

Abstract
Marfan syndrome (MFS) is an autosomal dominant, age-related but highly penetrant condition with substantial intrafamilial and interfamilial variability. MFS is caused by pathogenic variants in FBN1, which encodes fibrillin-1, a major structural component of the extracellular matrix that provides support to connective tissues, particularly in arteries, pericondrium and structures in the eye. Up to 25% of individuals with MFS have de novo variants. The most prominent manifestations of MFS are asymptomatic aortic root aneurysms, aortic dissections, dislocation of the ocular lens (ectopia lentis), and skeletal abnormalities that are characterized by overgrowth of the long bones. MFS is diagnosed based on the Ghent II nosology; genetic testing confirming the presence of a FBN1 pathogenic variant is not always required for diagnosis but can help to distinguish MFS from other heritable thoracic aortic disease syndromes that can present with skeletal features similar to MFS. Untreated aortic root aneurysms can progress to life-threatening acute aortic dissections. Management of MFS requires medical therapy to slow the rate of growth of aneurysms and decrease the risk for dissection. Routine surveillance with imaging techniques such as transthoracic echocardiography, CT or MRI is necessary to monitor aneurysm growth and determine when to perform prophylactic repair surgery to prevent an acute aortic dissection.
Toc blurb
Marfan syndrome (MFS) is a genetic disorder affecting the connective tissue, caused by mutations in FBN1 (encoding fibrillin-1, a structural component of the extracellular matrix); individuals with MFS usually present with cardiovascular (aortic aneurysms and dissections), skeletal and ocular manifestations.
INTRODUCTION
Marfan syndrome (MFS) is an autosomal dominant, age-related (that is, progressing with age) genetic disorder of the connective tissue with prominent manifestations in the skeletal, ocular and cardiovascular systems. The major pleiotropic manifestations of MFS are aortic root aneurysm, acute aortic dissection, disproportionate long bone overgrowth, and ectopia lentis (that is, the displacement or malposition of the crystalline lens of the eye). MFS is a highly penetrant condition that demonstrates substantial intrafamilial and interfamilial variability. In the early 1990s, pathogenetic variants in FBN1 (encoding the extracellular matrix glycoprotein fibrillin-1) were identified as the cause of MFS1–4 Up to 25% of FBN1 pathogenetic variants are de novo5 , that is, the mutation is new in the affected individual. Gonad mosaicism, in which unaffected parents harbour the pathogenetic variant in some of their germline cells and, therefore, can have multiple affected offspring, is rare but has been documented 6. FBN1 missense variants, insertions and deletions, and variants associated with loss of expression from one allele lead to MFS and are demonstrated to result in decreased levels of fibrillin-14,7, which is secreted and incorporated into the extracellular matrix. Fibrillin-1 is a major component of extracellular matrix structures called microfibrils, which are found in tissues alone or closely associated with elastin fibers.
In 1955, Victor McKusick established the first classification of connective tissue disorders, which included MFS8. One of the cardinal manifestations of MFS is aortic complications (Figure 1)9. In the majority of MFS patients, thoracic aortic disease begins as an asymptomatic enlargement of the aortic root, which progressively enlarges over time to form an aneurysm (a weakening of the artery wall leading to a bulge or distention). Medications can slow the rate of enlargement but do not prevent it. The aortic aneurysm becomes unstable as it enlarges and ultimately may lead to an acute ascending aortic dissection (known as the type A dissection based on Stanford classification), which is a life-threatening complication of MFS and can lead to decreased life expectancy 10,11. A dissection is a tear in the inner lining (intima) of the aorta that allows blood to enter the wall of the aorta and split through the middle layer (media) of the wall, causing the layers of the aorta to separate or dissect. (Figure 1) Type A aortic dissections are associated with high morbidity and mortality, and prior to the availability of aortic surgery, a large proportion of individuals with MFS died of complications of aortic dissection or rupture, with the majority dying by the age of 45 years.11,12 Currently, proper diagnosis and management of thoracic aortic aneurysms in individuals with MFS can prevent most acute type A aortic dissections, and since the 1970s patients with MFS have a life expectancy that approaches that of the general population13–15. It is important to note that <10% of MFS patients present with type B dissections, which originate just distal to the take of the left subclavian artery and typically propagate down the descending aorta16. These dissections are less acutely deadly than type A dissections but are associated with substantial morbidity and mortality. Importantly, type B dissections typically occur without substantial enlargement of the descending aorta where the dissections originate, and these patients commonly have aortic root enlargement at the time of type B dissection17.
Figure 1. Aortic root aneurysm and acute aortic dissections in patients with MFS.
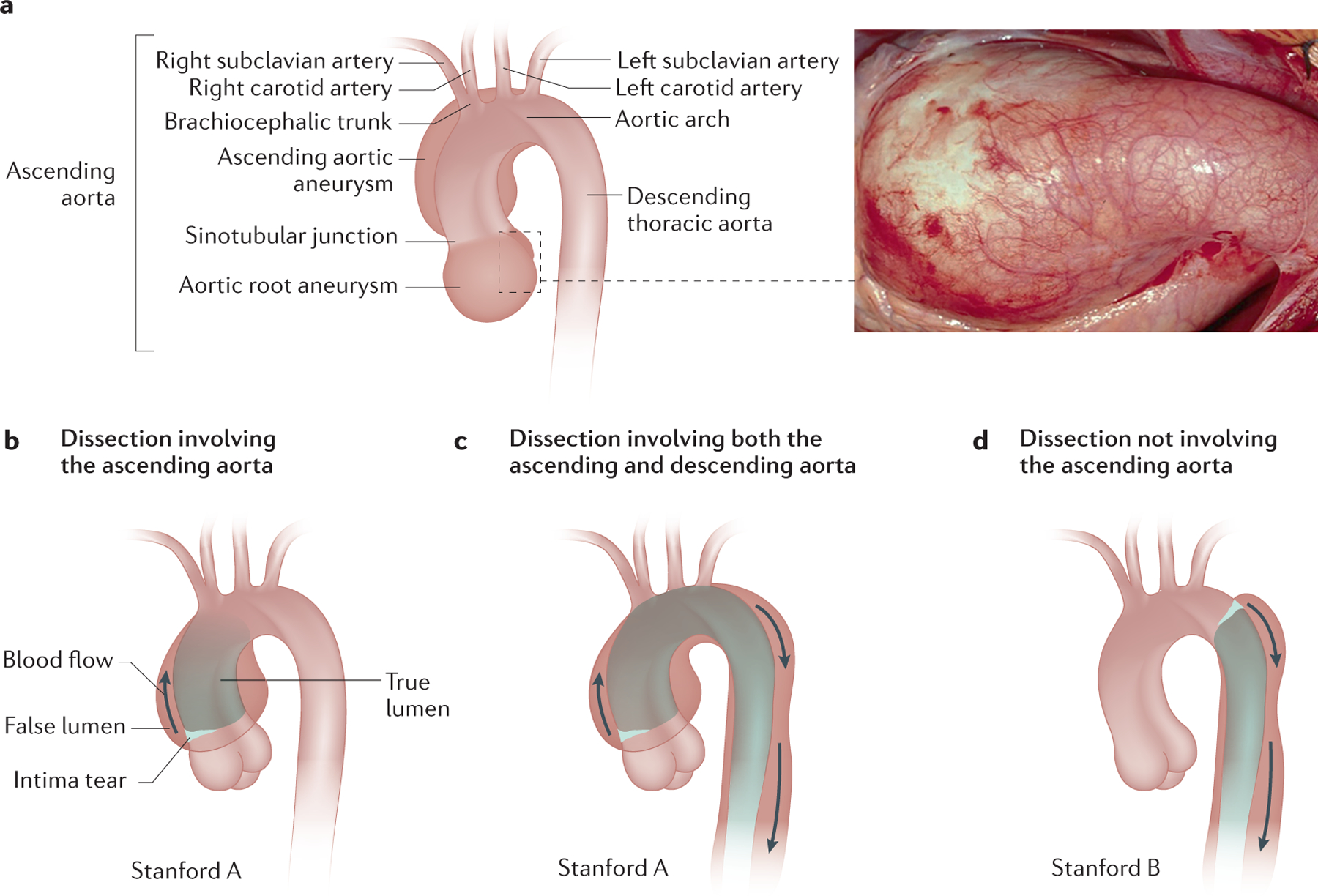
A. Illustration of the anatomy of the aorta, with a photo of an aortic root aneurysm. B, C. Patients with MFS present with aortic root aneurysms, which predispose to type A aortic dissections. D. Type B aortic dissection are also part of the disease spectrum. [CE: credit lines needed]
In addition to thoracic aortic disease, MFS affects multiple other organs and tissues in the majority of patients. In fact, a clinical diagnosis of MFS requires identifying features present throughout the body and can be made with or without genetic testing for FBN1 pathogenetic variants18. The most prominent MFS systemic features include ectopia lentis and skeletal abnormalities, including tall stature, disproportionately long arms and legs, abnormally flexible joints (including fallen arches of the feet), abnormal curvature of the spine (scoliosis), and protrusion (pectus carinatum) or indentation (pectus excavatum) of the sternum19 Over the past two decades, other features associated with MFS have been recognized, including a collapsed lung (pneumothorax, that is, abnormal collection of air in the pleural space between the lungs and the chest), abnormally indented hip sockets (protrusio acetabulae), enlargement of the lumbar segment of the spinal canal (dural ectasia), and stretch marks (striae).15
Because most bodily organs and structures are affected by MFS, understanding the pathophysiology of how the alteration of a single protein, fibrillin-1, can lead to such pleiotropic effects is essential to defining appropriate management. Importantly, basic research on mouse models of MFS, followed by clinical trials of molecularly targeted drugs, has provided additional therapies for all patients with thoracic aortic disease20,21.
In this Primer, we review our current understanding of the underlying pathophysiology of MFS, along with the diagnosis and management of individuals with MFS. The focus of the Primer is primarily on the aortic complications of MFS, owing to their association with premature death if not treated in an appropriate manner and to the fact that the majority of basic and clinical research is focused on these particular complications of MFS, but discussion of the ocular and skeletal systems is also provided.
EPIDEMIOLOGY
An early estimate of the prevalence of MFS was based on patients in the Baltimore-Washington region and evaluated at the Johns Hopkins Hospital. This estimate produced an obviously biased perspective and a prevalence of ~1 per 4–6,00022. Beginning in 1986, international experts have proposed criteria for diagnosis of MFS. The first set of criteria were developed in 1986 in Berlin23. These criteria were developed primarily to aid clinicians in determining which patients should be classified as having the condition. However, several studies used these criteria to attempt to determine prevalence with the following results: 1.5 per 100,000 persons in Northern Ireland, 4.6 per 100,000 persons in Denmark, and as high as 6.8 per 100,000 persons in northeastern Scotland24. All of these figures potentially overestimated the prevalence because the clinical criteria continued to evolve and molecular confirmation did not exist. The prevalence of MFS has not been estimated in many regions of the world, but MFS has been reported in every country and ethnicity.
With the identification of FBN1 as the gene that, if mutated, predisposes to MFS, new diagnostic criteria were established in 1996, referred to as the Ghent criteria (Ghent I)25. These more stringent criteria focused on clarifying the contribution of molecular analysis, diagnosing family members when a relative was affected, and quantifying the pleiotropic features. An effort was made to separate MFS from other heritable conditions with partially overlapping features. As FBN1 sequencing became widely available, it was recognized that rare variants in FBN1 predispose to a range of conditions, including but not limited to autosomal dominant Weill-Marchesani syndrome and acromelic dysplasia (rare syndromes characterized by short stature, short hands and stiff joints), along with isolated familial ectopia lentis and skeletal features of MFS in the absence of ectopia lentis and thoracic aortic disease26–30. The Ghent criteria were modified in 2010 to emphasize the importance of thoracic aortic disease and these modified criteria are those currently in use (Ghent II) 18. In 2015, prevalence of MFS was reassessed in Denmark on a nationwide registry and healthcare system using the Ghent II criteria.22 A total of 412 patients with MFS were identified in the population, of which 196 had genetic testing and 193 had a documented FBN1 mutation, suggesting a prevalence of 6.5 per 100,000 persons. A drawback of this analysis is that individuals and families can present with thoracic aortic disease due to an underlying FBN1 pathogenetic variant, thereby meeting the Ghent II criteria for MFS, but lack skeletal or ocular features and, therefore, often remain undiagnosed with MFS31,32. An additional issue with current prevalence estimates is that individuals can meet the Ghent II criteria of MFS but have mutations in other genes that lead to heritable thoracic aortic disease33,34.;
About one in four people with MFS who meet the Ghent II criteria do not have an affected parent.5 The phenotype of such ‘sporadic’ cases is often more severe than that of those who inherited a pathogenetic variant. Most instances of the severe, neonatal form of MFS result from de novo mutations in FBN135. The improved precision of diagnostic criteria of MFS has resulted in some conditions that were originally classified as MFS being recognized as different entities. In some families with skeletal and aortic complications similar to those found in MFS, no FBN1 mutations were identified, suggesting genetic heterogeneity for MFS36. Subsequently, TGFBR2 (encoding transforming growth factor receptor 2) missense mutations were identified in these families and tentatively classified as causing ‘Marfan syndrome 2’ (MFS2)33. Young children with either TGFBR2 mutations or TGFBR1 mutations were found to have additional systemic features, including craniosynostosis (a congenital defect in which the bones of the skull are fused together too early in development), developmental delay, and risk for aneurysm in other arteries.37 This syndrome of Marfan-like habitus with craniosynostosis and aortic disease was originally described in 1987 and termed Furlong syndrome38. The syndrome was subsequently renamed as Loeys-Dietz syndrome and expanded to include patients with mutations in genes encoding proteins involved in canonical TGFβ signalling39–42.
Numerous ‘biobanks’ of thousands of individuals, either based on patient samples or unbiased population registries, will permit estimating of the frequency of pathogenetic or likely pathogenetic variants in FBN1. However, estimating the prevalence of MFS based on such data is complicated by the numerous phenotypes associated with FBN1 variants.
MECHANISMS/PATHOPHYSIOLOGY
FBN1 pathogenetic variants
FBN1 is located on the long arm of chromosome 15 and has 65 coding exons. Rare pathogenic variants predisposing to MFS are distributed throughout the gene (Figure 2). Almost 2000 rare variants have been identified in FBN1 to date, many of which predispose to MFS. Fibrillin-1 is the major constituent of extracellular matrix microfibrils2,43. Fibrillin-1 has a modular domain structure with two different cysteine-rich domains repeated throughout its sequence. Fibrillin-1 contains 47 repetitions of the first cysteine-rich domain, which contains 6 conserved cysteine residues, that have homology with epidermal growth factors, termed EGF-like domains; of these 47 repeated EGF-like domains, 43 are predicted to bind calcium44. Disulfide bonds form between the 6 cysteines, thereby providing a rigid structure to these domains. In addition, there are 7 repeated domains of the second cysteine-rich domain, which contains 8 cysteine residues that share homology with domains found in latent transforming growth factor beta binding proteins (TGFBP modules). Finally, fibrillin-1 has a proline-rich domain and unique amino-terminal and carboxyl-terminal domains44. Missense variants are the most common type of disease-causing mutations and typically disrupt the repetitive EGF-like domains in the protein by the following mechanisms: substituting or inserting cysteines crucial for the proper folding of the EGF domains; altering the residues involved in calcium binding to the EGF-domain; or changing a glycine in a standard position in the EGF domain that does not have an identified function7. Approximately 10% of disease-causing variants disrupt canonical splice donor or acceptor sites and cause splicing errors, which can lead to in-frame deletion of an entire EGF-like domain. Variants leading to splicing errors can also cause a frameshift in translation and, along with small insertions, deletions and stop codons, lead to haploinsufficiency (loss of expression of protein from one allele) through degradation of the mutant transcripts with premature stop codons; fibrillin-1 haploinsufficiency is the cause of MFS in 10–15% of cases7. Up to 7% of MFS-causing mutations are large or complete deletions of FBN145.
Figure 2. Pathogenetic variants in FBN1.
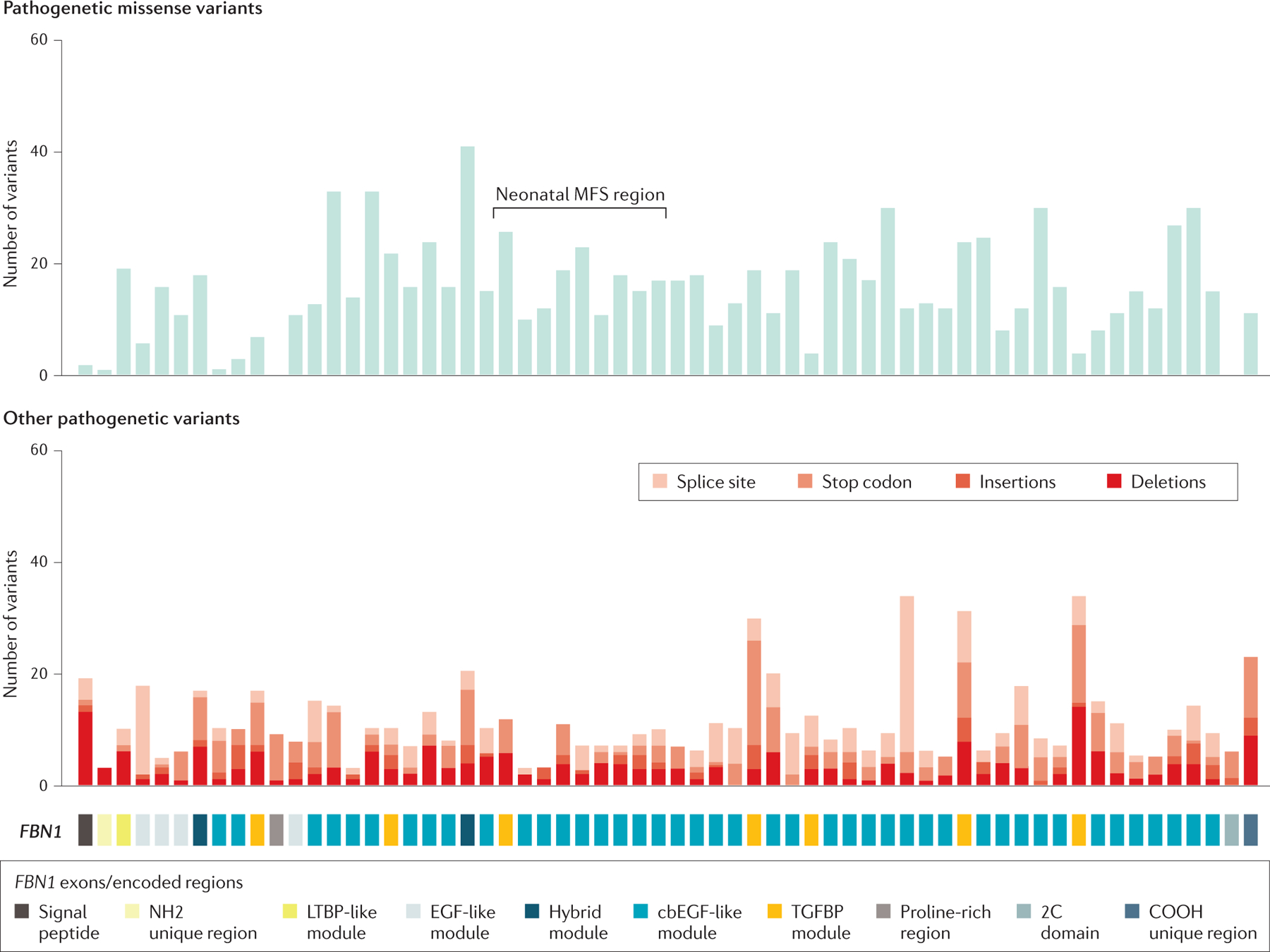
Schematic of the structure of the protein fibrillin-1, with numbers and location of mutations in FBN1, which encodes fibrillin-1, identified in patients with MFS; the location of de novo pathogenetic variants leading to neonatal MFS is also shown. Mutation data were extracted in 2020 from the last update of the UMD-FBN1 database (Ref 289)
Identification of FBN1 genotypes dictating a specific MFS phenotype has been complicated by the inter-familial and intra-familial variation in the clinical features of MFS. The most consistent and robust genotype-phenotype association is the association of de novo missense mutations in exons 24 through 32 with a severe, early onset form of MFS, termed neonatal or infantile MFS (Box 1)7,46–48. Although neonatal MFS variants cluster in this region, the majority of pathogenetic variants (>75%) in this region do not cause neonatal MFS49. Additional phenotype-genotype associations for FBN1 variants and MFS phenotype are the following: FBN1 pathogenic variants that disrupt cysteines are more common in patients with MFS presenting with ectopia lentis49,50and ectopia lentis was less common and skeletal features were more pronounced in patients with MFS with pathogenic variants leading to premature termination of translation7,51.
Box 1: Neonatal or Infantile MFS: The Severe End of the Phenotypic Spectrum of MFS.
Neonatal or infantile MFS represents the most severe end of the phenotypic spectrum of MFS. Cardiovascular features often present at birth or developing before 1 year of age are severe mitral valve prolapse with moderate to severe mitral regurgitation, considerable aortic root dilation, left ventricular dilation, possible ventricular dysfunction, symptoms of congestive heart failure, and failure to thrive264,265. Other features of neonatal MFS include an abnormal progeroid (resembling premature aging) and cachectic (physical wasting with loss of weight and muscle mass) appearance, marked arachnodactyly (“spidery fingers”, that is, long and thin fingers), dolichocephaly, loose skin, crumpled ears, highly arched palate, micrognathia (or mandibular hypoplasia, a condition in which the lower jaw is smaller than usual), camptodactyly (permanently bent fingers), flexion contractures (bent joints that cannot be straightened), hyperextensible joints, anterior chest deformities, muscle hypoplasia, pulmonary disease (blebs (pockets of air), pulmonary cysts, and overt emphysema), megalocornea (enlarged cornea), and ectopia lentis. Many of these features of MFS can be present at birth, and it is proposed that the two clinical features that are uncommon in classic MFS but common in neonatal MFS, congenital emphysema and mitral and/or tricuspid valve regurgitation, be used to define neonatal MFS266. A third feature that is used to confirm neonatal MFS is that affected children have de novo mutations (missense and in frame deletions) in a limited region of FBN1 defined by exons 24 through 3247,267. In these children, the valve problems relentlessly progress and lead to congestive heart failure. Although case reports and very small series suggest a grim prognosis in this population with a high likelihood of mortality typically due to heart failure, a better prognosis results from early mitral valve surgery and heart transplant268–272.
Fibrillin-1 function in tissues
Fibrillin-1 is a large, extracellular matrix (ECM) structural protein that polymerizes to form structures called microfibrils. Microfibrils adopt a tissue-specific architecture and provide strength and stability to tissues that undergo constant stretch and recoil, such as arteries, lung, and skin, but are also found in deformable tissues, such as perichondrium, sclera and cornea. Elastin is deposited onto bundles of microfibrils during development and growth, and, therefore, microfibrils in some tissues are intimately associated with elastin fibers.
