

Abstract
Cross-electrophile coupling (XEC) reactions of aryl and alkyl electrophiles are appealing but limited to specific substrate classes. Here, we report electroreductive XEC of previously incompatible electrophiles including tertiary alkyl bromides, aryl chlorides, and aryl/vinyl triflates. Reactions rely on the merger of an electrochemically active complex that selectively reacts with alkyl bromides through 1e– processes and an electrochemically inactive Ni0(phosphine) complex that selectively reacts with aryl electrophiles through 2e– processes. Accessing Ni0(phosphine) intermediates is critical to the strategy but is often challenging. We uncover a previously unknown pathway for electrochemically generating these key complexes at mild potentials through a choreographed series of ligand-exchange reactions. The mild methodology is applied to the alkylation of a range of substrates including natural products and pharmaceuticals.
Better C–C coupling through ligand swaps
The development of cross-coupling catalysis for carbon-carbon bond formation revolutionized pharmaceutical synthesis. Nonetheless, one drawback of the original reactions is the need to activate one of the coupling partners ahead of time. Recently, chemists have focused on direct coupling of two halocarbons, which is efficient but poses a selectivity challenge. Hamby et al. report a nickel catalyst for alkyl-aryl coupling that relies on ligand exchanges in concert with electrochemistry to react with each partner consecutively and thereby avoid alkyl-alkyl, aryl-aryl, or isomerized by-products. —JSY
Advances in Ni-based catalysis have enabled new C–C bond-forming methodologies that directly couple two C electrophiles in a net-reductive process without the need to preform a nucleophilic coupling partner (1–4). The ubiquity of simple organohalides or other C electrophiles has caused Ni-catalyzed cross-electrophile coupling (XEC) to become one of the most common strategies for C–C coupling in industry, particularly to form C(sp3)–C(sp2) bonds (5–11). Although chemical (3), photoredox (12), and electrochemical (13) approaches have been developed to deliver the reducing equivalents needed for XEC, all three reductive strategies are limited to couplings of similar classes of alkyl and aryl halides. An example of this substrate-specific reactivity is highlighted in Fig. 1A from our own work on electrochemical XEC (eXEC), in which coupling reactions of aryl bromides and primary (1°) or secondary (2°) alkyl bromides are often quantitative, whereas those of tertiary (3°) alkyl bromides fail to form any cross-products (14). This substantial difference in yield is not unique to eXEC because XEC reactions of 3° alkyl bromides are rare (15) and the few known examples require aryl iodides or activated (e-deficient) aryl bromides as the coupling partner (16–18). More broadly, a survey of organohalides that are successfully coupled under any of the three reductive approaches reveals a narrow chemical space of electrophiles that can be paired. XEC of aryl iodide/bromide + 1°/2° alkyl bromide (Fig. 1B, bottom left, dark blue) can be reliably performed in high yield with a wide range of catalysts. By contrast, reactions of substrate combinations that deviate from this constraint, such as those of 3° alkyl bromides or e-rich aryl bromides, are challenging (Fig. 1B, light red and light blue). In addition to the underdeveloped couplings at the boundary of known XEC reactions, more than half of the combinations in Fig. 1B lie beyond the current chemical space for XEC. Namely, couplings of widely available electrophiles such as aryl chlorides or triflates are currently unknown with any alkyl bromide. This work circumvents the limitations of alkyl-aryl XEC. The developed methodology enables couplings of a wide range of unknown or low-yielding combinations of electrophiles (summarized in Fig. 1B, red).
Fig. 1. Background, limitations, and design for C(sp2)-C(sp3) XEC.
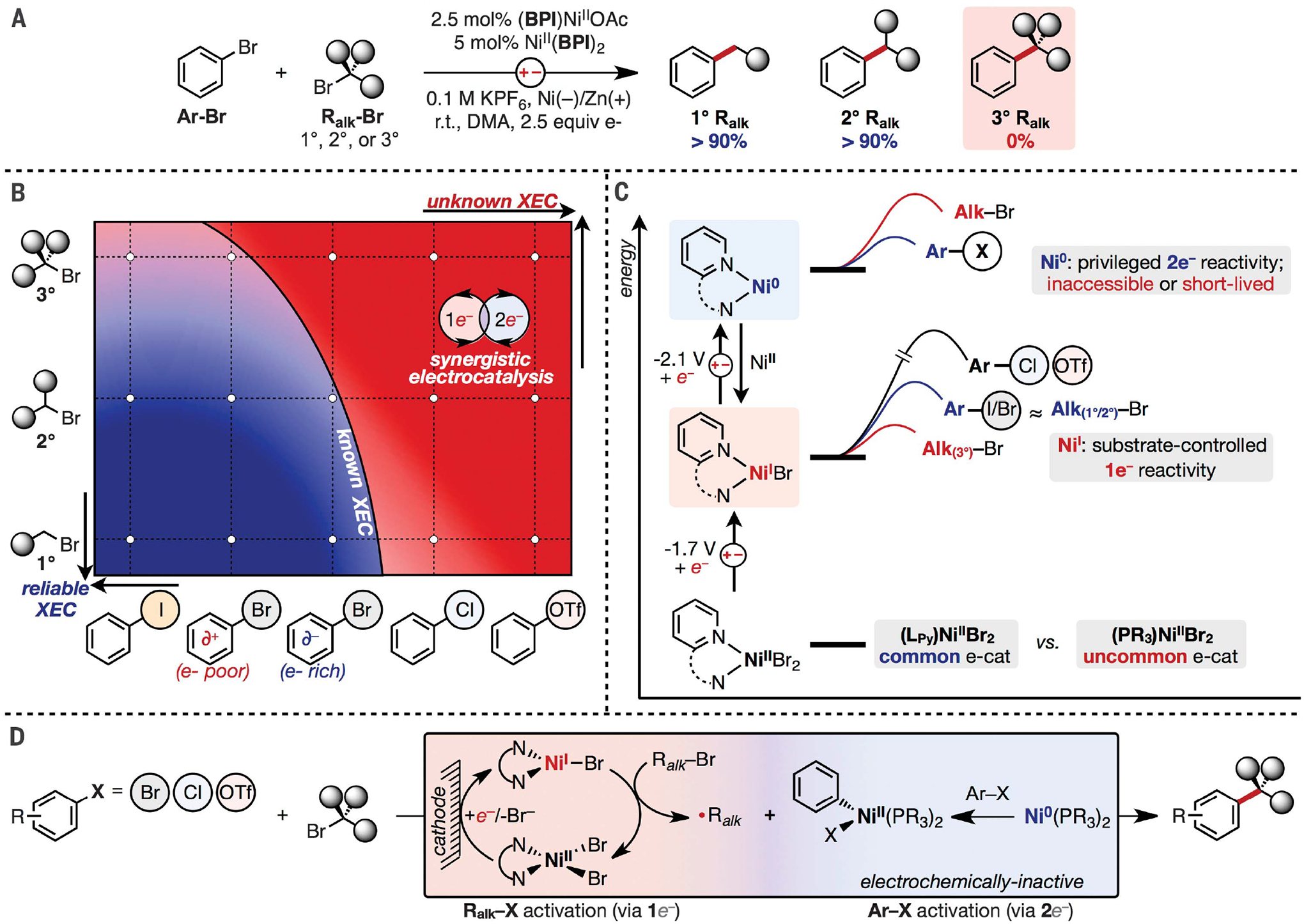
(A) Example of substrate-controlled limitations in XEC. DMA, dimethylacetamide; r.t., room temperature; BPI, bis(2-pyridylimino)isoindoline. (B) Reported outcomes of XEC reactions of varying classes of alkyl and aryl electrophiles. Blue indicates high-yielding/reliable combinations; red, low-yielding/unknown combinations. (C) Qualitative energy diagram illustrating the reductive accessibility of NiI or Ni0 complexes and their relative reactivities with various aryl or alkyl electrophiles. Reported E1/2 values (14) are for the electrocatalyst in (A) and referenced to ferrocene, Fc/Fc+. (D) Proposed strategy for XEC that decouples substrate activation to catalysts with dedicated 1e– or 2e– reactivity.
Conceptually, cross-product formation in XEC relies on the sequential activation of each electrophile at a low-valent metal complex, most often a Ni complex of pyridyl-based ligands (e.g., 2,2′-bipyridine) (3, 19). Aryl electrophiles are activated by 2e– oxidative addition at Ni, and alkyl electrophiles react through 1e– processes to form alkyl radicals (20, 21). Although it initially seemed that activation of each electrophile occurred at a distinct oxidation state of Ni (Ni0 or NiI), a growing body of evidence from electrochemical (22), photoredox (23), and chemical (24) studies suggests that only NiI(pyridyl) intermediates are accessible under reductive conditions. On the basis of these reports, we hypothesized that XEC is restricted to electrophiles that react with comparable rates at NiI, whereas electrophiles that are exceedingly reactive (3° alkyl halides) or unreactive (Ar–Cl/OTf) at NiI are incompatible coupling partners (Fig. 1C) (20, 25, 26). This competition between activation of alkyl and aryl electrophiles is circumvented in conventional Suzuki or Negishi methodologies that preactivate aryl halides in separate synthetic sequences as organoboron or organozinc reagents, respectively (27–30). However, even these methodologies remain underdeveloped for reactions of 3° alkyl bromides. Alternatively, alkyl-aryl Suzuki couplings with pre-formed 3° alkyl boron reagents suffer from similar limitations as XEC reactions, in which only e-deficient aryl electrophiles that readily react with NiI are compatible (31, 32).
One potential solution to the limitations pervading both XEC and conventional cross-coupling reactions is to access Ni0 complexes that could preferentially undergo 2e– reactions with aryl electrophiles over 1e– reactions with alkyl bromides. However, electrochemically generating Ni0(pyridyl) complexes while bypassing the NiI state that rapidly reacts with 3° alkyl bromides is challenging. Specifically, (pyridyl)NiII complexes often exhibit discrete 1e– redox couples, rather than 2e– redox couples, and require reduction to NiI before a second reduction forms Ni0 at a more negative potential (Fig. 1C) (33, 34). Even when Ni0 is formed, the complex undergoes rapid comproportionation with remaining NiII in solution to form the undesired NiI intermediate (22, 35, 36). By contrast, Ni(phosphine) complexes are known to react through Ni0/II processes with a wide range of aryl electrophiles, including aryl chlorides and ethers (37–40). Schoenebeck’s group recently contrasted the reactivities of phosphine- versus bipyridineligated Ni complexes for C–S coupling reactions, noting that Ni0(phosphine) complexes are accessible and catalyze reactions of aryl chlorides. Conversely, reactions catalyzed by complexes of pyridyl analogs react through the NiI state and are limited to couplings of aryl iodides or bromides (41). Despite the prevalence of phosphine-based complexes in Ni-catalyzed Suzuki- and Negishi-type catalysis, they are rarely used in electrosynthesis or even photoredox catalysis (Fig. 1C, right) (13, 42, 43). Electroreduction of neutral Ni(phosphine) complexes in polar solvents can be challenging (see mechanistic investigation below), necessitating stabilizers (e.g., hexamethylphosphoramide) or redox promoters (44, 45). Moreover, the rare examples of XEC catalyzed by phosphine-ligated complexes require Grignard reagents as reductants (46), which further highlights the challenge of reductively activating non-pyridyl Ni complexes. We viewed the poor electrochemical activity of Ni(phosphine) complexes under certain conditions as an opportunity to evade 1e– electrochemical events at Ni and possibly promote a Ni0/II manifold that preferentially activates aryl over alkyl electrophiles. Furthermore, an electrochemically active (pyridyl)Ni complex would generate alkyl radicals through complementary 1e– reactions upon electroreduction. Activation of one electrophile is thereby decoupled from activation of the other (Fig. 1D).
Our initial studies sought to establish the feasibility of a dual-catalyst approach by targeting XEC reactions of 3° alkyl bromides. Pseudo-stoichiometric reactions were performed at high Ni loadings (30 mol%) in the one-pot, two-step sequence illustrated in Fig. 2. First, Ni(COD)2 and a phosphine were combined with a mixture of 4-butylbromobenzene and tert-butyl bromide with the aim of selectively forming the NiII(aryl) intermediate. The mixture was then electrolyzed in the presence of (bpp)NiBr2 (1, bpp = 2,6-bispyrazolylpyridine), which was identified as an electrocatalyst that rapidly reacts with 3° alkyl bromides upon reduction (see the supplementary materials, figs. S6 and S11). Yields <30% from this assay would indicate only stoichiometric reactivity, and those >30% would indicate turnover of the Ni complex. Although reactions performed with most of the tested phosphines formed the target product in yields <30%, promising yields from reactions with PHOX and Quinap led us to evaluate ligands with a similar architecture. In particular, reactions with the isopropyl analog of quinazolinap (iPrQ) generated products in 61% yield, indicating that catalytic turnover was possible.
Fig. 2. Identification of catalyst combinations for dual XEC.
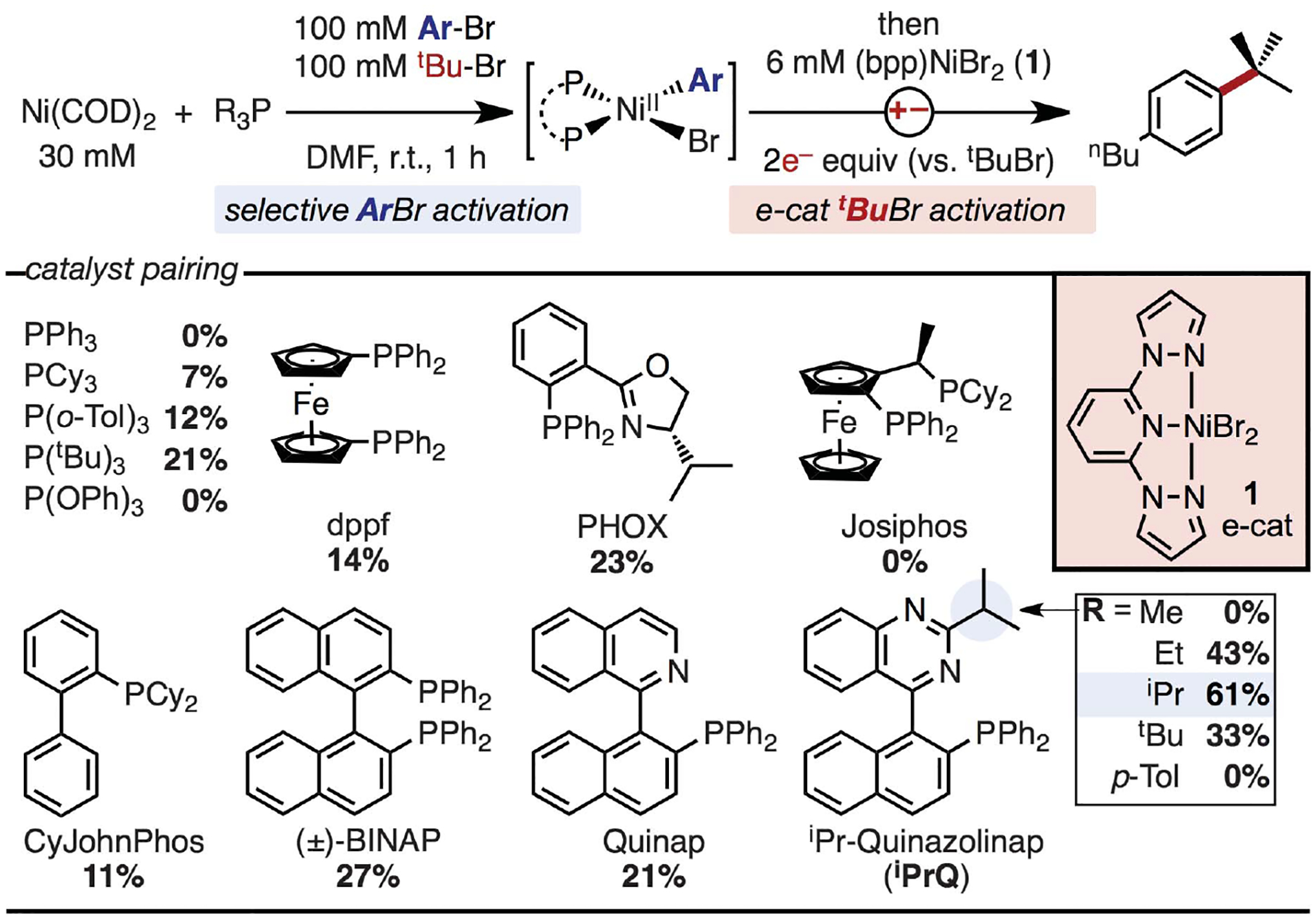
Evaluation of Ni-phosphine combinations paired with electrocatalyst 1 for XEC. Calibrated gas chromatography (GC) yields based on tBuBr are reported. DMF, dimethylformamide COD, cyclooctadiene.
The combination of phosphine iPrQ and radical generator 1 was further developed for catalytic eXEC, as summarized in Table 1. Reactions were performed under constant-current electrolysis at room temperature with a Ni-foam cathode and a Zn anode in DMF/KPF6 as electrolyte. We discovered that reactions performed with the Ni complex of bpp (1) were high yielding for the coupling of an electron-rich aryl bromide and tert-butyl bromide to form product 5 but also formed a small yet problematic quantity of the inseparable by-product 5-iso (entry 2). Such isomeric by-products are common in the rare examples of XEC reactions of 3° alkyl electrophiles after alkyl isomerization at Ni through β-hydride elimination and reinsertion to form a stable primary alkyl-Ni complex (15). By contrast, reactions performed with a combination of (bpp)MnCl2, NiCl2•dme, and iPrQ resulted in the exclusive formation of product 5 in 75% yield (entry 1). Cyclic voltammetry (CV) studies revealed that reduced (bpp)MnCl2 does not react with alkyl bromides and that Mn is readily displaced by NiII to form complex 1 in situ (see the supplementary materials, figs. S4, S5, and S18). Although the Ni analog 1 is responsible for alkyl radical formation upon electroreduction, the low concentration of remaining (bpp)MnCl2 may serve as an additional source of bpp ligand to promote the formation of key catalytic intermediates that mediate C–C coupling of alkyl radicals without isomerization (see below).
Table 1.
Development of catalytic reaction conditions.
| ||||
|---|---|---|---|---|
| Entry | Deviation from standard conditions | %conv ArBr | %yield 5 | %yield 5-iso |
| 1 | Standard conditions | 86 | 75 | 0 |
| 2 | (bpp)NiCl2 instead of [(bpp)MnCl2 + NiCl2•dme] | 92 | 83 | 8 |
| 3 | (bpp)Co(OAc)2 instead of (bpp)MnCl2 | 72 | 35 | 6 |
| 4 | (bpp)FeBr2 instead of (bpp)MnCl2 | 74 | 29 | 11 |
| 5 | Constant current: 6 mA | 82 | 49 | 1 |
| 6 | Constant current: 1.5 mA | 62 | 45 | 0 |
| 7 | Constant voltage: Ecell = 0.4 V | 81 | 47 | 0 |
| 8 | Constant voltage: Ecell = 0.6 V | 89 | 53 | 1 |
| 9 | No NiCl2•dme | 0 | 0 | 0 |
| 10 | No (bp)MnCl2 | 7 | 0 | 0 |
| 11 | No iPrQ ligand | 6 | 5 | 1 |
| 12 | Zn0 anode, no e-chem | 24 | 17 | 1 |
| 13 | Zn0 powder (3 equiv), no e-chem | 74 | 60 | 5 |
| 14 | 5 mol% iPrQ | 78 | 43 | 1 |
| 15 | 15 mol% iPrQ | 84 | 68 | 0 |
| 16 | 200 mM ArBr and 200 mM tBuBr | 100 | 57 | 0 |
Calibrated GC yields against an internal standard are reported. CCE, constant current electrolysis.
Other redox-active complexes that could activate 3° alkyl bromides were similarly evaluated, but reactions formed 5 in low yields and with poor selectivity over 5-iso (entries 3 and 4). Entries 5 to 8 summarize results from reactions performed at varying currents or constant potentials at the observed cell voltage of the standard conditions (Eanode-cathode = 0.4 to 0.6 V). Control experiments confirmed that Ni, (bpp) MnCl2, and phosphine are all necessary for product formation, and the omission of any one component resulted in only trace conversion of the aryl bromide (entries 9 to 11). However, electrolysis is not essential to the reaction. Reactions performed with a Zn anode but without an applied potential formed 5 in 17% yield (entry 12), and higher yields of 60% could be obtained from reactions with an excess of activated Zn powder after 24 hours (entry 13). Nonetheless, these reactions formed substantial quantities of 5-iso, which highlights the importance of electrochemically controlling the rate of catalyst activation through applied current. Finally, reactions were highest yielding when performed with an equimolar loading of iPrQ to Ni (entries 14 and 15) and a slight excess of aryl bromide (1.5 equivalents). Reactions conducted with a 1:1 ratio of aryl to alkyl electrophile resulted in yields of only 57% (entry 16). This low yield stems from the complete consumption of the aryl bromide rather than the 3° alkyl bromide. The relative reactivity of the electrophiles under these conditions contrasts conventional XEC reactions, in which the alkyl halide is preferentially consumed over the aryl halide.
We next investigated the mechanism of the reaction (Fig. 3) before evaluating the full scope of the methodology because it was unclear how aryl activation could occur beyond the stoichiometric approach outlined in Fig. 2. Control experiments and CV studies revealed that (bpp)NiBr2 1 is reduced at a mild potentials (E1/2 = –1.4 V versus Fc/Fc+; Fig. 3B, black trace) compared with other XEC electrocatalysts (47), but the resulting complex was only reactive toward alkyl bromides (see the supplementary materials, fig. S6). By contrast, aryl bromides are readily activated by the combination of iPrQ and a Ni0 precursor, but such low-valent phosphine complexes are not accessible by direct electroreduction. As demonstrated by the blue trace in Fig. 3B, no significant redox event was detected above –2 V from CVs of iPrQ and a NiII salt, highlighting the electrochemical inactivity of this metal-ligand combination under the reaction conditions. However, we noted an unusual effect of added iPrQ on the electroreduction of complex 1. CVs of this mixture exhibited the same onset potential for reduction as 1 alone, but the amplitude of the reductive current nearly doubled (red versus black traces). This result suggests that 2e–, rather than 1e–, are transferred at the reduction potential of 1 when phosphine is added. We performed spectroelectrochemical analysis to gain insight into the intermediates formed during reduction. As illustrated in Fig. 3C, the ultraviolet-visible (UV-vis) spectra acquired at points p1 to p4 during the CV scan revealed the growth of an absorbance at 370 nm. The UV-vis spectrum acquired at p4 almost perfectly matched that of the Ni0(iPrQ) bisphosphine (2) that we isolated and characterized from a reaction of Ni(COD)2 and iPrQ.
Fig. 3. Mechanistic investigation.
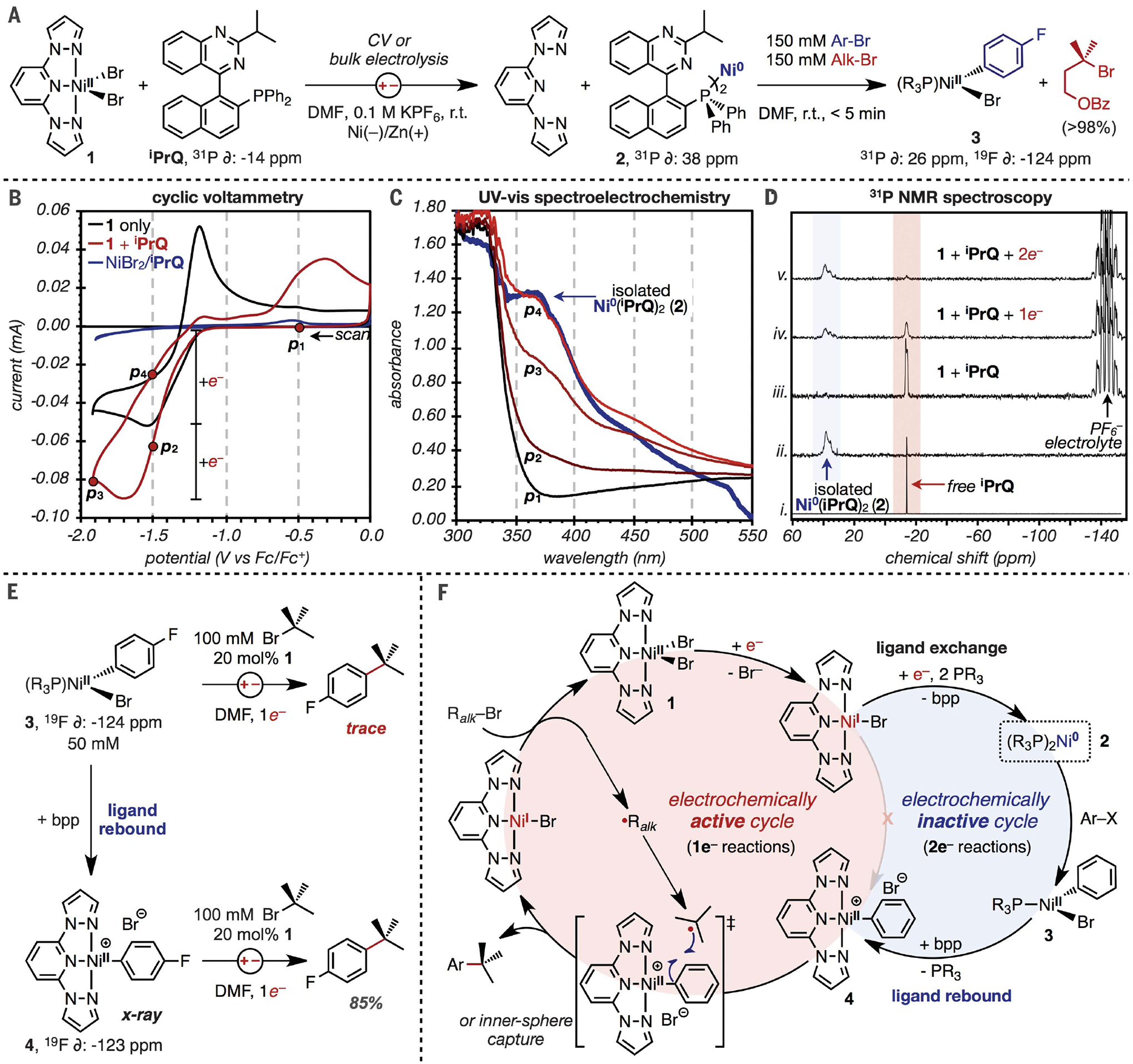
(A) Conditions for electrolysis of 1 in the presence of iPrQ to form Ni0(iPrQ)2 (left). Subsequent reactions of isolated 2 with an alkyl or aryl electrophile to form the aryl complex 3 exclusively (right). (B) CVs of 1 (10 mM) with and without iPrQ (10 mM). Conditions: 0.1 M KPF6/DMF, glassy C WE, Pt CE, 100 mV/s. (C) Spectroelectrochemical analysis from CVs in (B). Only four points from the CV are plotted for clarity along with the UV-vis spectrum of 2. (D) Conversion of 1 to 2 during bulk electrolysis as monitored by 31P NMR spectroscopy. (E) Electrolysis reactions of isolated Ni(aryl) intermediates 3 and 4 with tert-butyl bromide. (F) Proposed mechanism.
Surprised that a Ni0(phosphine) complex might be generated at such mild potentials, we conducted bulk electrolysis of complex 1 in the presence of iPrQ and analyzed solution aliquots by 31P nuclear magnetic resonance (NMR) spectroscopy throughout the reaction (Fig. 3D). As a reference, free iPrQ exhibited a 31P resonance at –14 ppm, whereas the ligated complex 2 exhibited a broad resonance at 38 ppm (spectra i and ii, respectively). Under the electrochemical conditions, the added iPrQ remained uncoordinated, indicating that a bpp-ligated NiII is more stable than a iPrQ-ligated NiII (spectrum iii). However, spectra acquired after reduction by 1e– and 2e– equivalents relative to 1 revealed a conversion of free iPrQ to complex 2 (spectra iv and v). These data provide further support that the Ni0(phosphine) complex is formed at mild potentials and that it persists in the bulk solution without undergoing comproportionation with remaining NiII complexes to form NiI. Most importantly, stoichiometric reactions of 2 with a combination of para-fluorobromobenzene and a 3° alkyl bromide revealed exclusive activation of the aryl bromide (>95% conversion versus Ni), whereas none of the 3° alkyl bromide reacted (Fig. 3A, right). The product from this reaction was characterized as 3 by 19F and 31P NMR spectroscopy (48). These studies underscore that the accessibility and persistence of a Ni0 intermediate is a critical feature that enables preferential activation of aryl over alkyl electrophiles.
Finally, we investigated the reactivity of NiII(aryl) 3 toward radical capture and C–C coupling with 3° alkyl bromides. Electroreduction of a solution containing 3, tert-butyl bromide, and 1 as the radical generator with 1e– equivalent (versus 3) formed only trace quantities of coupled product (Fig. 3E). This unexpected result indicates that the phosphine complex 3 is incompetent toward radical capture and product formation. We realized that electroreductive formation of the phosphine complexes in catalytic reactions releases one equivalent of the bpp ligand, whereas the stoichiometric studies from 3 bypass those steps and lack any bpp. Therefore, we added one equivalent of bpp to complex 3 and observed the immediate displacement of iPrQ in 31P NMR spectra along with a new 19F resonance at –123 ppm. The resulting species was isolated in 68% yield and characterized as the [(bpp)NiII(aryl)]Br complex 4 by x-ray diffraction (refer to fig. S22 for ORTEP). In contrast to C–C coupling attempts from 3, the analogous experiment from 4 formed the coupled product in 85% yield. Phosphine-bpp exchange to form 4 similarly occured when (bpp)MnCl2 was added to 3. The weakly bound Mn-bpp complex likely serves as a reservoir for bpp to promote the second ligand-exchange reaction before radical capture (see the supplementary materials, figs. S18 and S19).
Collectively, these data are summarized as the proposed catalytic cycle in Fig. 3F. The reaction is initiated by electroreduction of (bpp)NiBr2 (1), followed by rapid ligand exchange with concomitant reduction to form a stable Ni0(phosphine) (2). This phosphine complex is electrochemically inactive at the operating potentials (Ecathode = –1.5 V) and does not comproportionate to form NiI intermediates as is common for Ni(pyridyl) complexes. Rather, 2 undergoes rapid 2e– oxidative addition with aryl electrophiles to form 3 even in the presence of highly activated alkyl bromides. The bpp ligand that was displaced upon electroreductive activation or that was weakly bound to MnII subsequently recoordinates the aryl complex 3 in a second ligand exchange reaction to form 4. This ligand-rebound event is a critical step because product formation only occurs from the bpp-ligated aryl complex 4. Overall, these studies reveal a complex series of ligand exchange reactions that facilitate electrochemical generation of highly reactive Ni0(phosphine) complexes, ultimately enabling XEC of 3° alkyl bromides. Through these exchange reactions, the XEC reaction is decoupled into one regime in which electrochemically active complexes undergo 1e– reactions with alkyl electrophiles (bpp complexes 1 and 4; Fig. 3F, red) and a second regime in which electrochemically inactive complexes preferentially undergo 2e– reactions with aryl electrophiles (phosphine complexes 2 and 3; Fig. 3F, blue).
These mechanistic insights suggested that XEC could in principle be performed with any aryl electrophile prone to activation by a Ni0 (phosphine). Therefore, we targeted reactions of widely available, but previously incompatible, electrophiles: 3° alkyl bromides, aryl chlorides, and aryl/vinyl triflates (Fig. 4). Products from reactions of tert-butyl bromide and a range of e-rich and e-deficient aryl bromides at room temperature were formed in high analytical yield (generally 70 to 85%) and were isolated in good yields as single constitutional isomers (5 to 11). The mild conditions of the electrocatalytic reaction are critically important because the 3° electrophiles are prone to elimination at elevated temperatures or protodehalogenation under highly reducing conditions. Reactions of more complex alkyl electrophiles that are susceptible to isomerization formed the desired products in good yields (13 to 16) with selectivities over isomers that exceeded 30:1. Additionally, alkyl bromides containing strained rings underwent XEC in high yield to form cyclobutanes or cyclopropanes with all-C quaternary centers (17 to 23). The mild methodology was also compatible with unprotected indoles (24), pyridyl-substituted acetamides (26), vinyl bromides (27), and substituted tetrahydropyrans (28). Overall, we found coupling reactions of 3° alkyl bromides and aryl bromides to be robust and highly reliable.
Fig. 4. Reaction scope.
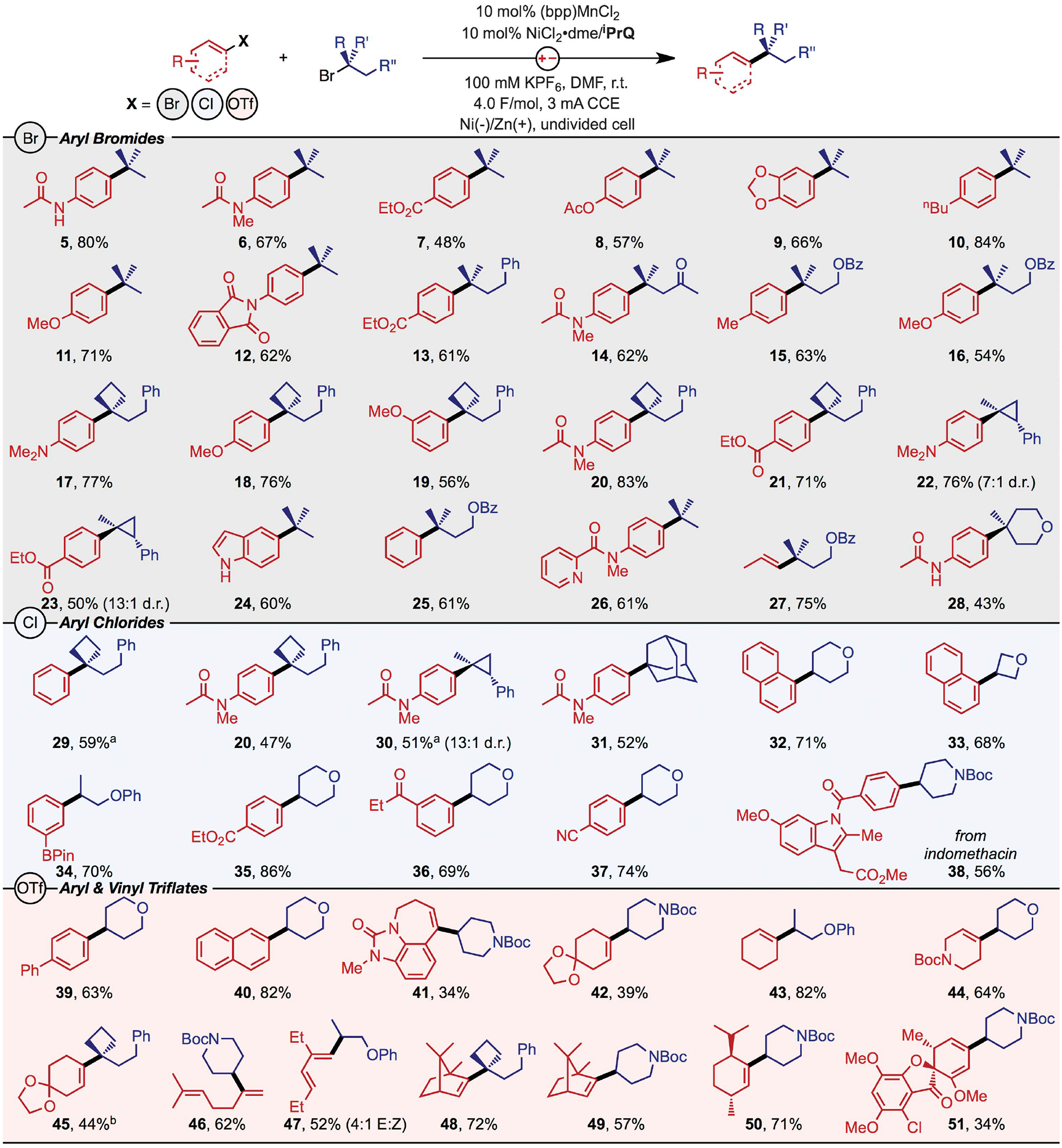
Isolated yields from XEC reactions are shown, organized by C(sp2) electrophile (Br, Cl, and OTf). Conditions: 200 mM alkyl bromide, 300 to 400 mM aryl electrophile, 2.5 ml solution, reaction temperature ranging from room temperature to 60°C, reaction time ranging from 12 to 18 hours. See the supplementary materials for specific reaction conditions. aCalibrated GC yield. bNMR yield.
We next targeted XEC reactions of aryl chlorides. Products from reactions of 3° alkyl bromides and aryl chlorides were all isolated in good yields (29 to 31, 20). We observed parallel conversion of both the aryl chloride and alkyl bromide throughout the reaction, rather than preferential consumption of just the alkyl bromide. Because XEC reactions of aryl chlorides are extremely rare and limited to couplings of unreactive alkyl electrophiles (49, 50), we also included coupling reactions with 2° alkyl bromides (32 to 38). A few noteworthy reactions from this combination include the chemoselective coupling at chloride to form the boryl-substituted product (34) in high yield and the modification of the chloroaryl fragment of indomethacin to install a BOC-protected piperidine (38).
Finally, we extended this methodology to reactions of aryl and vinyl triflates. Although reactions of aryl triflates formed products in high yield (39 and 40), we primarily focused our efforts toward developing reactions with vinyl triflates. These electrophilic substrates are attractive coupling partners because they can be prepared in a single step from a wide range of naturally occurring or pharmaceutically relevant ketones and aldehydes. As an example, the vinyl triflate of the zilpaterol precursor was prepared in a single step and directly coupled with an alkyl bromide to form the piperidyl-modified urea 41. Other vinyl triflates derived from cyclic ketones underwent coupling with a range of alkyl bromides (42 to 44), including 3° analogs (45 and 48). Reactions with vinyl triflates derived from acyclic ketones (46) and aldehydes (47) enabled the direct synthesis of highly substituted alkenes and dienes. These olefinic products could be subsequently hydrogenated to form products of a formal alkyl-alkyl coupling reaction (see the supplementary materials, fig. S20). Reactions with vinyl triflates of highly functionalized natural products could also be performed to generate C–C coupled products from a range of alkyl bromides (48 to 51).
Overall, the generality of this methodology highlights how an unusual mechanism involving dynamic ligand exchange can be used to control the redox states of Ni and ultimately expand C–C bond-forming methodologies as a whole. XEC reactions of organohalides are thus no longer limited to the canonical couplings of 1°/2° alkyl bromides and aryl iodides/bromides. Beyond the advances to XEC, these findings represent a fundamental solution to reductively accessing highly reactive Ni0 complexes in preference to the NiI intermediates that currently dominate Ni-redox catalysis.
Supplementary Material
ACKNOWLEDGMENTS
Funding:
This work was supported by the National Institutes of Health (grant R35 GM138373).
Footnotes
Competing interests: C.S.S. and T.B.H. are co-inventors on the application filed by The Ohio State University for US patent application number 63/308,319 related to this work.
SUPPLEMENTARY MATERIALS
Data and materials availability:
All experimental data, analytical procedures, and copies of spectra are available in the supplementary materials.
