

Abstract
Cortical spreading depolarizations (CSDs) are characterized by waves of diminished electroencephalography activity that propagate across the cortex with subsequent loss of ionic homeostasis. CSDs have been found in many pathological conditions, including migraine, traumatic brain injury, and ischemic stroke. Because of CSD-associated ionic and metabolic disturbances at the peri-infarct area after ischemic stroke, it is thought that CSDs exacerbate tissue infarction and worsen clinical outcomes. Microglia, the main innate immune cells in the brain, are among the first responders to brain tissue damage. Recent studies demonstrated that microglia play a critical role in CSD initiation and propagation. In this article, we discuss the significance of CSD in the setting of ischemic stroke and how microglia may modulate peri-infarct CSDs, also known as iso-electric depolarizations. Finally, we discuss the significance of microglial Ca2+ and how it might be used as a potential therapeutic target for patients with ischemic stroke.
Keywords: Microglia, Calcium signaling, In vivo imaging, 2-Photon, Ischemic stroke, Iso-electric depolarizations, Cortical spreading depolarization
Introduction
In response to brain injury, neurons undergo synchronized depolarization that initiates at the site of injury and propagates across the cortex. This phenomenon is known as a cortical spreading depolarization (CSD) [1, 2]. After a window of recovery, characterized by depression of electrical activity, neurons slowly repolarize and electrical homeostasis is reestablished. CSDs have been shown to occur after a variety of cerebral insults, including migraine, trauma, cortical chemical exposure, and ischemia [2–4]. The molecular hallmark of CSD is a near-complete breakdown of the transmembrane ion gradients, with subsequent increases in extracellular glutamate, adenosine triphosphate (ATP), and K+ and intracellular Ca2+, Na+, and Cl− [5–7]. This phenomenon was first inferred from the examination and analysis of scotoma during migraine aura [8, 9]. The researchers surmised that visual migraine auras might be caused by electrical disturbances in the cortex and began attempting to correlate visual auras with the speed of cortical spreading depression of activity recorded by Leao, who pioneered electrophysiological measurements of CSD [8–10]. Similar waves of depolarization activity have been observed in other cranial pathologies, such as subarachnoid hemorrhage and traumatic brain injury [11, 12]. Although tissue with reversible injury can recover electrical homeostasis and resume normal function, tissue facing permanent damage, as in the setting of ischemic stroke, is unable to return to baseline, and neurologic deficits result [13, 14].
Ischemia-Induced Mechanisms Driving Spreading Depolarizations
Ischemic stroke is characterized by vascular occlusion resulting in diminished cerebral blood flow, creating a hypoxic environment. Without sufficient levels of oxygen, ATP production decreases, energy deficit increases, and neurons within the affected tissue are unable to maintain the plasma membrane potential [15, 16]. Such an insult triggers progressive CSDs, which radiate across the cortex at a speed of 3–5 mm/s [3, 17]. Cell membrane depolarization leads to K+ and neurotransmitter egress out of the cell, whereas Na+, Cl−, and Ca2+ rapidly enter the cell [6, 7]. Because of the influx of positively charged electrolytes, neuron cell bodies begin to swell from their resulting hyperosmolar state, and higher intracellular Ca2+ levels trigger apoptotic processes [16]. The damaged neuron releases glutamate and free radicals into the extracellular space, which activate N-methyl-D-aspartate (NMDA) receptors on the surrounding neurons and induce further release of inflammatory cytokines [7, 15, 16]. Matrix metallopeptidases are subsequently released and contribute to the breakdown of the blood–brain barrier [18, 19]. These cumulative processes produce an acidic, hypoxic, and hyperkalemic environment that stimulates vasoconstriction, exacerbating the oxygen and energy deficits [6, 16]. This leads to a cyclical progression of neuronal damage and expansion of the ischemic tissue volume, as the peri-infarct region cannot adequately repolarize [14, 20].
