTL+ cells fractions were purified from single cell suspension of PT cells by using biotinylated lotus tetragonolobus lectin antibody (LTL) (L-132; Vector Laboratories) and anti-biotin microbeads (MACS Miltenyi Biotec). LTL+ cells were grown in primary cell culture media (RPMI 1640 supplemented with 10% FBS, 20 ng ml−1 EGF, 20 ng ml−1 bFGF and 1% penicillin-streptomycin).
Kidney organoids differentiation
ES[4] human embryonic stem cells were grown on vitronectin coated plates (1001–015, Life Technologies). Cells were incubated in 0.5mM EDTA (Merck) at 37°C for 3 minutes for disaggregation. To avoid the separation of the stem cell clusters, cells were then carefully collected into 12 ml supplemented Essential 8 Basal medium. For cell counting, 1 mL cell suspension was centrifuged for 4 minutes at 400 g and the pellet was resuspended in 200 μl of AccumaxTM (StemCell Technologies) to obtain single cells. Cells were incubated in AccumaxTM at 37°C for 3 minutes and next, 800 μL of FBS were added to stop the disaggregation. After cell counting (Countess ® Automated Cell Counter), 100,000 cells/well were plated on a 24 multi-well plate coated with 5μl/ml vitronectin. Cells were incubated in supplemented Essential 8 Basal medium at 37°C overnight. The next day (day 0), the differentiation was initiated by treating the cells with 8 μM CHIR (Merck) in Advanced RPMI 1640 basal medium (ThermoFisher) supplemented with 1% Penicillin-Streptomycin and 1% of GlutaMAXTM (ThermoFisher) for 3 days and changing the medium every day. On day 3, CHIR treatment was removed and cells were cultured in 200 ng·ml−1 FGF9 (Peprotech), 1 μg·ml−1 heparin (Merck) and 10 ng·ml-1 activin A (Act A) (Vitro) in supplemented Advanced RPMI for 1 day. On day 4, spheroid organoids were generated. Cells were rinsed twice with PBS, collected using supplemented Advanced RPMI and plated at 100,000 cells/well on a V-shape 96 multi-well plate. They were treated with 5 μM CHIR, 200ng·ml-1 FGF9 and 1 μg·ml-1 Heparin in supplemented Advanced RPMI. Organoids were incubated for 1hour at 37°C, CHIR induction was removed and they were incubated in 200 ng·ml-1 FGF9 and 1 μg·ml-1 Heparin in supplemented Advanced RPMI for 7 days with medium change every other day. From day 11, factors were eliminated, and cells were incubated only in supplemented Advanced RPMI for 5 days, medium was changed every other day.
METHOD DETAILS
Preparation of single-cell suspension
Euthanized mice were perfused with chilled 1x PBS via the left ventricle. Kidneys were harvested, minced into approximately 1 mm3 cubes and digested using Multi Tissue dissociation kit (Miltenyi, 130-110-201). The tissue was homogenized using 21G and 26 1/2G syringes. Up to 0.25 g of the tissue was digested with 50ul of Enzyme D, 25 ul of Enzyme R and 6.75 μl of Enzyme A in 1 ml of RPMI and incubated for 30mins at 37°C. Reaction was deactivated by 10% FBS. The solution was then passed through a 40 μm cell strainer. After centrifugation at 400 g for 5mins, cell pellet was incubated with 1ml of RBC lysis buffer on ice for 3 mins. Cell number and viability were analyzed using Countess AutoCounter (Invitrogen, C10227). This method generated single cell suspension with greater than 80% viability.
Single-cell RNA sequencing
Single cell RNA sequencing was performed as described in our previous study (Park et al., 2018). Briefly, the single cell suspension was loaded onto a well of a 10x Chromium Single Cell instrument (10x Genomics). Barcoding and cDNA synthesis were performed according to the manufacturer’s instructions. Qualitative analysis was performed using the Agilent Bioanalyzer High Sensitivity assay. The cDNA libraries were constructed using the 10x Chromium™ Single cell 3’ Library Kit according to the manufacturer’s original protocol. Libraries were sequenced on an Illumina HiSeq or NextSeq 2×150 paired-end kits using the following read length: 26bp Read1 for cell barcode and UMI, 8bp I7 index for sample index and 98bp Read2 for transcript.
Alignment and generation of data matrix
Cell Ranger 2.0 (http://10xgenomics.com) was used to process Chromium single cell 3’ RNA-seq output. First, “cellranger count” aligned the Read2 to the mouse reference genome (mm10) and exons of protein coding genes (Ensembl GTFs GRCm38.p4). Sequencing reads that were marked by multiple mapping were removed by adjusting the cellranger to unique mapping (marked MM:i:1 in the bam files). Third, the fastq files extracted from bam files of the first run were used again for “cellranger count” to generate data matrix. Finally, the output files for 6 normal and 2 FAN samples were aggregated into one gene-cell matrix using “cellranger aggr” with read depth normalization by total number of mapped reads.
Data quality control, preprocessing and dimension reduction
Seurat R package (version 2.3.4) was used for data QC, preprocessing and dimension reduction analysis. Once the gene-cell data matrix was generated, poor quality cells were excluded, such as cells with < 200 or > 3,000 expressed genes. Genes that were expressed in less than 10 cells, mitochondrial genes, ribosomal protein genes and HLA genes, that were reported to induce unwanted batch effects, were removed for further analysis (Smillie et al., 2019). Cells were also discarded if their mitochondrial gene percentages were over 50%. The data were natural log transformed and normalized for scaling the sequencing depth to a total of 10,000 molecules per cell, followed by regressing-out the number of UMI and genes. Batch effect was corrected by using removeBatchEffect function of edgeR. The expression values after batch correction were only used for PCA, t-Distributed Stochastic Neighbor Embedding (tSNE) visualization and clustering, and the original expression values before batch correction were used for all downstream analyses such as identification of marker genes and differentially expressed genes. For the dimension reduction, highly variable genes across the single cells were identified using 0.0125 low cutoff and 0.3 high cutoff. PCA was performed using the variable genes as input and top 20 PCs were used for initial tSNE projection.
