

Abstract
Slack channels are sodium-activated potassium channels that are encoded by the KCNT1 gene. Several KCNT1 gain of function mutations have been linked to malignant migrating partial seizures of infancy. Quinidine is an anti-arrhythmic drug that functions as a moderately potent inhibitor of Slack channels; however, quinidine use is limited by its poor selectivity, safety and pharmacokinetic profile. Slack channels represent an interesting target for developing novel therapeutics for the treatment of malignant migrating partial seizures of infancy and other childhood epilepsies; thus, ongoing efforts are directed toward the discovery of small-molecules that inhibit Slack currents. This review summarizes patent applications published in 2020–2021 that describe the discovery of novel small-molecule Slack inhibitors.
Keywords: : 1,2,4-oxadiazole; 5-chlorindanyl; EIMFS; KCNT1; KNa1.1; MMPSI; Slack; Slo2.2
Malignant migrating partial seizure of infancy (MMPSI), also known as epilepsy of infancy with migrating focal seizures, is a rare and devastating form of childhood epilepsy [1]. MMPSI is characterized by pharmacoresistant seizure onset in the first 6 months of life, with continuous multifocal partial seizures migrating to different lobes and between hemispheres. The disease leads to deterioration of psychomotor and cognitive development, progressive microcephaly, cortical visual impairment hypotonia and 25% of the patients die in the first year of life. Several studies have linked the etiology of approximately half of MMPSI cases to multiple gain of function (GOF) de novo heterozygous missense mutations in the KCNT1 gene [2–4]. This gene encodes the weakly voltage-dependent Slack (sequence like a calcium-activated K+) channel, a sodium-activated potassium channel and a member of Slo family (Slo2.2) of K+ channels along with Slo1 (Maxi-K, BK or KCa1.1), Slo2.1 (Slick) and Slo3 [5–7]. Slack channels are distributed throughout the CNS, including the olfactory bulb, brainstem, hippocampus and cortical embryonic neurons, as well as peripheral tissues such as muscle, gonads, pituitary glands and cardiomyocytes [3,5,6,8]. They are essential regulators of electrical activity in the CNS, responsible for potassium outward current and after hyperpolarization following repetitive firing, which controls the rate of action potentials [3–5,9,10]. Slack channels are composed of four subunits with six hydrophobic transmembrane domains (S1–S6) and a pore-forming domain between S5–S6, an extended C-terminal cytoplasmic domain, which includes regulator of potassium conductance domains responsible for conferring sodium sensitivity to Slack currents, and a nicotinamide adenine dinucleotide-binding (NAD+) domain [2,7,10–14]. Intracellular sodium levels primarily regulate their activity [13,15]; however, other signaling pathways contribute to this process, including direct phosphorylation of the channel by PKC, indirect modulation by PKA, cytoplasmic ATP and NAD+ levels, estradiol and others [13,16].
Over 30 mutations in Slack have been reported, and most mutations associated with MMPSI are GOF mutations that lead to a 3- to 22-fold increase in Slack current [3,17–20]. Previous research has shown that these mutations tend to cluster at the C-terminal region, including both the regulator of potassium conductance and NAD+ binding domains, and the transmembrane pore-forming region, which is essential for channel activity [3,7,9,14,21,22]. It has been found that a number of KCNT1-related mutations increase the channel’s maximal open probability [15]. The Slack channel also has nonconducting functions, and its cytoplasmic domain interacts with downstream signaling pathways and developmentally relevant proteins [23] such as FMRP [24], Phactr1 and Cyfip1 [25]. As a result, KCNT1 GOF mutations are associated with several epilepsy phenotypes other than MMPSI, such as autosomal dominant sleep-related hypermotor epilepsy [21], early-onset epileptic encephalopathies such as West syndrome and Ohtahara syndrome [22], myoclonic-atonic epilepsy [26], temporal lobe epilepsy with intellectual disability [27] and autosomal dominant nocturnal frontal lobe epilepsy [28,29]. Several mechanisms by which GOF mutations in Slack channels lead to increased neuronal excitability and rate of firing have been proposed. First, channel activation leads to increased amplitude of afterhyperpolarization, which results in shorter action potentials, and an increased neuronal firing frequency [17,30]. Second, suppression of inhibitory interneurons leads to prolonged hyperpolarization, decreased inhibitory interneuronal activity, and decreased inhibitory tone [14,18]. Furthermore, the physiologic interactions between Slack channels and cell signaling pathway proteins such as Phactr1 and FMRP might be disrupted due to Slack mutations [30]. Regardless of the precise mechanism, the link between MMPSI and GOF Slack mutations is conclusive [3,17–20] and the disease severity strongly implicates increased Slack activity leading to increase network excitability in MMPSI [3,9,17–20,30].
No broadly effective and safe drug therapy option has been identified for the treatment of MMPSI. Quinidine, bepridil and cofilium modulate Slack currents in vitro and were used in clinical settings as investigational treatments; however, their efficacy was limited and they were associated with toxicity due to a lack of selectivity and poor pharmacokinetic properties [4,9,14,19,29,31–37]. Recent efforts have focused on discovering selective Slack channel inhibitors. For example, a high-throughput screening campaign utilizing a thallium (TI+) flux assay led to the discovery of VU0606170 (1), a selective Slack channel inhibitor with low micromolar potency (Figure 1). VU0606170 effectively decreased the firing rate in an overexcited, spontaneously firing cortical neuronal culture and provided evidence that small-molecules can selectively inhibit Slack channels. While such inhibition in neurons provided efficacy consistent with an anti-epileptic effect, more experiments would be required to conclusively prove that the observed effects are entirely due to Slack inhibition. Of note, a Slack-inactive and structurally related compound from the same chemical series was used as a negative control in these studies [7]. Another discovery effort utilized a cryo-electron microscopy-derived structure of chicken KNa1.1 and mutational analysis to identify Slack inhibitor compounds. Specifically, this work employed a virtual docking screen of 100,000 drug-like molecules at the minimal pore structure of the open channel conformation. Six of these compounds inhibited Slack channels at micromolar potencies in a whole-cell patch clamp assay (compounds 2, 3 and 4 are representative examples, Figure 1) [38].
Figure 1. . Inhibitors of Slack channels published in the peer-reviewed literature.

Inhibitor VU0606170 (1), discovered via high-throughput screening, inhibitors 2, 3 and 4, discovered via virtual screening and in vivo tool inhibitor 5, discovered via hit optimization.
