

Summary
Prime editors (PEs) are clustered regularly interspaced short palindromic repeats (CRISPR)-based genome engineering tools that can introduce precise base-pair edits. We developed an automated pipeline to correct (therapeutic editing) or introduce (disease modeling) human pathogenic variants from ClinVar that optimizes the design of several RNA constructs required for prime editing and avoids predicted off-targets in the human genome. However, using optimal PE design criteria, we find that only a small fraction of these pathogenic variants can be targeted. Through the use of alternative Cas9 enzymes and extended templates, we increase the number of targetable pathogenic variants from 32,000 to 56,000 variants and make these pre-designed PE constructs accessible through a web-based portal (http://primeedit.nygenome.org). Given the tremendous potential for therapeutic gene editing, we also assessed the possibility of developing universal PE constructs, finding that common genetic variants impact only a small minority of designed PEs.
Subject areas: Biotechnology, Computational bioinformatics
Graphical abstract
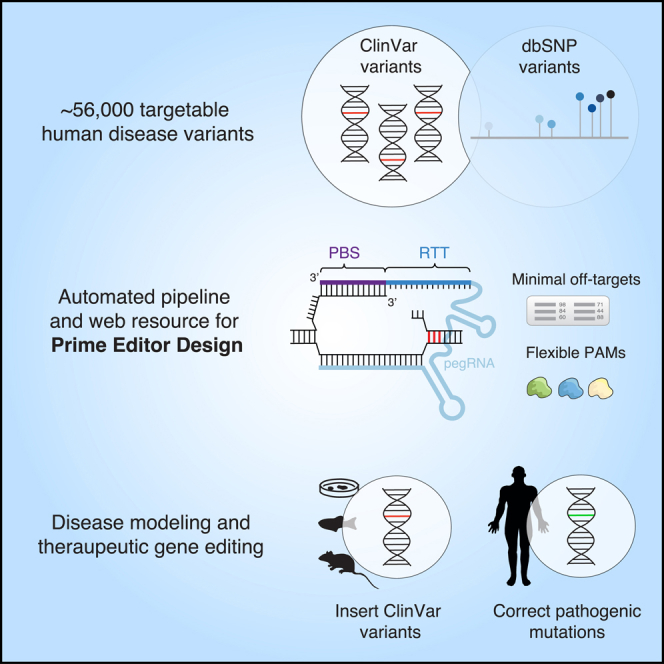
Highlights
-
•
CRISPR prime editors for therapeutic gene editing and disease modeling
-
•
Increase targetable variants with template extension and alternative Cas9 design
-
•
Optimize prime editors with off-target avoidance and integration of common variants
-
•
Web-tool for rapid prime editor design using gene name or ClinVar identifier
Biotechnology; Computational bioinformatics
Introduction
Recently, Anzalone et al. (2019) developed prime editors (PEs) for introducing precise edits with base-pair resolution using a Cas9 nickase tethered to reverse transcriptase. PEs are targeted via a PE guide RNA (pegRNA) to a specific genomic locus, where they make a targeted single-stranded break (nick) in the DNA and reverse transcribe a repair template on the 3′ end of the pegRNA with the desired base-pair substitution, insertion or deletion. Through an analysis of the ClinVar database of human genetic variants (Landrum et al., 2018), it was suggested that prime editing may be able to correct up to 89% of known genetic variants associated with human diseases (Anzalone et al., 2019). Here, we developed a computational pipeline to design PEs to correct pathogenic variants in ClinVar for therapeutic gene editing and to introduce these variants into wild-type cells to create disease models (Figure 1A). We then present PE design considerations to further improve the number of targetable variants and PE reagents per variant.
Figure 1.

Prime editing to insert or repair human pathogenic variants
(A) Structure of the prime editing guide RNA (pegRNA) and applications. Prime editing reagents are comprised of a single-guide RNA (PE2 guide RNA), the primer binding site (PBS), and the reverse transcription template (RTT). The PBS and PE2 guide are complementary to the target sequence and a single-stranded nick is induced three nucleotides from the protospacer adjacent motif (PAM). The RTT contains the desired edit to be introduced. PE3(b) single-guide RNA is not shown.