Removal of doublet-like cells
Doublet-like cells were identified using DoubletFinder which is a computational doublet detection tool with following parameters: proportion.artificial = 0.25 and proportion.NN = 0.01(McGinnis et al., 2019). Then, the number of expected doublets were calculated for each sample based on expected rates of doublets, which are provided by 10x Genomics. After removing the doublet-like cells, all steps including normalization, regressing out variables, batch effect removal, and dimension reduction were performed again.
Cell clustering analysis
Density-based spatial clustering algorithm, DBSCAN, was used to identify cell clusters on the tSNE plot with the eps value 0.4. Clusters were removed if their number of cells was less than 20. Proximal tubule clusters expressing a proximal tubule marker, Slc27a2, were separated from the rest of cell clusters in order to identify subgroups. PCA and UMAP (Uniform Manifold Approximation and Projection) were performed only for the remaining cells. DBSCAN was used to identify cell clusters on the UMAP plot with initial setting for the eps value 0.5. Each of the resulting clusters was subjected to sub-clustering by a shared nearest neighbor (SNN) modularity optimization-based clustering algorithm, which is implemented in Seurat package. Resolution 0.5 was used for sub-clustering of the clusters except T lymphocytes which required higher resolution (0.7) to identify T lymphocyte subgroups. Post-hoc differential expression analysis was performed for every pair of sub-clusters. Sub-clusters were merged when they had 15 or less than 15 (10 differential genes for T lymphocytes) differentially expressed genes (average expression difference > 1 natural log with an FDR corrected p<0.01). This clustering analysis resulted in 30 cell clusters. PT cell clusters were also subjected to sub-clustering. With same procedure used for other clusters, PT cells from control and FAN samples were subclustered into 5 and 9 sub-cell types, respectively.
Mouse bulk RNA-sequencing analysis
Total RNAs were isolated using the RNeasy mini kit (Qiagen). Sequencing libraries were constructed using the Illumina TruSeq RNA Preparation Kit. High-throughput sequencing was performed using Illumina HiSeq4000 with 100bp single-end according to the manufacturer’s instruction. Adaptor and lower-quality bases were trimmed with Trim-galore. Reads were aligned to the Gencode mouse genome (GRCm38) using STAR-2.4.1d. The aligned reads were mapped to the genes (GRCm38, version 7 Ensembl 82) using HTSeq-0.6.1. Differentially expressed genes between control and disease groups were identified using DESeq2 version 1.10.1. To examine the enrichment of the differentially expressed genes in single cell clusters, a z-score of normalized expression value was first obtained for every single cell. Then, we calculated the mean z-scores for individual cells in the same cluster, resulting in 30 values for each gene. The z-scores were visualized by heatmap showing the enrichment patterns of the genes across the cell types.
Estimation of cell proportions
From single cell datasets, the numbers of cells in each cluster were enumerated and normalized by total number of cells for each condition (6 control and 2 disease samples). Since a number of PT cells in FAN samples showed higher expression of apoptosis markers, we removed the cells that express Bax, Bad or Dap from all samples only for the cell proportion test. Deconvolution of bulk RNA sequencing data was performed to validate the cell proportion changes that were detected in single cell data. CellCODE package was used for deconvolution using 30 cell type-specific marker genes (Chikina et al., 2015).
Identification of marker genes and differentially expressed genes
Conserved marker genes between control and UUO samples were identified using FindConservedMakers function of Seurat with default options. Average expression difference > 0.5 natural log and FDR corrected p value < 0.01 were applied. Cell type-specific differentially expressed genes were identified using MAST, which is implemented in Seurat package with log fold change threshold = 0.2, minimum percent of cells expressing the genes=0.05 and adjusted p value < 0.05.
Cell trajectory analysis
RNA Velocity:
To calculate RNA velocity, Velocyto.R package was used as instructed (La Manno et al., 2018). We used Velocyto to impute the single-cell trajectory/directionality using the spliced and the unspliced reads. Resulting loom files were merged and loaded into R following the instructions. Furthermore, RNA velocity was estimated using gene-relative model with k-nearest neighbor cell pooling (k = 25). To visualize RNA velocity, we performed Principle Component Analysis and used the top 20 principle components to calculate UMAP embedding. The parameter n was set at 200, when visualizing RNA velocity on the UMAP embedding.
Monocle2:
To construct single cell pseudotime trajectory and to identify genes that change as the cells undergo transition, Monocle2 (version 2.4.0) algorithm was applied to the cells from proximal tubules and proliferating proximal tubules (Trapnell et al., 2014). To show the cell trajectory from the small cell population (proliferating proximal tubules) to predominant cell type (proximal tubules), 6,000 randomly selected PT cells and proliferating proximal tubules were used for Monocle analysis. Genes for cell ordering were selected if they were expressed in ≥ 10 cells, their mean expression value was ≥ 0.05 and dispersion empirical value was ≥ 2. Highly variable genes along the pseudotime were identified using differential GeneTest function of Monocle2 with q-value < 0.01. The trajectory analysis was also performed for precursors and PST cells from the control and FAN samples separately. DAVID GO term analysis was performed for the highly variable genes along the control and FAN trajectory, and then genes in the lipid metabolism GO term were visualized by heatmap.
Single cell ATAC sequencing analysis
Data matrix for PCT and PST cells-specific open chromatin regions was downloaded (Cao et al., 2018). HOMER package was used to identify known transcription factor binding motifs that are highly enriched in the PCT and PST-specific open chromatin regions. GSEA package was used to determine the enrichment patterns of ESRRA binding genes in differentiated PT cells and to identify core enrichment genes.