Characterization and in vivo studies with small-molecule Slack inhibitor 5 was published recently in the peer-reviewed literature (Figure 1) [39], and it was reported as active against mouse and human Slack. Whole-cell patch clamp electrophysiology studies revealed IC50 values of 49 and 545 nM versus wild-type (WT) cynomolgus monkey and rat Slack, respectively. In addition, further studies with 5 found IC50 values ranging from 221 to 1768 nM versus several human and mouse Slack variants. In vivo mouse pharmacokinetic studies with 5 revealed low clearance, almost complete oral bioavailability and good CNS penetration. Compound 5 was an inhibitor of the hERG channel (IC50 = 11.9 μM), demonstrating that Slack and hERG may be pharmacologically resolved to some degree in this series. Compound 5 was evaluated in brain slices from WT mice and mice homozygous for Slack variant P905L (Kcnt1L/L), and a significant reduction in firing was observed in neurons from Kcnt1L/L mice at 10 μM 5. Furthermore, compound 5 produced a decrease in interictal spike frequency that persisted over 24 h in the Kcnt1L/L mouse model [39]. Of note, the animal model utilized in these studies were homozygous for the P905L mouse Slack variant, which is orthologous to the human P924L variant associated with some MMPSI cases. Still, this homozygous genotype differs from the heterozygous nature of the disease in humans [30]. A control compound lacking Slack activity from the same chemical series was not included to address the potential for off-mechanism effects in these studies. On a related note, a binding assay showed 74% displacement at GABAA Cl- channels by compound 5; however, results in a corresponding functional assay were not reported, raising a question about potential contribution of GABAergic activity to the observed antiepileptic effect [40].
In summary, none of the clinical or preclinical compounds studied to date can provide conclusive proof that inhibition of Slack channels will produce the desired anti-epileptic therapeutic effect. Thus, there is a need to continue the discovery efforts directed toward the identification of novel Slack channel inhibitors with optimized pharmacological and pharmacokinetic profiles to establish a firm correlation between Slack inhibition and seizure control. At this early stage of the discovery process, it is difficult to predict all of the precise characteristics of an ideal small-molecule drug to be employed as a therapeutic for the treatment of KCNT1-related epilepsies. With that being said, it is reasonable to postulate that a potent and selective small-molecule that targets the active/open channel and has sustained exposure in the CNS in excess of its functional activity is required for a tool compound to provide confidence in results obtained from its use in animal models of KCNT1 GOF-related epilepsies.
To date, Praxis Precision Medicine is the only pharmaceutical company that has disclosed an advanced discovery program for the development of Slack-related therapeutics. According to their most recent annual report, they possess nine patent families for small-molecule Slack inhibitors [41]. At the end of 2021, four Patent Cooperation Treaty international patent applications have been published [42–45], while applications describing remaining patent families have yet to be published. This review presents the results of an analysis of the the four published applications from Praxis Precision Medicine and discusses the novel small-molecule Slack inhibitors disclosed therein. Selected compounds from each patent are highlighted along with information on their reported functional activity.
Small-molecule Slack inhibitors
In addition to disorders related to GOF mutations in the KCNT1 gene, potential therapeutic applications cited by the inventors include preventing and/or treating neurological disorders associated with abnormal increases in neuronal excitability. The functional assay employed in each of the patents utilized HEK-TREX cell lines stably expressing WT Slack channels. Activities at Slack channels were assessed utilizing current recordings obtained on a SyncroPatch 384 PE automated patch clamp system. Compounds were categorized into three categories based on observed potency in this assay: IC50 ≤1 μM, which will be referred to here as ‘potent’; 1 μM <IC50 <20 μM, which will be referred to here as ‘moderately potent’; IC50 ≥20 μM, which will be referred to here as ‘low or loss of potency’.
The first patent application from Praxis disclosed the biological activity for 123 compounds [42]. Of those compounds, 60 were potent Slack inhibitors, 36 compounds were moderately potent and the remaining 27 compounds had low potency. Most compounds share either a 5-chloroindanyl or 6-chlortetraline ring linked via an amide to different ring systems; the latter was the basis upon which this patent was organized into three different series. Within the first series, the amide was attached to different five-membered heterocycle rings (6–15, Figure 2). Notably, a thiophene ring (6–12) gave potent analogs in a variety of different orientations and substitution patterns and was found in 35 exemplified compounds. A sulfonamide was among the most frequent substituents, and both the mono and di-methyl sulfonamide were potent analogs (7 and 8). In addition, cyclic sulfonamide substituents such as pyrrolidine 10 were likewise potent, as were 3-hydroxylated pyrrolidine enantiomers 11 and 12. Moreover, piperazine and morpholine sulfonamide analogs were also potent. Additionally, thiophene ring substitution with other polar groups such as sulfone, methoxymethylene and simple amides was well tolerated. Beyond the thiophene, other 5-membered rings such as substituted pyrazole and thiazole (13) yielded potent compounds; however, pyrrole and furan analogs exhibited moderate to low potency.
Figure 2. . Representative examples of related 5-chloroindanyl and 6-chlorotetraline Slack inhibitors from Praxis Precision Medicine (WO 2020/227097).
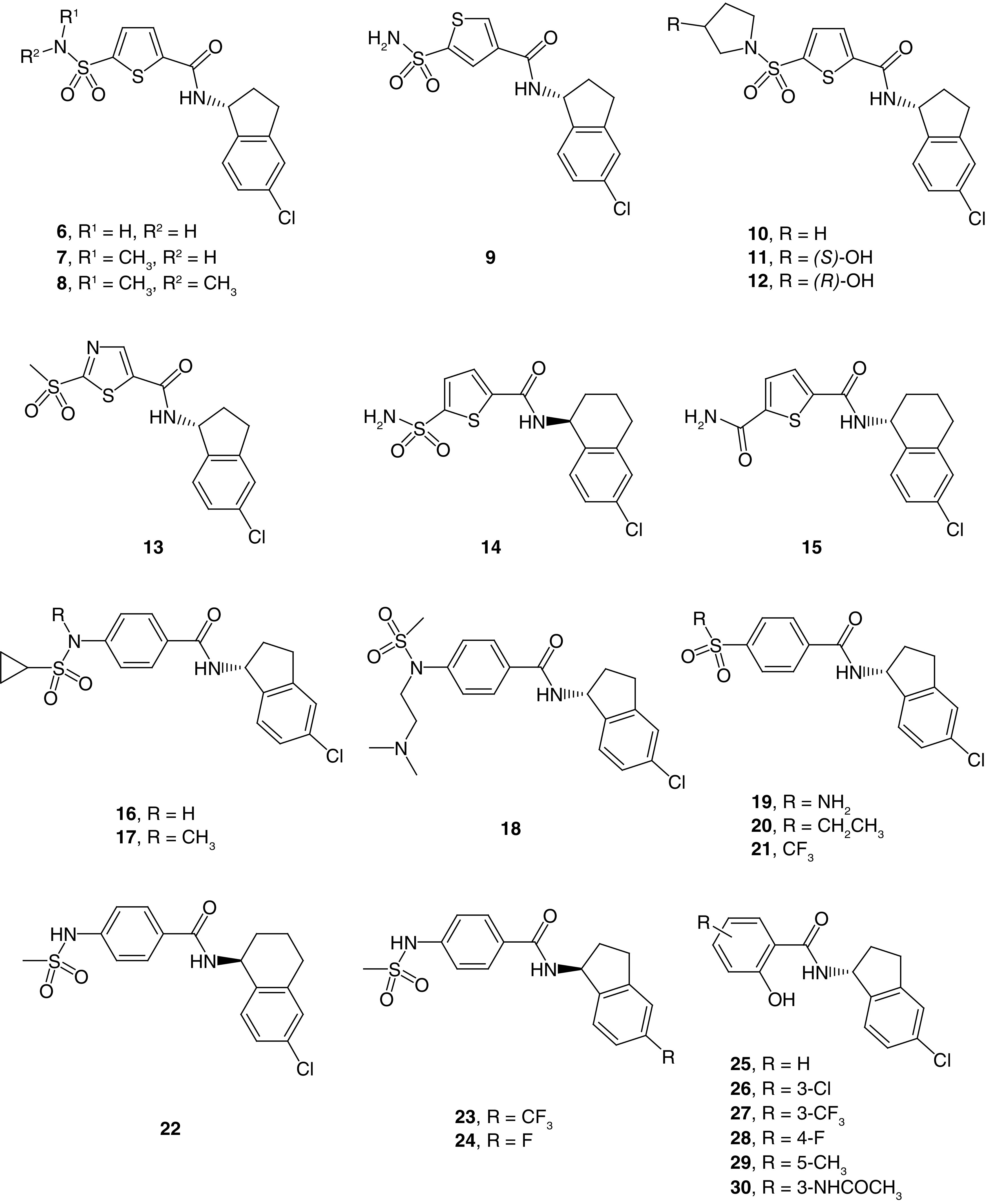
Depicted are 5-membered heteroaryl ring (6–15), para-substituted phenyl (16–24) and 2-phenol (25–30) analogs.
