

Graphical abstract

Keywords: Tubo-ovarian cancer, High grade serous carcinoma, Carcinoogenesis, Molecular profiling, Methylation profiling, Interactive data browser, Biomarkers, Integromics
Abstract
Introduction
Cancers presenting at advanced stages inherently have poor prognosis. High grade serous carcinoma (HGSC) is the most common and aggressive form of tubo-ovarian cancer. Clinical tests to accurately diagnose and monitor this condition are lacking. Hence, development of disease-specific tests are urgently required.
Methods
The molecular profile of HGSC during disease progression was investigated in a unique patient cohort. A bespoke data browser was developed to analyse gene expression and DNA methylation datasets for biomarker discovery. The Ovarian Cancer Data Browser (OCDB) is built in C# with a.NET framework using an integrated development environment of Microsoft Visual Studio and fast access files (.faf). The graphical user interface is easy to navigate between four analytical modes (gene expression; methylation; combined gene expression and methylation data; methylation clusters), with a rapid query response time. A user should first define a disease progression trend for prioritising results. Single or multiomics data are then mined to identify probes, genes and methylation clusters that exhibit the desired trend. A unique scoring system based on the percentage change in expression/methylation between disease stages is used. Results are filtered and ranked using weighting and penalties.
Results
The OCDB’s utility for biomarker discovery is demonstrated with the identified target OSR2. Trends in OSR2 repression and hypermethylation with HGSC disease progression were confirmed in the browser samples and an independent cohort using bioassays. The OSR2 methylation biomarker could discriminate HGSC with high specificity (95%) and sensitivity (93.18%).
Conclusions
The OCDB has been refined and validated to be an integral part of a unique biomarker discovery pipeline. It may also be used independently to aid identification of novel targets. It carries the potential to identify further biomarker assays that can reduce type I and II errors within clinical diagnostics.
1. Introduction
Epithelial ovarian cancer (EOC) is one of the most common causes of cancer death in women. The most common and most deadly type being high grade serous carcinoma (HGSC). It makes up ∼ 70 % of all EOCs and ∼ 90 % of advanced stage. From a molecular perspective, the presence of p53 mutations is ubiquitous and germline mutations in the BRCA1 or BRCA2 genes are present in 6.5–19 % [[1], [2], [3]]. In sporadic cases the presence of BRCA dysfunction and loss of function (BRCAness) is observed at relatively high frequency [[5], [6]].
Until recently, the underlying molecular mechanisms of HGSC development remained unknown. The development of HGSC was previously attributed to errors in cell replication associated with the repair of the recurrent trauma endured by the ovarian surface epithelium (OSE) incurred by ovulation [[7], [8], [9], [10], [11], [12], [13]]. In recent years, compelling pathological evidence has emerged that supports the theory that the distal fallopian tube is the origin of most extra-uterine HGSC [14]. Extensive pathological research, initially of risk-reducing salpingo-oophorectomy (RRSO) specimens in women at high risk of hereditary breast and tubo-ovarian cancers, has revealed most HGSCs arise from the distal fallopian tube from a precursor referred to as serous tubal intraepithelial carcinoma (STIC) [[15], [16]]. The presence of identical p53 mutations in STIC and its adjacent HGSC confirms clonality and a link between STIC and HGSC [17]. Previously, we have performed gene expression profiling, subsequent bioinformatic analysis and in-vitro validation, of a unique six-patient HGSC dataset [18]. This study provided further strong evidence that extrauterine HGSC arises from the fimbria of the distal fallopian tube.
Currently there is no effective screening method for EOC. Ovarian Cancer screening trials, in both the United States (US) and United Kingdom (UK), with pelvic ultrasound scanning and, the biomarker, serum CA125 were inconclusive [[19], [20], [21]]. Pelvic ultrasound in expert hands is a highly sensitive diagnostic method for EOC [22]. Unfortunately, because it relies heavily on individual expertise, discrimination between benign and malignant pelvic masses in routine clinical practice is challenging. Serum CA125 is most effective as a marker of disease status in patients undergoing chemotherapy treatment for EOC. It is not particularly specific to malignancy and is elevated by several benign conditions including endometriosis, pelvic infection, and uterine leiomyomata. CA125 is elevated in only 50 % of early stage EOCs and using it for screening can cause unnecessary medical intervention and significant patient distress [23]. Early diagnosis of HGSC is complicated by the fact that small tubal lesions can disseminate widely without the formation of a large tumour mass.
A novel approach, to combat this, would be the development of disease-specific molecular assays as an alternative, or complementary, diagnostic tool to radiological or serum biomarkers. A fast emerging area of tubo-ovarian cancer diagnostics is that of the liquid biopsy [24]. One novel methodology is the use of circulating free DNA (cfDNA) quantification and/or molecular profiling. Interrogating tumour specific cfDNA, known as circulating tumour DNA (ctDNA), for disease-specific genomic alterations carries the potential to significantly improve EOC diagnostics [24].
Expression profiling of genes at a transcriptional level, in a specific cell at a specific time can provide a global picture of cellular function [25]. Gene expression data has identified novel biomarkers that molecularly classify several cancers according to stage, recurrence potential, prognostic outcome and response to therapy [[26], [27], [28], [29]]. Similarly, DNA methylation profiling can identify disease-specific aberrations and provide a better understanding of the molecular events that promote disease survival/progression. DNA methylation at CpG sites is not evenly distributed throughout the genome. Regions with a higher frequency of CpG sites are termed as CpG islands [30] and most methylation occurs close by in “shores” or more distantly in “shelves” [[31], [32], [33]]. CpG islands often reside within the promoter region of genes. Large genome-wide methylation studies have shown that CpG methylation close to the transcriptional start site of a gene may result in repression of the gene [34]. Methylation within the region of the gene body usually results in stimulation/overexpression of the gene [35]. Methylation data can also be used to identify biomarkers.
The identification of disease-specific genetic aberrations requires in-depth mining of multiomics data. Whilst a wealth of big data has been collated for many cancers using third generation sequencing approaches, biomarkers are still lacking. Analytical tools to easily, and rapidly, analyse these large datasets are not readily available. New tools could identify genetic aberrations with disease-specific biomarker potential. Subsequent bioassay validation could be progressed on multiple markers concurrently with focussed confirmatory bioinformatics carried out on those with the most promise. A pipeline like this is needed to facilitate biomarker discovery in EOC.
Herein, the Ovarian Cancer Data Browser (OCDB) was developed to analyse multiomics data (gene expression, methylation) from a unique HGSC patient cohort [18]. The aim was to gain greater understanding of the underlying molecular biology that defines the HGSC carcinogenic pathway and, ultimately, refine a biomarker discovery pipeline. The OCDB was built in C# with a.NET framework using an integrated development environment of Microsoft Visual Studio. The OCDB allows users to rapidly and easily mine multiomics datasets. Trends of gene expression and methylation for a probe over the course of disease progression for each patient can be explored. The OCDB automatically calculates a score for each probe to reflect how consistently it shows an increasing or decreasing trend in expression or methylation between each disease stage. After scores are calculated, a sorted array is created which ranks results for probes, genes or methylation clusters. Results can be further filtered using weightings and penalties to adjust probe scores. The development of the OCDB and its performance and utility in a biomarker discovery pipeline is described. Validation of an example biomarker, OSR2, identified by the OCDB using bioinformatic, wet lab, and analysis of an independent dataset is also outlined.
2. Methods
2.1. Patient sample preparation, data collection and analytical pre-processing of data for the OCDB browser
The Northern Ireland Biobank provided six cases of sporadic, stage III + HGSC, who underwent primary cytoreductive surgery at the Northern Ireland Gynaecological Cancer Centre, for the study (Ethical approval: NIB11:005, NIB13:0094). The cases were chosen on the basis of the availability of the following tissue within their resectional specimen: normal OSE, normal FT, STIC, primary HGSC, and omental metastases. All cases had fully anonymised, matched clinico-pathological data [18].
