

Abstract
INTRODUCTION.
Physical activity (PA) is widely recommended for age-related brain health, yet its neurobiology is not well understood. Animal models indicate PA is synaptogenic. We examined the relationship between PA and synaptic integrity markers in older adults.
METHODS.
404 decedents from Rush Memory and Aging Project completed annual actigraphy monitoring (M visits=3.5±2.4) and postmortem evaluation. Brain tissue was analyzed for presynaptic proteins (synaptophysin, synaptotagmin-1, VAMP, syntaxin, complexin-I, and complexin-II), and neuropathology. Models examined relationships between late-life PA (averaged across visits), and timing-specific PA (time to autopsy) with synaptic proteins.
RESULTS.
Greater late-life PA associated with higher presynaptic protein levels (0.14<β<0.20), except complexin-II (β=0.08). Relationships were independent of pathology but timing-specific; participants who completed actigraphy within 2-years of brain tissue measurements showed largest PA-to-synaptic protein associations (0.32<β<0.38). Relationships between PA and presynaptic proteins were comparable across brain regions sampled.
DISCUSSION.
PA associates with synaptic integrity in a regionally-global, but time-linked nature in older adults.
Keywords: actigraphy, exercise, presynaptic protein, cognitive resilience
Introduction
Late life physical activity (PA) is one of the most consistently recommended lifestyle modifications to support brain and cognitive aging[1]. PA is associated with a reduced incidence of Alzheimer’s dementia[2], and inactivity alone is estimated to account for >4 million dementia cases[3]. Although not synonymous with free-living activity, human exercise trials also demonstrate a positive effect on cognition[4] and gray matter growth[5], potentially supporting directionality of PA effects. Yet, the field does not fundamentally understand the biological mechanisms linking PA to brain health in humans. Identification of these pathways will not only inform risk-stratification and monitoring tools for more precise exercise recommendations, but potentially identify biological targets of cognitive resilience for future dementia prevention therapies.
Pioneering but decades-old studies have established the causal impact of PA on synaptogenesis in animals[6], an effect consistently replicated[7–10]. Voluntary PA (e.g., access to running wheel) in early, middle, or aged rodents demonstrates robust increases in dendritic tree complexity, synaptophysin[11], Ca+ signaling[12], and trophic factors (e.g., BDNF)[13]. Furthermore, the beneficial effects of PA on synaptic-related functioning generalizes across disease models (e.g., wildtype aging, amyloidosis, APOEe4, tauopathy), and corresponds with improved behavioral outcomes[9,11,14,15].
Synaptic functioning is an appealing therapeutic target given its proximity to cognition. Regardless of pathology presence, cognition cannot occur without integrity of the synaptic unit. Furthermore, preservation of synaptic structure, protein levels, and gene expression uniquely differentiates adults who are clinically resilient to AD pathology burden compared to non-resilient AD peers[16–18]. Therefore, understanding the link between PA and markers of synaptic health in humans will both begin to translate a fundamental biology observed in animal models, and build support for a readily accessible approach to support synaptic health with age.
Exercise training is associated with gray matter growth in humans[5] and the synapse is a structural component of gray matter, supporting further study of these relationships. However, the molecular balance of synaptic functioning is highly dynamic, follows non-linear patterns, and is not fully captured by brain volume metrics[19,20]. In the one human study to our knowledge, Jensen and colleagues (2017) tested the effect of a 16-week exercise intervention on cerebrospinal fluid (CSF) synaptic protein levels (neurogranin and VILIP-1) in a small cohort of adults with mild-to-moderate Alzheimer’s disease[21]; significant changes were not reached, though there was a trend for increased protein levels with high intraindividual variability. Further work is needed to carefully dissect these relationships.
We aimed to bridge this translational gap and determine the relationship between objectively measured free-living PA and synaptic markers in brain tissue of older adults. We analyzed 404 decedents from the Rush Memory and Aging Project (MAP) who completed accelerometer-based actigraphy monitoring in late life and went to autopsy with brain tissue analyzed for presynaptic proteins (synaptophysin, synaptotagmin-1, VAMP, SNAP-25, syntaxin, complexin-I, complexin-II). Building on prior works from Rush MAP that have demonstrated significant associations between actigraphy monitoring and cognition[2,22], as well as between synaptic proteins and cognition in late life[23–26], we hypothesized that late life PA would relate to higher abundances of presynaptic proteins in human brain tissue.
Methods
Participants.
404 decedents from the Rush Memory and Aging Project (MAP) were included[27]. All participants included underwent comprehensive neuropathological evaluations with available synaptic protein markers and completed actigraphy monitoring. Exclusion criteria was inability to sign an informed consent and Anatomical Gift Act.
Actigraphy Monitoring.
An activity monitor was worn on the nondominant wrist and measured rest/activity continuously (24 hours a day) for up to 10 days (Actical; Mini Mitter, Bend, OR). Activity counts were extracted for each 15-second epoch yielding 5760 data points/day. Incomplete days of data were excluded from analyses. Incomplete data were determined based on inspection of the recordings via an automated program that flagged average daily counts at the extremes: ~0/day or >500/day. Only participants with valid data for 1+ days were included in analyses. Both weekday and weekend time were captured in the 10-day monitoring period. Daily physical activity, which summarized both exercise and non-exercise activities, was calculated as the average sum of all daily activity counts for each 15-second epoch for full days of data[2].
Actigraphy monitoring was completed annually. All participants completed one visit and 73% completed 2+ visits (M visits= 3.5, SD=2.4). We examined overall late life PA levels by averaging daily activity counts across all available visits per participant, as well as visit-specific daily activity counts.
Synaptic protein quantification.
Frozen gray matter samples were obtained from six brain areas (hippocampus, middle frontal cortex, inferior temporal cortex, calcarine cortex, ventromedial caudate, and posterior putamen), and used to prepare homogenates at a consistent protein concentration, followed by serial dilution for ELISA[28]. We evaluated both a regionally global index of synaptic protein levels (aggregated across all regions sampled, for each protein), consistent with prior MAP studies,[24,28] as well as clustered, region-specific synaptic protein levels. We examined the following regions: hippocampus, cortical (middle frontal cortex and inferior temporal cortex), striatal (ventromedial caudate and posterior putamen), and calcarine cortex. Region selection was based on conceptual links with PA (i.e., striatal regions associated with movement), prior literature implicating the region with PA (i.e., hippocampus[5]), and to determine specificity against regions less commonly affected by pathology (i.e., calcarine).
Monoclonal antibodies quantified synaptophysin, synaptotagmin-1, SNAP-25, syntaxin, VAMP, complexin-I, and complexin-II levels (Supplemental Table 1). Values were expressed in log10 units, standardized, and averaged across regions within each participant. Individual protein levels provide information regarding integrity of the presynaptic compartment, higher values indicate more protein available.
Neuropathological Evaluation. Neuropathological Evaluation.
Brain removal, tissue sectioning and preservation, and uniform gross and microscopic examination followed a standard protocol previously reported[27]. All staff were blinded to clinical diagnosis. Nine common age-related pathology indicators were quantified: global burden of Alzheimer’ disease (AD) pathology, hippocampal sclerosis, Lewy Body disease (LBD), TDP-43, cerebral amyloid angiopathy (CAA), severity of arteriosclerosis and atherosclerosis, and macro- and micro-infarct counts.
Other Covariates.
See Supplemental Methods for measure details. We adjusted all models for objective motor function performances (motor 10)[2] (i.e., lifestyle PA levels accounting for motor function limitations), and conducted sensitivity analyses additionally adjusting for reported late life cognitive[29] and social activities[30] and depressive symptoms[31].
Averaged covariates.
Covariates were averaged across participant visits when included in models testing the relationship between “late life physical activity” (i.e., averaged PA across visits) and synaptic protein level. Analyses examining visit-specific actigraphy effects included visit-specific covariate values (not averaged across visits).
Statistical Analyses.
Average Late Life PA.
