

Summary
Background
Statins remain one of the most prescribed medications worldwide. While effective in decreasing atherosclerotic cardiovascular disease risk, statin use is associated with adverse effects for a subset of patients, including disrupted metabolic control and increased risk of type II diabetes.
Methods
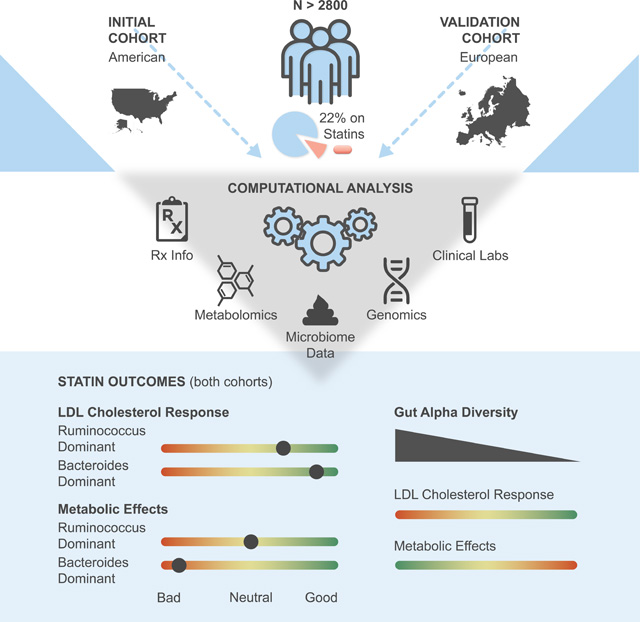
We investigated the potential role of the gut microbiome in modifying patient responses to statin therapy across two independent cohorts (Discovery N=1848, Validation N=991). Microbiome composition was assessed in these cohorts using stool 16S rRNA amplicon and shotgun metagenomic sequencing, respectively. Microbiome associations with markers of statin on-target and adverse effects were tested via a covariate-adjusted interaction analysis framework, utilizing blood metabolomics, clinical laboratory tests, genomics, and demographics data.
Findings
The hydrolyzed substrate for 3-hydroxy-3-methylglutarate-CoA (HMG-CoA) reductase, HMG, emerged as a promising marker for statin on-target effects in cross-sectional cohorts. Plasma HMG levels reflected both statin therapy intensity and known genetic markers for variable statin responses. Through exploring gut microbiome associations between blood-derived measures of statin effectiveness and adverse metabolic effects of statins, we find that heterogeneity in statin responses was consistently associated with variation in the gut microbiome across two independent cohorts. A Bacteroides-enriched and diversity-depleted gut microbiome was associated with more intense statin responses, both in terms of on-target and adverse effects.
Conclusions
With further study and refinement, gut microbiome monitoring may help inform precision statin treatment.
eTOC blurb
Prior work has indicated that stain therapy impacts gut microbiome composition and that gut bacteria can metabolize statins, but the clinical implications of these interactions remain unknown. Wilmanski et al. identify robust associations between microbiome composition and on-target and adverse responses to statins, which could prove useful in drug personalization.
Graphical Abstract
Introduction:
Between 25% and 30% of older adults across the United States and Europe take statins regularly for the purpose of treating or preventing atherosclerotic cardiovascular disease (ACVD), making statins one of the most commonly prescribed medications in the developed world 1,2. While statins have proven to be highly effective in decreasing ACVD-associated mortality, considerable heterogeneity exists in terms of efficacy (i.e. lowering low density lipoprotein (LDL) cholesterol) 3. Importantly, although LDL cholesterol is not the only factor contributing to ACVD progression 4, the extent to which statins decrease LDL cholesterol is directly proportional to their effectiveness in decreasing cardiovascular events 3. Furthermore, statin use can give rise to a number of adverse effects in a subset of patients, including myopathy, disrupted glucose control, and increased risk of developing type II diabetes (T2D) 5–9. Several guidelines exist for which at-risk populations should be prescribed statins and at what intensity 10. However, despite considerable progress in identifying pharmacological 11 and genetic factors 12 contributing to heterogeneity in statin response, personalized approaches to statin therapy remain limited. Many times, treatment decisions are made through trial and error between the clinician and patient to obtain an optimal tolerable dose 13. Avoiding this trial-and- error phase through individualized analysis of genetic, physiological, and health parameters has the potential to improve drug tolerance, adherence, and long-term health benefits, as well as guide complementary therapies aimed at mitigating adverse effects of statin therapy.
A number of studies have recently demonstrated a link between the gut microbiome and statin use 14,15, as well as between the gut microbiome and ASCVD risk 16. Similar to other prescription medications, statins are readily metabolized by gut bacteria into secondary compounds 17,18. This indicates that the gut microbiome may impact statin bioavailability or potency to its host, contributing to the interindividual variability in LDL response seen among statin users 19. Additionally, biochemical modification of statins by gut bacteria could potentially contribute to adverse effects of the drug 20. Independent of statins, the gut microbiome has a well characterized role in contributing to host metabolic health through regulating insulin sensitivity, blood glucose, and inflammation, hence sharing considerable overlap with the adverse effects of statin therapy 21,22.
Statin intake has also been implicated in shifting gut microbiome composition, where primarily obese individuals taking statins were less likely to be classified into a putative gut microbiome compositional state, or ‘enterotype’, defined by high relative abundance of Bacteroides and a depletion of short-chain fatty acid (SCFA) producing Firmicutes taxa 23.However, contradictory findings in animal models have also been reported, where a statin intervention decreased abundance of SCFA-producing taxa and, consequently, the gut ecosystem’s capacity to produce butyrate 24.
Given the numerous documented interactions between the gut microbiome and statins, and the established effect of the gut microbiome on metabolic health, we sought to explore the potential role of the gut microbiome in modifying the effect of statins on inhibiting their target enzyme 3-hydroxy-3-methylglutarate-CoA (HMG-CoA) reductase, as well as influencing the adverse effects of statins on metabolic health parameters. We analyzed data from over 1840 deeply-phenotyped individuals with extensive medication histories, clinical laboratory tests, plasma metabolomics, whole genome and stool 16S rRNA gene amplicon sequencing data. We found that the hydrolyzed product of HMG-CoA (i.e., HMG) may serve as a novel blood-derived marker of statin response. We further show that heterogeneity in statin on-target effects and metabolic disruption could be explained by variation in the composition of the gut microbiome. We replicated our findings in an independent European cohort consisting of individuals recruited to capture the full spectrum of cardiometabolic disease progression. Overall, our results suggest that, with further study and refinement, the taxonomic and/or functional composition of the gut microbiome may be used to inform personalized statin therapies.
Results:
Cohort
The study population is presented in Fig. 1A and Table S1 (see also Methods). Briefly, a total of 1848 American adults were included in the present analysis. Individuals in this cohort were self-enrolled in a now closed scientific wellness company (Arivale, Inc), had available plasma metabolomics and clinical laboratory data, and provided detailed information on prescription medication use. Of these 1848 Arivale participants, 244 were confidently identified as statin users, of which 97 provided detailed information on both dosage and type of statin prescribed. Demographics information was either collected through online questionnaires or one-on-one interviews with health coaches. In addition, we validated our main findings in a subset of an independent European cohort (n=688), consisting of individuals at various stages of cardiometabolic disease progression (Metacardis study)25, which collected stool shotgun metagenomics sequencing for gut microbiome analyses with paired medication use data, clinical laboratory test data, and serum metabolomics (see Methods).
Figure 1. Plasma HMG correlates with statin use and statin-associated LDL-response.

A) Frequency of statin use, type of statin taken, and number of participants with available data for each ‘omics for each participant included in the present analysis. B) Diagram of de novo cholesterol synthesis pathway, with HMG and the rate-limiting enzyme inhibited by statins highlighted. C) Scatterplots of LDL-cholesterol and plasma HMG in statin non-users (blue) and users (red) separately, across two different clinical laboratory test vendors used in the cohort. The lines shown are the y~x regression lines, and the shaded regions are 95% confidence intervals for the slope of each line. Below each scatter plot is the Spearman correlation coefficient and corresponding p-value. Adj. β(95%CI) corresponds to the β-coefficient for LDL cholesterol from GLMs predicting plasma HMG, adjusted for sex, age, and BMI. Also shown to the right of each scatter plot are kernel density plots for plasma HMG in statin users and non- users. The black lines indicate the mean of each group, and the p-value corresponds to the effect size of the difference between statin users and non-users from GLMs adjusted for the same covariates as above. D) Relationship between statin intensity therapy and plasma HMG as well LDL cholesterol levels for the subset of participants in the cohort who had available dosage intensity data (n=97) combined across both clinical lab test vendors. The lines shown are the y~x regression lines, and the shaded regions are 95% confidence intervals for the slope of each line. P-value corresponds to the dose-response relationship between therapy intensity and either plasma HMG (top box plot) or LDL cholesterol (bottom box plot). Values on the y-axis are analyte levels (residuals) adjusted for covariates (sex, age, BMI and clinical lab test vendor). Box plots represent the interquartile range (25th to 75th percentile, IQR), with the middle line denoting the median; whiskers span 1.5 × IQR, points beyond this range are shown individually.
Plasma HMG is a marker of statin use and on-target effects
The mechanism of action of statins is to inhibit the rate-limiting enzyme of de novo cholesterol synthesis, HMG-CoA reductase 26. Thus, we first sought to explore whether elevated plasma levels of the hydrolyzed substrate for this enzyme, HMG (measured in our broad untargeted metabolomics panel), could serve as a reliable marker of statin use (Fig. 1B). Plasma HMG levels were significantly higher in statin users than in non-users, consistent with our initial hypothesis and the drug’s well-established mechanism of action (Fig. 1C, generalized linear models (GLM) adjusted for sex, age, and BMI). HMG levels further showed a negative correlation with blood LDL-cholesterol across two independent clinical laboratory test vendors, but exclusively in statin users, indicating that plasma HMG may not only reflect statin use but also the extent to which statins inhibit their target enzyme (Fig. 1C).
