

Abstract
There is interest in the development of drugs to treat fungal infections due to the increasing threat of drug resistance, and here, we report the first crystallographic structure of the catalytic domain of a fungal squalene synthase (SQS), Aspergillus flavus SQS (AfSQS), a potential drug target, together with a bioinformatics study of fungal, human, and protozoal SQSs. Our X-ray results show strong structural similarities between the catalytic domains in these proteins, but, remarkably, using bioinformatics, we find that there is also a large, highly polar helix in the fungal proteins that connects the catalytic and membrane-anchoring transmembrane domains. This polar helix is absent in squalene synthases from all other lifeforms. We show that the transmembrane domain in AfSQS and in other SQSs, stannin, and steryl sulfatase, have very similar properties (% polar residues, hydrophobicity, and hydrophobic moment) to those found in the “penultimate” C-terminal helical domain in squalene epoxidase, while the final C-terminal domain in squalene epoxidase is more polar and may be monotopic. We also propose structural models for full-length AfSQS based on the bioinformatics results as well as a deep learning program that indicate that the C-terminus region may also be membrane surface-associated. Taken together, our results are of general interest given the unique nature of the polar helical domain in fungi that may be involved in protein–protein interactions as well as being a future target for antifungals.
Introduction
There is a need for novel antifungal drugs with novel mechanisms of action to combat antibiotic resistance. The sterol biosynthesis pathway is of interest in this context since the end-product is ergosterol (1; Figure 1), not the cholesterol (2) found in humans, and many current antifungals target ergosterol biosynthesis. The first step in sterol (both ergosterol and cholesterol) biosynthesis is the condensation of two molecules of farnesyl diphosphate (FPP, 3) by squalene synthase (SQS) to form presqualene diphosphate (4), followed by a rearrangement and NADH/NADPH-mediated reductive step to form squalene (5), which is then converted to squalene 2,3-epoxide (6) by squalene epoxidase followed by cyclization to lanosterol (7), catalyzed by oxidosqualene cyclase.
Figure 1.

Structures of compounds involved in sterol biosynthesis. Squalene synthase (SQS) catalyzes the conversion of 3 to 5 via 4.
After numerous additional steps, cholesterol (or ergosterol) is formed. To be useful as an antifungal drug, it would be desirable that the target be found only in the pathogen of interest—although this is not mandatory since, for example, squalene epoxidase is used in both cholesterol and ergosterol biosynthesis and is the target for antifungals such as terbinafin,1 and a cytochrome P450 14α-demethylase is the target for the azole antifungals such as clotrimazole and posaconazole, the latter also being of interest as an antiprotozoal drug against Trypanosoma cruzi, the causative agent of Chagas disease. Squalene synthase has been the target of several drug discovery programs aimed at the development of cholesterol-lowering drugs,2 though these were not commercialized. Since in previous work, it has been shown that there are functional differences between fungal and animal SQSs, there is interest learning more about the structure of fungal SQSs. For example, in a very early work, Kribii et al.3 investigated SQSs from the plant, Arabidopsis thaliana, finding that there was a sequence of ∼30 amino acids present in the C-terminal region of two yeasts: Saccharomyces cerevisiae and Schizosaccharomyces pombe, which were absent in plant, rat, mouse, and human SQSs and that “squalene-synthesized by Arabidopsis SQS1 in the yeast endoplasmic reticulum could not be used as a substrate for the following enzymes of the pathway”.3 However, they found that a chimeric derivative of A. thaliana SQS1 containing a yeast C-terminus did produce squalene 2,3-epoxide and lanosterol (from FPP) in microsomal fractions from yeast transformed with the chimeric enzyme, leading to the suggestion that there might be an important interaction between SQS and squalene epoxidase, consistent with earlier work4 in which it was shown that, in yeast microsomes, exogenous squalene was a poor substrate for a yeast squalene epoxidase (SQLE)—in contrast to endogenous squalene from farnesyl diphosphate—the suggestion being that there is an SQS/SQLE complex formed in microsomes.
More recently, Linscott et al.5 carried out an extensive series of studies of a fungal SQS using a wide variety of chimeric enzymes and proposed that there might be a “hinge domain” in the C-terminus of fungal SQSs that might be of importance in interacting with other proteins involved in ergosterol biosynthesis. Here, we report the first crystallographic structure of the catalytic domain of a fungal SQS, Aspergillus flavus SQS (AfSQS), together with results of bioinformatics investigations of the non-catalytic C-terminal domains in AfSQS as well as other SQSs and some comparisons with the membrane-binding domains in three other proteins: squalene epoxidase, stannin, and a steryl sulfatase.
Results and Discussion
Crystallographic Structure of the A. flavus SQS Catalytic Domain in Apo- and FSPP-Bound Forms
The structure of the catalytic domain of AfSQS with (PDB ID code 7WGH, cyan) or without (PDB ID code 7WGI, green) the FPP analog FSPP (S-thiolo-farnesyl diphosphate) is shown in Figure 2a. The structures are very similar with a Cα root mean squared deviation (rmsd) over ∼330 residues of ∼0.2 Å since the ligand-bound structure was obtained via soaking. Full data acquisition and structure refinement details are given in Table 1.
Figure 2.

Structures of AfSQS and comparisons with other proteins. (a) Superposition of the catalytic domain of AfSQS with (PDB ID code 7WGH, cyan) or without (PDB ID code 7WGI, green) the FPP analog, FSPP (the two ligands are shown in stick form). (b) Surface view of the two FSPP ligands in AfSQS showing the open pocket structure. Diphosphate groups in the two FSPP ligands are shown as red spheres. (c, d) Two views of the superposition of the AfSQS/2FSPP structure (green; PDB ID code 7WGH) with that of TcSQS/FSPP/2Mg2+ (cyan; PDB ID code 3WCA; Chain B). Mg2+ is shown as magenta spheres. (e, f) Two views of the superposition of AfSQS/2FSPP (PDB ID code 7WGH, cyan) with S. aureus CrtM (PDB ID code 3W7F, green) structures. The A and B helices are only seen in SQS. The arrows indicate the last residue in the catalytic domain, F384, adjacent to a proline.