First, we explored bivariate associations between late life PA (averaged across visits) with demographic and clinical factors. We next evaluated the overall relationship between late life PA with global synaptic protein levels at death using multivariable regression models. All models adjusted for age at death, sex, education, and average late life motor function (motor10 averaged across visits). Next, we probed the robustness of the observed relationship between late life PA and synaptic proteins by entering additional covariates, including average late life cognitive activity, social activity, and depression levels, post-mortem interval, as well as nine common neuropathologies (i.e., AD, TDP-43, hippocampal sclerosis, LBD, macro and micro-infarcts, CAA, arteriolosclerosis, atherosclerosis). After plotting the models, a small subset of participants demonstrated disproportionately high PA levels. To confirm our results were not driven by this small subset, we conducted sensitivity analyses excluding participants with actigraphy >90th%ile (average daily count>3.5; n=30).
Temporality.
Given putatively dynamic nature between PA and synaptic functioning in animal models showing both acute and long-term effects[32], we aimed to determine how time between measurement of in-vivo PA and post-mortem synaptic protein may affect the observed relationships. We identified participants’ last actigraphy visit and identified those who fell within ≤1 year (n=93), 1–2 years (n=119), 3–4 years (n=134), 5–6 years (n=101), or 9–10 years (n=33) to death (i.e., synaptic protein measurement; Table 3). We conducted parallel multivariable regression models examining visit-specific PA with synaptic protein levels stratified by time to death, adjusting for age at visit, education, sex, and motor function at visit. To determine whether the effects of visit were specific to the synaptic proteins versus an artifact common to other brain tissue metrics at death, we conducted the same multivariable models but instead estimated the relationship between visit-specific PA and nine common neuropathologies.
Table 3.
The relationship between in-vivo physical activity and brain tissue synaptic protein levels at death depends on time between measurements.
| Synaptophysin | Synaptotagmin1 | Syntaxin | VAMP | SNAP-25 | Complexin-I | Complexin-II | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Coeff (se) | Std Beta | Coeff (se) | Std Beta | Coeff (se) | Std Beta | Coeff (se) | Std Beta | Coeff (se) | Std Beta | Coeff (se) | Std Beta | Coeff (se) | Std Beta | |
| Average Physical Activity (n=404) | 0.11 (0.04) | 0.13* | 0.11 (0.05) | 0.13* | 0.10 (0.12) | 0.12* | 0.12 (0.05) | 0.15** | 0.10 (0.04) | 0.12* | 0.05 (0.03) | 0.07 | 0.02 (0.03) | 0.04 |
| Actigraphy ≤1yr to death (n=93) | 0.37 (0.11) | 0.36** | 0.35 (0.11) | 0.34** | 0.33 (0.11) | 0.32** | 0.36 (0.11) | 0.34** | 0.39 (0.11) | 0.38** | 0.28 (0.08) | 0.35** | 0.03 (0.08) | 0.04 |
| Actigraphy 1–2yr to death (n=119) | 0.24 (0.08) | 0.31** | 0.26 (0.08) | 0.32** | 0.19 (0.08) | 0.24* | 0.22 (0.08) | 0.26** | 0.21 (0.08) | 0.27** | 0.15 (0.06) | 0.26* | −0.06 (0.16) | −0.1 |
| Actigraphy 3–4yr to death (n=134) | 0.07 (0.06) | 0.11 | 0.06 (0.06) | 0.09 | 0.04 (0.06) | 0.06 | 0.07 (0.07) | 0.10 | 0.05 (0.06) | 0.07 | 0.02 (0.06) | 0.03 | 0.01 (0.05) | 0.03 |
| Actigraphy 5–6yr to death (n=101) | −0.003 (0.05) | −0.005 | −0.01 (0.05) | −0.02 | 0.01 (0.05) | 0.02 | −0.01 (0.05) | −0.02 | −0.46 (0.31) | −0.09 | −0.01 (0.05) | −0.16 | −0.09 (0.12) | −0.15 |
| Actigraphy 9–10yr to death (n=33) | −0.29 (0.17) | −0.37 | −0.24 (0.18) | −0.31 | −0.12 (0.18) | −0.16 | −0.22 (0.17) | −0.28 | −0.14 (0.16) | −0.2 | −0.18 (0.13) | −0.31 | −0.17 (0.11) | −0.31 |
Note.
p<0.05;
p<0.01;
each column represents a regression model. Average Late Life Actigraphy models adjusted for age at death, sex, education, and average late life motor10 performance. All other models adjusted for age at visit, sex, education and motor10 performance at visit.
To help determine if effects were related to pathology accumulation (e.g., reverse causality), we tested the relationship between PA and synaptic proteins adjusting for the interactions between PA with clinical severity at death (normal vs. MCI vs. dementia) or with individual neuropathologies on synaptic proteins.
Regionality.
Lastly, we evaluated the relationship between average late life PA and regionally-specific synaptic protein levels using parallel multivariable regression models, adjusting for age at death, sex, education, and motor function. Four a priori regions were selected (hippocampus, cortical, striatal, and calcarine).
A priori hypotheses used p-value < 0.05 to indicate statistical significance. Standardized and unstandardized beta coefficients with standard error are reported.
Results
Participants completed the actigraphy monitoring for an average of 3.5 visits (range=1–10), and averaged 90-years-old at death (Table 1). At autopsy, the cohort was roughly evenly split across clinical neurocognitive diagnoses (i.e., normal, MCI, or dementia), and 65% meet Reagan criteria for neuropathological AD.
Table 1.
Participant clinical and pathological characteristics (N=404).
| 1 | 404 (100%) |
| 2 | 298 (73.8%) |
| 3 | 224 (55.4%) |
| 4 | 164 (40.6%) |
| 5 | 111 27.5%) |
| 6 | 67 (16.5%) |
| 7 | 49 (12.1%) |
| 8 | 27 (6.7%) |
| 9 | 9 (2.2%) |
| 10 | 4 (1.0%) |
| mean = 3.5 (SD=2.4) | |
| AGE AT DEATH (YEARS; M, SD) | 90.6 (5.8) |
| AGE AT LAST ACTIGRAPHY VISIT (YEARS; M, SD) | 88.5 (6.1) |
| SEX (% FEMALE, N) | 72.1% (280) |
| EDUCATION (YEARS; M, SD) | 14.6 (2.8) |
| COGNITIVE DIAGNOSIS AT DEATH (N, %) | |
| CLINICALLY NORMAL | 36.3% (141) |
| MCI-AD ☨ | 24.2% (94) |
| MCI-OTHER | 1.8% (7) |
| DEMENTIA-AD ☨ | 33.2% (129) |
| DEMENTIA-OTHER | 4.4% (17) |
| AVERAGE LATE LIFE LIFESTYLE FACTORS | |
| ACTIGRAPHY: AVERAGE DAILY (COUNTS; M, SD) | 1.97 (1.1) |
| MOTOR FUNCTION COMPOSITE (Z-SCORE; M, SD) | 0.87 (0.17) |
| DEPRESSIVE SYMPTOMS (CES-D; M, SD) | 1.4 (1.3) |
| COGNITIVE ACTIVITY (POSSIBLE RANGE 1 TO 5; M, SD) | 2.98 (0.63) |
| SOCIAL ACTIVITY (POSSIBLE RANGE 1 TO 5; M, SD) | 2.34 (0.46) |
| NEUROPATHOLOGICAL EXAMINATION | |
| POSTMORTEM INTERVAL (HOURS; M, SD) | 8.2 (5.1) |
| ALZHEIMER’S DISEASE PATHOLOGY (%, REAGAN CRITERIA, N) | 64.6% (250) |
| BRAIN TISSUE SYNAPTIC PROTEINS (GLOBAL Z-SCORE)☨☨ | |
| COMPLEXIN-I (M, SD) | −0.20 (0.74) |
| COMPLEXIN-II (M, SD) | −0.17 (0.67) |
| SYNAPTOPHYSIN (M, SD) | −0.19 (0.92) |
| SNAP-25 (M, SD) | −0.18 (0.92) |
| SYNTAXIN (M, SD) | −0.18 (0.93) |
| VAMP (M, SD) | −0.18 (0.95) |
| SYNAPTOTAGMIN-1 (M, SD) | 0.02 (0.95) |
NINCDS consensus criteria for possible or probable AD
Brain synaptic protein values are expressed in log10 units, standardized to the MAP sample, and averaged across six regions within each participant (z-score units reported).
