

Abstract
Recombination contributes to the genetic diversity found in coronaviruses and is known to be a prominent mechanism whereby they evolve. It is apparent, both from controlled experiments and in genome sequences sampled from nature, that patterns of recombination in coronaviruses are non-random and that this is likely attributable to a combination of sequence features that favour the occurrence of recombination break points at specific genomic sites, and selection disfavouring the survival of recombinants within which favourable intra-genome interactions have been disrupted. Here we leverage available whole-genome sequence data for six coronavirus subgenera to identify specific patterns of recombination that are conserved between multiple subgenera and then identify the likely factors that underlie these conserved patterns. Specifically, we confirm the non-randomness of recombination break points across all six tested coronavirus subgenera, locate conserved recombination hot- and cold-spots, and determine that the locations of transcriptional regulatory sequences are likely major determinants of conserved recombination break-point hotspot locations. We find that while the locations of recombination break points are not uniformly associated with degrees of nucleotide sequence conservation, they display significant tendencies in multiple coronavirus subgenera to occur in low guanine-cytosine content genome regions, in non-coding regions, at the edges of genes, and at sites within the Spike gene that are predicted to be minimally disruptive of Spike protein folding. While it is apparent that sequence features such as transcriptional regulatory sequences are likely major determinants of where the template-switching events that yield recombination break points most commonly occur, it is evident that selection against misfolded recombinant proteins also strongly impacts observable recombination break-point distributions in coronavirus genomes sampled from nature.
Keywords: Coronavirus, Phylogenetics, Evolution, Recombination, Selection
Introduction
Coronaviruses are a family of vertebrate-infecting single-stranded, positive-sense RNA viruses with genomes ∼27–32 kb in length. The family has four genera—Alphacoronavirus, Betacoronavirus, Gammacoronavirus, and Deltacoronavirus—each of which has been further subdivided into a number of subgenera such as Pedacovirus in the genus Alphacoronavirus, and Merbecovirus, Embecovirus, Nobecovirus, and Sarbecovirus in the genus Betacoronavirus, and Igacovirus in the genus Gammacoronavirus (Coronaviridae—Positive Sense RNA Viruses—Positive Sense RNA Viruses (2011)—ICTV 2011). Besides Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), the Sarbecovirus member that causes Coronavirus Disease 2019 (COVID-19), there are four other known Betacoronavirus lineages and two known Alphacoronavirus lineages that either cause—or have caused—epidemiologically significant disease outbreaks in humans.
Coronavirus genomes generally contain seven to ten genes, often with varying arrangements and compositions (Fig. 1) (Lai 1996). The largest gene, ORF1ab, encodes multiple non-structural proteins which are involved in viral transcription, replication, proteolytic processing, modulation of host gene expression, and the suppression of host immune responses (Emam et al. 2021). Directly downstream of ORF1ab in most known coronavirus genomes is the Spike (S) gene (although in Embecoviruses, for example, a haemagglutinin-esterase gene separates ORF1ab and the S gene). Spike is the structural glycoprotein found on the outside of coronavirus particles that gives them their iconic crown-like protrusions. Spike binds to cell membrane receptors and mediates virus entry into host cells. It is considered a class I fusion protein in that it contains both a receptor-binding domain (called S1) and a domain for mediating the membrane fusion process (called S2) (White et al. 2008; Xia et al. 2020). The Spike of SARS-CoV-2 has become a target in the development of vaccines and therapeutic drugs for COVID-19, due both to its importance during the viral infection cycle and it being the primary target of host immune responses (Krumm et al. 2021).
Figure 1.
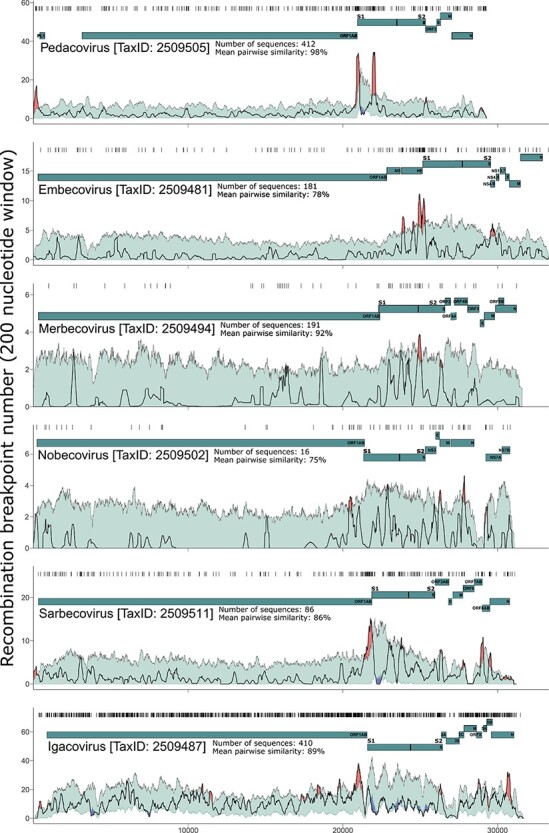
Variation across coronavirus genomes in the densities of detectable recombination break points. All detected break-point positions are indicated directly above each graph with vertical lines. A gene-map is shown as lines beneath the densities. The grey-lined, green areas indicate 99 per cent bounds of expected degrees of break-point clustering under random recombination. Areas where the dark/black lines (break-point number per 200 nucleotide window) have emerged above the green areas, are considered potential recombination hotspots, and are marked (brightly) in red. Areas where the black lines drop below the green areas are considered potential recombination cold-spots, and are marked in (cold) blue.
Although coronaviruses have low mutation rates relative to those of other single-stranded RNA viruses (Denison et al. 2011; Jaroszewski et al. 2021), coronavirus populations are characterized by high degrees of genetic diversity (Liu et al. 2017). Much of this genetic diversity is likely generated and maintained by high rates of within-species (Dudas and Rambaut 2016; Su et al. 2016; Anthony et al. 2017; Forni, Cagliani and Sironi 2020) and between-species genetic recombination (Wesley 1999; Decaro, Mari, and Elia et al. 2015; Wang, Lin, and Guo et al. 2015; Wang, Lin, and Zhang et al. 2020). The first credible reports of recombination in coronaviruses were made in the mid-to-late 1980s and focused on mixed in vitro and in vivo infections of different Murine mouse hepatitis virus strains (Makino, Keck, and SA et al. 1986; Keck, Matsushima, and Makino et al. 1988a; Keck et al. 1988b; Banner and Lai 1991). By the year 2000, comparative analyses of coronavirus genomes sampled from natural infections had yielded convincing evidence that recombination, particularly between divergent coronaviruses within individual subgenera, is a major contributor to coronavirus evolution (Kusters et al. 1990; Wang, Junker, and Collisson 1993; Jia et al. 1995; Lee and Jackwood 2000). For example, a complex recombinant history is evident between the Alphacoronavirus-1 species involving canine coronavirus, which primarily infects dogs (however, see recent exceptions of canine CoV infections in humans: (Lednicky et al. 2021; Vlasova et al. 2021; Zehr, Kosakovsky Pond, and Martin 2021)), transmissible gastroenteritis virus, infecting pigs and feline coronavirus from cats (Herrewegh et al. 1998; Decaro et al. 2009).
The most common mechanism of recombination in coronaviruses (and many other RNA viruses too) is known as copy-choice, where a viral RNA-dependent RNA polymerase (RdRp) is interrupted during replication, drops off the RNA template that it was copying, and re-engages with a different RNA template at a homologous position before resuming replication (Cheng and Nagy 2003). Such template switches during replication yield recombinant daughter genomes with different regions of sequence being derived from two different ‘parental’ genomes. The genome sites at which template switches occur are referred to as recombination break points.
Recombination likely provides viruses with more evolutionary options than would be available to them by mutation alone (Crameri et al. 1998; Simon-Loriere et al. 2009). While it is expected that many newly arising mutations within genetically compact viral genomes (such as those of coronaviruses) will have negative fitness consequences, so too will many of the recombination events that occur between genetically divergent genomes (Banner and Lai 1991; Drummond, Silberg, and Meyer et al. 2005). By transferring pieces of genomes into genomic backgrounds with which they did not co-evolve, recombination will frequently run the risk of disrupting favourable co-evolved intra-genome interactions (commonly referred to as epistatic interactions). Examples of favourable co-evolved intra-genome interactions that could be disrupted by recombination include those between nucleotides that base-pair to form biologically functional genomic secondary structures, those between pairs of amino acids that interact to mediate protein folding, those between the binding domains on protein surfaces that mediate multi-protein complex formation, and those between sequence-specific nucleic acid binding domains and nucleotide sequence motifs that mediate gene regulation and genome replication (Martin et al. 2005b). However, since recombination generally occurs between fully functioning genomes, the range of potential negative fitness consequences of recombination are, in general, expected to be less extreme than those that might occur due to newly arising mutations (Drummond et al. 2005). In fact, genetic recombination between closely related viruses almost certainly helps defend against the accumulation within genomes of mildly deleterious mutations that, in high enough numbers, might otherwise have serious fitness consequences (Goldstein et al. 2021; Muller 1964; Woo et al. 2010; Hussin et al. 2015).
Here we analyse patterns of recombination evident in whole-genome datasets drawn from one Alphacoronavirus subgenus, one Gammacoronavirus subgenus, and four Betacoronavirus subgenera. We confirm previous reports that natural recombination between genetically divergent coronaviruses is common and find strong evidence that detectable recombination break-point sites are not randomly distributed across coronavirus genomes. Specifically, we demonstrate the likely occurrence of break-point hot- and cold-spots, some of which are conserved across multiple coronavirus groups. Further, we find detectable associations across multiple different coronavirus subgenera between recombination break-point locations and various sequence features that might impact the mechanistic predisposition of certain genome sites to recombine more than others (such as decreased guanine-cytosine content and the locations of transcriptional regulatory sequences). Concordant with observations made in some of the earliest reported coronavirus recombination experiments (Banner and Lai 1991), we also find evidence across multiple subgenera that selection differentially favours the survival of recombinants based on the genome sites at which break points occur (such as at the edges of genes or in intergenic regions relative to the middle portions of genes).