Human bulk gene profiling data analysis
Kidney samples were collected from nephrectomies. Samples were permanently deidentified and clinical information was collected by an honest broker, therefore the study was deemed exempt by the institutional review board (IRB) of the University of Pennsylvania. Two datasets were used, one dataset included 91 human kidney samples and gene expression analysis was performed using Affymetrix U133A arrays (E-MTAB-2502) (Table S7) Raw expression levels of microarray data sets were normalized using the RMA algorithm and log transformed. The identified marker genes were used as an input for CellCODE deconvolution analysis to estimate the cell proportion changes in human patient kidney samples. To assess the effect of cell proportions changes on the correlation between gene expression and fibrosis score, we implemented linear regression models using age, gender, race and diabetes and hypertension status as covariates with and without cell proportions of PCT, PST, myeloid and lymphoid cells. The second dataset included 431 samples and gene expression was analyzed using RNAseq (Table S7).
Single Cell Suspension from Kidney organoid
Day 20 mature kidney organoids were washed twice with PBS and incubated first with AccumaxTM for 10 min at 37°C, followed by Trypsin-EDTA 0.25% incubation in order to dissociate into single cells. Cells were spun down at 400 g for 5 min resuspended in ADV RPMI and checked for viability using Countess Automated Cell Counter.
Mitotracker Green FM Flow Cytometric Analysis
Developing kidney organoids on day 14 of differentiation were cultured in EGM or REGM media for 4 additional days. In order to assess mitochondrial mass, organoids were stained with MitoTracker Green FM (100 nM), a mitochondrial specific fluorescent dye at 37°C for 30 min. After incubation, kidney organoids were washed twice with PBS and disaggregated into single cell suspension using AccumaxTM for 10 minutes followed by Trypsin-EDTA 0.25% (ThermoFisher) incubation for at least 10 minutes at 37°C. Once cells dissociated, FACS buffer (PBS supplemented with 5% of FBS) was added to cease the trypsin activity and samples were centrifuged for 5 minutes at 1800 rpm. After removing the supernatant, the pellet was resuspended in 300 μl of FACS buffer and the suspension was filtered into FACS tubes. Nuclei were stained with DAPI (ThermoFisher). Cells were counted using FACS Aria Fusion Instrument (BD Biosciences). FlowJo software version 10 was used for data analysis.
Protein extraction and western blot analysis in kidney organoids
Protein was extracted from kidney organoids cultured in EGM or REGM media for 4 days using RIPA buffer (Thermofisher) supplemented with complete protease inhibitor cocktail (Thermofisher), and centrifuged at 13,000 g for 15 mins at 4°C. The supernatant was collected, and protein concentration was measured using a bicinchoninic acid (BCA) protein quantification kit (Thermo Scientific). For western blot analyses, 25 ug of protein were separated in 10% sodium dodecyl sulphate-polyacrylamide gel (SDS-PAGE) and blotted onto nitrocellulose membranes. Membranes were blocked at room temperature for 1 hour with TBS 1X- 5% BSA. Membranes were then incubated in primary antibody (dilution 1:1000) Total OXPHOS cocktail (Abcam) overnight at 4°C. The membranes were then washed with PBST (PBS1X + 0.05% Tween20; Merck) for 5 minutes three times and incubated with anti-mouse secondary antibody (dilution 1:10,000) (IRDye®680RD Goat anti-Mouse; LI-COR). After washing with PBST for 5 minutes twice and with PBS for 5 minutes once, membrane-bound antibodies were detected by fluorescence with the Odyssey ® Fc Imaging System. Alpha-tubulin (1:5000; Sigma) was used as a loading control for normalization and quantification. Images were analyzed with Image Studio Lite Version 5.2 software.
Immunofluorescence
Kidney organoids were cultured in EGM or REGM media were transferred to 96 well plates. Fixation was performed with paraformaldehide 4% (ThermoFisher) for 20 minutes followed by 10 minutes washing in three changes PBS. Kidney organoids were incubated in Streptavidin/Biotin Blocking Kit (Vector Laboratories) and TBS – 1% triton + 6% donkey serum for 2 h at room temperature. Podocalyxin (PODXL, Dilution 1:250) and LTL (Dilution 1:200) antibodies were diluted in TBS-0.5% Triton + 1% BSA. Kidney organoids were then treated overnight at 4°C with primary antibodies. The next day, organoids were washed with TBS-0.5% triton + 1% BSA for 5 minutes three times and incubated with secondary antibody (Dilution 1:500) in TBS-0.5% Triton + 1% BSA at room temperature for 2 hours. Subsequently, organoids were washed with TBS twice for 5 minutes and nuclei were stained with DAPI (Dilution 1:5000) for 10 minutes. Organoids were collected with special wide-end tips, and placed on slides and mounted with Fluoromount-G (Southern Biotech). Confocal images were acquired using Leica SP5 microscope and LTL positive cells were analyzed using Image J.
Mitochondrial DNA analysis
MtDNA copy number is represented by the ratio of mitochondrial DNA to nuclear DNA (mtDNA/nDNA). Total DNA was isolated from LTL+ PT cells 48 hours after transfection using DNeasy Blood & Tissue Kit. The mtDNA/nDNA ratio was determined by quantifying two mitochondrial genes (16S rRNA and ND1) and two nuclear genes (HK2 or 18S rRNA) using qPCR (Quiros et al., 2017). The primer sequences are listed in Table S6.
PT Cell transfection
PT cells were transfected with Non-targeted (NT) siRNA or siEsrra (purchased from Dharmacon) and pcDNA3.0 (vector) or pAd-Track-Esrra/pcDNA3.1-Ppara/pcDNA3.1-HNF1A/pAd-track-HNF4A (overexpression constructs) (kind gift from Dr. Liming Pei, University of Pennsylvania). siRNA and plasmid transfections were performed using Lipofectamine 3000. For transfection, cells were seeded in 6-well plates, grown for overnight until 60–70% confluent, and then transfected with 100nM (final concentration) siRNA/siEsrra and 5μg of vector/ESRRA/PPARA/HNF1B/HNF4A overexpression plasmids. Transfection efficiency was determined under fluorescence microscope by the presence of Cy3 transfection control (data not shown). Cells were harvested and scraped off 48 hours post transfection under different condition.