Note. All lifestyle factors represent an average of participants values across visits to capture illustrate robust indicators of late life behaviors. MCI = Mild Cognitive Impairment; AD = Alzheimer’s disease; CES-D = Center for Epidemiologic Studies – Depression scale. M = mean; SD = standard deviation.
Does average late life physical activity relate to synaptic protein levels at death?
On average, participants wore the Actical device for 9.3 days (SD= 1.3) over the annual 10-day monitoring periods. Length of monitoring did not differ by proximity to death (M= 9.4 days for those ≤1 year to death and M= 9.2 days for those 7+ years to death). Average late life PA demonstrated small-to-minimal associations with age at death (r=−0.13, p=0.008), education (r=−0.06, p=0.17), and sex (t(402)=0.43, p=0.65). Greater PA related to better late life motor function performances (r=0.39, p<0.001).
Adjusting for demographics and average motor function, greater late life PA was associated with higher synaptophysin (B=0.13, p=0.017), VAMP (B=0.11, p<0.001), SNAP-25 (B=0.12, p=0.026), synaptotagmin-1 (B=0.13, p=0.014), and syntaxin (B=0.12, p=0.029) levels at death (Figure 1). Average PA did not significantly relate to complexins (complexin-I B=0.07, p=0.18; complexin-II B=0.04, p=0.45). No interactions between sex and PA were found (p-values>0.05).
Figure 1.
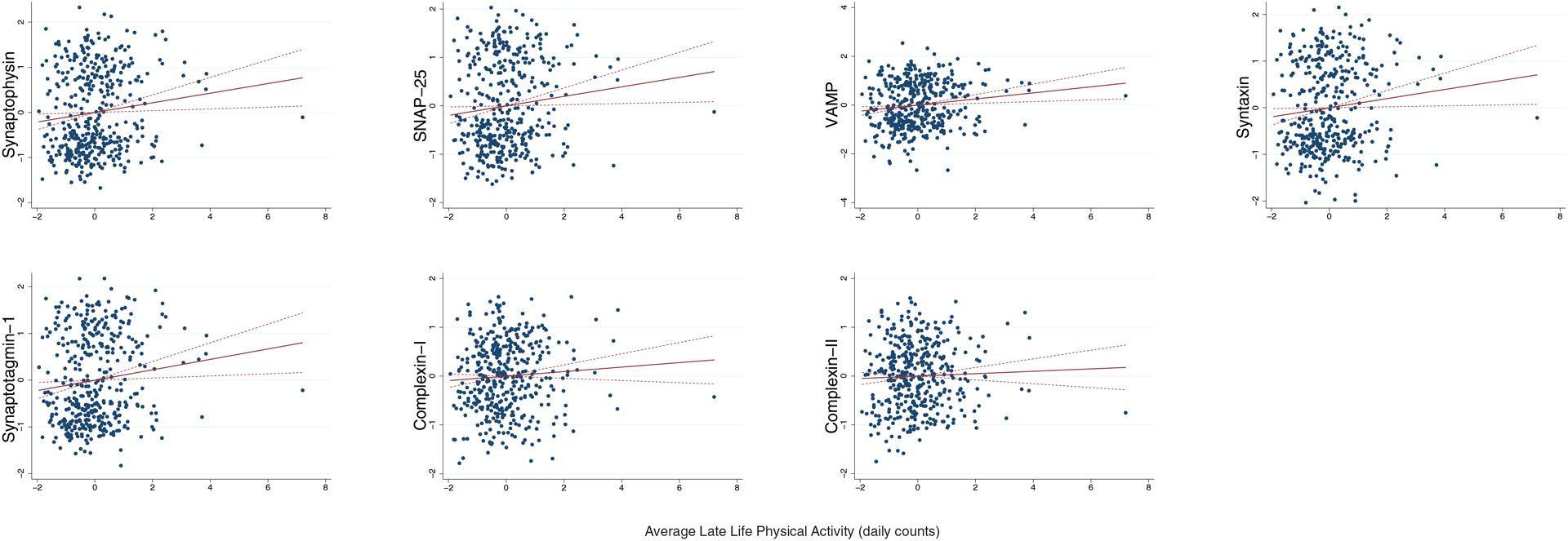
Greater average late life physical activity relates to higher synaptic protein in brain tissue at death, with the exception of complexin-II.
Note. Dashed lines represent 95%CI. Average late life physical activity represents actigraphy levels averaged across available study visits. All models adjusted for age at death, sex, education, and average late life motor10 performances. Synaptic protein levels are expressed in log10 units, standardized, and averaged across six brain regions within each participant (z-score units).
To test specificity of PA compared to other modifiable factors, we adjusted models for average late life social activity, cognitive activity, and depression symptomology. Only daily PA continued to significantly relate to the same synaptic proteins to a similar degree (Table 2). Given published associations between synaptic integrity and pathology burden[28], we further adjusted for nine common neuropathologies and post-mortem interval. The PA model coefficients became slightly stronger and now greater late life PA additionally significantly related to higher complexin-I, but not complexin-II levels (Table 2).
Table 2.
Average late life physical activity is independently associated with brain tissue synaptic protein levels at death (n=404).
| Estimate | Synaptophysin | Synaptotagmin-1 | Syntaxin | VAMP | SNAP-25 | Complexin-I | Complexin-II | |
|---|---|---|---|---|---|---|---|---|
| Avg Physical Activity | Coeff (se) | 0.15 (0.04) | 0.16 (0.05) | 0.14 (0.04) | 0.17 (0.05) | 0.14 (0.04) | 0.09 (0.03) | 0.05 (0.03) |
| Std Beta | 0.18** | 0.19** | 0.17** | 0.20** | 0.17** | 0.14* | 0.08 | |
| Age at death | Coeff (se) | −0.01 (0.01) | −0.01 (0.01) | −0.004 (0.01) | 0.002 (0.01) | −0.003 (0.01) | −0.02 (0.01) | −0.01 (0.01) |
| Std Beta | −0.05 | −0.04 | −0.03 | 0.01 | −0.02 | −0.13* | −0.09 | |
| Sex | Coeff (se) | 0.09 (0.11) | 0.10 (0.11) | 0.90 (0.11) | 0.04 (0.11) | 0.13 (0.11) | −0.03 (0.09) | −0.17 (0.08) |
| Std Beta | 0.003 | 0.05 | 0.05 | 0.02 | 0.06 | −0.02 | −0.11 | |
| Education | Coeff (se) | 0.001 (0.02) | −0.01 (0.02) | −0.01 (0.02) | −0.001 (0.02) | −0.01 (0.02) | 0.0001 (0.01) | 0.002 (0.01) |
| Std Beta | 0.003 | −0.02 | −0.02 | −0.002 | −0.03 | 0.003 | 0.001 | |
| Avg Motor10 | Coeff (se) | −0.44 (0.31) | −0.34 (0.31) | −0.38 (0.31) | −0.25 (0.32) | −0.46 (0.31) | −0.02 (0.24) | −0.11 (0.24) |
| Std Beta | −0.08 | −0.06 | −0.07 | −0.05 | −0.09 | −0.005 | −0.03 | |
| Avg Cognitive Activity | Coeff (se) | 0.02 (0.08) | 0.04 (0.09) | 0.01 (0.09) | 0.03 (0.09) | 0.03 (0.08) | 0.02 (0.07) | 0.08 (0.06) |
| Std Beta | 0.01 | 0.03 | 0.01 | 0.02 | 0.02 | 0.02 | 0.08 | |
| Avg Social Activity | Coeff (se) | −0.07 (0.12) | −0.11 (0.12) | −0.11 (0.12) | −0.11 (0.12) | −0.09 (0.12) | −0.25 (0.09) | −0.20 (0.09) |
