

Abstract
A 3.6-kb endogenous plasmid was isolated from a Propionibacterium freudenreichii strain and sequenced completely. Based on homologies with plasmids from other bacteria, notably a plasmid from Mycobacterium, a region harboring putative replicative functions was defined. Outside this region two restriction enzyme recognition sites were used for insertion of an Escherichia coli-specific replicon and an erythromycin resistance gene for selection in Propionibacterium. Hybrid vectors obtained in this way replicated in both E. coli and P. freudenreichii. Whereas electroporation of P. freudenreichii with vector DNA isolated from an E. coli transformant yielded 10 to 30 colonies per μg of DNA, use of vector DNA reisolated from a Propionibacterium transformant dramatically increased the efficiency of transformation (≥108 colonies per μg of DNA). It could be shown that restriction-modification was responsible for this effect. The high efficiency of the system described here permitted successful transformation of Propionibacterium with DNA ligation mixtures.
The genus Propionibacterium can be divided into two groups, a group containing the classical (or dairy) propionibacteria and a group containing the cutaneous propionibacteria (6). Members of the first group, especially Propionibacterium freudenreichii, play an essential role in the manufacture of Swiss and related types of cheeses (12). Other industrial applications are found in the production of propionic acid and vitamin B12 (5, 28). Of growing interest, but less well documented, are the probiotic properties ascribed to some propionibacterial strains (16, 21).
Strain improvement and, in general, study of this economically important group of bacteria would be greatly facilitated by the availability of a system for genetic modification. This report describes isolation and characterization of a 3.6-kb plasmid and successful use of this plasmid in the construction of a set of Escherichia coli-Propionibacterium shuttle vectors. Reproducible transformation of P. freudenreichii strains with these shuttle vectors was achieved by means of electroporation.
MATERIALS AND METHODS
Bacterial strains and plasmids.
Propionibacterium strains were obtained from the Belgian Coordinated Collections of Microorganisms/LMG (Ghent, Belgium), from the American Type Culture Collection (Rockville, Md.), and from the Deutsche Sammlung von Mikroorganismen (Braunschweig, Germany). P. freudenreichii subsp. freudenreichii VTB1 was obtained from DSM Food Specialties' industrial collection. For amplification of newly constructed shuttle vector DNA E. coli DH5α was used. pBluescript SKII+ was obtained from Stratagene (La Jolla, Calif.).
Media and growth conditions.
E. coli DH5α was cultivated at 37°C in L medium (24) supplemented with 50 μg of ampicillin per ml if necessary. Propionibacteria were cultivated anaerobically at 30°C in MRS (7) or SLB medium (8) supplemented with an appropriate antibiotic when plasmid isolation was to be performed or in SLB medium when electroporation was to be performed.
Isolation of plasmid DNA from propionibacteria.
Propionibacteria were cultivated in 5 ml of medium for 48 h. Plasmid DNA was extracted from the bacteria by a modified E. coli plasmid isolation procedure (4). Briefly, cells were washed in 25% sucrose–50 mM Tris-HCl (pH 8) and resuspended in 250 μl of TENS (25% sucrose, 50 mM NaCl, 50 mM Tris-HCl, 5 mM EDTA; pH 8) containing 10 mg of lysozyme per ml. After 20 to 30 min of incubation at 37°C, cells were lysed by adding 500 μl of 0.2 N NaOH–1% sodium dodecyl sulfate and incubating the preparation on ice for 2 to 5 min. Then 400 μl of 3 M sodium acetate (pH 4.8) was added, and this was followed by 5 min of incubation on ice and extraction with phenol-chloroform. DNA was precipitated by adding isopropanol.
General methods.
The molecular biological techniques used in this study were described by Sambrook et al. (24). Restriction enzymes and T4 DNA ligase were purchased from New England Biolabs and GIBCO BRL. Taq polymerase was obtained from SphaeroQ (Leiden, The Netherlands). All enzymes were used according to the manufacturers' instructions.
Sequence analysis.