Clinical Evidence for Iso-Electric Spreading Depolarizations and Depressions
CSDs have been measured in neurocritical care patients with large middle cerebral artery ischemic strokes using subdural electrocorticography strips placed over the affected area [21, 22]. Although infarcted tissue does not exhibit electrical activity, electrocorticography strips placed over the peri-infarct region capture depressed baseline electrical activity and slow depolarization waves as ischemic time progresses [12]. CSDs are commonly seen in ischemic strokes, and more frequent depolarization events appear to be directly associated with increased infarct size [2, 4, 21, 23, 24]. Compared with CSDs, which have a normal or near-normal initial baseline function, iso-electric spreading depressions have lower baseline activity and are slower to repolarize [14]. Patients with ischemic stroke who experience a transition from CSD to iso-electric spreading depression typically have more significant post-stroke neurological deficits compared with those with sustained CSD [25].
Microglial Roles in the Initiation and Amplification of Spreading Depolarizations
Microglia are a principal component of the cerebral immune system, which is readily activated in response to noxious stimuli. In the resting state, microglia constantly surveil the surrounding brain parenchyma with long ramified processes [26–28]. If an insult is detected, the microglia change morphology and become more amoeboid in shape to facilitate reactive responses in diverse activation states [29, 30]. Microglia may detect ischemia from signals such as damage-associated molecular patterns, elevated extracellular ATP and adenosine levels, or abnormal electrolyte concentrations [31–33]. These hypoxic microglia respond by either activating proinflammatory processes and releasing neurotoxic cytokines, such as tumor necrosis factor-α, interleukins 1β, and interferon-γ, or executing anti-inflammatory programs to encourage tissue repair and neuroprotection [34–37]. This dichotomous activity has made it challenging to clarify the role of microglia in ischemic stroke [38–41]. Remarkably, recent studies have suggested that microglia are important players in the induction and progression of CSD [41, 42]. For example, Pusic and coauthors [42] examined microglia-depleted organotypic hippocampal slice cultures and were unable to induce CSD at all. Conversely, restoration of microglia to previously depleted slice cultures enabled repeated CSD [42]. Similarly, animal studies with selectively depleted microglia in the brain exhibited diminished CSD occurrence and frequency [43]. Therefore, microglia appear to be required for CSD initiation and progression. It is somewhat surprising that microglia, which account for only about 10% of total brain cells, have such a profound impact on CSD. One important mechanism in microglia–neuron communication appears to be operating via NMDA receptors. It has been shown that NMDA receptors are required for CSD induction and propagation, and blockade of NMDA receptors inhibits CSD [44–46]. Further, Moriguchi et al. [47] have demonstrated that microglia can potentiate NMDA-receptor-mediated synaptic current in neurons. The same group showed that NMDA-receptor-mediated current in neurons increased 10-fold after application of microglia-conditioned medium [48]. This effect was mediated through activation of the glycine site on NMDA receptors by microglia secreting soluble factors [48]. Together, these findings support the notion that activated microglia affect NMDA currents and consequently increase neuronal excitability [49, 50].
Microglial Calcium as a Therapeutic Target in Ischemic Stroke
CSD-associated depolarization of neurons causes dramatic increase of the extracellular levels of K+, ATP, and adenosine. Adenosine and adenosine diphosphate, produced via ATP breakdown, activate purinergic receptors on microglia, elevating intracellular Ca2+ levels [51–57]. The Ca2+ influx is at least partially mediated by the combined action of the cell surface purinergic receptors and subsequently the calcium release-activated calcium (CRAC) channels. Next, elevated Ca2+ levels in microglia promote expression of genes encoding several proinflammatory factors, including tumor necrosis factor α. These inflammatory factors can lower the CSD threshold in neurons by initiating the flux of charged ions through the plasma membrane. This ionic flux changes the homeostatic membrane potential, contributing to increased susceptibility for the next depolarization event [58, 59]. As discussed above, activated microglia can, for example, stimulate NMDA-receptor-mediated Ca2+ influx into neurons by activating NMDA receptors [48, 60]. Consequently, neuronal membrane depolarization is prolonged, ionic dyshomeostasis is further aggravated, and the initiation and propagation of CSD is amplified. Therefore, we propose that neurons and microglia in the setting of CSD engage in a self-amplifying feedback loop that can increase infarct size (Fig. 1).