Other proteins associate with fibrillin-1-containing microfibrils include latent TGF-β binding proteins (LTBPs)52. Fibrillin-1-containing microfibrils store and regulate growth factors in the TGFβ family, including TGFβ and bone morphometric proteins (BMPs) 52–54. LTBPs are a family of proteins that regulate TGF-β activity by enabling its secretion, directing it to specific sites in the ECM, and participating in its activation55,56. TGFβ is secreted from cells bound to a complex that includes its dimeric pro-peptide (termed latency-associated peptide, or LAP) and one of the three LTBPs57. In the ECM, the C-terminal regions of LTBP-1 or LTBP-4, with inactive TGFβ covalently attached, interact with the four domains near the N terminus of fibrillin-158,59. Bound to the fibrillin-microfibril scaffold, LAP bound to LTBP requires an activation step to release the active TGFβ peptide. For example, myofibroblasts in wounds activate TGF β in the ECM through integrin-mediated myofibroblast contraction60.
Thoracic Aortic Disease
The aorta is an elastic artery that is uniquely designed to withstand a lifetime of biomechanical forces due to pulsatile blood flow from the heart. The thick medial layer of the human thoracic aorta is composed of >50 alternating layers of elastic laminae (which are predominantly composed of elastin) and smooth muscle cells (SMCs), which confers elasticity and strength to the aortic wall provide the structural support to withstand these biomechanical forces (Fig. 3)61. In individuals with MFS, the aorta shows fragmentation and loss of elastin fibres, decreased density of SMCs and an increase in proteoglycan deposition62. Fibrillin-1 is the major protein in the microfibril extensions from the elastic lamellae, which are anchored obliquely to focal adhesions (also termed dense plaques) on the cell surface of SMCs and, therefore, link the SMCs to the elastin fibers of the lamellae (Fig. 3)63. The integrin receptors in focal adhesions then link to the actin and myosin-containing contractile units within the SMCs, enabling the propagation of mechanical forces between elastin and SMCs. Thus, this elastin-contractile unit design is predicted to coordinate SMC contractile and elastic tensions in response to mechanical stresses from the pulsatile blood imposed on the aorta. Interestingly, genes with mutations that predispose to heritable thoracic aortic disease commonly disrupt components of the elastin-contractile unit61,64–66. FBN1 mutations in patients with MFS lead to decreased synthesis or secretion of fibrillin-1 by SMCs and other cells, or disrupt the polymerization of fibrillin-1 into microfibrils, thereby potentially decreasing the connections between microfibrils in the elastin fibers and the SMC contractile unit1,4.
Figure 3. Role of fibrillin-1 in the SMCs.
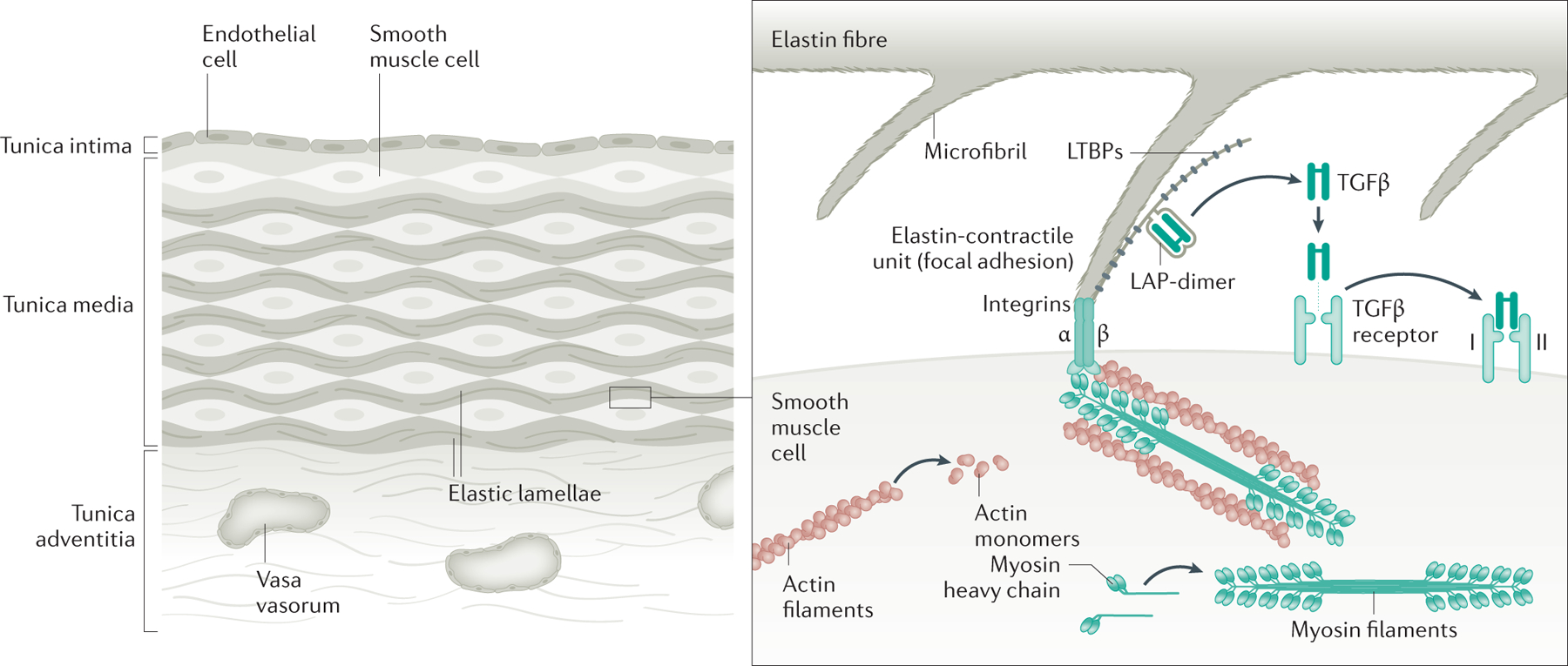
The human aorta is composed of over 50 layers of elastic lamellae and SMCs (note that for simplicity only a few layers are shown). Elastin fibers have oblique extensions that have microfibrils at the tips, which connect to focal adhesions on the SMC cell surface, and then to SMC contractile units; these structures are called elastin-contractile units. Fibrillin-1 is the major protein in the connecting microfibrils. Genes that are altered to predispose to heritable thoracic aortic disease disrupt major proteins in the elastin-contractile unit. Research has focused on the role of excessive TGF-β and angiotensin II signalling as drivers of thoracic aortic disease. More recent research has indicated that loss of TGF-β signalling is a primary driver of thoracic aortic disease, but increased signaling may have a role in late stages of the disease process.
Insights from animal models
Genetically engineered mouse models of MFS have been used to understand the link between the mutant FBN1 and thoracic aortic disease. MFS mouse models include but are not limited to Fbn1−/− mice, which lack fibrillin-1, Fbn1mgR/mgR mice, which produce 20% of normal levels of fibrillin-1, and Fbn1 C1041G/+ mice, which produce equal amounts of wild-type fibrillin-1 and a mutant fibrillin-1 encoded by an Fbn1 allele harbouring a missense mutation disrupting a cysteine in an EGF-like domain that has been identified in a patient with MFS ()20,67–69. In these mouse models, the degree of disruption of fibrillin-1 production correlates with the severity of the aortic disease. For example, Fbn1−/− mice die within the first two weeks of postnatal life, whereas Fbn1mgR/mgR mice die by 6 months of age, both owing to thoracic aortic dissection70. By contrast, Fbn1 C1041G /+ mice exhibit slowly enlarging aortic root aneurysms, but these aneurysms seldom progress to dissection or rupture, whereas the aortic root aneurysms in patients with MFS do progress to type A dissections in the absence of surgical repair. In the Fbn1mgR/mgR aorta, electron microscopic analyses shows loss of connections between the SMCs and elastin fibers associated with fibrillin-1 deficiency71.
Initial studies in Fbn1 C1041G /+ mice identified excessive TGF-β signaling based on increased levels of its downstream target, that is, activated, phosphorylated Smad2 (pSmad2), in the lung and aorta (Figure 3).20,72 TGF-β inhibition by a pan-TGF-β neutralizing antibody (TGF-β-NAb) prevented aneurysm formation and normalized pSmad2 levels in medial SMCs. In animal models of chronic renal insufficiency and cardiomyopathy, Losartan (an angiotensin II receptor blocker used to treat hypertension) had been shown to block TGF-β signaling and, therefore, was used to block TGF-β signaling in the Fbn1 C1041G /+ mice73,74. A study showed that Losartan was as effective as TGF-β-Nab in preventing aortic aneurysms in Fbn1 C1041G /+ mice and more effective than the standard of care, β-adrenergic receptor blockade (β-blockers)20.
Further studies disputed the hypothesis that excessive TGF-β activation is the primary driver of aortic disease in the MFS mouse model, and whether Losartan effectiveness is exclusively the result of TGF-β inhibition. First, SMAD2 may be activated by either TGF-β or Angiotensin II signaling75,76 and the MFS mice treated with either Losartan or TGF-β-NAb do not discriminate between the contributions of TGF-β versus type 1 angiotensin II receptor (AT1R) activation. Second, neutralization of TGF-β signaling starting at a young age (postnatal day 16) in Fbn1mgR/mgR mice accelerated rather than mitigated aortic aneurysm formation, leading to earlier dissection and death77. Similarly, genetic knockdown of TGF-β signaling in SMCs of Fbn1C1039G/+ mice, along with other mouse models of aneurysms, exacerbates rather than mitigates aortic pathology78–80.Third, losartan treatment does not prevent aneurysm formation and aortic ruptures in Fbn1mgR/mgR mice, suggesting that other AT1R-independent signaling pathways are activated77. Lastly and perhaps most importantly, loss-of-function mutations in genes involved in the canonical TGF-β signaling pathway, including mutations in the TGF-β receptors (TGFBR2 and TGFBR1), downstream signaling molecules (SMAD3), and TGF-β ligands(TGFB2), cause heritable thoracic aortic disease in humans81–84. Thus, studies of genes that, when mutated, predispose to thoracic aortic disease and mouse studies strongly support the hypothesis that lost rather than excessive TGFβ signaling is a driver of thoracic aortic disease.
Subsequent studies have further delineated a role for TGF-β and Ang II signaling in thoracic aortic disease in Fbn1mgR/mgR mice. Blocking TGF-β signaling in young animals at an early stage of disease worsened aortic outcomes, as mentioned above, whereas treatment at later stages attenuates disease77. Based on these observations, the treatment strategy was altered so that AT1R antagonists were administered early and continuously, followed by TGF-β-NAb administration at later timepoints. This combined strategy was effective to prevent aortic complications, and the results linked aberrant AT1R and TGF-β signaling with distinct stages of the disease progression. Further studies of the role of angiotensin II signaling determined that genetic inactivation of angiotensin II type I receptor, At1ar, which is blocked by losartan, in Fbn1C1039G/+ mice does not mitigate aneurysm formation and this finding has raised the intriguing possibility that losartan might exert its therapeutic effects independently of the targeted At1r receptor85. The fact that aneurysm formation is mitigated in Fbn1mgR/mgR mice with endothelial At1ar inactivation has implied that angiotensin II receptor signalling is a prominent determinant of aortic disease, most likely acting through intima-to-media communication76.
With additional mutant genes identified for heritable thoracic aortic disease, aberrant mechanosensing by SMCs has emerged as a possible mechanism for disease. Fibrillin-1 is the major protein in the microfibril extensions of elastic laminae to the SMCs in the aortic media, and these connections are disrupted in the Fbn1mgR/mgR mice71. Consistent with the hypothesis that the architecture of the SMC contractile-elastin unit is a functional and structural element important for the structural integrity of the aorta, many of the altered genes that predispose to thoracic aortic disease disrupt other components of this unit (Figure 3)66,86,87. Additionally, focal adhesion signaling in SMCs is predicted to be disrupted by genetic alterations affecting components of the elastin-contractile unit, and focal adhesion signaling is the most altered pathway in SMCs in Fbn1C1039G/+ mice when compared to wildtype mice, on the basis of single cell RNA sequencing of the aorta88. Thus, mechanosignaling is emerging to have a role in the aortic disease, and studies in MFS mouse models also suggest that the aetiology of the cardiomyopathy is altered mechanosignaling owing to the underlying fibrillin-1 defect 89–91.
The thoracic aortic disease in the mouse models of MFS is associated with alterations in other signalling pathways that have not been as fully explored as TGF-β and Angiotensin II signaling. Increased reactive oxygen species (ROS) is present in both SMCs explanted from patients with MFS and in the aortas of mouse models of MFS92–94. Interestingly, both MFS mouse models and another model of heritable thoracic aortic disease, Acta2−/− mice, present increased ROS levels and NADPH oxidase 4 (NOX4) activity, along with disruption of the elastin contractile unit93,95. ROS can activate the p38MAPK signaling pathway, which has been shown to be activated in both Fbn1mgR/mgR mice and SMCs derived from induced pluripotent stem cells harbouring a FBN1 mutation96,97. Aortic enlargement in the Fbn1C1039G/+ mice can also be attenuated by blocking metalloproteinase activity or caspase-driven SMC apoptosis98,99. Altering the expression of microRNAs, specifically blocking miR29b, which regulates apoptosis and ECM remodelling, can reduce aneurysm formation in a MFS mouse model100. Moderate aerobic exercise has been shown to attenuate aortic growth in the Fbn1C1039G/+ mice compared with Fbn1C1039G/+ mice with normal activity101. Potential therapeutic targets that mitigate thoracic aortic disease in preclinical studies in MFS mouse models need to be further explored through clinical studies in MFS patients86,102.
In summary, despite intense research on MFS disease mechanisms, the exact molecular pathogenetic pathway for thoracic aortic disease remains unknown and may involve complex interactions between cells in the aorta and biomechanical forces on the aorta86. Additionally, therapeutic efficacy in the mouse does not predict therapeutic success in patients with MFS. It is important to note that pursuing such studies may benefit the entire population of patients with thoracic aortic disease, as common genetic alterations in FBN1 increase the risk for thoracic aortic disease in the general population, suggesting that FBN1-driven pathways contribute to disease in all patients103.
Skeletal abnormalities
The severe skeletal abnormalities associated with MFS highlight the crucial role that fibrillin-1 and microfibrils have in bone formation and function, despite representing a low abundance component of skeletal matrices. The Fbn1mgR/mgR mouse model was the first in which severe kyphosis (forward rounding of the upper back) and overgrowth of the ribs, which are skeletal features associated with MFS were described 68. Studies of MFS mice have demonstrated a correlation between the skeletal phenotypes of these mutant animals and distinct pathophysiological mechanisms that reflect the contextual contribution of fibrillin-1 scaffolds to TGFβ signaling during growth and metabolism104,105.
The role of the mouse genetic backgrounds on kyphosis was assessed using heterozygotes for the mutant Fbn1 allele with an internal deletion of exons 19–24, with kyphosis more pronounced on a 129/Sv background than on C57Bk/6 one106. Interesting, the levels of Fbn1 expression in tissues inversely correlated with kyphosis in this mouse model, and the degree of kyphosis correlated with thoracic aortic aneurysms and dissections106.
Ocular manifestations
Fibrillin-1-containing microfibrils are ubiquitous in the normal eye. However, the amount of fibrillin-1 differs among human ocular structures. Fibrillin-1 is prominently found in the ciliary zonules, which extend from the ciliary body to the equatorial region of the lens, centering the lens in the eye and transmitting contracting forces from the ciliary muscle to the lens for accommodation107. The lack of fibrillin-1 in these zonules due to an underlying FBN1 mutation is most likely the cause of ectopia lentis in patients with MFS.
Fibrillin-1 is also found in the iris, the walls of Schlemm’s canal, throughout the sclera and in the subepithelial region of the peripheral cornea, and corneal stroma, but is absent in the vitreous. The lack of fibrillin-1 in patients with MFS can also lead to the enlarged corneal diameter, miosis (excessive constriction of the pupil), and hypoplasia of the iris 107–110.
Studies in mice sought to identify the fibrillin-1 producing cells contributing to ocular manifestations. Conditional knockout of Fbn1 in non-pigmented ciliary epithelium (NPCE) cells profoundly affected the ciliary zonule111. Deleting Fbn1 from just these NPCE cells lead to ectopia lentis by 3 months of age in the mouse model, increased length and volume of the eyes, and later, cataracts. Thus, deleting Fbn1 from one cell type in the eye recapitulates key aspects of MFS ocular complications.
DIAGNOSIS, SCREENING AND PREVENTION
Presentation
Patients with MFS are typically referred for diagnosis based on the presence of one or more of the following situations: skeletal features, ectopia lentis, thoracic aortic disease, or cascade testing for a disease-causing variant in a family member. Skeletal features often lead to the diagnosis. If the skeletal or ocular complications fail to lead to a timely diagnosis, asymptomatic and undetected aortic root aneurysms can eventually evolve into an acute aortic dissection, and presentation with a dissection is another feature that leads to diagnosis. In some cases, MFS is not diagnosed until an individual presents with an acute aortic dissection, which prompts sequencing of FBN1 and the subsequent identification of a pathogenic variant.32,112
MFS affects all ethnicities but may have a variable presentation depending on ethnic and racial specific features. The skeletal manifestations have been primarily described in adults of European descent but not extensively for other ethnicities. Importantly, Hispanic and Asian patients with MFS have been reported to lack substantial skeletal manifestations despite exhibiting ocular and aortic complications to the same extent as individuals with MFS of European descent113,114. Thus, these individuals are less likely to be referred for possible MFS owing to skeletal features.
The diagnostic criteria for MFS were most recently revised in 2010 and termed Ghent II nosology [Box 2]. The revised criteria emphasize the presence of the cardiovascular manifestations and incorporate FBN1 sequencing18,115. The criteria emphasize that individuals with features suggestive of other syndromes, such as Loeys-Dietz syndrome, Shprintzen-Goldberg syndrome, congenital contractural arachnodactyly, familial thoracic aortic aneurysms and dissection, and vascular Ehlers-Danlos syndrome, need to have these diagnoses excluded through genetic testing (Table 1).116 Of note, the diagnostic features for MFS in the Ghent II criteria that assess skeletal overgrowth, such as pectus deformities and scoliosis, may not be fully expressed until an individual completes bone growth18.
Box 2. Ghent II criteria.
Systemic features excluding aortic disease, ectopia lentis and family history for the diagnosis of MFS.
Wrist and thumb signs (3 points)
Wrist or thumb sign (1 point)
Anterior chest deformity (2 points)
Hind foot deformity (2 points)
Pneumothorax (2 points)
Dural ectasia (2 points)
Protrusion acetabuli (2 points)
Reduced upper segment or lower segment and increased arm span to height ratio (1 point)
Reduced elbow extension (1 point)
Facial features: dolichocephaly, enophthalmos, downslanting palpebral fissures, malar hypoplasia, and retrognathia (1 point if 3 out 5 features are present)
Skin striae other than due to pregnancy or obesity (1 point)
Myopia >3 diopters (1 point)
Mitral valve prolapse (1 point)
The total score of the systemic features is used in the diagnostic criteria.
Requirement for the diagnosis of Marfan syndrome
Aortic root dilatation & ectopia lentis
Aortic root dilatation & a FBN1 mutation
Aortic root dilatation & ≥7 systematic points (see above)
Ectopia lentis with a FBN1 mutation known to cause ascending aorta dilation
Family history of MFS & ectopia lentis
Family history of MFS & ≥7 systematic points (see above)
Family history of MFS & aortic root dilatation
Table 1.
Differential Diagnoses of Marfan Syndrome.
| Condition | Overlapping features | Distinguishing features | Altered gene(s) |
|---|---|---|---|
| Congenital contractural arachnodactyly257 (MIM 121050) | Arachnodactyly, Scoliosis and (rarely) aortic root dilatation | Congenital contractures of the digits, elbows and knees and crumpled ears | FBN2 |
| Loeys-Diez syndrome258 (MIM 609192, 610168, 615582, 614816, 613795) | Aortic root aneurysms, Dissections, Joint hypermobility, Mitral valve prolapse and MFS skeletal features | Aneurysms and dissections of other arteries, Bifid (split or forked) uvula, Craniosynostosis, Hypertelorism (increased distance between the eyes), Blue sclera, Easy bruising And Thin, translucent skin that has clearly visible veins |
TGFBR1
TGFBR2 SMAD3 TGFB2 TGFB3 |
| Vascular Ehlers Danlos syndrome259,260 (MIM 130050) | Ascending aortic aneurysms, Dissections And Joint laxity | Aneurysms and dissections of other arteries; Thin, translucent skin; Easy bruising; Blue sclera; Ptosis (drooping of the upper eyelid) and Spontaneous rupture of bowel and gravid uterus | COL3A1 |
| Hereditary thoracic aortic disease (HTAD)a261 | Root and ascending thoracic aortic aneurysms and Dissections | No associated features or features distinct from MFS | FBN1, TGFBR1, TGFBR2, SMAD3, TGFB2, COL3A1, LOX, ACTA2, MYH11, PRKG1, MYLK |
Pathogenic variants in FBN1, TGFBR1, TGFBR2, SMAD3, and TGFB2 can cause heritable thoracic aortic disease in the absence of MFS systemic features.
Genetic testing panels for the genes leading to heritable thoracic aortic disease are often the most useful and economical option to diagnose a patient with MFS. These panels sequence all genes established to predispose to thoracic aortic disease, including FBN1, and often also include evaluation for gene duplications and deletions. Since features of MFS overlap considerably with features of Loeys-Dietz syndrome, these panels can confirm the correct diagnosis of MFS through the identification of a pathogenetic variant in FBN1.34
Aortic manifestations
The most frequent aortic event associated with MFS is the dilatation of the aortic root, the aortic segment closest to the heart. Dilatation of the aortic root is usually symmetrical and limited to the aortic root, at least at the beginning of disease progression. Aortic root dilatation has a diagnostic value, but the normal aortic root diameter has to be adjusted using nomograms that include age, sex, height and weight (see Aortic Imaging below). Aortic root dilatation is typically present at the first echocardiography in patients who are eventually diagnosed with MFS, even if it is performed in infancy48,117,118. Thus, any patient, children or adult, being evaluated for MFS should undergo an echocardiography. The expected increase of the aortic root in children on medical therapy is an average of ~0.5–0.8 mm/year, although in a single year it may be as high as 3 mm21,119,120. For adults, mean aortic root growth is lower, ranging from 0.3 to 0.7 mm/year, with men and those with larger aortic roots at baseline having the fastest rates of growth, and with rates varying by treatment regimen.121–125. Thus, the growth rate of the aorta in an individual with MFS is greater than the normal growth rate of the aorta in an unaffected individual (0.1 mm per year) in the general population126.