Table 1. Data Collection and Refinement Statistics for AfSQS Crystalsa.
| AfSQS | AfSQS + FSPP | |
|---|---|---|
| data collection | ||
| space group | F 2 3 | F 2 3 |
| unit-cell | ||
| a, b, c [Å] | 206.46, 206.46, 206.46 | 208.34, 208.34, 208.34 |
| α/β/γ (°) | 90/90/90 | 90/90/90 |
| resolution (Å) | 25.00–2.50 (2.59–2.50) | 25.00–2.36 (2.44–2.36) |
| unique reflections | 25,222 (2508) | 30,779 (3031) |
| redundancy | 6.9 (6.9) | 7.5 (7.5) |
| completeness (%) | 100 (100) | 100 (100) |
| average I/σ(I) | 31.1 (2.07) | 38.5 (2.9) |
| CC 1/2 | 0.922 (0.626) | 0.946 (0.765) |
| refinement | ||
| Rwork (95% data) | 0.203 (0.307) | 0.193 (0.257) |
| Rfree (5% data) | 0.223 (0.325) | 0.223 (0.295) |
| rmsd bonds (Å) | 0.004 | 0.008 |
| rmsd angles (°) | 0.66 | 0.98 |
| dihedral angles | ||
| most favored (%) | 98.56 | 99.14 |
| allowed (%) | 1.44 | 0.86 |
| disallowed (%) | 0.00 | 0.00 |
| no. of non-H atoms/average B [Å2] | ||
| protein | 2864/63.76 | 2871/54.86 |
| water | 76/56.41 | 104/56.15 |
| ligand | 14/85.99 | 52/79.86 |
| PDB ID code | 7WGI | 7WGH |
Values in parentheses are for the outer-most resolution shells.
The SQS mechanism of action involves two reactions. The first involves FPP ionizing in the so-called S1 site to form a primary carbocation, which then moves down to react with the C2,3 double bond in the FPP in the S2 site to form (after H+ abstraction) PSPP, which is then reduced by NAD(P)H.6 Since FPP is expected to be reactive, we used the less reactive S-thiolo analog FSPP for crystallization and the structure of the FSPP ligand-bound protein is shown as a surface view (colored yellow) in Figure 2b with the ligands shown as red/cyan spheres. The structure is an “open” one in which two FSPP ligands (seen also in Figure 2a) are bound, one binding to the S1 site and the other to the S2 site. Both structures are very similar to the structures of human SQS (HsSQS) as well as T. cruzi SQS (TcSQS) with a ∼1.2 Å Cα rmsd over ∼330 residues (as determined by using the PDBefold/SSM server, http://www.ebi.ac.uk/msd-srv/ssm). These structures are the first of the catalytic domain of any fungal SQS, and we show in Figure 2c,d superpositions of the AfSQS/2FSPP structure (PDB ID code 7WGH) with that of TcSQS/FSPP/2Mg2+ (PDB ID code 3WCA)7 showing the strong similarity. A comparison with human SQS (PDB ID code 1EZF, cyan)8 is shown in Figure 3a. In each structure, the polar headgroups bind close to the two highly conserved (Figure S1) DxxED domains involved in Mg2+ binding and catalysis.
Figure 3.

AfSQS and HsSQS structures. (a) AfSQS (PDB ID code 7WGH, green) and HsSQS (PDB ID code 1EZF, cyan). (b) HsSQS (PDB ID code 3VJA) displayed using a B-factor putty view. The red helix X is generally not seen in other SQS structures. (c) Superposition of the AfSQS/FSPP structure (green; PDB ID code 7WGH) with that of the HsSQS/2FSPP/3Mg2+ structure (cyan; PDB ID code 3WEG) showing similar ligand and catalytic Asp/Glu residue conformations. The magenta spheres are the three Mg2+ seen in the HsSQS structure. S1 = allylic binding site; S2 = homoallylic binding site.
The structures in the ligand-binding regions are similar to those found in other head-to-head prenylsynthases such as dehydrosqualene synthase from Staphylococcus aureus (SaCrtM)9 and from Enterococcus hirae (PDB ID code 5IYS) and are also similar to other bacterial proteins such as Bacillus subtilis Yisp (PDB ID code 3WE9)10 and HpnC (15-hydroxysqualene synthase; PDB ID code 4HD1) from Alicyclobacillus acidocaldarius. A superposition of the AfSQS (green) and SaCrtM (PDB ID code 3W7F,11 cyan) structures is shown in Figure 2e,f in which it can be seen that there is a large domain into which ligands bind in both the bacterial and fungal structures as well as three additional helices in AfSQS, here called X, A, and B, which are more distant from the ligands. The “X” helical region is generally not observable in most X-ray structures of the human protein, HsSQS, although it is present in at least one structure as a helix with a large B-factor (Figure 3b) (PDB ID code 3VJA).12
In our AfSQS/FSPP structure, we did not observe any Mg2+, and in the TcSQS structure, there are four chains but none have the three Mg2+ and two FSPP ligands seen in the human SQS structure (PDB ID code 3WEG(6)), with three Mg2+ and two (FPP) ligands being required for product formation. What is interesting to note here then is that—even in the absence of Mg2+—there is a strong similarity in the ligand conformations as well as the conformations of the active site Asp and Glu residues, as shown in Figure 3c, making it less likely that selective inhibitors that bind to this site in AfSQS will be found.