Notably, the 5-chlorindanyl ring was only used in the form of its R-stereoisomer, which could indicate a potency advantage for that configuration; however, a definite conclusion cannot be drawn as data for corresponding S-stereoisomer analogs were not reported. In the case of 6-chlortetraline analogs, both configurations afforded potent analogs (14 and 15); however, substitution of the thiophene proved important. Specifically, the S-stereoisomer was associated with higher potency than the R-stereoisomer counterpart when the thiophene was substituted with a sulfonamide (14). On the other hand, when the thiophene ring was substituted with a primary amide, the R-enantiomer was associated with higher potency (15) (Figure 2).
Perhaps due to potential toxicity concerns related to the thiophene ring [46], the inventors prepared 76 bioisosteric phenyl analogs with a geometrically equivalent substitution pattern [47,48]. Here enhanced activity was observed for R-stereoisomers with the 5-chloroindanyl group. Masking the sulfonamide hydrogen with a methyl group also enhanced potency (16 vs 17), which could potentially be attributed to improved cell membrane permeability. Installation of a flexible, tertiary amine-containing group was associated with moderate potency (18). Reversing the sulfonamide orientation maintained potency (19), as did its replacement by a sulfone with extended alkyl (20) and fluoroalkyl (21) side chains. Interestingly, a methylene spacer between the phenyl ring and sulfone moiety was associated with moderate to low potency. The 6-chlortetraline 22 showed higher potency with the S-stereoisomer compared with its R-stereoisomer counterpart. Replacement of the chlorine on the indanyl ring with trifluoromethyl (23) yielded potent compounds, while fluorine analogs (24) yielded compounds with moderate to low potency.
Within the third series, an amide linker attached to a 2-phenol was used (25–30). The majority of compounds presented were in the R configuration in spite of the fact that S-stereoisomers showed higher potency initially. Still, potent R-enantiomer analogs were discovered with further optimization. Both electron-donating and electron-withdrawing functional groups were found at positions 3, 4 and 5 of the 2-phenol in potent compounds (Figure 2); however, substitution at position 6 led to compounds with moderate to low potency.
The second patent application disclosed the pharmacological activity for 310 distinct analogs from a 1,2,4-oxadiazole series, of which 161 are categorized as potent [43]. The Markush structure for the compounds disclosed in this patent is depicted in Figure 3A. The R1 group is an aromatic ring such as a substituted or unsubstituted phenyl or pyridine ring, the R2 and R3 groups are alkyl side chains, and the R4 group is a substituted or unsubstituted pyrazole ring. A carbamate linker was employed instead of an amide in a few examples. In the R1 region, unsubstituted phenyl gave moderate potency, while variation of substitution patterns and electronic properties yielded potent compounds (Figure 3B). Substitution at the 3-position of the phenyl ring was frequently employed with methyl (31) and fluoro (32) substituents and often afforded potent analogs. Other functional groups and substitution patterns such as 3-trifluoromethyl (33), 4-chloro (34) and 3,4-difluoro (35) yielded potent compounds as well. Ester analog 36 was a potent analog; however, the corresponding carboxylic acid had low potency, likely rendering 36 an unsuitable candidate for further optimization as rapid hydrolysis would be expected in vivo. The 3-cyano (37), 3-chloro-5-fluoro (38), 3,5-difluoro (39) and 3-chloro (40) analogs were also potent compounds. Analog 41 was a potent analog containing a 3-difluoromethyl group, which is less common in drugs. Other substitution patterns such as 2,6-difluoro were associated with low potency.
Figure 3. . Oxadiazole Slack inhibitors from Praxis Precision Medicine (WO 2020/227101).