An H&E stained slide was prepared from each of the relevant formalin fixed paraffin embedded (FFPE) blocks from all six cases. The cohort slides were pathologically reviewed and annotated for each of the five tissue types by a specialist Gynaecological Pathologist (WGM). Subsequently, ten 5 µm sections were taken from each tissue block for macrodissection and RNA preparation. Finally, a further H&E stained slide was prepared to confirm the annotated regions were still present and therefore, present throughout the sections for RNA preparation. The process was repeated for DNA preparation.
RNA preparation and gene expression profiling, using the Xcel® array (ALMAC, Craigavon, UK), was performed on the dataset as previously described [[18], [36]]. The array was validated using Quantitative reverse transcription PCR (RqPCR) [18]. The raw gene expression data in.CEL files was processed using the makecdfenv R package. This reads the AffymetriX array and creates a chip description file (.cdf). The.cdf file consists of a hash table environment containing the location/probe set membership mapping information. Gene expression data was background corrected and normalised using the justRMA function from the affy Bioconductor R package [37] and saved as a CSV file.
DNA preparation for bisulphite conversion and DNA methylation profiling was also performed on the same sample set using standard approaches and the Infinium HumanMethylation 450 K BeadChip array (Illumina® Inc., California, USA) [36]. Methylation arrays were validated using pyrosequencing [36]. The DNA methylation.idat files underwent a vanilla analysis using the RnBeads R package [38]. The methylation data passed quality control assessment for bisulphite conversion. The Greedycut algorithm was employed to filter out unreliable probes. Following this, the background was subtracted using the methylumi package (method “noob”) and the methylation β-values were normalized using the BMIQ normalization method [39]. The remaining probes were assessed for batch effects and corrected where necessary. Methylation values for probes were saved as a CSV file. The descriptor files for the probe genomic location within promoter, genebodies and CpG islands were saved as CSV files, as well as a sample identification file (sample_annotation.csv).
3. Data transformation and loading
Data for the three disease progression stages, normal FT (NFT), STIC and HGSC, from each of the six patients, was transformed into fast access files (.faf). Fast access files have a binary format suitable for high performance access and data processing efficiency. To transform data, a custom CSVSplitter program was written specifically to handle the normalised gene expression and methylation data as CSV files as well as the output descriptor files. The .cdf file and the sample_annotation.csv descriptor files were necessary for correct sample identification and interpretation of results. CSV files for the annotation of methylation probes location within gene bodies, islands and promoters were also processed. In all, expression values for 110,961 gene transcripts and methylation values for 424,583 CpG probes for each patient and disease stage were compiled as fast access files. Given the small dataset, patients were arbitrarily named patients 1 to 6. After the data has been loaded, the OCDB interface opens and calculations are automatically implemented. Some browser features require the internet, therefore the user will be alerted at this stage if the PC is not connected to the internet.
3.1. Data mining for expression/methylation probes associated with disease progression
3.1.1. Score calculations and ranking of probes
The OCDB aims to identify probes with consistently increasing or decreasing gene expression/methylation with disease progression. The approach used to estimate a probe’s score was consistent for gene transcript expression and methylation data and involved three stages of calculations. Firstly, the percentage change in score is estimated between disease stages. To estimate this, the gene transcript expression/ methylation values for each probe for the six patients are summed and a total is also calculated for each of the three disease stages. The percentage change in score (+/-) from NFT to STIC, from STIC to HGSC and from NFT to HGSC is then calculated. The final score is determined by adding the percentage changes together. The default browser setting is to treat each of these percentage changes equally. However, using two additional stages scores can be further calculated to prioritise probes that exhibit a particular directional change in expression/methylation and/or to filter results from the analysis.
3.1.2. Adjusting probe scores using weightings and penalties to filter results for reliability
In the “Sort Criteria” interface, users can specify to apply weightings to each of the disease progression changes to prioritise probes for that transition (Fig. 1a, 2a). Using the “Points per % change” settings, weightings can be applied as multipliers to each of the three scores independently. Consider a probe whose percentage change in expression/methylation increased by 8 % between NFT to HGSC disease transition. To prioritise identifying probes with this trend, the weighting setting for “Points per % change NFT to HGSC” is increased from the default of 1 to 4. The weightings for the “Points per % change” for the other disease transitions are not adjusted so they remain at 1. In this case, the 8 % percentage change is multiplied by 4 to give a score of 32 points for NFT to HGSC. Points for each of the three percentage changes are estimated using the weightings. If the “Direction of Progression” is left at the default of “Either” and if no “Patient Inconsistency Penalty” is applied, then the final score for a probe is simply the points for the three weighted percentage changes added together as a total. In this case, probes for NFT to HGSC will have higher scores and hence will be prioritised and ranked higher.
Fig. 1.

The Gene Sort criteria interface for mining the Gene Expression Data (a). Options are available to filter probe results to prioritise a disease stage using the “Points per % change” and a disease “Direction of Progression” as expression increasing, decreasing or either. Results can be further filtered for consistency by applying the “Patient inconsistency penalty”. The Gene Expression Data interface displaying results at the probe level (b). The ranked gene transcript probes are presented on the left as a scrollable list and results for the selected probe ADXECCRS.32699_at are displayed. The “Apply Filter” feature can be used to search the ranked list of probes. Expression values are provided for each patient at each disease stage as a data matrix. Expression trends in relation to Progression across disease stages are displayed for patients as line graphs and boxplots. The boxplot represents the median and interquartile range with the maximum and minimum values shown. The percentage change in expression between disease stages NFT to STIC and STIC to HGSC are also displayed. Above the boxplots panel, the ranking score for the selected probe is displayed as 376.798.
Next, probes can be further filtered using the “Direction of Progression” option, which can omit scores between disease stages depending on the setting. If the “Direction of Progression” is set to “Increasing only”, then decreasing scores are ignored from the probes total. A probe with a percentage decrease between stages will have those scores omitted from the total score. Similarly, if the “Direction of Progression” is set to “Decreasing only”, then a probe with an increase in percentage change between stages will have those scores omitted from the total score. In the special case of “Unchanging”, then ordering is effectively reversed and the gene/methylation probes that changed the least in relation to disease stage transition are ranked highest enabling the lowest-scoring probes to be ranked highest. This feature is useful to identify probes whose expression /methylation patterns are not associated with disease change that would be excluded as potential biomarkers.
Finally, probes can be further filtered for their consistency in trends with disease progression using the “Patient Inconsistency Penalty” option. The Patient Inconsistency Penalty is a value (not a percentage) that is deducted from the probes score for each patient, and for each of the three disease stage transitions for progression (NFT to STIC, STIC to HGSC, NFT to HGSC). This feature allows probes that show consistent increases or decreases in gene expression or methylation with disease progression to be prioritised. The penalty is only deducted if the patient's interim change in stage progression is opposite to the overall direction for an “increasing” or “decreasing” trend. Thus, the value of change from NFT to HGSC will determine whether the direction of change is considered “inconsistent” for the interim NFT to STIC and STIC to HGSC stages.
Consider an example where probes with a consistent “increasing” trend in expression/methylation with disease progression should be prioritised in the results. Following assessment for the three disease stages, Probe A has total score values of 1.8, 1.4, 3.9 and Probe B has scores of 1.8, 2.5, 3.9. In this case, Probe B expression/methylation values rise steadily with disease progression therefore it is considered to show greater evidence of association. Implementing the “Patient Inconsistency Penalty” option here would cause a value set to be deducted from Probe A’s score, thereby allowing Probe B to be prioritised in the ranking system. Note that the “Patient Inconsistency Penalty” does not apply when the “Direction of Progression” is set to “Unchanging”. Once the probes total score is calculated, the probes are sorted, ranked, and provided as a scrollable list for the user to select and evaluate. The score’s value will highly depend on the settings used for an analysis and their values are not necessarily of interest they are for comparative purposes only. Of greater importance is the relative ranking of results produced by the collated array of scores, provided in the scrollable list.