Fig. 1.

Neuron–microglia interactions in the setting of cortical spreading depolarization (CSD). After ischemic injury, neurons and microglia can engage in a self-propagating feedback loop, potentially worsening stroke outcome. Initial CSDs, occurring during prolonged depolarization of neurons at around − 10 mV, cause increase of extracellular purines, such as adenosine triphosphate (ATP), adenosine diphosphate (ADP), and adenosine, as well as potassium (K+). ATP/ADP activate both ionotropic purinergic receptors, P2X, and metabotropic purinergic receptors, P2Y, on microglia. Activation of P2X channels mediates Ca2+ influx in microglia, whereas the activation of P2Y receptors triggers Ca2+ release from microglial endoplasmic reticulum (ER) through phospholipase C–inositol 1,4,5-trisphosphate (PLC-IP3) signaling pathways. The depletion of ER store activates the calcium release-activated calcium (CRAC) channels, mediating additional Ca2+ influx into microglia. These events converge on the major elevation of intracellular Ca2+. High Ca2+ levels stimulate the inflammatory cytokine production through the calcineurin–nuclear factor of activated T cells (NFAT) pathway. Cytokines affect the CSD threshold in the nearby neurons by modulating N-methyl-D-aspartate (NMDA) currents. Sustained activation of NMDA receptors (NMDAR) further increases K+ leak to the extracellular space, provoking the next CSD initiation and propagation. This positive feedback loop may exacerbate neuronal damage in the periinfarct penumbra after stroke
Microglial purinergic receptors elevate intracellular Ca2+ through both ionotropic and metabotropic pathways. Upon activation, the P2X7 ionotropic receptors increase their plasma membrane channel conductance, mediating Ca2+ influx into the cell [37, 61–63]. The metabotropic P2Y receptors, including P2Y12, trigger inositol 1,4,5-trisphosphate (IP3) activity and allow Ca2+ release from the microglial intracellular stores, such as the endoplasmic reticulum [51, 64, 65]. The P2Y–IP3 pathway has been shown to contribute to microglial morphology changes, phagocytosis, chemotaxis toward the site of injury, and the formation of purinergic junctions between microglia and neurons [26, 66, 67]. These junctions appear to decrease Ca2+ influx into damaged neurons and have a neuroprotective effect by preventing cytotoxic edema and apoptosis [66].
Although this initial P2Y–IP3 pathway is initially neuroprotective and speaks to the beneficial role of microglia in neuronal recovery, as ischemic time progresses, extracellular calcium begins to enter the microglia via CRAC channels. After the intracellular Ca2+ stores become depleted by the initial signaling processes, the CRAC channels in the plasma membrane open to mediate a major influx of extracellular Ca2+ into the cell [68]. Within the hypoxic microglia, Ca2+ affects an incompletely understood set of downstream processes, including the calcineurin pathway, which is involved in modulating gene expression in the immune cells [69–71]. This delayed influx of calcium into microglia contributes to a persistent production of inflammatory cytokines that transitions the effect of microglia from neuroprotective to neurotoxic. A recent study by Mizuma and colleagues [69] reported the utility of the CRAC channel inhibitor, CM-EX-137, in the treatment of traumatic brain injury. They found that CM-EX-137 reduced the effect of nitric oxide and decreased intracellular microglial Ca2+ accumulation and the transcription of inflammatory cytokines. In their model, mice treated with CM-EX-137 after traumatic brain injury had smaller lesion sizes, less frequent hemorrhages, and improved overall neurological function compared with controls [69]. A recent study from our laboratory demonstrated that blockade of CRAC channels with CM-EM-137 partially decreased CSD-associated microglial Ca2+ influx [54]. Thus, CRAC channel inhibitors emerge as promising, well-tolerated, and effective antagonists of microglial activation, and prospective clinical studies are warranted to evaluate the benefits of CRAC channel inhibition in the treatment of ischemic stroke (Fig. 2).