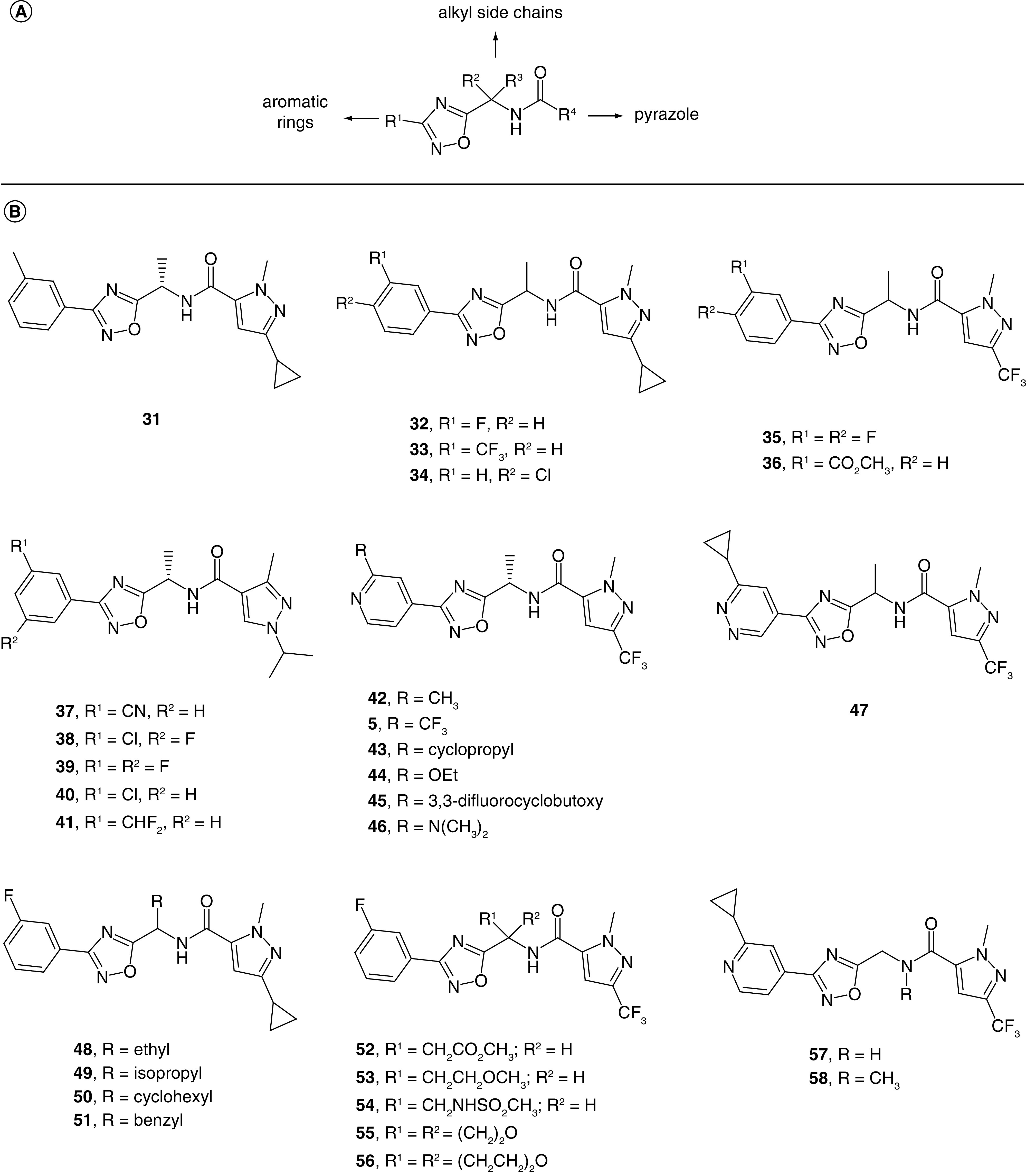
(A) Markush structure for claimed compounds. (B) Representative examples of exemplified oxadiazole Slack inhibitors.
Beyond the R1 phenyl ring, a pyridin-4-yl group was employed in over 100 examples, and substitution on the carbon adjacent to the pyridine nitrogen provided potent compounds. Examples of functional groups found in potent compounds included alkyl groups such as methyl (42), trifluoromethyl (5) and cyclopropyl (43). Alkoxy groups such as ethoxy (44) and 3,3-difluorocyclobutoxy (45) as wells as tertiary amine groups (46) also led to potent analogs. Finally, a pyridazine ring at the R1 position was well tolerated, leading to potent analog 47. In contrast, pyridin-2-yl, pyridyin-3-yl, and pyrimidin-4-yl groups at R1 were associated with moderate to low potency.
The methylene linker with substituents R2 and R3 represents a stereocenter in most of the examples; however, no clear preference pattern for either stereoisomer was evident. Several examples presented pairs of enantiomers falling into the same potency category, while other pairs showed a preference for one enantiomer over the other. Nonetheless, in a recent peer-reviewed publication describing structure–activity relationships (SAR) for a subset of compounds disclosed in this patent, the authors have reported a potency preference for the S-stereoisomer [39]. In some instances, potency was reported for the racemates (32–36, 47–54). A simple methyl group was found in the majority of potent analogs (5, 31–47); likewise, extending the alkyl chain to ethyl (48), isopropyl (49) and cyclohexyl (50) led to potent compounds, while benzyl analog 51 was moderately potent. While analogs such as ester 52, ether 53 and oxetane 55 were potent, sulfonamide 54 and tetrahydropyran 56 were moderately potent. Unsubstituted methylene linker analogs 57 and 58 were potent compounds, which was notable as a tertiary amide led to a loss of potency in other examples. Importantly, the authors have subsequently stated that the removal of the methyl group at the methylene linker led to a greater than tenfold loss in activity [39].
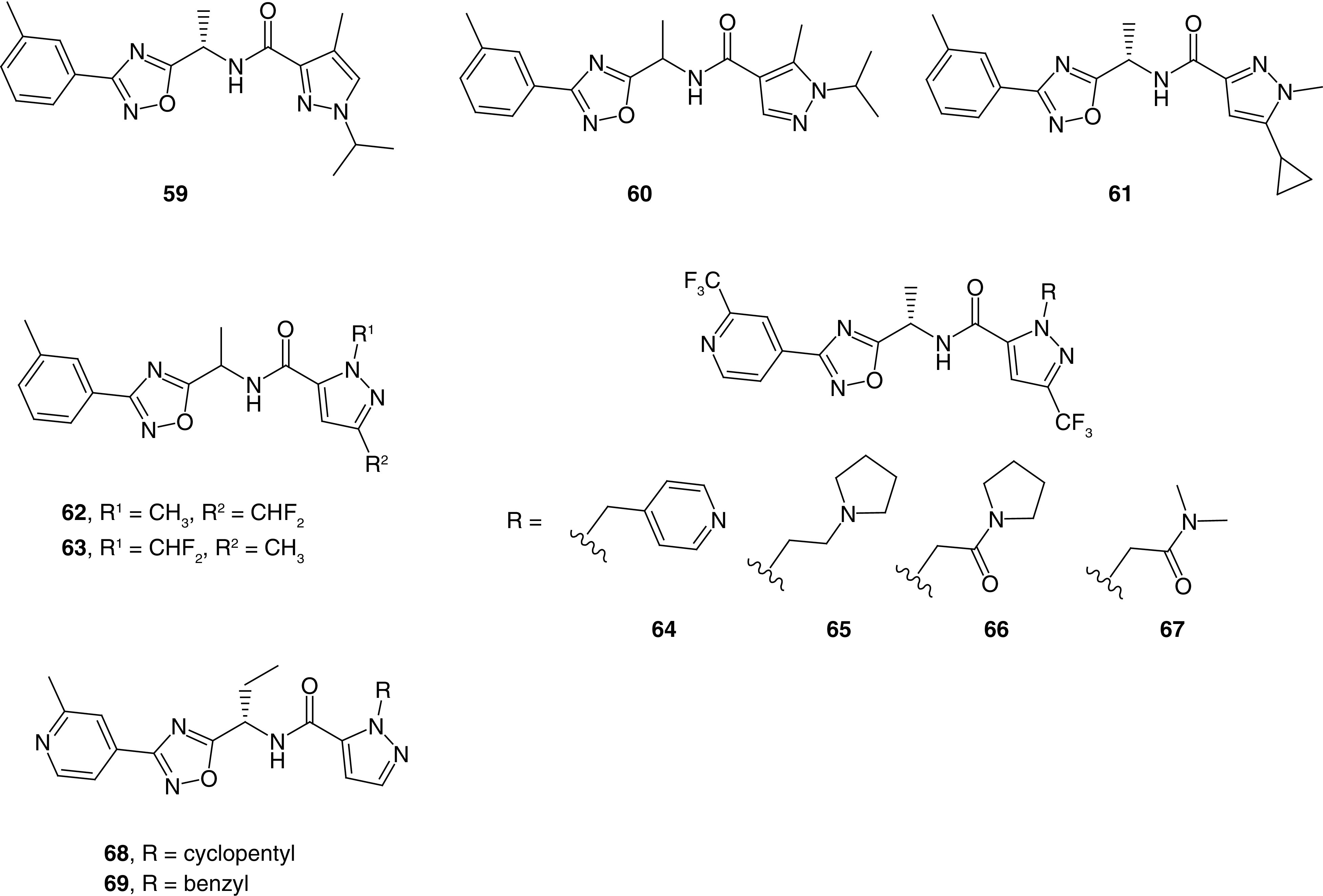
Turning to the R4 region, a pyrazole ring with different orientations and substitution patterns was utilized throughout the patent. The 1,3 substitution pattern was optimal for Slack activity as illustrated in analogs 5 and 31–58; moreover, changes to the pyrazole orientation and substitution pattern led to moderately potent compounds such as analog 59 and low potency in other examples such as analogs 60 and 61 (Figure 4). Alkyl and fluorinated alkyl groups were prepared at positions 1 and 3 of the pyrazole ring and yielded potent compounds in most instances. The bioisosteres methyl and difluoromethyl were used interchangeably and also yielded potent compounds (62 and 63). The combination of a methyl and trifluoromethyl group was frequently found in potent analogs as illustrated previously. Larger substituents were prepared at the pyrazole ring, leading to potent compounds such as analogs 64, 65 and 66. Interestingly, the small dimethyl amide analog 67 was only a moderately potent compound. A simple N-substituted pyrazole was generally associated with high potency (68 and 69), especially when it was combined with an ethyl substituent at the methylene linker. Finally, replacing the amide with carbamate in a few examples also yielded potent compounds.