(B) Workflow for the design of PE2, PE3, and PE3b classes of prime editors to target human pathogenic variants from ClinVar. After generating pegRNAs and PE3(b) sgRNAs, optional steps include scoring for potential off-targets, identifying sequences that may overlap common human genetic variants, and designing pegRNAs and PE3(b) sgRNAs that recognize a flexible NG PAM sequence.
(C) Screenshot of the prime editing design tool for ClinVar variants mapping to the FTO locus. As indicated by circle (1), the user first selects whether to search by Gene Symbol or by ClinVar Variant ID, which is entered into (2). With Circle (3), the user selects prime editors to introduce a pathogenic variant, to remove a pathogenic variant or both types. Circle (4) is used to toggle on/off reagents without off-target scores (e.g. no off-target scoring is available for flexible PAM variants). Circle (5) allows for only showing PE3(b) sgRNAs based on filters for the corresponding pegRNAs. Circle (6) selects pegRNAs based on a minimum off-target score cut-off. Circle (7) allows for PAM-sequence filtering of reagents. Circle (8) switches between the display of all pegRNAs or those filtered by criteria in the drop-down menu. The user downloads the displayed pegRNAs as a plain text file with (9).
PE design consists of four components: the primary single-guide RNA (sgRNA) that will target a specific site for inducing a nick, the primer binding site (PBS), the reverse transcription template (RTT) that includes the desired edit and a secondary sgRNA to improve editing efficiency. The first three components together form the pegRNA (Figure 1A), which when used alone is termed the PE2 approach. To further boost PE efficiency, the PE3 and PE3b approaches utilize a secondary sgRNA that is distinct from the pegRNA, to either induce a nick 40–90 bp downstream of the target or within 3 bp downstream of the target, respectively. Importantly, the PE3b approach requires sgRNA sequence complementarity with the edited strand of DNA sequence. Design consideration specifics for PE3 and PE3b both serve to improve the editing efficiency of prime editing threefold over PE2, with PE3b greatly reducing indel formation (Anzalone et al., 2019).
Results
Targeting human pathogenic variants with prime editing
Given the number of locus-specific reagents required for prime editing, we developed an optimized pipeline for PE design (Figure 1B) and made all pegRNAs discussed below available through a user-friendly web-based application (http://primeedit.nygenome.org; Figure 1C). To ensure ClinVar variants would be targetable within the default PE parameters (see STAR Methods), we only examined single base-pair substitutions and insertions and deletions of 10 base-pairs or less, resulting in 66,580 ClinVar variants (39% transition mutations, 25% transversion mutations, 11% insertions, and 24% deletions) (Figure 2A). As an initial consideration, we looked for how many pathogenic variants have a Cas9-targetable site; specifically, Cas9 requires a protospacer-adjacent motif (PAM) at the target site to bind and cut. Of these variants, 78% had PAMs within an appropriate distance for PE2 pegRNAs, 72% for PE3 pegRNAs, and 16% for PE3b pegRNAs (Figure 2A). When considering PE2 pegRNA design, the least restrictive of the three classes, most variants had at least 1 or 2 available PAMs (Figure 2B) and we designed PE2, PE3, and PE3b pegRNAs wherever possible to correct pathogenic variants (Tables S1, S2, and S3).
Figure 2.

Targetable pathogenic variants with prime editing
(A) Total number of ClinVar pathogenic variants by mutation type (transition, transversion, insertion, or deletion) (left) and the percentage targetable with prime editing after various filtering steps in the design pipeline (right). Over 70% of pathogenic variants have protospacer adjacent motifs (PAMs) within a suitable distance for PE2 and PE3 prime editing; however, only 16% of pathogenic variants have PAMs within a suitable distance for PE3b prime editing. These percentages decrease as we consider which of the prime editors meet the basic requirements for usability (e.g., no Pol3 terminator motif) and filtering for no predicted off-targets in the genome.