In addition to diagnosis, the degree of aortic root enlargement also carries crucialprognostic information, specifically the risk of aortic dissection127. Aortic root diameter is the main criterion used to consider prophylactic aortic root surgery to prevent acute ascending aortic dissection128,129. In addition to the diameter of aortic root (also termed the sinuses of Valsalva), criteria for recommending prophylactic aortic surgery include the rate of dilatation in the aorta, a family history of aortic dissection with minimal enlargement of the aortic root, and the severity of aortic regurgitation (the leaking of the aortic valve of the heart that causes blood to flow in the reverse direction during ventricular diastole). In adults, aortic regurgitation typically becomes an issue when the aortic root diameter is larger than 5.0 cm and, therefore, aortic regurgitation may be observed in patients whose diagnosis is delayed, who are lost to follow up and are found to have a large aortic root diameter, or who do not comply with recommendations for at least annual imaging examination. Other potential risk factors for aortic dissections that require further study are increased aortic stiffness and increased arterial tortuosity, which is abnormal lengthening of an artery that causes twisting or distortion of arteries130–133. The importance of confirming the molecular diagnosis in individuals with MFS skeletal features and aortic root enlargement is emphasized by the fact that the underlying mutated gene also informs the risk for dissection at a given aortic diameter, as, for example, patients with Loeys-Dietz syndrome with TGFBR2 mutations are at a higher risk for aortic dissection at lower diameters of the aortic root132,134,135 In the future, a patient’s specific FBN1 mutation might be a criterion for recommending early surgery, but the data are insufficient to currently make this recommendation136,137. Importantly, pregnancy in women with MFS can augment the rate of growth of aortic root and increase the risk for aortic dissections (Box 3).
Box 3: Pregnancy and MFS.
Pregnancy is recognized as a risk factor for aortic dissection in women in the general population and in women with MFS128,129,273. The risk for aortic dissection is reported both during pregnancy, primarily during the third trimester, and up to several months postpartum273–275.However, pregnancy-associated dissections in the general population are rare 273,276–278. In women with MFS, the risk of dissection is associated with an increased aortic root dilatation rate in pregnancy279. In the majority of reported dissections, the diagnosis of MFS had not been made prior to the aortic dissection273. Thus, a missed diagnosis of MFS is an important risk factor for pregnancy-associated dissections and underscores the crucial role for diagnosis and cascade assessment of family members274. Importantly, women with MFS should have genetic counselling and a cardiovascular assessment prior to pregnancy. Aortic diameter remains a risk factor for dissection, and current recommendations for the care of women with MFS who are considering conceiving are based on aortic root diameter. However, type B dissections associated with pregnancy may occur without substantial aortic dilation273,275The risk of pregnancy-associated type A dissection is considered low when maximal diameter of the aortic root is < 4.0 cm, provided β-adrenergic receptor blockers are used throughout the pregnancy and the postpartum period. When the aortic root diameter is > 4.5 cm, prophylactic valve-sparing aortic root replacement surgery should be considered before the pregnancy280. When the diameter is between 4.0 and 4.5 cm, the decision is made on a case-by-case basis. Importantly, aortic root replacement does not completely eliminate the risk of dissections during preganacy275,281. Vaginal delivery is possible when aortic root diameter is < 4.0 cm, and, in addition to the usual indications, C-section should be mainly considered , when the aortic diameter is > 4.5 cm. In some cases, elective delivery before full term is recommended to lessen hemodynamic stresses, and an expedited second stage of labor with the use of regional anesthesia should be considered to prevent blood pressure spikes280. Lastly, although β-adrenergic receptor blockers are compatible with pregnancy, angiotensin-receptor blockers should not be used during pregnancy because of potential harm to the developing fetus282.
Dilatation of the tubular ascending aorta can be observed in patients with MFS and is almost always associated with substantial dilatation of the aortic root. The additional dilatation of the tubular aorta is considered as indicating a higher risk for aortic dissection than dilatation of the aortic root alone138. In individuals with MFS, the aortic dilatation is very rarely greater at the level of the tubular ascending aorta than at the aortic root, whereas this presentation is more common in patients with aortic enlargement associated with a bicuspid aortic valve (BAV; aortic valve with two leaflets instead of three)139. Primary (nondissected) aneurysms involving the arch, descending, thoracoabdominal and abdominal aorta are relatively uncommon in patients with MFS 140,141
The largest diameter of the ascending aorta, regardless of whether it is in the root or in the tubular ascending aorta, is used to determine the timing of prophylactic repair of the aneurysm to prevent an acute type A aortic dissection. Type A dissections occur when there is a tear in the inner layer of the aorta (the intima) above the aortic root, and the blood enters the wall and extends into the middle layer of the aorta (the medial layer), establishing blood flow through a false lumen in the aortic wall. In most cases, the dissection can propagate further up to the distal parts of the ascending aorta and continues down the descending aorta. Alternatively, the blood can dissect proximally and rupture into the pericardial sac, and the majority of people who die suddenly owing to dissections die of pericardial tamponade142.
Individuals with MFS are also at risk for aortic dissections originating at a different aortic location, just distal to the orgin of the left subclavian artery (a branch of aortic arch going to the brain) and progressing down the descending aorta, termed type B dissections (Figure 1). These dissections less commonly cause sudden death and occur with little to no enlargement at the site of origin of the dissection. Complications requiring surgery can occur with type B dissection and include malperfusion of spinal arteries leading to paresis and paraplegia, malperfusion of visceral arteries leading to abdominal pain, and aortic rupture. In individuals with MFS, initial presentation with a type B dissection is less frequent than presentation for prophylactic aneurysm repair or type A dissection7. However, the observed rate of type B aortic dissection after prophylactic repair is increasing143. Some risk factors for type B aortic dissections that have been recognized include previous prophylactic surgery for the root and/or ascending aorta16,117, aortic diameter of the descending aorta > 27 mm16, and dilatation of the pulmonary artery144. Additionally, an aortic tortuosity index >1 (the ratio of the length of the aorta to the linear distance between the start and end of the aorta) raised the risk of type B dissection by 12.1-fold131. As type B dissections can occur in patients with MFS with normal aortic root diameter or after prophylactic repair of the aortic root, recommendations to prevent aortic dissections (avoidance of isometric exercise and treatment with β-adrenergic receptor blockers as discussed below) should be pursued in all patients with MFS 145.
Cardiac manifestations
MFS is also associated with complications involving the heart. Stretching of the aortic valve annulus (Figure 1) due to aortic root enlargement may give rise to leaflet malcoaptation, in which the leaflets fail to join together at the closure of the valve, and aortic valve regurgitation. This presentation is often combined with aortic valve cusp prolapse and valve cusp commissural fenestrations (ovoid apertures). A study in paediatric patients with MFS (< 18 years of age), moderate to severe aortic valve regurgitation was found to be an independent predictor of aortic root growth and cardiovascular events (for example, death, aortic dissection and cardiac valve or aortic root surgery)119. With the introduction of valve sparing aortic root replacement techniques, aortic valve regurgitation has become an important feature to take into account when defining the threshold for prophylactic surgery146.
Mitral valve prolapse (MVP) and mitral valve regurgitation (MVR) are established complications in patients with MFS. The prevalence of MVP in adults with MFS is estimated between 40–68% (compared with 1–2% in the general population)147,148. MVP is present in ~32–38% of children with MFS (<18 years of age), and prevalence increases with age149,150. In a large cohort of children and young adults with MFS, MVP was more prevalent in females than in males151. In the neonatal MFS, severe mitral valve prolapse with moderate to severe MVR is a major complication present at birth47,152.
A population-based study found that patients with MFS have an increased risk of 28% for mitral valve-related clinical events (endocarditis, surgery, and heart failure), compared with 13% in idiopathic MVP153. The age at the time of the event was also significantly lower in individuals with MFS than in individuals with MVP not associated with MFS (35 versus 65 years of age). Severe mitral regurgitation due to degenerative mitral valve disease may be successfully repaired in MFS, but it is often a more complex procedure than the same procedure the general population154. More recently, mitral annulus disjunction (MAD) was found to be highly prevalent in patients with MFS. MAD was a marker of severe disease, including aortic events at younger ages and mitral valve disease requiring repair155. Thus, detection of MAD may infer close clinical follow-up.
Pulmonary artery dilatation occurs in children and in adults with MFS and is correlated with aortic root dilatation, previous aortic root surgery, reduced left ventricular ejection fraction and increased pulmonary artery systolic pressure120,150,156–159. Clinical complications of pulmonary root dilatation are rare and may occur only with associated increased pulmonary artery pressure.
Heart failure is the cause of death in 5–30% of patients with MFS160,161. Underlying causes are severe valvular dysfunction and an intrinsic myocardial dysfunction. The reported prevalence of ‘Marfan cardiomyopathy’ ranges from 3% to 68% across different series162. Mild cardiomyopathy usually does not evolve with time but can lead to an unfavourable course in the event of an additional hemodynamic trigger, such as valve dysfunction and / or aortic root replacement, 159,163. Several studies have reported end-stage heart failure necessitating heart transplantation in patients with MFS163,164.
Children and adults with MFS have a predisposition for supraventricular as well as ventricular arrhythmias (that is, irregular heartbeat), which are not always related to valvular abnormalities165,166. Three studies report life-threatening ventricular arrhythmias in 7–9% of individuals in MFS cohorts, along with sudden cardiac arrest in up to 4% of individuals, which is most likely due to arrhythmia 166–168. A possible association with underlying intrinsic myocardial dysfunction, as described above, is supported by the finding that serum NT-proBNP levels (which increase as a result to damage to the myocardium) are the strongest independent predictor of arrhythmogenic events in patients with MFS166,167.
Skeletal manifestations
MFS is associated with substantial musculoskeletal abnormalities due to three pathophysiological processes. First, the growth of tubular bones is exaggerated, which results in elongation of the digits (arachnodactyly), legs (leading to disproportionate tall stature (dolichostenomelia)) and ribs (leading to pectus excavatum or carinatum) (Figure 4). Second, ligamentous laxity leads to joint hypermobility, especially of the digits, shoulders, knees, and ankles, and progressive vertebral column deformity (scoliosis). Third, progressive deformity of some areas leads to depressed hip joints (protrusion acetabulae) and thinning and widening of the lumbar vertebrae and neural foraminae (dural ectasia). Additionally, most patients develop degenerative arthritis earlier than expected15.
Figure 4. Clinical manifestations of the Marfan syndrome.
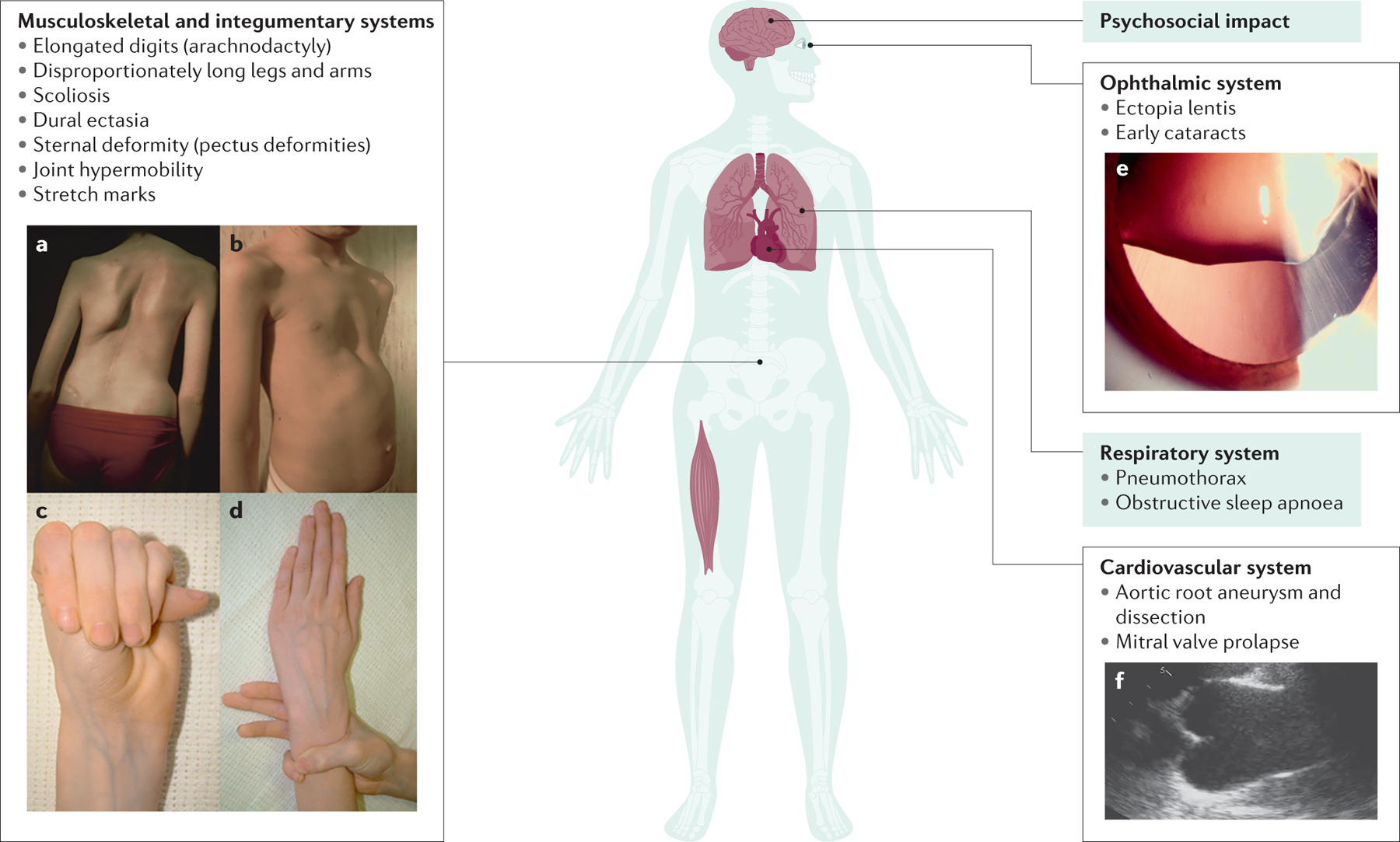
The major manifestations of MFS are illustrated. A. Scoliosis or curvature of the spine. B. Chest wall deformities, such as pectus excavatum. Arachnodactyly as evident by a positive thumb (C) and wrist (D) signs. E. Ectopia lentis. F. Aortic root aneurysm as seen by transthoracic echocardiography in parasternal long-axis view. Panels A and B adapted from https://www.elsevier.com/books/emery-and-rimoin-s-principles-and-practice-of-medical-genetics-and-genomics/pyeritz/978-0-12-812537-3; panels C and D adapted from https://www.nature.com/articles/5201851; [CE: credit lines needed]
Ocular manifestations
The ocular manifestations in patients with MFS vary by mutation and resulting severity of the disease. Patients with typical features of MFS develop lenticular and/or axial myopia prior to ten years of age and should be referred to an ophthalmologist for assessment of their nearsightedness. If the lens is dislocated in one or both eyes, the diagnosis of MFS should be suspected and confirmed based on the Ghent II criteria (Box 2)18. In the absence of lens dislocation, enlarged corneal diameter, corneal astigmatism, miosis (excessive constriction of the pupil) and hypoplasia of the iris may suggest a diagnosis of MFS. About 60% of MFS patients will develop lens dislocation in their lifetime, with the majority of patients being diagnosed in their teens, when the growth of the ocular globe is complete, but lens dislocation may occur late into their seventies (Figure 4). Both corneal and lenticular astigmatism are common and severe159. Strabismus secondary to amblyopia (lazy eye) may arise in the first decade of life, because the lens dislocation is typically asymmetric leading to preference of the less severely affected eye. The resulting amblyopia in the fellow eye is rarely deep seated and can be reversed with careful refraction assessment and glasses or contact lenses109. Presenile cataracts are a common complication in patients with MFS169. Open angle glaucoma and retinal detachments are the complications that lead to vision loss in patients with MFS170.
In patients with the most severe form of MFS, neonatal MFS, the globe is typically enlarged at birth with an increased corneal diameter. In rare patients, open angle glaucoma is observed in the first few years of life. The pupil will be miotic preventing the microspheric lens from prolapsing into the anterior chamber, and pupillary block is rare. Very high myopia up to thirty diopters may be observed; it is usually composed of both lenticular and axial myopia171.
Age-associated manifestations
In addition to the typical consequences of aging, ‘new’ manifestations of MFS have become apparent as the life expectancy of people with MFS increases15. The underlying pathogenesis of some of these manifestations remains unclear. People with MFS (and their physicians) are often surprised when these ‘new’ problems arise. In many instances, the prevalence and rate of progression of such issues have not been studied adequately, and for most, no effective therapies have been tested. With age, fluid-filled cysts can develop in the kidneys or liver in individuals with MFS. Typically, these cysts are painless and cause no functional problems, and they are often incidental findings on radiologic imaging of the aorta172. Due in part to laxity of the oropharyngeal musculature and a receded mandible, obstructive sleep apnoea is common 173. It typically presents with snoring, but sleep-disordered breathing can contribute to fatigue and difficulty with mental functions. In women, urogenital laxity occurs at younger ages and produces all of the typical problems, such as incontinence. Management can be quite inadequate and frustrating. Other abnormalities have been suggested to have increased frequency in older patients with MFS, but data to confirm these observations lack. These abnormalities include biliary tract disease, diaphragmatic hernia, premature labour, skeletal myopathy, reduced bone mineral density, atrophic scars, caries, craniomandibular dysfunction, migraine headaches, cognitive dysfunction, schizophrenia, depression, fatigue, and generalized pain174.
Finally, individuals with MFS across the age spectrum have psychosocial issues that can substantially affect their work-life balance; their relationships with relatives, friends and co-workers; and their general sense of well-being175–177. As with most of these emerging features, their prevalence and severity have not been well quantified. No specific management is recommended at this time, but counselling can be effective178. Any pharmacological treatment must account for medications already being used for cardiovascular complications.
Diagnosis
Assessment of the systemic features of MFS, along with the status of the thoracic aorta and FBN1 genetic testing, are the basis for the Ghent II nosology for the diagnosis of MFS (Box 2)18. The systemic features that are present in an individual, excluding thoracic aortic enlargement, result in a composite score up to 20 points. In practical terms, the radiologic imaging to identify protrusio acetabulae or dural ectasia is rarely performed when the diagnosis is being considered. If this systemic score is ≥7, it is combined with findings that are common in MFS but rare in the general population, that is, ectopia lentis, thoracic aortic dilatation, and/or a positive family history of MFS, and a diagnosis MFS is achieved. Sequencing of FBN1 to identify pathogenic variants is not required to make the diagnosis of MFS. However, FBN1 sequencing ensures that the MFS systemic features and thoracic aortic disease are not the result of pathogenic variants in another gene, for example, TGFBR2 or TGFBR1 (Table 1). A crucial caveat for making a clinical diagnosis of MFS is that the presence of both thoracic aortic disease and ectopia lentis is sufficient to make the diagnosis of MFS and is essentially always due an underlying FBN1 mutation. In the absence of family history of MFS, aortic dilatation or the presence of a FBN1 mutation known to cause aortic disease is required to make the diagnosis of MFS.
Cryptic pathogenic rare variants (variants not identified by standard genetic diagnostic studies) in FBN1 have been determined to cause MFS, including rare variants in the middle of introns (that is, far removed from splice donor and acceptor sites) that lead to intronic sequences being spliced into the FBN1 transcript and haploinsuffiency due to nonsense mediated decay of the mutant transcript179. Functional studies are necessary to identify these cryptic mutations but are not available except in a research setting. Homozygous or compound heterozygous FBN1 mutations have been identified that may or may not lead to earlier onset and more severe complications of the MFS180,181.
Aortic imaging
Assessment of the diameter of the aortic root is crucial to both make the diagnosis of MFS in many cases and to prevent acute aortic dissection once the diagnosis is made. Transthoracic echocardiography (TTE) has a crucial role in the diagnosis, follow-up and management of patients with MFS. Owing to its availability, reliability and lack of need for radiation or contrast material, TTE is the initial imaging tool used for the identification and serial follow-up of growth of the root and ascending aorta. The key echocardiographic measurement is the aortic root diameter (at the level of the sinuses of Valsalva). In adults, this measurement is standardly performed using the leading edge to leading edge convention at end-diastole and perpendicular to the long axis of the aorta by the parasternal long-axis view182 (Figure 5). This segment is dilated in 85–90% of patients with MFS183, and the aortic diameter in the root and ascending aorta is the best risk factor to predict a type A dissection (Fig. 5). Aortic root dilation is diagnosed if maximum aortic diameter is larger than the reference values obtained in a normal population when age, sex and body surface area are considered. Various nomograms with a normal upper limit or z-score equations (a score that reflects the two standard deviation from the mean) have been developed,184–188 but the consensus in the clinical community is to use the nomogram proposed by Campens,184 and the one proposed by Devereux 185 when the BMI is above or under the normal range. A z-score ≥ 2 in adults or ≥ 3 in children is considered abnormal129.
Figure 5. Imaging for thoracic aortic disease in Individuals with MFS.
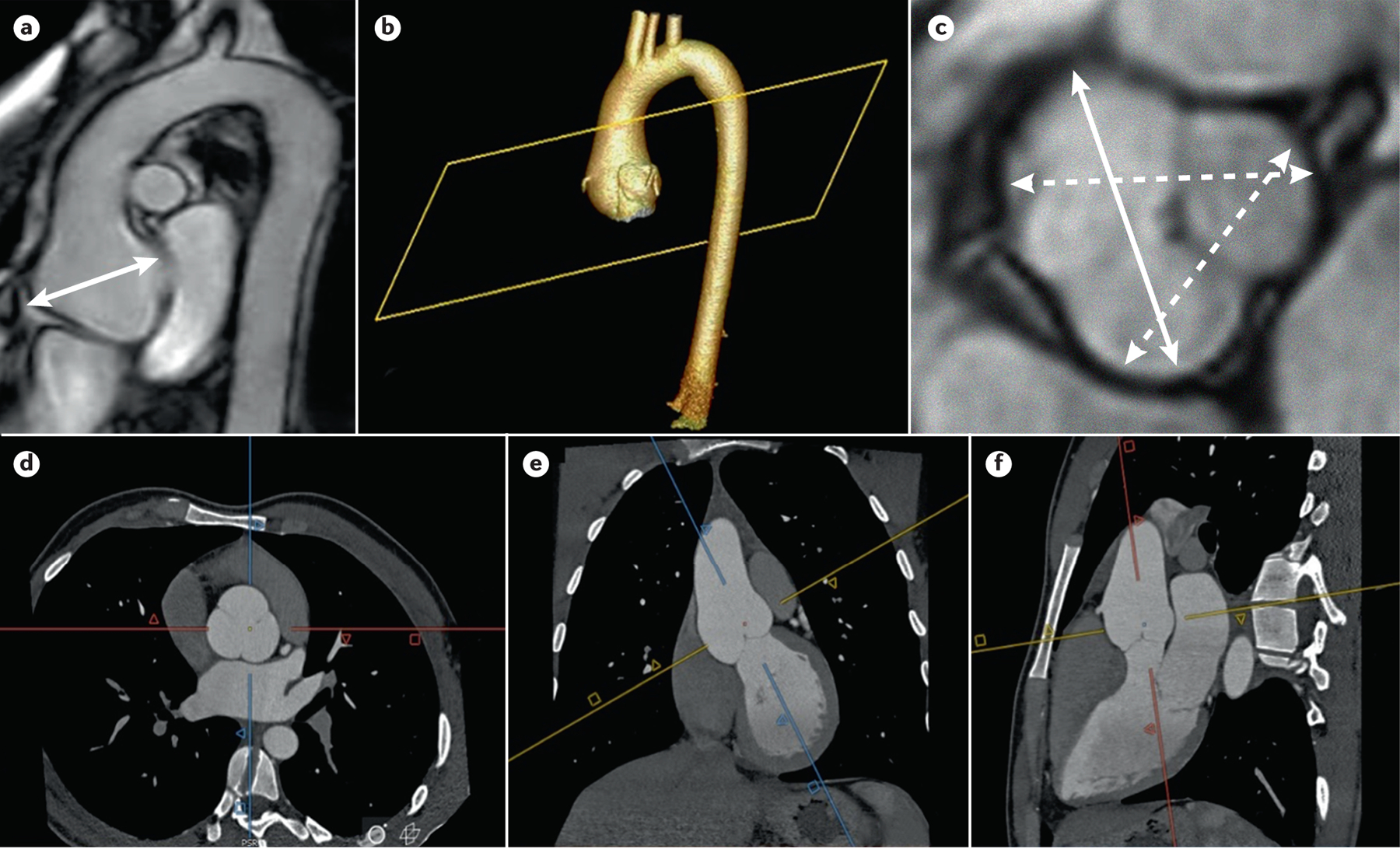
Imaging for thoracic aortic disease in MFS patients. A) MRI of the thoracic aorta shows an aortic root aneurysm (double arrow). 3D reconstruction (b) of CTA imaging (d, e, and f) of an aortic root aneurysm. The methodology of acquiring double oblique aortic images using the sagittal and coronal images to achieve perpendicularity to the aortic flow results in a corrected true transversal image of the aortic lumen; C) measurement of aortic root aneurysm by MRI using cusp to cusp diameters at end-diastole. Solid double arrow line shows the maximum aortic root diameter.