Fig. 2.
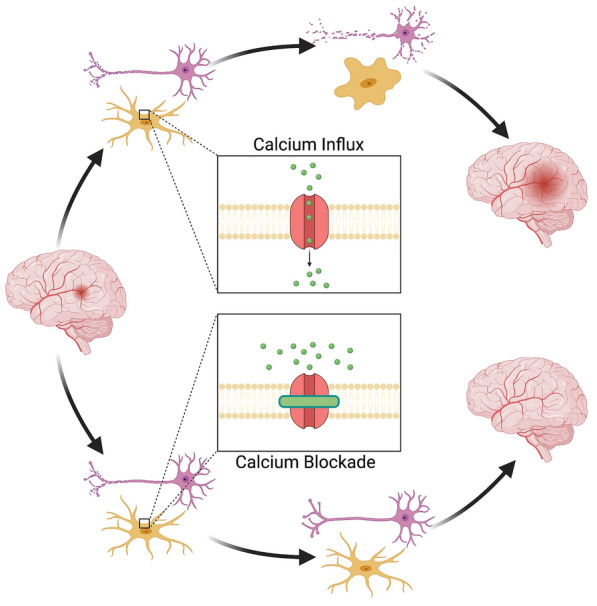
Calcium influx as an emerging treatment target for ischemic stroke. Blockade of Ca2+ influx through the calcium release-activated calcium (CRAC) channels may be a new therapeutic strategy for the treatment of ischemic stroke. Pharmacological inhibition of the CRAC-mediated Ca2+ current in the ischemic brain could facilitate significant benefits, without adverse side effects
Conclusions
Despite the considerable amount of research on pathophysiology of ischemic stroke, effective treatments are lacking. Several therapeutic targets have been identified, but clinical validation has yet to be obtained. Currently, one of the promising targets appears to be Ca2+ influx in ischemic brain cells, especially in microglia. Further research is needed to fully elucidate the significance of microglial Ca2+ overload during the acute phase of ischemic injury, and to identify the optimal approaches to limit its harmful consequences.
Author contributions
KNK and LL wrote the manuscript and designed the figures, SS and KAS edited the manuscript, MES and MSP read and approved the manuscript, and PT edited the manuscript and figures and approved the submission.
Source of support
This work was supported by National Institutes of Health grant R21NS116431 to PT.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval/informed consent
This article does not contain any studies with human participants or animals performed by any of the authors.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Dreier JP. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med. 2011;17(4):439–447. doi: 10.1038/nm.2333. [DOI] [PubMed] [Google Scholar]
- 2.Lauritzen M, Dreier JP, Fabricius M, et al. Clinical relevance of cortical spreading depression in neurological disorders: Migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J Cereb Blood Flow Metab. 2011;31(1):17–35. doi: 10.1038/jcbfm.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Somjen GG. Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol Rev. 2001;81(3):1065–1096. doi: 10.1152/physrev.2001.81.3.1065. [DOI] [PubMed] [Google Scholar]
- 4.Mies G, Iijima T, Hossmann KA. Correlation between peri-infarct dc shifts and ischaemic neuronal damage in rat. NeuroReport. 1993;4(6):709–711. doi: 10.1097/00001756-199306000-00027. [DOI] [PubMed] [Google Scholar]
- 5.Ayata C, Lauritzen M. Spreading depression, spreading depolarizations, and the cerebral vasculature. Physiol Rev. 2015;95(3):953–993. doi: 10.1152/physrev.00027.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chuquet J, Hollender L, Nimchinsky EA. High-resolution in vivo imaging of the neurovascular unit during spreading depression. J Neurosci. 2007;27(15):4036–4044. doi: 10.1523/JNEUROSCI.0721-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Enger R, Tang W, Vindedal GF, et al. Dynamics of ionic shifts in cortical spreading depression. Cereb Cortex. 2015;25(11):4469–4476. doi: 10.1093/cercor/bhv054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lashley KS. Patterns of cerebral integration indicated by the scotomas of migraine. Arch Neurol Psychiatry. 1941;46(2):331–339. [Google Scholar]
- 9.Milner PM. Note on a possible correspondence between the scotomas of migraine and spreading depression of Leão. Electroencephalogr Clin Neurophysiol. 1958;10(4):705. doi: 10.1016/0013-4694(58)90073-7. [DOI] [PubMed] [Google Scholar]