(B) The number of prime editing sites (PAMs for pegRNA) per pathogenic variant for PE2 prime editors. The majority of pathogenic variants have at least 1 or 2 sites for designing prime editors.
(C) The increase in the percentage of targetable variants when extending the RTT from 16 nucleotides to 50 nucleotides, and the increase in percentage when allowing for flexible NG PAM recognition instead of SpCas9 NGG PAM recognition.
Although the PAM site is a requirement, there are several other aspects of pegRNA design, such as suitable GC-content and avoidance of Pol3 terminator motifs that further constrain the space of targetable sites in the genome (“usable pegRNAs”). The percentages of targetable variants are reduced to 48%, 42%, and 8% for PE2, PE3, and PE3b, respectively, when only considering usable pegRNAs (Figure 2A). Given the importance of assessing off-target effects when performing genome editing experiments and for therapeutic gene editing, we also analyzed the guide RNAs (both the pegRNAs and PE3/3b sgRNAs) to identify potential off-targets genome-wide. Although the use of a Cas9 nickase should minimize off-target genome modification, others have found that nickases can, indeed, result in indel mutation at a low but detectable frequency (Cho et al., 2014). If we only retain guide RNAs with minimal predicted off-targets (Hsu-Scott off-target score >50, see STAR Methods) (Hsu et al., 2013), it reduces the number of targetable variants by an additional 7–17%, depending on the PE type (Figure 2A). Given the low number of targetable variants with usable pegRNAs (with or without the off-target optimization), we wondered if we could increase this number through alternate design strategies.
One method to increase the number of targetable variants is to extend the length of the RTT. Therefore, we re-designed pegRNAs with 50 nucleotide RTTs and, as expected, observed an increase in the number of PAMs for PE2 pegRNAs that were sufficiently close to ClinVar variants (Figure 1A). The percentage of targetable variants when considering usable pegRNAs with the RTT extension increased by 55% for PE2 and PE3 and by 129% for PE3b (Figure 2C). Similar to before, only selecting guide RNAs with minimal predicted off-targets (Hsu-Scott off-target score >50) reduces the number of targetable variants by 3–15%, depending on the PE type (Figure S1C).
An alternative strategy is to take advantage of recently engineered Cas9 variants with a less-restrictive PAM sequence, such as Cas9-NG, xCas9, xCas9-NG, or SpRY (near PAM-less) (Hu et al., 2018; Legut et al., 2020; Nishimasu et al., 2018; Walton et al., 2020). These variants require only an NG PAM instead of the NGG PAM for SpCas9, or NRN/NYN in the case of SpRY, and have been recently described and optimized in a variety of settings. We re-designed pegRNAs with flexible NG PAM recognition and a standard RTT length and provided additional documentation on how to design PAM-less pegRNAs (see Data and code availability). In comparison to the RTT extension, we observed even more targetable variants for PE2 pegRNAs when using NG PAMs (Figure S1B). When restricting to targetable variants with usable pegRNAs, we observed the highest increases in targetable variants from all strategies: 76%, 99%, and 553% for PE2, PE3, and PE3b, respectively (Figure 2C). Requiring a Hsu-Scott off-target score >20 (to avoid the most promiscuous guide RNAs) reduced the number of targetable variants by 24–57%, depending on the PE type (Figure S1C). It appears that allowing for flexible PAM recognition overcomes restrictions with designing secondary sgRNAs for PE3 and PE3b PEs, but that the increase in targetable variants comes with a trade-off in increasing potential off-target activity. This trade-off is an important consideration for prime editing with recent Cas9 variants. However, given the increased number of targetable variants, it may be worth considering even guide RNAs with potential off-targets, as long as careful analysis is performed to ensure minimal off-target modification after prime editing.