In children, aortic measurements are performed using the “paediatric method”, which measures the inner wall to inner wall distance across a structure in systole, as recommend by the American Society for Echocardiography in the pediatric population189. To determine if a pediatric patient has aortic root dilation, validated z-score algorithms should be used, with a z-score ≥ 2 defined as dilation186,190,191. Of note, z-scores for children may vary by model, and attention must be paid to the reference population. For example, some models including young children include very few children <10 years of age and rely on extrapolation. Also, z-scores may not be accurate in patients who have obesity or are severely underweight, as the algorithms are based on body surface area192. In these cases, practitioners should consider also calculating the z-score using ideal body weight, or calculating the aortic root ratio as aortic root diameter (mm) divided by the patient’s height (cm) multiplied by 100, with a ratio of ≥18.0 suggesting aortic root dilation, although this approach has not been validated in young children128.
At the time of initial diagnosis of MFS, additional imaging with CT or MRI is generally recommended to confirm that aorta size measured by TTE is accurate and to assess the distal ascending aorta, aortic arch and descending aortic segments, which rarely may be enlarged or chronically dissected. TTE provides suboptimal images in some patients with MFS, mainly when a substantial thorax deformation is present, and CT and MRI may be required. CT and/or MRI are not universally acquired at diagnosis in children, but are recommended if TTE is suboptimal, aortic dimensions are near surgical threshold, aortic growth is rapid, or disease outside the proximal aorta is suspected. After initial surveillance, imaging at 6 months is recommended to assess the rate of aortic root enlargement. If the aortic diameter remains stable and is < 45 mm, annual aortic imaging is reasonable. Patients with rapid enlargement or aortic diameter >45 mm should undergo repeat aortic imaging more frequently182. When TTE cannot be used to annual imaging of the aorta, MRI is preferred over CT to limit radiation exposure.
Distinct from elective aortic imaging and surveillance, when acute aortic dissection is suspected, urgent contrast CT is the most commonly performed imaging study and has a >95% accuracy in diagnosing dissection193. Additionally, a combination of CT and TTE provides the best information in aortic dissection diagnosis and its complications194.
It is important to be aware of the differences in aortic measurements between different imaging modalities, particularly with multimodality imaging follow-up and before surgical intervention. As mentioned above, leading edge-to-leading edge measurements of diameter in end-diastole (immediately before the heart contracts) is recommended in adults by the American Society of Echocardiography182; however, inner edge-to-inner edge diameter in mid-systole (the time in the cardiac cycle during which the ventricles contract) is recommended in children189. Nevertheless, measurement differences between these methods of measurement of the aortic diameter are small188. These differences should be considered when caring for patients undergoing transition from a paediatric-based clinic to an adult-based program (Box 4).
Box 4. Transitioning from Pediatric Care to Adult Care in MFS.
Successful transition from pediatric to adult care for patients with MFS is crucial. Major topics of consideration for the transitioning patient include change in responsibility from parent to patient; change in provider from pediatric to adult provider; need for health insurance; risk for mental health disorders, especially depression and anxiety; exercise guidance (transition from school-based limitations to lifestyle modification); increased risk for non-compliance with medical therapy; alcohol, tobacco, cannabis, and other controlled or illegal substances use; family planning, including safe contraception, reproductive risks for women, and risks of MFS in offspring.
These topics should be considered by parents and practitioners far before the actual transition to adult care. Pediatric practices should have stable ties with adult practices or have a facility that provides continuous care throughout the age range with structures to facilitate management differences in a consistent way. Pediatric practices should document how aortic measurements are obtained from imaging studies and document initial diagnostic criteria, including results of genetic testing. Adult practices receiving a new teen or young adult should closely review pediatric records and account for differences in the measurement of the aorta.
Self-management is defined as the individual’s self-directed participation in lifelong surveillance and self-care to promote health. The patient should be involved in discussions of the above topics starting in early teenage years to optimize self-management of their chronic condition283,284. Formal transition programmes have been developed for patients with chronic illness and have been shown to improve adherence and frequency of visits, but their efficacy over time still needs to be studied285,286. These transition programmes are now becoming more common for patients with MFS and hopefully will reduce the increased illness burden noted in the transition period287,288..
The recommendation for obtaining maximum aortic diameter by CT and cardiac MRI throughout the age range is to measure by inner edge-to-inner edge in end diastole182,195. Several studies demonstrated that “cusp to commissure” diameters on CT and cardiac MRI systematically underestimate aortic dilation by a mean value of 2–3mm when TTE is the reference196,197. Thus, most groups use maximum “cusp to cusp” diameter as it is closest to the maximum aortic diameter obtained by TTE198. When measuring distally to the aortic root, it is also important to avoid oblique imaging of the aorta as it will overestimate the maximum diameter199.
Screening
The diagnosis of MFS should trigger assessment of family members for MFS, which is accomplished most efficiently by testing for the presence of the causative FBN1 pathogenic variants (that is, site specific testing). Alternatively, physical exam, imaging of the aorta and ocular examination can be pursued in family members. The autosomal dominant inheritance of MFS predicts that 50% of offspring of a MFS patient will be similarly affected. The majority of patients with MFS have one affected parent and, therefore, their siblings have a 50% risk of being affected. If both parents are unaffected, then the siblings are at a very low risk for having MFS, but this risk is not zero owing to the possibility of germline mosaicism for FBN1 pathogenic variants. Additional cascade testing for MFS is based on the pedigree of the family.
The American Heart Association recommendations on screening for cardiovascular abnormalities in competitive athletes includes specific segments to identify the presence of MFS in the family history (as well as any family history of unexpected sudden death before age 50) and to evaluate for the presence of typical physical signs of MFS on the pre-participation physical examination200. In some US states, the physical examination forms for participation to high school sports specifically list the presence of some MFS characteristic signs (kyphoscoliosis, high-arched palate, pectus excavatum, arachnodactyly, hyperlaxity, myopia, mitral valve prolapse and aortic insufficiency). Cardiovascular screening in major North American Professional sports teams typically involves the history, physical examination and electrocardiography201. Only some colleges and universities in the United States perform screening echocardiograms on collegiate athletes. In major professional sports in North America, only the National Basketball Association and Major League Soccer mandate echocardiograms on players (personal communication, Jonathan Kim, MD, Emory University, 3/25/2021). Some individuals discovered to have a dilated aortic root during screening echocardiogram in the NBA have ultimately been diagnosed with MFS.
MANAGEMENT
It is crucial to manage the risk for acute aortic dissections in patients with MFS, which requires routine imaging of the aorta, medications to slow the growth of the aorta, and timely surgical repair of the enlarged aorta or aneurysm when the diameter reaches 5.0 cm in adults. Routine eye exams are also required to prevent ocular complications, and skeletal complications are treated as they arise.
Skeletal complications
The principal skeletal issues that require frequent intervention are deformities of the anterior chest and the spine. Pectus excavatum (depression of the sternum) is common and often asymmetrical. As the ribs grow, the deformity can progress and becomes irreversible once growth is complete. The indications for surgical repair are clinically important diminished lung capacity (dyspnea on exertion), compression of cardiovascular structures, or need for ascending aortic repair.202 A minimally invasive approach (Nuss procedure) is successful in most patients, but in the most severe deformity a substernal bar needs to be inserted until bone healing is complete203.
In people without a connective tissue disorder, deformity of the spine (scoliosis, abnormal kyphosis or lordosis) progresses during skeletal growth and typically stabilizes with maturity. In Marfan syndrome, spinal deformity is common and may progress after skeletal maturity204,205.This progression is especially probable in individuals with severe deformity (for example, scoliosis curves >30 degrees). One consequence is discrepancy in leg length. External bracing should be considered in children with severe or rapidly progressive curvature. Surgical stabilization of spinal deformity should be considered when the curve progresses beyond 40 degrees and can now be performed in children and adolescents using expandable rods205.
Ocular Complications Management
The diagnosis and management of the ocular features of MFS have markedly improved over the last decades owing to DNA diagnosis of MFS and refinement of ocular technology. Patients diagnosed with or suspected of having MFS ought to be examined at annual intervals or more often if complications have developed. Lens dislocation in MFS is often asymmetrical, leading to differences in acuity and refraction in the two eyes and development of amblyopia. This complication can be prevented if diagnosed early and an appropriate refraction is prescribed. Contact lenses are not contraindicated in individuals with MFS. If the lens is dislocated to the extent that the vision cannot be corrected through the lens, the risk, benefit and timing of removal of a dislocated lens have to be carefully weighed. An aphakic (that is, without lenses) prescription is usually well tolerated. Lensectomy and insertion of an artificial lens should be considered. This surgery aims for functional uncorrected vision and can usually be delayed until the eye is fully grown. Danger to the eye does not occur because of a dislocated lens; pupillary block is very rare and a totally dislocated lens in the vitreous cavity is well tolerated, however, phacolytic (caused by a leaking mature or hypermature cataract) glaucoma may develop if a totally dislocated lens remains in the vitreous cavity for many decades and will require lens removal. Many patients go through life without ever having their lenses removed, but their refraction shifts from phakic to aphakic (with and without lenses) with progression of the lens dislocation. Surgical removal of the dislocated lens and insertion of an intraocular implant should be performed by experienced surgeons.
Up to 10% of all patients with MFS develop a retinal detachment170, which may be secondary to the elongation of the globe due to decreased fibrillin-1 in the sclera. Retinal detachments need to be diagnosed early and can be managed with laser surgery, vitrectomy or scleral buckle according to the surgeon’s indications. Patients ought to be aware of the symptoms (flashes of light, sudden appearance of floaters and/or blurred vision) and seek consultations. The success rate of retinal reattachment surgery is high (>85%)206–208.
Another serious ocular complication is the development of glaucoma, which is seen in 30% of the patients during their lifetime171. The suspicion of glaucoma should always be present at annual examinations, and glaucoma needs to be aggressively managed. It can develop at all ages, and most often it is accompanied by an open angle. The exact pathogenesis remains unknown. Phacolytic glaucoma is also common. If signs of intraocular inflammation are seen, the lens ought to be removed as it may be the cause of phakolytic glaucoma207,209.
Planned cataract surgery with placement of an intraocular lens may become complicated in patients who have mild manifestations of MFS and, therefore, had not been diagnosed with MFS. The zonules may break, the capsule may rupture or the lens implant may progressively dislocate in the months following the surgery. Refractive corneal surgery may probably be safely performed in mildly affected patients. No long-term clinical data are available. The vitreous is normal, in contrast to the vitreous in individuals with Stickler syndrome (another genetic syndrome), a finding which may help in the differential diagnosis210.
Thoracic Aortic Disease
Lifestyle modifications.
Aortic aneurysms progressing to acute type A aortic dissections are the leading cause of mortality and morbidity in MFS. Regular imaging and lifestyle modifications are the first step to protect the aorta. Recreational exercise that includes low to moderate levels of aerobic exercise are important for physical and mental health for all people, including for those with MFS. In general, most individuals with MFS should exercise regularly through low-intensity (aerobic), low-impact physical activities, which can be adapted to meet their specific needs. Non-competitive exercise and physical activity performed at a non-strenuous pace or at about 50% of capacity are suggested. It is recommended to avoid contact sports, intense weight training or isometric exercises. Because shear stresses on the aorta may have a role in the development of an acute aortic dissection, individuals with MFS are restricted from participating in most competitive athletics, as well as heavy weight lifting129,211.
Pharmacological therapy
Medical therapy to slow aortic growth and prophylactic aortic aneurysm surgery to prevent type A dissections have led to improved lifespan in MFS13,14,212. Medications, either affecting myocardial inotropy (the strength of contraction) and chronotropy (heart rate) or targeting signalling pathways that have been implicated in the pathogenesis of MFS in mouse models, can affect the natural history of this condition21,213.
Beta-adrenergic receptor blockers have haemodynamic effects potentially beneficial in thoracic aortic aneurysm disease129. These agents reduce the inotropic state of the heart, decrease the impact force of ejected blood on the aorta, and are primarily used to lower heart rate and blood pressure. Thus, it was theorized that treatment with β-adrenergic receptor blockers may benefit patients with MFS and reduce the risk of aortic rupture213. Early studies of these agents in severe hypertension supported this line of evidence214. When patients with malignant hypertension (that is, extremely high blood pressure) were treated with agents that reduced blood pressure but did not change the rate of rise of blood pressure (dP/dt, or the ratio of pressure change in the left ventricular cavity during contraction), the risk of aortic dissection was not improved215,216. Furthermore, models of dissection in turkeys or mice had improved survival and reduced aortic events when the β-blocker propranolol was added to their feed217.
In patients with MFS, studies examining acute or chronic effects of β-blockers on the biomechanical properties of the aorta, measured with both invasive and non-invasive methods, have reported mixed results21,123,124,218–222. In a long-term open-label randomized trial, propranolol was compared with no therapy in 70 young patients with MFS (mean age of 15 years at the time of enrolment)213. After a 10-year follow-up, individuals who received propranolol had a lower rate of aortic root dilatation than individuals who did not receive the drug. A retrospective, nonrandomized study of β-blockers use in children with MFS also demonstrated a slowing of aortic dilatation in treated patients compared with individuals who did not receive the drug213. In a study of 417 patients with MFS, those treated with β-blockers had a longer survival than those who did not receive β-blockers223. The 2010 AHA/ACC Thoracic Aortic Disease guidelines recommended that β-blockers therapy be administered to patients with MFS with aortic aneurysms to reduce the rate of aortic dilatation, and β-blockers therapy have been recommended to individuals with hypertension and thoracic aortic aneurysm disease129.
In a MFS mouse model, losartan prevented aneurysm growth20. This finding led to clinical trials worldwide using angiotensin II receptor blockers (ARB)s to block aneurysm growth in patients with MFS (Table 2). Initially, a nonrandomized study of a small number of young patients with MFS with advanced aortic root dilation despite β-blockers therapy showed that ARB therapy (losartan or irbesartan) led to a dramatic reduction in the rate of aortic dilatation (3.5 mm/yr to 0.5 mm/yr)224. These encouraging results lead to multiple trials of ARB therapy in patients with MFS (Table 2)21,123,124,219–221. Randomized trials comparing an ARB to a β-blocker in patients with MFS found no statistically significant difference in the rate of aortic root dilation or clinical events, including aortic surgery or aortic dissection, among treatment groups21,221 Three large trials have compared the addition of an ARB to baseline therapy (which included a β-blocker in 50–86% of patients) in patients with MFS123,124,220 In two studies, the addition of an ARB led to a reduction of aortic growth rates over a 3 to 5-year follow-up123,124, whereas one study demonstrated no difference in aortic growth rate when ARB was added to baseline therapy220. These trials have confirmed a role of ARBs in preventing aortic growth in MFS, both alone and in combination with β-blockers, but found no evidence of a dramatic decrease or prevention of aortic growth with ARBs as it was observed in the MFS mouse model225.
Table 2.
Randomized Clinical Trials of ARBs in Patients with Marfan Syndrome
| Trial name | Trial type | Drugs tested | Patients age (years); number | Follow-up time | Primary outcome | Results | Notes |
|---|---|---|---|---|---|---|---|
| Pediatric Heart Network21 | Double-blind, stratified | Atenolol and Losartan | 0.5–25; 608 | 3.0±0.1 y | Rate of change in root Z-score per year | Primary outcome NS (−0.139 vs −-0.107 Z/y, p=0.08) | Pharmacogenomics substudy262; Aortic stiffness associated with aortic enlargement133 |
| COMPARE123 | Open-label | Losartan | >18; 233 | 3.1±0.4 y | Change in absolute root diameter | Primary outcome NS (0.77 with losartan vs 1.35 mm with no therapy, p=0.01) | Significant only for FBN1 haploinsufficiency263 |
| Spanish221 | Double-blind | Atenolol And Losartan | 5–60; 140 | 3.0 y | Change in absolute diameter or Z-score of root and asc | Primary outcome NS (−-0.04 vs −0.01 Z, P=0.193) | Ambulatory blood pressures not different between groups |
| Marfan Sartan220 | Double-blind | Losartan And Placebo | >10; 299 | 3.5 y | Rate of change in root Z-score/y | Primary outcome NS (−-0.01 vs −-0.03 Z/y, p=0.68) | 100% sequenced: 78% of cases had an FBN1 mutation |
| Taiwanese219 | Open-label | Losartan and a BB compared with a BB alone | None specified; 29 | ~3 y | Rate of change in absolute root diameter/y | Primary outcome significant (0.10 vs 0.89 mm/y, p=0.02) | None |
| Aortic Irbesartan Marfan Study124 | Double-blind | Irbesartan and Placebo | 6–40; 490 | 5 y | Rate of change in absolute root diameter/y | Primary outcome significant (0.53 vs 0.74mm/y, p=0.03) | None |
Abbreviations: asc, ascending aorta, BB, β-blocker; d, day; m, month; y, year; NS, not significant.
Medical treatment is indicated in children, as studies demonstrate medication slows aortic dilation, and the earlier medication is started, the greater the effect21,118,119. Titration of medical therapy in children is important to reach a therapeutic level of the medication. For β-blockers, titration goals may include a 20% heart rate reduction or goal heart rates (70s bpm in younger children, 60s bpm in older children and adolescents), while avoiding substantial adverse side effects. For ARBs, doses are usually titrated to a goal dose while avoiding adverse side effects. Some strategies for titrating these medications prior to puberty also include increasing medications until a decline in aortic root z-score is noted.
In summary, both β-blockers and ARBs are well tolerated in randomized trials. When one of these agents is chosen to treat a patient with MFS, it is important to titrate the dose to the maximally tolerated dose. Although some experts may choose to initiate therapy with a β-blocker first, others choose to initiate with an ARB. Many also utilize both a β-blockers and an ARB together to lessen hemodynamic stress on the aortic wall and potentially affect signalling pathways that are implicated in the pathogenesis of disease. It is important to emphasize that management of hypertension is a central target for all patients with thoracic aortic disease.
Aortic Imaging
The entire aorta needs to be assessed at the time of diagnosis and then routinely thereafter to monitor the growth of the aortic root. TTE can be used for serial imaging follow-up of patients with MFS and dilated aortic root when the correlation between the dimensions measured by CT or MRI has been documented182. However, reproducibility of maximum aortic diameter measurement is better by ECG-gated CT (synchronized data acquisition with the ECG waveforms) and MRI than by TTE when image quality is not optimal. Diameter changes that are < 4 mm by CT, MRI or by TTE might not reflect a real change in the aorta size (Fig. 4)192,196,226. The European Society of Cardiology guidelines recommend surgery when aortic diameters increase > 3mm per year128, whereas AHA guidelines specify >5mm per year129. In clinical practice, these indications rarely support aortic surgical repair, as aortic root diameter dilation rate over time in patients with MFS is < 0.5 mm per year125.
CT angiography (CTA) is the technique of choice for measuring aorta diameters and requires intravenous contrast medium to provide a volumetric data set that enables multiplanar and 3D reformatting182,195. Radiation is the main drawback of CTA for patients with MFS, who require lifelong surveillance. ECG-gated low-voltage techniques substantially reduce ionizing radiation exposure and minimize potential long-term harm. MRI does not utilize ionizing radiation and may be a better option in some patients with MFS (Fig. 5). Although spatial resolution and acquisition time are inferior to those of CTA, MRI offers high-quality assessment of the aorta, morphologically and dynamically, even without intravenous contrast medium227,228 In clinical practice, repeat CT or MRI is recommended at least every 3 years in adults as part of surveillance, because patients with MFS can have descending aorta or branch vessel involvement, and to confirm that measurement of the ascending aorta by TTE remains reliable16,145,229. Aortic branch aneurysms are present in patients with MFS, are related to age and aortic dilation, and independently predict the need for aortic surgery230. (Figure 5). Patients with MFS and aneurysmal dilation of the descending thoracic aorta require regular CT or MRI to monitor aortic stability, as TTE does not provide reliable imaging of this region.
Other imaging biomarkers are being assessed as predictors for adverse outcomes in MFS. Aorta biomechanics impairment, assessed by aortic distensibility, has been identified in MFS during the early stages of aortic dilation and associates with progressive aortic dilation135,231. An emerging technique, 4D flow MRI, provides advanced flow information, such as wall shear stress, vascular stiffness (based on pulse wave velocity), flow eccentricity, pulse wave reflection, and turbulent kinetic energy122,132,232–234. Such assessment of hemodynamic parameters could provide valuable information on both the presurgical and postsurgical aorta232,233. Longitudinal strain of the ascending aorta may be a useful independent predictor of aortic root expansion and aortic events (such as elective surgery and dissection)122. Finally, vertebral or aortic tortuosity is a marker of adverse cardiovascular outcomes in patients with MFS130,131.
When elective surgery is indicated, ECG-gated CTA is performed to obtain accurate aortic morphometric data, which include accurate aortic annulus and root diameter measurements, identification of chest wall abnormalities, determination of surgical access, and an assessment for coronary artery disease. In addition, TTE is recommended for monitoring surgical treatment, especially with David or Yacoub valve sparing aortic surgery, to rule out substantial residual aortic regurgitation and to assess the presence and severity of aortic regurgitation, left ventricular function, and other abnormalities, such as mitral valve prolapse or substantial mitral regurgitation235,236. After elective aortic root replacement, TTE and CT or MRI are generally performed to establish a baseline aortic assessment within 6 months or a year. The frequency of subsequent aortic imaging is individualized according to patient characteristics, such as the type of operation performed and the extent of aortic dilatation elsewhere. Serial, postoperative follow-up imaging should focus on disease progression affecting the native aorta and common postoperative complications, including the development of pseudoaneurysm and coronary anastomotic aneurysms237.