- 10.Leao AA. The slow voltage variation of cortical spreading depression of activity. Electroencephalogr Clin Neurophysiol. 1951;3(3):315–321. doi: 10.1016/0013-4694(51)90079-x. [DOI] [PubMed] [Google Scholar]
- 11.Drenckhahn C, Winkler MKL, Major S, et al. Correlates of spreading depolarization in human scalp electroencephalography. Brain. 2012;135(3):853–868. doi: 10.1093/brain/aws010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hartings JA, Wilson JA, Hinzman JM, et al. Spreading depression in continuous electroencephalography of brain trauma. Ann Neurol. 2014;76(5):681–694. doi: 10.1002/ana.24256. [DOI] [PubMed] [Google Scholar]
- 13.Harriott AM, Barrett KM. Dissecting the association between migraine and stroke. Curr Neurol Neurosci Rep. 2015;15(3):5. doi: 10.1007/s11910-015-0530-8. [DOI] [PubMed] [Google Scholar]
- 14.Hartings JA, Shuttleworth CW, Kirov SA, et al. The continuum of spreading depolarizations in acute cortical lesion development: Examining Leão’s legacy. J Cerebral Blood Flow Metab. 2017;37(5):1571–1594. doi: 10.1177/0271678X16654495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karsy M, Brock A, Guan J, et al. Neuroprotective strategies and the underlying molecular basis of cerebrovascular stroke. Neurosurg Focus. 2017;42(4):E3. doi: 10.3171/2017.1.FOCUS16522. [DOI] [PubMed] [Google Scholar]
- 16.Kramer DR, Fujii T, Ohiorhenuan I, Liu CY. Cortical spreading depolarization: Pathophysiology, implications, and future directions. J Clin Neurosci. 2016;24:22–27. doi: 10.1016/j.jocn.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 17.Takano T, Tian G-F, Peng W, et al. Cortical spreading depression causes and coincides with tissue hypoxia. Nat Neurosci. 2007;10(6):754–762. doi: 10.1038/nn1902. [DOI] [PubMed] [Google Scholar]
- 18.Asahi M, Wang X, Mori T, et al. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J Neurosci. 2001;21(19):7724–7732. doi: 10.1523/JNEUROSCI.21-19-07724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gursoy-Ozdemir Y, Qiu J, Matsuoka N, et al. Cortical spreading depression activates and upregulates MMP-9. J Clin Investig. 2004;113(10):1447–1455. doi: 10.1172/JCI21227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wainsztein N, Lucci Rodríguez F. Cortical Spreading Depression and Ischemia in Neurocritical Patients. Neurosurg Clin N Am. 2018;29(2):223–229. doi: 10.1016/j.nec.2017.11.003. [DOI] [PubMed] [Google Scholar]
- 21.Dohmen C, Sakowitz OW, Fabricius M, et al. Spreading depolarizations occur in human ischemic stroke with high incidence. Ann Neurol. 2008;63(6):720–728. doi: 10.1002/ana.21390. [DOI] [PubMed] [Google Scholar]
- 22.Dreier JP, Fabricius M, Ayata C, et al. Recording, analysis, and interpretation of spreading depolarizations in neurointensive care: review and recommendations of the COSBID research group. J Cerebral Blood Flow Metab. 2017;37(5):1595–1625. doi: 10.1177/0271678X16654496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hartings JA, Rolli ML, Lu X-CM, Tortella FC. Delayed secondary phase of peri-infarct depolarizations after focal cerebral ischemia: relation to infarct growth and neuroprotection. J Neurosci. 2003;23(37):11602–11610. doi: 10.1523/JNEUROSCI.23-37-11602.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Strong AJ, Fabricius M, Boutelle MG, et al. Spreading and synchronous depressions of cortical activity in acutely injured human brain. Stroke. 2002;33(12):2738–2743. doi: 10.1161/01.str.0000043073.69602.09. [DOI] [PubMed] [Google Scholar]