The A and B helices are present in AfSQS, TcSQS, and HsSQS, but ∼40% of the C-terminal region that is seen in the B-helix in AfSQS and TcSQS is absent in HsSQS, as shown in Figure 3a, due to the truncated species used for crystallization. In the AfSQS and TcSQS proteins, there are ∼4 additional residues present in the C-terminus region, as shown for example in Figure 3a. Helices A and B are essential for catalytic activity, at least in TcSQS and HsSQS. More specifically, a 31-370 residue HsSQS (full-length 416 residues) had 200% the activity of a rat 1-416 (full length) clone, while 31-319 and 31-335 truncated HsSQSs (corresponding to loss of X, A, and B helices) had <5% the activity of the full in length rat protein.13 Likewise, in TcSQS, removal of half of the C-terminal helix (of the catalytic domain) did not result in any active protein, while a truncation at the end of the B-helix did result in active protein.14 It thus appears that both the A and B helices in AfSQS, TcSQS, and HsSQS are essential for catalytic activity and that these helices may play a role in stabilizing the protein as for example the catalytically inactive β-domain in αβ prenylcyclases is thought to stabilize the structures of these proteins.15 The 26-residue “hinge” domain investigated earlier by Linscott et al.5 is now seen to be composed of the loop connecting the A and B helices, together with the B-helix, and is found in both the fungal and protozoal SQSs, so it is not kingdom-specific, though it is essential for catalytic activity. The role of the X-helix in all three proteins is unclear, but given its disorder in most structures, one possibility is that it is involved in closing the open form of the protein (in which Mg2+ and ligands are visible in the X-ray structures, e.g., Figure 2b) to the catalytically competent, closed form (not yet observed crystallographically in any such prenylsynthase). That is, the X-helix might adopt multiple conformations in some systems—though is clearly seen in AfSQS as well as in TcSQS.
A Comparison between Fungal, Human and Protozoal SQS Structures
The question then arises as to what might be the structure and function of the additional domains in the C-terminus of the fungal, human, and protozoal SQSs since, based on the truncation work, they are not required for catalytic activity. Clearly, based on the earlier work, it appears that there is a transmembrane domain in all SQSs that is thought to anchor SQS to the endoplasmic reticulum, with membrane insertion being a highly complex process involving chaperones (binding to these “TM domains”) as well as the endoplasmic reticulum membrane protein complex (EMC).16 As noted previously, in early work, Kribii et al. reported the presence of a sequence of ∼30 amino acids in two yeast SQSs that was absent in plant and animal SQSs. This domain is also absent in protozoal SQSs, e.g., in TcSQS, and in fungi it could be of importance in protein–protein interactions. To try and determine what the structure of the C-terminal region in the fungal SQSs might be, we first used the JPRED4 server (http://www.compbio.dundee.ac.uk/jpred4)17 to determine the helix content and whether residues were buried (i.e., not solvent-exposed) in full-length AfSQS and then compared these results with the crystallographic structure of the catalytic domain. Figure 4 shows helix-prediction results for the C-terminal regions in AfSQS (the top two panels; Figure 4a,b), as well as for the C-terminal regions in Candida albicans SQS (CaSQS; Figure 4c), HsSQS (Figure 4d), and TcSQS (Figure 4e). Note that, in Figure 4a, residues that are shown in the bold black font (the top line) correspond to residues seen in the X-ray structures reported here. A Clustal Omega18 sequence alignment of AfSQS, CaSQS, HsSQS, and TcSQS is shown in the Supporting Information, Figure S1, in which the highly conserved DxxED domains in the first and second aspartate-rich domains in the catalytic site (Figure 3c) are shown in boxes. The boxed P residues in Figure S1 are either immediately adjacent to or are one residue removed from the last residue found in the crystal structures: Q368 in TcSQS (PDB 3WCA); N370 in HsSQS (PDB 1EZF); F384 in AfSQS (PDB 7WGH) and define the end of the B-helix. For the proteins whose structures are known, we found remarkably good accord between the helix predictions (shown as red “Hs” in Figure 4) and those seen in the X-ray structures, the loop positions typically being predicted with a 1–2 residue error, giving confidence in use of the JPRED4 program to detect helical (and non-helical) regions.
Figure 4.

JPRED4 results for the C-terminal regions of four SQSs. The codes are H = helix, B = buried, bold font = residues in the catalytic domain, cyan = polar residues, and yellow = non-polar residues. The numbers indicate the reliability of the prediction (9 is best). (a) AfSQS (primarily the C-terminal end of the catalytic domain). (b) AfSQS (polar and TM helical regions). (c) C. albicans SQS (C-terminal end of the catalytic domain and polar and TM helical regions). (d) Human SQS (C-terminal end of the catalytic domain and TM helical regions). (e) T. cruzi SQS (C-terminal end of the catalytic domain and TM helical regions). The abbreviations are Jnet = final secondary structure prediction; jhmm = Jnet hmm profile prediction; jpssm = Jnet PSIBLAST pssm profile prediction; Jnet_25 = Jnet prediction of burial, less than 25% solvent accessibility; Jnet_5 = Jnet prediction of burial, less than 5% exposure; Jnet_0 = Jnet prediction of burial, 0% exposure; and Jnet Rel = Jnet reliability of prediction accuracy, ranges from 0 to 9, bigger is better.
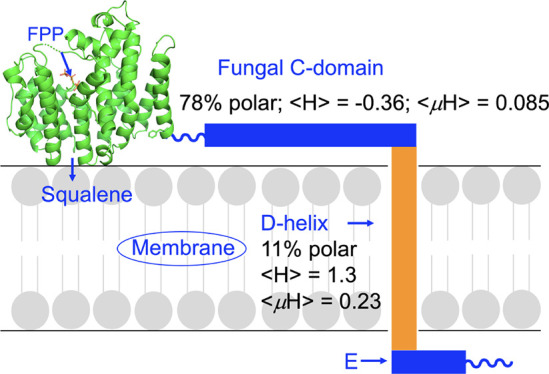
Next, we observed that the C-terminus in the AfSQS crystal structure (F384; the end of the B-helix) is connected via a proline-containing ∼4 residue (non-helical) loop to a ∼28 residue, polar, solvent-exposed (i.e., not buried, shown by a “B” in Figure 4a) primarily helical region, called here the C-helical domain, to a TM domain, called here the D-helix. These P are found in all four proteins (Figure S1) and are the last conserved residues before the highly variable C, D, and E-domains.