| Std Beta | −0.04 | −0.05 | −0.05 | −0.05 | −0.04 | −0.16** | −0.14* | |
| Avg CES-D | Coeff (se) | −0.04 (0.04) | −0.04 (0.04) | −0.06 (0.04) | −0.06 (0.04) | −0.04 (0.04) | 0.03 (0.03) | −0.01 (0.03) |
| Std Beta | −0.06 | −0.06 | −0.08 | −0.08 | −0.07 | −0.05 | −0.02 | |
| Global AD | Coeff (se) | −0.19 (0.08) | −0.17 (0.09) | −0.20 (0.08) | −0.17 (0.08) | −0.16 (0.08) | −0.24 (0.07) | −0.14 (0.06) |
| Std Beta | −0.12* | −0.11* | −0.13* | −0.11* | −0.10 | −0.19** | −0.12* | |
| Hippocampal sclerosis | Coeff (se) | 0.25 (0.17) | 0.31 (0.18) | 0.22 (0.18) | 0.23 (0.18) | 0.20 (0.17) | −0.02 (0.14) | −0.31 (0.13) |
| Std Beta | 0.08 | 0.09 | 0.07 | 0.07 | 0.06 | −0.007 | −0.13* | |
| DLB | Coeff (se) | −0.02 (0.04) | −0.01 (0.04) | −0.02 (0.04) | −0.04 (0.04) | −0.02 (0.04) | −0.02 (0.03) | −0.01 (0.03) |
| Std Beta | −0.03 | −0.01 | −0.03 | −0.04 | −0.02 | −0.03 | −0.02 | |
| TDP-43 | Coeff (se) | 0.02 (0.04) | 0.004 (0.04) | 0.04 (0.04) | 0.05 (0.04) | 0.03 (0.04) | −0.02 (0.03) | 0.06 (0.03) |
| Std Beta | 0.02 | 0.01 | 0.05 | 0.06 | 0.04 | −0.03 | 0.11* | |
| Arteriolosclerosis | Coeff (se) | 0.18 (0.06) | 0.17 (0.06) | 0.14 (0.05) | 0.17 (0.06) | 0.16 (0.06) | 0.04 (0.04) | 0.06 (0.04) |
| Std Beta | 0.17** | 0.16** | 0.13* | 0.15* | 0.15** | 0.05 | 0.08 | |
| CAA | Coeff (se) | 0.04 (0.05) | 0.01 (0.05) | 0.03 (0.05) | 0.03 (0.05) | 0.04 (0.05) | 0.03 (0.04) | 0.03 (0.04) |
| Std Beta | 0.04 | 0.01 | 0.03 | 0.03 | 0.04 | 0.04 | 0.04 | |
| CVDA | Coeff (se) | 0.24 (0.06) | 0.26 (0.06) | 0.23 (0.06) | 0.20 (0.06) | 0.20 (0.06) | 0.24 (0.05) | 0.05 (0.04) |
| Std Beta | 0.22** | 0.23** | 0.20** | 0.17** | 0.18** | 0.27** | 0.06 | |
| Macroinfarcts | Coeff (se) | −0.01 (0.10) | 0.02 (0.10) | −0.001 (0.10) | 0.05 (0.10) | 0.06 (0.10) | −0.11 (0.08) | −0.06 (0.07) |
| Std Beta | 0.003 | 0.12 | −0.0004 | 0.02 | 0.03 | −0.07 | −0.05 | |
| Microinfarcts | Coeff (se) | −0.14 (0.10) | −0.14 (0.10) | −0.23 (0.10) | −0.15 (0.10) | −0.17 (0.10) | −0.14 (0.08) | −0.03 (0.08) |
| Std Beta | −0.07 | −0.07 | −0.12* | −0.08 | −0.09 | −0.09 | −0.02 |
Note.
p<0.05;
p<0.01;
columns represent individual regression models. Avg = averaged across study visits. AD = Alzheimer’s disease; CES-D = Center for Epidemiologic Studies – Depression scale; DLB = dementia with Lewy Body pathology staging; TDP-43 = TAR DNA-binding protein-43; CAA = cerebral amyloid angiopathy; CVDA = cerebral atherosclerosis.
To test veracity of the models excluding possible outliers, we re-ran models excluding participants with >90th%ile of daily movement. Model estimates remained similar (B range=0.13 to 0.17; p<0.03).
Temporality: Does time between physical activity and synaptic measurement affect the relationship?
Given animal models suggesting the relationship between PA and synaptic outcomes may show both acute and longer-term effects, we stratified the cohort by years to death to better understand the temporal dynamics of this relationship in humans. Adjusting for demographics and motor function at visit, the relationship between PA and synaptic protein levels demonstrated a dose-dependent increase in size as participants neared the time of synaptic protein measurement (i.e., death). There was a positive, medium-sized relationship between PA and all synaptic protein markers, with the exception of complexin-II, in participants 0–2 years from death that attenuated the further out the actigraphy measurement was taken. Together, these data suggest that proximity between actigraphy and synaptic protein level measurement may be an important factor (Table 3; Figure 2).
Figure 2.
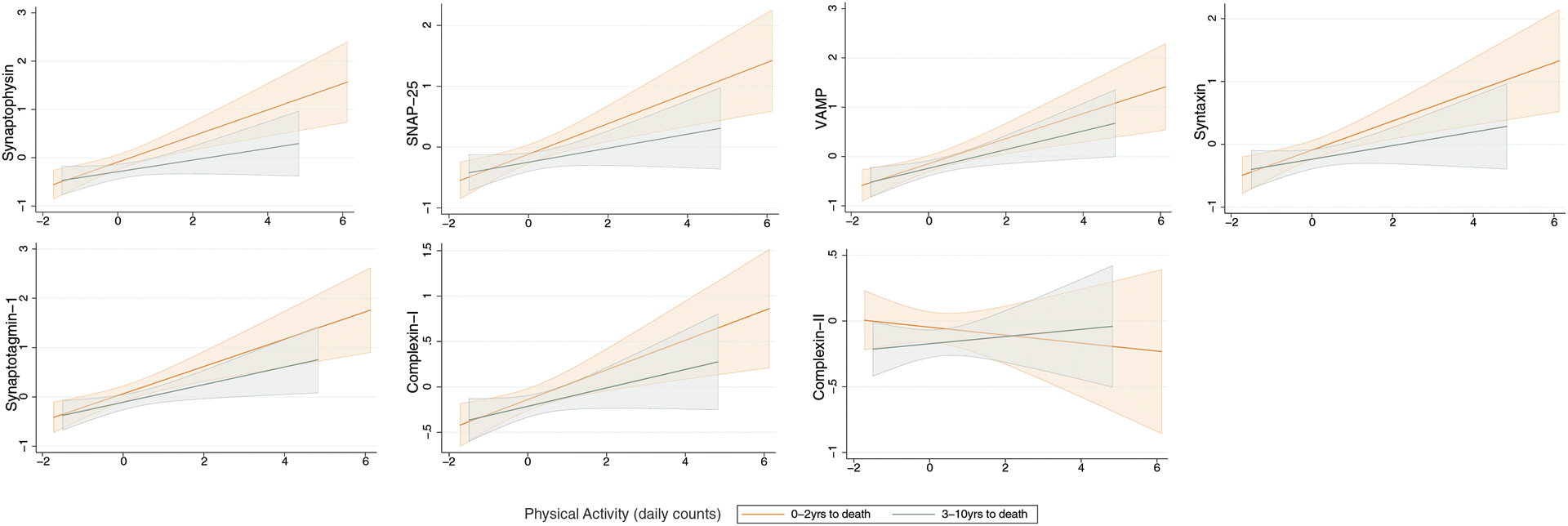
The relationship between physical activity and brain tissue synaptic protein levels is stronger the more proximate the measures were taken in time (i.e., time between actigraphy monitoring and death).
Note. N=150 participants 0–2 years to death and n=154 participants 3–10 years to death. Fitted regression lines adjusting for age at visit, sex, education, and motor10 performance at visit are plotted with 95% CI.