Oxygen consumption rate (OCR)
The measurement of OCR in PT cells transfected with siEsrra or ESRRA plasmid were performed using XFe96 extracellular flux analyzer (Seahorse Bioscience) as previously described (Kang et al., 2015). Briefly, PT cells were plated at density of 10,000 cells/well in a Seahorse cell culture microplate and transfected with siEsrra/ESRRA OE. Cellular OCR was measured 48 hours post-transfection and normalized to protein quantity in each well. The final concentration for oligomycin, FCCP, rotenone and antimycin used was 2 μM, 1 μM, 1 μM and 1 μM, respectively.
Chromatin immunoprecipitation qPCR
ChIP was performed to evaluate enrichment of ESRRA binding regions in targets SLCs genes in PT cells following the manufacturer’s instructions (492024, Invitrogen). Briefly, 107 PT cells were cross-linked with 1% formaldehyde for 10 min at room temperature. Then the reaction was stopped by adding glycine (final concentration, 0.125M). The cells were sonicated in lysis buffer to achieve a chromatin size of 100–500 bp. The sonicated chromatin was diluted by using dilution buffer. 5 μg antibody was coupled with Dynabead protein A and G (1:1 mixed), the mixture was incubated with chromatin lysates overnight at 4 °C with rotation. Immune complexes were washed with IP buffers. Antibody-bound chromatin was reverse-cross-linked, and the CHIP DNA samples were purified for PCR reaction. Primers used for CHIP-qPCR are shown in Table S6.
Fenofibrate treatment
LTL+ PT cells isolated from mouse kidneys were cultured in either presence of 1μM fenofibrate (PPARA agonist) to activate PPARA or DMSO in primary culture media from Day 0. Cells were harvested for RNA and protein isolation for Western Blot on Day 7.
XCT790 treatment
Kidney organoids cultured in REGM media for 7 days were treated with 10μM of XCT790 for 48 hours followed by Immunostaining or RNA isolation. Human proximal tubule cells (HPTC) and LTL+ mouse PT cells were cultured in REGM media and treated with 10 μM of XCT790 or DMSO for 48 hours and 24 hours respectively.
qRT-PCR
RNA was isolated from cells, kidney organoids and kidneys tissue using Trizol (Invitrogen). 2 μg RNA was reverse transcribed using the cDNA archival kit (Life Technology), and qRT-PCR was run in the ViiA 7 System (Life Technology) machine using SYBRGreen Master Mix (Applied Biosystem) and gene-specific primers. The data were normalized and analyzed using the ΔΔCt method. The primers sequences used are shown in Table S6.
Western Blot
Cells were lysed in radioimmunoprecipitation assay buffer (RIPA; Cell Signaling Technology) and protein was quantified by BCA method (Thermo Fisher Scientific). Protein samples (10 to 30 μg) were separated by SDS-PAGE and then transferred to PVDF membranes. After blocking, for 30 min with 5% milk in TBST and three times washing, membranes were incubated overnight with primary antibody (COX IV 1:1000, OXPHOS 1:250, GAPDH 1:2000, PGC1a 1:500, SLC6A13 1:1000, SMA 1:1000, FN 1:1000, ESRRA 1:2000, β-actin 1:20,000) in TBST (see KST). After three washes for 5 min, membranes were incubated for 45 min at RT to 1 hour with secondary HRP-conjugated antibody (1:20,000) in TBS-T. The signal was developed with Immobilon forte western HRP substrate (Milipore) and measured using Odyssey®Fc Imaging System (LICOR) equipment and software. The following antibodies were used in this study are listed in key resource table.
Histological Analysis
Kidneys samples were fixed in 10% neutral formalin and paraffin-embedded sections were stained Periodic acid–Schiff (PAS) and hematoxylin and eosin (H&E) to analyze the histology of samples. Sirius-red staining (Boekel Scientific, #147122) was performed to determine the degree of fibrosis. We performed immunocyto- and -histochemistry on paraformaldehyde fixed cells and formalin- fixed, paraffin-embedded kidney sections. We used the following primary antibodies: (ESRRA (1:200), SLC6A13 (1:100), SLC34A1 (1:100), and SLC7A13 (1:100). Staining was visualized using peroxidase-conjugated antibodies to mouse immunoglobulin using the Vectastain Elite kit and 3,3-diaminobenzidine (DAB) (Vector Labs). For cell proliferation, we used Ki67 (1:150) primary antibody and Alexa Fluor 555 as secondary antibody, and nuclei was stained by Hoechst dye.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data Representation and Statistical Analysis
Student’s t-test was used to analyze differences between two groups, and One-way or Two-way ANOVA was used to analyze intergroup differences (tukey’s multiple comparisons test). P-values less than 0.05 were considered statistically significant. The analysis was performed using GraphPad Prism 5 (GraphPad software). Densitometry results of Western Blots were quantified using ImageJ software. All data are presented as mean ± SEM and other details such as the number of replicates and the level of significance is mentioned in figure legends and supplementary tables. For mice experiments, animals were randomly allocated to different groups prior the experiments. No samples or animals were excluded from analysis. Sample size estimation was not performed, and sample size was determined based on the number of available age and gender matched animals in the colony. For single-cell data analysis, statistical details are provided in designated method section. All samples were processed in blinded fashion.
Supplementary Material
Supplementary table 1(Related to Figure 1). List of single cell cluster markers
Supplementary table 2 (Related to Figure 2). List of differential expressed genes in control and FAN mice samples
Supplementary table 3 (Related to Figure 3). List of PT subcluster markers in control and FAN samples
Supplementary table 4 (Related to Figure 4). The percent of cells that express EMT and SASP markers across the PT cell sub-clusters in FAN kidneys and all the cell clusters
Supplementary table 5 (Related to Figure 6). List of predicted ESRRA target genes in PT cells.
Supplementary table 6 (Related to Key Resource Table). List of primers used in this study.