Methods
Data collection
All publicly available near full-length genomic sequences for viruses in six well-sampled coronavirus subgenera (Igacovirus, Embecovirus, Merbecovirus, Nobecovirus, Pedacovirus, and Sarbecovirus) were downloaded from the NCBI Virus (Hatcher et al. 2017), CNCB (Song et al. 2021), and CoVDB (Zhu et al. 2021) databases between February and May 2021. Each of the six subgenus-level datasets was aligned with MAFFT using default settings (Katoh and Standley 2013). All but one sequence in groups of sequences sharing more than 99 per cent nucleotide sequence identity were removed to yield datasets for recombination analysis containing between 16 and 412 genome sequences sharing ≥75 per cent similarity (Supplementary Table S1; Supplementary data).
Recombination detection
Recombination was detected and analysed using Recombination detection program 5 (RDP5) (Martin et al. 2021) with default settings except that sequences were treated as linear. Each of the six coronavirus datasets were analysed for recombination using a fully exploratory automated scan with the RDP (Martin and Rybicki 2000), GENECONV (Sawyer 1989), and MaxChi (Maynard Smith 1992) methods to detect recombination signals (i.e. these were used as ‘primary scanning methods’), and the Bootscan (Martin et al. 2005a), Chimaera (Pettersen et al. 2004), SiScan (Gibbs, Armstrong, and Gibbs 2000), and 3Seq (Lam, Ratmann, and Boni 2018) methods to verify the signals (these latter four methods being used as ‘secondary scanning methods’). From among the individual recombination signals that were each detectable by four or more of these methods, RDP5 refined the positions of detected recombination break points using a hidden Markov model (HMM)-based approach (described in detail in the RDP manual at http://web.cbio.uct.ac.za/∼darren/RDP4Manual.pdf) and determined a plausible near-minimal subset of unique recombination events that would be needed to account for all of the detected recombination signals. Each of the unique recombination events detected by RDP5 in each of the six analysed coronavirus subgenera datasets was characterized by: (1) a 5ʹ and 3ʹ pair of maximum likelihood break-point locations and their associated probability distributions, (2) a list of one or more sequences carrying evidence of the recombination event (multiple sequences can have evidence of the same recombination event if the event occurred in a common ancestor), and (3) a list of analysed sequences that are closely related enough to the actual parents of the recombinant that they could be used as proxies for the actual parents to detect the recombination events. The overall-recombination patterns in the six subgenera datasets were visualized using recombination region count matrices produced using RDP5. These matrices indicate the numbers of detected recombination events that separated individual pairs of genome sites from one another.
Recombination break point hot- and cold-spot tests
For each of the subgenera, a recombination break-point distribution map was constructed from the lists of 5ʹ and 3ʹ break-point probability distributions associated with each detected recombination event. This was done by sliding a 200-nt window, one nucleotide at a time, along the full length of the analysed alignment, summing the probabilities of all identified break points falling within the window, and plotting these counts at the nucleotide coordinate at the centre of the window. A previously used (Heath et al. 2006; Lytras et al. 2022) permutation test implemented in RDP5 was then used to identify recombination break-point clustering patterns that varied significantly from expectations under random recombination. This test involved:
Randomly shuffling the break-point locations of each of the observed recombination events in the order in which they were ranked by RDP5 (primarily from most to least probable) while maintaining the spacing between 5ʹ and 3ʹ break-point pairs (all detected recombination events have two called break points) with respect to the numbers of polymorphic nucleotide sites separating the break-point pairs within the triplet of analysed sequences used to detect the recombination event. Specifically, in the context of the isolated triplet of sequences used to detect a recombination event, the 5ʹ and 3ʹ break-point pairs of the detected event will be separated by a particular number of polymorphic nucleotide sites (d). If the number of sites that are polymorphic between members of the triplet is p then RDP5 chooses a random number between 1 and p-d to place the 5ʹ break-point location, f (i.e. the break point is placed in relation to the number of polymorphic nucleotide sites). The 3ʹ break-point location, t, is placed at site f + d. The break points for this event in the permuted dataset are then tentatively placed at F and T, the sites in the original alignment that respectively fall midway between the coordinates in the alignment corresponding to polymorphic sites f and f-1 for the 5ʹ break point and midway between the coordinates in the alignment corresponding to polymorphic sites t and t + 1 for the 3ʹ break point. Therefore, while the spacing of the break points is maintained with respect to the string of polymorphic nucleotide sites that were used by RDP5 to originally detect a recombination signal, the spacing of the 5ʹ and 3ʹ break points in the alignment will not necessarily be maintained. This break-point randomization approach accounts for varying frequencies across alignments of polymorphic nucleotide sites that could potentially reveal evidence of recombination and therefore also accounts for the fact that recombination events can be more easily detected, and the recombination break-point sites involved can be more accurately located, in genome regions containing higher frequencies of polymorphic nucleotide sites.
Ensuring that in instances where individual recombinant sequences contained evidence of multiple independent recombination events, the regions bounded by 5ʹ and 3ʹ break-point pairs for those events did not overlap to a greater or lesser degree than those observed in the actual recombinants (i.e. the spacings of all the 5ʹ and 3ʹ break-point locations of all overlapping events detected within a single sequence were maintained). This meant that with each successive randomized recombination event within a sequence containing evidence of multiple recombination events, the valid locations of 5ʹ and 3ʹ break-point locations became more constrained. Specifically, if in a given sequence the genome region bounded by the 5ʹ and 3ʹ break points of a randomly placed recombination event overlapped with the region bounded by the 5ʹ and 3ʹ break points of a previous randomly placed recombination event, then the current randomized 5ʹ break-point location was deemed to be invalid and other tentative random 5ʹ break-point locations were repeatedly chosen (as in (1) above) until a valid location was found.
Ensuring that in instances where break points were flagged as having undetermined positions in the actual dataset (such as break points called at the start/end of the alignment or at sites that were overprinted by subsequent recombination events), these were excluded from break-point counts. This was necessary because coronavirus genomes are linear and it is possible that recombination events between them will involve just one break point. In such instances a second ‘uncalled’ break point is placed at the start or end of the alignment (for accounting purposes). These ‘uncalled’ break-point positions were labelled as such in the permuted datasets and did not contribute to break-point counts. Similarly, in instances where a break-point was left uncalled in the actual dataset because a detected recombination event was immediately adjacent to the break point of another detected recombination event in the same sequence (indicating that one of the break points for one of the events was likely overprinted by a subsequent recombination event), these break points were also labelled as uncalled in the permuted datasets.
Making recombination break-point distribution maps for each permuted dataset using exactly the same approach as that used for the actual dataset.
-
Identifying unusually high or low degrees of break-point clustering in the actual dataset as those window coordinates where the break-point probability sums of the actual dataset fell outside the bounds of those determined at that coordinate for 99 per cent of the permuted datasets. With this test, unusually high degrees of break-point clustering (i.e. greater than 99 per cent of the permuted datasets at a given genome site) are suggestive of recombination hotspots, whereas unusually low degrees of clustering (i.e. lower than 99 per cent of the permuted datasets at a given genome site) are suggestive of recombination cold-spots.
It is important to stress, that this break-point clustering test is not conservative; because of an unavoidable multiple testing issue, given the lengths and degrees of diversity of the analysed coronavirus genomes, it is expected that between one and three hotspot-like clusters of break points would be detectable in each of the datasets even under completely random recombination (Lytras et al. 2022). We, therefore, referred to hotspots detected by this test in individual datasets as ‘potential hotspots’ and required that for a particular genome site to be defined as an actual statistically-supported hotspot, potential hotspots needed to be detectable at a homologous site in two or more of the different analysed subgenus datasets.
Comparing recombination break-point counts between pairs of pre-defined genome regions
We used a version of the break-point clustering hot- and cold-spot test that compared observed break-point numbers in two preselected groups of sites in an analysed alignment (Lefeuvre et al. 2009). Since the original recombination break-point distribution test determined whether the numbers of break points observed in 200-nt sliding windows were greater or lesser than chance under random recombination, the test relied on the detection of sufficient break points for statistically implausible clusters of break points to emerge. As the number of detected recombination break points varied widely between the different coronavirus datasets (ranging from 65 for the Merbecoviruses and 1703 for the Igacoviruses), the power of the test varied substantially. In an adapted version of the test, we partitioned the sites in each of the six datasets into two large subsets and directly compared observed break-point numbers in each of the site subsets to those expected under random recombination. We specifically compared densities of break points falling at: (1) non-protein-coding sites vs protein-coding sites; (2) the beginning and ending 5 per cent of sites within individual protein-encoding regions vs the middle 90 per cent of these regions (in the case of ORF1ab we defined protein-encoding-regions as those encoding individual post-translational protein cleavage products), (3) genome sites encoding a particular protein vs those encoding all other proteins within the genome and (4) sites within a specified number of nucleotides (2, 9, 21, 46) of a transcriptional regulatory sequence vs those in the remainder of the genome.
Testing for associations between GC content or pairwise sequence similarity and recombination break-point sites
A further modification of the break-point clustering hot- and cold-spot test was used to test for associations between break-point sites (specifically break-point probability distributions) and: (1) guanine + cytosine (GC) content and (2) pairwise sequence similarity (Simon-Loriere et al. 2010). In this test average GC proportions or pairwise sequence similarities of sites between a specified number of nucleotides (either 10 or 20) of every site in the genome across all possible sequence pairs were determined. Break-point probabilities at each site were multiplied with the GC proportion or pairwise similarity associated with that site and summed across all sites. These sums for the real datasets were then compared with the corresponding sums from the permuted datasets. For each analysed subgenus dataset the proportion of permuted datasets with sums higher than or equal to the real dataset were reported as the probability that there was no association between break-point positions and either higher GC proportions or higher degrees of pairwise similarity. Conversely, the proportion of permuted datasets with sums lower than or equal to those determined for the real dataset was reported as the probability that there was no association between break-point positions and either lower GC proportions or lower degrees of pairwise sequence similarity.
Identification of potential transcriptional regulatory sequences
SuPER was used to detect transcriptional regulatory sequence leader (TRS-L) sites and a custom Python (Rossum and Drake 2010) script was used to infer transcriptional regulatory sequence body (TRS-B) sites (Yang et al. 2021). For the algorithm implemented in SuPER to infer the subgenomic mRNA positions without RNA-seq data, annotation files and reference sequence files were downloaded from NCBI in September 2021 for the best-sampled species in each of the six coronavirus subgenus datasets. A Python script (https://github.com/phillipswanepoel/trsb-finder) was used to search for potential TRS-B sites in each subgenus dataset, following the methodology used in SuPER, which involved searching for all occurrences of sub-sequences with a Levenshtein distance of one or zero from the TRS-Leader sequence (Yang et al. 2021). These potential TRS-B sites were then filtered, removing all the sites not conserved across at least 75 per cent of the analysed sequences and removing upstream sites when multiple sites were found in close proximity 5ʹ of the start of the same ORF. This filtered siteset was then tested for association with break-point positions in each of the six analysed subgenera datasets using RDP5.