Combination of CT and TTE provides the best information in the diagnosis of aortic dissection and its complications 194 MFS patients with repaired type A aortic dissection should undergo serial aortic imaging with CT or MRI to monitor the remainder of the aorta, and similarly individuals with type B aortic dissection require routine follow-up imaging of the chest, abdomen and pelvis129. In patients with MFS with type A dissection, long-term outcome after surgery is related to the extent of the dissection and degeneration of the chronically dissected segments, leading to aneurysmal enlargement. In addition to MFS and maximum aorta diameter, a large entry tear (> 10mm) located in the proximal part of the dissection identifies a high-risk subgroup of patients who may benefit from a more aggressive surgical treatment238. Thus, exclusion of a large entry tear or large proximal secondary communications is fundamental to avoid progressive and substantial enlargement of the false lumen during follow-up. Enlargement of the false lumen requires surgical treatment of the descending aorta. Intraoperative transesophageal echocardiogram (TEE) helps in the identification of large communications between true and false lumen, and aids the indication of extension of aorta replacement - including the arch or proximal descending aorta239.
Aortic Surgery
Prophylactic surgery of the aortic aneurysm to prevent acute type A dissection is a major factor associated with improvement in lifespan in MFS13,14,160. The long-term survival is reduced in individuals who survive the acute dissection, and repeat aortic surgical interventions are more common after an aortic dissection17,160. Thus, aortic aneurysm surgery before aortic dissection or rupture is the goal of treatment. Many factors influence the surgical threshold for aortic root aneurysm repair in MFS, with aortic diameter being paramount. The 2010 ACC/AHA/AATS Thoracic Aortic Disease guidelines recommend surgery for aortic root aneurysm when the aortic diameter reaches or exceeds 5 cm129. These guidelines are based upon expert opinion and observational studies. Elective aortic root replacement in MFS performed by experienced surgeons is highly successful and carries a low surgical risk160. There are multiple indications for considering prophylactic aortic root aneurysm surgery at an aortic diameter < 5 cm (but ≥4.5 cm). These indications may include a family history of aortic dissection at a relatively small aortic size (< 5.0 cm), rapid aortic growth (>3 mm per year if measurements are obtained using the same imaging technique), substantial aortic regurgitation, the requirement for mitral valve surgery, a prior type B aortic dissection, the desire for pregnancy, and patient or surgeon wishes (especially when considering valve-sparing root replacement). The patient’s age, sex, and body size and height may also be important factors to consider when sharing a decision about the timing of aortic root replacement. The Thoracic Aortic Disease guidelines recommend surgery if the maximal cross-sectional area (in square centimeters) of the aortic root divided by the patient’s height (in meters) is >10129.
The strategy of prophylactic surgery at an aortic diameter of ≥50 mm was evaluated in a population of 732 patients with MFS127. The risk of pre-operative aortic dissection rose with increasing aortic diameters: 0.09% with aortic diameter < 40 mm; 0.1% with aortic diameter 40–44 mm; 0.3% with aortic diameter 45–49 mm; 1.33% with aortic diameter 50–54 mm; and 8.14% with aortic diameter 55–59 mm. In an observational study of MFS patients, 11 aortic events (death and aortic dissection) occurred during 2195 patient-years of follow-up240. The risk of aortic events was related to aortic diameter: 0.2% per year with diameter <40 mm; 0.3% per year between 40 and 44 mm; 1.3% per year between 45 and 49 mm; and 5.2% with diameters ≥ 50 mm240 240. The aortic event rate was also related to body size, with a higher event rate noted when the aortic size index (aortic size divided by body surface area) increased240.
There are no current guidelines on when to perform aortic root replacement in children, although some experts suggest the same guidelines as adults for most children241. Intervention may be indicated at < 50 mm in any of the following situations: another cardiac surgery is planned, a family history of dissection at an aortic root dimension <50 mm, or the patient reaches 40 mm at a very young age (that is, < 5 years of age)129. Most families of young patients seem to opt for valve-sparing surgery to avoid bleeding complication and lifetime anticoagulation therapy.
Surgery to replace the aortic root in MFS typically includes one of two main procedures: the composite aortic valve graft (CVG; also known as Bentall procedure), and the valve-sparing root replacement (VSRR) procedure using the reimplantation (David) technique160,242. In some cases, the decision to perform a VSRR or a CVG replacement is determined when the aortic valve leaflets are inspected during surgery. Ideal patients for VSRR have a relatively small aortic root (<55 mm), no more than mild aortic regurgitation, and normal appearing cusps on surgical inspection242. Aortic valve replacement may be preferable when leaflets have large fenestrations or heavy calcification and scarring. If VSRR is not deemed not appropriate, CVG is pursued.
CVG involves replacing the aortic valve with a prosthetic valve (typically a mechanical valve) and a prosthetic polyethylene terephthalate (brand name Dacron) graft, with re-implantation of the mobilized coronary arteries into the graft. The aorta is typically replaced from the valve to the mid-to-distal ascending aorta. The major advantage of this procedure is the outstanding, long-term durability of the valve and graft160.When a mechanical aortic valve is used, lifelong anticoagulation therapy with warfarin is required to manage long-term risks such as valve-related thrombosis or embolism, bleeding risks related to anticoagulation, and infective endocarditis. VSRR utilizing the reimplantation (David) technique is a surgical procedure in which the aorta is replaced and the prosthetic graft sewn into the left ventricular outflow track extending to the ascending aorta, and the native aortic valve is re-implanted within the graft242. The coronary arteries are mobilized and sewn into the graft. An advantage of VSSR is that anticoagulation is not required. The main concern about VSRR is the durability of the repair and the potential for development of aortic valve regurgitation requiring reoperation243. Single center short-term and mid-term results report a relatively low rate of substantial aortic valve regurgitation and requirement for reoperation, especially when VSRR is performed by expert surgeons242. The Aortic Valve Operative Outcomes in Marfan Patients Study registry enrolled 316 nonrandomized patients with MFS between 2005 and 2010 at 19 centers in North and South America and Europe. In this study, 7% of patients in the VSRR group developed moderate or severe aortic regurgitation by one-year follow-up243. Late follow-up will be important to assess the natural history in this cohort.
A novel technique to reinforce (rather than replace) the aortic root aneurysm in MFS known as the personalized external aortic root support (PEARS) has been developed244. The PEARS procedure involves surgical implantation of a patient-specific individualized mesh support around the aortic root and ascending aorta to prevent further enlargement and dissections or ruptures. The graft is secured proximal to the coronary arteries and to the ascending aorta, and cardiopulmonary bypass is not typically required. This procedure has been performed on 162 patients with MFS with aortic diameters between 40 and 55 mm and no more than mild aortic regurgitation244. In a study of the first 30 patients with MFS undergoing PEARS (mean age 33 ± 13 years, mean aortic diameter 45 ± 2.8 mm, at an average follow-up of 6-years), there was no statistically significant change in aortic diameter or degree of aortic regurgitation245. Additional long-term outcome data are needed before this procedure can be recommended for patients with MFS.
After elective root replacement in MFS, long-term management includes continuation of pharmacotherapy and routine imaging surveillance of the entire aorta. Patients remain at risk for distal aortic disease, both type B dissection and aneurysm formation. In a series of 258 individuals with MFS with acute aortic dissection, 64% had a type A dissection, and 36% had acute type B dissection17. Notably, 44% of patients had undergone a prior cardiac surgery. In a series of 600 patients with MFS, there were 54 type B dissections at a mean age of 36 ± 14 years16. In 30 of these 54 individuals, (56%), a type B dissection was the first aortic complication. Importantly, prior elective aortic root replacement is a risk factor for subsequent acute type B dissection, which occurs at a rate of about 10% during a 6-to-7-year follow-up after root replacement16,246.
After an acute type A or B aortic dissection, long-term management includes therapy with a β-blocker and routine imaging surveillance of the aorta. It is common for MFS patients to require multiple aortic repairs after a type A dissection extends down the descending aorta, or after an isolated type B dissection. These aortic surgeries are required owing to the chronically dissected aorta degenerating and progressively enlarging14,136,247. In the absence of aortic dissection, multiple aortic surgeries are usually not required in individuals with MFS, a finding that supports the use of prophylactic aortic root surgery of the entire ascending aorta to prevent type A dissections. Open surgery of the descending aorta is often required and is associated with relatively high morbidity and mortality. Endovascular stent grafting is associated with complications in MFS patients and is still contraindicated in the more recent recommendations129. However, progress is being made, and a combined strategy is more and more frequently performed in high-risk patients with MFS with chronic aortic dissection248.
QUALITY OF LIFE
Living with MFS can affect the quality of life (Box 5, 6) A 2002 study of 174 adults with MFS found that overall quality of life was adequate but substantially lower in psychological domain249. The majority of individuals reported that MFS affected their reproductive decisions because of concerns about having an affected child and the cardiovascular risks to the pregnant woman with MFS. Data from the GenTAC (Genetically Triggered Thoracic Aortic Aneurysms and Cardiovascular Conditions Registry) of 389 adults with confirmed MFS found that the condition did decrease quality of life compared with that of the general population250. In multivariate analyses, insurance status and employment were significant predictors of quality of life. A 2019 review of all publications studying psychosocial factors in MFS identified a negative effect of the condition on an individual’s formative years, quality of life, reproductive decision-making, work participation and satisfaction with life251. The Marfan Foundation conducted a survey of their members to identify the quality of life issues in the community of people with MFS and related disorders, and 1051 individuals completed the survey (ref 290). Nearly 85% of the respondents self-reported as having MFS with Loeys-Dietz syndrome and Ehlers Danlos syndromebeing the other diagnoses most often present in respondents (29 and 24 individuals, respectively). Nearly 65% of respondents were 20–59 years of age, with the highest percentage (18%) being 30–39 years of age. The majority of respondents reported that pain (56%) and physical limitations (56%) were the greatest obstacles that affected their quality of life. Other reported challenges were lack of stamina (43%), vision issues (38%), sleep issues (36%), feeling “down” (31%), feeling anxious or restless (31%), financial insecurity (28%), gastrointestinal issues (26%), breathing issues (23%), and being underweight (20%). In a study of Norwegian patients, quality of life was reduced by physical but not mental limitations252.
Box 5. Living with Marfan syndrome (1).
On March 26th 2012 my life was forever altered. It was a Sunday morning, and like most Sunday mornings I got up early to get ready for church. My sister-in-law called me to tell me she was riding in an ambulance with my older brother and they were headed to the Medical Center. My brother had been experiencing significant back pain for the past few days, and it had now intensified to the point where they were extremely concerned. She took him to a small local hospital by their home. It was determined that his aorta was dilating and had a tear, and he needed immediate surgery. He was on his way to a major hospital in the Houston Medical Center that could handle such a complicated procedure. I actually live near the Medical Center so I arrived before they did. They rushed him back to surgery quickly. My sister-in-law hugged and kissed him. With tears in my eyes he squeezed my hand and just looked at me as if to say everything will be fine. Shortly after my pastor arrived and prayed with our family. My brother passed away around 2AM the next morning. The tear to his aorta was too difficult to repair, and the surgeon was unable to gain control of the bleeding. He was only 32.
My brother and I inherited MFS from our mother. A connective tissue disorder that can significantly affect the connective tissue in the aorta, eyes and even the tissue surrounding the joints. We were both diagnosed as children by our pediatrician when the lenses dislocated in our eyes.
Living with MFS has changed my perspective on life. We don’t know how much time we have, and no day is promised to us. I live my life to the fullest, enjoying every day surrounded with love, peace, and happiness. I thank MFS for teaching me to appreciate everything and every moment.
Box 6. Living with Marfan syndrome (2).
Living with MFS means giving up some control (which, for a Type A person, is hard to admit). I learned at 13 years old, when a friend passed, that we can be educated about our diagnosis, and we can do all the right things, but when push comes to shove, we can’t control how our body responds to medication, or how medical professionals respond to us. Some days that still feels scary.
There’s a sense of loss sometimes, because MFS limits my options. For example, when I decided to get pregnant, there was no real choice of hospital or provider or birth plan. When it was time for college and graduate school, I picked universities, in part, by their proximity to a MFS clinic. It’s my kids worrying when I get sick, whether my body will betray me and I’ll end up in the hospital again. And, it’s the feeling that I can’t grieve the ‘what might have beens’ because hey, I’m alive and I got to have kids, and too many of my friends can’t say the same.
Living with MFS is strength, too: a key part of my identity. I push back a little when unaffected parents say, “I won’t let MFS define my kid.” Why? Lots of things define me. I’m a mother, a writer, a student, and I’m also a ‘Marf’. I’ve faced more than any of my ‘non-Marf’ peers, and I’m still here. I have to be more flexible, empathetic, and determined. My experiences have shaped my career path. Even with the negatives of my diagnosis, I wouldn’t change anything.
At the end of the day, life with MFS is a duality of pride in who I am, and advocating for a world that is more appreciative of and accommodating to disability, while at the same time looking forward to the research that will someday stave off the worst aspects of the syndrome and make life physically more comfortable for the next generation.
OUTLOOK
Pathogenesis
To date, the research on thoracic aortic disease pathogenesis has focused extensively on the roles of TGF-β and Angiotensin II signaling primarily in aneurysm formation. Although both signalling have an established role in thoracic aortic disease in MFS, many other potential pathways need to be explored, including nitric oxide signaling253,254 and activation of stress pathways in SMCs96, along with other pathways255. Importantly, the MFS mouse model (Fbn1C1039G/+ mice) used for the majority of studies develop aneurysms but rarely progress to dissection. Thus, the molecular triggers for dissection have not been extensively studied.
A major unanswered question is how loss of fibrillin-1 drives long bone overgrowth. It was originally argued that loss of epiphyseal constraint by a structurally impaired perichondrial matrix might be responsible for the disproportionately long limbs. As a functional relationship was proposed between fibrillin-1 assembly and modulation of local TGF-β bioavailability, a new theory has emerged, postulating that TGFβ hyperactivity in skeletal tissues might account for MFS-related long bone overgrowth256. Future studies are needed to determine the pathogenesis of the long bone overgrowth.
As indicated by the Marfan Foundation survey discussed above, chronic pain and physical limitations are the major issues that decrease the quality of life for individuals with MFS. Although overuse associated with the joint laxity is the most probable contributor to the chronic pain, further studies are needed to address these major issues. In addition, little is known about the pathogenesis or complications of ‘new’ manifestations of MFS that arise as patients live longer.
Advances in Treatment
The treatment for aortic disease has rapidly evolved with the introduction of endovascular treatment for subacute and chronic type B dissections. Although only graft-to-graft endovascular repair is currently recommended for non-emergent aortic repair in patients with MFS, further modifications may enable more extensive use of endovascular repair.
Studies are in progress to further refine the timing of thoracic aortic aneurysm repair to prevent dissection. Functional imaging to understand the pathophysiological process in the aortic wall and four-dimensional imaging to assess the flow patterns and wall stress are currently being pursued. Biomarkers, such as vertebral artery tortuosity and the presence of branch aneurysms130,230, need to be assessed in combination with aortic diameters to improve the timing of aortic aneurysm repair to prevent dissections. Genetic modifiers, including further delineation of the phenotype associated with specific FBN1 pathogenetic variants and other variants in the human genome that modify the phenotype, need to be included in this assessment as they are identified.
Acknowledgements
NIH R01HL109942 and R01HL146583, American Heart Association Merit Award, John Ritter Foundation, Marfan Foundation to D.M.M. Thank you to the patients with Marfan syndrome who shared their story.
Footnotes
Competing interest
The authors declare no competing interests
Peer review information
Nature Reviews Disease Primers thanks M. Groenink, D. Reinhardt, who co-reviewed with R. Zhang, P. Robinson, L. Sakai and Y. Von Kodolitsch for their contribution to the peer review of this work.
References Cited:
- 1.Hollister DW, Godfrey M, Sakai LY & Pyeritz RE Immunohistologic abnormalities of the microfibrillar-fiber system in the Marfan syndrome. N. Engl. J. Med 323, 152–159 (1990). [DOI] [PubMed] [Google Scholar]
- 2.Sakai LY, Keene DR & Engvall E Fibrillin, a new 350-kD glycoprotein, is a component of extracellular microfibrils. J. Cell Biol 103, 2499–2509 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dietz HC et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature 352, 337–339 (1991 [DOI] [PubMed] [Google Scholar]
- 4.Milewicz DM, Pyeritz RE, Crawford ES & Byers PH Marfan syndrome: defective synthesis, secretion, and extracellular matrix formation of fibrillin by cultured dermal fibroblasts. J. Clin. Invest 89, 79–86 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chiu HH, Wu MH, Chen HC, Kao FY & Huang SK Epidemiological profile of Marfan syndrome in a general population: a national database study. Mayo Clin Proc 89, 34–42, doi: 10.1016/j.mayocp.2013.08.022 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Arnaud P et al. Unsuspected somatic mosaicism for FBN1 gene contributes to Marfan syndrome. Genet Med 23, 865–871, doi: 10.1038/s41436-020-01078-6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arnaud P et al. Clinical relevance of genotype-phenotype correlations beyond vascular events in a cohort study of 1500 Marfan syndrome patients with FBN1 pathogenic variants. Genet Med, doi: 10.1038/s41436-021-01132-x (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mc KV The cardiovascular aspects of Marfan’s syndrome: a heritable disorder of connective tissue. Circulation 11, 321–342, doi: 10.1161/01.cir.11.3.321 (1955). [DOI] [PubMed] [Google Scholar]
- 9.McKusick VA Heritable Disorders of Connective Tissue fourth edn, (The C.V. Mosby Company, 1972). [Google Scholar]
- 10.McKusick VA The cardiovascular aspects of Marfan’s syndrome; A heritable disorder of connective tissue. Circulation 11, 321–342 (1955). [DOI] [PubMed] [Google Scholar]
- 11.Murdoch JL, Walker BA, Halpern BL, Kuzma JW & McKusick VA Life expectancy and causes of death in the Marfan syndrome. N. Engl. J Med 286, 804–808 (1972). [DOI] [PubMed] [Google Scholar]
- 12.Pyeritz RE & McKusick VA The Marfan syndrome: diagnosis and management. N. Engl. J. Med 300, 772–777 (1979). [DOI] [PubMed] [Google Scholar]
- 13.Silverman DI et al. Life expectancy in the Marfan syndrome. Am. J Cardiol 75, 157–160 (1995). [DOI] [PubMed] [Google Scholar]
- 14.Finkbohner R, Johnston D, Crawford ES, Coselli J & Milewicz DM Marfan syndrome. Long-term survival and complications after aortic aneurysm repair. Circulation 91, 728–733 (1995). [DOI] [PubMed] [Google Scholar]