3.2. Data mining for reliable biomarkers associated with disease progression using combined expression and methylation probes score calculations
A combined score is also calculated for a gene based on both the gene expression and the methylation data. The aim of this measure is to try to identify the key genes involved in driving carcinogenesis during disease progression. The combined score is based on the total scores for all of the gene expression and methylation probes associated with a gene. Thus, the combined score measure should reflect the strength of the gene’s association with and between each stage during carcinogenesis. The combined score is estimated using the same approach as that previously described for individual probes. All probes associated with a gene are automatically identified by using their genomic location information from the descriptor files. For methylation probes, its location within a gene promoter, body or CpG Island is also noted. The scores for the identified probes are estimated individually and then added together for the combined score for a gene. Similarly, using the “Sort Criteria” a user can also specify whether to prioritise only increasing or decreasing trends associated with disease progression (Fig. 3a). Scores can also be adjusted using the “Points per % change” settings to apply weightings in order to prioritise probes for specific disease progression stages. Lastly the user can decide whether to include gene probes only, methylation probes only, or both in the calculation for the combined scoring of a gene.
Fig. 3.

The Combined Sort criteria for mining the Combined Gene Data (a). Options are available to filter probe results to prioritise a disease stage using the “Points per % change” and a disease “Direction of Progression” as expression/methylation increasing, decreasing or either. Results can be further filtered to include/exclude Gene Probes and Methylation probes based on increasing or decreasing trends in their expression/methylation. The Combined Gene Data interface displaying results for genes based on cumulative scores for all associated probes (b). The ranked genes are presented on the left as a scrollable list. The “Apply Filter” feature can be used to search the ranked list of genes. All of the associated expression and methylation probes included in the scoring for the PTPRN2 gene are displayed in the lower left panel. The “Gene Exp” line displays the expression probes, and other lines display the methylation probes listed based on their genomic location within Promoter, Gene Body and CpG Islands 1, 2 etc. The panel is interactive and each of the probes listed can be selected to examine their results. A probes score is indicated by the intensity of its shading in green or red. A green probe indicates it is positively related to disease progression, and a red one means the probe is negatively related to disease progression. By default, results for the first probe associated “Gene Exp Probe: ADXECAD.11185_at” with a probe score of 19.11234 are initially displayed. Probes listed with a zero score in parenthesis will have been excluded from the combined score based on the Sort Criteria. Expression/methylation trends in relation to Progression across disease stages are displayed for patients as line graphs. In the upper right panel, an interactive methylation map is provided for the region. This map illustrates the gene’s promotor initially followed by 2,000 genomic positions arranged as 40 consecutive rows of 50 positions each and the methylated positions are shaded green. The percentage change in methylation between disease stages NFT to STIC and STIC to HGSC are also displayed. Specific probes can be explored further individually using the Gene Expression Data and Methylation Data screens. The “UCSC” button can be selected to visualise the genomic region for the CpG probe of interest utilising the online UCSC Genome browser. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.3. Data mining for methylation clusters associated with disease progression to identify target regions for assay design
Methylation clusters are identified and ranked using a scoring calculation that sums the total change values for all of the methylation probes that occur within the target region. Using the “Sort Criteria”, the user can specify the particular trend of interest including “Direction of Progression” (increasing, decreasing, either), disease Progression stage (i.e. NFT to STIC, STIC to HGSC, or NFT to HGSC) and a “Minimum change threshold (%)” (Fig. 4a). Once the selection is made, only the relevant probes are included in the scoring calculation using their total change values. Target regions can be further restricted to occur within promoter, gene bodies or CpG islands, or alternatively outside of these regions. To facilitate assay development, target regions can be specified to be 250, 200, 150 or 100 bp in “Cluster length”. Threshold cut-off values for a probe to be considered as “non-methylated” or “methylated” can also be specified. The current default values for these are <0.1 and >0.2, respectively, however the methylated threshold could be raised to 0.6 or higher to provide greater confidence (see 39). An option to deduct points for non-conforming sites is also available. This feature imposes a penalty for probes inconsistent with the specified trend. The scoring calculation and ranking of methylation clusters by default is based on the summed total methylation values. An alternative ranking method is available that is based on scoring Methylation Clusters using the total number of probes within a target region. This method can be selected using the “Look only at Methylated/Non-Methylated” option in the “Sort Criteria”.
Fig. 4.

The Sort Criteria for mining for Methylation clusters (a). Options are available to filter probe results to prioritise particular disease “Progression Stages”, the “Direction of Progression” as the “Minimum change threshold (%)” required. To facilitate assay development, methylation target regions can be specified to a particular “Cluster Length” between 100 and 250 bp. Methylation probes included can be restricted to Promoter, Gene Body, CpG Island regions or none of these. Upper and lower thresholds for considering a probe as Non-methylated/Methylated can be specified. The scoring calculation can include all probes or only those that are Methylated/Non-methylated. Probes from certain patients can be included or excluded from the scoring calculation. The Methylation clusters interface displaying results for methylation probes “From cg16265906 to cg23180938” (b). The ranked Methylation clusters and their genomic locations are presented on the left as a scrollable list. The “Apply Filter” feature can be used to search the ranked list. The “Summary of methylation progression” displays as a line, a probes methylation status across each of the patients. The methylation status of a probe during disease progression is indicated with shading that is explained in the “Methylation Colour Guide” legend. Overall mean values for the methylation cluster for disease stages for each patient are provided in the table. All probes associated with a Methylation cluster are displayed in the lower right panel and the intensity of the green shading is reflective of a probes’ score and how positively its trends in methylation related to disease progression. The panel is interactive, and a probe can be selected to examine its results in detail. After probe selection, results in the “Methylation Clusters” interface are updated with probe specific results (c). The “UCSC” button can be selected to visualise the genomic region for the CpG probe of interest utilising the online UCSC Genome browser. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.4. Example of performance of OCDB pipeline: identification and validation of potential biomarker using pyrosequencing and RqPCR assays
To evaluate the performance of the OCDB, and the associated biomarker discovery pipeline, a CpG site, and its associated gene, were identified from the OCDB using the methodology described above. To confirm trends for the marker, expression values for the transcript and methylation values for the CpG site, for each patient sample and disease progression stage were plotted as line graphs. Mean values (and interquartile range) for all patients for each disease stage were also plotted as boxplots. The specific genomic sequence and gene of interest identified using the OCDB was further evaluated using bioassays. These were performed on FFPE tissue samples from the six patient cohort in the OCDB as well as on a larger validation cohort (N = 100), which consisted of 50 cases with advanced HGSC and 50 unmatched controls with no current or previous history of cancer. The validation cohort was also provided by the Northern Ireland Biobank (Ethical approval: NIB11:005, NIB13:0094).
Expression patterns of the gene of interest were validated using RqPCR as previously described [18]. A RealTime® Ready Custom assay (Roche, UK) was designed for the gene of interest and performed on a LightCycler 96 platform (Roche, UK) according to manufacturers’ guidelines. Pre-amplified cDNA was prepared following RNA extraction and cDNA synthesis of normal FT, STIC, and HGSC samples. The RqPCR assay assessed the gene of interest and two controls in duplicate using 200 ng cDNA as input. Relative gene expression was calculated from the mean RqPCR cycle threshold data using the δδCt method. NFT was used as the calibrator in results calculations. Statistical analysis of gene expression between disease stages was performed using GraphPad Prism version 5 software (La Jolla, California, USA).
Methylation patterns of the CpG sites of interest were validated using pyrosequencing. Initially, the relevant DNA sequence containing the CpG site was visualised using the UCSC feature in the OCDB. The genomic region was further examined using the Integrative Genomics Viewer (IGV®, Broad Institute, Massachusetts, USA) [[40], [41]]. Next, site-specific primers were designed for the identified target sequence using the Pyromark Assay Design Software 2.0 (Qiagen UK, Manchester, UK). All FFPE samples underwent DNA preparation, quantification and bisulphite conversion using standard approaches. The bisulphite converted DNA samples were first amplified using a standard PCR reaction, and prior to proceeding, the products were electrophoresed through a 1 % agarose gel to confirm successful amplification. Following confirmation, the pyrosequencing assay was carried out using a Pyromark Q-24 Instrument (Qiagen UK, Manchester, UK) according to manufacturer’s guidelines. The mean percentage methylation across the target sites for each tissue type was calculated and analysed. As with the validation of gene expression data, NFT was used as the calibrator in results calculations. Statistical analysis of DNA methylation between disease stages was performed using GraphPad Prism version 5 software (La Jolla, California, USA).