The negative association between HMG and LDL-cholesterol, observed exclusively in statin users, indicates that this compound may serve as a proxy for statin efficacy. However, it is also possible HMG simply reflects patient adherence, where individuals who take the drug as prescribed have higher HMG and lower LDL-cholesterol than those who do not. To further evaluate the reliability of HMG as a marker for statin on-target effects, we explored its correspondence to variable doses of statins prescribed in a subset of statin users where this information was available (n=97). Different statins (atorvastatin, simvastatin, etc.) exhibit different potencies and are often prescribed at variable doses. In order to synchronize medical practices in terms of statin therapy, the American Heart Association (AHA) released guidelines for adjusting statin doses across all types of statins, which cluster into one of three intensity categories (low, moderate, and high) aimed at achieving desired decreases in LDL-cholesterol of <30%, 30–49%, ≥50%, respectively 10. Based on these AHA guidelines, a daily 40mg dose of rosuvastatin would place a patient in the high intensity category, while the same dose of fluvastatin would place a patient in the low intensity group. Hence, we re-classified participants into their respective therapy intensity groups based on the AHA guidelines (Fig. 1A) and evaluated the associations between therapy intensity, plasma HMG, and blood LDL-cholesterol levels. Therapy intensity showed a positive dose response relationship with HMG, independent of sex, age, and BMI (GLM adj. β(95% CI):0.15(0.12–0.17), P=1.1e-22)). Consistently, an inverse relationship was observed between therapy intensity and blood LDL-cholesterol, adjusted for the same covariates as well as for clinical laboratory test vendor (Fig. 1D, adj. β(95% CI):−15(−18 - −12), P=6.7e-20).
Previous large-scale pharmacogenomic studies of statin users have identified a number of single nucleotide polymorphisms (SNPs) predisposing patients to variable responses to statin therapy. To evaluate if plasma HMG captures known genetic variability in statin response, we tested associations between HMG and 9 SNPs most strongly associated with statin-mediated decrease in LDL-cholesterol in previous studies 12, using GLMs with a statin-by-genetic variant interaction term while adjusting for sex, age, BMI and genetic ancestry (see Methods). Of the 9 SNPs tested, 2 SNPs in close linkage disequilibrium (rs445925 and rs7412 mapping to the APOC1 and APOE genes, respectively, r > 0.80 in Caucasians) showed significant associations with HMG, that were dependent on statin intake (i.e., the effect was only present in statin users, FDR<0.05), in the directions consistent with the previously described associations of the same variants with statin response (Fig. S1, Table S2). Interestingly, running the same analysis with LDL-cholesterol instead of plasma HMG as an outcome variable (both measured from the same blood draw) did not reveal the same statin-dependent interactions (Table S2). In the case of both rs445925 and rs7412, carrying at least one copy of the minor allele was associated with a decrease in LDL-cholesterol across statin users and non-users alike, hence providing no additional insight into statin-mediated effects when measured cross-sectionally (Fig. S1). Together, our combined analyses of statin use, statin therapy intensity and genetic variants known to modify statin response indicate that HMG may provide additional insight into statin on-target effects, not captured by a snapshot measurement of LDL-cholesterol in a cross-sectional study.
Statin use is associated with subtle shifts in the gut microbiome
Given the previously established associations between the gut microbiome and statin use, we next investigated whether statin intake is associated with changes in gut microbiome composition. Consistent with previous research, statin use showed a significant association with interindividual variability in gut microbiome composition, using the Bray-Curtis dissimilarity metric (PERMANOVA models adjusted for microbiome vendor, sex, age, and BMI) (Fig. 2A, Fig. S1). We further tested the association between statin use and measures of gut α-diversity. We calculated observed Amplicon Sequence Variants (ASVs), a measure of species richness reflecting the number of unique taxa in the ecosystem, and Shannon diversity, a correlated measure that captures both richness and evenness in the abundances of taxa present. Statin intake was associated with a significant, but modest, decrease in one of the two α-diversity metrics calculated (linear regression models predicting Shannon diversity adjusted for the same covariates as PERMANOVA, adj. β(95% CI):−0.095 (−0.16 - −0.028), P=0.0051) (Fig. 2B). When looking at specific statin therapy intensity for a subset of participants where this information was available, there was no monotonic dose-response relationship between gut α-diversity, with only individuals receiving moderate intensity statin therapy demonstrating a significant decrease in measures of gut α-diversity relative to non-users (Fig. 2C, Fig. S1).
Figure 2. Gut microbiome composition is associated with markers of statin efficacy.

A) Proportion of variance explained by statin use, plasma HMG levels, and a statin-by-HMG interaction term from unadjusted PERMANOVA models or models adjusted for sex, age, BMI, and microbiome vendor using the Weighted UniFrac genus-level dissimilarity matrix. Grey area corresponds to the cumulative R-squared of variables added to the model prior to the variable indicated on the x-axis, while the colored areas of the bars represent the additional variance explained by that variable. B) Measures of gut α-diversity in statin users compared to non-users. The β-coefficient, 95%CI and p-value shown is derived from OLS models predicting each of the α-diversity measures adjusted for microbiome vendor, sex, age, and BMI. Values on the y-axis are diversity measures adjusted for these covariates (residuals). C) Measures of Observed ASVs in statin users and non-users with known therapy intensity (low, moderate, high). P-values shown correspond to β-coefficients from OLS models predicting Observed ASVs comparing each intensity group to the no statin control group, adjusted for the same covariates as in B). D) Plasma HMG levels among statin users and non-users across tertiles of gut α-diversity. Interaction P corresponds to the statin-by-α-diversity interaction term p-value from GLM predicting plasma HMG adjusted for the same covariates as in B) and C). Values on the y-axis are diversity measures adjusted for these covariates (residuals). Box plots represent the interquartile range (25th to 75th percentile, IQR), with the middle line denoting the median; whiskers span 1.5 × IQR, points beyond this range are shown individually. E) Scatter plots of Observed ASVs (x-axis) and covariate adjusted plasma HMG levels (residuals) (y-axis) in statin users with known dosage therapy intensity as well as statin non-users. Levels were adjusted for the same covariates as in B), as well as dosage intensity.
Gut microbiome α- and β-diversity correlate with markers of statin efficacy
Next, we investigated whether gut microbiome β-diversity may explain interindividual heterogeneity in response to statin therapy. Using HMG as a proxy for statin inhibition of its target enzyme, we modeled correspondence between statin on-target effects and interindividual variability in gut microbiome β-diversity using PERMANOVA and including a statin-by-HMG interaction term. The interaction terms had permutation-based P-values of 0.0070 (R2=0.0017) and 0.0013 (R2=0.0032) for Bray-Curtis and Weighted Unifrac metrics, respectively, which remained significant after adjusting for microbiome vendor, BMI, sex, and age (Fig. 2A, Fig. S1). These results indicate that HMG associations with gut microbiome composition are dependent on statin intake, similar to the HMG-SNP associations reported earlier (Fig. S1). Very similar patterns were observed for gut α-diversity, where, once again, the association between our proxy for statin efficacy, HMG, and gut α-diversity was dependent on statin intake (Fig. 2D). Plotting the association between gut α-diversity and HMG stratified by statin use revealed that, among statin users, higher α-diversity corresponded to lower plasma HMG levels, indicating decreased on-target effects of the drug in individuals with more diverse microbiomes (Fig. 2D). The negative association between HMG and α-diversity in statin users was also orthogonal to genetic variants predisposing individuals to variable statin responses. Running a stepwise forward regression model predicting HMG levels using the 9 SNPs previously associated with statin response explained an additional 3.2% of variance in HMG, on top of age (i.e., the base model). Including observed ASVs as a measure of gut α-diversity in the model, in addition to age and the chosen SNPs, increased the percent variance explained by an additional 3.9% (complete model R2=0.185).
To further exclude the possibility that individuals with higher α-diversity are generally healthier and simply prescribed less potent statin therapies to begin with, thus leading to lower levels of HMG, we further adjusted our models for dosage intensity in the subset of participants with microbiome data where this information was available (n=75). In this smaller group of individuals, associations between gut α-diversity and HMG were not impacted by correcting for statin intensity (Fig. 2E & Fig. S2). Similar results were observed when investigating statin dependent associations between LDL-cholesterol and gut α-diversity, although to a weaker extent (OLS models predicting LDL-cholesterol adjusted for clinical laboratory and microbiome vendors, sex, age, and BMI, Fig. S2). A weaker interaction effect with LDL cholesterol is to be expected, given the cross-sectional nature of our study and our inability to capture the percent decrease in LDL-cholesterol from baseline following the initiation of statin treatment, one of the most common and direct measures of statin effectiveness 3,19.
As another measure of gut microbiome correspondence to statin response, we tested the association between measures of gut α-diversity and the likelihood of having reached pre-defined target LDL-cholesterol levels for statin users (<70mg/dL and <100mg/dL). These are clinically relevant targets, as clinicians are recommended to adjust dosage and type of statin prescribed to reach these particular levels of LDL-cholesterol depending on the presence of specific ASCVD risk factors in their patients 27. Both Shannon diversity and Observed ASVs showed negative associations with likelihood of having reached target LDL-levels among statin users (Multivariable logistic regression adjusted for clinical lab vendor, sex, age, BMI, and T2D status [a common criteria, in combination with one or more CVD risk factors, where more aggressive LDL-lowering therapy is pursued]: Odds Ratios (OR) ranging from 0.60–0.69, Table 1). Together, these results indicate that gut microbiome composition is able to explain a significant proportion of variability in statin on-target effects in a generally healthy community-dwelling population.
Table 1. Gut microbiome measures correlate with having reached LDL-cholesterol target levels among statin users.