Overlap of common human genetic variants with prime editing target sites
Given that there are a large number of genetic variants that are relatively common in the general population (minor allele frequency ≥1%), PEs may overlap variants that are not disease-causing but that may disrupt the PAM or their sequence complementarity. As we designed all pegRNAs reported in this study using a human genome reference (GRCh37), we also assessed whether the sequences of the pegRNAs that require complementarity (the primary sgRNA, the PBS, and the secondary sgRNA) or respective PAMs mapped to sites with common human genetic variation using the dbSNP catalog (Kitts and Sherry, 2011). We found that the majority of usable prime editing reagents (>95%) did not overlap common genetic variation (Figure 3), suggesting that they would not need to be modified further and allow for the development of universal therapeutic PEs for most variants.
Figure 3.

The number of prime editing reagents that map to sites of common human genetic variation
Over 95% of prime editing reagents do not map to sites of common human genetic variation, represented by single-nucleotide polymorphisms (SNPs) occurring in over 1% of the gene population. The data are represented on a log scale to allow for the visualization of the number of prime editing reagents that do map to sites of common human genetic variation, as these are relatively low in number. However, it should be noted that some prime editors may have to be modified if they map to multiple sites of common human genetic variation.
Discussion
We have made all designed pegRNAs available through a user-friendly web-based application (http://primeedit.nygenome.org; Figure 1C). Using this application, users can search for pegRNAs targeting specific ClinVar variants by their variant ID or by gene ID (if they map to a gene), or use the design program to customize their own pegRNAs (Kweon et al., 2021). The pegRNAs discussed in this study were designed to replace ClinVar variants with the human reference allele; however, we also designed pegRNAs to introduce ClinVar variants for creating genetic models of disease (Figure S2, Tables S4, S5, and S6). These alternate pegRNAs for introducing ClinVar variants are also accessible through our web-based application. Two additional PE design tools have been developed, PrimeDesign (Hsu et al., 2021) and pegFinder (Chow et al., 2020), and we provide a comparison of the tools in Table S7. Briefly, our tool is able to design pegRNAs with diverse PAM recognition sites (like pegFinder) but can also provide additional metrics to inform pegRNA selection (like PrimeDesign), such as GC content or on/off target scores.
Prime editing has tremendous potential for therapeutic gene editing and model organism research. Here, we have shown not only that thousands of known human disease-causing variants are targetable with prime editing reagents, but that thousands more can be targeted with further developments in prime editing and nuclease technology. We have developed an automated pipeline to design prime editing reagents for specific base-pair edits and created a web-based portal to make these reagents readily accessible. Notably, our designed prime editing reagents are for human pathogenic variants from the ClinVar database. Research is still needed to carefully test these prime editing reagents, to determine optimal parameters, but we are now able to design these reagents with ease and at high throughput.
Limitations of the study
In this study, we designed prime editing reagents to introduce or correct human pathogenic variants, providing a web-based portal for easy accessibility. As our study focused on the computational aspects of prime editing reagent design, experimental validation, and potential further optimization based on those results will be an important future step for the widespread adoption of these tools.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| ClinVar database | NCBI | http://ftp.ncbi.nlm.nih.gov/pub/clinvar |
| Human genome build (H. sapiens, GRCh37, hg19) | NCBI | http://hgdownload.soe.ucsc.edu/goldenPath/hg19/bigZips/hg19.fa.gz |
| dbSNP (build 151) | NCBI | http://hgdownload.cse.ucsc.edu/goldenpath/hg19/database/snp151Common.txt.gz |
| Software and algorithms | ||
| Prime editor design algorithm and analysis scripts | This paper | http://gitlab.com/sanjanalab/primeediting |
| Prime editing design tool | This paper | http://primeedit.nygenome.org/ |
| FlashFry | McKenna and Shendure, 2018 | http://github.com/mckennalab/FlashFry/ |
| Bedtools v2.29.2 | Quinlan and Hall, 2010 | http://github.com/arq5x/bedtools2/ |
| R v3.4.4 | R-Project | http://www.r-project.org/ |
| Biostrings v2.46.0 | Bioconductor | http://bioconductor.org/packages/release/bioc/html/Biostrings.html |
| Python v3.7.0 | Python Software Foundation | http://python.org/ |
Resource availability
Lead contact
Further requests for information should be directed to the lead contact, Neville E. Sanjana (neville@sanjanalab.org).