Figure 4. . Additional representative examples of related oxadiazole Slack inhibitors from Praxis Precision Medicine (WO 2020/227101).
The third patent application disclosed Slack inhibitory activity for 122 compounds, from a similar 1,2,4-oxadiazole series (Figure 5) [44]. Among these derivatives, 73 compounds fall into the potent category. While the core and overall chemotype are very similar to the previously discussed application, substituents other than pyrazole were explored at the R4 region in this instance. Unsubstituted and substituted phenyl were associated with potent compounds (70–76). Both electron-withdrawing substituents such as chloro (71–73) and electron-donating substituents such as methoxy (74–76) were found in potent compounds. A 2-phenylpropanamide group results in a stereocenter (77A & B), and both the S- and R-stereoisomers were potent analogs. A variety of different and diverse heterocycles were prepared and evaluated as well. A thiazole ring in different orientations led to potent compounds such as 78, where the trifluoromethyl group was key for potency. A variety of substituted thiophen-2-yl analogs were potent; however, substituted imidazole, triazole and isoxazole analogs displayed moderate to low potency. Six-membered heterocycles such as 5-trifluoromethyl pyridin-2-yl, 2-trifluoromethyl pyridine-4-yl, and pyridine-3-yl were associated with high to moderate potency; however, unsubstituted pyridin-2-yl and pyridin-4-yl led to a loss of potency. Fused heterocycles such as benzothiophene 79 were also able to produce potent compounds in some instances; however, imidazo[1,2-a]pyridine and 2,3-dihydro-1,4-benzodioxine were associated with low potency.
Figure 5. . Representative examples of related oxadiazole Slack inhibitors from Praxis Precision Medicine (WO 2021/173930).
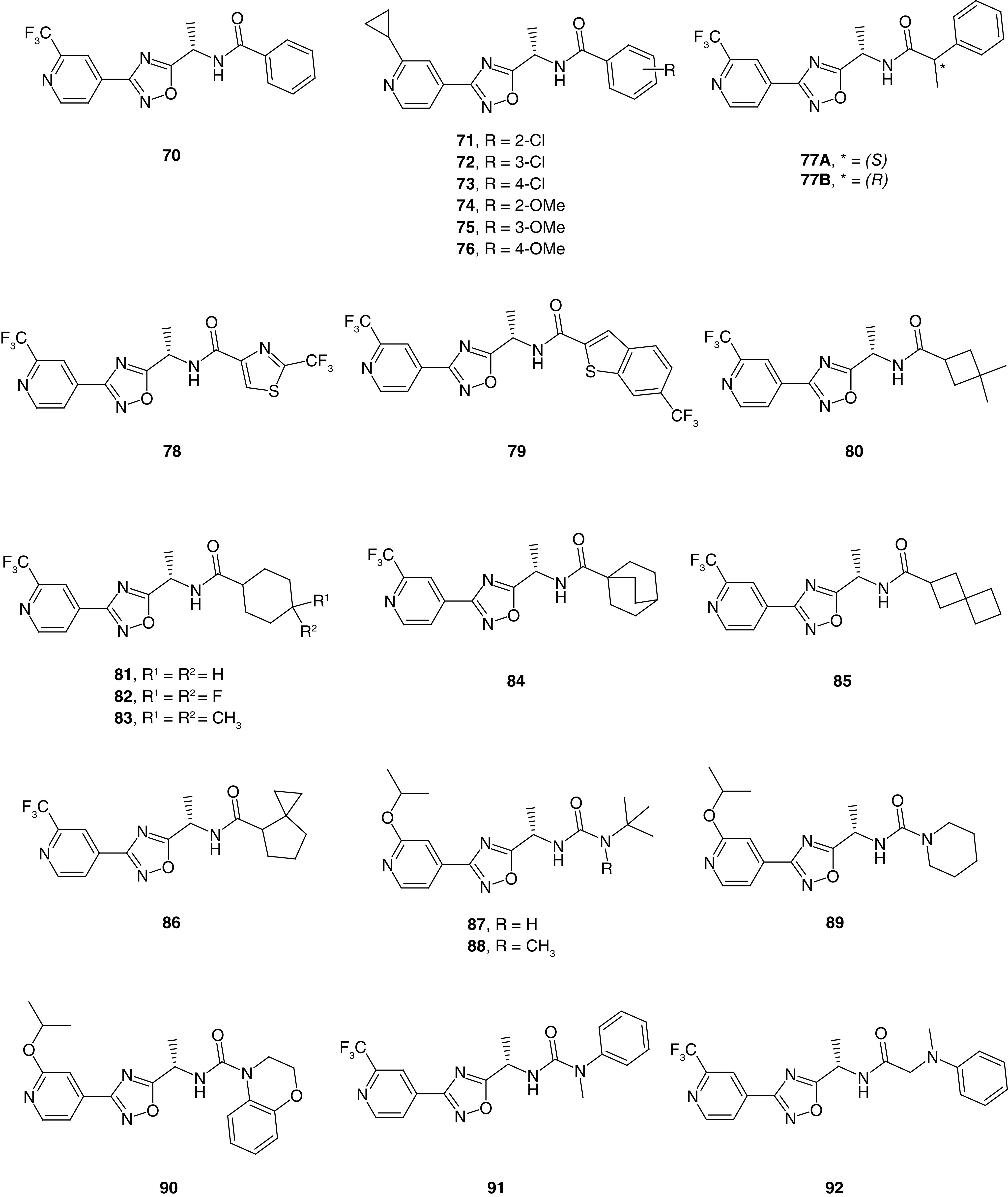
Saturated side chains also yielded potent compounds. Examples include alicyclic rings such as 3,3-dimethylcyclobutane (80) and substituted cyclohexanes (81–83). Interestingly, while 4,4-difluorocyclohexane 82 was potent, a 2,2-difluorocyclobutane analog was only moderately potent. Bicyclic ring analogs such as bicyclo[2.2.2]octane (84) proved potent compounds; however, other similar compounds such as a bicyclo[1.1.1.]pentane analog was moderately potent. Similarly, spirocyclic side chains including spiro[3.3]heptane 85 and spiro[2.4]heptane 86 also were potent compounds. Within this patent, 12 compounds with a urea in place of the amide linker were disclosed, and 11 of these were potent. It was noted that alkylation of one of the urea nitrogen atoms maintained the potency. Functional groups attached to the urea linker included simple amines such as tert-butyl analogs 87 and 88 and piperidine analog 89. Bicyclic amine 90 and N-methyl aniline derived 91 were well tolerated; likewise, a methylene spacer (92) also maintained the potency.
The fourth published patent application disclosed the biological activity for 47 compounds, and in this case the 1,2,4-oxadiazole core has been replaced with 1,2,4 thiadiazole (93 and 94) and isoxazole (95–97) cores (Figure 6) [45]. Both cores produced potent analogs with SAR patterns similar to those described for the previous patents above [43,44]. In these examples, enantiomers produced by the methyl group on the methylene linker were separated via chiral chromatography and tested separately; however, absolute stereochemistry was not determined. Both stereoisomers showed similar potency within the 1,2,4-thiadiazole series, while there was a clear preference for one isomer within the isoxazole series. A urea linker was used in place of the amide linker in six analogs and yielded compounds of varying potency. Among these derivatives, analog 97 was potent. Though not included in this application, analogs with several alternative cores, such as 1,3,4-oxadiazole and pyrazole, have since been published; however, these alternatives demonstrated moderate to low potency [39].
Figure 6. . Representative examples of related Slack inhibitors from Praxis Precision Medicine (WO 2021/195066).
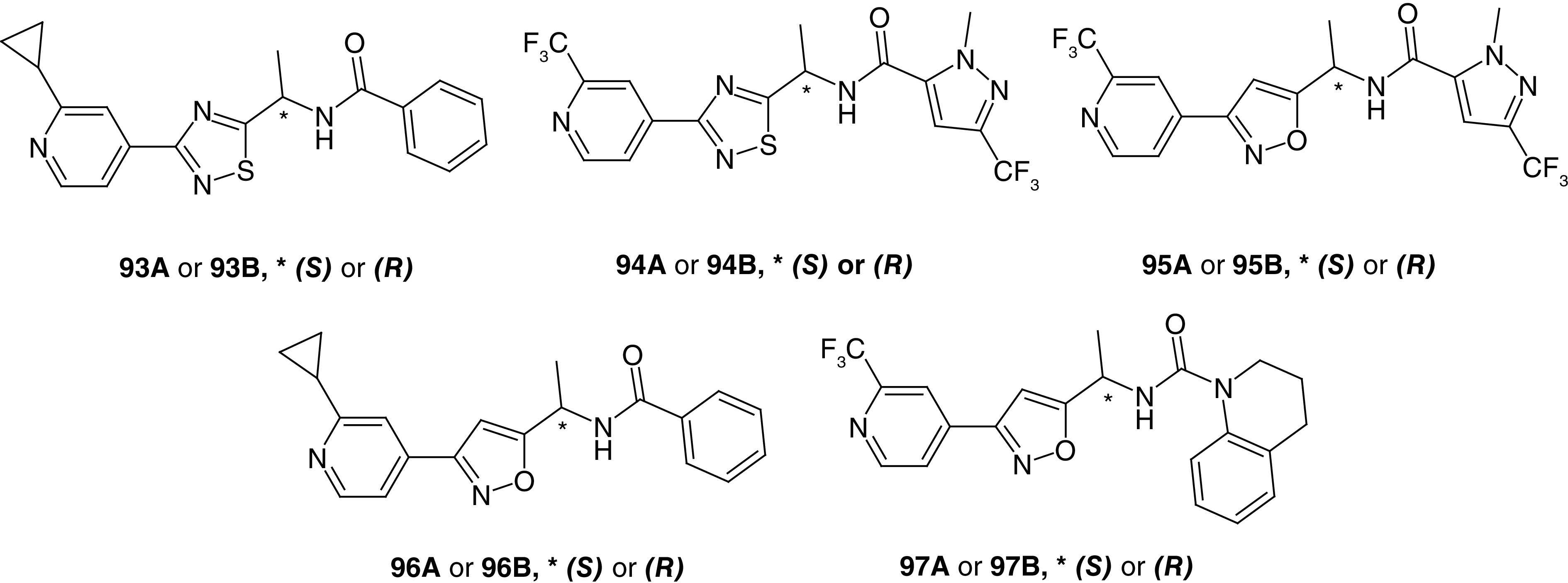
Depicted are thiadiazole (93 and 94) and isoxazole (95–97) analogs.
Conclusion
A number of Slack GOF mutations are associated with MMPSI and other childhood epilepsies refractory to conventional anti-epileptic drugs. Importantly, recent discovery efforts have demonstrated that these channels can be modulated by drug-like small-molecules [7,38,39,42–45]. As a result, Slack channels represent an interesting drug discovery target for the potential treatment of these conditions. Praxis Precision Medicine has been active in this space, leading to the first known patent applications claiming small-molecule Slack inhibitors. Herein is summarized the SAR of the four patent applications published to date from this organization. The disclosed compounds are mainly centered around two scaffolds: a 5-chlorindanyl chemotype and a 1,2,4-oxadiazole chemotype. Within the 5-chlorindanyl application, three series were explored: 5-membered heterocycles such as thiophene, which was of particular interest and generally afforded potent analogs; phenyl, which yielded potent compounds when substituted at the para position; and 2-phenol, which tolerated substitution with different electron-donating and electron-withdrawing groups and yielded potent compounds. Two patent applications focused on the 1,2,4-oxadiazole scaffold and SAR was studied extensively. This scaffold was tolerant to many structural changes at different portions of the molecule; most of which yielded potent analogs. Finally, in the fourth application, the 1,2,4-oxadiazole ring was successfully replaced with isoxazole and 1,2,4-thiadiazole rings.