Odds Ratios (OR) for each gut microbiome measure from logistic regression models predicting having achieved either <100 mg/dL or <70 mg/dL target LDL-cholesterol level among statin users. The Bac.2 enterotype was compared against all other enterotypes. Measures of α-diversity were scaled and centered prior to analysis for easier comparison of effect sizes. Models were adjusted for clinical laboratory and microbiome vendors, age, sex and BMI. Further adjustment for T2D status was done in participants where this information was available (n=174). Significant OR (P<0.05) are bolded.
| <100 mg/dL (n cases=132, N total=197) | <100 mg/dL (n cases=132, N total=197) | <70 mg/dL (n cases=44, N total=197) | <70 mg/dL (n cases=44, N total=197) | |
|---|---|---|---|---|
| Cov. adj. OR(95%CI) | Cov. & T2D adj. OR(95%CI) | Cov. adj. OR(95%CI) | Cov. & T2D adj. OR(95%CI) | |
| Shannon diversity | 0.69 (0.49–0.97) | 0.72 (0.50–1.03) | 0.67 (0.48–0.95) | 0.60 (0.41–0.87) |
| Observed ASVs | 0.67 (0.47–0.95) | 0.67 (0.45–0.98) | 0.66 (0.45–0.95) | 0.62 (0.40–0.96) |
| Bac.2 enterotype | 2.19 (1.04–4.60) | 2.11 (0.95–4.66) | 3.61 (1.68–7.77) | 4.33 (1.83–10.25) |
Statin-associated changes in on-target and adverse effects are dependent on gut microbiome compositional states
Prior work on the gut microbiome and statins has relied on clustering individuals into microbiome-based compositional states called ‘enterotypes’ 28,29. A recent study revealed that statin intake among obese individuals was associated with lower prevalence of the Bacteroides 2 (Bac.2) enterotype, which is generally considered to be less healthy than other broad enterotype groupings common to cohorts in the United States and Europe 23. To evaluate the extent to which these coarse ecological groupings might help explain interindividual variation in statin on-target and adverse effects, we stratified our cohort into enterotypes. Using a previously established method for enterotype identification, Dirichlet multinomial mixture (DMM) modeling 30, the participants in the Arivale cohort separated optimally into four groups, according to the Bayesian Information Criterion (BIC), consistent with some, but not all, previous human gut microbiome studies (Bacteroides 1 (Bac.1), Bac.2, Ruminococcaceae (Rum.), and Prevotella (Prev.) clusters) 23,29–31 (Fig. 3A, Fig. S2). The four enterotypes identified showed very similar characteristics to those described previously in European cohorts, with two Bacteroides-dominated enterotypes (Bac.1 and Bac.2), with the Bac.2 enterotype being further characterized by decreased α-diversity and a depletion of SCFA-producing commensals like Faecalibacterium and Subdoligranulum (Fig. 3B, Fig. S2). The Rum. enterotype was enriched for taxa primarily from the Firmicutes phylum, as well as Akkermansia (Fig. S2, Table S2), consistent with previous findings 28. The Prev. enterotype was the smallest in size and characterized by high relative abundance of the Prevotella genus (Fig. 3D, Data S1).
Figure 3. Statin associations with markers of efficacy and metabolic adverse effects are microbiome-dependent.

A) Principle Coordinate Analysis (PCoA) plot of the genus-level Bray-Curtis Dissimilarity matrix color-coded by enterotypes. B-D) Relative abundance of Bacteroides (B), Prevotella (C), and Faecalibacterium (D) genera across the four enterotypes identified in the cohort. E) Proportion of each enterotype in statin users and non-users across the whole cohort (left) and stratified by obesity (right). F) Plasma HMG levels among statin users and non-users stratified by enterotype. Interaction P corresponds to the statin*enterotype interaction term p-value from unadjusted ANOVA models, while the cov. Adj. interaction P corresponds to the statin-by-enterotype interaction term p-value from covariate adjusted ANCOVA models. Plasma HMG levels shown on the y-axis are values adjusted for the same covariates (residuals). G) HOMA-IR measures among statin users and non-users stratified by enterotype. Interaction P corresponds to the statin-by-enterotype interaction term p-value from unadjusted ANOVA models, while the cov. Adj. interaction P corresponds to the statin-by-enterotype interaction term p-value from ANCOVA models adjusted for covariates. P-values above the box plots in F) and correspond to tests of significance between statin non-users and statin users within each enterotype using two-samples t-test. Differences with Bonferroni corrected P<0.05 were considered statistically significant and are highlighted in red. Box plots represent the interquartile range (25th to 75th percentile, IQR), with the middle line denoting the median; whiskers span 1.5 × IQR, points beyond this range are shown individually.
We first attempted to replicate previous findings 23 documenting an observed lower prevalence of the Bac.2 enterotype in obese individuals taking statins. Consistent with previous results, obesity itself was associated with a higher likelihood of being assigned to the Bac.2 enterotype (Multivariable logistic regression adjusted for microbiome vendor, sex, and age, OR(95%CI): 1.8 (1.4–2.3), P=5.0e-5). However, contrary to the original findings, we actually observed a higher prevalence of the Bac.2 enterotype among statin users compared to non- users, particularly among obese individuals (Fig. 3E). This association among obese individuals was further confirmed using multivariable logistic regression adjusting for sex, age, and microbiome vendor (OR(95%CI): 2.1 (1.2–3.7), P=0.013, n=462).
We next set out to explore whether an individual’s enterotype was associated with their response to statin therapy. Focusing on statin on-target effects, we observed a significant enterotype-by-statin interaction when predicting plasma HMG levels (analysis of covariance (ANCOVA) adjusted for microbiome vendor, clinical lab vendor, sex, age, and BMI, P=0.034). Stratifying the cohort by enterotypes and comparing statin users to non-users revealed that the Bac.2 enterotype displayed the greatest increase in HMG with statin use (37% mean increase), followed by the Bac.1 (24%) and Rum. enterotypes (18%). Interestingly, individuals with a Prev. enterotype showed no significant increase in HMG while on statins, although our sample size for this particular enterotype was small and thus this result needs to be interpreted with caution (Fig. 3F). Similar results were obtained when evaluating statin-by-enterotype interaction effects on LDL-cholesterol levels (ANCOVA adjusted for same covariates as HMG models, P=0.0032), with statin users within the Bac.2 enterotype demonstrating the lowest LDL-cholesterol levels (- 33%) relative to non-users within the same enterotype (Fig. S3). Statin users who were assigned the Bac.2 enterotype were also two to four-times more likely to have reached common LDL-cholesterol target levels for statin-users at higher risk for ASCVD (Table 1). Collectively, these results suggest that microbiome enterotypes may potentially reflect the extent to which statins inhibit HMG-CoA reductase and reduce LDL-cholesterol levels across individuals.
Statin use has previously been associated with disrupted glucose control and increased risk of developing T2D in a subset of patients 6,8,32. Given the known role of the gut microbiome in contributing to metabolic homeostasis, and the variable metabolic profiles previously observed across different microbiome enterotypes 23,33, we investigated whether enterotypes may modify the association between statin use and markers of insulin resistance. Focusing initially on Homeostatic Model Assessment for Insulin Resistance (HOMA-IR) 34,35, we tested for an enterotype-by-statin interaction effect while adjusting for microbiome vendor, clinical lab vendor, sex, age, BMI, LDL-cholesterol, and plasma HMG using ANCOVA. Individuals showed variable responses to statin therapy based on their microbiome enterotype, with Bac.2 individuals on statins demonstrating the highest levels of HOMA-IR relative to non-statin users, while Rum. individuals showed no significant difference in HOMA-IR between statin users and non-users (Interaction term P=0.0495, Fig. 3G, Table 2). It is worth noting that in the subset of participants where dosage intensity information was available, all three intensities (low, moderate, high) were associated with comparable measures of HOMA-IR, suggesting that differences in therapy intensity are likely not the main driver behind the observed statin-enterotype interaction (Fig. S2).
Table 2. Gut microbiome enterotypes modify the association between statin use and markers of glucose homeostasis.
Percentage in the first four columns corresponds to the percent median increase in each marker between statin users and non-users within each enterotype. P-values in these columns correspond to t-tests comparing covariate adjusted values between statin users and non-users. Values shown are raw p-values, and those that remained significant after correcting for type-1-error (Bonferroni P<0.05) are bolded. The last three columns in the table show the F- and P-values for the statin-by-enterotype interaction term from ANOVA (unadjusted) and ANCOVA (covariate adjusted) models predicting each of the specified markers of glucose homeostasis. Covariate adjusted models were adjusted for microbiome vendor, clinical lab vendor, sex, age, BMI, LDL cholesterol and plasma HMG. Second to last column corresponds to models adjusted for the same covariates as well as T2D status (yes/no, N=1385, T2D n=64), while the last column corresponds to models adjusted for the same covariates as well as metformin use. P-values<0.05 are bolded. Abbreviations: HOMA-IR: Homeostatic Model Assessment for Insulin Resistance; HbA1c: Glycated Hemoglobin A1c.
| Bac.1 | Rum. | Bac.2 | Prev. | Unadjusted model N=1512 | Covariate adj. model N=1512 | Covariate and diabetes adj. model N=1385 | Covariate and Metformin adj. model N=1512 | |
|---|---|---|---|---|---|---|---|---|
| HOMA-IR | 73%, P=7.2e-07 | 21% P=0.27 | 99% P=1.2e-04 | 29% P=0.33 | F=4.5, P=0.0037 | F=2.6, P=0.0495 | F=2.6, P=0.049 | F=2.5, P=0.0595 |
| Insulin | 63% P=5.6e-06 | 19% P=0.17 | 89% P=9.1e-04 | 22% P=0.25 | F=3.0, P=0.032 | F=1.4, P=0.23 | F=1.5, P=0.22 | F=1.4, P=0.24 |
| Glucose | 6.6% P=9.7e-04 | 4.5% P=0.51 | 9.3% P=8.1e-04 | 7.6% P=0.84 | F=6.4, P=0.00025 | F=4.4, P=0.0041 | F=3.9, P=0.0092 | F=3.9, P=0.0083 |
| HbA1c | 5.6% P=2.0e-03 | 1.9% P=0.16 | 7.3% P=1.2e-04 | 1.8% P=0.57 | F=8.1, P=2.3E-05 | F=6.3, P=0.00030 | F=3.4, P=0.017 | F=5.6, P=0.00086 |
We next expanded our analysis into additional markers of metabolic health, including fasting insulin and blood glucose, as well as glycated hemoglobin A1c (HbA1c). There was a significant enterotype-by-statin interaction across all tested metabolic parameters, which remained significant after adjusting for covariates across all markers other than insulin (Table 1, Fig. S3). As individuals with T2D are often recommended to take statins, we further adjusted all models for T2D status in participants where this information was available (N=1691, T2D n=66), which did not change the significance of enterotype-by-statin interaction effects observed (Table 2). Because a subset of individuals on statins is often concurrently treated with glucose-controlling medication, we further adjusted ANCOVA models for metformin use (the most commonly reported glucose-controlling drug in our cohort; Fig.S3), which did not drastically change the significance of the enterotype-by-statin interaction effects observed (Table 2). Collectively, these results provide strong preliminary evidence that gut microbiome composition may modify how statins influence off-target physiology, particularly glucose homeostasis.
Statin-microbiome interactions are confirmed in an independent cohort
To evaluate the robustness of the microbiome associations with markers of statin on-target and adverse effects reported in the Arivale cohort, we attempted to validate our main results in an independent European cohort of adults recruited to capture various stages of the cardiometabolic disease spectrum (the MetaCardis cohort, see Methods) 25. Consistent with our original findings, serum HMG was markedly increased in MetaCardis individuals on statins compared to non-statin users (Fig.4A), further pointing to its utility as a readily-available biomarker of statin efficacy. Using metagenomics species (MGS) count calculated by the authors of the original study as a measure of gut α-diversity, we observed a significant MGS count-by-statin interaction effect when predicting serum HMG levels, consistent with our original results (covariate adjusted ANCOVA, P=0.035) (Fig. 4B–C). Similar to the individuals in the Arivale cohort, MetaCardis participants with higher gut alpha-diversity demonstrated lower levels of serum HMG compared to individuals with low alpha-diversity, with this relationship being present exclusively in statin users, This interaction was independent of sex, age, BMI, nationality of the participant and microbial load, the latter being a covariate we were not able to adjust for in our discovery cohort study. This sheds some light on potential mechanisms underlying the observed associations, where the primary driver of the observed phenomenon is likely not the difference in the total number of microbes present in the ecosystem, but rather the differences in the taxonomic and functional composition of the gut microbiome.
Figure 4. Statin-HMG associations and statin-dependent microbiome associations are replicated in a validation cohort.

A) Serum HMG levels in statin users versus non-users in a subset of the MetaCardis cohort. P-value is derived from a linear regression model adjusted for sex, age, BMI, and participant nationality. HMG values plotted on the y-axis are adjusted for these covariates (residuals). B) Serum HMG levels among statin users and non-users across tertiles of gut α-diversity, assessed using metagenomics species count per sample. “Cov. Adj. Interaction P” corresponds to the statin-by-α-diversity interaction term p-value from linear models predicting serum HMG, adjusted for the same covariates as in A), as well as for microbial load. C) Percent variance explained by the base model (same covariates as in B)), and full model, where the MGS count measure of alpha-diversity is included. Additional variance explained in serum HMG by MGS count, on top of the base model containing covariates, is shown in red. D) Serum HMG levels among statin users and non-users stratified by enterotype. “Cov. Adj. interaction P” corresponds to the statin-by-enterotype interaction term P-value from covariate adjusted ANCOVA models (same covariates as in B). E) Associations between Bacteroides genus counts and markers of glycemic control (HbA1c) and statin effectiveness (HMG) in statin users and non-users. Bacteroides counts have been obtained after rarefaction and adjusting for microbial cell count, as described in the original study by Fromentin et al. 25. Box plots represent the interquartile range (25th to 75th percentile, IQR), with the middle line denoting the median; whiskers span 1.5 × IQR, points beyond this range are shown individually.
Given that the MetaCardis study collected stool shotgun metagenomics sequencing data to characterize the gut microbiome, we next explored possible functional characteristics of the gut metagenome associated with markers of statin efficacy. To this end, we tested for associations between microbiome functions (gut metabolic modules (GMMs) and Kyoto Encyclopedia of Genes and Genomes (KEGG) modules) calculated in the original study, and serum HMG, specifically in statin-users, adjusted for age, sex, BMI, and participant nationality utilizing a beta-binomial regression approach (corncob) 36. A total of 5 modules remained significantly associated with serum HMG among statin users after multiple-hypothesis correction (Bonferroni P<0.05), including a negative association between HMG and a mucin degradation module (MF0103; Data S2). These functional associations are still preliminary, and will require further validation across additional cohorts to assess their robustness. Once validated, these functional hits could provide useful targets for mechanistic studies in vitro or in non-human animal models.
We next evaluated the statin-dependent associations between gut microbiome enterotypes and measures of statin on-target effects (serum HMG) and adverse effects (Hba1c, the sole marker of glucose homeostasis available in the validation dataset). MetaCardis participants separated into four enterotype groups, similar in taxonomic composition to the Arivale dataset, and consistent with previous studies on the same study population (Fig. S4). Once again, we attempted to replicate previous associations between statin use and the likelihood of having a Bac.2 enterotype. Consistent with previously reported findings by Veira-Silva et al., individuals with ischemic heart disease within the MetaCardis cohort demonstrated a lower likelihood of having a Bac.2 enterotype while on statins (OR(95%CI):0.4 (0.2–0.9),n=303,p=0.022, models adjusted for sex and age). However, non-IHD (i.e., the remainder of the cohort) obese individuals from the MetaCardis cohort demonstrated a trend more consistent with what was observed in the Arivale dataset (i.e., higher odds of Bac.2 enterotype with statin use, adj. OR(95%CI): 1.9(0.8–4.8), P=0.16), indicating the need to further refine the context and identify subpopulations where statin-enterotype associations are most consistent (Fig.S4).
We next proceeded to validate our main findings of statin-dependent associations between gut microbiome enterotypes and markers of statin on-target and adverse effects. There was a significant enterotype-by-statin interaction when modelling serum HMG, independent of age, sex, BMI, nationality, and microbial load, with results strikingly similar to those originally obtained in the Arivale cohort (P=0.035, Fig.3D, Fig.4D). Similarly, HbA1c levels were significantly higher in statin users versus non-users across both the Bac.1 and Bac.2 enterotypes, while this increase was absent in the Rum. enterotype. This once again suggests that the risk of metabolic adverse effects may be modulated by an individual’s gut microbiome compositional state. However, the P-value for the interaction term did not reach statistical significance (covariate-adjusted interaction term P=0.195) in the validation cohort, partially due to the smaller sample size compared to the Arivale dataset (Arivale N=1512, MetaCardis N=688) (Fig. S4). Because Bac.1 and Bac.2 enterotypes are both enriched for the genus Bacteroides and show similar associations with HbA1c based on statin use, we further examined the association between this marker of glycemia and rarefied (i.e., even subsampling of counts without replacement across samples) Bacteroides abundance counts adjusted for total microbial cell count, calculated by the authors of the original MetaCardis study25. Consistent with the enterotype analysis, we found significant associations between Bacteroides abundance and markers of statin on-target efficacy and metabolic health parameters in statin users, which were entirely absent in non-users (Fig.4E). Collectively, these results show a high degree of consistency across geographically distinct populations and different gut microbiome sequencing methods (16S rRNA amplicon sequencing in the Arivale cohort versus shotgun metagenomic sequencing in the MetaCardis cohort), converging on strong evidence for the potential clinical applicability of the reported findings.
Discussion:
There is considerable heterogeneity in response to statin therapy among individuals, both in terms of on-target effects (lowering LDL-cholesterol) and likelihood of experiencing unwanted adverse effects 3,8,32. Herein, we report that variation in gut microbiome taxonomic composition can explain interindividual variability in statin responses. The main findings of our analyses are as follows: 1) HMG measured in plasma is a potential biomarker of both statin use and statin on-target effects, which also reflects known genetic variability in statin responses; 2) Gut α-diversity negatively correlates with HMG exclusively in statin users, independent of dose intensity and genetic predisposition, indicating a more diverse microbiome may interfere with statin on-target effects; 3) Enterotype analysis further confirms similar patterns of microbiome modification of statin response, with the Bacteroides dominant, α-diversity-depleted Bac.2 enterotype showing the highest plasma HMG and lowest LDL-cholesterol levels among statin users; and 4) Of the four enterotypes identified, individuals with the Bac.2 followed by Bac.1 enterotypes experience greatest disruption to glucose control with statin use, while the Firmicutes rich Rum. enterotype appears most protective, indicating variable risk of statin- mediated adverse metabolic effects based on gut microbiome composition. Our results are further strengthened by the strong agreement between data coming from independent American and European cohorts. Collectively, our findings indicate that the gut microbiome may influence statin activity within the human host. With further refinement, knowledge of these effects may inform statin therapy guidelines and help personalize ASCVD treatment.
To the best of our knowledge, measuring HMG in large observational studies for the purpose of exploring statin-mediated effects has not been previously proposed. The conversion of HMG-CoA to HMG is dependent on the hydrolysis of the thioester bond linking HMG to its Coenzyme-A moiety, which has been previously shown to be facilitated by at least one known thioesterase (peroxisomal acyl-CoA thioesterase 2) 37. Relatively little is known about the accumulation of HMG with statin therapy and the pathways involved, which warrants further research. Nevertheless, there are several advantages for including HMG along with LDL-cholesterol measurements when evaluating statin effects. For one, given the limitations of a cross-sectional study design like ours, HMG may provide more time-invariant insight into statin efficacy, as opposed to LDL-cholesterol, which would require knowledge of pre-statin cholesterol levels to calculate the percent decrease in LDL over time 3. This seemed to be the case in our genetics analysis, where cross-sectional measurements of plasma HMG were able to capture previously reported genetic variability in statin response while LDL-cholesterol measurements from the same blood draw were less sensitive.