- 25.Fabricius M, Fuhr S, Bhatia R, et al. Cortical spreading depression and peri-infarct depolarization in acutely injured human cerebral cortex. Brain J Neurol. 2006;129(Pt 3):778–790. doi: 10.1093/brain/awh716. [DOI] [PubMed] [Google Scholar]
- 26.Davalos D, Grutzendler J, Yang G, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8(6):752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- 27.Liu YU, Ying Y, Li Y, et al. Neuronal network activity controls microglial process surveillance in awake mice via norepinephrine signaling. Nat Neurosci. 2019;22(11):1771–1781. doi: 10.1038/s41593-019-0511-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science (New York, N.Y.) 2005;308(5726):1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 29.Ransohoff RM. A polarizing question: Do M1 and M2 microglia exist. Nat Neurosci. 2016;19(8):987–991. doi: 10.1038/nn.4338. [DOI] [PubMed] [Google Scholar]
- 30.Kim CC, Nakamura MC, Hsieh CL. Brain trauma elicits non-canonical macrophage activation states. J Neuroinflamm 2016;13(1). [DOI] [PMC free article] [PubMed]
- 31.Benakis C, Garcia-Bonilla L, Iadecola C, Anrather J. The role of microglia and myeloid immune cells in acute cerebral ischemia. Front Cell Neurosci. 2015;8(JAN):461. doi: 10.3389/fncel.2014.00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Inoue K. The function of microglia through purinergic receptors: Neuropathic pain and cytokine release. Pharmacol Ther. 2006;109(1–2):210–226. doi: 10.1016/j.pharmthera.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 33.Ma Y, Wang J, Wang Y, Yang GY. The biphasic function of microglia in ischemic stroke. Prog Neurobiol. 2017;157:247–272. doi: 10.1016/j.pneurobio.2016.01.005. [DOI] [PubMed] [Google Scholar]
- 34.Franco ECS, Cardoso MM, Gouvêia A, Pereira A, Gomes-Leal W. Modulation of microglial activation enhances neuroprotection and functional recovery derived from bone marrow mononuclear cell transplantation after cortical ischemia. Neurosci Res. 2012;73(2):122–132. doi: 10.1016/j.neures.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 35.Nimmervoll B, White R, Yang J-W, et al. LPS-induced microglial secretion of TNFα increases activity-dependent neuronal apoptosis in the neonatal cerebral cortex. Cerebral Cortex (New York, N.Y.:1991) 2013;23(7):1742–1755. doi: 10.1093/cercor/bhs156. [DOI] [PubMed] [Google Scholar]
- 36.Patel AR, Ritzel R, Mccullough LD, Liu F. Microglia and ischemic stroke: a double-edged sword. Int J Physiol Pathophysiol Pharmacol. 2013;5(2):73–90. [PMC free article] [PubMed] [Google Scholar]
- 37.Witting A, Walter L, Wacker J, Möller T, Stella N. P2X7 receptors control 2-arachidonoylglycerol production by microglial cells. Proc Natl Acad Sci USA. 2004;101(9):3214–3219. doi: 10.1073/pnas.0306707101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fernández-López D, Faustino J, Klibanov AL, et al. Microglial cells prevent hemorrhage in neonatal focal arterial stroke. J Neurosci. 2016;36(10):2881–2893. doi: 10.1523/JNEUROSCI.0140-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guruswamy R, ElAli A. Complex roles of microglial cells in ischemic stroke pathobiology: new insights and future directions. Int J Mol Sci 2017;18(3). [DOI] [PMC free article] [PubMed]