Materials availability
This study did not generate unique reagents.
Method details
Automated prime editor design software
We developed a pipeline in R v3.4.4 for designing prime editor guide RNAs (pegRNAs) to induce specific base-pair substitutions, insertions or deletions. This R pipeline uses dplyr v0.8.5, ggplot2 v3.3.0, data.table v1.12.8, BSgenome.Hsapiens.UCSC.hg19 v1.4.0, Biostrings v2.46.0 and future.apply v1.4.0 libraries to design all four components of pegRNAs: the primary sgRNA, the primer binding site (PBS), the reverse transcription template (RTT) and the secondary sgRNA for the PE3 and PE3b classes of prime editors. While we used the human genome build hg19 to design our prime editing reagents, this can be changed to any input genome by the user. We have successfully tested the automated prime editor design software with the mouse genome build mm10 and zebrafish genome build danRer10 and provide directions on using these genomes and other alternate genomes in a tutorial available on our GitLab repository. In addition, we have tested the automated prime editor design software with PAM-less design criteria and included it in the tutorial (see Data and code availability).
To design canonical 20 nucleotide (nt) sgRNAs for Cas9, several protocols already exist (Meier et al., 2017). Briefly, we reimplemented standard sgRNA design considerations to target NGG PAMs. Initial studies recommend a PBS length of 13 nt, but this can be modified for a more favorable GC sequence content depending on local sequence context. The PBS end site is explicitly determined by the location of the nick site (3 nt downstream from the PAM), as the RTT must begin with the nick site. We allowed for the length of the PBS to be determined by user input but set 13 nt as the recommended default. If the default length for the PBS is modified, it is extended from the 3’ end of the RTT. GC content is reported in the web-based interface so that the user is aware of this parameter throughout the design process.
Since Anzalone et al. (2019) suggest not using PBS sequences if the GC content is outside of approximately 40-60%, we flag any PBS with a GC content less than 35% and greater than 65% as potentially problematic. The default RTT size was set to 16 nt and allows for the targeted edit to be in any position except for the last nt. We compare 16 and 50 nt RTT sequences to demonstrate the increase in targetable sites when making the RTT larger. The PBS and RTT are packaged together and provided as the complete 3’ extension. If secondary sgRNAs are required for PE3 or PE3b prime editors, these are separately generated and can be matched with a corresponding pegRNA complementary to the opposite strand, to ensure nicks are made on opposite strand.
The PE2, PE3 and PE3b design programs were written separately as R functions and can be imported into a user’s R environment. The functions were wrapped in modified apply functions from the future.apply v1.4.0 package to parallelize prime editor reagent design, as the task for designing sets of prime editors will increase if allowing for more flexible PAM recognition sites designing genome-scale libraries. Packaging each design program separately also allows for users to selectively design specific classes of prime editors, and ignore other classes if not desired.
Pathogenic variants for prime editing
We identified human pathogenic variants for prime editing using the catalog of variants reported by the ClinVar database (modified 12/2/2019), which was downloaded from the NCBI website (see Data and code availability). First, we examined only variants in ClinVar with the “pathogenic” identifier. We explicitly selected for single base-pair substitutions, insertions less than 10 base-pairs and deletions less than 10 base-pairs, to ensure the full size of any desired edit could be contained with a 16 nt RTT. Larger variants can also be targeted with the automated prime editor design program: The user can modify either the scripts or the web-tool to specify an appropriately large RTT size.