An intriguing finding in the present analysis was an absence of statin-associated metabolic disruption in individuals with a Rum. enterotype (Fig. 3G, Fig. S4). Statin use in this group was still associated with increased plasma HMG and decreased LDL-cholesterol levels (Fig. 3F, Fig. S3), indicating that patients with this microbiome composition type may benefit from statin therapy without an increased risk of unwanted metabolic complications. There are several possible explanations for this observation. For example, the Rum. enterotype is enriched in the genus Akkermansia, as well as several butyrate-producing taxa, which are known to positively impact host metabolism through multiple mechanisms (Data S1, Fig. S2) 28,38, potentially serving as a buffer against statin off-target effects on glucose homeostasis. In addition, statins and other prescription drugs have been previously shown to be most readily metabolized by species within the Bacteroides genus, of which the Rum. enterotype is most depleted. The lower degree of drug metabolism by Firmicutes taxa comprising the Rum. enterotype may therefore be potentially protective from statin off-target effects. Consistently, both Bacteroides rich Bac.1 and Bac.2 enterotypes showed greatest increases in markers of insulin resistance with statin use.
Statin use in individuals with a Bacteroides-dominated gut microbiome was associated with the strongest on-target effects (i.e. high plasma HMG and low LDL-cholesterol levels) but also greatest metabolic disruption (Fig. 3F–G, Fig. S3, Fig.4E, Fig.S4). This is consistent with previous observational studies that have identified an association between the magnitude of decrease in LDL-cholesterol with statin use and risk of developing T2D (i.e. the greater the percent decrease in LDL-cholesterol with statin therapy, the higher the risk of new onset T2D) 7,39. One possible mechanism behind the reported association is the previously mentioned ability of Bacteroides species to metabolize prescription drugs, including statins 17. Bacteroides dominance within both the Bac.1 and Bac.2 enterotypes may modify drug activity, impacting both potency and potential adverse effects. Paired with depletion of several major butyrate-producing taxa within the Bac.2 enterotype (Fig. 3D, Fig. S1, Data S1), this bacterial composition may put patients at particularly high risk of metabolic complications. If this were indeed the case, individuals with a Bac.2 enterotype could benefit most from lower intensity therapy, which might still achieve the desired percent decrease in LDL-cholesterol while mitigating potential metabolic disruptions. Complementary pro- and prebiotic interventions could also be potentially pursued in these individuals. However, further experimental work is needed to fully elucidate the microbiome-statin interactions that may be driving the reported associations. In addition, the adoption of microbiome sequencing data to inform therapy personalization will further require effective dissemination of this information to clinicians to inform their decisions, a likely challenge in the years to come.
While our present investigation identified very similar enterotype structure to a previous study on statin use and the gut microbiome by Veiera-Silva et al. 23, our analysis also showed conflicting results in terms of prevalence of the putatively dysbiotic Bac.2 enterotype among obese statin users (Fig. 3E, Fig.S4). One possible explanation for this discrepancy is that in the original study individuals were primarily prescribed Simvastatin (48% of statin users), which is a lower intensity HMG-CoA reductase inhibitor than the most commonly prescribed Atorvastatin in our cohort (53% of all statin users). However, further analysis of the MetaCardis cohort shows instances where Veiera-Silva et al.’s findings are confirmed (i.e., in individuals with IHD) and instances where they are not (i.e., in non-IHD obese individuals; consistent with results reported herein from the Arivale cohort), suggesting the dependence of these associations on heart disease and other factors may require further investigation. In any case, while the prior study 23 focused on how statins might influence the composition of the microbiome, our study focused on how the composition of the microbiome impacts the on- and off-target effects of statins in the host. Our analyses indicate that statins may have a detectable, but very weak effect on the composition of the gut microbiome, while the gut microbiome appears to have a more sizable impact on host responses to statin treatment.
Growing evidence suggests a bidirectional interaction between prescription medication use and the gut microbiome, which may inform drug treatment for hundreds of millions of people worldwide. Here we present a proof-of-concept study on how gut microbiome composition may be used to stratify patients to inform statin therapy. As our understanding of microbe-drug interactions deepens, gut microbiome modification and monitoring hold promise for informing pharmacological treatment optimization.
Limitations of study:
While our analysis shows several promising and potentially translational associations between the ecology of the human gut microbiome and host statin responses, it is not without limitations. A major limitation of the study is its cross-sectional design. Although the rich multi-omics data collected from Arivale and MetaCardis participants allows us to adjust our statistical models for a number of potential confounders, we are unable to capture the percent change in LDL-cholesterol and markers of insulin resistance since statin initiation. Furthermore, our cohorts consist of predominantly Caucasian (>80%) individuals from the west coast of the U.S. and Western Europe, which limits the broader applicability of our findings to other populations. This is particularly important to note given prior reports of variable statin responses based on race and ethnicity 40. Finally, although we reference prior work where certain gut bacterial taxa have been directly implicated in metabolizing statins, we are unable to point to a specific mechanism responsible for the observed associations. We hope this work inspires future experimental and clinical studies to build towards a more mechanistic understanding of the reported phenomenon.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Sean Gibbons (sgibbons@isbscience.org).
Data and code availability
Plasma HMG and clinical laboratory tests, along with demographics and statin information on Arivale participants included in this study, are available in Data S3. 16S amplicon sequencing data from the Arivale data set, in the form of FASTQ files, has been uploaded to the Sequence Read Archive (SRA) with the following accession numbers: PRJNA826530, PRJNA826648.
Qualified researchers can further access the full Arivale deidentified dataset supporting the findings in this study for research purposes through signing a Data Use Agreement (DUA). Inquiries to access the data can be made at data-access@isbscience.org and will be responded to within 7 business days. Data from the MetaCardis cohort used for validation of our results is freely available to download from the original study under the following link: https://www.nature.com/articles/s41591–022-01688–4#data-availability (supplementary tables 9–14).
Code used for data analysis has been deposited in GitHub (https://github.com/PriceLab/Statins_microbiome_project). It is also available on Figshare under the following DOI: 10.6084/m9.figshare.19579234.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Materials availability
This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Study Participants
Procedures for the Arivale cohort study were run under the Western Institutional Review Board (WIRB) with Institutional Review Board (IRB) study number 20170658 at the Institute for Systems Biology and 1178906 at Arivale. The research was performed entirely using deidentified and aggregated data of individuals who had signed a research authorization allowing the use of their anonymized data in research. Per current U.S. regulations for use of deidentified data, informed consent was not required. The Arivale cohort consists of adults who self-enrolled in a now closed lifestyle intervention program (Arivale, Inc. 2015–2019) 41–44. To be eligible to join the program, participants had to be over 18 years of age, not pregnant, and a resident of any U.S. state except New York. The participants analyzed in this study are the 92% of Arivale participants who agreed to research use as of 19 June 2018 and enrolled in the program between July 2015 and March 2018. The lifestyle intervention was designed to improve a number of key outcomes based on longitudinal profiling of clinical biomarkers and individualized coaching by registered nurses and dietitians. For the present study, only individuals who filled out medication questionnaires, and/or reported their prescription medication information directly to their coach during a 1-on-1 session, were included. Participants further had to have available fasting plasma metabolomics and clinical laboratory test data (N=1848). Only baseline measurements and corresponding medication doses at the start of the program were considered, i.e. before any lifestyle interventions were recommended. Of the 1848 participants originally included, after excluding individuals who reported taking antibiotics in that last 3 months, 1512 had available stool 16S rRNA gene sequencing data. Similar to the larger Arivale population, the majority of participants of this study were residents of Washington and California when in the program. Although the cohort tends to be leaner than the general U.S. population (prevalence of obesity is 31% relative to the national prevalence of 42%45), it is representative of the populations in the states where the majority of the participants were located. The cohort is further predominantly female (63%) and is skewed towards Caucasians (81%). Additional demographic information on the cohort age, BMI and other measures is provided in Table S1. Participants information on sex, age, medication use, and race was self-reported. Information on socioeconomic status was not collected.
Primary findings from our study were further validated in a European cohort which included 1241 individuals across the spectrum of cardiometabolic disease progression (MetaCardis cohort)25. Briefly, the MetaCardis project recruited adults from Denmark, France and Germany with increasing stages of ischemic heart disease (IHD), including 275 healthy controls (HC) matched based on demographics, 222 untreated metabolically matched controls (UMMC), 372 metabolically matched controls (MMC) and 372 individuals with IHD. Most of the individuals in the study had paired medication history, stool shotgun metagenomics sequencing data, serum metabolomics, and a subset of clinical laboratory tests. Because the overwhelming majority of IHD patients reported taking statins or other lipid lowering drugs (~87%), we validated our results specifically in the combined HC, MMC, and UMMC groups (N=688 with the necessary data available in supplementary tables 9–14 of the original study), excluding IHD patients, to discern the primary statin-microbiome interactions of interest from other potential drug interactions and demographic/lifestyle factors that are enriched in IHD patients and cannot be easily adjusted for in statistical models. Further validation was also performed using strictly MMC and UMMC groups, where participants were matched based on sex, age, BMI, and metabolic syndrome features to IHD patients, with UMMC being further not treated with any lipid lowering medication (Fig.S4). Analysis was performed using the processed dataset available in the supplementary tables (9–14) of the original study (see Data Availability statement).
METHOD DETAILS
Microbiome Analysis
Stool samples in the Arivale cohort were collected using kits developed by two microbiome vendors (DNAGenotek or Second Genome), and processed as described previously 42,46. Briefly, stool sample collection kits with proprietary chemical DNA stabilizers to maintain DNA integrity at ambient temperatures were shipped directly to participants’ homes and then shipped back to the vendors. Gut microbiome sequencing data in the form of FASTQ files were then obtained from the vendors on the basis of either the 300-bp paired-end MiSeq profiling of the 16S V3 + V4 region (DNAgenotek) or 250-bp paired-end MiSeq profiling of the 16S V4 region (Second Genome). Downstream analysis was performed using a denoise workflow from mbtools (https://github.com/gibbons-lab/mbtools) that wraps functions from DADA2. DADA2 47 error models were first trained separately for each sequencing run and subsequently used to obtain amplicon sequence variants (ASVs) for each sample. Next, chimera removal was performed using the de novo DADA2 algorithm, which removed ~17% of all reads. Taxonomy assignment was performed using the RDP classifier with the SILVA database (version 132). In summary, 99% of the reads could be classified to the family level, 89% to the genus level and 32% to the species level. Sequence variants were aligned to each other using DECIPHER 48 and multiple sequence alignment was trimmed by removing each position that consisted of more than 50% gaps. The resulting core alignment was then used to reconstruct a phylogenetic tree using FastTree 49. Downstream gut microbiome analysis was conducted using the Phyloseq Package in R 50. Gut microbiome samples were first rarefied to an even sampling depth of 25596 reads, corresponding to the minimum number of reads per sample in the dataset. Bray- Curtis 51 and Weighted UniFrac 52 dissimilarity matrices were calculated at the genus-level using the Phyloseq package. α-diversity measures were calculated at the ASV-level using the Phyloseq Package.
Clinical Laboratory Tests
Blood draws for all assays were performed by trained phlebotomists at LabCorp (n=1309) or Quest (n=553) service centers, and assaying was performed in CLIA-certified laboratory facilities by the vendors. Blood samples for clinical laboratory tests were obtained at the same time as the metabolomics blood draw, and only the baseline sample prior to any lifestyle coaching intervention was considered. Prior to the blood draw, Arivale participants were advised to avoid alcohol, vigorous exercise, aspartame and monosodium glutamate for 24 hours, and to begin fasting 12 hours in advance.
Plasma Metabolomics
Plasma HMG was measured as part of the metabolomics data generated by Metabolon, Inc.( North Carolina, USA), on the same blood draws as clinical laboratory tests, and has been described previously 42. Briefly, EDTA-plasma samples were thawed on ice, after which a recovery standard was added to each sample for quality control. Aqueous methanol extraction was performed to remove the protein fraction while retaining the maximum amount of small molecular weight compounds in the sample. Sample extract was next aliquoted into five separate fractions, one for each of the four methods used for metabolite quantification, as well as one aliquot as a potential backup. Excess organic solvent was removed from the aliquoted samples by placing the samples on a TurboVap® (Zymark). Aliquoted sample extracts were stored overnight under nitrogen before analysis. All samples were run on the Waters ACQUITY ultra-performance liquid chromatography (UPLC) and a Thermo Scientific Q-Exactive high resolution/accurate mass spectrometer interfaced with a heated electrospray ionization (HESI-II) source and Orbitrap mass analyzer operated at 35,000 mass resolution. The four aliquoted sample extracts were dried then reconstituted in solvents compatible with each of the four methods used for downstream metabolite quantification. To ensure injection and chromatographic consistency, each solvent further contained a series of standards at fixed concentrations. Two of the four aliquots were analyzed using acidic positive ion conditions chromatographically optimized for either more hydrophobic (solvent consisting of water, methanol, acetonitrile, 0.05% perfluoropentanoic acid (PFPA) and 0.01% formic acid (FA)) or hydrophilic compounds (water and methanol, containing 0.05% PFPA and 0.1% FA). Both of these aliquots were eluted using a C18 column (Waters UPLC BEH C18–2.1×100 mm, 1.7 μm). Elution for aliquot 3 was performed using a dedicated C18 column in solvent containing methanol and water under basic negative ion optimized conditions, with 6.5mM Ammonium Bicarbonate at pH 8. The fourth and final aliquot was analyzed via negative ionization following elution from a HILIC column (Waters UPLC BEH Amide 2.1×150 mm, 1.7 μm) using a gradient consisting of water and acetonitrile with 10mM Ammonium Formate, pH 10.8. Mass spectrometry (MS) analysis was performed using dynamic exclusion and alternating between MS and data-dependent MSn scans. The scan range varied slightly between the four methods used, and covered 70–1000 m/z. Process blanks and EDTA-plasma technical replicates were run intermittently throughout the study run-days to account for potential run and day variability. A biochemical library of over 3300 purified standards based on chromatographic properties and mass spectra was used for identification of known chemical entities. Raw metabolomics data was next normalized as described previously 41,42. Values were median scaled within each batch, such that the median value for each metabolite was 1. To adjust for possible batch effects, further normalization across batches was performed by dividing the median-scaled value of each metabolite by the corresponding average value for the same metabolite in technical control samples processed in the same batch. The same technical control samples were used to ensure the comparability of abundance estimates obtained across batches.
Genetics Analysis
Participant DNA was extracted from whole blood and, following quality control and purification, as needed, underwent 150 PE whole genome sequencing (WGS) using Illumina’s HiSeq X at 30x coverage as described previously 53. Variant calling was performed by the vendor using the pipeline that follows GATK’s Best Practices, using Haplotype Caller and hg19 build as the reference genome. A total of 1747 participants (~94% of the present cohort) had available WGS data and were used in the present analysis. Following extensive quality control and assurance, genetic ancestry was calculated as principal components (PCs) using a set of ~100,000 ancestry-informative SNP markers as described previously 54. SNPs chosen for testing associations with HMG were based on prior studies investigating genetic predisposition to statin efficacy defined as percent decrease in LDL-cholesterol from baseline, and included the following variants: rs10455872, rs2199936, rs2900478, rs4420638, rs445925, rs5908, rs646776, rs7412, and rs8014194 12.
QUANTIFICATION AND STATISTICAL ANALYSIS
Depending on the statistical approach, analysis was conducted using either R (v 3.6) or Python (v 3.7). Of the 1848 participants included in our study, 73 had missing data on sex and age, 66 on BMI, 81 on HMG and 6 on LDL-cholesterol. These missing values were imputed using plasma metabolomics data and the K nearest neighbor algorithm implemented through the sklearn.impute module in Python.
Metabolomic & Clinical marker analysis
The associations of plasma HMG levels with LDL-cholesterol, statin intensity, and measures of gut α-diversity were all tested using Generalized Linear Models (GLM) with a Gamma distribution and a log-link function within the statsmodels module in Python, with HMG as the dependent variable. OLS regression (Python) was used whenever LDL-cholesterol or measures of gut α-diversity were the dependent variables. Testing for associations between variables and interindividual variability in gut microbiome composition was conducted using permutational multivariate analysis of variance (PERMANOVA) through the Adonis package in R using both the genus-level Bray-Curtis and Weighted UniFrac dissimilarity matrices. The number of permutations to obtain P-values was set to 3000.
For assessing dose-response relationships between HMG/LDL-cholesterol and dosage intensity (Fig. 1D), dosage was recoded into an ordinal variable (0(none/no statins), 1(low), 2(moderate), 3(high)), and the significance of the β-coefficient for that variable from covariate adjusted models predicting either HMG (GLM adjusted for sex, age, and BMI) or LDL-cholesterol (OLS adjusted for sex, age, BMI, and clinical lab vendor) was reported. Wherever associations were visualized using box plots or scatter plots, the residuals (values adjusted for covariates from either GLM or OLS models) were plotted instead of the original values. For comparing the differences in prevalence of the four enterotypes among statin users and non-users, the χ2 test was performed using the chisq.test function in R. When evaluating the association between obesity and Bac.2 enterotype, as well as statin use and Bac.2 enterotype among obese participants, multivariable logistic regression models were generated through the statsmodels module in Python with Bac.2 membership (versus all other enterotypes) as the dependent variable.
When testing for significant enterotype-by-statin interactions, HMG and metabolic parameters (blood glucose, blood insulin, HOMA-IR, and HbA1c) were log transformed prior to fitting the models. Analysis of Variance (ANOVA) or covariance (ANCOVA) models were then used to test for significant interactions using the statsmodels module in Python. If a significant interaction was present, post-hoc comparisons were performed between statin users and non-users within each enterotype on the covariate adjusted values (residuals) using two-sample t-tests, with Bonferroni corrected P<0.05 considered statistically significant.
Genetics Analysis
To model the association between SNPs and HMG in statin users, individuals homozygous and heterozygous for the minor allele were grouped together. Statistical analysis was performed on each SNP individually using GLM with a Gamma distribution and a log-link function within the statsmodels module in Python, with HMG as the dependent variable and a statin-by-SNP interaction term. The interaction term tests for a significant association between HMG and statin use, that is modified by the SNP of interest (i.e. the effect of statins on HMG are variable based on the genetic variant). Models were further adjusted for sex, age, BMI and the first 7 ancestry PCs. Ordinary Least Square (OLS) regression models with the same covariates and interaction term were also run with LDL-cholesterol as the dependent variable. Type-1 error was controlled using the Benjamini-Hochberg method (FDR<0.05).
Enterotype Analysis
Enterotype analysis was performed using Dirichlet Multinomial Mixture (DMM) modeling on the rarefied genus-level count data, which utilizes a combination of dirichlet multinomial mixtures and Expectation Maximization 30. For selecting the optimal number of DMM groups in our cohort (i.e. enterotypes), we used the Bayesian information criterion (BIC).
However, BIC as a model penalization metric is not without limitations and tends to err on the side of underfitting (i.e. estimating a smaller number of clusters). The Laplace approximation for model penalization 30, on the other hand, did not identify an optimal number of clusters in this particular dataset (out to a maximal number of eight clusters tested), indicating limited statistical evidence for a small number of coarse-grained compositional states within our cohort (Fig. S2). Nevertheless, the main enterotype groupings tend to be relatively consistent from study-to-study in large U.S. and European populations, even if the statistical evidence for such states is somewhat limited 29. Given that the four BIC-identified enterotypes in our cohort show strikingly similar taxonomic signatures to those identified in prior work on statins 23, we moved forward with an analysis of these compositional states and how they relate to statin response.
Supplementary Material
List of genera significantly differing across enterotypes, identified using a Kruskal-Wallis test (Bonferroni P<0.05). Each enterotype column corresponds to the median relative abundance of a particular genus for that enterotype. The last six columns present pairwise comparisons between enterotypes for each genus using the Wilcoxon Rank Sum test, with significant values after correcting for type 1 error shown (Bonferroni P<0.05). Abbreviations: ns = not significant.
Results of beta-binomial regression models testing the association between serum HMG and microbiome functional modules, adjusted for sex, age, BMI and participant Nationality. “Feature” corresponds to the module being tested, “coef” to the Beta-Coefficients, “se” to the standard error of the coefficient, “pvalue” to the unadjusted p-value and “padj” to the Bonferroni adjusted p-value.
Plasma HMG, clinical laboratory tests, demographics, statin and vendor information on Arivale participants analyzed in the present study.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bacterial and virus strains | ||
| Biological samples | ||
| Stool and blood samples from the Arivale Cohort | This paper | |
| Chemicals, peptides, and recombinant proteins | ||
| Critical commercial assays | ||
| Deposited data | ||
| Stool 16S rRNA sequencing data | This paper | Sequence Read Archive (SRA) Accession numbers: PRJNA826530 PRJNA826648 |
| Demographics, clinical, and metabolomics data | This paper | Data S3 |
| Experimental models: Cell lines | ||
| Experimental models: Organisms/strains | ||
| Oligonucleotides | ||
| Recombinant DNA | ||
| Software and algorithms | ||
| DADA2 (version 1.20.0) | Callahan et al. 2016 | https://benjjneb.github.io/dada2/ |
| DECIPHER (version 2.20.0) | Wright 2015 | http://www2.decipher.codes/AlignSequences.html |
| mbtools (version 0.38.0) | This paper | https://github.com/Gibbons-Lab/mbtools |
| FastTree (version 2.1.11) | Price et al. 2010 | http://www.microbesonlineorg/fasttree/ |
| Phyloseq (version 1.30.0) | McMurdie & Holmes 2013 | https://joey711.github.io/phyloseq/ |
| DirichletMultinomial (version 1.28.0) | Holmes et al. 2012 | https://microbiome.github.io/tutorials/DMM.html |
| vegan (version 2.5–7) | Oksanen et al. 2020 | https://cran.r-project.org/web/packages/vegan/index.html |
| RDP | Wang et al. 2007 | http://rdp.cme.msu.edu/classifier/classifier.jsp |
| Statsmodels (version 0.10.2) | Skipper & Perktold 2010 | https://www.statsmodels.org/stable/index.html# |
| Other | ||
| SILVA Database (version 132) | Quast et al. 2013 | https://www.arb-silva.de/ |
| Metacardis Cohort dataset | Fromentin et al. 2022 | https://www.nature.com/articles/s41591–022-01688–4#data-availability |
Highlights.
HMG in blood identified as a potential cross-sectional biomarker for statin responses
Gut microbiome alpha-diversity negatively associated with on-target statin responses
Bacteroides-enriched individuals at higher risk of statin-induced metabolic disruption
Firmicutes-dominant individuals at lower risk of statin-induced metabolic disruption
Context and Significance.
Despite the undeniable cholesterol lowering benefits of statin therapy, considerable heterogeneity exists in individual responses to the same treatment. Human gut bacteria are known to metabolize statins in vitro, but there is limited information on how microbiome composition may contribute to statin on-target and/or adverse effects. Here, the authors identify a novel blood-based biomarker for monitoring statin effects in two large, independent human cohorts. They identify gut microbiome features robustly associated with variable statin responses, both in terms of on-target (cholesterol lowering) and adverse (insulin resistance) effects. Furthermore, these microbiome-statin associations are independent of human genetic variation associated with statin response variability. These results support the potential clinical utility of monitoring the gut microbiome for optimizing drug therapy.
Acknowledgments:
We thank Arivale participants who consented to let their deidentified data be used for research purposes. This work was supported by the M.J. Murdock Charitable Trust (to L.H. and N.D.P.), and Arivale. S.M.G., and C.D. were supported by a Washington Research Foundation Distinguished Investigator Award and by start-up funds from the Institute for Systems Biology. Further support came from the National Academy of Medicine Catalyst Award (to E.S.O, S.M.G., and L.H.) and a National Institutes of Health (NIH) grant (no. U19AG023122) awarded by the National Institute on Aging (NIA) (to P.S., E.S.O, and N.R.).
Funding
This research was supported by the M.J. Murdock Charitable Trust, WRF, NAM Catalyst Award, and the NIH grant U19AG023122 awarded by the NIA.
Footnotes
Inclusion and diversity: One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community. The author list of this paper includes contributors from the location where the research was conducted who participated in the data collection, design, analysis, and/or interpretation of the work.
Declaration of interests: Arivale, which closed in May 2019, partially funded this study. At the time this study was conceived and designed, J.L., S.K., A.M., N.P., and L.H. held positions and/or held stock options in the company. The authors declare no ongoing financial interests in Arivale. The authors declare no other competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Byrne P, Cullinan J, Murphy C, and Smith SM (2018). Cross-sectional analysis of the prevalence and predictors of statin utilisation in Ireland with a focus on primary prevention of cardiovascular disease. BMJ Open 8, e018524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Salami JA, Warraich H, Valero-Elizondo J, Spatz ES, Desai NR, Rana JS, Virani SS, Blankstein R, Khera A, Blaha MJ, et al. (2017). National Trends in Statin Use and Expenditures in the US Adult Population From 2002 to 2013: Insights From the Medical Expenditure Panel Survey. JAMA Cardiol 2, 56–65. [DOI] [PubMed] [Google Scholar]
- 3.Ridker PM, Mora S, and Rose L. (2016). Percent reduction in LDL cholesterol following high-intensity statin therapy: potential implications for guidelines and for the prescription of emerging lipid-lowering agents. European Heart Journal 37, 1373–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spence JD, and Solo K. (2017). Resistant Atherosclerosis: The Need for Monitoring of Plaque Burden. Stroke 48, 1624–1629. [DOI] [PubMed] [Google Scholar]
- 5.Ridker PM, Danielson E, Fonseca FAH, Genest J, Gotto AM Jr, Kastelein JJP., Koenig W., Libby P., Lorenzatti AJ., MacFadyen JG., et al. (2008). Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N. Engl. J. Med. 359, 2195–2207. [DOI] [PubMed] [Google Scholar]
- 6.Carter AA, Gomes T, Camacho X, Juurlink DN, Shah BR, and Mamdani MM (2013). Risk of incident diabetes among patients treated with statins: population based study. BMJ 346, f2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang S, Cai R, Yuan Y, Varghese Z, Moorhead J, and Ruan XZ (2017). Association between reductions in low-density lipoprotein cholesterol with statin therapy and the risk of new-onset diabetes: a meta-analysis. Sci. Rep. 7, 39982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Engeda JC, Stackhouse A, White M, Rosamond WD, Lhachimi SK, Lund JL, Keyserling TC, and Avery CL (2019). Evidence of heterogeneity in statin-associated type 2 diabetes mellitus risk: A meta-analysis of randomized controlled trials and observational studies. Diabetes Research and Clinical Practice 151, 96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bruckert E, Hayem G, Dejager S, Yau C, and Bégaud B. (2005). Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients--the PRIMO study. Cardiovasc. Drugs Ther. 19, 403–414. [DOI] [PubMed] [Google Scholar]
- 10.Arnett DK, Blumenthal RS, Albert MA, Buroker AB, Goldberger ZD, Hahn EJ, Himmelfarb CD, Khera A, Lloyd-Jones D, McEvoy JW, et al. (2019). 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 74, 1376–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wiggins BS., Saseen JJ., Page RL 2nd., Reed BN., Sneed K., Kostis JB., Lanfear D., Virani S., Morris PB., and American Heart Association Clinical Pharmacology Committee of the Council on Clinical Cardiology; Council on Hypertension; Council on Quality of Care and Outcomes Research; and Council on Functional Genomics and Translational Biology (2016). Recommendations for Management of Clinically Significant Drug-Drug Interactions With Statins and Select Agents Used in Patients With Cardiovascular Disease: A Scientific Statement From the American Heart Association. Circulation 134, e468–e495. [DOI] [PubMed] [Google Scholar]
- 12.Postmus I, Trompet S, Deshmukh HA, Barnes MR, Li X, Warren HR, Chasman DI, Zhou K, Arsenault BJ, Donnelly LA, et al. (2014). Pharmacogenetic meta-analysis of genome-wide association studies of LDL cholesterol response to statins. Nat. Commun. 5, 5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reiter-Brennan C, Osei AD, Iftekhar Uddin SM, Orimoloye OA, Obisesan OH, Mirbolouk M, Blaha MJ, and Dzaye O. (2020). ACC/AHA lipid guidelines: Personalized care to prevent cardiovascular disease. Cleve. Clin. J. Med. 87, 231–239. [DOI] [PubMed] [Google Scholar]
- 14.Kim J, Lee H, An J, Song Y, Lee C-K, Kim K, and Kong H. (2019). Alterations in Gut Microbiota by Statin Therapy and Possible Intermediate Effects on Hyperglycemia and Hyperlipidemia. Front. Microbiol. 10, 1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaddurah-Daouk R, Baillie RA, Zhu H, Zeng Z-B, Wiest MM, Nguyen UT, Wojnoonski K, Watkins SM, Trupp M, and Krauss RM (2011). Enteric microbiome metabolites correlate with response to simvastatin treatment. PLoS One 6, e25482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bogiatzi C, Gloor G, Allen-Vercoe E, Reid G, Wong RG, Urquhart BL, Dinculescu V, Ruetz KN, Velenosi TJ, Pignanelli M, et al. (2018). Metabolic products of the intestinal microbiome and extremes of atherosclerosis. Atherosclerosis 273, 91–97. [DOI] [PubMed] [Google Scholar]
- 17.Zimmermann M, Zimmermann-Kogadeeva M, Wegmann R, and Goodman AL (2019). Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature 570, 462–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao C, Hu Y, Chen H, Li B, Cao L, Xia J, and Yin Y. (2020). An in vitro evaluation of the effects of different statins on the structure and function of human gut bacterial community. PLOS ONE 15, e0230200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karlson BW, Wiklund O, Palmer MK, Nicholls SJ, Lundman P, and Barter PJ (2016). Variability of low-density lipoprotein cholesterol response with different doses of atorvastatin, rosuvastatin, and simvastatin: results from VOYAGER. Eur Heart J Cardiovasc Pharmacother 2, 212–217. [DOI] [PubMed] [Google Scholar]
- 20.Tuteja S, and Ferguson JF (2019). Gut Microbiome and Response to Cardiovascular Drugs. Circulation: Genomic and Precision Medicine 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tungland B. (2018). Human Microbiota in Health and Disease: From Pathogenesis to Therapy (Academic Press; ). [Google Scholar]
- 22.Tilg H, Zmora N, Adolph TE, and Elinav E. (2020). The intestinal microbiota fuelling metabolic inflammation. Nat. Rev. Immunol. 20, 40–54. [DOI] [PubMed] [Google Scholar]
- 23.Vieira-Silva S., Falony G., Belda E., Nielsen T., Aron-Wisnewsky J., Chakaroun R., Forslund SK., Assmann K., Valles-Colomer M., Nguyen TTD., et al. (2020). Statin therapy is associated with lower prevalence of gut microbiota dysbiosis. Nature 581, 310–315. [DOI] [PubMed] [Google Scholar]
- 24.Caparrós-Martín JA, Lareu RR, Ramsay JP, Peplies J, Jerry Reen F, Headlam HA, Ward NC, Croft KD, Newsholme P, Hughes JD, et al. (2017). Statin therapy causes gut dysbiosis in mice through a PXR-dependent mechanism. Microbiome 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fromentin S, Forslund SK, Chechi K, Aron-Wisnewsky J, Chakaroun R, Nielsen T, Tremaroli V, Ji B, Prifti E, Myridakis A, et al. (2022). Microbiome and metabolome features of the cardiometabolic disease spectrum. Nat. Med. 28, 303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Istvan ES, and Deisenhofer J. (2001). Structural mechanism for statin inhibition of HMG- CoA reductase. Science 292, 1160–1164. [DOI] [PubMed] [Google Scholar]
- 27.Grundy SM, Stone NJ, Bailey AL, Beam C, Birtcher KK, Blumenthal RS, Braun LT, de Ferranti S, Faiella-Tommasino J, Forman DE, et al. (2019). 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol. Journal of the American College of Cardiology 73, e285–e350. [DOI] [PubMed] [Google Scholar]
- 28.Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto J-M, et al. (2011). Enterotypes of the human gut microbiome. Nature 473, 174–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Costea PI, Hildebrand F, Arumugam M, Bäckhed F, Blaser MJ, Bushman FD, de Vos WM, Ehrlich SD, Fraser CM, Hattori M, et al. (2018). Enterotypes in the landscape of gut microbial community composition. Nat Microbiol 3, 8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holmes I, Harris K, and Quince C. (2012). Dirichlet multinomial mixtures: generative models for microbial metagenomics. PLoS One 7, e30126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vandeputte D, Kathagen G, D’hoe K, Vieira-Silva S, Valles-Colomer M, Sabino J, Wang J, Tito RY, De Commer L, Darzi Y, et al. (2017). Quantitative microbiome profiling links gut community variation to microbial load. Nature 551, 507–511. [DOI] [PubMed] [Google Scholar]
- 32.Boekholdt SM, Hovingh GK, Mora S, Arsenault BJ, Amarenco P, Pedersen TR, LaRosa JC, Waters DD, DeMicco DA, Simes RJ, et al. (2014). Very low levels of atherogenic lipoproteins and the risk for cardiovascular events: a meta-analysis of statin trials. J. Am. Coll. Cardiol. 64, 485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Molinaro A, The MetaCardis Consortium, Lassen PB., Henricsson M., Wu H, Adriouch S., Belda E., Chakaroun R., Nielsen T., Bergh P-O., et al. (2020). Imidazole propionate is increased in diabetes and associated with dietary patterns and altered microbial ecology. Nature Communications 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Katsuki A, Sumida Y, Gabazza EC, Murashima S, Furuta M, Araki-Sasaki R, Hori Y, Yano Y, and Adachi Y. (2001). Homeostasis model assessment is a reliable indicator of insulin resistance during follow-up of patients with type 2 diabetes. Diabetes Care 24, 362–365. [DOI] [PubMed] [Google Scholar]
- 35.Wallace TM, Levy JC, and Matthews DR (2004). Use and abuse of HOMA modeling. Diabetes Care 27, 1487–1495. [DOI] [PubMed] [Google Scholar]
- 36.Martin BD., Witten D., and Willis AD. (2020). MODELING MICROBIAL ABUNDANCES AND DYSBIOSIS WITH BETA-BINOMIAL REGRESSION. Ann. Appl. Stat. 14, 94–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hunt MC, Solaas K, Kase BF, and Alexson SEH (2002). Characterization of an acyl- coA thioesterase that functions as a major regulator of peroxisomal lipid metabolism. J. Biol. Chem. 277, 1128–1138. [DOI] [PubMed] [Google Scholar]
- 38.Depommier C, Everard A, Druart C, Plovier H, Van Hul M, Vieira-Silva S, Falony G, Raes J, Maiter D, Delzenne NM, et al. (2019). Supplementation with Akkermansia muciniphila in overweight and obese human volunteers: a proof-of-concept exploratory study. Nat. Med. 25, 1096–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cai R, Yuan Y, Zhou Y, Xia W, Wang P, Sun H, Yang Y, Huang R, and Wang S. (2014). Lower Intensified Target LDL-c Level of Statin Therapy Results in a Higher Risk of Incident Diabetes: A Meta-Analysis. PLoS ONE 9, e104922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liao JK (2007). Safety and efficacy of statins in Asians. Am. J. Cardiol. 99, 410–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilmanski T, Rappaport N, Earls JC, Magis AT, Manor O, Lovejoy J, Omenn GS, Hood L, Gibbons SM, and Price ND (2019). Blood metabolome predicts gut microbiome α-diversity in humans. Nat. Biotechnol. 37, 1217–1228. [DOI] [PubMed] [Google Scholar]
- 42.Wilmanski T, Diener C, Rappaport N, Patwardhan S, Wiedrick J, Lapidus J, Earls JC, Zimmer A, Glusman G, Robinson M, et al. (2021). Gut microbiome pattern reflects healthy ageing and predicts survival in humans. Nat Metab 3, 274–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Earls JC, Rappaport N, Heath L, Wilmanski T, Magis AT, Schork NJ, Omenn GS, Lovejoy J, Hood L, and Price ND (2019). Multi-Omic Biological Age Estimation and Its Correlation With Wellness and Disease Phenotypes: A Longitudinal Study of 3,558 Individuals. J. Gerontol. A Biol. Sci. Med. Sci. 74, S52–S60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Manor O, Dai CL, Kornilov SA, Smith B, Price ND, Lovejoy JC, Gibbons SM, and Magis AT (2020). Health and disease markers correlate with gut microbiome composition across thousands of people. Nat. Commun. 11, 5206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hales CM (2020). Prevalence of Obesity and Severe Obesity Among Adults: United States, 2017–2018. [PubMed] [Google Scholar]
- 46.Diener C, Qin S, Zhou Y, Patwardhan S, Tang L, Lovejoy JC, Magis AT, Price ND, Hood L, and Gibbons SM (2021). Baseline Gut Metagenomic Functional Gene Signature Associated with Variable Weight Loss Responses following a Healthy Lifestyle Intervention in Humans. mSystems, e0096421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, and Holmes SP (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wright ES (2015). DECIPHER: harnessing local sequence context to improve protein multiple sequence alignment. BMC Bioinformatics 16, 322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Price MN, Dehal PS, and Arkin AP (2010). FastTree 2 – Approximately Maximum- Likelihood Trees for Large Alignments. PLoS ONE 5, e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McMurdie PJ., and Holmes S. (2013). phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 8, e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bray JR, Roger Bray J, and Curtis JT (1957). An Ordination of the Upland Forest Communities of Southern Wisconsin. Ecological Monographs 27, 325–349. [Google Scholar]
- 52.Lozupone CA, Hamady M, Kelley ST, and Knight R. (2007). Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 73, 1576–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zubair N, Conomos MP, Hood L, Omenn GS, Price ND, Spring BJ, Magis AT, and Lovejoy JC (2019). Genetic Predisposition Impacts Clinical Changes in a Lifestyle Coaching Program. Scientific Reports 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Conomos MP, Laurie CA, Stilp AM, Gogarten SM, McHugh CP, Nelson SC, Sofer T, Fernández-Rhodes L, Justice AE, Graff M, et al. (2016). Genetic Diversity and Association Studies in US Hispanic/Latino Populations: Applications in the Hispanic Community Health Study/Study of Latinos. Am. J. Hum. Genet. 98, 165–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of genera significantly differing across enterotypes, identified using a Kruskal-Wallis test (Bonferroni P<0.05). Each enterotype column corresponds to the median relative abundance of a particular genus for that enterotype. The last six columns present pairwise comparisons between enterotypes for each genus using the Wilcoxon Rank Sum test, with significant values after correcting for type 1 error shown (Bonferroni P<0.05). Abbreviations: ns = not significant.
Results of beta-binomial regression models testing the association between serum HMG and microbiome functional modules, adjusted for sex, age, BMI and participant Nationality. “Feature” corresponds to the module being tested, “coef” to the Beta-Coefficients, “se” to the standard error of the coefficient, “pvalue” to the unadjusted p-value and “padj” to the Bonferroni adjusted p-value.
Plasma HMG, clinical laboratory tests, demographics, statin and vendor information on Arivale participants analyzed in the present study.
Data Availability Statement
Plasma HMG and clinical laboratory tests, along with demographics and statin information on Arivale participants included in this study, are available in Data S3. 16S amplicon sequencing data from the Arivale data set, in the form of FASTQ files, has been uploaded to the Sequence Read Archive (SRA) with the following accession numbers: PRJNA826530, PRJNA826648.
Qualified researchers can further access the full Arivale deidentified dataset supporting the findings in this study for research purposes through signing a Data Use Agreement (DUA). Inquiries to access the data can be made at data-access@isbscience.org and will be responded to within 7 business days. Data from the MetaCardis cohort used for validation of our results is freely available to download from the original study under the following link: https://www.nature.com/articles/s41591–022-01688–4#data-availability (supplementary tables 9–14).
Code used for data analysis has been deposited in GitHub (https://github.com/PriceLab/Statins_microbiome_project). It is also available on Figshare under the following DOI: 10.6084/m9.figshare.19579234.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.