- 40.Li M, Li Z, Ren H, et al. Colony stimulating factor 1 receptor inhibition eliminates microglia and attenuates brain injury after intracerebral hemorrhage. J Cerebral Blood Flow Metab 2016:0271678X16666551. [DOI] [PMC free article] [PubMed]
- 41.Szalay G, Martinecz B, Lénárt N, et al. Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat Commun 2016;7. [DOI] [PMC free article] [PubMed]
- 42.Pusic KM, Pusic AD, Kemme J, Kraig RP. Spreading depression requires microglia and is decreased by their M2a polarization from environmental enrichment. Glia. 2014;62(7):1176–1194. doi: 10.1002/glia.22672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Varga DP, Menyhárt Á, Pósfai B, et al. Microglia alter the threshold of spreading depolarization and related potassium uptake in the mouse brain. J Cerebral Blood Flow Metab 2020;40(1_suppl):S67-S80. [DOI] [PMC free article] [PubMed]
- 44.Lauritzen M, Hansen AJ. The effect of glutamate receptor blockade on anoxic depolarization and cortical spreading depression. J Cereb Blood Flow Metab. 1992;12(2):223–229. doi: 10.1038/jcbfm.1992.32. [DOI] [PubMed] [Google Scholar]
- 45.Marrannes R, Willems R, De Prins E, Wauquier A. Evidence for a role of the N-methyl-D-aspartate (NMDA) receptor in cortical spreading depression in the rat. Brain Res. 1988;457(2):226–240. doi: 10.1016/0006-8993(88)90690-7. [DOI] [PubMed] [Google Scholar]
- 46.Sakowitz OW, Kiening KL, Krajewski KL, et al. Preliminary evidence that ketamine inhibits spreading depolarizations in acute human brain injury. Stroke. 2009;40(8):e519–e522. doi: 10.1161/STROKEAHA.109.549303. [DOI] [PubMed] [Google Scholar]
- 47.Moriguchi S, Mizoguchi Y, Tomimatsu Y, et al. Potentiation of NMDA receptor-mediated synaptic responses by microglia. Brain Res Mol Brain Res. 2003;119(2):160–169. doi: 10.1016/j.molbrainres.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 48.Hayashi Y, Ishibashi H, Hashimoto K, Nakanishi H. Potentiation of the NMDA receptor-mediated responses through the activation of the glycine site by microglia secreting soluble factors. Glia. 2006;53(6):660–668. doi: 10.1002/glia.20322. [DOI] [PubMed] [Google Scholar]
- 49.Klapal L, Igelhorst BA, Dietzel-Meyer ID. Changes in neuronal excitability by activated microglia: differential Na(+) current upregulation in pyramid-shaped and bipolar neurons by TNF-alpha and IL-18. Front Neurol. 2016;7:44. doi: 10.3389/fneur.2016.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vezzani A, Viviani B. Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology. 2015;96(Pt A):70–82. doi: 10.1016/j.neuropharm.2014.10.027. [DOI] [PubMed] [Google Scholar]
- 51.Färber K, Kettenmann H. Functional role of calcium signals for microglial function. Glia. 2006;54(7):656–665. doi: 10.1002/glia.20412. [DOI] [PubMed] [Google Scholar]
- 52.Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10(11):1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- 53.Lindquist BE, Shuttleworth CW. Spreading depolarization-induced adenosine accumulation reflects metabolic status in vitro and in vivo. J Cereb Blood Flow Metab. 2014;34(11):1779–1790. doi: 10.1038/jcbfm.2014.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu L, Kearns KN, Eli I, et al. Microglial calcium waves during the hyperacute phase of ischemic stroke. Stroke 2021. [DOI] [PMC free article] [PubMed]
- 55.McLarnon JG. Purinergic mediated changes in Ca2+ mobilization and functional responses in microglia: effects of low levels of ATP. J Neurosci Res. 2005;81(3):349–356. doi: 10.1002/jnr.20475. [DOI] [PubMed] [Google Scholar]