PE2, PE3 and PE3b reagents to target variants
After we selected human pathogenic variants for prime editing, we generated human reference genome sequences surrounding each variant and designed PE2, PE3 and PE3b reagents to introduce the pathogenic allele. We then generated human genome pseudo-reference sequences with the pathogenic allele instead of the reference allele and designed PE2, PE3 and PE3b reagents to correct these variants. We discuss the reagents used to correct variants but provide reagents in the web-tool to both introduce (disease model) and correct (therapeutic) pathogenic variants. In practice, all PE3 and PE3b pegRNAs are subsets of PE2 pegRNAs, as the former classes of prime editors include a secondary sgRNA to assist the pegRNA’s genome editing efficiency. The number of designable PE3 secondary sgRNAs will be much greater than the number of designable PE3b secondary sgRNAs, given the restrictive nature of the latter class of prime editors.
Common human genetic variation overlap
One concern when performing genome editing is the occurrence of mismatches between the target genome and the reference from which sgRNAs are designed. To address this, we used dbSNP build 151 (Kitts and Sherry, 2011) to generate a list of all known single nucleotide polymorphisms (SNPs) with minor allele frequencies (MAF) ≥ 1% in the human population. We used bedtools v2.29.2 (Quinlan and Hall, 2010) to perform an intersection between our designed prime editing reagents and common human genetic variation.
Off-target prediction
To mitigate off-target effects by our designed prime editing reagents, we analyzed the 20 nt sgRNA sequences with FlashFry (McKenna and Shendure, 2018) to generate their Hsu et al. off-target scores (“Hsu-Scott score”). FlashFry can generate a variety of quality control metrics for sgRNAs, but we focused on the Hsu-Scott scores, as mitigating off-target effects would be a higher priority for prime editing reagents designed to target human pathogenic variation, over other scores such as on-target cutting frequency. Briefly, Hsu-Scott scores range from 0 to 100 for each scored sgRNA, where the higher the score, the lower the predicted number of off-targets that sgRNA will have. Given the Hsu-Scott experimental dataset was generated using only standard Cas9 and that FlashFry does not score sgRNAs with an NG PAM recognition, we modified FlashFry for scoring NG PAMs and report these on our website (see Data and code availability). The scored sgRNAs we provide includes both the sgRNA component of the PE2 pegRNA and the secondary sgRNAs for PE3 and PE3b classes of prime editors.
Quantification and statistical analysis
Designed PE reagents or targetable variants are given as exact numbers or percentages (Figures 2, 3, S1, and S2).
Additional resources
The web-tool described in the paper containing all the designed prime editing reagents is accessible at: http://primeedit.nygenome.org. The code used to design and optimize all the prime editors is accessible at: http://gitlab.com/sanjanalab/primeediting.
Acknowledgments
We thank the entire Sanjana laboratory for support and advice and thank M. Zaran and S. Brock for assistance with the web-tool server. We thank A. McKenna for help in modifying the FlashFry source code. J.A.M. is supported by a Banting Postdoctoral Fellowship and the Canadian Institutes of Health Research. N.E.S. is supported by New York University and New York Genome Center startup funds, National Institutes of Health (NIH)/National Human Genome Research Institute (grant nos. R00HG008171, DP2HG010099), NIH/National Cancer Institute (grant no. R01CA218668), Defense Advanced Research Projects Agency (grant no. D18AP00053), the Sidney Kimmel Foundation, the Melanoma Research Alliance, and the Brain and Behavior Foundation.
Author contributions
J.A.M and N.E.S. conceived the project and designed the study. J.A.M. wrote the design pipeline and J.R. built the web-tool. J.A.M. and J.R. performed analyses. X.G. helped with data presentation. All authors contributed to drafting and reviewing the manuscript, provided feedback, and approved the manuscript in its final form.
Declaration of interests
N.E.S. is an advisor for Vertex and Qiagen.
Published: November 19, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103380.
Supplemental information
Data and code availability
-
•
Code and software to reproduce our analyses are available for download from our GitLab repository (http://gitlab.com/sanjanalab/primeediting).