4. Results
4.1. Overall OCDB design and analytical modes
The OCDB has four modes for data mining: 1) Gene Expression Data; 2) Methylation Data; 3) Combined Gene Data; and 4) Methylation Clusters. Biomarkers can be mined from gene expression data alone and methylation data alone in modes 1 and 2, respectively, or using the combination of both data together in mode 3. Mode 4 focusses on identifying clusters of methylated CpG sites in genes to assist with bioassay design. The “View Data” interface is for displaying results, whereas the “Sort Criteria” is for specifying the desired trends in disease progression using the settings, weighting and penalties. The radio buttons at the top of the screen allow the user to toggle between the four modes and also between the “View Data” and “Change Sort Criteria” interfaces (Fig. 1a). Each mode has its own “Sort Criteria” interface with particular options. After a mode is selected, the results output are calculated according to the “Sort Criteria” settings page and ranked with the highest score first. Results can be explored in more detail by selecting them from the ranked list, at which point the OCDB updates to display more detail for the selected entry.
4.2. Mining gene transcript expression and methylation data for probes associated with disease progression
Fig. 1b illustrates the “Gene Expression Data” interactive screen displaying results at the probe level obtained using default settings in the “Sort Criteria”. The ranked gene transcript probes are presented on the left-hand side as a scrollable list. The “Apply Filter” feature can be used to search the list of probes. The panels display information for the selected probe, ADXECCRS.32699_at. Trends in expression of the gene transcript probe during disease progression are visualised for each patient as a line graph in the upper left panel. The expression of ADXECCRS.32699_at decreases during disease progression for five patients. For patient six, expression increases slightly between STIC and HGSC disease stages, as evidenced by the orange line graph. The raw gene expression values for ADXECCRS.32699_at at each disease stage for each patient are presented in the table below. Boxplots representing the median expression across all patients for each disease stage are displayed in the upper right panel. The interquartile range with the maximum and minimum values are also shown. ADXECCRS.32699_at had median expression values of 9 to ∼ 4 to ∼ 2 for NFT, STIC and HGSC, respectively. Thus, expression of the ADXECCRS.32699_at probe appears to decrease with disease progression. Above the boxplots panel, the ranking score for the selected probe is displayed as 376.798. If the user selects another result in the scrollable list on the left-hand side, then they will notice that this ranking score increases or decreases depending on the probe being higher or lower in the list. The increase/decrease percentage change in expression between disease stages NFT to STIC and STIC to HGSC is displayed in the lower right panel. For NFT to STIC and STIC to HGSC disease stages transition there is a median change in expression of ∼ -2% and ∼ -4%, respectively for ADXECCRS.32699_at.
Fig. 2b illustrates the “Methylation Data” interface displaying results at the probe level obtained using default settings in the “Sort Criteria”. The ranked methylation probes are presented on the left as a scrollable list. The “Apply Filter” feature can be used to search the ranked list of probes. The panels display information for the selected probe, which in this example is cg06825142. The trends in methylation of a probe during disease progression are visualised for each patient as a line graph in the upper left panel.
Fig. 2.

The Meth Sort criteria interface for mining the Methylation Data (a). Options are available to filter probe results to prioritise a disease stage using the “Points per % change” and a disease “Direction of Progression” as methylation increasing, decreasing or either. Results can be further filtered for consistency by applying the “Patient inconsistency penalty”. The Methylation Data interface displaying results at the probe level (b). The ranked methylation probes are presented on the left as a scrollable list and results for the probe cg06825142 are displayed. The “Apply Filter” feature can be used to search the ranked list of probes. Methylation values are provided for each patient at each disease stage as a data matrix. Methylation trends in relation to Progression across disease stages are displayed for patients as line graphs and boxplots. The boxplot represents the median and interquartile range with the maximum and minimum values shown. The percentage change in methylation between disease stages NFT to STIC and STIC to HGSC are also displayed. Above the boxplots panel, the ranking score for the selected probe is displayed as 1722.603.
Patients display differing trends in methylation of cg06825142 with disease progression. Methylation seems to increase with disease progression for patients 1, 3 and 5 only. Patients 2 and 4 show no association of methylation with disease progression, while methylation is highest for STIC for patient 6. The raw methylation values are presented for each patient at each disease stage in the table below. Boxplots representing the median methylation values for all patients at each disease stage are displayed in the upper right panel. The interquartile range with the maximum and minimum values are also shown. Above the boxplots panel, the ranking score for the selected probe is displayed as 1722.603. Similarly, if the user selects another result in the scrollable list on the left-hand side, then they will notice that this ranking score increases or decreases depending on the probe being higher or lower in the list. Probe cg06825142 is relatively unmethylated for NFT for all patients and then increases to a median methylation value of ∼ 0.25 for both STIC and HGSC disease stages. The increase/decrease percentage change in methylation between disease stages NFT to STIC and STIC to HGSC is displayed in the lower left panel. For NFT to STIC and STIC to HGSC transition in disease stages there is a median change in methylation of ∼ 0.2 % and ∼ 0.1 %, respectively, for probe cg06825142.
For both “Gene Expression Data” and “Methylation Data” modes, the user can alter the “Sort criteria” to prioritise results for probes that exhibit specific trends in disease progression (Fig. 1a, 2a). Altering the “Points per % change” (0–100) from the default of 1 to a higher number for one of the different disease stage transitions of NFT to STIC, STIC to HGSC and NFT to HGSC is useful for prioritising probes that show the greatest change depending on that disease transition stage. The “Patient inconsistency penalty” can be increased or decreased. This feature is useful to exclude any outlier patients that have expression/methylation patterns that behave differently to the rest of the cohort. This feature will be particularly useful when larger patient cohorts are introduced to the platform. The direction of the disease progression can also be set to increasing, decreasing or either. This is useful to identify probes most associated with a particular disease progression trend.
4.3. Mining genes associated with disease progression using multiomics
Fig. 3b illustrates the “Combined Gene Data” interactive screen displaying results for genes based on cumulative scores for all their associated probes. Results were obtained using default settings in the Sort Criteria. The ranked genes are presented on the left as a scrollable list. The “Apply Filter” feature can be used to search the ranked list of genes. The gene PTPRN2 is selected and the 30,285 indicates the combined data score for this gene. In the lower left panel, all of gene’s associated probes are displayed as well as its Ensembl identifier and chromosome location. Each of these probes listed were included in the combined scoring of PTPRN2. Probes comprised of seven transcript expression probes and 17 + methylation probes located within promoter [4], gene body (8 + ), CpG Island 1 [1] and Island 2 [4] regions. Three methylation probes had green shading indicating a strong association with the desired trend in disease progression. All the transcript expression probes were shaded grey indicating no association. No probes were shaded red, which would have indicated that they were negatively related to the specified trend in disease progression. The lower left panel is interactive and each of the probes can be selected to examine their result summaries. Probes shaded green would be worthy of further investigation within this mode and also individually using the “Gene Expression Data” and “Methylation Data” modes.
Results for the first probe listed are displayed by default initially in the other three panels. In this case, results for “Gene Exp Probe: ADXECAD.11185_at” with a probe score of 19.11234 are displayed. Expression of ADXECAD.11185_at seems to decrease with disease progression for patients 2, 3, 4 and 5, while expression remains equivalent between NFT and HGSC stages for patients 1 and 6 as seen in the upper left panel. The median change in expression from NFT to STIC and STIC to HGSC disease stages transition for probe ADXECAD.11185_at is −0.5 % and 0.1 %, respectively.
In the top right panel, a Methylation map displays the first 2,000 genomic positions (from left to right) of a region of the PTPRN2 gene with the location of overlapping methylation probes identified. By default the Methylation map displays the promoter region initially, followed by 40 rows of 50 positions, with each row immediately following the row above. The methylation map is useful to identify clusters of methylation probes by eye. Those shaded green would be indicative of a positive association with the increasing or decreasing trend with disease progression trend as specified in the “Sort Criteria”. The Methylation map panel is interactive and the other regions for Gene Body, CpG Island1 or Island2 can be selected to jump to their genomic location to view overlapping methylation probes. This map provides a guide for the level of methylation of a gene. For a more in-depth analysis, the “UCSC” button can be selected to visualise the genomic region of interest online utilising the UCSC Genome browser (https://genome.ucsc.edu/). This call-out feature requires the PC to have internet connectivity.
4.4. Data mining for methylation clusters associated with disease progression to identify target regions for assay design
Fig. 4b illustrates the “Methylation Clusters” interactive screen displaying results for methylation probes “From cg16265906 to cg23180938”. The ranked Methylation clusters and their genomic locations are presented on the left as a scrollable list. The “Apply Filter” feature can be used to search the ranked list. Overall mean values for the methylation cluster for disease stages for each patient are provided in the table in the lower left panel. The “Summary of methylation progression” displays as a line, a probes methylation status summary for each of the patients. In this example, the five lines represent the five probes associated with this methylation cluster. The summary status of a probe during disease progression is indicated with shading as explained by the “Methylation Colour Guide” legend. Probes have nine possible outcomes based on a disease progression transition from the normal state NFT, which may be methylated or de-methylated. Methylation status summaries for a probe include: 1) Methylated at STIC plus; 2) Methylated at HGSC-STIC non; 3) Methylated at HGSC-STIC partial; 4) Methylated temporarily at STIC; 5) De-Methylated at STIC plus; 6) De-Methylated at HGSC-STIC meth; 7) De-Methylated at HGSCSTIC partial; 8) De-Methylated temporarily at STIC; 9) None of the above. Table 1 provides further explanation of the methylation summary status for a probe during disease progression. Determination relied on thresholds to define a probe as methylated or non-methylated in the “Sort Criteria”. If a probe’s methylation value fell between the two thresholds it was deemed to be a partial methylated probe.
Table 1.
The methylation status summary of a cluster during disease progression is summarised using the “Methylation Colour Guide” legend. Classification is based on the clusters probes methylation status compared to the NFT disease stage.
| Methylated at NFT | Methylated at STIC | Methylated at HGSC | Summary for disease progression |
|---|---|---|---|
| N | Y | Y | Methylated at STIC plus |
| N | N | Y | Methylated at HGSC-STIC non |
| N | Partial | Y | Methylated at HGSC-STIC partial |
| N | Y | N | Methylated temporarily at STIC |
| Y | N | N | De-Methylated at STIC plus |
| Y | Y | N | De-Methylated at HGSC-STIC |
| Y | Partial | N | De-Methylated at HGSC-STIC partial |
| Y | N | Y | De-Methylated temp at STIC |
| Any other combination | None of the above |
All probes associated with a Methylation cluster are displayed in the lower right panel. Probes shaded green are positively associated with the desired methylation trend in disease progression. This panel is interactive and a probe can be selected to examine its results in more detail. After selection for probe cg02792792, results in the “Methylation Clusters” interface panels are updated (Fig. 4c). The summary of methylation progression for the probe is now highlighted with an arrow. The table now displays the methylation values for probe cg02792792 for each patient at each disease stage. A summary of the probes methylation status as methylated or de-methylated at each of the disease stages is provided as shading in both the table and the upper right hand panel. Green shading represents a methylated probe status, while red shading represents de-methylated status. Probes shaded grey were deemed neither methylated nor de-methylated because their methylation values were between the threshold cut-offs.
4.5. Example of performance of the OCDB pipeline to identify OSR2
The OCDB was utilised to identify probes that exhibited decreasing gene expression and increasing methylation with disease progression. Sort Criteria were specified accordingly to prioritise probes displaying underexpression and hypermethylation from NFT through to HGSC. Amongst the top ranked genes identified for the specified trend was OSR2 (Odd-Skipped Related Transcription Factor 2; ENSG00000164920).
Examination of the microarray data confirmed that gene expression of the OSR2 transcript (ADXEC7060C1_s_at) decreases with disease progression from NFT to HGSC consistently for each patient (Fig. 5a). Overall mean values at each disease stage also decreased, and the variation in expression values for STIC and HGSC was relatively high compared to NFT (Fig. 5b). Examination of the methylation array data for the CpG site associated with OSR2 (cg08202494) confirmed that methylation increased with disease progression consistently for each patient (Fig. 5c). Overall, mean methylation values for each disease stage also increased (Fig. 5d).
Fig. 5.

Validation of the OCDB results for the OSR2 gene expression and methylation array data. The trend of OSR2 repression with tubo-ovarian cancer disease progression for the associated probe ADXEC7060C1_s_at. Each line represents one patient from the cohort and there is an obvious downward trend from NFT to HGSC (a,c). Conversely, the trend towards global hypermethylation of the OSR2 associated CpG site with disease progression is equally obvious (b,d). The lack of overlap between confidence intervals of HGSC and NFT in both boxplots is indicative of statistical significance.
The RqPCR bioassay of OSR2 expression in the six patient cases included in the OCDB confirmed repression of OSR2 from NFT to HGSC (P-value = 0.0008; Fig. 6a). The DNA methylation of OSR2 of the six patient cases included in the OCDB was assessed using a pyrosequencing assay that examined five ‘target’ CG dinucleotides across the genomic region (∼50 bp; Fig. 6b). The mean percentage methylation across the target sites for each tissue type was calculated and analysed. The first three target dinucleotides (positions 66 – 68) were consistently reliable across all samples compared to the fifth target site (position 70) which was more variable. Results indicated that OSR2 was progressively hypermethylated between NFT and HGSC (P-value < 0.0001; Fig. 6c). STIC samples were statistically significant compared to both NFT and HGSC, suggesting this intermediary disease stage may have potential for biomarker testing of early-stage disease.
Fig. 6.

RqPCR confirmed repression of OSR2 across the carcinogenic pathway (a) and the hypermethylation of its associated CpG site was confirmed using pyrosequencing (c). A sample pyrogram from an OSR2 methylation assay in normal FT sample (b). Sequence analysed is along the bottom of figure. The blue bars represent the target CpG dinucelotides and the figures above these represent the percentage methylation at that position. A figure highlighted in red indicates a result that needs review and may constitute a failed reading. In the initial validation within the “study cohort” all values were included, and a mean methylation calculated for each sample. However, within the larger validation sample set combinations of the methylation position scores were assessed to confirm which was most discriminatory. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The consistency of OSR2 hypermethylation in HGSC was further validated in a larger sample set comprising 50 cases of HGSC and 50 unmatched cases with no current or previous history of cancer. The pyrosequencing assay was performed as previously outlined. Prior to bisulphite conversion, two samples from each cohort were excluded due to having extremely low DNA concentrations. Prior to sequencing, three samples from the normal FT group and four samples from the HGSC group were excluded because of inadequate bisulphite conversion and/or failed PCR. Forty-five normal FT and forty-four HGSC samples were included in the final comparison. Mean methylation score from each sample was calculated and showed accurate discrimination between normal FT (31.49 %) and HGSC tissue (69.38 %; Table 2). As with the six patient cohort results, the fourth and fifth target sites were unreliable. Hence, the mean methylation of target dinucleotides 66, 67, and 68 (Fig. 7A) was calculated for each sample, and compared to the total score and again for dinucleotide 66 alone (Fig. 7B). It is evident that results of the pyrosequencing assay of OSR2 methylation focussing on sites 66–68 is the most precise (P-value < 0.0001) and also has the narrowest range (Table 2).
Table 2.
Comparison of mean methylation scores (%) of all, 66–68, or 66 only positions of the OSR2 assay in Normal FT versus HGSC samples of the larger validation cohort.
| Target CpG Dinucleotide | NORMAL FT (n = 45) |
HGSC (n = 44) |
||||||
|---|---|---|---|---|---|---|---|---|
| Mean | SEM (+/-) | Range | CV (%) | Mean | SEM (+/-) | Range | CV (%) | |
| All | 31.49 | 0.7374 | 23.2 – 48.4 | 15.71 | 69.38 | 2.605 | 26.2 – 90.8 | 24.9 |
| 66 – 68 | 25.09 | 0.7351 | 13.33–38 | 19.65 | 67.63 | 3.137 | 18.67 – 95.67 | 30.77 |
| 66 Only | 22.73 | 1.105 | 12 – 38 | 32.61 | 69.20 | 3.730 | 10 – 100 | 35.75 |
Fig. 7.
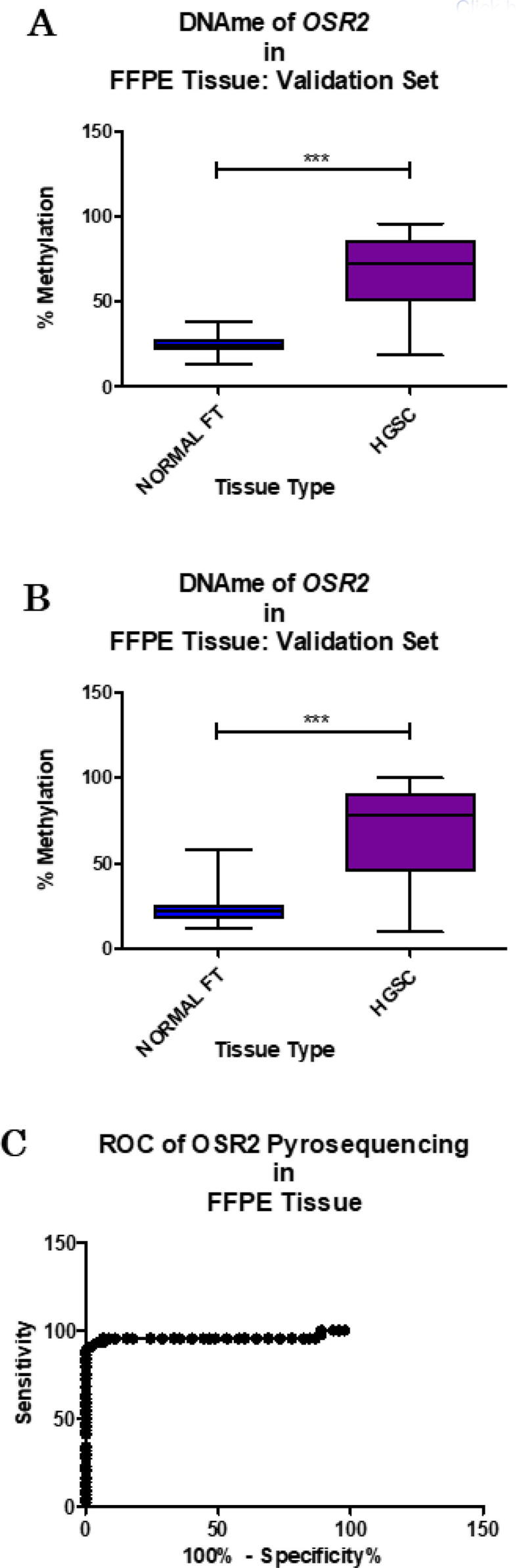
Further validation in a larger validation cohort, comprising 50 cases of HGSC and 50 unmatched cases with no current or previous history of cancer. Results of the pyrosequencing assay for OSR2 can consistently discriminate between normal FT and HGSC tissue across the first three methylated positions (A; P-value < 0.0001, t-test) and also with only the first methylated position (B; P-value < 0.0001, t-test). ROC analysis shows an AUC of 0.9573 (C; P-value < 0.0001).
A receiver operator characteristic (ROC) analysis of OSR2 methylation (dinucelotides 66 – 68) and normal FT was performed. It returned an area under the curve (AUC) of 0.9573 (P-value < 0.0001) (Fig. 7c). The sensitivity and specificity of the OSR2 methylation assay was calculated on an upper limit diagnostic threshold (Diagnostic Threshold = Mean Methylation of Normal Cohort + (2 × Standard Deviations of Mean). This equates to a diagnostic threshold of 34.952 %. At a clinically relevant specificity (95 %), sensitivity was 93.18 % at a diagnostic threshold of 36.33 % (see Table 3). Serum CA125 results were available for all 44 HGSC cases but only 43 % of the normal cases. Serum CA125 level below 35 U/ml is deemed “normal” in clinical practice. A review of pathologically “normal” cases with borderline/high (>35 U/ml) CA125 (false positives) showed all cases were OSR2 methylation assay “negative” (i.e. < 36.33 %). This indicates the potential of OSR2 assay at reducing type I and II error within clinical diagnostics.
Table 3.
ROC analysis of the OSR2 methylation biomarker (AUC 0.9573, P < 0.0001) for reported sensitivity and 629 specificity of methylation score thresholds ∼ 35 %.
| Methylation Score (%) | Sensitivity | Specificity |
|---|---|---|
| >34.33 | 93.18 % | 93.33 % |
| >36.33 | 93.18 % | 95.56 % |
| >37.67 | 90.91 % | 97.78 % |
| >38.5 | 88.64 % | 100 % |
5. Discussion
Multiomics approaches can provide greater insight into the underlying biology of malignant processes.
Analyses can identify disease-specific oncogenes and the key pathways involved in disease progression. Aberrant genomic sequences can also be identified as potential druggable targets and/or biomarkers of therapeutic response, minimal residual disease, or even early-warning diagnosis. However, there is a need for bespoke tools to interrogate data to gain valuable knowledge for translational research, especially for diseases with poor prognosis, such as HGSC. In this study, a novel genomic discovery pipeline for biomarkers is presented. The pipeline was facilitated by the collection of a unique dataset for an aggressive cancer, HGSC, as well as the development of the OCDB a bespoke multiomics analytical tool, and the optimisation of validation bioassays. Results are providing a better understanding of the carcinogenic pathway of HGSC and the underlying mechanisms that are the hallmarks of this disease.
This is the first study to gather matched genomic and epigenomic profiles of patients over the course of HGSC disease progression. The samples, spanning from normal FT to HGSC, were identified with highly accurate pathological annotation. Whilst the study cohort is small, the gene expression and DNA methylation datasets are unique and allowed for the development of the OCDB. The OCDB can quickly interrogate multiomics datasets to identify HGSC-specific gene targets associated with disease progression. The graphical user interface is easy to navigate and has rapid query response time due to the file format of the data as fast access. Data mining can be carried out using four analytical modes: 1) Gene Expression Data; 2) Methylation Data; 3) Combined Gene Data; and 4) Methylation Clusters. These provide alternative options for probe identification correlated to progression through the three disease stages. Single omics with the gene expression and the methylation data can be analysed independently or using multiomics with both data types together in the combined data mode. Results from the Gene Expression Data mode lists the genes whose expression correlate (positively/negatively) with progression from NFT to STIC to HGS. The Methylation Data mode is extremely similar, except that it lists methylation points, and changes in methylation in place of genes and gene expression. The Combined Gene Data mode is more complex. For each gene, it takes the gene expression data changes, together with methylation changes for methylation points within the gene to give a combined correlation against progression through disease stages. The details of this and the weightings applied can be adjusted by the user. The combined mode has the potential to suggest genes which may be relevant to the disease mechanisms that would perhaps not be identified otherwise using gene expression or methylation data alone. The Methylation Clusters mode should identify sets of CpG methylation sites that were correlated to disease progression and were located close enough together in a genomic region to be suitable as the basis for a biomarker assay. This could be used for patient diagnosis and stratification to guide their treatment. The methylation cluster mode is therefore used to identify a set of candidate marker regions, and then the other analytical modes can be used for further investigations of those regions for example.
The OCDB implements a scoring system that was developed to identify and rank probes and or genes that show consistency with the specified trends. The scoring system involves summing either for or across probes and disease stages and ranking results. The combined score is based on the percentage change in expression /methylation between disease stages. The combined score is therefore scaled to the gene’s own expression or methylation. Thus, the strength of the expression or methylation would not influence the results, only the difference in change. Moreover, results for genes are not biased by some sort of filter for minimum thresholds such as those applied in differential expression analyses, for example. For the combined score, all the transcript expression and methylation probes associated are considered and summed. Whilst the scoring system is cumulative it isn’t necessarily biased towards ranking genes or methylation clusters with a greater number of probes higher. This is because scores can be positive or negative depending on whether they are consistent with the desired trend. So, if a gene has many probes but some are not related to HGSC cancer progression, the average change of those should be zero. Thus, a gene with more associated probes would not have an “advantage” in the ranking if probes vary in their trends. However, probes and or genes with a greater number of probes showing consistent trends would have greater evidence and hence be prioritised. The scoring system of the OCDB is relatively simple. Despite its simplicity, many considerations have been given to the criteria available for filtering results using weighting and penalty options. The score values for probes, genes or methylation clusters will depend on the user’s settings and are only of interest to compare and rank results from individual analyses. The ranked results obtained by the OCDB are very specific to the desired trends defined in the “Sort Criteria” options. The user can prioritise results from early disease transition stages using the options, providing an ability to identify biomarkers for early intervention. To our knowledge, other data browsers or softwares do not exist that analyse cancer disease progression multiomics datasets for biomarker identification or employ similar scoring systems for analysis.