To test if this was an artifact common to other brain tissue metrics at death, we evaluated the relationship between last PA measurement and neuropathologies, similarly stratified by years to death. We did not observe a consistent change in the size of relationship between PA and neuropathology as a function of time to death (Supplementary Table 2), suggesting our visit-related findings were more specific to synaptic protein levels.
We further probed if the stronger relationship between PA and synaptic protein in participants 0–2 years from death reflected effects of pathology. In participants 0–2 years from death (n=150), the size of the relationship between PA and synaptic protein levels did not substantially change when adjusting for nine common neuropathologies (Synaptophysin B=0.29; Synaptotagmin-1 B=0.29; Syntaxin B=0.24; VAMP B=0.25; SNAP-25 B=0.28; Complexin-I B=0.25; Complexin-II B=0.08), and there were no significant interactions between PA and individual pathologies (e.g., not driven by individuals with AD pathology; all p-values >0.05). There was, however, a significant interaction between PA and clinical diagnosis at death on levels of synatotagmin-1, syntaxin, SNAP-25, and complexin-I (interaction term p-values <0.04). Stratified analyses showed positive associations between PA and these synaptic proteins in all three diagnosis, though the effect size was strongest in participants diagnosed with MCI (Supplemental Figure 1).
Regional specificity: Does the relationship between physical activity and synaptic proteins differ across the brain?
Adjusting for demographics and average motor function, greater average late life PA related to higher synaptic protein levels relatively equally across brain regions (Table 4). Given the important time of measurement effects observed, we also evaluated these relationships stratified by participants 0–2 years to death (n=150) compared to those 3+ years from death (n=154). Again, we found greater PA related to higher synaptic protein levels most strongly in those 0–2 years to death, an effect that appeared similar across brain regions for all synaptic proteins (Table 4). In other words, no brain region appeared to be driving or susceptible to the observed timing effects.
Table 4.
Models demonstrating the size of the relationship between physical activity and brain tissue synaptic protein levels appears similar across brain regions.
| Synaptophysin | Synaptotagmin1 | Syntaxin | VAMP | SNAP-25 | Complexin-I | Complexin-II | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Coeff (se) | Std Beta | Coeff (se) | Std Beta | Coeff (se) | Std Beta | Coeff (se) | Std Beta | Coeff (se) | Std Beta | Coeff (se) | Std Beta | Coeff (se) | Std Beta | |
| Average Late Life Actigraphy (all participants, n=404) | ||||||||||||||
| Striatal | 0.14 90.05) | 0.16** | 0.15 (0.05) | 0.17** | 0.10 (0.05) | 0.12* | 0.16 (0.05) | 0.18** | 0.12 (0.05) | 0.14* | 0.05 (0.04) | 0.06 | 0.02 (0.04) | 0.03 |
| Hippo | 0.10 (0.05) | 0.12* | 0.11 (0.05) | 0.13* | 0.10 (0.05) | 0.11* | 0.11 (0.05) | 0.12* | 0.09 (0.05) | 0.11 | 0.08 (0.05) | 0.09 | 0.01 (0.05) | 0.01 |
| Cortical | 0.10 (0.05) | 0.12* | 0.10 (0.05) | 0.11* | 0.09 (0.05) | 0.10 | 0.11 (0.05) | 0.13* | 0.10 (0.05) | 0.12* | 0.06 (0.04) | 0.07 | 0.02 (0.04) | 0.03 |
| Occipital | 0.12 (0.05) | 0.14* | 0.13 (0.05) | 0.15** | 0.11 (0.05) | 0.13* | 0.13 (0.05) | 0.15** | 0.12 (0.05) | 0.14* | 0.05 (0.05) | 0.05 | 0.07 (0.05) | 0.08 |
| Actigraphy 0–2yr to death (n=150) | ||||||||||||||
| Striatal | 0.27 (0.07) | 0.33** | 0.27 (0.07) | 0.32** | 0.22 (0.07) | 0.28** | 0.26 (0.08) | 0.30** | 0.22 (0.07) | 0.27** | 0.12 (0.07) | 0.16 | −0.07 (0.06) | −0.10 |
| Hippo | 0.29 (0.07) | 0.34** | 0.32 (0.08) | 0.37** | 0.26 (0.07) | 0.31** | 0.25 (0.08) | 0.31** | 0.29 (0.08) | 0.34** | 0.30 (0.08) | 0.33** | −0.01 (0.07) | −0.01 |
| Cortical | 0.26 (0.07) | 0.31** | 0.26 (0.08) | 0.31** | 0.22 (0.07) | 0.27** | 0.23 (0.08) | 0.27** | 0.25 (0.07) | 0.30** | 0.18 (0.07) | 0.23** | −0.03 (0.07) | −0.04 |
| Occipital | 0.28 (0.07) | 0.33** | 0.29 (0.08) | 0.33** | 0.24 (0.08) | 0.30** | 0.27 (0.07) | 0.32** | 0.28 (0.08) | 0.32** | 0.17 (0.08) | 0.20* | 0.01 (0.08) | 0.01 |
| Actigraphy 3+yr to death (n=154) | ||||||||||||||
| Striatal | 0.17 (0.08) | 0.21* | 0.20 (0.08) | 0.25** | 0.13 (0.07) | 0.17 | 0.23 (0.07) | 0.30** | 0.15 (0.08) | 0.19 | 0.13 (0.07) | 0.16 | 0.02 (0.07) | 0.03 |
| Hippo | 0.13 (0.08) | 0.15 | 0.21 (0.08) | 0.26** | 0.14 (0.08) | 0.17 | 0.17 (0.07) | 0.22* | 0.17 (0.08) | 0.20* | 0.08 (0.09) | 0.09 | 0.001 (0.08) | 0.001 |
| Cortical | 0.14 (0.08) | 0.17 | 0.17 (0.08) | 0.21* | 0.11 (0.08) | 0.13 | 0.19 (0.08) | 0.23* | 0.15 (0.08) | 0.18 | 0.11 (0.07) | 0.15 | 0.02 (0.07) | 0.03 |
| Occipital | 0.09 (0.08) | 0.10 | 0.17 (0.08 | 0.20* | 0.10 (0.08) | 0.12 | 0.16 (0.08) | 0.18 | 0.09 (0.08) | 0.11 | 0.11 (0.08) | 0.14 | 0.08 (0.08) | 0.10 |
Note.
p<0.05;
p<0.01;
Average Late Life Actigraphy models adjusted for age at death, sex, education, and average late life motor10 performance. All other models adjusted for age at visit, sex, education and motor10 performance at visit. “Cortical” represents middle frontal and inferior temporal gyri. Hippo = hippocampal.
Discussion
Physical activity (PA) is consistently linked to brain health, yet the biological pathways supporting this relationship in humans are not well understood. We demonstrate that greater free-living PA levels, measured by objective actigraphy monitoring in life, associate with presynaptic protein levels of synaptophysin, synaptotagmin-1, syntaxin, VAMP, SNAP-25, and complexin-I in brain tissue at death. Although this relationship was initially small, it was robust and statistically independent from overall motor function (i.e., ability to engage in PA), other modifiable factors (i.e., cognitive and social activities, depression), and neuropathology burden. Interestingly, the association between PA and presynaptic proteins became strikingly larger the closer the measurements were taken in time. The strongest effect sizes were observed in participants 0–2 years to death, a relationship disproportionately evident among individuals diagnosed with MCI before death. Additionally, this appeared to be a regionally global phenomenon with PA relating to synaptic protein levels relatively equally across brain regions sampled. Despite adjusting for confounding factors, due to the observational study design, we cannot fully determine directionality of effects; it is likely the relationship between PA and synaptic integrity markers are bidirectional with at least some contribution from reverse causality (i.e., declines in synaptic functioning leading to lower PA engagement). Nonetheless, these are the first data to demonstrate a relationship between PA and markers of synaptic integrity in human brain tissue and suggest: 1) PA may support and/or build synaptic health in a temporally dynamic nature, 2) PA interventions may have optimal therapeutic windows for synaptic outcomes and be particularly effective in individuals in transitional cognitive states (MCI), and 3) PA may be beneficial across brain networks supporting cross-diagnostic utility.