Given that neither the TRS distributions nor the break-point distributions were random in any of the analysed datasets and that both TRS sites and recombination break-point clusters occurred at the edges of coronavirus genes, we anticipated that the association test could have a high false-positive rate. To estimate the false discovery rate (FDR) of the break-point association test, a custom Python script (https://github.com/phillipswanepoel/trsb-finder) was used to generate randomly permuted versions of TRS-B site locations, for each of the subgenera alignments. As input, the script takes a coronavirus subgenus alignment and associated TRS-B sites, then outputs an RDP5 readable siteset file containing the permuted nucleotide positions. These positions are calculated by collectively shifting all the TRS-B sites by some number of nucleotides (which preserves their spacing), varied randomly between one and the length of the analysed alignment. If a new shifted TRS position was beyond the end of the genome, the position was ‘wrapped’ around to the other end of the genome. Two hundred permuted TRS-B site-sets were tested and the average estimated FDR across all datasets for the association between break point and TRS sites was 17.27 per cent (i.e. 17.27 per cent of the analyses with ‘shifted’ TRS-B sites yielded a significant association—with a P-value < 0.05—between these sites and observed break-point positions). Given that the FDRs for individual subgenera datasets ranged from 5 per cent to 29 per cent, we only considered associations detected between TRS-B and break-point sites as being significant if they were detected in multiple datasets.
Protein folding disruption test
To test whether the observed recombination events were less disruptive of protein folding than would be expected if recombination break points were randomly distributed, the SCHEMA test (Meyer et al. 2003; Lefeuvre et al. 2007), implemented in RDP5, was used to examine all protein-coding regions with associated publicly available high-resolution atomic coordinate data (obtained from the Protein Data Bank; https://www.rcsb.org/ (Berman, Henrick, and Nakamura 2003)) and within which more than ten recombination break points were detected. These stipulations were required to ensure that the test would have sufficient power to detect whether observed recombinants displayed significantly lower degrees of protein folding disruption with the SCHEMA test than would be expected under random recombination. Of all 56 unique encoded proteins for which structural data was available (across all subgenera), only Spike was amenable to further analysis. Specifically, four subgenera (Pedacovirus, Merbecovirus, Sarbecovirus, and Igacovirus) had both available Spike atomic coordinate structural data and >10 detected recombination break points in the portion of the S gene corresponding to the structural data.
The SCHEMA test involves identifying potential interactions that occur between amino acid residues within folded proteins (in our case pairs of non-hydrogen atoms from different amino acids within 4.5 Å of one another) and counting the numbers of interacting amino acid pairs within a chimaera of two parental amino acid sequences, where the chimaera has a different pair of amino acids than both parents. The 4.5 Å interaction cut-off (the default setting) corresponds to approximately five to eight potential pairwise interactions per residue. The counts of potentially altered pairwise amino acid interactions (called the disruption or E-score) that the SCHEMA method calculates have been shown to strongly correlate with observed degrees of fold disruption within chimaeric proteins (Meyer et al. 2003). To determine whether observed recombinants expressed chimaeric proteins with significantly lower E-scores than expected under random recombination, we used the permutation-based recombinant protein simulation approach of Lefeuvre et al. (2009).
Results and discussion
Conserved recombination break point hot- and cold-spots within coronavirus genomes
Using a combination of recombination detection methods implemented in RDP5, we identified 416 unique recombination events in the Pedacovirus dataset, 255 in Embecovirus, 65 in Merbecovirus, 107 in Nobecovirus, 282 in Sarbecovirus, and 1703 in Igacovirus. The variable numbers of detected recombination events between datasets should not be considered evidence that the viruses in some subgenera recombine more than others. Rather, the variable numbers reflect differences in both the numbers of analysed sequences in each dataset (e.g. the Igacovirus and Pedacovirus datasets had the most sequences and the Nobecovirus dataset the fewest) and the genetic diversity of the sequences in the different datasets (e.g. the Pedacovirus dataset had the least diverse sequences and the Nobecovirus and Embecovirus datasets the most; Supplementary Table S1).
To visualize the recombination break points associated with these events in each subgenus, break-point distribution plots (Fig. 1) and recombination region count matrices (Fig. 2) were constructed. The break-point distribution plots revealed clusters of break points that were either more or less dense at individual genome sites than those observed at corresponding sites in 99 per cent of permuted datasets where recombination break-point positions were randomly distributed (Fig. 1). Potential recombination hotspots were detected in all of the analysed subgenera (indicated by red shading in Fig. 1) and recombination cold-spots in three of them (indicated by blue shading in Fig. 1).
Figure 2.
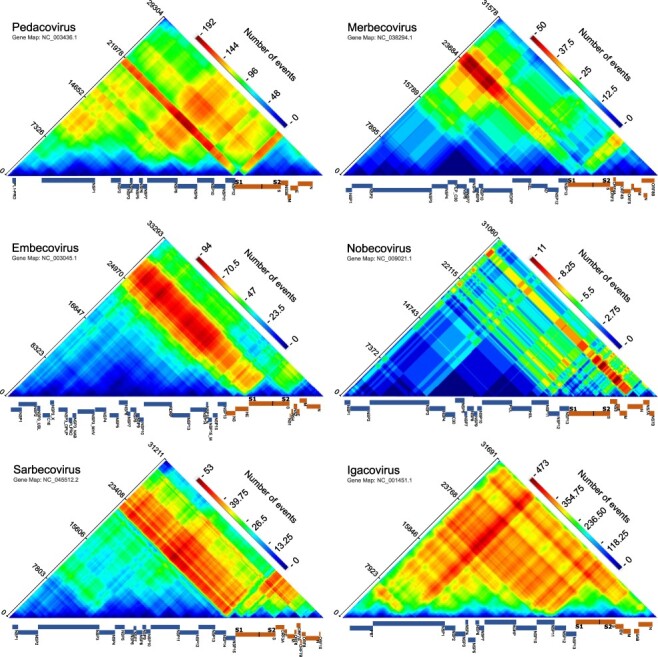
Recombination region count matrices indicating genome regions that are most and least commonly transferred during detectable coronavirus recombination events. Unique recombination events for six coronavirus subgenera, mapped onto recombination region count matrices based on determined break-point positions. Each cell in the matrix represents a pair of genome sites with the colours (heat) of cells indicating the number of times recombination events separated the represented pairs of sites. Reference sequence gene maps of the most prevalent virus in each subgenus were obtained from the NCBI nucleotide database (https://www.ncbi.nlm.nih.gov/nuccore) and are plotted alongside each matrix. Nucleotide positions are plotted according to full analysed nucleotide sequence alignment (Supplementary material). Genome maps indicate the coding regions of individual protein products. Non-structure proteins encoded by ORF1ab are indicated in blue (cold) and other genes are indicated in orange (warm).
In all the subgenera other than Embecovirus and Merbecovirus, potential recombination hotspots were detected within 300 nucleotides of the 5ʹ end of the genome. This non-coding region is upstream of ORF1ab, where the transcription and replication initiation sites are. These initiation sites prime the transcription of subgenomic mRNAs and contain extensive secondary structures that are partially conserved amongst the viruses belonging to a given coronavirus genus (reviewed in (Yang and Leibowitz 2015) (Siegfried et al. 2014; Manfredonia et al. 2020)). In various other viruses such as HIV, recombination break points tend to colocalize with highly structured genome regions (Simon-Loriere et al. 2010) and it is therefore plausible that recombination hotspots detected at the 5ʹ end of Pedacovirus, Nobecovirus, Sarbecovirus, and Igacovirus genomes might also be attributable to secondary-structure induced template-switching during replication.
Multiple other potential recombination hotspots were detected near the boundaries of various different genes in the 3ʹ genome regions of Sarbecoviruses, Nobecoviruses, and Igacoviruses. In Sarbecoviruses, four potential recombination hotspots were detected in the 3ʹ genome regions, between the M gene and ORF6, between ORF7AB and ORF8AB, and between ORF8AB and N. In Nobecoviruses, potential recombination hotspots were detected in the 3ʹ genome regions, between the E gene and the M gene, and near the centre of the N gene. In Igacoviruses there were several potential hotspots in the 3ʹ genome region, the clearest of which fell towards the 3ʹ end of N.
Break-point distributions in and around the S gene are consistent with recombination facilitating host adaptation and/or immune evasion
Most noteworthy of all the detected potential break-point hotspots were those falling within 800 nucleotides upstream of the S gene start codon in all subgenera other than the Merbecoviruses. The conserved arrangement of recombination break-point clusters in relation to the S gene likely underlies the observation that, according to our analyses, the S gene has been frequently transferred in its entirety during recombination events in Igacoviruses, Sarbecoviruses, and Embecoviruses (note red diagonals associated with the S genes of these subgenera in Fig. 2). However, in all analysed coronavirus groups other than the Embecoviruses and Igacoviruses, potential recombination hotspots were also detected within the 5ʹ half of the S gene (S1 domain), further suggesting that the 3ʹ half of the gene (S2 domain) is the portion that is most commonly transferred during recombination events as a complete module (note the red diagonals associated with the 3ʹ part of the S gene in Fig. 2). The locations of the detected recombination hotspots in, and immediately adjacent to, the S gene suggest that either the complete S gene or its 3ʹ half, have been frequently transferred during recombination.
The Spike proteins that are encoded by the S gene are composed of an amino-terminal subunit 1 (S1) and a carboxyl-terminal subunit 2 (S2) (Wrapp et al. 2020) (Supplementary Diagram 1; Supplementary data). The S1 contains the N-terminal domain (NTD) and a receptor-binding domain (RBD) which mediates the binding of viral particles to host cell surface receptors. Different coronaviruses bind to different receptors. For example, the Merbecovirus, MERS-CoV, binds dipeptidyl peptidase-4 (DPP4), the Pedacovirus, PEDV, binds aminopeptidase N, and the Sarbecoviruses SARS-CoV and SARS-CoV-2 bind to angiotensin-converting enzyme 2 (ACE2) (Wan et al. 2020; Yeager et al. 1992; Li, Ge, and Li 2007; Belouzard et al. 2012; Raj et al. 2013; Reusken et al. 2016). It is also likely that in some coronaviruses the NTD of Spike also interacts with cell surface receptors. For example, the NTD of the SARS-CoV-2 Spike interacts with the tyrosine-protein kinase receptor UFO (AXL) which appears to function as a co-receptor for human cell entry (Wang et al. 2021). The S2 subunit contains a heptad repeat region (including subregions HR1 and HR2) which mediate the fusion of the virion envelope with the host cell membrane during viral entry (Liu et al. 2004; Cui, Li, and Shi 2019).
Being responsible for receptor binding and cellular entry, the evolution of the S gene is, therefore, key to host adaptation. It may be beneficial for coronaviruses to exchange either entire S genes, S1 subunit encoding portions of S genes, or smaller subdomains within the N-terminal domains of S1 during recombination, both because Spike is the main target of neutralizing antibodies (Ou et al. 2020) and because the S gene is the main determinant of host species and host cell-type specificity (Lu, Wang, and Gao 2015). Although recombination frequently transfers the entire S1-encoding region of the gene it is not uncommon in particular groups of viruses for it to transfer smaller subsections of the S1 (as can be seen with the red diagonals associated with the S genes of Pedacoviruses and Merbecoviruses in Fig. 2). In the Alphacoronaviruses, for example, recombination has involved a transfer of the 5ʹ half of the NTD of S1 from transmissible gastroenteritis virus into canine coronavirus (type CCoV2b) (Decaro et al. 2009; Licitra, Duhamel, and Whittaker 2014).
In all subgenera other than the Igacoviruses, the S gene is also the only gene in which recombination cold-spots were detected in our break-point distribution analyses. Most noteworthy is that the 5ʹ 500 nucleotides of the S gene is the site of a conserved cold-spot detected in the Igacoviruses, Pedacoviruses, and Sarbecoviruses. In the Igacoviruses, the coronavirus group with the richest full genome dataset in terms of both numbers of analysed sequences and their diversity, and within which the highest numbers of recombination break points were detected (n = 1703), our power to detect recombination cold-spots was greatest. Accordingly, recombination cold-spots were additionally detectable in the region of the S gene encoding the RBD, dispersed throughout the 3ʹ half of the gene encoding the S2 subunit, and at two sites in the ORF1a corresponding to the coding regions of non-structural proteins 3 and 4.
It must be stressed that our inability to detect such cold spots in the other subgenera is clearly due to our break-point clustering test generally lacking sufficient power to detect these: note that the lower 99 per cent CI is at zero for >90 per cent of genome sites in all datasets other than that of the Igacoviruses.
With this caveat in mind, we note that the arrangement of recombination cold-spots in the S gene suggests that either basal recombination rates are suppressed within the NTD- and S2-encoding regions of this gene or that recombination break points falling within these regions tend to yield S genes that encode defective chimaeric Spike proteins. The NTD-encoding region of the S gene is among the most genetically variable regions of coronavirus genomes and this alone might explain the relative absence of recombination break points near the 5ʹ end of the S genes of Igacoviruses, Nobecoviruses, Pedacoviruses, and Sarbecoviruses (Archer et al. 2008; Boni et al. 2020). Similarly, the S2-encoding region of the S gene also tends to be more variable than most other coronavirus genome regions. However, the S2 subunit of Spike also contains multiple co-evolved intra-protein amino acid interactions that are crucial for the cell-fusion functions of Spike (Bosch et al. 2003; Tang et al. 2020). It is also plausible, therefore, that the relative absence of detectable recombination break points in the 3ʹ half of the S gene might be because recombinants carrying break points falling within this region commonly express defective Spike proteins. In this regard, the S2-encoding region of the S gene may be a functional module that, while tending to retain its functionality when transferred by recombination as a complete unit into divergent genomic backgrounds (Wege et al. 1998), might be highly sensitive to recombination-induced disruptions of co-evolved amino acid interactions within S2 whenever recombination break points fall within its boundaries (Supplementary Fig. S1; Supplementary data).
Selection likely disfavours recombinants expressing Spike proteins with disrupted folds
We used the SCHEMA method (Voigt et al. 2002; Lefeuvre et al. 2007) to more directly test for evidence of the inferred coronavirus recombination break-point distributions in the S gene having been impacted by natural selection disfavouring the survival of recombinants that express chimeric Spike proteins with disrupted folds. The only coronavirus proteins for which high-resolution atomic coordinate data were available, and for which sufficient recombination break-point numbers were detected within their associated genome sites to perform the SCHEMA folding disruption test, were those of sequences in the Merbecovirus, Sarbecovirus, Pedacovirus, and Igacovirus datasets.
We found that in the Igacoviruses and Sarbecoviruses, potential amino acid interactions within the Spike proteins expressed by observed recombinants have significantly fewer predicted structural impacts than would be expected under random recombination (P < 0.05; SCHEMA permutation test). It is noteworthy that the test result for the Pedacoviruses also approached significance (P = 0.079) but that for the Merbecoviruses displayed no such tendencies (P = 0.875; although it should be noted that, of the four datasets tested, this dataset had the lowest number of detected break points in the S gene). This implies that, as has been suggested previously with in vitro recombination experiments involving the Embecovirus, murine coronavirus (Banner and Lai 1991), the Igacoviruses, and Sarbecoviruses (and possibly also the Pedacoviruses) display lower degrees of predicted recombination-induced protein folding disruption in their expressed Spike proteins than would be expected under random recombination in the absence of selection. It should be stressed that our power to detect such ‘avoidance of protein folding disruption’ signals was restricted to Spike and that it remains plausible that, given enough additional sequence data and more extensive atomic-resolution 3D structure information for other coronavirus proteins, many of these proteins might also display such signals.
Indirect evidence that selection against protein misfolding impacts observable break-point distributions throughout coronavirus genomes
It would be expected that if natural selection tended to disfavour recombinants with misfolded proteins then break points would tend to be found more frequently per non-coding nucleotide site than per amino acid encoding nucleotide site (Drummond et al. 2005). Also, it might be expected that, of the recombination break points falling at amino acid encoding sites within genes, those falling at the edges of genes (for example, in the first and last 5 per cent of the coding sequence of a particular protein) might be less disruptive of co-evolved intra-protein amino acid contacts that were crucial for correct folding than break points falling within the middle regions of genes (Lefeuvre, Lett, and Reynaud et al. 2007). If selection against misfolded proteins was impacting the distributions of recombination break points throughout coronavirus genomes we would, therefore, expect that observed break points might tend to fall more commonly: (1) in non-coding regions than in coding regions and (2) at the edges of genes than in the middle parts of genes.
Accordingly, we found that the intergenic regions of the Pedacoviruses, Embecoviruses, Nobecoviruses, and Sarbecoviruses all had significantly higher break-point densities (P < 0.05; permutation test; Table 1) than those in the protein-coding regions. Similarly, we detected that in the Pedacoviruses, Embecoviruses, Sarbecoviruses, and Igacoviruses, detectable break-point densities were significantly higher in the beginning and ending 5 per cent of coding regions than in the middle 90 per cent of these regions (P < 0.05; permutation test; Table 2) with marginal significance observed in Nobecoviruses (P = 0.054; permutation test; Table 2).
Table 1.
Comparison of detectable break-point numbers in non-coding regions and coding regions with rows in bold indicating subgenera with significantly more break points in non-coding regions than would be expected under random recombination.
| Subgenus | BPsa in non-coding regions | BPs in coding regions | Permutation P-val |
|---|---|---|---|
| Pedacovirus | 32 | 392 | <0.001 |
| Embecovirus | 11 | 73 | <0.001 |
| Merbecovirus | 1 | 66 | 0.660 |
| Nobecovirus | 4 | 79 | 0.012 |
| Sarbecovirus | 7 | 307 | <0.001 |
| Igacovirus | 30 | 1683 | 0.650 |
BPs = Break points.
Table 2.
Break-point densities falling in the end 10 per cent (5 per cent each end) of genes vs the middle 90 per cent of genes with rows in bold indicating subgenera with significantly higher numbers of detectable break points in the ending 10 per cent of genes than would be expected under random recombination.
| Subgenus | BPsa in the end 10% of genes | BPs in the middle 90% of genes | Permutation P-val |
|---|---|---|---|
| Pedacovirus | 68 | 507 | <0.001 |
| Embecovirus | 25 | 195 | 0.003 |
| Merbecovirus | 5 | 127 | 0.810 |
| Nobecovirus | 12 | 112 | 0.054 |
| Sarbecovirus | 47 | 612 | 0.007 |
| Igacovirus | 191 | 3369 | 0.004 |
BPs = Break points.
Taken together the lower densities of break points both within genes than in intergenic regions and within the middle parts of genes than in the ends of genes are reminiscent of similar break-point distribution patterns detected in HIV (Simon-Loriere et al. 2010) and the members of various single-stranded DNA virus families (Lefeuvre et al. 2009) and is consistent with the hypothesis that in coronaviruses natural selection generally disfavours the survival of recombinants that express chimeric proteins with disrupted folds.
ORF1a genome regions generally have lower break-point densities than other coding regions
There was a significantly lower density of break points detected in ORF1a than in other coding regions of the genome for all six of the analysed subgenera (P < 0.05; permutation test; Table 3). The relatively low number of recombination events involving transfers of sequence fragments within this region is most notable in three of the Betacoronaviruses subgenera: Embecovirus, Nobecovirus, and Sarbecoviruses (note the blue/cyan/green triangles associated with most of ORF1ab in these subgenera in Fig. 2). Our results here are therefore consistent with previous observations that there is a significant tendency for recombination break points to fall outside ORF1a in the human-infecting coronaviruses OC43 (an Embecovirus) and NL63 (an Alphacoronavirus in the subgenus Setracovirus) (Pollett et al. 2021).
Table 3.
Individual genes and sub-gene regions with significantly lower numbers of detectable break points than would be expected under random recombination.
| Subgenus | Genome region | BPsa inside region | BPs outside region | Permutation P-val |
|---|---|---|---|---|
| Pedacovirus | ORF1a | 114 | 278 | 0.001 |
| Embecovirus | ORF1a | 43 | 130 | 0.001 |
| Merbecovirus | ORF1a | 43 | 130 | 0.001 |
| Nobecovirus | ORF1a | 14 | 65 | <0.001 |
| Sarbecovirus | ORF1a | 94 | 213 | <0.001 |
| Igacovirus | ORF1a | 667 | 1016 | 0.024 |
| Nobecovirus | plpro (nsp3) | 7 | 72 | 0.039 |
| Sarbecovirus | plpro (nsp3) | 49 | 258 | 0.031 |
| Igacovirus | plpro (nsp3) | 282 | 1401 | 0.016 |
| Merbecovirus | nsp4 | 0 | 66 | 0.035 |
| Igacovirus | nsp4 | 78 | 1605 | 0.002 |
BPs = Break points.
Within ORF1a the regions encoding the non-structural proteins (nsps) nsp3 (a papain-like cysteine protease), and nsp4 have particularly low densities of identified break points in multiple different subgenera (Nobecovirus, Sarbecovirus, and Igacovirus for nsp3 and Merbecoviruses and Igacoviruses for nsp4). Together with nsp6, nsp3 and nsp4 cooperatively modify the endoplasmic reticulum (ER) of coronavirus-infected cells into vesicles with double membranes to which viral replication complexes are tethered (Knoops et al. 2008; Hartenian et al. 2020; Klein et al. 2020; Mohan and Wollert 2021).
It is plausible that the relatively low numbers of recombination events detectable in ORF1a are attributable to the high degree to which these components interact with one another (Stark et al. 2006; Li et al. 2021). It is expected that these interactions might rely on co-evolved interaction motifs and that these proteins might therefore not function optimally if transferred into a genomic background within which they did not co-evolve (Jain, Rivera, and Lake 1999; Martin et al. 2005b).
Break points tend to fall at sites with lower than average GC content
To further our understanding of why, irrespective of selection, some coronavirus genomic sites might be more mechanistically predisposed to recombination than others, we tested break-point positions detected in each of the six analysed coronavirus datasets for associations with local GC contents (i.e. calculated proportions of all nucleotide residues that were G or C between 10 and 20 nucleotide sites up and downstream of detected break-point locations). High GC content is expected to potentially impact the frequencies at which recombination break points occur in various ways such as (1) predisposing genome regions to form stable secondary structures that could cause pausing of RNA-Dependent RNA polymerase (RdRP) (Stark et al. 2006) (Experimental Evidence Codes| BioGRID 2021), (2) increasing the energy needed to break base-pairs during replication, and increasing the amount of time taken for RdRP to traverse these regions (Petes and Merker 2002; Sershen et al. 2011) and, if RdRPs disengages during replication, (3) increasing the probability of re-engagement through annealing with the same or a different template molecule (Lai 1990).
Contrary to expectations, but consistent with a recent report on recombination in coronaviruses (Pollett et al. 2021), we found that GC content within twenty nucleotides of break-point positions (Table 4) tended to be lower than expected under random recombination in the Pedacovirus, Embecovirus, Merbecovirus and Sarbecovirus datasets: significantly so in the case of the Sarbecovirus and Pedacovirus datasets (P < 0.05; permutation test). When we repeated the test only considering GC contents within 10 nucleotides of recombination break points (20-nt window in Table 4), the significant associations between break-point positions and lower GC content in Sarbecoviruses and Pedacoviruses were strengthened, and additionally, marginally significant associations with lower GC contents (0.05 < P < 0.1; permutation test) were detected in Merbecoviruses and Embecoviruses (Fig. 3, Supplementary Fig. S2 and Supplementary Fig. S3; Supplementary data).
Table 4.
Associations between decreased GC content and detected recombination break-point sites with rows in bold indicating subgenera displaying average GC contents in the vicinity of break-point sites that are significantly lower than what would be expected under random recombination.
| Within 20 nt of break-point site | WIthin 10 nt of break-point site | |||
|---|---|---|---|---|
| Subgenus | P-val. | Significant | P-val | Significant |
| Pedacovirus | 0.047 | Yes | 0.019 | Yes |
| Embecovirus | 0.322 | No | 0.080 | Marginal |
| Merbecovirus | 0.590 | No | 0.051 | Marginal |
| Nobecovirus | 0.791 | No | 0.693 | No |
| Sarbecovirus | 0.005 | Yes | 0.004 | Yes |
| Igacovirus | 0.948 | No | 0.911 | No |
Figure 3.
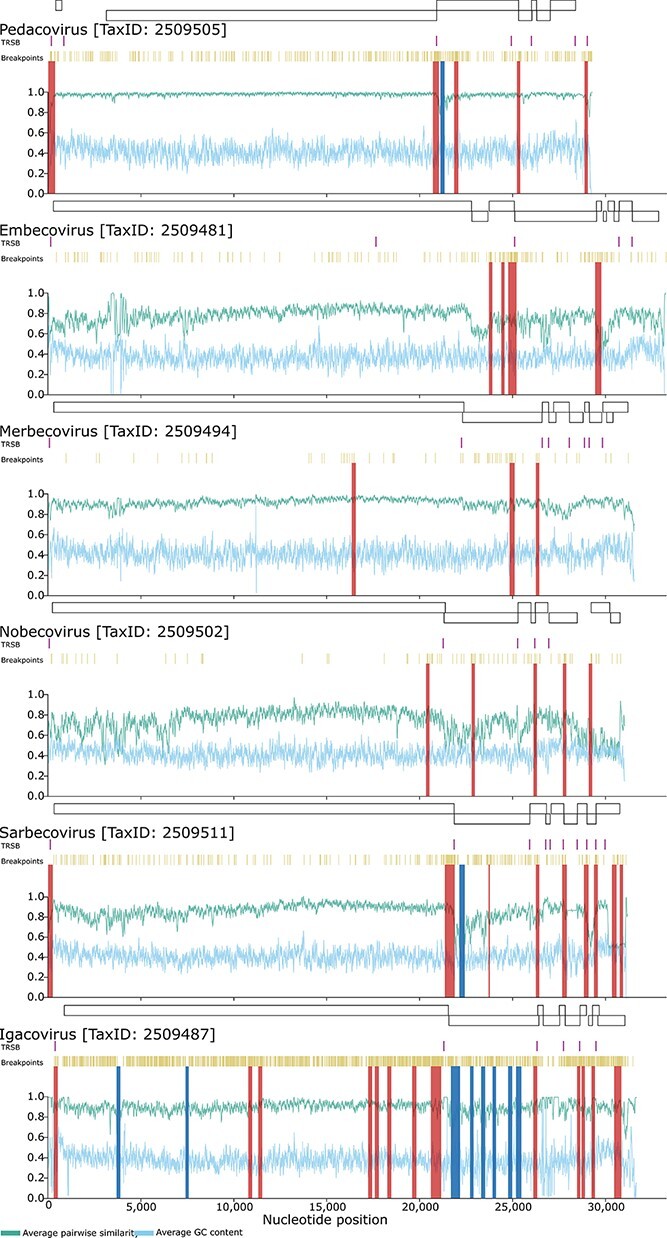
Regional variations in average pairwise sequence similarity (green/ top x-axis parameter) and GC content (blue/ bottom horizontal X-axis parameter) across coronavirus genomes. The plotted values indicate the pairwise sequence similarity and GC proportions within a moving 40-nucleotide window. Also indicated are the locations of the main genes (above each graph), transcriptional regulatory sequences (TRDs; in purple/ top stripes beneath gene boxes), identified break-point locations (in mustard/ beneath TRSB locations), potential recombination hotspots (in red/ Y-axis bright stripes through graphs) and potential recombination cold-spots (in blue/ Y-axis cold stripes through graphs).
It is unclear whether sequence similarity directly influences the locations of recombination break points
It has been previously found in other viruses that recombination break-point sites tend to occur more commonly at genome sites with elevated degrees of sequence conservation (van Vugt et al. 2001; Dazza et al. 2005; Archer et al. 2008). We, therefore, tested whether this pattern held for the six analysed coronavirus subgenera.
Although recombination break points in the Sarbecovirus and Igacoviruses datasets displayed a significant tendency to occur in genome regions displaying elevated degrees of average pairwise similarity among the analysed sequences (P < 0.007; permutation test; Table 5), for the Pedacoviruses and Embecoviruses the opposite was the case. In these subgenera, detectable recombination break points have tended to fall in genome regions with lower degrees of average pairwise sequence similarity (P < 0.005; permutation test; Table 5). It is therefore unclear from our test whether pairwise sequence similarity within 10 or 20 nucleotides of prospective recombination break-point sites is a direct determinant of where break points occur within coronavirus genomes.
Table 5.
Association of break-point locations with higher/lower degrees of average pairwise sequence similarity with rows in bold indicating significant associations.
| Within 20 nt of break-point site | Within 10 nt of break-point site | |||
|---|---|---|---|---|
| Subgenus | Association with higher/lower similarity | P-val | Association with higher/lower similarity | P-val |
| Pedacovirus | Lower | <0.001 | Lower | <0.001 |
| Embecovirus | Lower | 0.006 | Lower | 0.007 |
| Merbecovirus | Higher | 0.184 | Higher | 0.192 |
| Nobecovirus | Higher | 0.465 | Higher | 0.475 |
| Sarbecovirus | Higher | 0.005 | Higher | 0.005 |
| Igacovirus | Higher | <0.001 | Higher | <0.001 |
It is noteworthy in this regard that there are substantial variations in degrees of sequence conservation across the analysed sequence datasets with, for example, the genome regions corresponding to the recombination break-point hotspot immediately upstream of the S gene in the Nobecovirus, Sarbecovirus, and Igacovirus datasets (all with a tendency for break points to fall at more conserved sites) displaying among the highest degrees of sequence conservation within these datasets (Fig. 3). Conversely, for the Pedacovirus and Embecovirus datasets (both with a tendency for break points to fall at less conserved sites) the corresponding recombination hotspots upstream of the S gene start codon fall at genome sites that have among the lowest degrees of genome-wide conservation in these datasets (Fig. 3). It is therefore likely that, for this conserved hotspot at least, sequence similarity has not been a primary determinant of where break points have occurred.
Besides the obscuring influence of the recombination hotspots 5ʹ of the S-gene start codon, the relationship between sequence similarity and break-point locations may have also been obscured by the fact that (1) recombination events are only detectable in genome regions with sufficient diversity to reveal alternating relationships between recombinants and their parental genomes, and (2) recombination break-point locations can be most accurately inferred when they occur between closely-spaced genome sites at which parental genomes differ from one another. This possibly underlies the discordant associations between degrees of pairwise sequence similarity and recombination break-point locations observed for the Sarbecovirus, Embecovirus, and Nobecovirus datasets (Table 5): this despite the ORF1a regions of viruses in these three genera all having both lower degrees of genetic diversity (Fig. 3) and lower numbers of detectable recombination break points (Fig. 2 and Table 3) than most other genome regions. Part of the reason for this may be that the overall diversity of the Embecovirus and Nobecovirus datasets (78 per cent and 75 per cent average pairwise identity, respectively) is substantially higher than that of the Sarbecovirus dataset (86 per cent average pairwise identity). As such, recombination events would likely have been more readily detectable in the lower diversity genome regions of Embecoviruses and Nobecoviruses than they were in the corresponding genome regions of Sarbecoviruses.
There is a strong association between recombination break-point locations and those of transcriptional regulatory sequences
Coronavirus transcription involves template switching at specific genome sites, called transcriptional regulatory sequences (TRSs) (Yang et al. 2021), previously called the intergenic sequence (Alonso et al. 2002). A possible link between template switching during gene expression and the genomic sites where recombination break points occur during genome replication has been noted previously for coronaviruses, in general, (Zúñiga et al. 2004; Sola et al. 2015) and SARS-CoV, specifically (Graham et al. 2018). Template switching is prone to occur during transcription of coronavirus negative genome strands whenever RdRp encounters the TRS sequences that are commonly found upstream of various genes. Because these ‘body TRS’ (or TRS-B) (Alonso et al. 2002; Sola et al. 2015) sites are involved in frequent template switching during transcription, it has been suggested that these sites might also promote template switching during genome replication (Graham et al. 2018) and, therefore, that they might colocalize with recombination hotspots (Yang et al. 2021).
We used the SuPER method (Yang et al. 2021) to detect potential TRS-B sites in each of our six coronavirus datasets. Whereas SuPER can use RNA-seq data to precisely locate TRS-B sites, in our case we used previously identified TRS-L sequences (Yang et al. 2021) to find and annotate likely TRS-B sites within the six analysed coronavirus datasets.
We found strong evidence for associations between the locations of conserved TRS-B sites (i.e. those detected in >75 per cent of the analysed sequences in each dataset) and the locations of detected recombination break points in the Pedacoviruses, Igacoviruses, Embecoviruses, and Sarbecoviruses (P < 0.05; permutation test; Table 6). These associations were detectable when we varied the required proximity between break points and potential TRS-B sites to be considered a match from between 2 and 46 nucleotides.
Table 6.
Associations between transcription regulatory sequence (TRS) sites and the locations of detected recombination break points with P-values in bold indicating significant associations of TRS sites with higher break-point numbers.
| Subgenus | Within 46 nts P-val | Within 21 nts P-val | Within 9 nts P-val | Within 2 nts P-val |
|---|---|---|---|---|
| Pedacovirus | <0.001 | <0.001 | <0.001 | <0.001 |
| Embecovirus | <0.001 | <0.001 | <0.001 | <0.001 |
| Merbecovirus | 0.117 | 0.178 | 0.210 | 0.806 |
| Nobecovirus | 0.014 | 0.039 | 0.020 | 0.478 |
| Sarbecovirus | <0.001 | <0.001 | <0.001 | 0.005 |
| Igacovirus | <0.001 | <0.001 | <0.001 | <0.001 |
Given the large detectable recombination break-point hotspots directly upstream of the S gene in most of the analysed subgenera datasets and the TRS-B sequences that map near these hotspots, it was possible that the associations detected between TRS locations and break-point positions could have been attributable entirely to the TRS-B sites upstream of the Spike gene. To determine if this was the case, we repeated the association test (25 nt window size) but this time with the TRS upstream of Spike removed from the analysis. We observed a minimal decrease in the significance of the association between TRS-B sites and recombination break-point positions, indicating that the initial result was not simply being driven by the coincidental colocalization of the S-gene-associated TRS-B site and the conserved recombination hotspot upstream of the S gene in most of the analysed datasets.
We reran the TRS-B association tests with the positions of the TRS-B sites randomly shifted along the genome. The script takes as input the alignment file for each of the six datasets and places five to ten ‘false’ TRS-B sites across each genome (the exact number corresponding for each subgenus dataset to the ‘true’ TRS-B number for that dataset). When considering break-point probability distributions and an analysis window of twenty-five nucleotides, there was a significant absence of break points within twelve nucleotides of TRS-B sites in the Embecovirus and Sarbecovirus datasets and neither significantly more nor less break points in close proximity to TRS-B sites in any of the other datasets. Both these results, along with our previous tests, are strong evidence that recombination break points in coronaviruses generally tend to cluster at TRS-B sites.
However, given that detectable recombination break points tend to fall near the edges of genes, this association between break-point locations and TRS-B sites might simply be attributable to the fact that TRS-B sites also tend to fall at the edges of genes. We, therefore, attempted to determine whether the association between break-point locations and TRS-B sites was still evident if we controlled for the colocalization of these sites at the edges of genes. We were specifically interested in whether the presence/absence of a TRS-B site immediately upstream of a gene was associated with the presence/absence of a recombination break-point hotspot upstream of the gene. Considering only the TRS-B sites and recombination break-point hotspots falling either in intergenic regions or within 300 nucleotides of the beginning of genes we found a significant association between the presence of a TRS-B site near the beginning of a gene and the presence of a hotspot near that location (P = 0.0392, Chi-square test with N−1 correction). Therefore suggesting that, for the Pedacovirus, Sarbecovirus, Igacovirus, and Embecovirus datasets at least, the significant association we found between TRS-B sites and recombination break-point locations was not merely attributable to a coincidental tendency for break points and TRS-B sites to colocalize near the edges of genes.
Conclusion
Across all of the tests that we performed, viruses in the different analysed coronavirus genera displayed similar patterns of recombination (Table 7). The most strikingly similar of these patterns were those observed in the Sarbecoviruses (members of the Betacoronavirus genus) and the Pedacoviruses (members of the Alphacoronavirus genus). These mostly concordant patterns indicate that the processes that yield and select recombinant coronaviruses are likely broadly conserved across the three analysed coronavirus genera.
Table 7.
Conserved patterns of recombination across various coronavirus subgenera. Rows each contain the result of either a statistical test or the presence/absence of a particular characteristic of recombination (such as the presence of a hotspot at a specific genome location): BP = break point; blue = significant association or presence of characteristic; light blue = marginally significant association; pink = no significant association or absence of characteristic; white = untested.
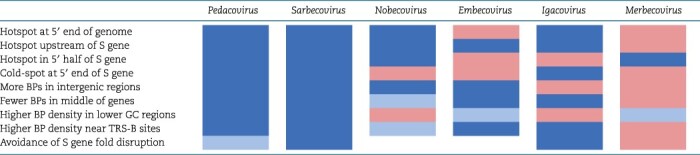
The subgenus dataset displaying the least concordant recombination patterns was that of the Merbecoviruses. It is unclear to us why the Merbecovirus dataset displays recombination break-point patterns that differ from the other analysed datasets: it is not an outlier among the datasets in terms of the numbers of sequences analysed or the average pairwise similarities of these sequences, but the dataset does have the lowest number of detectable recombination events. It is therefore possible that either the processes that generate recombinant genomes, the genetic factors that determine the viability of recombinants, or the epidemiological and evolutionary processes that impact the survival of recombinants, might differ somewhat between the Merbecoviruses and most other coronaviruses.
Nevertheless, the non-random and mostly conserved recombination patterns that we and others have detected in various coronavirus subgenera are likely shaped both by evolutionarily conserved variations in the mechanistic predispositions of different genome regions to recombination and by shared selective processes disfavouring the survival of recombinants that express improperly folded proteins. There are two non-exclusive explanations for why coronavirus genome sites that are mechanistically predisposed to recombination (such as those of TRS-B sequences) tend to coincide with sites where recombination seems to have had a minimal impact on protein folding: (1) negative selection over the short-term may be so efficient at purging all viral variants with recombination-induced protein misfolding that such variants are only rarely sequenced and/or (2) longer-term selection, possibly acting since the most recent common ancestor of all known coronaviruses, may have yielded coronavirus genomes that are configured such that they are mechanistically predisposed to only recombine at sites where recombination break points are minimally disruptive of protein folding. When high-resolution maps of amino acid contacts within coronavirus protein complexes become available, and when the conserved nucleotide interactions within biologically functional RNA structural elements in a diverse enough array of coronavirus genomes have been identified, it should also be possible to determine the degree to which selection acting over the short- and/or long-terms to preserve these other categories of co-evolved intra-genome interactions have impacted observable coronavirus recombination patterns.
Supplementary Material
Contributor Information
Arné de Klerk, Institute of Infectious Diseases and Molecular Medicine, Division Of Computational Biology, Department of Integrative Biomedical Sciences, University of Cape Town, Cape Town 7701, South Africa.
Phillip Swanepoel, Institute of Infectious Diseases and Molecular Medicine, Division Of Computational Biology, Department of Integrative Biomedical Sciences, University of Cape Town, Cape Town 7701, South Africa.
Rentia Lourens, Division of Neurosurgery, Neuroscience Institute, Department of Surgery, University of Cape Town, Cape Town, 7701, South Africa.
Mpumelelo Zondo, Institute of Infectious Diseases and Molecular Medicine, Division Of Computational Biology, Department of Integrative Biomedical Sciences, University of Cape Town, Cape Town 7701, South Africa.
Isaac Abodunran, Institute of Infectious Diseases and Molecular Medicine, Division Of Computational Biology, Department of Integrative Biomedical Sciences, University of Cape Town, Cape Town 7701, South Africa.
Spyros Lytras, MRC-University of Glasgow Centre for Virus Research, University of Glasgow, Glasgow G61 1QH, UK.
Oscar A MacLean, MRC-University of Glasgow Centre for Virus Research, University of Glasgow, Glasgow G61 1QH, UK.
David Robertson, MRC-University of Glasgow Centre for Virus Research, University of Glasgow, Glasgow G61 1QH, UK.
Sergei L Kosakovsky Pond, Department of Biology, Temple University, Institute for Genomics and Evolutionary Medicine, Philadelphia, PA 19122, USA.
Jordan D Zehr, Department of Biology, Temple University, Institute for Genomics and Evolutionary Medicine, Philadelphia, PA 19122, USA.
Venkatesh Kumar, Department of Microbiology, Tumor and Cell Biology, Karolinska Institutet, Stockholm, 14186, Sweden.
Michael J Stanhope, Department of Population and Ecosystem Health, College of Veterinary Medicine, Cornell University, Ithaca, NY, 14853, USA.
Gordon Harkins, South African National Bioinformatics Institute, University of the Western Cape, Cape Town, 7535, South Africa.
Ben Murrell, Department of Microbiology, Tumor and Cell Biology, Karolinska Institutet, Stockholm, 14186, Sweden.
Darren P Martin, Institute of Infectious Diseases and Molecular Medicine, Division Of Computational Biology, Department of Integrative Biomedical Sciences, University of Cape Town, Cape Town 7701, South Africa.
Supplementary data
Supplementary data are available at Virus Evolution online.
Funding
ADK and PS were supported by a University of Cape Town Masters Research Scholarship [960000000760]. RL was supported by the South African National Research Foundation. DPM was supported by the Wellcome Trust [222574/Z/21/Z]. SLKP was supported by the U.S. National Institutes of Health [R01 AI134384 and AI140970] and the US National Science Foundation [RAPID 2027196 NSF/DBI,BIO]. SL was supported by the Medical Research Council of the United Kingdom [MC_UU_12014/12]. OAM was supported by the Wellcome Trust [206369/Z/17/Z]. JDZ was supported by the U.S. National Institutes of Health [R01 AI134384 and AI140970]. GH was supported by a US National Institutes of Health grant (1U01Al152151-01), MZ, IA, DR, VK, MJS, and BM were not supported.
Conflict of interest:
None declared.
References
- Alonso S. et al. (2002) ‘Transcription Regulatory Sequences and mRNA Expression Levels in the Coronavirus Transmissible Gastroenteritis Virus’, Journal of Virology, 76: 1293–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony S. J. et al. (2017) ‘Further Evidence for Bats as the Evolutionary Source of Middle East Respiratory Syndrome Coronavirus Schultz-Cherry, S. Ed’, mBio, 8: e00373–17.doi: 10.1128/mBio.00373-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer J. et al. (2008) ‘Identifying the Important HIV-1 Recombination Breakpoints’, PLoS Computational Biology, 4: e1000178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banner L. R., and Lai M. M. (1991) ‘Random Nature of Coronavirus RNA Recombination in the Absence of Selection Pressure’, Virology, 185: 441–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belouzard S. et al. (2012) ‘Mechanisms of Coronavirus Cell Entry Mediated by the Viral Spike Protein’, Viruses, 4: 1011–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H., Henrick K., and Nakamura H. (2003) ‘Announcing the Worldwide Protein Data Bank’, Nature Structural & Molecular Biology, 10: 980. [DOI] [PubMed] [Google Scholar]
- Boni M. F. et al. (2020) ‘Evolutionary Origins of the SARS-CoV-2 Sarbecovirus Lineage Responsible for the COVID-19 Pandemic’, Nature Microbiology, 5: 1408–17. [DOI] [PubMed] [Google Scholar]
- Bosch B. J. et al. (2003) ‘The Coronavirus Spike Protein Is a Class I Virus Fusion Protein: Structural and Functional Characterization of the Fusion Core Complex’, Journal of Virology, 77: 8801–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C.-P., and Nagy P. D. (2003) ‘Mechanism of RNA Recombination in Carmo- and Tombusviruses: Evidence for Template Switching by the RNA-Dependent RNA Polymerase in Vitro’, Journal of Virology, 77: 12033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coronaviridae - Positive Sense RNA Viruses - Positive Sense RNA Viruses . (2011) ICTV. ICTV 2011.
- Crameri A. et al. (1998) ‘DNA Shuffling of a Family of Genes from Diverse Species Accelerates Directed Evolution’, Nature, 391: 288–91. [DOI] [PubMed] [Google Scholar]
- Cui J., Li F., and Shi Z.-L. (2019) ‘Origin and Evolution of Pathogenic Coronaviruses’, Nature Reviews Microbiology, 17: 181–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dazza M.-C. et al. (2005) ‘Characterization of a Novel Vpu -harboring Simian Immunodeficiency Virus from a Dent’s Mona Monkey (Cercopithecus Mona Denti )’, Journal of Virology, 79: 8560–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decaro N. et al. (2009) ‘Recombinant Canine Coronaviruses Related to Transmissible Gastroenteritis Virus of Swine are Circulating in Dogs’, Journal of Virology, 83: 1532–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2015) ‘Full-length Genome Analysis of Canine Coronavirus Type I’, Virus Research, 210: 100–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denison M. R. et al. (2011) ‘Coronaviruses: An RNA Proofreading Machine Regulates Replication Fidelity and Diversity’, RNA Biology, 8: 270–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond D. A. et al. (2005) ‘On the Conservative Nature of Intragenic Recombination’, Proceedings of the National Academy of Sciences, 102: 5380–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudas G. (2016) ‘Rambaut A. MERS-CoV Recombination: Implications about the Reservoir and Potential for Adaptation’, Virus Evolution, 2: vev023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emam M. et al. (2021) ‘Positive Selection as a Key Player for SARS-CoV-2 Pathogenicity: Insights into ORF1ab, S and E Genes’, Virus Research, 302: 198472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Experimental Evidence Codes | BioGRID . (2021) BioGRID.
- Forni D., Cagliani R., and Sironi M. (2020) ‘Recombination and Positive Selection Differentially Shaped the Diversity of Betacoronavirus Subgenera’, Viruses, 12: 1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rossum G., and Drake F. L. (2010) The Python Language Reference. Python Software Foundation: Hampton, NH. [Google Scholar]
- Gibbs M. J., Armstrong J. S., and Gibbs A. J. (2000) ‘Sister-Scanning: A Monte Carlo Procedure for Assessing Signals in Recombinant Sequences’, Bioinformatics, 16: 573–82. [DOI] [PubMed] [Google Scholar]
- Goldstein S. A., Brown J., and Pedersen B. S. et al. (2021) Extensive Recombination-Driven Coronavirus Diversification Expands the Pool of Potential Pandemic Pathogens, BioRxiv, 1–24. 2021.02.03.429646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham R. L. et al. (2018) ‘Evaluation of a Recombination-resistant Coronavirus as a Broadly Applicable, Rapidly Implementable Vaccine Platform’, Communications Biology, 1: 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartenian E. et al. (2020) ‘The Molecular Virology of Coronaviruses’, The Journal of Biological Chemistry, 295: 12910–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatcher E. L. et al. (2017) ‘Virus Variation Resource - Improved Response to Emergent Viral Outbreaks’, Nucleic Acids Research, 45: D482–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath L. et al. (2006) ‘Recombination Patterns in Aphthoviruses Mirror Those Found in Other Picornaviruses’, Journal of Virology, 80: 11827–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrewegh A. A. P. M. et al. (1998) ‘Feline Coronavirus Type II Strains 79-1683 and 79-1146 Originate from a Double Recombination between Feline Coronavirus Type I and Canine Coronavirus’, Journal of Virology, 72: 4508–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussin J. G. et al. (2015) ‘Recombination Affects Accumulation of Damaging and Disease-associated Mutations in Human Populations’, Nature Genetics, 47: 400–4. [DOI] [PubMed] [Google Scholar]
- Jain R., Rivera M. C., and Lake J. A. (1999) ‘Horizontal Gene Transfer among Genomes: The Complexity Hypothesis’, Proceedings of the National Academy of Sciences, 96: 3801–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaroszewski L. et al. (2021) ‘The Interplay of SARS-CoV-2 Evolution and Constraints Imposed by the Structure and Functionality of Its Proteins. Punta M (Ed.)’, PLOS Computational Biology, 17: e1009147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia W. et al. (1995) ‘A Novel Variant of Avian Infectious Bronchitis Virus Resulting from Recombination among Three Different Strains’, Archives of Virology, 140: 259–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Standley D. M. (2013) ‘MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability’, Molecular Biology and Evolution, 30: 772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck J. G. et al. (1988a) ‘In Vivo RNA-RNA Recombination of Coronavirus in Mouse Brain’, Journal of Virology, 62: 1810–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (1988b) ‘RNA Recombination of Murine Coronaviruses: Recombination between Fusion-positive Mouse Hepatitis Virus A59 and Fusion-negative Mouse Hepatitis Virus 2’, Journal of Virology, 62: 1989–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein S. et al. (2020) ‘SARS-CoV-2 Structure and Replication Characterized by in Situ Cryo-electron Tomography’, Nature Communications, 11: 5885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoops K., and Kikkert M. (2008) ‘Worm SHE van den Et Al. SARS-Coronavirus Replication Is Supported by a Reticulovesicular Network of Modified Endoplasmic Reticulum’, PLoS Biology, 6: e226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumm Z. A. et al. (2021) ‘Precision Therapeutic Targets for COVID-19’, Virology Journal, 18: 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusters J. G. et al. (1990) ‘Sequence Evidence for RNA Recombination in Field Isolates of Avian Coronavirus Infectious Bronchitis Virus’, Vaccine, 8: 605–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M. M. (1990) ‘Coronavirus: Organization, Replication and Expression of Genome’, Annual Review of Microbiology, 44: 303–303. [DOI] [PubMed] [Google Scholar]
- Lai M. M. C. (1996) Recombination in Large RNA Viruses: Coronaviruses. [DOI] [PMC free article] [PubMed]