Supplementary table 7 (Related to Figure 7 and S7). Demographic and clinical of human kidney samples.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Primary antibodies for Immunoblots | ||
| Anti-COX IV antibody | Abcam | Cat#Ab16056 |
| Total OXPHOS Rodent WB Antibody Cocktail | Abcam | Cat#ab110413 |
| GAPDH | CST(14C10) | Cat#2118 |
| PGC1a | Calbiochem | Cat#KP9803 |
| SLC6A13 | Invitrogen | Cat#PA5-68331 |
| α-Tubulin | Sigma | Cat# T9026 |
| SMA | Sigma | Cat# A5228 |
| FN | Abcam | Cat#Ab2413 |
| ESRRA | CST | Cat#CS 13826 |
| β-actin | Millipore | Cat#A3854 |
| Secondary antibodies for Immunoblots | ||
| Anti-mouse IgG, HRP-linked Antibody | CST | Cat#CS 7076S |
| Anti-rabbit IgG, HRP-linked Antibody | CST | Cat# CS 7074S |
| Antibodies for Immunohistochemistry (IHC) | ||
| ESRRA | CST | Cat#CS 13826 |
| SLC34A1 | Novus Biologicals | Cat#NBP2-42216 |
| SLC7A13 | Creative Diagnostics | Cat#CABT-BL3359 |
| SLC6A13 | Invitrogen | Cat#PA5-68331 |
| Antibodies for Immunofluorescence (IF) | ||
| LTL | Vector laboratories | Cat#FL-1321-2 |
| PODXL | Thermofischer | Cat# 39-3800 |
| IRDye®680RD | LI-COR | Cat# 926-68070 |
| Ki67 | CST | Cat# CS12202 |
| A555 | Life technology | Cat# A31572 |
| Antibodies for FACS | ||
| LTL | Vector laboratories | Cat#B-1325 |
| Biological Samples | ||
| Human kidney samples | Chung et al., 2019 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| MitoTracker Green | Life Technology | Cat#M7514 |
| Collagenase IV | Life Technologies | Cat#17104019 |
| Trypan blue solution | Sigma | Cat#T8154 |
| Protease inhibitor cocktail | Roche | Cat#11836153001 |
| EDTA solution | Life Technologies | Cat#15575-038 |
| Accumax | Stem Cell Technologies | Cat#07921 |
| Phosphate buffered saline (PBS) pH 7.4 (1x) | Life Technologies | Cat#1001-015 |
| Essential 8 medium | Life Technologies | Cat#A1517001 |
| Vitronectin | Life Technologies | Cat#A14700 |
| Fenofibrate | Sigma | CAS#49562-28-9 |
| RPMI 1640 | Gibco | Cat#21875-034 |
| EGM media | Lonza | Cat#CC-3162 |
| REGM media | Lonza | Cat#CC-4127 |
| RIPA buffer | Cell signaling | Cat#9806 |
| SYBR Green PCR Master Mix | Applied Biosystem | Cat#KK4605 |
| Fluoromount-G | Southern Biotech | Cat# 0100-01 |
| XCT790 | Tocris | Cat#3928 |
| Folic Acid | Fisher Scientific | Cat#AC216630500 |
| Lipofectamine 3000 | ThermoFisher | Cat#11668027 |
| CHIR99021 | Merck | Cat#SML1046; CAS: 252917-06-9 |
| Recombinant human FGF9 | PeproTech | Cat#100-23 |
| Heparin | Merck | Cat#H3149; CAS: 9041-08-1 |
| Activin A | Vitro | Cat#338-AC-050 |
| Dimethyl Sulfoxide (DMSO) | Merck | Cat#D2650; CAS: 67-68-5 |
| Cell culture grade distilled water | Life Technologies | Cat#15230-089 |
| Paraformaldehyde solution 4% in PBS | Santa Cruz | Cat#sc-281692 |
| Hoechst | Molecular probes | Cat# H-1399 |
| Critical Commercial Assays | ||
| BCA Protein Assay Kit | Thermo Scientific | Cat#23225 |
| Multi Tissue dissociation kit | Miltenyi | Cat#130-110-201 |
| Anti-Biotin microbeads | Miltenyi | Cat#130-090-485 |
| cDNA Reverse Transcription Kit | Applied Biosystems | Cat#4368813 |
| Rneasy Mini Kit | Qiagen | Cat#74106 |
| MAGnify™ ChIP Kit | Thermo Scientific | Cat# 492024 |
| DNeasy Blood & Tissue Kits | Qiagen | Cat#69506 |
| Seahorse XF Cell Mito Stress Test kit | Agilent Technologies | Cat#103708-100 |
| Seahorse XFe96 FluxPak mini | Agilent Technologies | Cat#102601-100 |
| VECTASTAIN® Elite ABC-HRP Kit | Vector laboratories | Cat# PK-6100 |
| Streptavidin/Biotin blocking kit | Vector laboratories | Cat#SP-2002 |
| Deposited Data | ||
| scATAC seq data | (Cao et al., 2018) | GSE117089 |
| scRNA-seq of FAN kidneys of mice | GEO | GSE156686 |
| scRNA seq data kidney organoids | GEO | GSE152765 |
| Experimental Models: Organisms/Strains | ||
| ESRRA Knock-out | Dr. Vincent Giguère Research Lab | McGill University |
| Oligonucleotides | ||
| Primers for qPCR, mtDNA copy no., and ChIP-qPCR, see Table S6 | This paper | N/A |
| Recombinant DNA | ||
| pAd-track-Esrra | Dr. Liming Pei lab | University of Pennsylvania |
| pcDNA3.1-Ppara | Dr. Liming Pei lab | University of Pennsylvania |
| pc-DNA3.1-HNF1B | Dr. Liming Pei lab | University of Pennsylvania |
| pAd-track-HNF4A | Dr. Liming Pei lab | University of Pennsylvania |
| Experimental Models: Cell Lines | ||
| ES[4] Human Embryonic Stem Cell line | The National Bank of Stem Cells (ISCIII,Madrid) | https://www.isciii.es/ |
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij |
| Prism 5 | Graphpad Software | https://www.graphpad.com/scientific-software/prism |
| Image Studio Lite Version 5.2 software | LICOR | https://www.licor.com/bio/image-studio-lite/d5 |
| FlowJo Software | FlowJo | N/A |
| Cell Ranger 2.0 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest |
| Seurat R package 2.3.4 | open source | https://satijalab.org/seurat/ |
| DoubletFinder | open source | https://github.com/chris-mcginnis-ucsf/DoubletFinder |
| STAR-2.4.1d | open source | https://github.com/alexdobin/STAR |
| HTSeq-0.6.1 | open source | https://htseq.readthedocs.io/en/release_0.11.1/history.html#version-0-6-1 |
| DESeq2 1.10.1 | open source | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| CellCODE | open source | https://github.com/mchikina/CellCODE/ |
| Velocyto | open source | https://github.com/velocyto-team/velocyto.R |
| Monocle2 2.4.0 | open source | http://cole-trapnell-lab.github.io/monocle-release/ |
| Other | ||
Acknowledgement