To give a perhaps better graphical illustration of the polar (hydrophilic) versus the hydrophobic domains, we show in Figure 4 charged residues (D, E, K, R, and H) highlighted in cyan while hydrophobic residues (A, V, L, J, F, and Y) are highlighted in yellow. As can also be seen, e.g., in Figure 4b, there is a highly hydrophobic helical region (418–445) in AfSQS that is essentially completely buried (B = buried). In addition, there is a small extra-membrane helix E, attached to a solvent-exposed non-helical region. In another fungal SQS, from C. albicans, the overall sequence is shorter than that in AfSQS since the E-helix is absent (Figure 4c). In HsSQS (Figure 4d), there is again good accord with the helix (and loop) predictions in the catalytic domain, and interestingly, the last residue in the B-helix (N370) seen in the X-ray structure (the last one in the truncated protein) also corresponds to the end of the helix predicted computationally and is followed by a ∼14 residue non-helical, non-buried segment, then the buried, helical TM region. Thus, as expected, HsSQS lacks the long, polar, solvent-exposed residues and it appears that the catalytic domain is directly connected to the TM domain (helix D) via the linker region (Figure 4d). Results with TcSQS are generally similar, with a linker connecting the catalytic domain to a TM helix (Figure 4e).
To help confirm the conclusion drawn above—that there is a large, polar, primarily helical domain connecting the catalytic and TM regions in AfSQS—we next used the HeliQuest program (http://heliquest.ipmc.cnrs.fr)21 that computes hydrophobicity ⟨H⟩ and hydrophobic moment ⟨μH⟩ values22 for helical sequences, in addition to providing useful graphical representations of polar/non-polar residues, on helical wheels. A large positive ⟨H⟩ (e.g., >1) means a very hydrophobic helix, and a negative value means a hydrophilic helix. A large ⟨μH⟩ means that the helix is amphiphilic perpendicular to its axis. The sequences that are of most interest here (since their structures have not been determined) are the polar helices seen in the fungi and the TM domains. We show some typical HeliQuest results in Figure 5.
Figure 5.

HeliQuest results for % polar residues, hydrophobicity ⟨H⟩, hydrophobic moment ⟨μH⟩, and charge in SQSs. (a) Polar helix C in AfSQS. (b) TM helix D in AfSQS. (c) TM helix D in HsSQS. (d) TM helix D in TcSQS.
For the proposed exposed, hydrophilic C-helical domain in AfSQS, as can be seen in Figure 5a, ∼78% of the polar residues (and Gly) map to an 18-residue α-helical wheel with a hydrophobicity ⟨H⟩ of −0.361 and a hydrophobic moment ⟨μH⟩ of 0.085. A very polar or hydrophilic helix. In the TM domain, the region (of 18 amino acids) with the least (11%) polar residues, the hydrophobicity ⟨H⟩ is 1.301 and the hydrophobic moment ⟨μH⟩ is 0.226 (Figure 5b), consistent with a TM domain. Similar results are obtained with CaSQS (not shown), with 61% polar residues in the polar helix C, a ⟨H⟩ of 0.213 and a hydrophobic moment ⟨μH⟩ of 0.226, and in the TM domain D, 17% polar residues, a ⟨H⟩ of 1.228 and a hydrophobic moment ⟨μH⟩ of 0.241 (for the last 18 residues in the protein; there is no E-domain). In the TM domain C in HsSQS (Figure 5c), there are 17% polar residues, a ⟨H⟩ of 1.097 and a hydrophobic moment ⟨μH⟩ of 0.111, while in the TM domain in TcSQS (Figure 5d), there are 33% polar residues, a ⟨H⟩ of 0.656 and a hydrophobic moment ⟨μH⟩ of 0.229. While there is a little variability in the values above, depending on where the sequences of the 18 residues that are scanned begin and there are typically 23 residues in a TM helix, the TM domain in the trypanosome SQS is clearly more polar than found in the fungal and human SQSs and may bind in a somewhat different membrane-associated manner. The same may be said of the N-terminal residues in SQS whose removal facilitates expression of soluble, active (and crystallizable) AfSQS, HsSQS, and TcSQS, but we found no evidence for N-terminal canonical TM helical domains in these proteins. Thus, our results indicate that SQSs are so-called “single-pass” membrane proteins.
Based on the results discussed above, it thus appears that the fungal protein AfSQS and the protozoal protein TcSQS have very similar catalytic domain structures with well-defined X, A, and B helices that (at least in TcSQS) are essential for activity. Notably, the A and B helices are not present in the homologous bacterial protein, CrtM, which is not a membrane protein. In AfSQS, there is, therefore, a large, hydrophilic helical domain, the C-domain, which is not found in any non-fungal SQSs. Based on the X-ray and bioinformatics results, we thus propose that the fungal SQSs are organized as illustrated in Figure 6a. What is attractive about the model is that the presence of the polar, non-buried (solvent-exposed) C-domain would appear to offer strong possibilities for SQS interacting with not only the membrane surface (acting as an in-plane membrane anchor) but also with other proteins involved in sterol biosynthesis, via electrostatic interactions.
Figure 6.

Proposed structural arrangement of the polar and non-polar (trans-membrane) helices in AfSQS based on JPRED4, HeliQuest, and RoseTTAFold results. (a) AfSQS model based on JPRED4 and HeliQuest results. (b) AfSQS model based on RoseTTAFold prediction, illustrated as an electrostatic charge surface computed using the PyMOL program. Blue = positively charged region, red = negatively charged region, and white = neutral (hydrophobic residues) region. Four other models are shown in Figure S2. (c) Error estimates in Ångström units for the model in panel (b) computed in the RoseTTAFold program.
We next attempted to see to what extent it might be possible to predict the 3D structure of AfSQS. We first used the Phyre2 program.23 There was an excellent prediction of the catalytic domain but no prediction of the other regions. We thus next used the deep learning-based modeling method, RoseTTAFold.24 There were five models predicted, and these are shown in Figure S2 together with their error estimates. We show the predicted structures as electrostatic potential surfaces (calculated in PyMOL25) in which red represents a negatively charged region, blue is a positively charged region, and white is a neutral (hydrophobic) region. The large catalytic domain was well predicted (1.08 Å/314 Cα atoms) and is connected via the polar helical domain to the (vertical, white) transmembrane region. The E-domain is helical and is connected to the C-terminus. All five predicted models have a very similar “L-shaped” TM/chain terminus arrangement, as proposed (Figure 6a) using the other computational methods, and one model is shown in Figure 6b (error estimates shown in Figure 6c) for comparison with the model shown in Figure 6a. There are large error estimates for the C-domain, the TM domain, and the C-terminus, presumably because the catalytic domain can rotate, as seen in Figure S2. It should also be noted that there is no common structure (such as a pocket) in the polar C-domain. That notwithstanding, there is general accord with the model shown in Figure 6a, with the L-shaped TM/E-helix region perhaps being an intrinsic feature that may facilitate binding to a membrane surface.