- Lam H., Ratmann O., and Boni M. (2018) ‘Improved Algorithmic Complexity for the 3SEQ Recombination Detection Algorithm’, Molecular Biology and Evolution, 35: 247–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lednicky J. A. et al. (2021) ‘Isolation of a Novel Recombinant Canine Coronavirus from a Visitor to Haiti: Further Evidence of Transmission of Coronaviruses of Zoonotic Origin to Humans’, Clinical Infectious Diseases, 28: ciab924.doi: 10.1093/cid/ciab924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. W., and Jackwood M. W. (2000) ‘Evidence of Genetic Diversity Generated by Recombination among Avian Coronavirus IBV’, Archives of Virology, 145: 2135–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefeuvre P. et al. (2007) ‘Avoidance of Protein Fold Disruption in Natural Virus Recombinants’, PLoS Pathogens, 3: e181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2009) ‘Widely Conserved Recombination Patterns among Single-Stranded DNA Viruses’, Journal of Virology, 83: 2697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B. X., Ge J. W., and Li Y. J. (2007) ‘Porcine Aminopeptidase N Is a Functional Receptor for the PEDV Coronavirus’, Virology, 365: 166–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. et al. (2021) ‘Virus-Host Interactome and Proteomic Survey Reveal Potential Virulence Factors Influencing SARS-CoV-2 Pathogenesis’, Med, 2: 99–112.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licitra B., Duhamel G., and Whittaker G. (2014) ‘Canine Enteric Coronaviruses: Emerging Viral Pathogens with Distinct Recombinant Spike Proteins’, Viruses, 6: 3363–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P. et al. (2017) ‘Prevalence and Genetic Diversity Analysis of Human Coronaviruses among Cross-border Children’, Virology Journal, 14: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S. et al. (2004) ‘Interaction between Heptad Repeat 1 and 2 Regions in Spike Protein of SARS-associated Coronavirus: Implications for Virus Fusogenic Mechanism and Identification of Fusion Inhibitors’, The Lancet, 363: 938–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G., Wang Q., and Gao G. F. (2015) ‘Bat-to-human: Spike Features Determining “host jump” of Coronaviruses SARS-CoV, MERS-CoV, and Beyond’, Trends in Microbiology, 23: 468–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lytras S. et al. (2022) ‘Exploring the Natural Origins of SARS-CoV-2 in the Light of Recombination’, Genome Biology and Evolution, 14/2, evac018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino S. et al. (1986) ‘High-frequency RNA Recombination of Murine Coronaviruses’, Journal of Virology, 57: 729–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfredonia I. et al. (2020) ‘Genome-wide Mapping of SARS-CoV-2 RNA Structures Identifies Therapeutically-relevant Elements’, Nucleic Acids Research, 48: 12436–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D. et al. (2005a) ‘A Modified Bootscan Algorithm for Automated Identification of Recombinant Sequences and Recombination Breakpoints’, AIDS Research and Human Retroviruses, 21: 98–102. [DOI] [PubMed] [Google Scholar]
- Martin D., and Rybicki E. (2000) ‘RDP: Detection of Recombination Amongst Aligned Sequences’, Bioinformatics, 16: 562–3. [DOI] [PubMed] [Google Scholar]
- Martin D. et al. (2005b) ‘The Evolutionary Value of Recombination Is Constrained by Genome Modularity’, PLOS Genetics, 1: e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2021) ‘RDP5: A Computer Program for Analyzing Recombination In, and Removing Signals of Recombination From, Nucleotide Sequence Datasets’, Virus Evolution, 7: veaa087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard Smith J. (1992) ‘Analyzing the Mosaic Structure of Genes’, Journal of Molecular Evolution, 34: 126–9. [DOI] [PubMed] [Google Scholar]
- Meyer M. M. et al. (2003) ‘Library Analysis of SCHEMA-guided Protein Recombination’, Protein Science, 12: 1686–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan J., and Wollert T. (2021) ‘Membrane Remodeling by SARS-CoV-2 – Double-enveloped Viral Replication’, Faculty Reviews, 10: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller H. (1964) ‘The Relation of Recombination to Mutational Advance’, Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, 106: 2–9. [DOI] [PubMed] [Google Scholar]
- Ou X. et al. (2020) ‘Characterization of Spike Glycoprotein of SARS-CoV-2 on Virus Entry and Its Immune Cross-reactivity with SARS-CoV’, Nature Communications, 11: 1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petes T. D., and Merker J. D. (2002) ‘Context Dependence of Meiotic Recombination Hotspots in Yeast: The Relationship between Recombination Activity of a Reporter Construct and Base Composition’, Genetics, 162: 2049–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. et al. (2004) ‘UCSF Chimera–a Visualization System for Exploratory Research and Analysis’, Journal of Computational Chemistry, 25: 1605–12. [DOI] [PubMed] [Google Scholar]
- Pollett S. et al. (2021) ‘A Comparative Recombination Analysis of Human Coronaviruses and Implications for the SARS-CoV-2 Pandemic’, Scientific Reports, 11: 17365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj V. S. et al. (2013) ‘Dipeptidyl Peptidase 4 Is a Functional Receptor for the Emerging Human coronavirus-EMC’, Nature, 495: 251–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reusken C. B. et al. (2016) ‘Cross Host Transmission in the Emergence of MERS Coronavirus’, Current Opinion in Virology, 16: 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer S. (1989) ‘Statistical Tests for Detecting Gene Conversion’, Molecular Biology and Evolution, 6: 526–38. [DOI] [PubMed] [Google Scholar]
- Sershen C. L. et al. (2011) ‘Superhelical Duplex Destabilization and the Recombination Position Effect. Lustig AJ (Ed.)’, PLoS ONE, 6: e20798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegfried N. A. et al. (2014) ‘RNA Motif Discovery by SHAPE and Mutational Profiling (SHAPE-map)’, Nature Methods, 11: 959–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon-Loriere E. et al. (2009) ‘Molecular Mechanisms of Recombination Restriction in the Envelope Gene of the Human Immunodeficiency Virus’, PLoS Pathogens, 5: 1000418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2010) ‘RNA Structures Facilitate Recombination-mediated Gene Swapping in HIV-1’, Journal of Virology, 84: 12675–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sola I. et al. (2015) ‘Continuous and Discontinuous RNA Synthesis in Coronaviruses’, Annual Review of Virology, 2: 265–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S. et al. (2021) ‘The Global Landscape of SARS-CoV-2 Genomes, Variants, and Haplotypes in 2019nCoVR’, Genomics, Proteomics & Bioinformatics, 18/6: 749–759.doi: 10.1016/j.gpb.2021.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark C. et al. (2006) ‘BioGRID: A General Repository for Interaction Datasets’, Nucleic Acids Research, 34: D535–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su S. et al. (2016) ‘Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses’, Trends in Microbiology, 24: 490–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang T. et al. (2020) ‘Coronavirus Membrane Fusion Mechanism Offers a Potential Target for Antiviral Development’, Antiviral Research, 178: 104792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Vugt J. J. F. A. et al. (2001) ‘High Frequency RNA Recombination in Porcine Reproductive and Respiratory Syndrome Virus Occurs Preferentially between Parental Sequences with High Similarity’, Journal of General Virology, 82: 2615–20. [DOI] [PubMed] [Google Scholar]
- Vlasova A. N. et al. (2021) ‘Novel Canine Coronavirus Isolated from a Hospitalized Patient with Pneumonia in East Malaysia’, Clinical Infectious Diseases, 74/3: 446–454.doi: 10.1093/cid/ciab456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt C. A. et al. (2002) ‘Protein Building Blocks Preserved by Recombination’, Nature Structural Biology, 9: 553–8. [DOI] [PubMed] [Google Scholar]
- Wan Y, Shang J, Graham R et al. (2020) ‘Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. Gallagher T (Ed.)’, Journal of Virology, 94: 221–224.doi: 10.1128/JVI.00127-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Junker D., and Collisson E. W. (1993) ‘Evidence of Natural Recombination within the S1 Gene of Infectious Bronchitis Virus’, Virology, 192: 710–6. [DOI] [PubMed] [Google Scholar]
- Wang S. et al. (2021) ‘AXL Is a Candidate Receptor for SARS-CoV-2 that Promotes Infection of Pulmonary and Bronchial Epithelial Cells’, Cell Research, 31: 126–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. et al. (2015) ‘Discovery, Diversity and Evolution of Novel Coronaviruses Sampled from Rodents in China’, Virology, 474: 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2020) ‘Extensive Genetic Diversity and Host Range of Rodent-borne Coronaviruses’, Virus Evolution, 6: veaa078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wege H. et al. (1998) ‘Coronavirus Infection and Demyelination: Sequence Conservation of the S-gene during Persistent Infection of Lewis-rats’, Advances in Experimental Medicine and Biology, 440: 767–73. [PubMed] [Google Scholar]
- Wesley R. D. (1999) ‘The S Gene of Canine Coronavirus, Strain UCD-1, Is More Closely Related to the S Gene of Transmissible Gastroenteritis Virus than to that of Feline Infectious Peritonitis Virus’, Virus Research, 61: 145–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White J. M. et al. (2008) ‘Structures and Mechanisms of Viral Membrane Fusion Proteins: Multiple Variations on a Common Theme’, Critical Reviews in Biochemistry and Molecular Biology, 43: 189–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P. C. Y. et al. (2010) ‘Coronavirus Genomics and Bioinformatics Analysis’, Viruses, 2: 1804–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrapp D. et al. (2020) ‘Cryo-EM Structure of the 2019-nCoV Spike in the Prefusion Conformation’, Science, 367: 1260–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S. et al. (2020) ‘Inhibition of SARS-CoV-2 (Previously 2019-nCoV) Infection by a Highly Potent Pan-coronavirus Fusion Inhibitor Targeting Its Spike Protein that Harbors a High Capacity to Mediate Membrane Fusion’, Cell Research, 30: 343–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D., and Leibowitz J. L. (2015) ‘The Structure and Functions of Coronavirus Genomic 3′ and 5′ Ends’, Virus Research, 206: 120–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y. et al. (2021) ‘Characterizing Transcriptional Regulatory Sequences in Coronaviruses and Their Role in Recombination’, Molecular Biology and Evolution, 38: 1241–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeager C. L. et al. (1992) ‘Human Aminopeptidase N Is a Receptor for Human Coronavirus 229E’, Nature, 357: 420–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehr J. D., Kosakovsky Pond S. L., and Martin D. P. et al. (2021) ‘Recent Zoonotic Spillover and Tropism Shift of a Canine Coronavirus Is Associated with Relaxed Selection and Putative Loss of Function in NTD Subdomain of Spike Protein’, Evolutionary Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z. et al. (2021) ‘A Database Resource and Online Analysis Tools for Coronaviruses on A Historical and Global Scale’, Database, 2020: 1–8 (baaa070).doi: 10.1093/DATABASE/BAAA070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zúñiga S. et al. (2004) ‘Sequence Motifs Involved in the Regulation of Discontinuous Coronavirus Subgenomic RNA Synthesis’, Journal of Virology, 78: 980–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.