Work in the Susztak lab is supported by NIH National Institute of Diabetes and Digestive and Kidney Diseases grants R01DK076077, R01 DK087635, and DP3 DK108220. We thank the University of Pennsylvania Diabetes Research Center (DRC) for the use of the Core services (P30-DK19525). J.P. is supported by the National Research Foundation of Korea funded by the Korea government (MSIP) (2019R1C1C1005403 and 2019R1A4A1028802). J.Z. and L.P. are supported by the Office of the Assistant Secretary of Defense for Health Affairs through the Peer Reviewed Medical Research Program under Award W81XWH-16-1-0400, NIH DK111495, and pilot awards from the Diabetes Research Center at the University of Pennsylvania NIH DK19525. M.S.B. is supported by German Research Foundation grant BA 6205/2-1. This work has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation Programe (StG-2014-640525_REGMAMKID to P.P., and N.M.). NM is also supported by the Spanish Ministry of Economy and Competitiveness/FEDER (SAF2017-89782-R), the Generalitat de Catalunya and CERCA Programme(2017 SGR 1306), Asociación Española contra el Cáncer (LABAE16006) and Institute of Health Carlos III (ACE2ORG). C.H.P. is supported by Marie Skłodowska-Curie Individual Fellowships (IF) grant agreement no. 796590. We thank Vincent Giguere (McGill University) for sharing the Esrra KO mice. N.M and C.H.P are supported by EFSD/Boehringer Ingelheim European Research Programme in Microvascular Complications of Diabetes. This work was supported in part by the ISCIII and FEDER through TERCEL RETIC RD16/0011/0005 and RD16/0011/0027. PJ.D. was supported by an American Heart Association postdoctoral fellowship (20POST35210738).
Declaration of Interests
The Susztak lab is supported by Boehringer Ingelheim, Lilly, Regeneron, GSK, Merck, Bayer and Gilead for work that is not related to the current manuscript.
References
- Angelin A, Gil-de-Gomez L, Dahiya S, Jiao J, Guo L, Levine MH, Wang Z, Quinn WJ 3rd, Kopinski PK, Wang L, et al. (2017). Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab 25, 1282–1293 e1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelotti ML, Ronconi E, Ballerini L, Peired A, Mazzinghi B, Sagrinati C, Parente E, Gacci M, Carini M, Rotondi M, et al. (2012). Characterization of renal progenitors committed toward tubular lineage and their regenerative potential in renal tubular injury. Stem Cells 30, 1714–1725. [DOI] [PubMed] [Google Scholar]
- Barnett AH, Mithal A, Manassie J, Jones R, Rattunde H, Woerle HJ, Broedl UC, and investigators E.-R.R.t. (2014). Efficacy and safety of empagliflozin added to existing antidiabetes treatment in patients with type 2 diabetes and chronic kidney disease: a randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol 2, 369–384. [DOI] [PubMed] [Google Scholar]
- Beckerman P, Qiu C, Park J, Ledo N, Ko YA, Park AD, Han SY, Choi P, Palmer M, and Susztak K (2017). Human Kidney Tubule-Specific Gene Expression Based Dissection of Chronic Kidney Disease Traits. EBioMedicine 24, 267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielesz B, Sirin Y, Si H, Niranjan T, Gruenwald A, Ahn S, Susztak K. Epithelial Notch signaling regulates interstitial fibrosis development in the kidneys of mice and humans. J Clin Invest. 2010; 120:4040–4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breyer MD, and Susztak K (2016). The next generation of therapeutics for chronic kidney disease. Nat Rev Drug Discov 15, 568–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Cusanovich DA, Ramani V, Aghamirzaie D, Pliner HA, Hill AJ, Daza RM, McFaline-Figueroa JL, Packer JS, Christiansen L, et al. (2018). Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 361, 1380–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang-Panesso M, and Humphreys BD (2017). Cellular plasticity in kidney injury and repair. Nat Rev Nephrol 13, 39–46. [DOI] [PubMed] [Google Scholar]
- Chikina M, Zaslavsky E, and Sealfon SC (2015). CellCODE: a robust latent variable approach to differential expression analysis for heterogeneous cell populations. Bioinformatics 31, 1584–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KW, Dhillon P, Huang S, Sheng X, Shrestha R, Qiu C, Kaufman BA, Park J, Pei L, Baur J, et al. (2019). Mitochondrial Damage and Activation of the STING Pathway Lead to Renal Inflammation and Fibrosis. Cell Metab 30, 784–799 e785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combes AN, Zappia L, Er PX, Oshlack A, and Little MH (2019). Single-cell analysis reveals congruence between kidney organoids and human fetal kidney. Genome Med 11, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lau W, Barker N, Low TY, Koo BK, Li VS, Teunissen H, Kujala P, Haegebarth A, Peters PJ, van de Wetering M, et al. (2011). Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 476, 293–297. [DOI] [PubMed] [Google Scholar]
- Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, Worley PF, Kozma SC, and Powell JD (2009). The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 30, 832–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edeling M, Ragi G, Huang S, Pavenstadt H, and Susztak K (2016). Developmental signalling pathways in renal fibrosis: the roles of Notch, Wnt and Hedgehog. Nat Rev Nephrol 12, 426–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garreta E, Prado P, Tarantino C, Oria R, Fanlo L, Marti E, Zalvidea D, Trepat X, Roca-Cusachs P, Gavalda-Navarro A, et al. (2019). Fine tuning the extracellular environment accelerates the derivation of kidney organoids from human pluripotent stem cells. Nat Mater 18, 397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez IG, MacKenna DA, Johnson BG, Kaimal V, Roach AM, Ren S, Nakagawa N, Xin C, Newitt R, Pandya S, et al. (2015). Anti-microRNA-21 oligonucleotides prevent Alport nephropathy progression by stimulating metabolic pathways. J Clin Invest 125, 141–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grande MT, Sanchez-Laorden B, Lopez-Blau C, De Frutos CA, Boutet A, Arevalo M, Nieto MA. (2015) Snail1-induced partial epithelial-to mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat Med. 21, 989–997. [DOI] [PubMed] [Google Scholar]