Future perspective
Slack channels have recently received attention as a novel target for drug discovery due to the considerable amount of evidence linking KCNT1 GOF mutations to increased neuronal firing and MMPSI development. As a result, there is a critical need to develop new therapeutics that modulate the activity of Slack channels. To date, there are only four published patent applications, all from Praxis Precision Medicine, describing the discovery of novel small-molecule Slack inhibitors. The scarcity of patent applications within this area is understandable as Slack channels were cloned and characterized relatively recently in 1998 [6]. Moreover, concerted efforts to develop new small-molecule ligands targeting Slack channels commenced only within the last decade. Although the studies summarized here strengthen the link between Slack inhibition and anti-epileptic effects, far more work is required to conclusively prove this connection. Indeed, several questions remain unanswered at present regarding Slack channels as a therapeutic target for MMPSI. For example, the ideal pharmacological profile that leads to optimal treatment outcomes has yet to be determined. The safety profile of small-molecule Slack inhibitors and their effects on developmental proteins such as FMRP also require further study. Additional Slack modulator tool compounds from diverse chemotypes and varying pharmacological profiles are required to better understand this target. For example, compounds with pharmacological profiles that target mutant Slack variants selectively over WT would be interesting to study to learn if such compounds have any potential safety advantages. On the other hand, should no such advantages exist, compounds with similar levels of inhibition for the WT and multiple mutants might be the more attractive profile for increasing the probability of having broad-based activity at variants that have yet to be discovered. Along these lines, it is worth noting that the patent applications reviewed here disclosed activity only at WT Slack; however, small-molecule Slack channel inhibitors may exhibit higher potency on one or more of the mutant channels compared with the WT. Such an observation was previously reported for quinidine [14], suggesting that at least some small-molecule Slack ligands favor binding to the open state of the channel. Thus, much work remains and more resources are needed to develop such compounds.
Executive summary.
Slack channels represent an interesting potential target for the treatment of malignant migrating partial seizure of infancy, which is resistant to conventional anti-epileptic drugs.
Praxis Precision Medicine has been quite active in this space, filing multiple patent applications on novel Slack inhibitors, four of which have been published to date.
The disclosed compounds are mainly centered around two scaffolds: 5-chlorindanyl and 1,2,4-oxadiazole; the latter was alternated with 1,2,4-thiadiazole and isoxazole.
Within the 5-chlorindanyl scaffold, three series were explored: 5-membered heterocycles, phenyl and 2-phenol.
Two applications focused on the 1,2,4-oxadiazole scaffold, and the structure-activity relationships (SAR) was studied extensively. This scaffold proved tolerant to many structural changes at different portions of the molecule.
The 1,2,4-oxadiazole core was successfully replaced with isoxazole and 1,2,4-thiadiazole rings in the fourth application.
Acknowledgments
The authors gratefully acknowledge the National Institute of Neurological Disorders and Stroke (NINDS) for their funding of our work on small-molecule inhibitors of Slack potassium channels (R33NS109521).
Footnotes
Author contributions
AM Qunies researched the patent applications and prepared the figures. AM Qunies and KA Emmitte collaboratively drafted and edited the manuscript.
Financial & competing interests disclosure
KA Emmitte receives funding from the National Institute of Health (R33NS109521 and R21MH125257) for research centered on small-molecule modulators of Slack potassium channels. KA Emmitte is a member of the Scientific Advisory Board at the KCNT1 Epilepsy Foundation. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Coppola G, Plouin P, Chiron C, Robain O, Dulac O. Migrating partial seizures in infancy: a malignant disorder with developmental arrest. Epilepsia 36(10), 1017–1024 (1995). [DOI] [PubMed] [Google Scholar]; • Reports the first characterized case of malignant migrating partial seizures of infancy (MMPSI).
- 2.McTague A, Nair U, Malhotra S et al. Clinical and molecular characterization of KCNT1-related severe early-onset epilepsy. Neurology 90(1), e55–e66 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barcia G, Fleming MR, Deligniere A et al. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat. Genet. 44(11), 1255–1259 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Reports the association of MMPSI to KCNT1 gain of function mutations.
- 4.Cole BA, Clapcote SJ, Muench SP, Lippiat JD. Targeting KNa1.1 channels in KCNT1-associated epilepsy. Trends Pharmacol. Sci. 42(8), 700–713 (2021). [DOI] [PubMed] [Google Scholar]
- 5.Yuan A, Santi CM, Wei A et al. The sodium-activated potassium channel is encoded by a member of the Slo gene family. Neuron 37(5), 765–773 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Joiner WJ, Tang MD, Wang LY et al. Formation of intermediate-conductance calcium-activated potassium channels by interaction of Slack and Slo subunits. Nat. Neurosci. 1(6), 462–469 (1998). [DOI] [PubMed] [Google Scholar]
- 7.Spitznagel BD, Mishra NM, Qunies AaM et al. VU0606170, a selective Slack channels inhibitor, decreases calcium oscillations in cultured cortical neurons. ACS Chem. Neurosci. 11(21), 3658–3671 (2020). [DOI] [PubMed] [Google Scholar]; • Reports the pharmacological profile of Slack inhibitor VU0606170, which was discovered through a high-throughput screen.