-
•
Predesigned prime editing reagents are available for download from a dedicated website (http://primeedit.nygenome.org/).
-
•
The latest release of the ClinVar database is available from the ClinVar FTP (http://ftp.ncbi.nlm.nih.gov/pub/clinvar/).
-
•
FlashFry can be downloaded from its GitHub repository (http://github.com/mckennalab/FlashFry).
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact.
References
- Anzalone A.V., Randolph P.B., Davis J.R., Sousa A.A., Koblan L.W., Levy J.M., Chen P.J., Wilson C., Newby G.A., Raguram A., Liu D.R. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019 doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S.W., Kim S., Kim Y., Kweon J., Kim H.S., Bae S., Kim J.-S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014;24:132. doi: 10.1101/gr.162339.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow R.D., Chen J.S., Shen J., Chen S. A web tool for the design of prime-editing guide RNAs. Nat. Biomed. Eng. 2020:1–5. doi: 10.1038/s41551-020-00622-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu J.Y., Grünewald J., Szalay R., Shih J., Anzalone A.V., Lam K.C., Shen M.W., Petri K., Liu D.R., Joung J.K., Pinello L. PrimeDesign software for rapid and simplified design of prime editing guide RNAs. Nat. Commun. 2021;12:1034. doi: 10.1038/s41467-021-21337-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J.H., Miller S.M., Geurts M.H., Tang W., Chen L., Sun N., Zeina C.M., Gao X., Rees H.A., Lin Z., Liu D.R. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature. 2018;556:57–63. doi: 10.1038/nature26155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitts A., Sherry S. The NCBI Handbook [Internet]. National Center for Biotechnology Information (US); 2011. The Single Nucleotide Polymorphism Database (dbSNP) of Nucleotide Sequence Variation. [Google Scholar]
- Kweon J., Yoon J.-K., Jang A.-H., Shin H.R., See J.-E., Jang G., Kim J.-I., Kim Y. Engineered prime editors with PAM flexibility. Mol. Ther. 2021;29:2001–2007. doi: 10.1016/j.ymthe.2021.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum M.J., Lee J.M., Benson M., Brown G.R., Chao C., Chitipiralla S., Gu B., Hart J., Hoffman D., Jang W., et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46:D1062–D1067. doi: 10.1093/nar/gkx1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legut M., Daniloski Z., Xue X., McKenzie D., Guo X., Wessels H.-H., Sanjana N.E. High-throughput screens of PAM-flexible Cas9 variants for gene knockout and transcriptional modulation. Cell Rep. 2020;30:2859–2868.e5. doi: 10.1016/j.celrep.2020.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A., Shendure J. FlashFry: A fast and flexible tool for large-scale CRISPR target design. BMC Biol. 2018;16:74. doi: 10.1186/s12915-018-0545-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier J.A., Zhang F., Sanjana N.E. GUIDES: sgRNA design for loss-of-function screens. Nat. Methods. 2017;14:831–832. doi: 10.1038/nmeth.4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimasu H., Shi X., Ishiguro S., Gao L., Hirano S., Okazaki S., Noda T., Abudayyeh O.O., Gootenberg J.S., Mori H., et al. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science. 2018;361:1259–1262. doi: 10.1126/science.aas9129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton R.T., Christie K.A., Whittaker M.N., Kleinstiver B.P. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science. 2020 doi: 10.1126/science.aba8853. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Code and software to reproduce our analyses are available for download from our GitLab repository (http://gitlab.com/sanjanalab/primeediting).
-
•
Predesigned prime editing reagents are available for download from a dedicated website (http://primeedit.nygenome.org/).
-
•
The latest release of the ClinVar database is available from the ClinVar FTP (http://ftp.ncbi.nlm.nih.gov/pub/clinvar/).
-
•
FlashFry can be downloaded from its GitHub repository (http://github.com/mckennalab/FlashFry).
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact.