

CONSPECTUS:
This Account describes fundamental chemistry that promoted the discovery of new antibiotics. Specifically, the NH acidity of simple hydroxamic acid derivatives facilitated the syntheses of novel β-lactams (oxamazins and monobactams), siderophore mimics that limit bacterial iron uptake and bacterially targeted sideromycins (siderophore-antibiotic conjugates). The development of resistance to our current limited set of antibiotic scaffolds has created a dire medical situation. As recently stated, “if you weren’t taking antibiotic resistance seriously before, now would be a good time to start.” A project commissioned by the British government (https://amr-review.org/) has released estimates of the near-future global toll of antibiotic resistance that are jaw-dropping in their seriousness and scale: 10 million deaths per year and at least $100 trillion in sacrificed gross national product. The 2020 COVID pandemic confirmed that infectious disease problems are no longer localized but worldwide. Many classical antibiotics, especially β-lactams, previously provided economical cures, but the evolution of antibiotic destructive enzymes (i.e., β-lactamases), efflux pumps, and bacterial cell wall permeability barriers has made many types of bacteria, especially Gram-negative strains, resistant. Still, and in contrast to other therapies, the public expectation is that any new antibiotic must be inexpensive. This creates market limitations that have caused most major pharmaceutical companies to abandon antibiotic research. Much needs to be done to address this significant problem.
The critical need for bacteria to sequester essential iron provides an Achilles’ heel for new antibiotic development. Although ferric iron is extremely insoluble, bacteria need micromolar intracellular concentrations for growth and virulence. To this end, they biosynthesize siderophores (Gr. iron bearer) and excrete them into their environment, where they bind iron with high affinity. The iron complexes are recognized by specific outer-membrane transporters, and once actively internalized, the iron is released for essential processes. To conserve biosynthetic energy, some bacteria recognize and utilize siderophores made by competing strains. As a counter-revolution in the never-ending fight for survival, bacteria have also evolved sideromycins, which are siderophores conjugated to warheads that are lethal to rogue bacteria. While none are now used therapeutically, natural sideromycins called albomycins have been used clinically, and others have been shown to be well tolerated and active in animal infection models. Herein we describe practical methods to synthesize new antibiotics and artificial sideromycins with the generalized structure shown above (siderophore-linker drug). Utilizing the molecular-recognition-based siderophore/sideromycin bacterial assimilation processes, it is possible to design both broad spectrum and exquisitely narrow spectrum (targeted) sideromycins and even repurpose older or more classical antibiotics. Relevant microbiological assays, in vivo animal infection studies, and the recent FDA approval of cefiderocol demonstrate their effectiveness.
Graphical Abstract
INTRODUCTION
The evolution of life, as we now know it, depended on the slow transition from a reducing to an oxidizing environment during what has been labeled the great oxidation event.5 Simultaneously, oxidized forms of many elements and minerals required for life became more prevalent with concomitant changes in both their reactivity and solubility that required many organisms, including bacteria and fungi, to adapt. The sequestration of essential iron is a critical process controlling microbial growth and virulence. As environmental oxygen concentrations increased, the dominant ionic form of iron changed from the more soluble ferrous (Fe2+) to the much less soluble ferric (Fe3+) form. Thus, bacteria began to exert significant energy for biosyntheses of complex, often highly oxygentated ligands, now called siderophores,6 that could be excreted into their medium, chelate ferric iron, and then be recognized and actively assimilated. Producing strains of bacteria thus had selective growth advantages while also avoiding random redox chemistry that would lead to reactive oxygen (ROS)-induced cellular damage. The importance of siderophores has been recognized not only for their role in providing iron to pathogenic and nonpathogenic bacteria but also more generally including bacterial virulence during infections (host–pathogen), and microbiome community interactions, plant pathology, marine microbiology, soil chemistry, for use as contrast agents, as potential antimalaria and anticancer agents, for use in classifying organisms (siderotyping), diagnositics, vaccines and even therapeutically for toxic metal regulation, especially for the treatment of iron overload diseases such as thalassemia.7 This Account will focus on the chemistry used to evaluate the potential of siderophore-based microbial iron transport processes for the design, syntheses, and studies of new antibiotics. The primary methodology is based on the NH aciditiy of O-alkyl hydroxamic acids that facilitates both inter- and intramolecular nitrogen alkylation. The results allowed the syntheses of three types of antibiotic compounds: siderophore analogs that limit iron uptake by pathogenic bacteria, heteroatom-activated monocyclic β-lactam antibiotics (oxamazins and monobactams), and synthetic siderophore-antibiotic conjugates (sideromycins) that target very specific multi-drug-resistant (MDR) bacteria and allow the repurposing of Gram-positive-only antibiotics for the treatment of MDR Gram-negative bacteria.
SIDEROPHORES AND SIDEROMYCINS
Siderophores are everywhere microbes grow.8 The structures, properties, and producing organisms of hundreds of siderophores are known.9 Representative siderophores are shown in Figure 1. They typically comprise a backbone appended with oxygenated bidentate iron-binding ligands combined to effectively coordinate and solubilize Fe3+ usually by forming hexacoordinate complexes. In many cases, the ligand types are the same: amino acid or amino alkane-based hydroxamic acids (ferrichrome and ferrioxamine, respectively), catechols (enterobactin), α-hydroxy acids (citrate-based schizokinens, aerobactin arthrobactin), ortho-hydroxy phenyl oxazolines (agrobactins), or combinations of ligand types (mixed ligands as in pyoverdines, fimsbactins, and mycobactins). The backbone and even the bound iron are often chiral, which in some cases affects the initial bacterial outer membrane (OM) reception and direct or shuttle-based transport.10
Figure 1.

Representative hydroxamate (1–13), catechol (14–16) and mixed-ligand (17–19) siderophores.
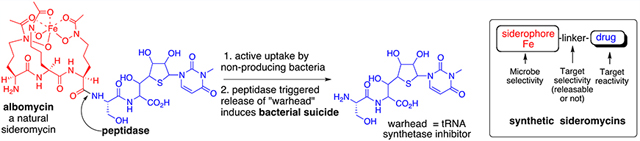
While some bacteria became remarkably selective and dependent on very specific self-generated siderophores, others could assimilate ionic iron in many forms or, more remarkably, evolved transport proteins to allow them to utilize siderophores made by other bacteria (xenosiderophores).11 To counter this iron thievery,12 some bacteria made Trojan horse siderophores (sideromycins), which are essentially conjugates of the requisite iron binding and bacterial recognition properties linked to a lethal component.13,14 Figure 2 shows structures of some of the few known natural sideromycins. Albomycins (20) contain the same tri-δ-N-hydroxy-l-ornithine peptide as the fungal siderophore, ferrichrome (1, Figure 1), but in linear form with an attached seryl thionucleoside t-RNA synthetase inhibitor.15 While the producing streptomyces utilize the iron sequestration properties of the albomycins, other microbes also recognize them, but after assimilation, a cytoplasmic peptidase (N) cleaves the N-terminal serine to release the now deadly payload, inducing the culprit bacteria to commit suicide. Salmycins (22)16 contain three N-hydroxylated diamino alkane-based hydroxamates linked to unusual amino glycosides that presumably are also released intracellularly by a cyclization after bacterial uptake of the natural conjugate and reductive removal of the iron. Fewer details are known about the ferrimycin (24)17 warhead, but the ability to actively transport large antibiotic peptides such as the 84mer peptide of the enterobactin-related sideromycins called microcins (25)18 is noteworthy. The size and structural limits of the ability of siderophores to carry cargo into bacterial cells are of considerable interest.19
Figure 2.

Natural sideromycins.
There has been skepticism about the use of natural and synthetic sideromycins for the treatment of infections in humans.20 The primary argument is that bacteria rapidly develop resistance by deletion of the cognate outer membrane transport proteins, thus limiting the ability of the sideromycins to deliver their toxic payload.21 However, the early and now more recent literature has repeatedly demonstrated the in vivo antibacterial effectiveness of sideromycins. In fact, in the 1950s, the albomycins were used extensively and successfully to treat patients with infections that were already resistant to the penicillins.22 The salmycins are effective in animals, and while siderophore transport mutants are frequently isolated, those “resistant” strains are iron-starved and typically not viable in a normal iron-deficient environment associated with an infection.23 The 1962 paper describing the isolation of ferrimycin also demonstrated that it was superbly active against Gram-positive bacteria and was “highly effective (more so than penicillin or erythromycin) against staphylococcal infection in mice. Although resistant mutants appear in the blood on the first day, they are replaced by sensitive organisms before eventually disappearing altogether.”24 As described later in this Account, properly designed synthetic sideromycins also are effective in vivo.
DEVELOPMENT OF COMMON METHODOLOGY FOR SIDEROPHORE SYNTHESES AND THE INVENTION OF NEW β-LACTAM ANTIBIOTICS
We and others anticipated that it should be possible to design synthetic sideromycins if the important siderophore components could be accessed.25,26 A number of siderophore iron-binding ligands are readily available, including catechols, dihydroxy benzoate, α-hydroxy carboxylic acids, and ortho-hydroxyphenyl oxazoles. Backbone scaffolds based on amino acids, peptides, or other cores are also accessible. However, access to N-alkyl hydroxamic acids (32), the key component of most fungal27 and many bacterial siderophores,28 is more synthetically challenging. A number of variably effective methods for chemical syntheses of siderophore N-alkyl hydroxylamines have been reported but will not be reviewed here. Early on, we attempted to use the NH acidity of O-alkyl N-acyl hydroxylamines (O-alkyl hydroxamic acids, 26, pKa ~9, Scheme 1)29 to form N-alkyl hydroxamates. However, O-alkyl hydroxamic acids are ambident nucleophiles, and alkylation often gives mixtures of N-alkyl (hydroxamate, 28) and O-alkyl (hydroximate, 29) products. Fortunately, the alkylation of carbamate (i.e., Boc, alloc, troc, Cbz, etc.)-protected O-alkylhydroxylamines (30) occurs exclusively on nitrogen (31). Although this then requires the removal of the carbamate protecting group and reacylation (31 → 28), the process is robust and enabled the syntheses of a number of siderophores and analogs.
Scheme 1.

In relevant examples, the reduction of the ω-carboxylic acid of α-amino- and carboxyl-protected l-glutamate and adipate to the alcohols (33, n = 0; 33, n = 1, Scheme 2) and then direct Mitsunobu reaction of the alcohols with carbamate-protected O-benzyl hydroxylamine provided the optically pure protected δ-N-hydroxy-l-ornithine (34, n = 0) and ε-N-hydroxy-l-lysine (34, n = 1) derivatives. The preparation of linear di- and tripeptides facilitated the syntheses of natural polyhydroxamate siderophores including diketopiperazine rhodotorulic acid 3, a bis-hydroxamate that binds Fe3+ with 3:2 stoichiometry,30 the ferrichrome (1) and albomycin trihydroxamate peptide,31 foroxymethine, (4)32 amamistatins,33 citrate-based hydroxamates awaitin A (8), aerobactin (11),34 and others (Figure 1).
Scheme 2.

Similar alkylation chemistry (Scheme 2) starting with simple protected amino alcohols (36) gave differentiated α-amino ω-hydroxylamino compounds 38 that were used for the syntheses of desferrioxamines, danoxamines,35 awaitin B (9), schizokinin (12), arthrobactin (13),36 and a series of synthetic trihydroxamic acid-containing siderophore mimetics.37
Application of the simple hydroxamate chemistry to syntheses of mycobactins and analogs was of special interest. Mycobactins are essential growth and virulence factors for many types of mycobacteria.38 Extensive isolation and structural studies of mycobacterial siderophores in the 1950s to 1970s revealed that many shared the same core with minor peripheral variation.39 Most amazingly, Snow reported that mycobactins isolated from one strain antagonized the growth of different strains of mycobacteria even though their structures were similar. Notable to us was that mycobactin T (19), the siderophore used by deadly Mycobacteria tuberculosis, and mycobactin S, the siderophore required by relatively nonpathogenic M. smegmatis, differ only by the stereochemistry at the β-carbon of the β-hydroxy butyrate that links the linear lysine hydroxamate to the cyclic terminal N-hydroxy lysine. We decided to separately synthesize representative forms of mycobactin T and S and test them for growth promotion or inhibitory activity of the producing strains of mycobacteria. Our first approach40 was designed to test the synthetic methodology and focused on the synthesis of the simple N-acetyl version of mycobatin S, which we called mycobactin S2 since it had a simple two-carbon acyl group rather than the long fatty acyl chains of natural mycobactins (Scheme 3). The linear N-hydroxy-l-lysine component was synthesized by the deprotection of 34 and acetylation to give suitably protected forms of 35 (R′ = Me). As expected on the basis of the intermolecular alkylation studies of O-alkyl hydroxamates 26, the intramolecular Mitsunobu reaction of amino acid hydroxamates 40 formed the desired seven-membered cyclic hydroxamate (41) along with E and Z isomers of corresponding hydroximates 42. Separation of the hydroxamates followed by stepwise assembly of the components provided mycobactin S2 and thus the first synthesis of the core of the mycobactins that contained the iron-binding components. As expected, since mycobactin S2 lacks the typical long acyl side chain, it had no effect on mycobacterial growth. Extensions to the syntheses of other mycobactins of interest are described after further discussion of intramolecular hydroxamate reactions.
Scheme 3.

Additional studies of cyclizations of O-substituted hydroxamates to form N-hydroxy lactams revealed that the amount of N-alkylation improved relative to O-alkylation as the ring size was reduced. Fortunately, four-membered ring formation proceeds exclusively on nitrogen and facilitates the syntheses of important β-lactams.41 Indeed, the Mitsunobu or Mitsunobu-like reaction of essentially any β-hydroxy hydroxamate (i.e., 43) produces the corresponding N-alkoxy β-lactam (44) with the retention of configuration at the α carbon and clean inversion at the β-carbon without the competitive formation of oxazoline (45) when the α-amino group is protected as a carbamate (Scheme 4).42 Alternate activation of the β-hydroxy group by mesylation, sulfation, or conversion to a halide followed by deprotonation of the acidic hydroxamate NH also is effective.43 Bromine-induced cyclization of β, γ unsaturated hydroxamic acids (52) also produces β-lactams with a pendant bromomethyl group (53) suitable for further functionalization.44 The facility of the hydroxamate-mediated cyclization allowed major β-lactam SAR advances. For example, deprotection of the N-hydroxy group of 46 and simple alkylation of the resulting free N–OH (49) with bromoacetates to incorporate an ionizable group required for β-lactam antibiotic activity led to the syntheses of what we called the oxamazins (50).45 Assays revealed that, for the first time, it was possible to synthesize monocyclic β-lactams with activity comparable to or in some cases even superior to that of classical bicyclic penicillins, cephalosporins, and carbapenems. During this time, the Squibb and Takeda groups independently discovered natural N-sulfated monocyclic β-lactams that are now called monobactams (48).46 This emphasized that monocyclic β-lactams could be potential antibiotics and that the substitution of the classical ionizable group with a sulfate could provide the required heteroatom-activation-induced activity. Though the natural products had only moderate activity, use of the N–C4 cyclization chemistry allowed the syntheses of more potent monocyclic-heteroatom-activated β-lactams. The only marketed monobactam, aztreonam (56), was readily derived from l-threonine by the hydroxamate route (46 → 47 → 48) and by cyclization of the sulfamate (55). The Squibb group47 and our laboratory48 also reported that O-sulfation of the N-hydroxy β-lactams (49) produced the monosulfactams (51) that had comparable or even superior activity relative to the monobactams.
Scheme 4.

The β-lactam ring (“the Enchanted Ring”)49 is the essential component of penicillins (57), cephalosporins (58), carbapenems (59), and other β-lactam antibiotics. Although thousands of analogs have been prepared, especially in the 1960s to 1980s, most efforts relied on the development of fermentation processes to generate core bicyclic structures for subsequent modification. In contrast, the hydroxamate method made it possible to synthesize the β-lactam core with complete control of peripheral functionality and stereochemistry, with the only requirement being access to appropriate β-hydroxy acids, many of which became available by the development of chemical (chiral-auxiliary-based)50 and enzymatic (serine hydroxymethyl transferase, SHMT) asymmetric aldol chemistry (Scheme 5).51
Scheme 5.

Further studies of substituted N-hydroxy β-lactams revealed more chemical and biological utility. N-Sulfonyloxy derivatives (65) were found to be potent β-lactamase inhibitors (Scheme 6).52 Interestingly, the reactions of 65 with nucleophiles, including amines, resulted in attack at the α-carbon of the β-lactams with the simultaneous loss of the N-sulfonyloxy group to give N-unsubstituted, α-functionalized β-lactams 66 suitable for the syntheses of a number of important β-lactam antibiotics.53 The heteroatom activation strategy has recently been utilized to design and synthesize important new bridged bicyclic β-lactamase inhibitors that incorporate N–O bonds (67–70, Scheme 6).54
Scheme 6.

Although hydroxamate cyclizations to give β-lactams are effective, the competitive N- and O-alkylation when forming larger rings prompted us to explore other methods for making important linear and cyclic siderophore hydroxamic acid-containing components. After reconsidering the extensive literature on methods to oxidize amines to hydroxylamines,55 we found that simple reaction of the ε-amine of lysine (71) with dimethyl dioxirane provided nitrone (72, Scheme 7). Hydrolysis then gave the hydroxylamines (73) that could then be acylated intermolecularly and intramolecularly to give the desired linear (74)56 and cyclic (75)57 hydroxamate components of the mycobactins and structurally related amamistatins. We then completed the syntheses of mycobactin T (2) and S (76).58 Assays showed that synthetic mycobactin S was a potent growth inhibitor of M. tuberculosis H37Rv, though it differs in only one stereogenic center from mycobactin T, the siderophore growth promoter of M. tuberculosis (Mtb). Thus, decades after Snow’s reports, we were able to confirm that changing even one stereogenic center in a complex mycobactin structure converted it from an essential growth promoter to a potent inhibitor of Mtb.
Scheme 7.

We also synthesized two types of analogs that include functionality suitable for preparing drug conjugates: one set that modified the most remote peripheral position by incorporating an amino group at the para position of the terminal oxazoline moiety (78–80)59 and another that changed the internal β-hydroxy butyrate to a β-amino acid (83, Scheme 8).60 The former incorporates a p-aminosalicylate (PAS, 81) analog into the mycobactin structure. PAS has long been known to have anti-TB activity.61 Even the simple acetylated (78, MIC = 0.09 μM) and Boc-protected (79, MIC = 0.02 μM) versions had impressive anti-TB activity. The maleimide derivative (80) prepared for future conjugation studies was also active by itself (MIC = 0.88 μM).62
Scheme 8.

Boc-protected diaminopropionate analog 84 (Scheme 8), in which diamino acid 83 was prepared using our previously described β-lactam chemistry, also was very active (MIC = 0.2–0.5 μM). However, the free amine obtained after Boc removal was devoid of activity as was the corresponding pivaloyl analog (85). Thus, what appeared to be a simple difference of an oxygen atom between 84 (with Boc) and 85 (with pivaloyl) had a remarkable influence. It is tempting to consider that once taken up by Mtb, the Boc form might be converted to the free amine that would then be positively charged and disrupt the binding and/or cell wall structure. Additional extensive SAR of differentially modified mycobactins has been summarized elsewhere.63,64
Removal of the Boc group from 84 and reacylation with a modified artemisinin gave conjugate 86. While artemisinin (87) itself is a potent antimalarial agent, it has no independent anti-TB activity. However, we anticipated that, once sequestered, the Mtb iron reductase or the resulting ferrous iron might cleave artemisinin’s peroxide to form a lethal reactive oxygen species (ROS). Indeed, conjugate 86 was active (MIC = 0.3 μM) against Mtb H37Rv and even more active against a wide range of multi-drug-resistant (MDR) and extensive-drug-resistant (XDR) strains. Electrochemical studies were consistent with the ferrous induction of Fenton-like chemistry of the peroxide, and studies with live Mtb incubated with the artemisinin conjugate revealed the generation of reactive oxygen species.2 Conjugate 86 was completely selective for Mtb because it was not active against other strains of mycobacteria or a broad panel of Gram-positive and Gram-negative bacteria. However, as an artemisinin derivative it did retain potent antimalarial activity. Thus, conjugate 86 was shown to be remarkably active against the agents responsible for two of the most deadly infectious diseases.
Although conjugate 86 targets both Mtb and malaria, its synthesis is not practical for drug development. With many protected and unprotected synthetic mycobactin components in hand, we performed fragment-based anti-TB screening. We quickly found that benzyl-protected oxazoline 88 (Figure 3) had moderate anti-TB activity (7.7 μM) and was not cytotoxic but was metabolically labile.65 Extended SAR of alternative heterocycles revealed that imidazopyridine carboxamides (IAPs), represented by an early example (89), have potent anti-TB activity, including against MDR TB, are not cytotoxic, and are metabolically stable.66 Additional studies indicated that IAPs have considerable activity against replicating MDR and XDR Mtb.67 The IAPs were found to be active in vivo,68 and they target Mtb cytochrome bc1:aa3 (QcrB) to deplete ATP levels.69 More recent structure–activity relationship (SAR) studies have produced IAP derivatives with nanomolar anti-TB activity that will be reported in due course.
Figure 3.

SAR from fragment-based screening to generate new anti-TB agents.
SYNTHETIC SIDEROMYCINS
Alternative amine oxidation chemistry also allowed us to improve the syntheses of δ-N-acyl-δ-N-hydroxy-l-ornithine (92, n = 1) and ornithine peptides (92, n = 3) of the ferrichromes, albomycins, and related antibiotic conjugates (Scheme 9).70 As indicated earlier, the albomycins represent a nearly ideal natural class of sideromycins. Since linkages to alternate drug components might not be recognized by bacterial peptidases to promote release once internalized, we decided to first conjugate our synthetic tri-δ-N-acyl-δ-N-hydroxy-l-ornithine peptides with β-lactam antibiotics since they need to cross only the outer membrane (OM) of Gram-negative organisms. Extensive peripheral modification around the β-lactam core is also well tolerated. Our initial independent and then collaborative studies with Prof. F. Malouin and scientists at Eli Lilly and Co. with strains of E. coli and other representative Gram-negative bacteria confirmed that the synthetic conjugate (94) with a carbacepahalosporin (95) was indeed recognized, actively assimilated, and inhibited growth without the need for a release mechanism. Additional kinetic and agar diffusion studies allowed the selection of resistant strains missing the OM FhuA transporter. However, in more relevant iron-deficient media, the mutant71 strains were unable to grow due to impaired iron uptake caused by a lack of FhuA. While the parent strains of E. coli were lethal in animal infection studies, the mutants were not.
Scheme 9.

For All Conjugates, the Siderophore Is Shown in Red and the Warhead/Drug Is Shown in Blue
Anticipating that they would utilize bacterial catechol transport proteins for active transport,72 we also prepared carbacephalosporin conjugates 98 and 99 of bis-catechol azotochelin (96) and an analog of the tricatechol agrobactin (97, Figure 4.). The conjugates were active against representative strains of E. coli, and again, we could select different outer membrane transport-deficient and thus resistant mutants that were also at a growth disadvantage in iron-limited media. A mixture of the hydroxamate and catechol conjugates was still more effective, and eventually we were able to isolate doubly resistant mutants missing both hydroxamate and catechol outer membrane transporters, which could only be grown in iron-rich media and were not pathogenic in vivo.
Figure 4.

Catechol siderophores and conjugates.
Since our hydroxamate and catechol siderophore analogs and corresponding conjugates utilized different OM proteins for active transport, we thought it would be interesting to make a mixed ligand (hydroxamate and catechol) siderophore-like compound and antibiotic conjugates. Both a carbacephalosporin with the usual d-phenylglycyl side chain (loracarbef) and the corresponding cephalosporin (ceclor) conjugates (100a,b) were synthesized (Figure 5). Gram-positive and Gram-negative antibacterial assays revealed selective and potent activity against strains of Acinetobacter baumannii.73 Further studies indicated that the synthetic mixed ligand mimicked fimsbactin (101),74 a subsequently discovered natural A. baumannii selective siderophore.75 Infections due to multi-drug-resistant (MDR) strains of A. baumannii have been listed as those of critical concern by the WHO.
Figure 5.

Mixed-ligand drug conjugate and natural siderophore fimsbactin.
The remarkable antibacterial selectivity of mixed-ligand conjugates 100a,b indicates that, like mycobactin conjugates (Schemes 7 and 8), it is possible to use the exquisite molecular recognition properties associated with siderophore transport to design narrow-spectrum sideromycins that target specific problematic MDR bacteria. In complementary fashion, synthetic sideromycins that are widely recognized by many strains of bacteria could be broad-spectrum antibiotics. The potential of selective targeting was also demonstrated by studies of penicillin conjugates of pyoverdin II (102, Figure 6) reported by Budzikiewicz.76 Pyoverdines are specific siderophores for different strains of Pseudomonas aeruginosa. In contrast to ampicillin, against which P. aeruginosa is resistant, pyoverdine II conjugate (102) exhibited excellent antibacterial activity, but only against the strain that can use the parent pyoverdin for iron uptake. Knowing that enterobactins are recognized and utilized by broader types of Gram-negative bacteria, the Nolan group performed studies to determine the extent that they could be used to transport variably sized cargos, and using that information, they designed β-lactam conjugates with enhanced activity against pathogenic strains of E. coli.77 Ji developed a practical synthesis of an enterobactin mimic as well as ampicillin and amoxicillin conjugates (103).78 Although the parent penicillin derivatives were not active, the conjugates were extraordinarily active (MIC values = 0.05–0.4 μM) against tested strains of P. aeruginosa, except those that were strong β-lactamase producers. Schalk very recently discussed the potential advantages of using catechol-based drug conjugates for Gram-negative bacteria.79
Figure 6.

Pyoverdin II and tricatechol ampicillin conjugates.
Möllmann reported that the tetraacetate prodrug form of biscatechol penicillin conjugate (104, Figure 7) based on a diaminobutane analog of the lysine-derived azotochelin core was broadly and potently active against Gram-negative bacteria80 with the added benefit of circumventing problematic β-lactam efflux pumps. The results encouraged us to expand our set of biscatechol-like β-lactam conjugates, including cephalosporin and carbacephalosporin derivatives 105 and 106. These conjugates were very potent against a large set of Gram-negative bacteria, especially E. coli and MDR strains of P. aeruginosa and A. baumannii. In vivo studies in mice showed that they were well tolerated at more than 250 mg/kg with no observed negative effects, and in A. baumannii-infected mouse studies, 100% that were treated with 105 or 106 survived whereas control mice did not.20 Thus, consistent with the earlier separate studies reported by Gause and Braun with albomycins, sideromycins can be effective in vivo. The clinical efficacy of sideromycins has been further demonstrated by the recent FDA approval of Shionogi’s catechol-cephalosporin, cefiderocol (107).81
Figure 7.

Catechol conjugates.
While catechol-based siderophore-penicillin and cephalosporin conjugates have broad-spectrum Gram-negative antibiotic activity, many are susceptible to β-lactamases. Since monobactams are much less prone to β-lactamase destruction, a number of groups have synthesized analogs, including monocarbam analogs of monobactams with pendant iron chelating ligands, mostly nonsiderophore-based hydroxypyridones that were intended to mimic catechols. Because of marketing considerations and other concerns, these industrial programs have been discontinued. We recently used our hydroxamate-mediated β-lactam methodology to prepare a biscatechol-monobactam (113, Scheme 10) that incorporates the usual and important ATMO (amino thiazole oxime) α-amino side chain.3 Important key steps included bromine-induced cyclization of the simple vinyl acetohydroxamate (109) to the 4-bromomethyl β-lactam (110) and subsequent conversion to the N-tosyloxy derivative (111) that upon reaction with azide provided the precursor (112) to the required α-amino-substituted β-lactam. The final conjugate (113) has superb activity against Gram-negative bacteria, including MDR- and β-lactamase-producing strains that are also resistant to aztreonam. Most noteworthy is the extraordinary activity against carbapenemase- and cephalosporinase-producing strains of A. baummannii (0.4 μM) that have been designated by the WHO as causing infections of most concern.
Scheme 10.

RELEASABLE LINKERS
Many non-β-lactam antibiotics, like ciprofloxacin, have cytoplasmic targets that do not tolerate extensive peripheral modification, and not all siderophores cross the cytoplasmic membrane.82 Thus, most related conjugates have been less active than the parent drug, reflecting the need for an intracellular bacterial release process such as the peptidase-triggered process of the natural albomycins. Attempts to synthesize randomly labile linkers have problems with premature release. The Nolan group exploited the enterobactin hydrolysis-induced iron release process by demonstrating that the esterase-mediated cleavage of an enterobactin-ciprofloxacin conjugate allowed the generation of fragments that were then antibacterially active.83 We synthesized desferrioxamine-ciprofloxacin conjugates (114) with linkers that rely on the rapid Thorpe-Ingold (trimethyl lock) accelerated lactonization of esterase/phosphatase- or reductase-generated phenols (115) to induce drug release after bacterial internalization (Scheme 11).84 While the low activity of the former relative to ciprofloxacin suggested the lack of or slow phenol generation, the reductase-triggered conjugates showed enhanced selective antibacterial activity.
Scheme 11.

While considering a number of other triggered linker possibilities, we decided to exploit the usually detrimental β-lactamase problem associated with siderophore β-lactam conjugates by designing a siderophore-cephalosporin-oxazolidinone conjugate (116, Scheme 12).1 The intent was that the cephalosporin of the conjugate would be the primary warhead in non-β-lactamase-producing bacteria, but in others, hydrolysis by a β-lactamase would release the oxazolidinone (118). Oxazolidinones are normally Gram-positive-only antibiotics with a cytoplasmic target. Assays of the separately synthesized oxazolidinone (119) and cephalosporin (120) conjugates revealed that the oxazolidinone-only conjugate was not active against Gram-negative bacteria and the cephalosporin-only conjugate was active only against non-β-lactamase-producing strains. However, the dual drug conjugate (116) was impressively active against both β-lactamase-producing and nonproducing strains. Separate studies with isolated enzyme confirmed the β-lactamase-triggered release of the oxazolidinone (118). Thus, this dual drug conjugate successfully demonstrated the ability to utilize the β-lactamase problem to deliver and repurpose alternative antibiotics.85
Scheme 12.

REPURPOSING ALTERNATIVE ANTIBIOTICS
Another way to avoid the β-lactamase and cytoplasmic targeting problems is to incorporate non-β-lactam warheads with different modes of action. With this in mind, our recent efforts have focused on repurposing other Gram-positive only antibiotics to make them active against Gram-negative bacteria by siderophore conjugation without the use of special linkers. Again, the consideration of natural sideromycins was helpful. The microcins (25) are enterobactin-based sideromycins with very large peptides (84 mer) as the warhead. The indication is that conjugation with siderophores will allow active transport of components even larger than the siderophore itself. To test this, we synthesized conjugates of large, complex, and Gram-positive-only antibiotics daptomycin (121)4 and the teicoplanins (122, Figure 8).86 Daptomycin binds to bacterial cell membranes, causing depolarization, while the teicoplanins are a mixture of glycopeptides that, like vancomycin, inhibit bacterial cell wall syntheses by binding to the d-ala-d-ala terminus of peptidoglycan. Daptomycin 123 and 128 and teicoplanin conjugates 124 and 129 (Figure 8) are remarkably and selectively potent in vitro against MDR strains of Acinetobacter baumannii, including carbapenemase- and cephalosporinase-producing strains (0.2–1.6 μM) that cause infections of greatest current concern. In vivo, studies of mixed ligand daptomycin conjugate 128 showed that it was well tolerated and efficacious in mice infected with A. bauamannii.
Figure 8.

Repurposing Gram-positive antibiotics to target Gram-negative bacteria.
These studies demonstrate that siderophore conjugation can repurpose normally Gram-positive-only antibiotics to extend their activity to targeted MDR Gram-negative bacteria by using active iron-mediated transport rather than size- and charge-limited passive diffusion. During the preparation of this Account, Boyce et al. reported the design of siderophore conjugates (125 and 127, Figure 8) with bacterial periplasmic protease-susceptible linkers.87 For comparison to our earlier described β-lactamase-triggered strategy, they used the same biscatechol siderophore linked by the WSPKYM peptide to two of the same antibiotics (amino eperezolid 118 and daptomycin 121) that we had used. Microbiological assays demonstrated that the large conjugates were recognized by targeted Gram-negative bacteria (i.e., A. baumannii), and after being actively assimilated, the periplasmic protease caused cleavage at the C-terminal amino acid of the peptide linker to release the pendant drug and induce antibacterial activity. This traceless drug-release process has considerable promise for the expansion of the development of sideromycin-based Trojan horse antibiotic development.
Overall, our goal in this Account is to demonstrate that fundamental chemistry, especially the NH acidity of simple hydroxamic acid derivatives, is useful for the design and syntheses of new antibiotics (β-lactams, including oxamazins and monobactams as well as siderophore mimics that limit bacterial iron uptake) and bacterially targeted sideromycins. Microbial adaptation to the oxidative consequences of our current environment created a critical balance of selective growth and virulence based on iron sequestration. While no antibiotic will ever be perfect, extended studies of this process have the potential to facilitate the design, syntheses, and eventual utilization of novel broad-spectrum and rationally engineered targeted narrow-spectrum antibiotics. Potential resistance mechanisms have generated skepticism, but syntheses and studies of synthetic sideromycins and appropriate consideration of earlier studies of the naturally occurring sideromycins indicate that the essential iron-transport-mediated process can provide new classes of antibiotics that are well tolerated, are active in vivo, and have clinical potential.
ACKNOWLEDGMENTS
M.J.M. gratefully acknowledges continuous funding from the NIH from 1975 (NIH postdoctoral fellowship) through the present NIH MERIT award (R37AI054193) as well as 37 years of consulting and collaborative research with Eli Lilly and Co (1979–2016) and current developmental support through Hsiri Therapeutics. This Account reflects tremendous scientific effort by co-workers at all levels (research associates, postdoctoral researchers, and graduate and undergraduate students) as well as valuable collaborators, many, but not all, of whom are co-authors in the references.
Biographies
Biographies
Marvin J. Miller was born and raised in Dickinson, ND. He received his B.S. degree in chemistry at North Dakota State University, where he was introduced to research by Prof. S. P. Pappas, and his Ph.D. at Cornell under the direction of Prof. G. Marc Loudon. After completing an NIH postdoctoral fellowship in Prof. Henry Rapoport’s group at Berkeley, Dr. Miller joined the faculty at Notre Dame in 1977. He is now the George and Winifred Clark Professor Emeritus. His research program focuses on the development of organic synthetic methodology to facilitate medicinal chemistry research. His wife, Patricia Miller, is the group microbiologist. They also have 4 children and 11 grandchildren.
Rui Liu was born in China. She received her Ph.D in medicinal chemistry at Peking Union Medical College in 2011. After 1 year as an assistant investigator at Shanghai Institute of Materia Medica Chinese Academy of Sciences, she joined Dr. Marvin J. Miller’s group at the University of Notre Dame as a postdoctoral researcher in 2013. Now she is a research scientist. Dr. Liu’s research focuses on the development of novel molecules with bioactivities to treat different diseases, including HBV, HCV, malaria, and TB. She also works on the design and synthesis of new sideromycins to target Gram-positive and Gram-negative bacteria.
KEY REFERENCES
- Liu, R.; Miller, P. A.; Vakulenko, S. B.; Stewart, N. K.; Boggess, W. C.; Miller, M. J. A Synthetic Dual Drug Sideromycin Induces Gram-Negative Bacteria to Commit Suicide with a Gram-Positive Antibiotic. J. Med. Chem. 2018, 61, 3845–3854.1
- Miller, M. J.; Walz, A. J.; Zhu, H.; Wu, C.; Moraski, G. C.; Möllmann, U.; Tristani, E. M.; Crumbliss, A. L.; Ferdig, M.; Checkley, L.; Edwards, R. L.; Boshoff, H. I. Design, Synthesis and Study of a Mycobactin-artemisinin Conjugate that has Selective and Potent Activity Against Tuberculosis and Malaria. J. Am. Chem. Soc. 2011, 133, 2076–2079.2
- Carosso, S.; Liu, R.; Miller, P. A.; Hecker, S. J.; Glinka, T.; Miller, M. J. Methodology for Monobactam Diversification: Syntheses and Studies of 4-Thiomethyl Substituted β-Lactams with Activity Against Gram-Negative Bacteria, Including Carbapenemase Producing Acinetobacter baumannii. J. Med. Chem. 2017, 60, 8933–8944.3
- Ghosh, M.; Miller, P. A.; Möllmann, U.; Claypool, W. D.; Schroeder, V. A.; Wolter, W. R.; Suckow, M.; Yu, H.; Li, S.; Huang, W.; Zajicek, J.; Miller, M. J. Targeted Antibiotic Delivery: Selective Siderophore Conjugation with Daptomycin Confers Potent Activity Against Multi-Drug Resistant Acinetobacter baumannii Both in vitro and in vivo. J. Med. Chem. 2017, 60, 4577–4583.4
The authors declare no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.accounts.1c00004
Contributor Information
Marvin J. Miller, Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana 46556, United States.
Rui Liu, Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana 46556, United States.
REFERENCES
- (1).Liu R; Miller PA; Vakulenko SB; Stewart NK; Boggess WC; Miller MJ A Synthetic Dual Drug Sideromycin Induces Gram-Negative Bacteria to Commit Suicide with a Gram-Positive Antibiotic. J. Med. Chem. 2018, 61, 3845–3854. [DOI] [PubMed] [Google Scholar]