Synapses are the biologic correlate of cognition and therefore an appealing therapeutic target. Our data show that greater PA relates to higher levels of presynaptic proteins, suggesting that PA may maintain or build brain resilience (i.e., more synaptogenesis, and/or increased synaptic protein reservoir) through processes involved in vesicle formation and trafficking, and regulation of neurotransmitter release. We observed some specificity, such that complexin-II did not demonstrate meaningful relationships with PA. Complexin-II is primarily present in glutamatergic terminals and prior analyses from our group suggest it is more affected and most strongly associated with cognition in later stages of disease[26]. This is in contrast to the association we did observe between PA and complexin-I, which more highly represented in inhibitory terminals, and more strongly relates to cognition, particularly in earlier disease states[26]. Interestingly, animal models have shown increases in excitatory (glutamatergic) and decreases in inhibitory (GABA) transcriptional pathways acutely following exercise[7]. Our findings indicating specific relevance of inhibitory terminals may reflect differences in the timing (acute vs. years), age, and/or species examined. Our data are also closely aligned with extant animal experiments demonstrating the causal benefit of exercise on synaptogenesis, as well as induction of protein or mRNA levels of the specific presynaptic proteins we examined (synaptophysin, SNAP-25, synaptotagmins, and syntaxin)[33–38]. For example, using a data-driven approach (microarray reflecting >1000 cDNAs), Molteni and colleagues (2002) demonstrated the largest changes following PA were observed for upregulation in presynaptic trafficking pathways, including synaptotagmin and syntaxin. Interestingly, blockade of TrkB (BDNF receptor) inhibits exercise-induced increases in cognition, as well as synaptic protein levels (e.g., synaptotagmin, synaptophysin, synapsin[36–38]), highlighting a critical role for BDNF coordinating PA-induced synaptic changes. Given prior exercise animal models have also implicated molecules in the postsynaptic compartment (e.g., PSD95, neurogranin)[35,39], a more comprehensive understanding of the other trans- and post-synaptic pathways that may mediate PA effects on the brain in humans is needed. Careful dissection of these molecular relationships will both improve our fundamental understanding of behavior-to-brain relationships and aid in identifying possible resilience-related pathways promotable via PA or other therapies.
Other outstanding questions regarding the effects of PA on the brain center around potential durability and regionality of this relationship. We found there was a dose-dependent increase in the size of the relationship between PA and synaptic proteins the more proximal the measurements were taken in time. Medium effect sizes (standardized betas ~0.30) were observed for participants within 0–2 years to death that was not driven by neuropathology burden but was most evident for individuals diagnosed with MCI. Therefore, the relationship between PA and synaptic regulation appeared relatively tightly coupled in time, potentially indicating that initiation and/or continuity of higher activity levels may be needed to support ongoing synaptic health. Additionally, individuals who are just beginning to display cognitive symptoms (MCI) may represent a particularly plastic therapeutic window of opportunity ideal for PA interventions targeting synaptic functioning. Consistent with this latter finding, prior works have demonstrated an initial “compensatory” increase in synaptic protein levels among individuals and animals in the earliest stages of neurodegeneration[40,41]. Again, it is important to note that given our observational design, it is not possible to determine if PA is driving synaptic health or vice versa. Nonetheless, these data help set the stage for the human intervention work that is needed to untangle the true temporal dynamics and person-specific predictors (e.g., disease stage) of those who stand to benefit most.
Although many prior animal (e.g.,[9,34,35]) and human exercise studies (e.g., [5]) have focused on the hippocampus, we show broadly comparable relationships between PA and synaptic protein levels across brain regions sampled. Perhaps this is not surprising given the systemic nature of PA. For example, at least some of the neurotrophic pathways of PA are likely through maintenance of cerebrovascular and glymphatic systems, as well as immune homeostasis, all of which are globally present throughout the brain[42]. The global nature of the PA-synaptic relationship observed is also consistent with data-driven voxel-based morphometry analyses and analyses examining whole-brain metrics that have demonstrated globally distributed relationships between PA and brain structural integrity[43,44]. Additionally, the lack of regional specificity suggests that PA may be a promising intervention across neurodegenerative syndromes (i.e., regardless of brain network affected), which is supported by an increasing array of PA studies across diagnostically diverse populations (e.g., aging, AD, FTLD, HIV, Parkinson’s disease).[5,45,46]
Our study is not without important limitations. As noted, PA and synapse relationships are likely highly dynamic and bidirectional in nature and our observational study design precludes conclusions regarding causality. This may be particularly relevant in our analyses showing temporal dependence of the PA-synapse relationship in which phenomena surrounding death may be a confounding factor (e.g., both PA and synaptic integrity may decrease closer to death). We demonstrate that PA relates to synaptic outcomes independent of chronological age at death and time to death (data not shown), and that PA did not show time-linked associations with other brain tissue outcomes and synaptic markers did not show time-linked associations with other in-vivo outcomes (data not shown). Together, this suggests specificity of temporal patterns to the PA-synaptic relationship. However, it is not possible to fully rule out effects of death on our results given our observational study design. Additionally, we focused on markers of synaptic functioning; there are likely many, diverse up- and down-stream mechanisms at play linking PA to the synapse and brain health, more broadly. Integration of molecular markers reflecting glial, immune, and endothelial functioning (among others) may be highly fruitful to understand the neurobiologic cascade of PA. Relatedly, our synaptic protein measurements are quantified in brain tissue supporting their specificity in CNS function, yet they are limited in truly capturing the time-linked dynamics of PA. Given the increasing availability and validity supporting measurement of synaptic protein levels via CSF or PET in living humans[19,47], in-vivo replication of these relationships will be an important future direction to help refine our understanding of the relevance and temporal dynamics between PA and synaptic markers. Additionally, our metric of physical activity was an aggregated composite of daily movement. We cannot determine which specific activities (e.g., structured vs. unstructured exercise, intensity) or patterns of activity may be most relevant in the observed relationships. Future studies further dissecting the characteristics of these relationships to determine the pattern(s) of physical activity most related to brain health are needed to inform more precise clinical recommendations. Lastly, it is important to note potential sample bias inherent in intensive clinicopathologic studies such as Rush MAP. Although this cohort is invaluable for discovery of potential targets and pathways of interest as measured directly in brain tissue, it may not inherently represent the broader older adult population and future works are needed to determine the generalizability of our findings.
In sum, our data are the first to demonstrate a link between a lifestyle behavior, PA, and markers of synaptic integrity in human brain tissue. We suggest PA may help build synaptic health, even at late ages (e.g., “brain reserve”)[48], independent from pathology, but that this is a potentially plastic process that may need to be sustained over time. Given its proximity to cognitive functioning and posited role in sustaining cognitive resilience to neuropathology[16–18], modifiable approaches to support synaptic health are of high relevance in our aging population. These data suggest that synaptic metrics may be particularly compelling and relevant outcomes for future behavioral intervention studies.
Supplementary Material
Research in Context.
Systemic review: We reviewed current literature using traditional search engines (e.g., PubMed, GoogleScholar). Physical activity (PA) is consistently linked with age-related brain structural and functional health; yet, its biological pathways are not well understood in humans. Decades of animal experiments demonstrate PA is synaptogenic. The fundamental relationship between PA and synaptic integrity has not been elucidated in humans.
Interpretation: Our data are the first to show PA relates to markers of synaptic integrity in human brain tissue. The relationship appeared regionally global and independent of pathology, but timing-dependent; the closer measurements were taken in time, the larger the relationship. PA may help build synaptic health, and this relationship may be a temporally dynamic process.
Future directions: Future works are needed to parse out in-vivo temporal dynamics of PA with synaptic markers, including intervention studies to support directionality.
Highlights.
Do free-living physical activity levels relate to synaptic integrity in humans?
Greater late life activity related to higher presynaptic protein levels in brain tissue.
Relationships were strongest the more proximate the measurements were taken in time.
Relationships were independent of pathology and comparable across brain regions.
Physical activity may be a modifiable tool to support synaptic health.
Acknowledgements/Funding/Other Disclosures.