- He W, Dai C, Li Y, Zeng G, Monga SP, and Liu Y (2009). Wnt/beta-catenin signaling promotes renal interstitial fibrosis. J Am Soc Nephrol 20, 765–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellwege JN, Velez Edwards DR, Giri A, Qiu C, Park J, Torstenson ES, Keaton JM, Wilson OD, Robinson-Cohen C, Chung CP, et al. (2019). Mapping eGFR loci to the renal transcriptome and phenome in the VA Million Veteran Program. Nat Commun 10, 3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Park J, Qiu C, Chung KW, Li SY, Sirin Y, Han SH, Taylor V, Zimber-Strobl U, and Susztak K (2018). Jagged1/Notch2 controls kidney fibrosis via Tfam-mediated metabolic reprogramming. PLoS Biol 16, e2005233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HM, Ahn SH, Choi P, Ko YA, Han SH, Chinga F, Park AS, Tao J, Sharma K, Pullman J, et al. (2015). Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med 21, 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HM, Huang S, Reidy K, Han SH, Chinga F, and Susztak K (2016). Sox9-Positive Progenitor Cells Play a Key Role in Renal Tubule Epithelial Regeneration in Mice. Cell Rep 14, 861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Gruenwald A, Suh JH, Miner JH, Barisoni-Thomas L, Taketo MM, Faul C, Millar SE, Holzman LB, and Susztak K (2011). Wnt/beta-catenin pathway in podocytes integrates cell adhesion, differentiation, and survival. J Biol Chem 286, 26003–26015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg MD, Bhargava P, Kim PM, Putluri V, Snowman AM, Putluri N, Calabresi PA, and Snyder SH (2018). Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity. Science 360, 449–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovesdy CP, Bleyer AJ, Molnar MZ, Ma JZ, Sim JJ, Cushman WC, Quarles LD, and Kalantar-Zadeh K (2013). Blood pressure and mortality in U.S. veterans with chronic kidney disease: a cohort study. Ann Intern Med 159, 233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lonnerberg P, Furlan A, et al. (2018). RNA velocity of single cells. Nature 560, 494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake BB, Chen S, Hoshi M, Plongthongkum N, Salamon D, Knoten A, Vijayan A, Venkatesh R, Kim EH, Gao D, et al. (2019). A single-nucleus RNA-sequencing pipeline to decipher the molecular anatomy and pathophysiology of human kidneys. Nat Commun 10, 2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin A, Tonelli M, Bonventre J, Coresh J, Donner JA, Fogo AB, Fox CS, Gansevoort RT, Heerspink HJL, Jardine M, et al. (2017). Global kidney health 2017 and beyond: a roadmap for closing gaps in care, research, and policy. Lancet 390, 1888–1917. [DOI] [PubMed] [Google Scholar]
- Li Y, Wen X, and Liu Y (2012). Tubular cell dedifferentiation and peritubular inflammation are coupled by the transcription regulator Id1 in renal fibrogenesis. Kidney Int 81, 880–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Krautzberger AM, Sui SH, Hofmann OM, Chen Y, Baetscher M, Grgic I, Kumar S, Humphreys BD, Hide WA, et al. (2014). Cell-specific translational profiling in acute kidney injury. J Clin Invest 124, 1242–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovisa S, LeBleu VS, Tampe B, Sugimoto H, Vadnagara K, Carstens JL, Kalluri R. (2015) Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med 21, 998–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa H, Inoue T, Ouchi H, Jao TM, Inoue R, Nishi H et al. (2019) Mitochondrial Damage Causes Inflammation via cGAS-STING Signaling in Acute Kidney Injury. Cell Rep. 29, 1261.e6–1273.e6 [DOI] [PubMed] [Google Scholar]
- Marable SS, Chung E, and Park J (2020). Hnf4a is required for the development of Cdh6-expressing progenitors into proximal tubules in the mouse kidney. bioRxiv preprint doi: 10.1101/2020.02.16.951731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis CS, Murrow LM, and Gartner ZJ (2019). DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst 8, 329–337 e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meersch M, Schmidt C, Van Aken H, Martens S, Rossaint J, Singbartl K, Gorlich D, Kellum JA, and Zarbock A (2014). Urinary TIMP-2 and IGFBP7 as early biomarkers of acute kidney injury and renal recovery following cardiac surgery. PLoS One 9, e93460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon R, Otto. EA, Hoover. P, et al. (2020). Single cell transcriptomics identifies focal segmental glomerulosclerosis remission endothelial biomarker. JCI Insight 5, e133267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG, and Rathmell JC (2011). Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol 186, 3299–3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Shrestha R, Qiu C, Kondo A, Huang S, Werth M, Li M, Barasch J, and Susztak K (2018). Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 360, 758–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu C, Huang S, Park J, Park Y, Ko YA, Seasock MJ, Bryer JS, Xu XX, Song WC, Palmer M, et al. (2018). Renal compartment-specific genetic variation analyses identify new pathways in chronic kidney disease. Nat Med 24, 1721–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiros PM, Goyal A, Jha P, Auwerx J. (2017). Analysis of mtDNA/nDNA Ratio in Mice. Curr Protoc Mouse Biol., 7(1):47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reidy K, Kang HM, Hostetter T, and Susztak K (2014). Molecular mechanisms of diabetic kidney disease. J Clin Invest 124, 2333–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinkevich Y, Montoro DT, Contreras-Trujillo H, Harari-Steinberg O, Newman AM, Tsai JM, Lim X, Van-Amerongen R, Bowman A, Januszyk M, et al. (2014). In vivo clonal analysis reveals lineage-restricted progenitor characteristics in mammalian kidney development, maintenance, and regeneration. Cell Rep 7, 1270–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh BK, Sinha RA, Tripathi M, Mendoza A, Ohba K, Sy JAC, … Yen PM (2018). Thyroid hormone receptor and ERRα coordinately regulate mitochondrial fission, mitophagy, biogenesis, and function. Science Signaling, 11(536), eaam5855. [DOI] [PubMed] [Google Scholar]