- 56.Pozner A, Xu B, Palumbos S, et al. Intracellular calcium dynamics in cortical microglia responding to focal laser injury in the PC::G5-tdT reporter mouse. Front Mol Neurosci 2015;8(MAY). [DOI] [PMC free article] [PubMed]
- 57.Tvrdik P, Kearns KN, Sharifi KA, et al., Calcium imaging of microglial network activity in stroke. In: Methods in molecular biology;2019. [DOI] [PubMed]
- 58.Grinberg YY, Dibbern ME, Levasseur VA, Kraig RP. Insulin-like growth factor-1 abrogates microglial oxidative stress and TNF-α responses to spreading depression. J Neurochem. 2013;126(5):662–672. doi: 10.1111/jnc.12267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grinberg YY, van Drongelen W, Kraig RP. Insulin-like growth factor-1 lowers spreading depression susceptibility and reduces oxidative stress. J Neurochem. 2012;122(1):221–229. doi: 10.1111/j.1471-4159.2012.07763.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Viviani B, Bartesaghi S, Gardoni F, et al. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci. 2003;23(25):8692–8700. doi: 10.1523/JNEUROSCI.23-25-08692.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hide I, Tanaka M, Inoue A, et al. Extracellular ATP triggers tumor necrosis factor-α release from rat microglia. J Neurochem. 2000;75(3):965–972. doi: 10.1046/j.1471-4159.2000.0750965.x. [DOI] [PubMed] [Google Scholar]
- 62.Skaper SD. Ion channels on microglia: therapeutic targets for neuroprotection. CNS Neurol Disord: Drug Targets. 2011;10(1):44–56. doi: 10.2174/187152711794488638. [DOI] [PubMed] [Google Scholar]
- 63.Virginio C, Church D, North RA, Surprenant A. Effects of divalent cations, protons and calmidazolium at the rat P2X7 receptor. Neuropharmacology. 1997;36(9):1285–1294. doi: 10.1016/s0028-3908(97)00141-x. [DOI] [PubMed] [Google Scholar]
- 64.Hidetoshi T-S, Makoto T, Inoue K. P2Y receptors in microglia and neuroinflammation. Wiley interdisciplinary reviews. Membrane Transp Signal. 2012;1(4):493–501. [Google Scholar]
- 65.Pedata F, Dettori I, Coppi E, et al. Purinergic signalling in brain ischemia. Neuropharmacology. 2016;104:105–130. doi: 10.1016/j.neuropharm.2015.11.007. [DOI] [PubMed] [Google Scholar]
- 66.Cserép C, Pósfai B, Lénárt N, et al. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science. 2020;367(6477):528–537. doi: 10.1126/science.aax6752. [DOI] [PubMed] [Google Scholar]
- 67.Haynes SE, Hollopeter G, Yang G, et al. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci. 2006;9(12):1512–1519. doi: 10.1038/nn1805. [DOI] [PubMed] [Google Scholar]
- 68.Parekh AB. Store-operated CRAC channels: function in health and disease. Nat Rev Drug Discovery. 2010;9(5):399–410. doi: 10.1038/nrd3136. [DOI] [PubMed] [Google Scholar]
- 69.Mizuma A, Kim JY, Kacimi R, et al. Microglial calcium release-activated calcium channel inhibition improves outcome from experimental traumatic brain injury and microglia-induced neuronal death. J Neurotrauma. 2019;36(7):996–1007. doi: 10.1089/neu.2018.5856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cahalan MD, Zhang SL, Yeromin AV, et al. Molecular basis of the CRAC channel. Cell Calcium. 2007;42(2):133–144. doi: 10.1016/j.ceca.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stauderman KA. CRAC channels as targets for drug discovery and development. Cell Calcium. 2018;74:147–159. doi: 10.1016/j.ceca.2018.07.005. [DOI] [PubMed] [Google Scholar]