Comparisons with Other Membrane Proteins
The question then arises as to how similar are the non-catalytic domains found in AfSQSs (as well as the other SQSs) to those found in other membrane proteins. There are only a small number of membrane protein structures with what appear to be primarily structural (i.e., non-catalytic) TM or helical domains. One example is that of estrone sulfatase (ES; 562 amino acids), responsible for maintaining high levels of estrogens in breast tumor cells, which as with SQS is associated with the ER.19 Another is stannin, shown to mediate neuronal cell apoptosis mediated by Me3SnCl20 and more recently to inhibit the entry of human papillomavirus into the trans-Golgi network.26 The structures of both proteins are shown in Figure 7a,b (estrone sulfatase PDB ID code 1P49;19 stannin PDB ID code 1ZZA(20)), and the HeliQuest results are shown in Figure 7c–e. Clearly, the ⟨H⟩ and ⟨μH⟩ values are very similar to those found with AfSQS with ⟨H⟩ ∼1.1–1.3 and ⟨μH⟩ ∼0.1–0.2. In stannin, there is also a membrane surface-associated domain that has ⟨H⟩ ∼0.3 and ⟨μH⟩ ∼0.2.
Figure 7.

Structures and HeliQuest results for two other membrane proteins with TM helices. (a) Estrone sulfatase (ES) structure (PDB ID code 1P49). (b) Stannin structure (PDB ID code 1ZZA). (c) HeliQuest result for the cyan helix in ES. (d) HeliQuest result for the yellow helix in ES. (e) HeliQuest result for the TM helix in stannin.
As we noted above, one of the potential partners for interacting with SQS would be squalene epoxidase, SQLE, as suggested in a very early work.3,4 In this context, it is of interest to note that the X-ray structure of a catalytically active N-terminally truncated human SQLE (118–574) was recently reported27 and reviewed.28 The protein contains ligand-binding domains (as shown in green in Figure 8a (PDB ID code 6C6P);27 the FAD and NB-598 ligands are omitted for clarity) together with two “membrane-associated” helical domains, suggested to both be buried in the endoplasmic reticulum.28
Figure 8.

Polarity, hydrophobicity, and hydrophobic moments for the two C-terminal helices in human squalene epoxidase and a structural model for binding to a cell membrane. (a) HsSQLE (PDB ID 6C6P). (b) HeliQuest results for the penultimate (yellow) helix. (c) HeliQuest results for the terminal (cyan) helix. (d) Charged residues present in the two C-terminal helices. (e) Proposed model for membrane-binding based on rotations of the C-terminal helices.
Interestingly, the penultimate C-terminal helix is very hydrophobic (11% polar residues) and has a ⟨H⟩ of 1.147 and a ⟨μH⟩ of 0.154 (Figure 8b), very similar to what we find for the D-helix in AfSQS and the other TM proteins (∼10% polar residues, hydrophobicity ⟨H⟩ ∼1.3, and hydrophobic moment ⟨μH⟩ ∼0.2). The actual C-terminal helix in HsSQLE has 33% polar residues, a net charge of +2, a ⟨H⟩ of 0.691, and a ⟨μH⟩ of 0.394 (Figure 8c), very similar to what we find with the D-helix in TcSQS (33% polar residues, a net charge of +2, a ⟨H⟩ of 0.656, and a ⟨μH⟩ of 0.229). There are a total of six charged residues in the HsSQLE terminal helix and turn region (Figure 8d), suggesting that this region might be membrane surface-associated (Figure 8e), in which the putative TM domain (in yellow) has been rotated to interact with the membrane bilayer. While this is a speculative proposal, the % polar residues and ⟨H⟩ and ⟨μH⟩ values in the HsSQLE penultimate helix are very similar to those seen in the single AfSQS TM domain, helix D (and in the other TM proteins) while the terminal helix is far less hydrophobic and indeed has multiple charged residues (Figure 8d). However, it is also possible that the C-terminal helix shown in Figure 8e might be rotated “up” and be a TM helix since five of the six charged residues are at either end of the helix and could be at the solvent-exposed membrane surfaces. This arrangement, though unusual, might then help explain the nature of the more polar TM helix in TcSQS.
Finally, it should be noted that it is not yet clear whether SQS is a good target for antifungal development, at least as a monotherapy. Although inhibition of ergosterol biosynthesis and cell growth in Candida glabrata by depletion of its SQS gene (ERG9) is observed,29 cell growth is rescued by cholesterol, both in vitro and in mice. Related partial rescue effects in vitro are also seen in A. fumigatus with azole antifungals,30 and cholesterol-dependent C. glabrata has been found in clinical specimens.31 On the other hand, compounds such as the bisphosphonate that inhibits both ASfSQS as well as human SQS32 could result in lowering of both host cholesterol as well as pathogen ergosterol. In addition, it has been found that inhibiting SQS in neutrophils leads to formation of neutrophil extracellular traps (NETS),33 and NETS have antifungal activity.34
Conclusions
The results we have presented above are of interest for several reasons. First, we have obtained the structure of a fungal squalene synthase catalytic domain, both as the apo- and as a ligand (FSPP) bound form. Second, the structure obtained is very similar to that of T. cruzi SQS and contains highly superimposable X, A, and B helices. Third, the structure is of interest when compared with human SQS, HsSQS, which appears (based on computational predictions) to have a shorter B-helix, something that might lead to differences in ligand (inhibitor or protein) binding between human and pathogen proteins. Fourth, we identified a very polar, solvent-exposed (i.e., non-buried) long (∼35 residue) highly helical domain, the C-helical domain, that is unique to the fungal proteins. This is of interest since the fungal SQSs are thought to interact with other proteins (such as squalene epoxidase) involved in ergosterol biosynthesis, and this domain may be a target for inhibitor development. Fifth, using a deep learning program, we find a similar L-shaped TM/E-helix structure to that proposed using simpler modeling methods, suggesting that the E-helix/chain terminus may be involved in binding to the membrane surface. Sixth, we find that the transmembrane domains in the fungal and human SQSs have very similar properties (% polar residues and ⟨H⟩ and ⟨μH⟩ values) to the TM domains in estrone sulfatase and stannin as well as to the penultimate helix in HsSQLE, leading to proposed models for SQLE in which the two C-terminal helices seen in the crystal structure may form one or two transmembrane helices. Overall, the crystallographic and computational results provide new perspectives on the structures of the fungal squalene synthases and should encourage renewed efforts in experimental structure determinations.
Materials and Methods
Cloning, Expression, and Purification
The gene encoding the AfSQS gene (GenBank accession number: KOC07597.1) from A. flavus was chemically synthesized by GENE ray Biotech Co. (Shanghai, China), ligated into the pET46 vector, and transformed into Escherichia coli strain BL21(DE3). Cells were grown in the LB medium at 37 °C to an OD600 of ∼0.8 and then induced by 0.6 mM isopropyl β-d-thiogalactopyranoside (IPTG) at 16 °C for 20 h. Cells were then harvested by centrifugation at 5000g for 15 min and then resuspended in lysis buffer containing 25 mM Tris–HCl, pH 8.0, 150 mM NaCl, and 20 mM imidazole followed by disruption with a French Press. Cell debris was removed by centrifugation at 17,000g for 1 h. The supernatant was then applied to a Ni-NTA column with an FPLC system (GE Healthcare). AfSQS was eluted at ∼150 mM imidazole when using a 20–250 mM imidazole gradient and was then dialyzed against a buffer containing 25 mM Tris–HCl, pH 8.0, passed through a DEAE column pre-equilibrated with the same buffer, and then eluted with 25 mM Tris–HCl, pH 8.0, 250 mM NaCl. Purity was checked by SDS-PAGE analysis and was >95%. The purified protein was then concentrated to 10 mg/mL for crystallization screening.
Crystallization, Data Collection, Structure Determination, and Refinement
All crystallization experiments were conducted at 25 °C using the sitting-drop vapor-diffusion method. In general, 1 μL of AfSQS containing solution (25 mM Tris, 150 mM NaCl; 40 mg/mL) was mixed with 1 μL of reservoir solution in 48-well Cryschem Plates and then equilibrated against 100 μL of the reservoir solution. The optimized crystallization conditions were 1.1 M NaH2PO4 and 0.5 M K2HPO4. Within 2–3 days, the crystals reached a size suitable for X-ray diffraction. The AfSQS crystals in complex with FSPP were obtained by soaking with mother liquor containing 10 mM FSPP for 72 h.
All of the X-ray diffraction data sets were collected at beam line BL15A1 at the National Synchrotron Radiation Research Center (NSRRC). The crystals were mounted in a cryo-loop and soaked with cryoprotectant solution (1.1 M NaH2PO4, 0.5 M K2HPO4, 10% glycerol) prior to data collection at 100 K. The diffraction images were processed by using HKL2000.35 The crystal structure of AfSQS was solved by using the molecular replacement method with the Phaser program36 from the Phenix37 suite, using the structure of HsSQS (PDB code 1EZF) as a search model. Further refinement was carried out by using the programs phenix.refine38 and Coot.39 Prior to structure refinement, 5% randomly selected reflections were set aside for calculating Rfree as a monitor. All figures were prepared by using the PyMOL program.25
Computational Aspects
We used the JPRED program17 for predicting helical, sheet, disordered, and solvent-exposed residues; the Clustal Omega program18 for sequence alignments; the HeliQuest program21 for ⟨H⟩ and ⟨μH⟩ predictions; and the RoseTTAFold program24 for 3D structure predictions.
Acknowledgments
This work was supported by the National Key Research and Development Program of China (2021YFC2102400), Tianjin Synthetic Biotechnology Innovation Capacity Improvement Project (TSBICIP-PTJJ-008, TSBICIP-KJGG-009-01, TSBICIP-KJGG-002-06, and TSBICIP-IJCP-003), and by the University of Illinois Foundation and a Harriet A. Harlin Professorship (to E.O.).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c01924.
Clustal Omega alignment of T. cruzi SQS, Homo sapiens SQS, A. flavus SQS, and C. albicans SQS; electrostatic surfaces and structural error estimates for five RoseTTAFold structure models of AfSQS (PDF)
Author Contributions
# S.R.M. and N.S. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Petranyi G.; Ryder N. S.; Stütz A. Allylamine derivatives: new class of synthetic antifungal agents inhibiting fungal squalene epoxidase. Science 1984, 224, 1239–1241. 10.1126/science.6547247. [DOI] [PubMed] [Google Scholar]
- a Magnin D. R.; Biller S. A.; Chen Y.; Dickson J. K. Jr.; Fryszman O. M.; Lawrence R. M.; Logan J. V.; Sieber-McMaster E. S.; Sulsky R. B.; Traeger S. C.; Hsieh D. C.; Lan S. J.; Rinehart J. K.; Harrity T. W.; Jolibois K. G.; Kunselman L. K.; Rich L. C.; Slusarchyk D. A.; Ciosek C. P. Jr. α-Phosphonosulfonic acids: potent and selective inhibitors of squalene synthase. J. Med. Chem. 1996, 39, 657–660. 10.1021/jm9507340. [DOI] [PubMed] [Google Scholar]; b Stein E. A.; Bays H.; O’Brien D.; Pedicano J.; Piper E.; Spezzi A. Lapaquistat acetate: development of a squalene synthase inhibitor for the treatment of hypercholesterolemia. Circulation 2011, 123, 1974–1985. 10.1161/CIRCULATIONAHA.110.975284. [DOI] [PubMed] [Google Scholar]
- Kribii R.; Arró M.; Del Arco A.; González V.; Balcells L.; Delourme D.; Ferrer A.; Karst F.; Boronat A. Cloning and characterization of the Arabidopsis thaliana SQS1 gene encoding squalene synthase--involvement of the C-terminal region of the enzyme in the channeling of squalene through the sterol pathway. Eur. J. Biochem. 1997, 249, 61–69. 10.1111/j.1432-1033.1997.00061.x. [DOI] [PubMed] [Google Scholar]
- M’Baya B.; Karst F. In vitro assay of squalene epoxidase of Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 1987, 147, 556–564. 10.1016/0006-291X(87)90967-3. [DOI] [PubMed] [Google Scholar]
- Linscott K. B.; Niehaus T. D.; Zhuang X.; Bell S. A.; Chappell J. Mapping a kingdom-specific functional domain of squalene synthase. Biochim. Biophys. Acta 2016, 1861, 1049–1057. 10.1016/j.bbalip.2016.06.008. [DOI] [PubMed] [Google Scholar]
- Liu C. I.; Jeng W. Y.; Chang W. J.; Shih M. F.; Ko T. P.; Wang A. H.-J. Structural insights into the catalytic mechanism of human squalene synthase. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 231–241. 10.1107/S1399004713026230. [DOI] [PubMed] [Google Scholar]
- Shang N.; Li Q.; Ko T. P.; Chan H. C.; Li J.; Zheng Y.; Huang C. H.; Ren F.; Chen C. C.; Zhu Z.; Galizzi M.; Li Z. H.; Rodrigues-Poveda C. A.; Gonzalez-Pacanowska D.; Veiga-Santos P.; de Carvalho T. M.; de Souza W.; Urbina J. A.; Wang A. H.; Docampo R.; Li K.; Liu Y. L.; Oldfield E.; Guo R. T. Squalene synthase as a target for Chagas disease therapeutics. PLoS Pathog. 2014, 10, e1004114 10.1371/journal.ppat.1004114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandit J.; Danley D. E.; Schulte G. K.; Mazzalupo S.; Pauly T. A.; Hayward C. M.; Hamanaka E. S.; Thompson J. F.; Harwood H. J. Jr. Crystal structure of human squalene synthase. A key enzyme in cholesterol biosynthesis. J. Biol. Chem. 2000, 275, 30610–30617. 10.1074/jbc.M004132200. [DOI] [PubMed] [Google Scholar]
- Lin F. Y.; Liu C. I.; Liu Y. L.; Zhang Y.; Wang K.; Jeng W. Y.; Ko T. P.; Cao R.; Wang A. H.; Oldfield E. Mechanism of action and inhibition of dehydrosqualene synthase. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 21337–21342. 10.1073/pnas.1010907107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X.; Hu Y.; Zheng Y.; Zhu W.; Li K.; Huang C. H.; Ko T. P.; Ren F.; Chan H. C.; Nega M.; Bogue S.; López D.; Kolter R.; Götz F.; Guo R. T.; Oldfield E. Structural and functional analysis of Bacillus subtilis YisP reveals a role of its product in biofilm production. Chem. Biol. 2014, 21, 1557–1563. 10.1016/j.chembiol.2014.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. I.; Liu G. Y.; Song Y.; Yin F.; Hensler M. E.; Jeng W. Y.; Nizet V.; Wang A. H.; Oldfield E. A cholesterol biosynthesis inhibitor blocks Staphylococcus aureus virulence. Science 2008, 319, 1391–1394. 10.1126/science.1153018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. I.; Jeng W. Y.; Chang W. J.; Ko T. P.; Wang A. H. Binding modes of zaragozic acid A to human squalene synthase and staphylococcal dehydrosqualene synthase. J. Biol. Chem. 2012, 287, 18750–18757. 10.1074/jbc.M112.351254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J. F.; Danley D. E.; Mazzalupo S.; Milos P. M.; Lira M. E.; Harwood H. J. Jr. Truncation of human squalene synthase yields active, crystallizable protein. Arch. Biochem. Biophys. 1998, 350, 283–290. 10.1006/abbi.1997.0502. [DOI] [PubMed] [Google Scholar]
- Sealey-Cardona M.; Cammerer S.; Jones S.; Ruiz-Pérez L. M.; Brun R.; Gilbert I. H.; Urbina J. A.; González-Pacanowska D. Kinetic characterization of squalene synthase from Trypanosoma cruzi: Selective inhibition by quinuclidine derivatives. Antimicrob. Agents Chemother. 2007, 51, 2123–2129. 10.1128/AAC.01454-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.-C.; Malwal S. R.; Han X.; Liu W.; Ma L.; Zhai C.; Dai L.; Huang J.-W.; Shillo A.; Desai J.; Ma X.; Zhang Y.; Guo R.-T.; Oldfield E. Terpene Cyclases and Prenyltransferases: Structures and Mechanisms of Action. ACS Catal. 2021, 11, 290–303. 10.1021/acscatal.0c04710. [DOI] [Google Scholar]
- Miller-Vedam L. E.; Bräuning B.; Popova K. D.; Schirle Oakdale N. T.; Bonnar J. L.; Prabu J. R.; Boydston E. A.; Sevillano N.; Shurtleff M. J.; Stroud R. M.; Craik C. S.; Schulman B. A.; Frost A.; Weissman J. S. Structural and mechanistic basis of the EMC-dependent biogenesis of distinct transmembrane clients. Elife 2020, 9, e62611 10.7554/eLife.62611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drozdetskiy A.; Cole C.; Procter J.; Barton G. J. JPred4: a protein secondary structure prediction server. Nucleic Acids Res. 2015, 43, W389–W394. 10.1093/nar/gkv332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F.; Wilm A.; Dineen D.; Gibson T. J.; Karplus K.; Li W.; Lopez R.; McWilliam H.; Remmert M.; Söding J.; Thompson J. D.; Higgins D. G. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Guzman F. G.; Higashiyama T.; Pangborn W.; Osawa Y.; Ghosh D. Structure of human estrone sulfatase suggests functional roles of membrane association. J. Biol. Chem. 2003, 278, 22989–22997. 10.1074/jbc.M211497200. [DOI] [PubMed] [Google Scholar]