The nucleotide sequence of plasmid p545, linearized by EcoRI and inserted in pBluescript SKII+, was determined by the primer walking strategy, starting from both ends. Overall, 25 primers were designed to cover the complete 3.5 kb, and each nucleotide was read at least two times in each direction. Finally, absence of a small EcoRI fragment could be ruled out by sequence analysis of noncloned p545 in the region surrounding the EcoRI site.
Sequencing was accomplished by using an Applied Biosystems model 373A automatic sequencer according to procedures provided by the supplier and fluorescent-dye-labeled dideoxyribonucleotides.
Isolation of a Propionibacterium-specific 16S rRNA promoter.
On the basis of the sequence of 16S rRNA from P. freudenreichii DSM 20271 (= ATCC 6207) (GenBank accession number X53217), we chose an appropriate restriction enzyme (HindIII) and designed primers that enabled us to amplify an approximately 3-kb region encompassing the promoter by inverse PCR (18). From the PCR product a 0.6-kb SphI-HindIII fragment directly upstream of the 16S rRNA coding sequence was isolated for further use.
Construction of E. coli-Propionibacterium shuttle vectors.
To construct E. coli-Propionibacterium shuttle vectors, we used pBR322 in which the EcoRI-AvaI fragment encompassing the tetracycline resistance gene was replaced by a polylinker. In pBR322ΔtetPL the sequence of this polylinker is as shown in Fig. 1, which includes restriction sites for EcoRI (restored), HindIII, SalI, HpaI, PstI, SphI, BamHI, Acc65I, EcoRV, and BglII (AvaI is not restored). In the Acc65I site a 1.7-kb Acc65I fragment was cloned that encompassed the erythromycin resistance gene (ermE) from Saccharopolyspora erythraea NRLL2338 (3, 27), which yielded pBRES1. In pBRES1 the genes for erythromycin and ampicillin resistance were transcribed in opposite directions. The EcoRV site of pBRES1 was used for insertion of plasmid p545 from P. freudenreichii LMG 16545 linearized with BsaBI, which resulted in pBRESP36B1 and pBRESP36B2; the difference between the latter two plasmids was the orientation of p545.
FIG. 1.

Sequence of the polylinker in pBR322ΔtetPL.
For insertion of AlwNI-linearized p545, a polylinker was introduced into pBRES1 between the BglII site and the proximal Acc65I site, and the sequence of this polylinker was as follows:
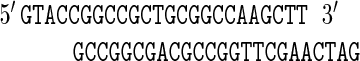 |
The polylinker supplied restriction sites for Acc65I (restored), SfiI, and HindIII (BglII was not restored). The resulting plasmid was designated pBRES2. Ligation of SfiI-linearized pBRES2 with AlwNI-linearized p545 yielded pBRESP36A.
To enable stepwise deletion of p545-specific parts from pBRESP36A DNA, this DNA was first digested with SstII and BclI, which resulted in 1.7- and 6.5-kb fragments. The 1.7-kb fragment was replaced by a synthetic duplex DNA. In this way, in effect, the 1.6-kb AlwNI-BclI fragment of plasmid p545 was deleted from the vector. The synthetic duplex DNA was designed to link SstII and BclI ends and to supply a number of unique restriction sites; its sequence was as follows:
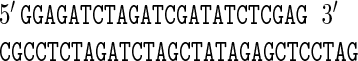 |
Thus, the following restriction enzyme recognition sites were supplied: SstII (restored), BglII, XbaI, ClaI, EcoRV, and XhoI (BclI was not restored). The ligation mixture was transferred to E. coli, and we selected a transformant that contained a vector having the expected composition. The vector was designated pBRESAΔS-B.
Electroporation.
P. freudenreichii strains cultivated to the stationary growth phase were diluted 1:50 in fresh SLB medium. After incubation for about 20 h, the cells, which were in the exponential growth phase, were harvested and washed extensively in ice-cold 0.5 M sucrose. Electroporation of P. freudenreichii strains was performed with a Gene Pulser apparatus (Bio-Rad) by using a modified protocol developed for electroporation of bifidobacteria (2). Briefly, cells were washed once in ice-cold electroporation buffer (0.5 M buffered sucrose) and resuspended in electroporation buffer (about 1/100 of the original culture volume). Then 80 to 100 μl of the suspension was mixed with DNA in a cooled electroporation cuvette, and an electric pulse was delivered at 200-Ω resistance and 25-μF capacitance. Optimal electroporation results were obtained in 0.5 M sucrose buffered with 1 mM potassium acetate (pH 5.5) at 20 kV/cm. Immediately after the pulse 900 μl of cold SLB medium containing 0.5 M sucrose was added, and after 2.5 to 3 h of incubation at 30°C, cells were plated on SLB agar plates containing 0.5 M sucrose and 10 μg of erythromycin per ml. After 5 to 7 days of incubation at 30°C under anaerobic conditions, transformants could be detected.
Nucleotide sequence accession number.
The nucleotide sequence of plasmid p545 has been deposited in the GenBank database under accession number AF291751.
RESULTS AND DISCUSSION
Initial transformation experiments.
Our initial attempts to transform propionibacteria were aimed at P. freudenreichii type strain ATCC 6207. We used electroporation procedures developed in our laboratory for lactobacilli (15, 22) and for bifidobacteria (2) with Corynebacterium-E. coli shuttle vectors pECM2 and pEBM3 (gifts from J. Kalinowski), Bifidobacterium-E. coli shuttle vector pDG7 (17), Lactobacillus-E. coli shuttle vector pLP825, Lactobacillus-specific vector pLPE323 (14, 22), and broad-host-range Lactococcus-derived plasmid pGK12 (10). None of these attempts yielded any transformants. Since one of the possible explanations for this was the inability of propionibacteria to support replication of the vectors used, a set of new shuttle vectors based on a Propionibacterium-specific replicon was constructed.
Screening Propionibacterium strains for endogenous plasmids.
Seventy-five Propionibacterium strains representing all four recognized species of dairy propionibacteria were screened for the presence of endogenous plasmids. In the majority of these strains no small endogenous plasmids could be found, in accordance with reports on other Propionibacterium strains (19, 20, 23). The following six strains were found to contain a 6- to 10-kb plasmid: P. acidipropionici ATCC 4875 (= DSM 20272) and LMG 16447, P. jensenii LMG 16453, P. freudenreichii LMG 16545, P. freudenreichii subsp. freudenreichii LMG 16546, and Propionibacterium sp. strain LMG 16550. All of these strains except ATCC 4875 also harbor one or more large (≥20-kb) plasmids. P. freudenreichii LMG 16545 and LMG 16546 were both found to contain a 3.6-kb plasmid; these strains were chosen for further study, and the plasmids which they harbor are designated p545 and p546, respectively. In Southern blot experiments in which cloned p545 (see below) was used as a probe, strong hybridization was observed with plasmid preparations from strains LMG 16545 and LMG 16546, indicating that plasmids p545 and p546 are closely related. No hybridization was observed with plasmid DNA from P. acidipropionici ATCC 4875 (results not shown); the plasmid of this strain has been described previously by Rehberger and Glatz as plasmid pRG01 (23).
Analysis of plasmids p545 and p546.
Restriction enzyme analysis of p545 and p546 DNA revealed identical restriction patterns (data not shown). Because of the assumed identity, only one of these plasmids, p545, was sequenced; the sequence was 3,555 bp long. A BLAST search in GenBank (1) revealed that two open reading frames (ORFs) (ORF1 and ORF2, comprising 303 and 85 amino acids, respectively) showed significant homology to (putative) replication proteins from a number of plasmids, including pAL5000 (11, 25), from Mycobacterium fortuitum. ORF1 and ORF2 were 28 to 30% identical and 34 to 38% similar to pAL5000 replication proteins repA and repB, respectively. As found for the other plasmids, the two replication proteins in p545 showed translational coupling. Such coupling was also suggested by L. Meile (personal communication) for Propionibacterium plasmid pLME108 (accession number AJ006662).
In pAL5000 a minimal replicon could be defined, and this replicon consisted of the translationally coupled repA and repB genes and a 435-bp “inc region” located upstream from repA and containing the origin of replication (25). Although a similar origin could not be found in p545, it was deemed likely that sites not interrupting the two ORFs or the approximately 500-bp upstream region would not interfere with replication of the plasmid. Analysis of p545 DNA for suitable unique restriction sites yielded two likely candidates, a BsaBI site and an AlwNI site (Fig. 2A). These sites were used to introduce an E. coli-specific replicon and a selection marker for Propionibacterium, as described in Materials and Methods.
FIG. 2.

Limited restriction map of plasmid p545 (A) and of one of its derivative shuttle vectors (B). The positions of the two translationally coupled ORFs are also indicated. The restriction enzyme recognition sites in boldface type indicate the 1.8-kb p545-specific region that can be deleted without disturbing replication. HindIII sites used for deletion of the pBR-specific part are indicated by asterisks.
Transformation of P. freudenreichii strains by electroporation.
By using P. freudenreichii ATCC 6207, LMG 16545, and VTB1 as host organisms, low but reproducible transformation efficiencies, 10 to 30 transformants per μg of plasmid DNA, were obtained (Table 1). Within limits, the type of buffer and the actual voltage applied had only modest effects. The sizes and restriction patterns of plasmid DNA isolated from P. freudenreichii transformants were indistinguishable from those of the input DNA, indicating that replication took place without detectable alteration of the plasmid DNA. Southern blot hybridization confirmed that the vectors were present as autonomously replicating DNA; chromosomal integration was never observed. Moreover, the assumption that BsaBI and AlwNI sites in p545 were located outside the replication region proved to be correct. Finally, the replicon was found to be active irrespective of the polarity relative to the selection marker and E. coli replicon (pBRESP36B1 versus pBRESP36B2). From Table 1 we also concluded that activity of the p545 replicon may be limited to P. freudenreichii strains, since electroporation of other Propionibacterium species did not yield any transformants.
TABLE 1.
Transformation efficiencies in Propionibacterium strains with DNA isolated from E. coli or P. freudenreichii ATCC 6207a
| Recipient strain | Transformation efficiency (no. of transformants/μg of DNA)
|
|
|---|---|---|
| DNA isolated from E. coli | DNA isolated from P. freudenreichii | |
| P. freudenreichii strains | ||
| ATCC 6207 | 10–30 | ≥108 |
| VTB1 | 10–30 | ≥108 |
| LMG 16545 | 10–30 | ≥108 |
| P. acidipropionici strains | ||
| DSM 13572 | 0 | 0 |
| DSM 20727 | 0 | 0 |
| P. jensenii DSM 20535 | 0 | 0 |
| P. thoenii DSM 20276 | 0 | 0 |
Electroporation was performed at 20 kV/cm, 200 Ω, and 25 μF. The vectors used for transformation were pBRESP36A, pBRESP36B1, and pBRESP36B2.
Restriction-modification.
In an attempt to increase the efficiency of transformation, an electroporation experiment was performed with plasmid DNA isolated from a P. freudenreichii ATCC 6207 transformant. A 106- to 107-fold greater transformation efficiency compared to that obtained with DNA isolated from E. coli DH5α was observed (Table 1); i.e., there were ≥108 transformants per μg of DNA, suggesting that one or more likely several restriction-modification systems were present in ATCC 6207. Indeed, when plasmid DNA isolated from an ATCC 6207 transformant was used to transform ATCC 6207 again after passage through E. coli DH5α, the same low frequency of transformation that was initially observed was obtained, ruling out the possibility that the high frequency of transformation obtained with ATCC 6207-derived plasmid DNA was caused by a mutation in the plasmid DNA.
In the same way the existence of restriction-modification systems in P. freudenreichii LMG 16545 and VTB1 could be shown. Moreover, since the same increase in efficiency was observed when ATCC 6207-derived plasmid DNA was used to transform ATCC 6207, LMG 16545, or VTB1, it is plausible that the restriction-modification systems in these strains are identical. Analysis of the type and specificity of the restriction-modification system is currently under way; preliminary results indicate that no type II restriction enzymes are present. Again, no transformants were obtained upon electroporation of other Propionibacterium species (Table 1).
Towards identification of a minimal replicon in p545.
Since a selectable derivative of plasmid p545 (i.e., pBRESP36A) (Fig. 2B) and a highly efficient transformation procedure were available, experiments were performed to determine which parts of the p545 plasmid are essential for replication in propionibacteria by transferring pBRESP36A and deletion derivatives of pBRESP36A to propionibacteria.
As might be expected, the E. coli-specific part of pBRESP36A was not involved, since deletion of this part from the shuttle vector by partial digestion with HindIII and religation (Fig. 2B) did not impair replication in propionibacteria.
Since P. freudenreichii VTB1 and ATCC 6207 could be successfully transformed with vector pBRESAΔS-B (see Materials and Methods), we concluded that the 1.6-kb region between AlwNI and BclI in p545 is not essential for replication of the plasmid. Further deletion of the 240-bp p545-specific SalI-BclI fragment (achieved by ligation of the 1.3-kb SalI-SstI fragment and the 6.6-kb SstI-XhoI fragment of pBRESAΔS-B, isolated from a Propionibacterium transformant) did not impair replication in propionibacteria either; transfer of the ligation mixture to P. freudenreichii ATCC 6207 yielded numerous transformants. Analysis of a number of transformants showed that they all carried the expected deletion variant of pBRESAΔS-B. The newly derived plasmid was designated pBRESAΔS-S. In effect, we showed that all essential information for replication of p545 in propionibacteria is located on a 1.7-kb fragment and that the other 1.8 kb can be deleted without obviously disturbing replication of the plasmid.
Electroporation of P. freudenreichii strains with vectors carrying other selection markers.
Vectors were constructed in which the erythromycin resistance gene present in the pBRESP36 series of shuttle vectors was replaced by a different selection marker or into which a second selection marker was introduced (Table 2). Vectors carrying as a single selection marker the erythromycin resistance gene from Enterococcus faecalis plasmid pAMβ1 (13) or the chloramphenicol resistance (cat) gene from Staphylococcus aureus plasmid pC194 (9) with either its own promoter or with a P. freudenreichii-specific rRNA promoter (see Materials and Methods) did not yield any transformants upon electroporation of P. freudenreichii strains. Given the high G+C content of ermE and the low G+C contents of the other selection markers (34% for E. faecalis and 29% for S. aureus), it is tempting to speculate that genes with low G+C contents are poorly expressed in propionibacteria if they are expressed at all. Therefore, the chloramphenicol resistance (cat) gene from pACYC184 (G+C content, 53%) was introduced as a second selection marker into pBRESP36B2, and this marker carried either its own promoter, the ermE promoter, or the P. freudenreichii-specific 16S rRNA promoter. The vectors obtained in this way all conferred chloramphenicol resistance to E. coli, but after introduction into P. freudenreichii by electroporation, primary selection of transformants with chloramphenicol proved to be impossible. Only after primary selection with erythromycin could chloramphenicol be used as a selective agent, and this occurred only when the 16S rRNA promoter was present.
TABLE 2.
Antibiotic resistance genes used in this study
| Gene | G+C content (%) | Origin | Promoter | Propionibacterium transformants |
|---|---|---|---|---|
| cat | 29 | pC194 (Staphylococcus aureus) | Original | − |
| 16S rRNA | − | |||
| cat | 53 | pACYC184 (Escherichia coli) | Original | − |
| ermE | − | |||
| 16S rRNA | (+)a | |||
| cml | 63 | pEBM3 (Corynebacterium striatum) | Original | + |
| ery | 34 | pAMβ1 (Enterococcus faecalis) | Original | − |
| ery | 71 | pIJ488 (Saccharopolyspora erythraea) | Original (= ermE) | + |
Only as a secondary selection marker.
Introduction of the chloramphenicol resistance gene (cml) from Corynebacterium striatum (26) into pBRESP36B2 yielded a vector that could be directly selected for with chloramphenicol. However, although the G+C content of cml (63%) is comparable to that of P. freudenreichii, this may not be the only reason, since cml provides resistance by expelling chloramphenicol, whereas cat provides resistance by acetylating chloramphenicol.
Analysis of vector stability.
P. freudenreichii transformants containing pBRESP36A, pBRESP36B1, or pBRESP36B2 were cultivated for about 25 generations in medium without erythromycin and subsequently plated on solidified medium with and without erythromycin. No gross differences in the number of colonies were observed, indicating that the vector is segregationally stably maintained (≤5% loss after 25 generations without selection). In addition, structural stability was studied by cultivation in selective medium for about 25 generations, plating on solidified medium, and analysis of plasmid DNA from a number of colonies. No deletions were observed, indicating that the vectors were also structurally stably maintained.
Conclusion.
A reproducible and highly efficient host-vector system for P. freudenreichii has been developed. To our knowledge, this is the first time that such a system has been described.
ACKNOWLEDGMENT
Valuable discussions with Rob Leer are greatly appreciated.
REFERENCES
- 1.Altschul S F, Gish W, Miller W, Meyers E W, Lipman D J. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 2.Argnani A, Leer R J, van Luijk N, Pouwels P H. A convenient and reproducible method to genetically transform bacteria of the genus Bifidobacterium. Microbiology. 1996;142:109–114. doi: 10.1099/13500872-142-1-109. [DOI] [PubMed] [Google Scholar]
- 3.Bibb M J, Janssen G R, Ward J M. Cloning and analysis of the promoter region of the erythromycin resistance gene (ermE) of Streptomyces erythraeus. Gene. 1985;38:215–226. doi: 10.1016/0378-1119(85)90220-3. [DOI] [PubMed] [Google Scholar]
- 4.Birnboim H C, Doly J. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 1979;7:1513–1523. doi: 10.1093/nar/7.6.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boyaval P, Corre C. Production of propionic acid. Lait. 1995;75:453–461. [Google Scholar]
- 6.Cummins C S, Johnson J L. Irregular, nonsporing Gram-positive rods. Genus I. Propionibacterium. In: Sneath P H A, Mair N S, Sharpe M E, Holt J G, editors. Bergey's manual of systematic bacteriology. Vol. 2. Baltimore, Md: Williams and Wilkins; 1984. pp. 1346–1353. [Google Scholar]
- 7.De Man J C, Rogosa M, Sharpe M E. A medium for the cultivation of lactobacilli. J Appl Bacteriol. 1960;23:130–135. [Google Scholar]
- 8.De Vries W, van Wijck-Kapteijn M C, Stouthamer A H. Influence of oxygen on growth, cytochrome synthesis and fermentation pattern in propionic acid bacteria. J Gen Microbiol. 1972;71:515–524. doi: 10.1099/00221287-71-3-515. [DOI] [PubMed] [Google Scholar]
- 9.Ehrlich S D. Replication and expression of plasmids from Staphylococcus aureus in Bacillus subtilis. Proc Natl Acad Sci USA. 1977;74:1433–1436. doi: 10.1073/pnas.74.4.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kok J, van der Vossen J M B M, Venema G. Construction of plasmid cloning vectors for lactic acid streptococci which also replicate in Bacillus subtilis and Escherichia coli. Appl Environ Microbiol. 1984;48:726–731. doi: 10.1128/aem.48.4.726-731.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Labidi A, Mardis E, Roe B A, Wallace R J., Jr Cloning and DNA sequence of the Mycobacterium fortuitum var. fortuitum plasmid pAL5000. Plasmid. 1992;27:130–140. doi: 10.1016/0147-619x(92)90013-z. [DOI] [PubMed] [Google Scholar]