- 15.Pyeritz RE Marfan syndrome: improved clinical history results in expanded natural history. Genet. Med 21, 1683–1690, doi: 10.1038/s41436-018-0399-4 [doi]; 10.1038/s41436-018-0399-4 [pii] (2019). [DOI] [PubMed] [Google Scholar]
- 16.den Hartog AW et al. The risk for type B aortic dissection in Marfan syndrome. J Am Coll Cardiol 65, 246–254, doi:S0735–1097(14)07007–7 [pii]; 10.1016/j.jacc.2014.10.050 [doi] (2015). [DOI] [PubMed] [Google Scholar]
- 17.de Beaufort HWL et al. Aortic dissection in patients with Marfan syndrome based on the IRAD data. Ann. Cardiothorac. Surg 6, 633–641, doi: 10.21037/acs.2017.10.03 [doi];acs-06–06-633 [pii] (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loeys BL et al. The revised Ghent nosology for the Marfan syndrome. J. Med. Genet 47, 476–485 (2010). [DOI] [PubMed] [Google Scholar]
- 19.Pyeritz RE The Marfan syndrome. Annu. Rev. Med 51, 481–510 (2000). [DOI] [PubMed] [Google Scholar]
- 20.Habashi JP et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 312, 117–121 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lacro RV et al. Atenolol versus losartan in children and young adults with Marfan’s syndrome. N. Engl. J. Med 371, 2061–2071 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Groth KA et al. Prevalence, incidence, and age at diagnosis in Marfan Syndrome. Orphanet. J Rare. Dis 10, 153, doi: 10.1186/s13023-015-0369-8 [doi]; 10.1186/s13023-015-0369-8 [pii] (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beighton P et al. International Nosology of Heritable Disorders of Connective Tissue, Berlin, 1986. Am. J. Med. Genet 29, 581–594 (1988). [DOI] [PubMed] [Google Scholar]
- 24.Fuchs J Marfan syndrome and other systemic disorders with congenital ectopia lentis. A Danish national survey. Acta Paediatr 86, 947–952, doi: 10.1111/j.1651-2227.1997.tb15176.x [doi] (1997). [DOI] [PubMed] [Google Scholar]
- 25.De Paepe A, Devereux RB, Dietz HC, Hennekam RC & Pyeritz RE Revised diagnostic criteria for the Marfan syndrome. Am. J. Med. Genet 62, 417–426 (1996). [DOI] [PubMed] [Google Scholar]
- 26.Faivre L et al. Clinical homogeneity and genetic heterogeneity in Weill-Marchesani syndrome. Am. J. Med. Genet. A 123A, 204–207 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Le GC et al. Mutations in the TGFbeta binding-protein-like domain 5 of FBN1 are responsible for acromicric and geleophysic dysplasias. Am. J. Hum. Genet 89, 7–14 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ades LC, Holman KJ, Brett MS, Edwards MJ & Bennetts B Ectopia lentis phenotypes and the FBN1 gene. Am J Med. Genet A 126, 284–289 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Milewicz DM et al. A mutation in FBN1 disrupts profibrillin processing and results in isolated skeletal features of the Marfan syndrome. J. Clin. Invest 95, 2373–2378 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Milewicz DM et al. Fibrillin-1 (FBN1) mutations in patients with thoracic aortic aneurysms. Circulation 94, 2708–2711 (1996). [DOI] [PubMed] [Google Scholar]
- 31.Faivre L et al. Pathogenic FBN1 mutations in 146 adults not meeting clinical diagnostic criteria for Marfan syndrome: further delineation of type 1 fibrillinopathies and focus on patients with an isolated major criterion. Am. J. Med. Genet. A 149A, 854–860 (2009). [DOI] [PubMed] [Google Scholar]
- 32.Guo DC et al. Heritable Thoracic Aortic Disease Genes in Sporadic Aortic Dissection. J. Am. Coll. Cardiol 70, 2728–2730, doi:S0735–1097(17)40988–0 [pii]; 10.1016/j.jacc.2017.09.1094 [doi] (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mizuguchi T et al. Heterozygous TGFBR2 mutations in Marfan syndrome. Nat. Genet 36, 855–860 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.LeMaire SA et al. Severe aortic and arterial aneurysms associated with a TGFBR2 mutation. Nat. Clin. Pract. Cardiovasc. Med 4, 167–171 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peng Q, Deng Y, Yang Y & Liu H A novel fibrillin-1 gene missense mutation associated with neonatal Marfan syndrome: a case report and review of the mutation spectrum. BMC Pediatr 16, 60, doi: 10.1186/s12887-016-0598-6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boileau C et al. Autosomal dominant Marfan-like connective-tissue disorder with aortic dilation and skeletal anomalies not linked to the fibrillin genes [see comments]. Am. J. Hum. Genet 53, 46–54 (1993). [PMC free article] [PubMed] [Google Scholar]
- 37.Loeys BL et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat. Genet (2005). [DOI] [PubMed] [Google Scholar]
- 38.Furlong J, Kurczynski TW & Hennessy JR New Marfanoid syndrome with craniosynostosis. Am J Med. Genet 26, 599–604 (1987). [DOI] [PubMed] [Google Scholar]
- 39.MacFarlane EG et al. Lineage-specific events underlie aortic root aneurysm pathogenesis in Loeys-Dietz syndrome. J. Clin. Invest 129, 659–675, doi:123547 [pii]; 10.1172/JCI123547 [doi] (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van de Laar IM et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat. Genet 43, 121–126 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Rienhoff HY Jr. et al. A mutation in TGFB3 associated with a syndrome of low muscle mass, growth retardation, distal arthrogryposis and clinical features overlapping with Marfan and Loeys-Dietz syndrome. Am. J. Med. Genet. A 161A, 2040–2046, doi: 10.1002/ajmg.a.36056 [doi] (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bertoli-Avella AM et al. Mutations in a TGF-beta ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J. Am. Coll. Cardiol 65, 1324–1336, doi:S0735–1097(15)00403–9 [pii]; 10.1016/j.jacc.2015.01.040 [doi] (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sakai LY, Keene DR, Glanville RW & Bachinger HP Purification and partial characterization of fibrillin, a cysteine-rich structural component of connective tissue microfibrils. J. Biol. Chem 266, 14763–14770 (1991). [PubMed] [Google Scholar]
- 44.Corson GM, Chalberg SC, Dietz HC, Charbonneau NL & Sakai LY Fibrillin binds calcium and is coded by cDNAs that reveal a multidomain structure and alternatively spliced exons at the 5’ end. Genomics 17, 476–484 (1993). [DOI] [PubMed] [Google Scholar]
- 45.Lerner-Ellis JP et al. The spectrum of FBN1, TGFbetaR1, TGFbetaR2 and ACTA2 variants in 594 individuals with suspected Marfan Syndrome, Loeys-Dietz Syndrome or Thoracic Aortic Aneurysms and Dissections (TAAD). Mol Genet Metab 112, 171–176, doi: 10.1016/j.ymgme.2014.03.011 (2014). [DOI] [PubMed] [Google Scholar]
- 46.Milewicz DM & Duvic M Severe neonatal Marfan syndrome resulting from a de novo 3-bp insertion into the fibrillin gene on chromosome 15. Am. J Hum. Genet 54, 447–453 (1994). [PMC free article] [PubMed] [Google Scholar]
- 47.Putnam EA et al. Delineation of the Marfan phenotype associated with mutations in exons 23–32 of the FBN1 gene. Am. J. Med. Genet 62, 233–242 (1996). [DOI] [PubMed] [Google Scholar]
- 48.Faivre L et al. Clinical and mutation-type analysis from an international series of 198 probands with a pathogenic FBN1 exons 24–32 mutation. Eur. J Hum. Genet 17, 491–501, doi:ejhg2008207 [pii]; 10.1038/ejhg.2008.207 [doi] (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Faivre L et al. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am. J. Hum. Genet 81, 454–466 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schrijver I, Liu W, Brenn T, Furthmayr H & Francke U Cysteine substitutions in epidermal growth factor-like domains of fibrillin-1: distinct effects on biochemical and clinical phenotypes. Am. J. Hum. Genet 65, 1007–1020 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schrijver I et al. Premature Termination Mutations in FBN1: Distinct Effects on Differential Allelic Expression and on Protein and Clinical Phenotypes. Am. J. Hum. Genet 71, 223–237 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sengle G & Sakai LY The fibrillin microfibril scaffold: A niche for growth factors and mechanosensation? Matrix Biol 47, 3–12, doi: 10.1016/j.matbio.2015.05.002 (2015). [DOI] [PubMed] [Google Scholar]
- 53.Sengle G et al. Targeting of bone morphogenetic protein growth factor complexes to fibrillin. J Biol Chem 283, 13874–13888, doi: 10.1074/jbc.M707820200 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ramirez F & Dietz HC Fibrillin-rich microfibrils: Structural determinants of morphogenetic and homeostatic events. J. Cell Physiol 213, 326–330 (2007). [DOI] [PubMed] [Google Scholar]
- 55.Chen Y, Dabovic B, Annes JP & Rifkin DB Latent TGF-beta binding protein-3 (LTBP-3) requires binding to TGF-beta for secretion. FEBS Lett 517, 277–280, doi:S0014579302026480 [pii] (2002). [DOI] [PubMed] [Google Scholar]
- 56.Robertson IB et al. Latent TGF-beta-binding proteins. Matrix Biol 47, 44–53, doi:S0945–053X(15)00104–3 [pii]; 10.1016/j.matbio.2015.05.005 [doi] (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zilberberg L et al. Specificity of latent TGF-beta binding protein (LTBP) incorporation into matrix: role of fibrillins and fibronectin. J. Cell Physiol 227, 3828–3836, doi: 10.1002/jcp.24094 [doi] (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Isogai Z et al. Latent transforming growth factor beta-binding protein 1 interacts with fibrillin and is a microfibril-associated protein. J Biol. Chem 278, 2750–2757 (2003). [DOI] [PubMed] [Google Scholar]
- 59.Ono RN et al. Latent transforming growth factor beta-binding proteins and fibulins compete for fibrillin-1 and exhibit exquisite specificities in binding sites. J Biol Chem 284, 16872–16881, doi: 10.1074/jbc.M809348200 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wipff PJ, Rifkin DB, Meister JJ & Hinz B Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J. Cell Biol 179, 1311–1323 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Humphrey JD, Milewicz DM, Tellides G & Schwartz MA Cell biology. Dysfunctional mechanosensing in aneurysms. Science 344, 477–479 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Waters KM, Rooper LM, Guajardo A & Halushka MK Histopathologic differences partially distinguish syndromic aortic diseases. Cardiovasc Pathol 30, 6–11, doi: 10.1016/j.carpath.2017.05.008 (2017). [DOI] [PubMed] [Google Scholar]
- 63.Davis EC Smooth muscle cell to elastic lamina connections in developing mouse aorta. Role in aortic medial organization. Lab Invest 68, 89–99 (1993). [PubMed] [Google Scholar]
- 64.Milewicz DM et al. Genetic Basis of Thoracic Aortic Aneurysms and Dissections: Focus on Smooth Muscle Cell Contractile Dysfunction. Annu. Rev. Genomics Hum. Genet 9, 283–302 (2008). [DOI] [PubMed] [Google Scholar]
- 65.Humphrey JD, Schwartz MA, Tellides G & Milewicz DM Role of Mechanotransduction in Vascular Biology: Focus on Thoracic Aortic Aneurysms and Dissections. Circ. Res 116, 1448–1461, doi:CIRCRESAHA.114.304936 [pii]; 10.1161/CIRCRESAHA.114.304936 [doi] (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Milewicz DM et al. Altered Smooth Muscle Cell Force Generation as a Driver of Thoracic Aortic Aneurysms and Dissections. Arterioscler. Thromb. Vasc. Biol 37, 26–34, doi:ATVBAHA.116.303229 [pii]; 10.1161/ATVBAHA.116.303229 [doi] (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pereira L et al. Targetting of the gene encoding fibrillin-1 recapitulates the vascular aspect of Marfan syndrome. Nat. Genet 17, 218–222 (1997). [DOI] [PubMed] [Google Scholar]
- 68.Pereira L et al. Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-1. Proc. Natl. Acad. Sci. U. S. A 96, 3819–3823 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Judge DP et al. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J. Clin. Invest 114, 172–181 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Carta L et al. Fibrillins 1 and 2 perform partially overlapping functions during aortic development. J Biol Chem 281, 8016–8023, doi: 10.1074/jbc.M511599200 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bunton TE et al. Phenotypic alteration of vascular smooth muscle cells precedes elastolysis in a mouse model of marfan syndrome. Circ. Res 88, 37–43 (2001). [DOI] [PubMed] [Google Scholar]
- 72.Neptune ER et al. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat. Genet 33, 407–411 (2003). [DOI] [PubMed] [Google Scholar]
- 73.Lavoie P et al. Neutralization of transforming growth factor-beta attenuates hypertension and prevents renal injury in uremic rats. J. Hypertens 23, 1895–1903, doi:00004872–200510000-00022 [pii] (2005). [DOI] [PubMed] [Google Scholar]
- 74.Lim DS et al. Angiotensin II blockade reverses myocardial fibrosis in a transgenic mouse model of human hypertrophic cardiomyopathy. Circulation 103, 789–791 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gibbons GH, Pratt RE & Dzau VJ Vascular smooth muscle cell hypertrophy vs. hyperplasia. Autocrine transforming growth factor-beta 1 expression determines growth response to angiotensin II. J. Clin. Invest 90, 456–461 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Galatioto J et al. Cell Type-Specific Contributions of the Angiotensin II Type 1a Receptor to Aorta Homeostasis and Aneurysmal Disease-Brief Report. Arterioscler. Thromb. Vasc. Biol 38, 588–591, doi:ATVBAHA.117.310609 [pii]; 10.1161/ATVBAHA.117.310609 [doi] (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cook JR et al. Dimorphic effects of transforming growth factor-beta signaling during aortic aneurysm progression in mice suggest a combinatorial therapy for Marfan syndrome. Arterioscler. Thromb. Vasc. Biol 35, 911–917, doi:ATVBAHA.114.305150 [pii]; 10.1161/ATVBAHA.114.305150 [doi] (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lindsay ME et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat. Genet 44, 922–927 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li W et al. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J. Clin. Invest 124, 755–767 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wei H et al. Aortopathy in a Mouse Model of Marfan Syndrome Is Not Mediated by Altered Transforming Growth Factor beta Signaling. J. Am. Heart Assoc 6, doi:JAHA.116.004968 [pii]; 10.1161/JAHA.116.004968 [doi] (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Boileau C et al. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat. Genet 44, 916–921 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Inamoto S et al. TGFBR2 Mutations Alter Smooth Muscle Cell Phenotype and Predispose to Thoracic Aortic Aneurysms and Dissections. Cardiovasc. Res 88, 520–529 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.van d. L., I et al. Phenotypic spectrum of the SMAD3-related aneurysms-osteoarthritis syndrome. J Med. Genet 49, 47–57 (2012). [DOI] [PubMed] [Google Scholar]
- 84.Regalado ES et al. Exome sequencing identifies SMAD3 mutations as a cause of familial thoracic aortic aneurysm and dissection with intracranial and other arterial aneurysms. Circ. Res 109, 680–686, doi:CIRCRESAHA.111.248161 [pii]; 10.1161/CIRCRESAHA.111.248161 [doi] (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sellers SL et al. Inhibition of Marfan Syndrome Aortic Root Dilation by Losartan: Role of Angiotensin II Receptor Type 1-Independent Activation of Endothelial Function. Am. J. Pathol 188, 574–585, doi:S0002–9440(17)30549–7 [pii]; 10.1016/j.ajpath.2017.11.006 [doi] (2018). [DOI] [PubMed] [Google Scholar]
- 86.Milewicz DM, Prakash SK & Ramirez F Therapeutics Targeting Drivers of Thoracic Aortic Aneurysms and Acute Aortic Dissections: Insights from Predisposing Genes and Mouse Models. Annu. Rev. Med 68, 51–67, doi: 10.1146/annurev-med-100415-022956 [doi] (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pinard A, Jones GT & Milewicz DM Genetics of Thoracic and Abdominal Aortic Diseases. Circ. Res 124, 588–606, doi: 10.1161/CIRCRESAHA.118.312436 [doi] (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pedroza AJ et al. Single-Cell Transcriptomic Profiling of Vascular Smooth Muscle Cell Phenotype Modulation in Marfan Syndrome Aortic Aneurysm. Arterioscler Thromb Vasc Biol 40, 2195–2211, doi: 10.1161/ATVBAHA.120.314670 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cook JR et al. Abnormal muscle mechanosignaling triggers cardiomyopathy in mice with Marfan syndrome. J. Clin. Invest 124, 1329–1339 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rouf R et al. Nonmyocyte ERK1/2 signaling contributes to load-induced cardiomyopathy in Marfan mice. JCI. Insight 2, doi:91588 [pii]; 10.1172/jci.insight.91588 [doi] (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wisler JW et al. The role of beta-arrestin2-dependent signaling in thoracic aortic aneurysm formation in a murine model of Marfan syndrome. Am. J. Physiol Heart Circ. Physiol 309, H1516–H1527, doi:ajpheart.00291.2015 [pii]; 10.1152/ajpheart.00291.2015 [doi] (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Crosas-Molist E et al. Vascular smooth muscle cell phenotypic changes in patients with Marfan syndrome. Arterioscler. Thromb. Vasc. Biol 35, 960–972, doi:ATVBAHA.114.304412 [pii]; 10.1161/ATVBAHA.114.304412 [doi] (2015). [DOI] [PubMed] [Google Scholar]
- 93.Jimenez-Altayo F et al. Redox stress in Marfan syndrome: Dissecting the role of the NADPH oxidase NOX4 in aortic aneurysm. Free Radic. Biol. Med 118, 44–58, doi:S0891–5849(18)30078–9 [pii]; 10.1016/j.freeradbiomed.2018.02.023 [doi] (2018). [DOI] [PubMed] [Google Scholar]
- 94.Yang HH, van BC & Chung AW Vasomotor dysfunction in the thoracic aorta of Marfan syndrome is associated with accumulation of oxidative stress. Vascul. Pharmacol 52, 37–45 (2010). [DOI] [PubMed] [Google Scholar]
- 95.Chen J et al. Loss of Smooth Muscle alpha-Actin Leads to NF-kappaB-Dependent Increased Sensitivity to Angiotensin II in Smooth Muscle Cells and Aortic Enlargement. Circ. Res 120, 1903–1915, doi:CIRCRESAHA.117.310563 [pii]; 10.1161/CIRCRESAHA.117.310563 [doi] (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Carta L et al. MAPKp38 is an early determinant of promiscuous Smad2/3 signaling in the aortas of fibrillin-1 (Fbn1) null mice. J. Biol. Chem (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Granata A et al. An iPSC-derived vascular model of Marfan syndrome identifies key mediators of smooth muscle cell death. Nat. Genet 49, 97–109, doi:ng.3723 [pii]; 10.1038/ng.3723 [doi] (2017). [DOI] [PubMed] [Google Scholar]
- 98.Chung AW, Yang HH, Radomski MW & van BC Long-term doxycycline is more effective than atenolol to prevent thoracic aortic aneurysm in marfan syndrome through the inhibition of matrix metalloproteinase-2 and −9. Circ. Res 102, e73–e85 (2008). [DOI] [PubMed] [Google Scholar]
- 99.Emrich FC et al. Enhanced caspase activity contributes to aortic wall remodeling and early aneurysm development in a murine model of Marfan syndrome. Arterioscler. Thromb. Vasc. Biol 35, 146–154 (2015). [DOI] [PubMed] [Google Scholar]
- 100.Merk DR et al. miR-29b Participates in Early Aneurysm Development in Marfan Syndrome. Circ. Res 110, 312–324 (2012). [DOI] [PubMed] [Google Scholar]
- 101.Mas-Stachurska A et al. Cardiovascular Benefits of Moderate Exercise Training in Marfan Syndrome: Insights From an Animal Model. J. Am. Heart Assoc 6, doi:JAHA.117.006438 [pii]; 10.1161/JAHA.117.006438 [doi] (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Milewicz DM & Ramirez F Therapies for Thoracic Aortic Aneurysms and Acute Aortic Dissections. Arterioscler. Thromb. Vasc. Biol 39, 126–136, doi: 10.1161/ATVBAHA.118.310956 [doi] (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.LeMaire SA et al. Genome-wide association study identifies a susceptibility locus for thoracic aortic aneurysms and aortic dissections spanning FBN1 at 15q21.1. Nat. Genet 43, 996–1000 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Smaldone S et al. Fibrillin-1 Regulates Skeletal Stem Cell Differentiation by Modulating TGFbeta Activity Within the Marrow Niche. J Bone Miner Res 31, 86–97, doi: 10.1002/jbmr.2598 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Smaldone S & Ramirez F Fibrillin microfibrils in bone physiology. Matrix Biol 52–54, 191–197, doi: 10.1016/j.matbio.2015.09.004 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lima BL et al. A new mouse model for marfan syndrome presents phenotypic variability associated with the genetic background and overall levels of Fbn1 expression. PLoS. ONE 5, e14136 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Beene LC et al. Nonselective assembly of fibrillin 1 and fibrillin 2 in the rodent ocular zonule and in cultured cells: implications for Marfan syndrome. Invest Ophthalmol Vis Sci 54, 8337–8344, doi: 10.1167/iovs.13-13121 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mir S, Wheatley HM, Hussels IE, Whittum-Hudson JA & Traboulsi EI A comparative histologic study of the fibrillin microfibrillar system in the lens capsule of normal subjects and subjects with Marfan syndrome. Invest Ophthalmol Vis Sci 39, 84–93 (1998). [PubMed] [Google Scholar]
- 109.Wheatley HM et al. Immunohistochemical localization of fibrillin in human ocular tissues. Relevance to the Marfan syndrome. Arch Ophthalmol 113, 103–109, doi: 10.1001/archopht.1995.01100010105028 (1995). [DOI] [PubMed] [Google Scholar]
- 110.Hanlon SD, Behzad AR, Sakai LY & Burns AR Corneal stroma microfibrils. Exp Eye Res 132, 198–207, doi: 10.1016/j.exer.2015.01.014 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jones W, Rodriguez J & Bassnett S Targeted deletion of fibrillin-1 in the mouse eye results in ectopia lentis and other ocular phenotypes associated with Marfan syndrome. Dis Model Mech 12, doi: 10.1242/dmm.037283 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tan L et al. FBN1 mutations largely contribute to sporadic non-syndromic aortic dissection. Hum Mol Genet 26, 4814–4822, doi: 10.1093/hmg/ddx360 (2017). [DOI] [PubMed] [Google Scholar]
- 113.Akutsu K et al. Characteristics in phenotypic manifestations of genetically proved Marfan syndrome in a Japanese population. Am J Cardiol 103, 1146–1148, doi: 10.1016/j.amjcard.2008.12.037 (2009). [DOI] [PubMed] [Google Scholar]
- 114.Villamizar C et al. Paucity of skeletal manifestations in hispanic families with FBN1 mutations. Eur. J. Med. Genet 53, 80–84 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Faivre L et al. The new Ghent criteria for Marfan syndrome: what do they change? Clin Genet 81, 433–442, doi: 10.1111/j.1399-0004.2011.01703.x (2012). [DOI] [PubMed] [Google Scholar]
- 116.Bombardieri E et al. Marfan syndrome and related connective tissue disorders in the current era in Switzerland in 103 patients: medical and surgical management and impact of genetic testing. Swiss Med Wkly 150, w20189, doi: 10.4414/smw.2020.20189 (2020). [DOI] [PubMed] [Google Scholar]
- 117.Roman MJ et al. Associations of Age and Sex With Marfan Phenotype: The National Heart, Lung, and Blood Institute GenTAC (Genetically Triggered Thoracic Aortic Aneurysms and Cardiovascular Conditions) Registry. Circ. Cardiovasc. Genet 10, doi:CIRCGENETICS.116.001647 [pii]; 10.1161/CIRCGENETICS.116.001647 [doi] (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ladouceur M et al. Effect of beta-blockade on ascending aortic dilatation in children with the Marfan syndrome. Am. J. Cardiol 99, 406–409 (2007). [DOI] [PubMed] [Google Scholar]
- 119.Hascoet S et al. Incidence of cardiovascular events and risk markers in a prospective study of children diagnosed with Marfan syndrome. Arch. Cardiovasc. Dis 113, 40–49, doi:S1875–2136(19)30191–3 [pii]; 10.1016/j.acvd.2019.09.010 [doi] (2020). [DOI] [PubMed] [Google Scholar]
- 120.Wozniak-Mielczarek L et al. Differences in Cardiovascular Manifestation of Marfan Syndrome Between Children and Adults. Pediatr. Cardiol 40, 393–403, doi: 10.1007/s00246-018-2025-2 [doi]; 10.1007/s00246-018-2025-2 [pii] (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Detaint D et al. Aortic dilatation patterns and rates in adults with bicuspid aortic valves: a comparative study with Marfan syndrome and degenerative aortopathy. Heart 100, 126–134, doi:heartjnl-2013–304920 [pii]; 10.1136/heartjnl-2013-304920 [doi] (2014). [DOI] [PubMed] [Google Scholar]
- 122.Guala A et al. Proximal aorta longitudinal strain predicts aortic root dilation rate and aortic events in Marfan syndrome. Eur. Heart J 40, 2047–2055, doi:5448881 [pii]; 10.1093/eurheartj/ehz191 [doi] (2019). [DOI] [PubMed] [Google Scholar]
- 123.Groenink M et al. Losartan reduces aortic dilatation rate in adults with Marfan syndrome: a randomized controlled trial. Eur. Heart J 34, 3491–3500, doi:eht334 [pii]; 10.1093/eurheartj/eht334 [doi] (2013). [DOI] [PubMed] [Google Scholar]
- 124.Mullen M et al. Irbesartan in Marfan syndrome (AIMS): a double-blind, placebo-controlled randomised trial. Lancet 394, 2263–2270, doi:S0140–6736(19)32518–8 [pii]; 10.1016/S0140-6736(19)32518-8 [doi] (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Teixido-Tura G et al. Losartan Versus Atenolol for Prevention of Aortic Dilation in Patients With Marfan Syndrome. J Am Coll Cardiol 72, 1613–1618, doi:S0735–1097(18)35818–2 [pii]; 10.1016/j.jacc.2018.07.052 [doi] (2018). [DOI] [PubMed] [Google Scholar]
- 126.Turkbey EB et al. Determinants and normal values of ascending aortic diameter by age, gender, and race/ethnicity in the Multi-Ethnic Study of Atherosclerosis (MESA). J Magn Reson Imaging 39, 360–368, doi: 10.1002/jmri.24183 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jondeau G et al. Aortic event rate in the Marfan population: a cohort study. Circulation 125, 226–232 (2012). [DOI] [PubMed] [Google Scholar]
- 128.Erbel R et al. 2014 ESC Guidelines on the diagnosis and treatment of aortic diseases: Document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. The Task Force for the Diagnosis and Treatment of Aortic Diseases of the European Society of Cardiology (ESC). Eur. Heart J 35, 2873–2926, doi:ehu281 [pii]; 10.1093/eurheartj/ehu281 [doi] (2014). [DOI] [PubMed] [Google Scholar]
- 129.Hiratzka LF et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with Thoracic Aortic Disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation 121, e266–e369 (2010). [DOI] [PubMed] [Google Scholar]
- 130.Morris SA et al. Increased vertebral artery tortuosity index is associated with adverse outcomes in children and young adults with connective tissue disorders. Circulation 124, 388–396 (2011). [DOI] [PubMed] [Google Scholar]
- 131.Franken R et al. Increased aortic tortuosity indicates a more severe aortic phenotype in adults with Marfan syndrome. Int. J. Cardiol 194, 7–12, doi:S0167–5273(15)01129–8 [pii]; 10.1016/j.ijcard.2015.05.072 [doi] (2015). [DOI] [PubMed] [Google Scholar]
- 132.Mortensen K et al. Augmentation index relates to progression of aortic disease in adults with Marfan syndrome. Am J Hypertens 22, 971–979, doi: 10.1038/ajh.2009.115 (2009). [DOI] [PubMed] [Google Scholar]
- 133.Selamet Tierney ES et al. Influence of Aortic Stiffness on Aortic-Root Growth Rate and Outcome in Patients With the Marfan Syndrome. Am. J. Cardiol 121, 1094–1101, doi:S0002–9149(18)30158–9 [pii]; 10.1016/j.amjcard.2018.01.016 [doi] (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jondeau G et al. International Registry of Patients Carrying TGFBR1 or TGFBR2 Mutations: Results of the MAC (Montalcino Aortic Consortium). Circ. Cardiovasc. Genet 9, 548–558, doi:CIRCGENETICS.116.001485 [pii]; 10.1161/CIRCGENETICS.116.001485 [doi] (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Teixido-Tura G et al. Aortic biomechanics by magnetic resonance: early markers of aortic disease in Marfan syndrome regardless of aortic dilatation? Int. J Cardiol 171, 56–61, doi:S0167–5273(13)02062–7 [pii]; 10.1016/j.ijcard.2013.11.044 [doi] (2014). [DOI] [PubMed] [Google Scholar]
- 136.Franken R et al. Relationship between fibrillin-1 genotype and severity of cardiovascular involvement in Marfan syndrome. Heart, doi:heartjnl-2016–310631 [pii]; 10.1136/heartjnl-2016-310631 [doi] (2017). [DOI] [PubMed] [Google Scholar]
- 137.Takeda N et al. Impact of Pathogenic FBN1 Variant Types on the Progression of Aortic Disease in Patients With Marfan Syndrome. Circ. Genom. Precis. Med 11, e002058, doi:CIRCGEN.117.002058 [pii]; 10.1161/CIRCGEN.117.002058 [doi] (2018). [DOI] [PubMed] [Google Scholar]
- 138.Roman MJ, Rosen SE, Kramer-Fox R & Devereux RB Prognostic significance of the pattern of aortic root dilation in the Marfan syndrome. J. Am. Coll. Cardiol 22, 1470–1476 (1993). [DOI] [PubMed] [Google Scholar]
- 139.Attias D et al. Comparison of clinical presentations and outcomes between patients with TGFBR2 and FBN1 mutations in Marfan syndrome and related disorders. Circulation 120, 2541–2549 (2009). [DOI] [PubMed] [Google Scholar]
- 140.LeMaire S & al., e. Spectrum of aortic operations in 300 patients with confirmed or suspected Marfan syndrome. Ann. Thorac. Surg (2006). [DOI] [PubMed] [Google Scholar]
- 141.Hagerty T, Geraghty P & Braverman AC Abdominal Aortic Aneurysm in Marfan Syndrome. Ann Vasc Surg 40, 294 e291–294 e296, doi: 10.1016/j.avsg.2016.07.067 (2017). [DOI] [PubMed] [Google Scholar]
- 142.Prakash SK, Haden-Pinneri K & Milewicz DM Susceptibility to acute thoracic aortic dissections in patients dying outside the hospital: An autopsy study. Am. Heart J 162, 474–479 (2011). [DOI] [PubMed] [Google Scholar]
- 143.Reutersberg B et al. Hospital Incidence and In-Hospital Mortality of Surgically and Interventionally Treated Aortic Dissections: Secondary Data Analysis of the Nationwide German Diagnosis-Related Group Statistics From 2006 to 2014. J Am Heart Assoc 8, e011402, doi: 10.1161/JAHA.118.011402 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Brouwer C et al. Progressive Pulmonary Artery Dilatation is Associated with Type B Aortic Dissection in Patients with Marfan Syndrome. J Clin. Med 8, doi:jcm8111848 [pii]; 10.3390/jcm8111848 [doi] (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mimoun L et al. Dissection in Marfan syndrome: the importance of the descending aorta. Eur. Heart J 32, 443–449, doi:ehq434 [pii]; 10.1093/eurheartj/ehq434 [doi] (2011). [DOI] [PubMed] [Google Scholar]
- 146.Baumgartner H et al. 2017 ESC/EACTS Guidelines for the management of valvular heart disease. Eur. Heart J 38, 2739–2791, doi:4095039 [pii]; 10.1093/eurheartj/ehx391 [doi] (2017). [DOI] [PubMed] [Google Scholar]
- 147.Pyeritz RE Marfan syndrome: current and future clinical and genetic management of cardiovascular manifestations. Semin. Thorac. Cardiovasc. Surg 5, 11–16 (1993). [PubMed] [Google Scholar]
- 148.Muhlstadt K et al. Case-matched Comparison of Cardiovascular Outcome in Loeys-Dietz Syndrome versus Marfan Syndrome. J Clin. Med 8, doi:jcm8122079 [pii]; 10.3390/jcm8122079 [doi] (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Mueller GC et al. Impact of age and gender on cardiac pathology in children and adolescents with Marfan syndrome. Pediatr. Cardiol 34, 991–998, doi: 10.1007/s00246-012-0593-0 [doi] (2013). [DOI] [PubMed] [Google Scholar]
- 150.Selamet Tierney ES et al. Echocardiographic methods, quality review, and measurement accuracy in a randomized multicenter clinical trial of Marfan syndrome. J Am Soc. Echocardiogr 26, 657–666, doi:S0894–7317(13)00142–9 [pii]; 10.1016/j.echo.2013.02.018 [doi] (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lacro RV et al. Characteristics of children and young adults with Marfan syndrome and aortic root dilation in a randomized trial comparing atenolol and losartan therapy. Am Heart J 165, 828–835, doi:S0002–8703(13)00146–4 [pii]; 10.1016/j.ahj.2013.02.019 [doi] (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Seo YJ et al. Infantile Marfan syndrome in a Korean tertiary referral center. Korean J. Pediatr 59, 59–64, doi: 10.3345/kjp.2016.59.2.59 [doi] (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Rybczynski M et al. Frequency of sleep apnea in adults with the Marfan syndrome. Am. J. Cardiol 105, 1836–1841 (2010). [DOI] [PubMed] [Google Scholar]
- 154.Helder MR et al. Management of mitral regurgitation in Marfan syndrome: Outcomes of valve repair versus replacement and comparison with myxomatous mitral valve disease. J Thorac. Cardiovasc. Surg 148, 1020–1024, doi:S0022–5223(14)00844–7 [pii]; 10.1016/j.jtcvs.2014.06.046 [doi] (2014). [DOI] [PubMed] [Google Scholar]
- 155.Chivulescu M et al. Mitral annulus disjunction is associated with adverse outcome in Marfan and Loeys-Dietz syndromes. Eur Heart J Cardiovasc Imaging, doi: 10.1093/ehjci/jeaa324 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Stark VC et al. The Pulmonary Artery in Pediatric Patients with Marfan Syndrome: An Underestimated Aspect of the Disease. Pediatr. Cardiol 39, 1194–1199, doi: 10.1007/s00246-018-1880-1 [doi]; 10.1007/s00246-018-1880-1 [pii] (2018). [DOI] [PubMed] [Google Scholar]
- 157.Nollen GJ et al. Pulmonary artery root dilatation in Marfan syndrome: quantitative assessment of an unknown criterion. Heart 87, 470–471, doi: 10.1136/heart.87.5.470 [doi] (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lundby R, Rand-Hendriksen S, Hald JK, Pripp AH & Smith HJ The pulmonary artery in patients with Marfan syndrome: a cross-sectional study. Genet. Med 14, 922–927, doi:gim201282 [pii]; 10.1038/gim.2012.82 [doi] (2012). [DOI] [PubMed] [Google Scholar]
- 159.Kinori M et al. Biometry Characteristics in Adults and Children With Marfan Syndrome: From the Marfan Eye Consortium of Chicago. Am J Ophthalmol 177, 144–149, doi: 10.1016/j.ajo.2017.02.022 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Gott VL et al. Replacement of the aortic root in patients with Marfan’s syndrome. New England Journal of Medicine 340, 1307–1313 (1999). [DOI] [PubMed] [Google Scholar]
- 161.Diller GP et al. Survival Prospects and Circumstances of Death in Contemporary Adult Congenital Heart Disease Patients Under Follow-Up at a Large Tertiary Centre. Circulation 132, 2118–2125, doi:CIRCULATIONAHA.115.017202 [pii]; 10.1161/CIRCULATIONAHA.115.017202 [doi] (2015). [DOI] [PubMed] [Google Scholar]
- 162.Alpendurada F et al. Evidence for Marfan cardiomyopathy. Eur. J Heart Fail 12, 1085–1091, doi:hfq127 [pii]; 10.1093/eurjhf/hfq127 [doi] (2010). [DOI] [PubMed] [Google Scholar]
- 163.Knosalla C et al. Orthotopic heart transplantation in patients with Marfan syndrome. Ann. Thorac. Surg 83, 1691–1695, doi:S0003–4975(07)00141–5 [pii]; 10.1016/j.athoracsur.2007.01.018 [doi] (2007). [DOI] [PubMed] [Google Scholar]
- 164.Audenaert T, De PM, Francois K & De BJ Type B aortic dissection triggered by heart transplantation in a patient with Marfan syndrome. BMJ Case. Rep 2015, doi:bcr-2015–211138 [pii]; 10.1136/bcr-2015-211138 [doi] (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Chen S, Fagan LF, Nouri S & Donahoe JL Ventricular dysrhythmias in children with Marfan’s syndrome. Am J Dis. Child 139, 273–276, doi: 10.1001/archpedi.1985.02140050067024 [doi] (1985). [DOI] [PubMed] [Google Scholar]
- 166.Aydin A et al. Observational cohort study of ventricular arrhythmia in adults with Marfan syndrome caused by FBN1 mutations. PLoS. ONE 8, e81281, doi: 10.1371/journal.pone.0081281 [doi];PONE-D-13–33425 [pii] (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Hoffmann BA et al. Prospective risk stratification of sudden cardiac death in Marfan’s syndrome. Int. J Cardiol 167, 2539–2545, doi:S0167–5273(12)00811-X [pii]; 10.1016/j.ijcard.2012.06.036 [doi] (2013). [DOI] [PubMed] [Google Scholar]
- 168.Yetman AT, Bornemeier RA & McCrindle BW Long-term outcome in patients with Marfan syndrome: is aortic dissection the only cause of sudden death? J Am Coll Cardiol 41, 329–332, doi:S0735109702026992 [pii]; 10.1016/s0735-1097(02)02699-2 [doi] (2003). [DOI] [PubMed] [Google Scholar]
- 169.Thompson ME et al. Differential regulation of chromogranin B/secretogranin I and secretogranin II by forskolin in PC12 cells. Brain Res Mol Brain Res 12, 195–202, doi: 10.1016/0169-328x(92)90084-o (1992). [DOI] [PubMed] [Google Scholar]
- 170.Sharma T et al. Retinal detachment in Marfan syndrome: clinical characteristics and surgical outcome. Retina 22, 423–428, doi: 10.1097/00006982-200208000-00005 (2002). [DOI] [PubMed] [Google Scholar]
- 171.Maumenee IH The eye in the Marfan syndrome. Trans. Am. Ophthalmol. Soc 79, 684–733 (1981). [PMC free article] [PubMed] [Google Scholar]
- 172.Chow K, Pyeritz RE & Litt HI Abdominal visceral findings in patients with Marfan syndrome. Genet. Med 9, 208–212 (2007). [DOI] [PubMed] [Google Scholar]
- 173.Muino-Mosquera L et al. Sleep apnea and the impact on cardiovascular risk in patients with Marfan syndrome. Mol Genet Genomic Med 7, e805, doi: 10.1002/mgg3.805 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.von KY et al. Features of Marfan syndrome not listed in the Ghent nosology - the dark side of the disease. Expert. Rev. Cardiovasc. Ther 17, 883–915, doi: 10.1080/14779072.2019.1704625 [doi] (2019). [DOI] [PubMed] [Google Scholar]
- 175.Speed TJ et al. Characterization of pain, disability, and psychological burden in Marfan syndrome. Am J Med Genet A 173, 315–323, doi: 10.1002/ajmg.a.38051 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Handisides JC et al. Health-Related Quality of Life in Children and Young Adults with Marfan Syndrome. J Pediatr 204, 250–255 e251, doi: 10.1016/j.jpeds.2018.08.061 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Warnink-Kavelaars J et al. Marfan syndrome in adolescence: adolescents’ perspectives on (physical) functioning, disability, contextual factors and support needs. Eur J Pediatr 178, 1883–1892, doi: 10.1007/s00431-019-03469-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Velvin G, Bathen T, Rand-Hendriksen S & Geirdal AO Systematic review of the psychosocial aspects of living with Marfan syndrome. Clin Genet 87, 109–116, doi: 10.1111/cge.12422 (2015). [DOI] [PubMed] [Google Scholar]
- 179.Guo DC et al. An FBN1 pseudoexon mutation in a patient with Marfan syndrome: confirmation of cryptic mutations leading to disease. J Hum. Genet 53, 1007–1011 (2008). [DOI] [PubMed] [Google Scholar]
- 180.Hilhorst-Hofstee Y et al. The clinical spectrum of missense mutations of the first aspartic acid of cbEGF-like domains in fibrillin-1 including a recessive family. Hum. Mutat 31, E1915–E1927, doi: 10.1002/humu.21372 [doi] (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Arnaud P et al. Homozygous and compound heterozygous mutations in the FBN1 gene: unexpected findings in molecular diagnosis of Marfan syndrome. J Med. Genet 54, 100–103, doi:jmedgenet-2016–103996 [pii]; 10.1136/jmedgenet-2016-103996 [doi] (2017). [DOI] [PubMed] [Google Scholar]
- 182.Goldstein SA et al. Multimodality imaging of diseases of the thoracic aorta in adults: from the American Society of Echocardiography and the European Association of Cardiovascular Imaging: endorsed by the Society of Cardiovascular Computed Tomography and Society for Cardiovascular Magnetic Resonance. J. Am. Soc. Echocardiogr 28, 119–182, doi:S0894–7317(14)00859–1 [pii]; 10.1016/j.echo.2014.11.015 [doi] (2015). [DOI] [PubMed] [Google Scholar]
- 183.Brown OR et al. Aortic root dilatation and mitral valve prolapse in Marfan’s syndrome: an ECHOCARDIOgraphic study. Circulation 52, 651–657, doi: 10.1161/01.cir.52.4.651 (1975). [DOI] [PubMed] [Google Scholar]
- 184.Campens L et al. Reference values for echocardiographic assessment of the diameter of the aortic root and ascending aorta spanning all age categories. Am. J. Cardiol 114, 914–920, doi:S0002–9149(14)01371-X [pii]; 10.1016/j.amjcard.2014.06.024 [doi] (2014). [DOI] [PubMed] [Google Scholar]
- 185.Devereux RB et al. Normal limits in relation to age, body size and gender of two-dimensional echocardiographic aortic root dimensions in persons >/=15 years of age. Am. J. Cardiol 110, 1189–1194 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Saura D et al. Two-dimensional transthoracic echocardiographic normal reference ranges for proximal aorta dimensions: results from the EACVI NORRE study. Eur. Heart J Cardiovasc. Imaging 18, 167–179, doi:jew053 [pii]; 10.1093/ehjci/jew053 [doi] (2017). [DOI] [PubMed] [Google Scholar]
- 187.Muraru D et al. Ascending aorta diameters measured by echocardiography using both leading edge-to-leading edge and inner edge-to-inner edge conventions in healthy volunteers. Eur. Heart J Cardiovasc. Imaging 15, 415–422, doi:jet173 [pii]; 10.1093/ehjci/jet173 [doi] (2014). [DOI] [PubMed] [Google Scholar]
- 188.Bossone E et al. Normal Values and Differences in Ascending Aortic Diameter in a Healthy Population of Adults as Measured by the Pediatric versus Adult American Society of Echocardiography Guidelines. J Am Soc. Echocardiogr 29, 166–172, doi:S0894–7317(15)00727–0 [pii]; 10.1016/j.echo.2015.09.010 [doi] (2016). [DOI] [PubMed] [Google Scholar]
- 189.Lopez L et al. Recommendations for quantification methods during the performance of a pediatric echocardiogram: a report from the Pediatric Measurements Writing Group of the American Society of Echocardiography Pediatric and Congenital Heart Disease Council. J Am Soc. Echocardiogr 23, 465–495, doi:S0894–7317(10)00266-X [pii]; 10.1016/j.echo.2010.03.019 [doi] (2010). [DOI] [PubMed] [Google Scholar]
- 190.Lopez L et al. Pediatric Heart Network Echocardiographic Z Scores: Comparison with Other Published Models. J Am Soc Echocardiogr 34, 185–192, doi: 10.1016/j.echo.2020.09.019 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Rutten DWE et al. Comparability of different Z-score equations for aortic root dimensions in children with Marfan syndrome. Cardiol Young, 1–7, doi: 10.1017/S1047951121001311 (2021). [DOI] [PubMed] [Google Scholar]
- 192.Veldhoen S et al. Exact monitoring of aortic diameters in Marfan patients without gadolinium contrast: intraindividual comparison of 2D SSFP imaging with 3D CE-MRA and echocardiography. Eur. Radiol 25, 872–882, doi: 10.1007/s00330-014-3457-6 [doi] (2015). [DOI] [PubMed] [Google Scholar]
- 193.Hagan PG et al. The International Registry of Acute Aortic Dissection (IRAD): new insights into an old disease. JAMA 283, 897–903 (2000). [DOI] [PubMed] [Google Scholar]
- 194.Evangelista A et al. Imaging modalities for the early diagnosis of acute aortic syndrome. Nat. Rev. Cardiol 10, 477–486, doi:nrcardio.2013.92 [pii]; 10.1038/nrcardio.2013.92 [doi] (2013). [DOI] [PubMed] [Google Scholar]
- 195.Freeman LA et al. CT and MRI assessment of the aortic root and ascending aorta. AJR Am J Roentgenol 200, W581–W592, doi: 10.2214/AJR.12.9531 [doi] (2013). [DOI] [PubMed] [Google Scholar]
- 196.Rodriguez-Palomares JF et al. Multimodality Assessment of Ascending Aortic Diameters: Comparison of Different Measurement Methods. J. Am. Soc. Echocardiogr 29, 819–826, doi:S0894–7317(16)30045–1 [pii]; 10.1016/j.echo.2016.04.006 [doi] (2016). [DOI] [PubMed] [Google Scholar]
- 197.Burman ED, Keegan J & Kilner PJ Aortic root measurement by cardiovascular magnetic resonance: specification of planes and lines of measurement and corresponding normal values. Circ. Cardiovasc. Imaging 1, 104–113, doi:CIRCIMAGING.108.768911 [pii]; 10.1161/CIRCIMAGING.108.768911 [doi] (2008). [DOI] [PubMed] [Google Scholar]
- 198.Amsallem M et al. Comparative assessment of ascending aortic aneurysms in Marfan patients using ECG-gated computerized tomographic angiography versus trans-thoracic echocardiography. Int. J Cardiol 184, 22–27, doi:S0167–5273(15)00113–8 [pii]; 10.1016/j.ijcard.2015.01.086 [doi] (2015). [DOI] [PubMed] [Google Scholar]
- 199.Mendoza DD et al. Impact of image analysis methodology on diagnostic and surgical classification of patients with thoracic aortic aneurysms. Ann. Thorac. Surg 92, 904–912 (2011). [DOI] [PubMed] [Google Scholar]
- 200.Maron BJ et al. Recommendations and considerations related to preparticipation screening for cardiovascular abnormalities in competitive athletes: 2007 update: a scientific statement from the American Heart Association Council on Nutrition, Physical Activity, and Metabolism: endorsed by the American College of Cardiology Foundation. Circulation 115, 1643–1455, doi: 10.1161/CIRCULATIONAHA.107.181423 (2007). [DOI] [PubMed] [Google Scholar]
- 201.Harris KM, Sponsel A, Hutter AM Jr. & Maron BJ Brief communication: Cardiovascular screening practices of major North American professional sports teams. Ann Intern Med 145, 507–511, doi: 10.7326/0003-4819-145-7-200610030-00008 (2006). [DOI] [PubMed] [Google Scholar]
- 202.Scherer LR, Arn PH, Dressel DA, Pyeritz RM & Haller JA Jr. Surgical management of children and young adults with Marfan syndrome and pectus excavatum. J Pediatr Surg 23, 1169–1172, doi: 10.1016/s0022-3468(88)80335-x (1988). [DOI] [PubMed] [Google Scholar]
- 203.Redlinger RE Jr. et al. Minimally invasive repair of pectus excavatum in patients with Marfan syndrome and marfanoid features. J Pediatr Surg 45, 193–199, doi: 10.1016/j.jpedsurg.2009.10.037 (2010). [DOI] [PubMed] [Google Scholar]
- 204.Erkula G, Jones KB, Sponseller PD, Dietz HC & Pyeritz RE Growth and maturation in Marfan syndrome. Am J Med Genet 109, 100–115, doi: 10.1002/ajmg.10312 (2002). [DOI] [PubMed] [Google Scholar]
- 205.Sponseller PD, Hobbs W, Riley LH 3rd & Pyeritz RE The thoracolumbar spine in Marfan syndrome. J Bone Joint Surg Am 77, 867–876, doi: 10.2106/00004623-199506000-00007 (1995). [DOI] [PubMed] [Google Scholar]
- 206.Loewenstein A, Barequet IS, De Juan E Jr. & Maumenee IH Retinal detachment in Marfan syndrome. Retina 20, 358–363, doi: 10.1097/00006982-200007000-00006 (2000). [DOI] [PubMed] [Google Scholar]
- 207.Xu W et al. Comparative data on SD-OCT for the retinal nerve fiber layer and retinal macular thickness in a large cohort with Marfan syndrome. Ophthalmic Genet 38, 34–38, doi: 10.1080/13816810.2016.1275017 (2017). [DOI] [PubMed] [Google Scholar]
- 208.Rahmani S, Lyon AT, Fawzi AA, Maumenee IH & Mets MB Retinal Disease in Marfan Syndrome: From the Marfan Eye Consortium of Chicago. Ophthalmic Surg Lasers Imaging Retina 46, 936–941, doi: 10.3928/23258160-20151008-06 (2015). [DOI] [PubMed] [Google Scholar]
- 209.Schou S, Holmstrup P, Hjorting-Hansen E & Lang NP Plaque-induced marginal tissue reactions of osseointegrated oral implants: a review of the literature. Clin Oral Implants Res 3, 149–161, doi: 10.1034/j.1600-0501.1992.030401.x (1992). [DOI] [PubMed] [Google Scholar]
- 210.Bard LA Genetic counseling of families with Marfan syndrome and other disorders showing a Marfanoid body habitus. Ophthalmology 86, 1764–1793, doi: 10.1016/s0161-6420(79)35344-1 (1979). [DOI] [PubMed] [Google Scholar]
- 211.Braverman AC, Harris KM, Kovacs RJ & Maron BJ Eligibility and Disqualification Recommendations for Competitive Athletes With Cardiovascular Abnormalities: Task Force 7: Aortic Diseases, Including Marfan Syndrome: A Scientific Statement From the American Heart Association and American College of Cardiology. J Am Coll Cardiol 66, 2398–2405, doi:S0735–1097(15)06575–4 [pii]; 10.1016/j.jacc.2015.09.039 [doi] (2015). [DOI] [PubMed] [Google Scholar]
- 212.Vanem TT et al. Survival, causes of death, and cardiovascular events in patients with Marfan syndrome. Mol. Genet. Genomic. Med 6, 1114–1123, doi: 10.1002/mgg3.489 [doi] (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Shores J, Berger KR, Murphy EA & Pyeritz RE Progression of aortic dilatation and the benefit of long-term beta- adrenergic blockade in Marfan’s syndrome [see comments]. N. Engl. J. Med 330, 1335–1341 (1994). [DOI] [PubMed] [Google Scholar]
- 214.Elefteriades JA & Farkas EA Thoracic aortic aneurysm clinically pertinent controversies and uncertainties. J. Am. Coll. Cardiol 55, 841–857 (2010). [DOI] [PubMed] [Google Scholar]
- 215.Beaven DW & Murphy EA Dissecting aneurysm during methonium therapy; a report on nine cases treated for hypertension. Br Med J 1, 77–80, doi: 10.1136/bmj.1.4958.77 (1956). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Amende I, Simon R, Hood WP Jr. & Lightlen PR The effects of the beta-blocker atenolol and nitroglycerin on left ventricular function and geometry in man. Circulation 60, 836–849, doi: 10.1161/01.cir.60.4.836 (1979). [DOI] [PubMed] [Google Scholar]
- 217.Simpson CF, Kling JM & Palmer RF The use of propranolol for the protection of turkeys from the development of beta-aminopropionitrile-induced aortic ruptures. Angiology 19, 414–418 (1968). [DOI] [PubMed] [Google Scholar]
- 218.Groenink M, de RA, Mulder BJ, Spaan JA & van der Wall EE Changes in aortic distensibility and pulse wave velocity assessed with magnetic resonance imaging following beta-blocker therapy in the Marfan syndrome. Am. J. Cardiol 82, 203–208, doi:S0002–9149(98)00315–4 [pii]; 10.1016/s0002-9149(98)00315-4 [doi] (1998). [DOI] [PubMed] [Google Scholar]
- 219.Chiu HH et al. Losartan added to beta-blockade therapy for aortic root dilation in Marfan syndrome: a randomized, open-label pilot study. Mayo Clin. Proc 88, 271–276, doi:S0025–6196(12)01093–2 [pii]; 10.1016/j.mayocp.2012.11.005 [doi] (2013). [DOI] [PubMed] [Google Scholar]
- 220.Milleron O et al. Marfan Sartan: a randomized, double-blind, placebo-controlled trial. Eur. Heart J 36, 2160–2166, doi:ehv151 [pii]; 10.1093/eurheartj/ehv151 [doi] (2015). [DOI] [PubMed] [Google Scholar]
- 221.Forteza A et al. Efficacy of losartan vs. atenolol for the prevention of aortic dilation in Marfan syndrome: a randomized clinical trial. Eur. Heart J 37, 978–985, doi:ehv575 [pii]; 10.1093/eurheartj/ehv575 [doi] (2016). [DOI] [PubMed] [Google Scholar]
- 222.Muino-Mosquera L et al. Efficacy of losartan as add-on therapy to prevent aortic growth and ventricular dysfunction in patients with Marfan syndrome: a randomized, double-blind clinical trial. Acta Cardiol 72, 616–624, doi: 10.1080/00015385.2017.1314134 [doi] (2017). [DOI] [PubMed] [Google Scholar]
- 223.Silverman DI et al. Family history of severe cardiovascular disease in Marfan syndrome is associated with increased aortic diameter and decreased survival. J. Am. Coll. Cardiol 26, 1062–1067 (1995). [DOI] [PubMed] [Google Scholar]
- 224.Brooke BS et al. Angiotensin II blockade and aortic-root dilation in Marfan’s syndrome. N. Engl. J. Med 358, 2787–2795, doi:358/26/2787 [pii]; 10.1056/NEJMoa0706585 [doi] (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Al-Abcha A et al. Meta-analysis Examining the Usefulness of Angiotensin Receptor blockers for the Prevention of Aortic Root Dilation in Patients With the Marfan Syndrome. Am J Cardiol 128, 101–106, doi: 10.1016/j.amjcard.2020.04.034 (2020). [DOI] [PubMed] [Google Scholar]
- 226.Quint LE, Liu PS, Booher AM, Watcharotone K & Myles JD Proximal thoracic aortic diameter measurements at CT: repeatability and reproducibility according to measurement method. Int. J Cardiovasc. Imaging 29, 479–488, doi: 10.1007/s10554-012-0102-9 [doi] (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Dormand H & Mohiaddin RH Cardiovascular magnetic resonance in Marfan syndrome. J Cardiovasc. Magn Reson 15, 33, doi:1532–429X-15–33 [pii]; 10.1186/1532-429X-15-33 [doi] (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Weinrich JM et al. Reliability of non-contrast magnetic resonance angiography-derived aortic diameters in Marfan patients: comparison of inner vs. outer vessel wall measurements. Int J Cardiovasc Imaging 36, 1533–1542, doi: 10.1007/s10554-020-01850-4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Mariucci EM et al. Dilation of peripheral vessels in Marfan syndrome: importance of thoracoabdominal MR angiography. Int. J Cardiol 167, 2928–2931, doi:S0167–5273(12)01000–5 [pii]; 10.1016/j.ijcard.2012.08.001 [doi] (2013). [DOI] [PubMed] [Google Scholar]
- 230.Lopez-Sainz A et al. Aortic Branch Aneurysms and Vascular Risk in Patients With Marfan Syndrome. J Am Coll Cardiol 77, 3005–3012, doi: 10.1016/j.jacc.2021.04.054 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Nollen GJ, Groenink M, Tijssen JG, van der Wall EE & Mulder BJ Aortic stiffness and diameter predict progressive aortic dilatation in patients with Marfan syndrome. Eur. Heart J 25, 1146–1152, doi: 10.1016/j.ehj.2004.04.033 [doi];S0195668X04002878 [pii] (2004). [DOI] [PubMed] [Google Scholar]
- 232.Guala A et al. Decreased rotational flow and circumferential wall shear stress as early markers of descending aorta dilation in Marfan syndrome: a 4D flow CMR study. J Cardiovasc. Magn Reson 21, 63, doi: 10.1186/s12968-019-0572-1 [doi];10.1186/s12968-019-0572-110.1186/s12968-019-0572-110.1186/s12968-019-0572-110.1186/s12968-019-0572-1 [pii] (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Cavalcante JL, Lima JA, Redheuil A & Al-Mallah MH Aortic stiffness: current understanding and future directions. J Am Coll Cardiol 57, 1511–1522, doi:S0735–1097(11)00278–6 [pii]; 10.1016/j.jacc.2010.12.017 [doi] (2011). [DOI] [PubMed] [Google Scholar]
- 234.Laurent S et al. Expert consensus document on arterial stiffness: methodological issues and clinical applications. Eur. Heart J 27, 2588–2605, doi:ehl254 [pii]; 10.1093/eurheartj/ehl254 [doi] (2006). [DOI] [PubMed] [Google Scholar]
- 235.Kunkala MR et al. Mitral valve disease in patients with marfan syndrome undergoing aortic root replacement. Circulation 128, S243–S247 (2013). [DOI] [PubMed] [Google Scholar]
- 236.JB, l. P. d. W. et al. Mechanisms of recurrent aortic regurgitation after aortic valve repair: predictive value of intraoperative transesophageal echocardiography. JACC Cardiovasc. Imaging 2, 931–939, doi:S1936–878X(09)00390–8 [pii]; 10.1016/j.jcmg.2009.04.013 [doi] (2009). [DOI] [PubMed] [Google Scholar]
- 237.Groner LK, Lau C, Devereux RB & Green DB Imaging of the Postsurgical Aorta in Marfan Syndrome. Curr. Treat. Options. Cardiovasc. Med 20, 80, doi: 10.1007/s11936-018-0675-2 [doi]; 10.1007/s11936-018-0675-2 [pii] (2018). [DOI] [PubMed] [Google Scholar]
- 238.Evangelista A et al. Long-term outcome of aortic dissection with patent false lumen: predictive role of entry tear size and location. Circulation 125, 3133–3141, doi:CIRCULATIONAHA.111.090266 [pii]; 10.1161/CIRCULATIONAHA.111.090266 [doi] (2012). [DOI] [PubMed] [Google Scholar]
- 239.Rylski B et al. Type A aortic dissection in Marfan syndrome: extent of initial surgery determines long-term outcome. Circulation 129, 1381–1386 (2014). [DOI] [PubMed] [Google Scholar]
- 240.Martin C et al. Aortic Complications in Marfan Syndrome: Should We Anticipate Preventive Aortic Root Surgery? Ann. Thorac. Surg, doi:S0003–4975(19)31488–2 [pii]; 10.1016/j.athoracsur.2019.08.096 [doi] (2019). [DOI] [PubMed] [Google Scholar]
- 241.Fraser CD III et al. Valve-sparing aortic root replacement in children: Outcomes from 100 consecutive cases. J Thorac. Cardiovasc. Surg 157, 1100–1109, doi:S0022–5223(18)33223–9 [pii]; 10.1016/j.jtcvs.2018.09.148 [doi] (2019). [DOI] [PubMed] [Google Scholar]
- 242.David TE et al. Outcomes of Aortic Valve-Sparing Operations in Marfan Syndrome. J. Am. Coll. Cardiol 66, 1445–1453, doi:S0735–1097(15)04597–0 [pii]; 10.1016/j.jacc.2015.07.041 [doi] (2015). [DOI] [PubMed] [Google Scholar]
- 243.Coselli JS et al. Early and 1-year outcomes of aortic root surgery in patients with Marfan syndrome: a prospective, multicenter, comparative study. J. Thorac. Cardiovasc. Surg 147, 1758–1766, 1767 (2014). [DOI] [PubMed] [Google Scholar]
- 244.Izgi C et al. External Aortic Root Support to Prevent Aortic Dilatation in Patients With Marfan Syndrome. J Am Coll Cardiol 72, 1095–1105, doi:S0735–1097(18)35445–7 [pii]; 10.1016/j.jacc.2018.06.053 [doi] (2018). [DOI] [PubMed] [Google Scholar]
- 245.Treasure T et al. Personalised external aortic root support (PEARS) in Marfan syndrome: analysis of 1–9 year outcomes by intention-to-treat in a cohort of the first 30 consecutive patients to receive a novel tissue and valve-conserving procedure, compared with the published results of aortic root replacement. Heart 100, 969–975, doi: 10.1136/heartjnl-2013-304913 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Song HK et al. Long-term implications of emergency versus elective proximal aortic surgery in patients with Marfan syndrome in the Genetically Triggered Thoracic Aortic Aneurysms and Cardiovascular Conditions Consortium Registry. J. Thorac. Cardiovasc. Surg 143, 282–286 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Shalhub S et al. Type B Aortic Dissection in Young Individuals With Confirmed and Presumed Heritable Thoracic Aortic Disease. Ann. Thorac. Surg 109, 534–540, doi:S0003–4975(19)31066–5 [pii]; 10.1016/j.athoracsur.2019.07.004 [doi] (2020). [DOI] [PubMed] [Google Scholar]
- 248.Pellenc Q et al. Optimising Aortic Endovascular Repair in Patients with Marfan Syndrome. Eur. J Vasc. Endovasc. Surg, doi:S1078–5884(19)32014–3 [pii]; 10.1016/j.ejvs.2019.09.501 [doi] (2019). [DOI] [PubMed] [Google Scholar]
- 249.Peters KF, Kong F, Hanslo M & Biesecker BB Living with Marfan syndrome III. Quality of life and reproductive planning. Clin. Genet 62, 110–120, doi:cge620203 [pii]; 10.1034/j.1399-0004.2002.620203.x [doi] (2002). [DOI] [PubMed] [Google Scholar]
- 250.Goldfinger JZ et al. Marfan Syndrome and Quality of Life in the GenTAC Registry. J Am Coll Cardiol 69, 2821–2830, doi:S0735–1097(17)37088–2 [pii]; 10.1016/j.jacc.2017.04.026 [doi] (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Nielsen C, Ratiu I, Esfandiarei M, Chen A & Selamet Tierney ES A Review of Psychosocial Factors of Marfan Syndrome: Adolescents, Adults, Families, and Providers. J Pediatr. Genet 8, 109–122, doi: 10.1055/s-0039-1693663 [doi];1800055 [pii] (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Vanem TT, Rand-Hendriksen S, Brunborg C, Geiran OR & Roe C Health-related quality of life in Marfan syndrome: a 10-year follow-up. Health Qual Life Outcomes 18, 376, doi: 10.1186/s12955-020-01633-4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Oller J et al. Nitric oxide mediates aortic disease in mice deficient in the metalloprotease Adamts1 and in a mouse model of Marfan syndrome. Nat. Med, doi:nm.4266 [pii]; 10.1038/nm.4266 [doi] (2017). [DOI] [PubMed] [Google Scholar]
- 254.Redondo A. d. l. F.-A. a. J. M. Aortic diseasein Marfan syndrome is caused by overactivation of sGC-PRKG signaling by NO. Nature Communications (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Hansen J et al. Systems pharmacology-based integration of human and mouse data for drug repurposing to treat thoracic aneurysms. JCI. Insight 4, doi:127652 [pii]; 10.1172/jci.insight.127652 [doi] (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Nistala H, Lee-Arteaga S, Siciliano G, Smaldone S & Ramirez F Extracellular regulation of transforming growth factor beta and bone morphogenetic protein signaling in bone. Ann N Y Acad Sci 1192, 253–256, doi: 10.1111/j.1749-6632.2009.05350.x (2010). [DOI] [PubMed] [Google Scholar]
- 257.Putnam EA, Zhang H, Ramirez F & Milewicz DM Fibrillin-2 (FBN2) mutations result in the Marfan-like disorder, congenital contractural arachnodactyly. Nat. Genet 11, 456–458 (1995). [DOI] [PubMed] [Google Scholar]
- 258.Maccarrick G et al. Loeys-Dietz syndrome: a primer for diagnosis and management. Genet. Med (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Pepin M, Schwarze U, Superti-Furga A & Byers PH Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N. Engl. J. Med 342, 673–680 (2000). [DOI] [PubMed] [Google Scholar]
- 260.Pepin MG et al. Survival is affected by mutation type and molecular mechanism in vascular Ehlers-Danlos syndrome (EDS type IV). Genet. Med 16, 881–888, doi:gim201472 [pii]; 10.1038/gim.2014.72 [doi] (2014). [DOI] [PubMed] [Google Scholar]
- 261.Renard M et al. Clinical Validity of Genes for Heritable Thoracic Aortic Aneurysm and Dissection. J. Am. Coll. Cardiol 72, 605–615, doi:S0735–1097(18)35141–6 [pii]; 10.1016/j.jacc.2018.04.089 [doi] (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Van Driest SL et al. Variants in ADRB1 and CYP2C9: Association with Response to Atenolol and Losartan in Marfan Syndrome. J Pediatr 222, 213–220 e215, doi: 10.1016/j.jpeds.2020.03.064 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Franken R et al. Beneficial Outcome of Losartan Therapy Depends on Type of FBN1 Mutation in Marfan Syndrome. Circ. Cardiovasc. Genet 8, 383–388, doi:CIRCGENETICS.114.000950 [pii]; 10.1161/CIRCGENETICS.114.000950 [doi] (2015). [DOI] [PubMed] [Google Scholar]
- 264.Stheneur C et al. Prognosis factors in probands with an FBN1 mutation diagnosed before the age of 1 year. Pediatr Res 69, 265–270, doi: 10.1203/PDR.0b013e3182097219 (2011). [DOI] [PubMed] [Google Scholar]
- 265.Geva T, Hegesh J & Frand M The clinical course and echocardiographic features of Marfan’s syndrome in childhood. Am. J Dis. Child 141, 1179–1182 (1987). [DOI] [PubMed] [Google Scholar]
- 266.Hennekam RC Severe infantile Marfan syndrome versus neonatal Marfan syndrome. Am. J. Med. Genet. A 139, 1, doi: 10.1002/ajmg.a.30979 [doi] (2005). [DOI] [PubMed] [Google Scholar]
- 267.Booms P et al. Novel exon skipping mutation in the fibrillin-1 gene: two ‘hot spots’ for the neonatal Marfan syndrome 1. Clin. Genet 55, 110–117 (1999). [DOI] [PubMed] [Google Scholar]
- 268.Morse RP et al. Diagnosis and management of infantile marfan syndrome. Pediatrics 86, 888–895 (1990). [PubMed] [Google Scholar]
- 269.Tognato E et al. Neonatal Marfan Syndrome. Am J Perinatol 36, S74–S76, doi: 10.1055/s-0039-1691770 [doi] (2019). [DOI] [PubMed] [Google Scholar]
- 270.Liu LH, Lin SM, Lin DS & Chen MR Losartan in combination with propranolol slows the aortic root dilatation in neonatal Marfan syndrome. Pediatr. Neonatol 59, 211–213, doi:S1875–9572(16)30130–9 [pii]; 10.1016/j.pedneo.2017.07.005 [doi] (2018). [DOI] [PubMed] [Google Scholar]
- 271.Carande EJ, Bilton SJ & Adwani S A Case of Neonatal Marfan Syndrome: A Management Conundrum and the Role of a Multidisciplinary Team. Case. Rep. Pediatr 2017, 8952428, doi: 10.1155/2017/8952428 [doi] (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Krasemann T et al. Cardiac transplantation in neonatal Marfan syndrome -- a life-saving approach. Thorac. Cardiovasc. Surg 53 Suppl 2, S146–S148, doi: 10.1055/s-2004-830455 [doi] (2005). [DOI] [PubMed] [Google Scholar]
- 273.Braverman AC et al. Clinical Features and Outcomes of Pregnancy-Related Acute Aortic Dissection. JAMA Cardiol 6, 58–66, doi: 10.1001/jamacardio.2020.4876 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Roman MJ et al. Aortic Complications Associated With Pregnancy in Marfan Syndrome: The NHLBI National Registry of Genetically Triggered Thoracic Aortic Aneurysms and Cardiovascular Conditions (GenTAC). J Am Heart Assoc 5, doi:JAHA.116.004052 [pii]; 10.1161/JAHA.116.004052 [doi] (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Cauldwell M et al. Maternal and fetal outcomes in pregnancies complicated by Marfan syndrome. Heart 105, 1725–1731, doi:heartjnl-2019–314817 [pii]; 10.1136/heartjnl-2019-314817 [doi] (2019). [DOI] [PubMed] [Google Scholar]
- 276.Rossiter JP, Repke JT, Morales AJ, Murphy EA & Pyeritz RE A prospective longitudinal evaluation of pregnancy in the Marfan syndrome. Am. J. Obstet. Gynecol 173, 1599–1606 (1995). [DOI] [PubMed] [Google Scholar]
- 277.Banerjee A, Begaj I & Thorne S Aortic dissection in pregnancy in England: an incidence study using linked national databases. BMJ Open 5, e008318, doi:bmjopen-2015–008318 [pii]; 10.1136/bmjopen-2015-008318 [doi] (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Meijboom LJ et al. Pregnancy and aortic root growth in the Marfan syndrome: a prospective study. Eur. Heart J 26, 914–920 (2005). [DOI] [PubMed] [Google Scholar]
- 279.Pacini L et al. Maternal complication of pregnancy in Marfan syndrome. Int. J. Cardiol 136, 156–161 (2009). [DOI] [PubMed] [Google Scholar]
- 280.Regitz-Zagrosek V et al. 2018 ESC Guidelines for the management of cardiovascular diseases during pregnancy. Eur Heart J 39, 3165–3241, doi: 10.1093/eurheartj/ehy340 (2018). [DOI] [PubMed] [Google Scholar]
- 281.Williams D et al. Pregnancy after Aortic Root Replacement in Marfan’s Syndrome: A Case Series and Review of the Literature. AJP Rep 8, e234–e240, doi: 10.1055/s-0038-1675347 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Bullo M, Tschumi S, Bucher BS, Bianchetti MG & Simonetti GD Pregnancy outcome following exposure to angiotensin-converting enzyme inhibitors or angiotensin receptor antagonists: a systematic review. Hypertension 60, 444–450, doi: 10.1161/HYPERTENSIONAHA.112.196352 (2012). [DOI] [PubMed] [Google Scholar]
- 283.Giarelli E, Bernhardt BA, Mack R & Pyeritz RE Adolescents’ transition to self-management of a chronic genetic disorder. Qual Health Res 18, 441–457, doi: 10.1177/1049732308314853 (2008). [DOI] [PubMed] [Google Scholar]
- 284.Modi AC et al. Pediatric self-management: a framework for research, practice, and policy. Pediatrics 129, e473–485, doi: 10.1542/peds.2011-1635 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Gauci J, Bloomfield J, Lawn S, Towns S & Steinbeck K Effectiveness of self-management programmes for adolescents with a chronic illness: A systematic review. J Adv Nurs, doi: 10.1111/jan.14801 (2021). [DOI] [PubMed] [Google Scholar]
- 286.Sattoe JNT et al. Value of an outpatient transition clinic for young people with inflammatory bowel disease: a mixed-methods evaluation. BMJ Open 10, e033535, doi: 10.1136/bmjopen-2019-033535 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Giarelli E, Bernhardt BA & Pyeritz RE Self-surveillance by adolescents and young adults transitioning to self-management of a chronic genetic disorder. Health Educ Behav 37, 133–150, doi: 10.1177/1090198109331670 (2010). [DOI] [PubMed] [Google Scholar]
- 288.Stark VC et al. The transition of pediatric Marfan patients to adult care: a challenge and its risks. Cardiovasc Diagn Ther 8, 698–704, doi: 10.21037/cdt.2018.09.13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Collod-Beroud G, Le Bourdelles S, Ades L, Ala-Kokko L, Booms P, Boxer M, Child A, Comeglio P, De Paepe A, Hyland JC, Holman K, Kaitila I, Loeys B, Matyas G, Nuytinck L, Peltonen L, Rantamaki T, Robinson P, Steinmann B, Junien C, Beroud C and Boileau C (2003) Update of the UMD-FBN1 mutation database and creation of an FBN1 polymorphism database. Hum Mutat, 22, 199–208. [DOI] [PubMed] [Google Scholar]
- 290.The Marfan Foundation. Survey Results Reveal Greatest Obstacles to Quality of Life The Marfan Foundation https://marfan.org/2017/11/01/survey-results-reveal-greatest-obstacles-to-quality-of-life/ (2017).