The genomic discovery pipeline employing the OCDB is particularly suited to identifying two trends associated with disease progression: (a) unique hypermethylated CpG sites and (b) genes transcriptionally silenced by DNA hypermethylation. Unique CpGs, if HGSC-specific, carry distinct potential as future biomarkers, whereas characterising the role of transcriptionally silenced genes in HGSC may identify new disease-specific drug targets. One example of this, presented in this paper, is OSR2, which is a mammalian homolog of the Drosophila odd-skipped family of transcription factors. Until now, OSR2 has not been identified as having an association with gynaecological malignancy. The OCDB identified OSR2 as becoming repressed and hypermethylated with disease progression to HGSC. Trends were confirmed in the browser samples and a larger patient cohort using RqPCR and pyrosequencing bioassays. Interestingly, results of the OSR2 methylation assay in the larger patient cohort revealed its potential as an HGSC specific biomarker. The assay could detect HGSC with high accuracy, sensitivity and specificity, indicating its utility for clinical diagnostics.
The future development for the OCDB is to revise and further update this version with more data. The newer version would be populated with more advanced multiomics data for a larger cohort of patients, with the same uniquely matched tumour samples. This would require some development and re-design of the OCDB framework and the graphical user interface (e.g. scrollbar, colour palette) to enable the import of new data sets including patient labels. Currently, the OCDB browser requires file input as a data matrix together with annotation and descriptor files all as CSV file format. Therefore, data from other sources that could also be provided in this format could in principle also be incorporated into the browser framework (e.g. GeneChip™ Human Transcriptome Array, Infinium Methylation EPIC array). These updates should allow a secondary layer of validation of novel transcripts identified from the initial six patient cohort and, consequently, more focussed discovery of disease-specific targets or biomarkers. Once identified, targets can be progressed into the laboratory validation stage of the pipeline. The identification of a more refined and focussed transcript will increase the likelihood of successfully translating such a discovery into a useful research or clinical tool; whether it be new knowledge on a particular biological pathway or identification of a transcript suitable to act as a biomarker of therapeutic response. Additional developments of the analytics of the OCDB could also be implemented. At present the combined method for a gene considers multiple transcripts. Depending on disease progression, it may be that different gene transcripts differ in their roles in carcinogenesis during disease progression. An alternative scoring system could be considered to assess transcripts individually. This may not be easily implemented given that transcripts can comprise of all or just a portion of gene exons. Nevertheless, a combined scoring system that appraises transcripts individually, considering their exonic structure with overlapping methylation probes, could provide superior results for revealing the underlying biology of carcinogenesis of HGSC at the transcript level. Results for transcripts could also provide more accurate targets for therapeutic interventions.
Declaration of Competing Interest
1. JPB, PBM, and LF are majority shareholders in GenoME Diagnostics Limited.
2. DMcA is a majority shareholder in Sonrai Analytics.
3. No author has received royalties, payments or personal funding from any aspect of this work.
Acknowledgments
Acknowledgements
The authors would like to acknowledge the support of the Northern Ireland Biobank throughout this project.
Authors' contributions
Conceptualization, James Beirne, W McCluggage, Ian Harley and Paul Mullan; Data curation, James Beirne and Alan Gilmore; Formal analysis, Aideen Roddy, Muhammed Alvi and Darragh McArt; Funding acquisition, James Beirne, Darragh McArt and Paul Mullan; Investigation, James Beirne, Aideen Roddy, W McCluggage, Ian Harley, Muhammed Alvi and Paul Mullan; Methodology, James Beirne, Alan Gilmore, Caitríona McInerney, Aideen Roddy, W McCluggage, Muhammed Alvi, Darragh McArt and Paul Mullan; Project administration, James Beirne, Caitríona McInerney, Darragh McArt and Paul Mullan; Resources, Ian Harley and Kevin Prise; Software, Alan Gilmore and Darragh McArt; Supervision, Kevin Prise, Darragh McArt and Paul Mullan; Validation, James Beirne; Visualization, Alan Gilmore; Writing – original draft, James Beirne, Alan Gilmore and Caitríona McInerney; Writing – review & editing, James Beirne, Alan Gilmore, Caitríona McInerney, Aideen Roddy, W McCluggage, Ian Harley, Kevin Prise, Darragh McArt and Paul Mullan.
Ethics approval and consent to participate
Ethical approval for the work presented in this manuscript was covered under the Northern Ireland Biobank scientific peer review (Ethical approval: NIB11:005, NIB13:0094).
References
- 1.Vang R., Shih Ie M., Kurman R.J. Ovarian low-grade and high-grade serous carcinoma: pathogenesis, clinicopathologic and molecular biologic features, and diagnostic problems. Adv Anat Pathol. 2009;16(5):267–282. doi: 10.1097/PAP.0b013e3181b4fffa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crum C.P., McKeon F.D., Xian W. The oviduct and ovarian cancer: causality, clinical implications, and “targeted prevention“. Clin Obstet Gynecol. 2012;55(1):24–35. doi: 10.1097/GRF.0b013e31824b1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beirne J.P., Irwin G.W., McIntosh S.A., Harley I.J.G., Harkin D.P. The molecular and genetic basis of inherited cancer risk in gynaecology. Obstet Gynaecol. 2015;17(4):233–241. [Google Scholar]
- 4.Gourley C., Michie C.O., Roxburgh P., Yap T.A., Harden S., Paul J., et al. Increased incidence of visceral metastases in scottish patients with BRCA1/2-defective ovarian cancer: an extension of the ovarian BRCAness phenotype. J Clin Oncol. 2010;28(15):2505–2511. doi: 10.1200/JCO.2009.25.1082. [DOI] [PubMed] [Google Scholar]
- 5.Weberpals J.I., Clark-Knowles K.V., Vanderhyden B.C. Sporadic epithelial ovarian cancer: clinical relevance of BRCA1 inhibition in the DNA damage and repair pathway. J Clin Oncol. 2008;26(19):3259–3267. doi: 10.1200/JCO.2007.11.3902. [DOI] [PubMed] [Google Scholar]
- 6.Bowtell D.D. The genesis and evolution of high-grade serous ovarian cancer. Nat Rev Cancer. 2010;10(11):803–808. doi: 10.1038/nrc2946. [DOI] [PubMed] [Google Scholar]
- 7.Fathalla M.F. Incessant ovulation–a factor in ovarian neoplasia? Lancet. 1971;2(7716):163. doi: 10.1016/s0140-6736(71)92335-x. [DOI] [PubMed] [Google Scholar]
- 8.Casagrande J.T., Louie E.W., Pike M.C., Roy S., Ross R.K., Henderson B.E. “Incessant ovulation” and ovarian cancer. Lancet. 1979;2(8135):170–173. doi: 10.1016/s0140-6736(79)91435-1. [DOI] [PubMed] [Google Scholar]
- 9.Whittemore A.S. Personal characteristics relating to risk of invasive epithelial ovarian cancer in older women in the United States. Cancer. 1993;71(2 Suppl):558–565. doi: 10.1002/cncr.2820710211. [DOI] [PubMed] [Google Scholar]
- 10.Whittemore A.S., Harris R., Itnyre J. Characteristics relating to ovarian cancer risk: collaborative analysis of 12 US case-control studies. IV. The pathogenesis of epithelial ovarian cancer. Collaborative Ovarian Cancer Group. Am J Epidemiol. 1992;136(10):1212–1220. doi: 10.1093/oxfordjournals.aje.a116429. [DOI] [PubMed] [Google Scholar]
- 11.Rosenblatt K.A., Thomas D.B. Lactation and the risk of epithelial ovarian cancer. The WHO Collaborative Study of Neoplasia and Steroid Contraceptives. Int J Epidemiol. 1993;22(2):192–197. doi: 10.1093/ije/22.2.192. [DOI] [PubMed] [Google Scholar]