- 12.Langsrud T, Sørhaug T, Vegarud G E. Protein degradation and amino acid metabolism by propionibacteria. Lait. 1995;75:325–330. [Google Scholar]
- 13.Leblanc D J, Lee L N. Physical and genetic analysis of streptococcal plasmid pAMβ1 and cloning of its replication origin. J Bacteriol. 1984;157:445–453. doi: 10.1128/jb.157.2.445-453.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leer R J, van Luijk N, Posno M, Pouwels P H. Structural and functional analysis of two cryptic plasmids from Lactobacillus pentosus MD353 and Lactobacillus plantarum ATCC 8014. Mol Gen Genet. 1992;234:265–274. doi: 10.1007/BF00283847. [DOI] [PubMed] [Google Scholar]
- 15.Leer R J, Posno M, van Rijn J M M, Lokman B C, Pouwels P H. Transformation of Lactobacillus plantarum by plasmid DNA. FEMS Microbiol Rev. 1987;46:P20. [Google Scholar]
- 16.Mantere-Alhonen S. Propionibacteria used as probiotics—a review. Lait. 1995;75:175–185. [Google Scholar]
- 17.Matteuzzi D, Brigidi P, Rossi M, Di D. Characterization and molecular cloning of Bifidobacterium longum cryptic plasmid pMB1. Lett Appl Microbiol. 1990;11:220–223. doi: 10.1111/j.1472-765x.1990.tb00165.x. [DOI] [PubMed] [Google Scholar]
- 18.Ochman H, Medhora M M, Garza D, Hartl D L. Amplification of flanking sequences by inverse PCR. In: Innis M A, Gelfand D A, Sninsky J J, White T J, editors. PCR protocols. A guide to methods and applications. San Diego, Calif: Academic Press; 1990. pp. 219–228. [Google Scholar]
- 19.Panon G. Presence of plasmids in propionic acid bacteria. Lait. 1988;68:103–107. [Google Scholar]
- 20.Pérez Chaia A, Sesma F, Pesce de Ruiz Holgado A, Oliver G. Screening of plasmids in strains of propionibacterium and mesophilic lactobacilli isolated from Swiss type cheeses. Microbiol Aliment Nutr. 1988;6:171–174. [Google Scholar]
- 21.Pérez Chaia A, Zárata G, Oliver G. The probiotic properties of propionibacteria. Lait. 1999;79:175–185. [Google Scholar]
- 22.Posno M, Leer R J, van Luijk N, van Giezen M J F, Heuvelmans P T H M, Pouwels P H. Incompatibility of Lactobacillus vectors with replicons derived from small cryptic Lactobacillus plasmids and segregational instability of the introduced vectors. Appl Environ Microbiol. 1991;57:1822–1828. doi: 10.1128/aem.57.6.1822-1828.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rehberger T G, Glatz B A. Characterization of Propionibacterium plasmids. Appl Environ Microbiol. 1990;56:864–871. doi: 10.1128/aem.56.4.864-871.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 25.Stolt P, Stoker N G. Functional definition of regions necessary for replication and incompatibility in the Mycobacterium fortuitum plasmid pAL5000. Microbiology. 1996;142:2795–2802. doi: 10.1099/13500872-142-10-2795. [DOI] [PubMed] [Google Scholar]
- 26.Tauch A, Zheng Z, Puhler A, Kalinowski J. Corynebacterium striatum chloramphenicol resistance transposon Tn5564: genetic organization and transposition in Corynebacterium glutamicum. Plasmid. 1998;40:126–139. doi: 10.1006/plas.1998.1362. [DOI] [PubMed] [Google Scholar]
- 27.Thompson C J, Kieser T, Ward J M, Hopwood D A. Physical analysis of antibiotic resistance genes from Streptomyces and their use in vector construction. Gene. 1982;20:51–62. doi: 10.1016/0378-1119(82)90086-5. [DOI] [PubMed] [Google Scholar]
- 28.Youngsmith B, Sonomoto K, Tanka S, Fukui S. Production of vitamin B12 by immobilized cells of propionic acid bacterium. Appl Microbiol Biotechnol. 1982;16:70–74. [Google Scholar]