This study was supported by NIH-NIA grants R01AG17917 (PI: DAB), and K23AG058752 and R01AG072475 (PI: KBC). Our work was also supported by the Alzheimer’s Association (AARG-20-683875, PI: KBC). WGH receives support from the Canadian Institutes of Health Research. MM receives support from the San Francisco VA. DAB receives consulting fees from AbbVie DSMB, Takeda adjudication committee, Origent SBIR, and Rush philanthropy. WGH receives consulting fees AbbVie and Translational Life Sciences, University of Hong Kong, and Polytechnical University of Hong Kong. WGH receives fees from the University of British Columbia for licensing of monoclonal antibodies for research purposes.
MAP data can be requested at https://www.radc.rush.edu.
Footnotes
Conflicts of interest: The authors have no relevant conflicts of interest to disclose.
References
- [1].Livingston G, Huntley J, Sommerlad A, Ames D, Ballard C, Banerjee S, et al. The Lancet Commissions Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 2020;396. doi: 10.1016/S0140-6736(20)30367-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Buchman AS, Boyle PA, Yu L, Shah RC, Wilson RS, Bennett DA. Total daily physical activity and the risk of AD and cognitive decline in older adults. Neurology 2012. doi: 10.1212/WNL.0b013e3182535d35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Norton S, Matthews FE, Barnes DE, Yaffe K, Brayne C. Potential for primary prevention of Alzheimer’s disease: An analysis of population-based data. Lancet Neurol 2014. doi: 10.1016/S1474-4422(14)70136-X. [DOI] [PubMed] [Google Scholar]
- [4].Northey JM, Cherbuin N, Pumpa KL, Smee DJ, Rattray B. Exercise interventions for cognitive function in adults older than 50: a systematic review with meta-analysis. Br J Sports Med 2017:bjsports-2016–096587. doi: 10.1136/bjsports-2016-096587. [DOI] [PubMed] [Google Scholar]
- [5].Erickson KI, Voss MW, Prakash RS, Basak C, Szabo A, Chaddock L, et al. Exercise training increases size of hippocampus and improves memory. Proc Natl Acad Sci U S A 2011;108:3017–22. doi: 10.1073/pnas.1015950108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Diamond MC, Krech D, Rosenzweig MR. The effects of an enriched environment on the histology of the rat cerebral cortex. J Comp Neurol 1964. doi: 10.1002/cne.901230110. [DOI] [PubMed] [Google Scholar]
- [7].Molteni R, Ying Z, Gómez-Pinilla F. Differential effects of acute and chronic exercise on plasticity-related genes in the rat hippocampus revealed by microarray. Eur J Neurosci 2002;16:1107–16. doi: 10.1046/j.1460-9568.2002.02158.x. [DOI] [PubMed] [Google Scholar]
- [8].Chen K, Zheng Y, Wei J an, Ouyang H, Huang X, Zhang F, et al. Exercise training improves motor skill learning via selective activation of mTOR. Sci Adv 2019. doi: 10.1126/sciadv.aaw1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Horowitz AM, Fan X, Bieri G, Smith LK, Sanchez-Diaz CI, Schroer AB, et al. Blood factors transfer beneficial effects of exercise on neurogenesis and cognition to the aged brain. vol. 369. American Association for the Advancement of Science; 2020. doi: 10.1126/SCIENCE.AAW2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].van Praag H, Kempermann G, Gage FH. Neural consequences of enviromental enrichment. Nat Rev Neurosci 2000;1:191–8. doi: 10.1038/35044558. [DOI] [PubMed] [Google Scholar]
- [11].Nichol K, Deeny SP, Seif J, Camaclang K, Cotman CW. Exercise improves cognition and hippocampal plasticity in APOE ϵ4 mice. Alzheimer’s Dement 2009. doi: 10.1016/j.jalz.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ambrogini P, Cuppini R, Lattanzi D, Ciuffoli S, Frontini A, Fanelli M. Synaptogenesis in adult-generated hippocampal granule cells is affected by behavioral experiences. Hippocampus 2010;20:799–810. doi: 10.1002/hipo.20679. [DOI] [PubMed] [Google Scholar]
- [13].Ambrogini P, Lattanzi D, Ciuffoli S, Betti M, Fanelli M, Cuppini R. Physical exercise and environment exploration affect synaptogenesis in adult-generated neurons in the rat dentate gyrus: Possible role of BDNF. Brain Res 2013. doi: 10.1016/j.brainres.2013.08.023. [DOI] [PubMed] [Google Scholar]
- [14].Belarbi K, Burnouf S, Fernandez-Gomez FJ, Laurent C, Lestavel S, Figeac M, et al. Beneficial effects of exercise in a transgenic mouse model of Alzheimer’s disease-like Tau pathology. Neurobiol Dis 2011;43:486–94. doi: 10.1016/j.nbd.2011.04.022. [DOI] [PubMed] [Google Scholar]
- [15].Zajac MS, Pang TYC, Wong N, Weinrich B, Leang LSK, Craig JM, et al. Wheel running and environmental enrichment differentially modify exon-specific BDNF expression in the hippocampus of wild-type and pre-motor symptomatic male and female Huntington’s disease mice. Hippocampus 2009;20:NA-NA. doi: 10.1002/hipo.20658. [DOI] [PubMed] [Google Scholar]
- [16].Boros BD, Greathouse KM, Gentry EG, Curtis KA, Birchall EL, Gearing M, et al. Dendritic spines provide cognitive resilience against Alzheimer’s disease. Ann Neurol 2017;82:602–14. doi: 10.1002/ana.25049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Pereda-Pérez I, Valencia A, Baliyan S, Núñez A, Sanz-García A, Zamora B, et al. Systemic administration of a fibroblast growth factor receptor 1 agonist rescues the cognitive deficit in aged socially isolated rats. Neurobiol Aging 2019. doi: 10.1016/J.NEUROBIOLAGING.2019.02.011. [DOI] [PubMed] [Google Scholar]
- [18].Arnold SE, Louneva N, Cao K, Wang LS, Han LY, Wolk DA, et al. Cellular, synaptic, and biochemical features of resilient cognition in Alzheimer’s disease. Neurobiol Aging 2013;34:157–68. doi: 10.1016/j.neurobiolaging.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Casaletto KB, Elahi FM, Bettcher BM, Neuhaus J, Bendlin BB, Asthana S, et al. Neurogranin, a synaptic protein, is associated with memory independent of Alzheimer biomarkers. Neurology 2017:10.1212/WNL.0000000000004569. doi: 10.1212/WNL.0000000000004569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mak E, Holland N, Jones PS, Savulich G, Low A, Malpetti M, et al. In vivo coupling of dendritic complexity with presynaptic density in primary tauopathies. MedRxiv 2020:2020.12.24.20248838. doi: 10.1101/2020.12.24.20248838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Jensen CS, Portelius E, Høgh P, Wermuth L, Blennow K, Zetterberg H, et al. Effect of physical exercise on markers of neuronal dysfunction in cerebrospinal fluid in patients with Alzheimer’s disease. Alzheimer’s Dement Transl Res Clin Interv 2017;3:284–90. doi: 10.1016/j.trci.2017.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Buchman AS, Dawe RJ, Yu L, Lim A, Wilson RS, Schneider JA, et al. Brain pathology is related to total daily physical activity in older adults. Neurology 2018;90:E1911–9. doi: 10.1212/WNL.0000000000005552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Honer WG, Ramos-Miguel A, Alamri J, Sawada K, Barr AM, Schneider JA, et al. The synaptic pathology of cognitive life. Dialogues Clin Neurosci 2019. doi: 10.31887/DCNS.2019.21.3/whoner. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Boyle PA, Wilson RS, Yu L, Barr AM, Honer WG, Schneider JA, et al. Much of late life cognitive decline is not due to common neurodegenerative pathologies. Ann Neurol 2013. doi: 10.1002/ana.23964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ramos-Miguel A, Jones AA, Sawada K, Barr AM, Bayer TA, Falkai P, et al. Frontotemporal dysregulation of the SNARE protein interactome is associated with faster cognitive decline in old age. Neurobiol Dis 2018;114:31–44. doi: 10.1016/j.nbd.2018.