- 8.The Human Protein Atlas. KCNT1. https://www.proteinatlas.org/search/KCNT1
- 9.Barcia G, Chemaly N, Kuchenbuch M et al. Epilepsy with migrating focal seizures: KCNT1 mutation hotspots and phenotype variability. Neurol. Genet. 5(6), e363 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhattacharjee A, Gan L, Kaczmarek LK. Localization of the Slack potassium channel in the rat central nervous system. J. Comp. Neurol. 454(3), 241–254 (2002). [DOI] [PubMed] [Google Scholar]
- 11.Bhattacharjee A, Joiner WJ, Wu M, Yang Y, Sigworth FJ, Kaczmarek LK. Slick (Slo2.1), a rapidly-gating sodium-activated potassium channel inhibited by ATP. J. Neurosci. 23(37), 11681–11691 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Z, Rosenhouse-Dantsker A, Tang QY, Noskov S, Logothetis DE. The RCK2 domain uses a coordination site present in Kir channels to confer sodium sensitivity to Slo2.2 channels. J. Neurosci. 30(22), 7554–7562 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaczmarek LK. Slack, slick and sodium-activated potassium channels. ISRN Neurosci. 2013(2013), 354262 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rizzo F, Ambrosino P, Guacci A et al. Characterization of two de novo KCNT1 mutations in children with malignant migrating partial seizures in infancy. Mol. Cell. Neurosci. 72, 54–63 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Tang QY, Zhang FF, Xu J et al. Epilepsy-related Slack channel mutants lead to channel over-activity by two different mechanisms. Cell Rep. 14(1), 129–139 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nuwer MO, Picchione KE, Bhattacharjee A. cAMP-dependent kinase does not modulate the slack sodium-activated potassium channel. Neuropharmacology 57(3), 219–226 (2009). [DOI] [PubMed] [Google Scholar]
- 17.Quraishi IH, Mercier MR, McClure H et al. Impaired motor skill learning and altered seizure susceptibility in mice with loss or gain of function of the Kcnt1 gene encoding Slack (KNa1.1) Na+-activated K+ channels. Sci. Rep. 10(1), 3213 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim Grace E, Kronengold J, Barcia G et al. Human Slack potassium channel mutations increase positive cooperativity between individual channels. Cell Rep. 9(5), 1661–1672 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Milligan CJ, Li M, Gazina EV et al. KCNT1 gain of function in 2 epilepsy phenotypes is reversed by quinidine. Ann. Neurol. 75(4), 581–590 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martin HC, Kim GE, Pagnamenta AT et al. Clinical whole-genome sequencing in severe early-onset epilepsy reveals new genes and improves molecular diagnosis. Hum. Mol. Genet. 23(12), 3200–3211 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heron SE, Smith KR, Bahlo M et al. Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat. Genet. 44(11), 1188–1190 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Ohba C, Kato M, Takahashi N et al. De novo KCNT1 mutations in early-onset epileptic encephalopathy. Epilepsia 56(9), e121–128 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Kessi M, Chen B, Peng J et al. Intellectual disability and potassium channelopathies: a systematic review. Front.Genet. 11, 614, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown MR, Kronengold J, Gazula VR et al. Fragile X mental retardation protein controls gating of the sodium-activated potassium channel Slack. Nat. Neurosci. 13(7), 819–821 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fleming MR, Brown MR, Kronengold J et al. Stimulation of Slack K+ channels alters mass at the plasma membrane by triggering dissociation of a phosphatase-regulatory complex. Cell Rep. 16(9), 2281–2288 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Routier L, Verny F, Barcia G et al. Exome sequencing findings in 27 patients with myoclonic-atonic epilepsy: is there a major genetic factor? Clin. Genet. 96(3), 254–260 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Hansen N, Widman G, Hattingen E, Elger CE, Kunz WS. Mesial temporal lobe epilepsy associated with KCNT1 mutation. Seizure 45, 181–183 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Moller RS, Heron SE, Larsen LH et al. Mutations in KCNT1 cause a spectrum of focal epilepsies. Epilepsia 56(9), e114–120 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Borlot F, Abushama A, Morrison-Levy N et al. KCNT1-related epilepsy: an international multicenter cohort of 27 pediatric cases. Epilepsia 61 (4), 679–692 (2020). [DOI] [PubMed] [Google Scholar]
- 30.Quraishi IH, Stern S, Mangan KP et al. An epilepsy-associated KCNT1 mutation enhances excitability of human iPSC-derived neurons by increasing slack KNa currents. J. Neurosci. 39(37), 7438–7449 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bearden D, Strong A, Ehnot J, DiGiovine M, Dlugos D, Goldberg EM. Targeted treatment of migrating partial seizures of infancy with quinidine. Ann. Neurol. 76(3), 457–461 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Dilena R, DiFrancesco JC, Soldovieri MV et al. Early treatment with quinidine in 2 patients with epilepsy of infancy with migrating focal seizures (EIMFS) due to gain-of-function KCNT1 mutations: functional studies, clinical responses, and critical issues for personalized therapy. Neurotherapeutics 15(4), 1112–1126 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jia Y, Lin Y, Li J et al. Quinidine therapy for Lennox-Gastaut Syndrome with KCNT1 mutation. A case report and literature review. Front. Neurol. 10, 64 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patil AA, Vinayan KP, Roy AG. Two South Indian children with KCNT1-related malignant migrating focal seizures of infancy - clinical characteristics and outcome of targeted treatment with quinidine. Ann. Indian Acad. Neurol. 22(3), 311–315 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Passey CC, Erramouspe J, Castellanos P, O'Donnell EC, Denton DM. Concurrent quinidine and phenobarbital in the treatment of a patient with 2 KCNT1 mutations. Curr. Ther. Res. Clin. Exp. 90, 106–108 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.El Kosseifi C, Cornet MC, Cilio MR. Neonatal developmental and epileptic encephalopathies. Semin. Pediatr. Neurol. 32, 100770 (2019). [DOI] [PubMed] [Google Scholar]
- 37.Yoshitomi S, Takahashi Y, Yamaguchi T et al. Quinidine therapy and therapeutic drug monitoring in four patients with KCNT1 mutations. Epileptic Disord. 21(1), 48–54 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Cole BA, Johnson RM, Dejakaisaya H et al. Structure-based identification and characterization of inhibitors of the epilepsy-associated KNa1.1 (KCNT1) potassium channel. iScience 23(5), 101100–101100 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Describes the discovery of small-molecule slack inhibitors utilizing a virtual screening approach.
- 39.Griffin AM, Kahlig KM, Hatch RJ et al. Discovery of the first orally available, selective KNa1.1 inhibitor: in vitro and in vivo activity of an oxadiazole series. ACS Med. Chem. Lett. 12(4), 593–602 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Summarizes the discovery of an orally available, selective slack inhibitor with efficacy in a mouse model of MMPSI.
- 40.Treiman DM. GABAergic mechanisms in epilepsy. Epilepsia 42(Suppl. 3), 8–12 (2001). [DOI] [PubMed] [Google Scholar]
- 41.Praxis Precision Medicines Annual Report 2021 (2021). https://investors.praxismedicines.com/static-files/90c829e3-7ea6-4106-87e1-a7ef0a922baf
- 42.Praxis Precision Medicine, Inc.: WO 2020/227097 (2020).
- 43.Praxis Precision Medicine, Inc.: WO 2020/227101 (2020).
- 44.Praxis Precision Medicine, Inc.: WO 2021/173930 (2021).
- 45.Praxis Precision Medicine, Inc.: WO 2021/195066 (2021).
- 46.Gramec D, Peterlin Mašič L, Sollner Dolenc M. Bioactivation potential of thiophene-containing drugs. Chem. Res. Toxicol. 27(8), 1344–1358 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Wallinder C, Sköld C, Botros M et al. Interconversion of functional activity by minor structural alterations in nonpeptide AT2 receptor ligands. ACS Med. Chem. Lett. 6(2), 178–182 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Flood A, Trujillo C, Sanchez-Sanz G et al. Thiophene/thiazole-benzene replacement on guanidine derivatives targeting α2-Adrenoceptors. Eur. J. Med. Chem. 138, 38–50 (2017). [DOI] [PubMed] [Google Scholar]