- Shen Y, Wen Z, Li Y, Matteson EL, Hong J, Goronzy JJ, and Weyand CM (2017). Metabolic control of the scaffold protein TKS5 in tissue-invasive, proinflammatory T cells. Nat Immunol 18, 1025–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smillie CS, Biton M, Ordovas-Montanes J, Sullivan KM, Burgin G, Graham DB, Herbst RH, Rogel N, Slyper M, Waldman J, et al. (2019). Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis. Cell 178, 714–730 e722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano FX, Liesa M, Bach D, Chan DC, Palacín M, Zorzano A (2006). Evidence for a mitochondrial regulatory pathway defined by peroxisome proliferator-activated receptor-gamma coactivator-1 alpha, estrogen-related receptor-alpha, and mitofusin. Diabetes 55,1783–1791. [DOI] [PubMed] [Google Scholar]
- Tran M, Tam D, Bardia A, Bhasin M, Rowe GC, Kher A, Zsengeller ZK, Akhavan-Sharif MR, Khankin EV, Saintgeniez M, et al. (2011). PGC-1alpha promotes recovery after acute kidney injury during systemic inflammation in mice. J Clin Invest 121, 4003–4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran MT, Zsengeller ZK, Berg AH, Khankin EV, Bhasin MK, Kim W, Clish CB, Stillman IE, Karumanchi SA, Rhee EP, et al. (2016). PGC1alpha drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 531, 528–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, and Rinn JL (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 32, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayan A, Faubel S, Askenazi DJ, Cerda J, Fissell WH, Heung M, Humphreys BD, Koyner JL, Liu KD, Mour G, et al. (2016). Clinical Use of the Urine Biomarker [TIMP-2] × [IGFBP7] for Acute Kidney Injury Risk Assessment. Am J Kidney Dis 68, 19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woroniecka KI, Park AS, Mohtat D, Thomas DB, Pullman JM, and Susztak K (2011). Transcriptome analysis of human diabetic kidney disease. Diabetes 60, 2354–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Kirita Y, Donnelly EL, and Humphreys BD (2019). Advantages of Single-Nucleus over Single-Cell RNA Sequencing of Adult Kidney: Rare Cell Types and Novel Cell States Revealed in Fibrosis. J Am Soc Nephrol 30, 23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Uchimura K, Donnelly EL, Kirita Y, Morris SA, and Humphreys BD (2018). Comparative Analysis and Refinement of Human PSC-Derived Kidney Organoid Differentiation with Single-Cell Transcriptomics. Cell Stem Cell 23, 869–881 e868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuttke M, Li Y, Li M, Sieber KB, Feitosa MF, Gorski M, Tin A, Wang L, Chu AY, Hoppmann A, et al. (2019). A catalog of genetic loci associated with kidney function from analyses of a million individuals. Nat Genet 51, 957–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Fujii H, Mohan SV, Goronzy JJ, and Weyand CM (2013). Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells. J Exp Med 210, 2119–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Choi SC, Xu Z, Perry DJ, Seay H, Croker BP, Sobel ES, Brusko TM, and Morel L (2015). Normalization of CD4+ T cell metabolism reverses lupus. Sci Transl Med 7, 274ra218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young MD, Mitchell TJ, Vieira Braga FA, Tran MGB, Stewart BJ, Ferdinand JR, Collord G, Botting RA, Popescu DM, Loudon KW, et al. (2018). Single-cell transcriptomes from human kidneys reveal the cellular identity of renal tumors. Science 361, 594–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg. M, Duffield JS (2010) EMT produces fibroblasts in the kidney. J Am Soc Nephrol., 21(8):1247–1253. [DOI] [PubMed] [Google Scholar]
- Zhang R, Boareto M, Engler A, Louvi A, Giachino C, Iber D, and Taylor V (2019). Id4 Downstream of Notch2 Maintains Neural Stem Cell Quiescence in the Adult Hippocampus. Cell Rep 28, 1485–1498 e1486. [DOI] [PubMed] [Google Scholar]
- Zhao J, Lupino K, Wilkins BJ, Qiu C, Liu J, Omura Y, Allred AL, McDonald C, Susztak K, Barish GD, et al. (2018). Genomic integration of ERRgamma-HNF1beta regulates renal bioenergetics and prevents chronic kidney disease. Proc Natl Acad Sci U S A 115, E4910–E4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M, Cai J, Liu Z, Shu S, Wang Y, Tang C, and Dong Z (2019). Nicotinamide reduces renal interstitial fibrosis by suppressing tubular injury and inflammation. J Cell Mol Med 23, 3995–4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary table 1(Related to Figure 1). List of single cell cluster markers
Supplementary table 2 (Related to Figure 2). List of differential expressed genes in control and FAN mice samples
Supplementary table 3 (Related to Figure 3). List of PT subcluster markers in control and FAN samples
Supplementary table 4 (Related to Figure 4). The percent of cells that express EMT and SASP markers across the PT cell sub-clusters in FAN kidneys and all the cell clusters
Supplementary table 5 (Related to Figure 6). List of predicted ESRRA target genes in PT cells.
Supplementary table 6 (Related to Key Resource Table). List of primers used in this study.
Supplementary table 7 (Related to Figure 7 and S7). Demographic and clinical of human kidney samples.
Data Availability Statement
Processed and raw data can be downloaded from NCBI GEO (GSE156686 and GSE152765). Furthermore, the data is available via an interactive web browser at http://susztaklab.com/VisCello/.