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ramos-Miguel A, Sawada K, Jones AA, Thornton AE, Barr AM, Leurgans SE, et al. Presynaptic proteins complexin-I and complexin-II differentially influence cognitive function in early and late stages of Alzheimer’s disease. Acta Neuropathol 2017;133:395–407. doi: 10.1007/s00401-016-1647-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bennett DA, Buchman AS, Boyle PA, Barnes LL, Wilson RS, Schneider JA. Religious Orders Study and Rush Memory and Aging Project. J Alzheimer’s Dis 2018;64:S161–89. doi: 10.3233/JAD-179939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Honer WG, Barr AM, Sawada K, Thornton AE, Morris MC, Leurgans SE, et al. Cognitive reserve, presynaptic proteins and dementia in the elderly. Transl Psychiatry 2012;2:e114–e114. doi: 10.1038/tp.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wilson RS, Boyle PA, Yu L, Barnes LL, Schneider JA, Bennett DA. Life-span cognitive activity, neuropathologic burden, and cognitive aging. Neurology 2013;81:314–21. doi: 10.1212/WNL.0b013e31829c5e8a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].James BD, Wilson RS, Barnes LL, Bennett DA. Late-life social activity and cognitive decline in old age. J. Int. Neuropsychol. Soc, 2011. doi: 10.1017/S1355617711000531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kohout FJ, Berkman LF, Evans DA, Cornoni-Huntley J. Two Shorter Forms of the CES-D Depression Symptoms Index. J Aging Health 1993. doi: 10.1177/089826439300500202. [DOI] [PubMed] [Google Scholar]
- [32].Cotman CW, Berchtold NC. Exercise: A behavioral intervention to enhance brain health and plasticity. Trends Neurosci 2002. doi: 10.1016/S0166-2236(02)02143-4. [DOI] [PubMed] [Google Scholar]
- [33].Kang CM, Lavoie PA, Gardiner PF. Chronic exercise increases SNAP-25 abundance in fast-transported proteins of rat motoneurones. Neuroreport 1995. doi: 10.1097/00001756-199502000-00035. [DOI] [PubMed] [Google Scholar]
- [34].Molteni R, Ying Z, Gómez-Pinilla F. Differential effects of acute and chronic exercise on plasticity-related genes in the rat hippocampus revealed by microarray. Eur J Neurosci 2002;16:1107–16. doi: 10.1046/j.1460-9568.2002.02158.x. [DOI] [PubMed] [Google Scholar]
- [35].Hu S, Ying Z, Gomez-Pinilla F, Frautschy SA. Exercise can increase small heat shock proteins (sHSP) and pre- and post-synaptic proteins in the hippocampus. Brain Res 2009;1249:191–201. doi: 10.1016/j.brainres.2008.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Liu YF, Chen HI, Wu CL, Kuo YM, Yu L, Huang AM, et al. Differential effects of treadmill running and wheel running on spatial or aversive learning and memory: Roles of amygdalar brain-derived neurotrophic factor and synaptotagmin I. J Physiol 2009. doi: 10.1113/jphysiol.2009.173088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Liu YF, Chen H ing, Yu L, Kuo YM, Wu F Sen, Chuang JI, et al. Upregulation of hippocampal TrkB and synaptotagmin is involved in treadmill exercise-enhanced aversive memory in mice. Neurobiol Learn Mem 2008;90:81–9. doi: 10.1016/j.nlm.2008.02.005. [DOI] [PubMed] [Google Scholar]
- [38].Vaynman SS, Ying Z, Yin D, Gomez-Pinilla F. Exercise differentially regulates synaptic proteins associated to the function of BDNF. Brain Res 2006;1070:124–30. doi: 10.1016/j.brainres.2005.11.062. [DOI] [PubMed] [Google Scholar]
- [39].Huang FL, Huang KP, Wu J, Boucheron C. Environmental enrichment enhances neurogranin expression and hippocampal learning and memory but fails to rescue the impairments of neurogranin null mutant mice. J Neurosci 2006. doi: 10.1523/JNEUROSCI.1182-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Höglund K, Schussler N, Kvartsberg H, Smailovic U, Brinkmalm G, Liman V, et al. Cerebrospinal fluid neurogranin in an inducible mouse model of neurodegeneration: A translatable marker of synaptic degeneration. Neurobiol Dis 2020;134:104645. doi: 10.1016/j.nbd.2019.104645. [DOI] [PubMed] [Google Scholar]
- [41].Mukaetova-Ladinska EB, Hurt J, Honer WG, Harrington CR, Wischik CM. Loss of synaptic but not cytoskeletal proteins in the cerebellum of chronic schizophrenics. Neurosci Lett 2002. doi: 10.1016/S0304-3940(01)02458-2. [DOI] [PubMed] [Google Scholar]
- [42].Hillman CH, Erickson KI, Kramer AF. Be smart, exercise your heart: Exercise effects on brain and cognition. Nat Rev Neurosci 2008. doi: 10.1038/nrn2298. [DOI] [PubMed] [Google Scholar]
- [43].Colcombe SJ, Erickson KI, Raz N, Webb AG, Cohen NJ, McAuley E, et al. Aerobic fitness reduces brain tissue loss in aging humans. Journals Gerontol - Ser A Biol Sci Med Sci 2003. doi: 10.1093/gerona/58.2.m176. [DOI] [PubMed] [Google Scholar]
- [44].Spartano NL, Davis-Plourde KL, Himali JJ, Andersson C, Pase MP, Maillard P, et al. Association of Accelerometer-Measured Light-Intensity Physical Activity With Brain Volume: The Framingham Heart Study. JAMA Netw Open 2019;2:e192745. doi: 10.1001/jamanetworkopen.2019.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Casaletto KB, Staffaroni AM, Wolf A, Appleby B, Brushaber D, Coppola G, et al. Active lifestyles moderate clinical outcomes in autosomal dominant frontotemporal degeneration. Alzheimer’s Dement 2020. doi: 10.1002/alz.12001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Dufour CA, Marquine MJ, Fazeli PL, Umlauf A, Henry BL, Zlatar Z, et al. A Longitudinal Analysis of the Impact of Physical Activity on Neurocognitive Functioning Among HIV-Infected Adults. AIDS Behav 2018. doi: 10.1007/s10461-016-1643-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Finnema SJ, Nabulsi NB, Eid T, Detyniecki K, Lin SF, Chen MK, et al. Imaging synaptic density in the living human brain. Sci Transl Med 2016;8. doi: 10.1126/scitranslmed.aaf6667. [DOI] [PubMed] [Google Scholar]
- [48].Stern Y, Arenaza-Urquijo EM, Bartrés-Faz D, Belleville S, Cantilon M, Chetelat G, et al. Whitepaper: Defining and investigating cognitive reserve, brain reserve, and brain maintenance. Alzheimer’s Dement 2018. doi: 10.1016/j.jmarsys.2011.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Honer WG, Kaufmann CA, Kleinman JE, Casanova MF, Davies P. Monoclonal antibodies to study the brain in schizophrenia. Brain Res 1989. doi: 10.1016/0006-8993(89)90335-1. [DOI] [PubMed] [Google Scholar]
- [50].Honer WG, Hu L, Davies P. Human synaptic proteins with a heterogeneous distribution in cerebellum and visual cortex. Brain Res 1993. doi: 10.1016/0006-8993(93)90848-H. [DOI] [PubMed] [Google Scholar]
- [51].Matthew WD, Tsavaler L, Reichardt LF. Identification of a synaptic vesicle-specific membrane protein with a wide distribution in neuronal and neurosecretory tissue. J Cell Biol 1981. doi: 10.1083/jcb.91.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Takahashi S, Yamamoto H, Matsuda Z, Ogawa M, Yagyu K, Taniguchi T, et al. Identification of two highly homologous presynaptic proteins distinctly localized at the dendritic and somatic synapses. FEBS Lett 1995. doi: 10.1016/0014-5793(95)00713-J. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.