- Buck-Koehntop B. A.; Mascioni A.; Buffy J. J.; Veglia G. Structure, dynamics, and membrane topology of stannin: a mediator of neuronal cell apoptosis induced by trimethyltin chloride. J. Mol. Biol. 2005, 354, 652–665. 10.1016/j.jmb.2005.09.038. [DOI] [PubMed] [Google Scholar]
- Gautier R.; Douguet D.; Antonny B.; Drin G. HELIQUEST: a web server to screen sequences with specific α-helical properties. Bioinformatics 2008, 24, 2101–2102. 10.1093/bioinformatics/btn392. [DOI] [PubMed] [Google Scholar]
- Schiffer M.; Edmundson A. B. Use of helical wheels to represent the structures of proteins and to identify segments with helical potential. Biophys. J. 1967, 7, 121–135. 10.1016/S0006-3495(67)86579-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley L. A.; Mezulis S.; Yates C. M.; Wass M. N.; Sternberg M. J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek M.; DiMaio F.; Anishchenko I.; Dauparas J.; Ovchinnikov S.; Lee G. R.; Wang J.; Cong Q.; Kinch L. N.; Schaeffer R. D.; Millán C.; Park H.; Adams C.; Glassman C. R.; DeGiovanni A.; Pereira J. H.; Rodrigues A. V.; van Dijk A. A.; Ebrecht A. C.; Opperman D. J.; Sagmeister T.; Buhlheller C.; Pavkov-Keller T.; Rathinaswamy M. K.; Dalwadi U.; Yip C. K.; Burke J. E.; Garcia K. C.; Grishin N. V.; Adams P. D.; Read R. J.; Baker D. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. 10.1126/science.abj8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC. [Google Scholar]
- Lipovsky A.; Erden A.; Kanaya E.; Zhang W.; Crite M.; Bradfield C.; MacMicking J.; DiMaio D.; Schoggins J. W.; Iwasaki A. The cellular endosomal protein stannin inhibits intracellular trafficking of human papillomavirus during virus entry. J. Gen. Virol. 2017, 98, 2821–2836. 10.1099/jgv.0.000954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padyana A. K.; Gross S.; Jin L.; Cianchetta G.; Narayanaswamy R.; Wang F.; Wang R.; Fang C.; Lv X.; Biller S. A.; Dang L.; Mahoney C. E.; Nagaraja N.; Pirman D.; Sui Z.; Popovici-Muller J.; Smolen G. A. Structure and inhibition mechanism of the catalytic domain of human squalene epoxidase. Nat. Commun. 2019, 10, 97. 10.1038/s41467-018-07928-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A. J.; Chua N. K.; Yan N. The shape of human squalene epoxidase expands the arsenal against cancer. Nat. Commun. 2019, 10, 888. 10.1038/s41467-019-08866-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama H.; Izuta M.; Nakayama N.; Arisawa M.; Aoki Y. Depletion of the squalene synthase (ERG9) gene does not impair growth of Candida glabrata in mice. Antimicrob. Agents Chemother. 2000, 44, 2411–2418. 10.1128/AAC.44.9.2411-2418.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Q.; Hassan S. A.; Wilson W. K.; Han X. Y.; May G. S.; Tarrand J. J.; Matsuda S. P. T. Cholesterol import by Aspergillus fumigatus and its influence on antifungal potency of sterol biosynthesis inhibitors. Antimicrob. Agents Chemother. 2005, 49, 518–524. 10.1128/AAC.49.2.518-524.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazen K. C.; Stei J.; Darracott C.; Breathnach A.; May J.; Howell S. A. Isolation of cholesterol-dependent Candida glabrata from clinical specimens. Diagn. Microbiol. Infect. Dis. 2005, 52, 35–37. 10.1016/j.diagmicrobio.2004.12.006. [DOI] [PubMed] [Google Scholar]
- Song J.; Shang N.; Baig N.; Yao J.; Shin C.; Kim B. K.; Li Q.; Malwal S. R.; Oldfield E.; Feng X.; Guo R. T. Aspergillus flavus squalene synthase as an antifungal target: Expression, activity, and inhibition. Biochem. Biophys. Res. Commun. 2019, 512, 517–523. 10.1016/j.bbrc.2019.03.070. [DOI] [PubMed] [Google Scholar]
- a Lin F. Y.; Zhang Y.; Hensler M.; Liu Y. L.; Chow O. A.; Zhu W.; Wang K.; Pang R.; Thienphrapa W.; Nizet V.; Oldfield E. Dual dehydrosqualene/squalene synthase inhibitors: leads for innate immune system-based therapeutics. ChemMedChem 2012, 7, 561–564. 10.1002/cmdc.201100589. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Chow O. A.; von Köckritz-Blickwede M.; Bright A. T.; Hensler M. E.; Zinkernagel A. S.; Cogen A. L.; Gallo R. L.; Monestier M.; Wang Y.; Glass C. K.; Nizet V. Statins enhance formation of phagocyte extracellular traps. Cell Host Microbe 2010, 8, 445–454. 10.1016/j.chom.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban C. F.; Nett J. E. Neutrophil extracellular traps in fungal infection. Semin. Cell Dev. Biol. 2019, 89, 47–57. 10.1016/j.semcdb.2018.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. [20] Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 76, 307–326. 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D.; Afonine P. V.; Baker M. L.; Bunkoczi G.; Chen V. B.; Croll T. I.; Hintze B.; Hung L.-W.; Jain S.; McCoy A. J.; Moriarty N. W.; Oeffner R. D.; Poon B. K.; Prisant M. G.; Read R. J.; Richardson J. S.; Richardson D. C.; Sammito M. D.; Sobolev O. V.; Stockwell D. H.; Terwilliger T. C.; Urzhumtsev A. G.; Videau L. L.; Williams C. J.; Adams P. D. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D Biol. Crystallogr. 2019, 75, 861–877. 10.1107/S2059798319011471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine P. V.; Grosse-Kunstleve R. W.; Echols N.; Headd J. J.; Moriarty N. W.; Mustyakimov M.; Terwilliger T. C.; Urzhumtsev A.; Zwart P. H.; Adams P. D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 352–367. 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.