REFERENCES AND NOTES
- 1.Conan A, Sibille S, D’Incan E, Périchon J, J. Chem. Soc. Chem. Commun 1, 48–49 (1990). [Google Scholar]
- 2.Wang X, Dai Y, Gong H, Top. Curr. Chem. (Cham) 374, 43 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Weix DJ, Acc. Chem. Res 48, 1767–1775 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harwood SJ et al. , Science 375, 745–752 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nimmagadda SK et al. , Org. Process Res. Dev 24, 1141–1148 (2020). [Google Scholar]
- 6.Le CC et al. , ACS Cent. Sci 3, 647–653 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duvadie R et al. , Org. Process Res. Dev 25, 2323–2330 (2021). [Google Scholar]
- 8.Perkins RJ, Hughes AJ, Weix DJ, Hansen EC, Org. Process Res. Dev 23, 1746–1751 (2019). [Google Scholar]
- 9.Beutner GL et al. , J. Org. Chem 86, 10380–10396 (2021). [DOI] [PubMed] [Google Scholar]
- 10.Grimm I et al. , Org. Process Res. Dev 24, 1185–1193 (2020). [Google Scholar]
- 11.Dombrowski AW et al. , ACS Med. Chem. Lett 11, 597–604 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang P, Le CC, MacMillan DWC, J. Am. Chem. Soc 138, 8084–8087 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Malapit CA et al. , Chem. Rev 122, 3180–3218 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Truesdell BL, Hamby TB, Sevov CS, J. Am. Chem. Soc 142, 5884–5893 (2020). [DOI] [PubMed] [Google Scholar]
- 15.Xue W et al. , Chem. Soc. Rev 50, 4162–4184 (2021). [DOI] [PubMed] [Google Scholar]
- 16.Wang X, Wang S, Xue W, Gong H, J. Am. Chem. Soc 137, 11562–11565 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Wang X et al. , J. Am. Chem. Soc 140, 14490–14497 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Liu J, Ye Y, Sessler JL, Gong H, Acc. Chem. Res 53, 1833–1845 (2020). [DOI] [PubMed] [Google Scholar]
- 19.Diccianni JB, Diao T, Trends Chem 1, 830–844 (2019). [Google Scholar]
- 20.Wagner CL, Herrera G, Lin Q, Hu CT, Diao T, J. Am. Chem. Soc 143, 5295–5300 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diccianni JB, Katigbak J, Hu C, Diao T, J. Am. Chem. Soc 141, 1788–1796 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Kawamata Y et al. , J. Am. Chem. Soc 141, 6392–6402 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Till NA, Oh S, MacMillan DWC, Bird MJ, J. Am. Chem. Soc 143, 9332–9337 (2021). [DOI] [PubMed] [Google Scholar]
- 24.Sun R, Qin Y, Nocera DG, Angew. Chem. Int. Ed 59, 9527–9533 (2020). [DOI] [PubMed] [Google Scholar]
- 25.Lin Q, Fu Y, Liu P, Diao T, J. Am. Chem. Soc 143, 14196–14206 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bajo S, Laidlaw G, Kennedy AR, Sproules S, Nelson DJ, Organometallics 36, 1662–1672 (2017). [Google Scholar]
- 27.Xu J, Bercher OP, Watson MP, J. Am. Chem. Soc 143, 8608–8613 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zultanski SL, Fu GC, J. Am. Chem. Soc 135, 624–627 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen TG et al. , Angew. Chem. Int. Ed 58, 2454–2458 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Z, Yang ZP, Fu GC, Nat. Chem 13, 236–242 2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Primer DN, Molander GA, J. Am. Chem. Soc 139, 9847–9850 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yuan M, Song Z, Badir SO, Molander GA, Gutierrez O, J. Am. Chem. Soc 142, 7225–7234 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palmer TC et al. , ChemSusChem 14, 1214–1228 (2021). [DOI] [PubMed] [Google Scholar]
- 34.Lin Q, Dawson G, Diao T, Synlett 32, 1606–1620 (2021). [Google Scholar]
- 35.Sandford C et al. , Chem. Sci 10, 6404–6422 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lappin AG, McAuley A, Adv. Inorg. Chem 32, 241–295 (1988). [Google Scholar]
- 37.Taylor BLH, Swift EC, Waetzig JD, Jarvo ER, J. Am. Chem. Soc 133, 389–391 (2011). [DOI] [PubMed] [Google Scholar]
- 38.Ge S, Green RA, Hartwig JF, J. Am. Chem. Soc 136, 1617–1627 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Somerville RJ, Hale LVA, Gómez-Bengoa E, Burés J, Martin R, J. Am. Chem. Soc 140, 8771–8780 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Rosen BM et al. , Chem. Rev 111, 1346–1416 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kalvet I, Guo Q, Tizzard GJ, Schoenebeck F, ACS Catal 7, 2126–2132 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yan M, Kawamata Y, Baran PS, Chem. Rev 117, 13230–13319 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Twilton J et al. , Nat. Rev. Chem 1, 0052 (2017). [Google Scholar]
- 44.Jutand A, Chem. Rev 108, 2300–2347 (2008). [DOI] [PubMed] [Google Scholar]
- 45.Walker BR, Sevov CS, ACS Catal 9, 7197–7203 (2019). [Google Scholar]
- 46.Lucas EL, Jarvo ER, Nat. Rev. Chem 1, 0065 (2017). [Google Scholar]
- 47.Zackasee JLS, Al Zubaydi S, Truesdell BL, Sevov CS, ACS Catal 12, 1161–1166 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kehoe R et al. , Organometallics 37, 2450–2467 (2018). [Google Scholar]
- 49.Sakai HA, Liu W, Le CC, MacMillan DWC, J. Am. Chem. Soc 142, 11691–11697 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim S, Goldfogel MJ, Gilbert MM, Weix DJ, J. Am. Chem. Soc 142, 9902–9907 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All experimental data, analytical procedures, and copies of spectra are available in the supplementary materials.