- 12.Adami H.O., Hsieh C.C., Lambe M., Trichopoulos D., Leon D., Persson I., et al. Parity, age at first childbirth, and risk of ovarian cancer. Lancet. 1994;344(8932):1250–1254. doi: 10.1016/s0140-6736(94)90749-8. [DOI] [PubMed] [Google Scholar]
- 13.Risch H.A., Marrett L.D., Howe G.R. Parity, contraception, infertility, and the risk of epithelial ovarian cancer. Am J Epidemiol. 1994;140(7):585–597. doi: 10.1093/oxfordjournals.aje.a117296. [DOI] [PubMed] [Google Scholar]
- 14.Singh N., McCluggage W.G., Gilks C.B. High-grade serous carcinoma of tubo-ovarian origin: recent developments. Histopathology. 2017;71(3):339–356. doi: 10.1111/his.13248. [DOI] [PubMed] [Google Scholar]
- 15.Gilks C.B., Irving J., Kobel M., Lee C., Singh N., Wilkinson N., et al. Incidental nonuterine high-grade serous carcinomas arise in the fallopian tube in most cases: further evidence for the tubal origin of highgrade serous carcinomas. Am J Surg Pathol. 2015;39(3):357–364. doi: 10.1097/PAS.0000000000000353. [DOI] [PubMed] [Google Scholar]
- 16.Morrison J.C., Blanco L.Z., Jr., Vang R., Ronnett B.M. Incidental serous tubal intraepithelial carcinoma and early invasive serous carcinoma in the nonprophylactic setting: analysis of a case series. Am J Surg Pathol. 2015;39(4):442–453. doi: 10.1097/PAS.0000000000000352. [DOI] [PubMed] [Google Scholar]
- 17.Kuhn E., Kurman R.J., Vang R., Sehdev A.S., Han G., Soslow R., et al. TP53 mutations in serous tubal intraepithelial carcinoma and concurrent pelvic high-grade serous carcinoma–evidence supporting the clonal relationship of the two lesions. J Pathol. 2012;226(3):421–426. doi: 10.1002/path.3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beirne J.P., McArt D.G., Roddy A., McDermott C., Ferris J., Buckley N.E., et al. Defining the molecular evolution of extrauterine high grade serous carcinoma. Gynecol Oncol. 2019;155(2):305–317. doi: 10.1016/j.ygyno.2019.08.029. [DOI] [PubMed] [Google Scholar]
- 19.Buys S.S., Partridge E., Black A., Johnson C.C., Lamerato L., Isaacs C., et al. Effect of screening on ovarian cancer mortality: the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Randomized Controlled Trial. JAMA. 2011;305(22):2295–2303. doi: 10.1001/jama.2011.766. [DOI] [PubMed] [Google Scholar]
- 20.Jacobs I.J., Menon U., Ryan A., Gentry-Maharaj A., Burnell M., Kalsi J.K., et al. Ovarian cancer screening and mortality in the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS): a randomised controlled trial. Lancet. 2016;387(10022):945–956. doi: 10.1016/S0140-6736(15)01224-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pinsky P.F., Yu K., Kramer B.S., Black A., Buys S.S., Partridge E., et al. Extended mortality results for ovarian cancer screening in the PLCO trial with median 15years follow-up. Gynecol Oncol. 2016;143(2):270–275. doi: 10.1016/j.ygyno.2016.08.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sayasneh A., Kaijser J., Preisler J., Smith A.A., Raslan F., Johnson S., et al. Accuracy of ultrasonography performed by examiners with varied training and experience in predicting specific pathology of adnexal masses. Ultrasound Obst Gynecol. 2015;45(5):605–612. doi: 10.1002/uog.14675. [DOI] [PubMed] [Google Scholar]
- 23.Kristjansdottir B., Levan K., Partheen K., Sundfeldt K. Diagnostic performance of the biomarkers HE4 and CA125 in type I and type II epithelial ovarian cancer. Gynecol Oncol. 2013;131(1):52–58. doi: 10.1016/j.ygyno.2013.07.094. [DOI] [PubMed] [Google Scholar]
- 24.Feeney L., Harley I.J., McCluggage W.G., Mullan P.B., Beirne J.P. Liquid biopsy in ovarian cancer: Catching the silent killer before it strikes. World J Clin Oncol. 2020;11(11):868–889. doi: 10.5306/wjco.v11.i11.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trevino V., Falciani F., Barrera-Saldaña H.A. DNA microarrays: a powerful genomic tool for biomedical and clinical research. Mol Med. 2007;13(9–10):527–541. doi: 10.2119/2006-00107.Trevino. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagaraja G.M., Othman M., Fox B.P., Alsaber R., Pellegrino C.M., Zeng Y., et al. Gene expression signatures and biomarkers of noninvasive and invasive breast cancer cells: comprehensive profiles by representational difference analysis, microarrays and proteomics. Oncogene. 2006;25(16):2328–2338. doi: 10.1038/sj.onc.1209265. [DOI] [PubMed] [Google Scholar]
- 27.Adib T.R., Henderson S., Perrett C., Hewitt D., Bourmpoulia D., Ledermann J., et al. Predicting biomarkers for ovarian cancer using gene-expression microarrays. Br J Cancer. 2004;90(3):686–692. doi: 10.1038/sj.bjc.6601603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Foukakis T., Lovrot J., Sandqvist P., Xie H., Lindstrom L.S., Giorgetti C., et al. Gene expression profiling of sequential metastatic biopsies for biomarker discovery in breast cancer. Mol Oncol. 2015;9(7):1384–1391. doi: 10.1016/j.molonc.2015.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mulligan J.M., Hill L.A., Deharo S., Irwin G., Boyle D., Keating K.E., et al. Identification and validation of an anthracycline/cyclophosphamide-based chemotherapy response assay in breast cancer. J Natl Cancer Inst. 2014;106(1) doi: 10.1093/jnci/djt335. djt335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma X., Wang Y.W., Zhang M.Q., Gazdar A.F. DNA methylation data analysis and its application to cancer research. Epigenomics. 2013;5(3):301–316. doi: 10.2217/epi.13.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones P.A. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13(7):484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 32.Doi A., Park I.H., Wen B., Murakami P., Aryee M.J., Irizarry R., et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nature Genet. 2009;41(12):1350–1353. doi: 10.1038/ng.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baylin S.B. DNA methylation and gene silencing in cancer. Nat Clin Pract Oncol. 2005;2(Suppl 1):S4–S. doi: 10.1038/ncponc0354. [DOI] [PubMed] [Google Scholar]
- 34.Jjingo D., Conley A.B., Yi S.V., Lunyak V.V., Jordan I.K. On the presence and role of human gene-body DNA methylation. Oncotarget. 2012;3(4):462–474. doi: 10.18632/oncotarget.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beirne JP. The Identification and Characterisation of Disease-Specific Biomarkers in Pelvic High Grade Serous Carcinomas. British Library https://ethos.bl.uk/OrderDetails.do?uin=uk.bl.ethos.705640: Queen's University, Belfast; 2016.
- 36.Gautier L., Cope L., Bolstad B.M., Irizarry R.A. affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics (Oxford, England) 2004;20(3):307–315. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- 37.Assenov Y., Muller F., Lutsik P., Walter J., Lengauer T., Bock C. Comprehensive analysis of DNA methylation data with RnBeads. Nat Methods. 2014;11(11):1138–1140. doi: 10.1038/nmeth.3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Teschendorff A.E., Marabita F., Lechner M., Bartlett T., Tegner J., Gomez-Cabrero D., et al. A betamixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics (Oxford, England) 2013;29(2):189–196. doi: 10.1093/bioinformatics/bts680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robinson J.T., Thorvaldsdottir H., Winckler W., Guttman M., Lander E.S., Getz G., et al. Integrative genomics viewer. Nat Biotech. 2011;29(1):24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thorvaldsdóttir H., Robinson J.T., Mesirov J.P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings Bioinf. 2013;14(2):178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., et al. The human genome browser at UCSC. Genome Res. 2002;12(6):996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]