- (2).Miller MJ; Walz AJ; Zhu H; Wu C; Moraski GC; Möllmann U; Tristani EM; Crumbliss AL; Ferdig M; Checkley L; Edwards RL; Boshoff HI Design, Synthesis and Study of a Mycobactin-artemisinin Conjugate that has Selective and Potent Activity Against Tuberculosis and Malaria. J. Am. Chem. Soc. 2011, 133, 2076–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Carosso S; Liu R; Miller PA; Hecker SJ; Glinka T; Miller MJ Methodology for Monobactam Diversification: Syntheses and Studies of 4-Thiomethyl Substituted β-Lactams with Activity Against Gram-Negative Bacteria, Including Carbapenemase Producing Acinetobacter baumannii. J. Med. Chem. 2017, 60, 8933–8944. [DOI] [PubMed] [Google Scholar]
- (4).Ghosh M; Miller PA; Möllmann U; Claypool WD; Schroeder VA; Wolter WR; Suckow M; Yu H; Li S; Huang W; Zajicek J; Miller MJ Targeted Antibiotic Delivery: Selective Siderophore Conjugation with Daptomycin Confers Potent Activity Against Multi-Drug Resistant Acinetobacter baumannii Both in vitro and in vivo. J. Med. Chem. 2017, 60, 4577–4583. [DOI] [PubMed] [Google Scholar]
- (5).Schirrmeister BE; de Vos JM; Antonelli A; Bagheri HC Evolution of multicellularity coincided with increased diversification of cyanobacteria and the Great Oxidation Event. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 1791–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Neilands JB Siderophores: Structure and Function of Microbial Iron Transport Compounds. J. Biol. Chem. 1995, 270, 26723–26726. [DOI] [PubMed] [Google Scholar]
- (7).Bergeron RJ; Wiegand J; McManis JS; Bharti N The Design, Synthesis, and Evaluation of Organ-Specific Iron Chelators. J. Med. Chem. 2006, 49, 7032–7043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Rai V; Fisher N; Duckworkth OW; Baars O Extraction and Detection of Structurally Diverse Siderophores in Soil. Frontiers Microbiol. 2020, 11, 581508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Hider RC; Kong XL Chemistry and biology of siderophores. Nat. Prod. Rep. 2010, 27, 637–657. [DOI] [PubMed] [Google Scholar]
- (10).Stintzi A; Barnes C; Xu J; Raymond KN Microbial iron transport via a siderophore shuttle: A membrane ion transport paradigm. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 10691–10696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Diarra MS; Lavoie MC; Jacques M; Darwish I; Dolence EK; Dolence JA; Ghosh A; Ghosh M; Miller MJ; Malouin F Species selectivity of new siderophore-drug conjugates that usespecific iron uptake for entry into bacteria. Antimicrob. Agents Chemother. 1996, 40, 2610–2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Griffin A; West SA; Buckling A Cooperation and competition in pathogenic bacteria. Nature 2004, 430, 1024–1027. [DOI] [PubMed] [Google Scholar]
- (13).Braun V; Pramanik A; Gwinner T; Köberle M; Bohn E Sideromycins: tools and antibiotics. BioMetals 2009, 22, 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Wencewicz TA; Miller MJ Sideromycins as Pathogen-Targeted Antibiotics. Top. Med. Chem. 2017, 26, 151. [Google Scholar]
- (15).Benz G; Schroeder T; Kurz J; Wuensche C; Karl W; Steffens G; Pfitzner J Konstitution der Desferriform der Albomycine δ1, δ2, ε. Angew. Chem., Int. Ed. Engl. 1982, 21, 1322–1335. [Google Scholar]
- (16).Vertesy L; Aretz W; Fehlhaber H-W; Kogler H Salmycin A–D, antibiotika aus Streptomyces violaceus, DSM 8286, mit siderophor-aminoglycosid-struktur. Helv. Chim. Acta 1995, 78, 46–60. [Google Scholar]
- (17).Bickel H; Mertens P; Prelog V; Seibl J; Walser A Stoffwechselprodukte von mikroorganismen – 53: ueber die konstitution von ferrimycin A1. Tetrahedron 1966, 22, 171–179. [Google Scholar]
- (18).Destoumieux-Garzon D; Peduzzi J; Thomas X; Djediat C; Rebuffat S Parasitism of iron-siderophore receptors of Escherichia coli by the siderophore-peptide microcin E492m and its unmodified counterpart. BioMetals 2006, 19, 181–191. [DOI] [PubMed] [Google Scholar]
- (19).Zheng T; Bullock JL; Nolan EM Siderophore-Mediated Cargo Delivery to the Cytoplasm of Escherichia coli and Pseudomonas aeruginosa: Syntheses of Monofunctionalized Enterobactin Scaffolds and Evaluation of Enterobactin–Cargo Conjugate Uptake. J. Am. Chem. Soc. 2012, 134, 18388–18400. [DOI] [PubMed] [Google Scholar]
- (20).Lin Y-M; Ghosh M; Miller P; Möllmann U; Miller MJ Synthetic sideromycins (skepticism and optimizm): selective generation of either broad or narrow spectrum Gram-negative antibiotics. BioMetals 2019, 32, 425–451. [DOI] [PubMed] [Google Scholar]
- (21).Tomaras AP; Crandon JL; McPherson CJ; Banevicius MA; Finegan SM; Irvine RL; Brown MF; O’Donnell JP; Nicolau DP Adaptation-based resistance to siderophore conjugated antibacterial agents by Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2013, 57, 4197–4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).(a) Gause GF Recent studies on albomycin, a new antibiotic. Br. J. Med. 1955, 2, 1177–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gamburg RL Use of albomycin in pneumonia in children. Pediatriia 1951, 5, 37–44. [PubMed] [Google Scholar]
- (23).Braun V; Braun M Active transport of iron and siderophore antibiotics. Curr. Opin. Microbiol. 2002, 5, 194–201. [DOI] [PubMed] [Google Scholar]
- (24).Sackmann W; Reusser P; Neipp L; Kradolfer F; Gross F Ferrimycin A, a New Iron-Containing Antibiotic. Antibiot. Chemother 1962, 12, 34–45. [PubMed] [Google Scholar]
- (25).Miller MJ; Malouin F Microbial Iron Chelators as Drug Delivery Agents: The Rational Design and Synthesis of Siderophore-Drug Conjugates. Acc. Chem. Res. 1993, 26, 241–249. [Google Scholar]
- (26).Miller MJ Syntheses and Therapeutic Potential of Hydroxamic Acid Based Siderophores and Analogs. Chem. Rev. 1989, 89, 1563–1579. [Google Scholar]
- (27).Winkelmann G Structures and functions of fungal siderophores containing hydroxamate and complex one type iron binding ligands. Mycol. Res. 1992, 96, 529–534. [Google Scholar]
- (28).(a) Van der Helm D; Winkelmann G Hydroxamates and Polycarboxylates as Ion Transport Agents (Siderophores). In Winkelmann G, Winge D, Eds.; Metal Ions in Fungi; Marcel Dekker: New York, 1994; Vol. 11, pp 39–98. [Google Scholar]; (b) Al Shaer D; Al Musaimi O; de la Torre BG; Albericio F Hydroxamate Siderophores: Natural Occurrence, Chemical Synthesis, Iron Binding Affinity and Use as Trojan Horses Against Pathogens. Eur. J. Med. Chem. 2020, 208, 112791. [DOI] [PubMed] [Google Scholar]
- (29).Codd R Traversing the coordination chemistry and chemical biology of hydroxamic acids. Coord. Chem. Rev. 2008, 252, 1387–1408. [Google Scholar]
- (30).(a) Atkin CL; Neilands JB Rhodotorulic acid, a diketopiperazine dihydroxamic acid with growth-factor activity. I. Isolation and characterization. Biochemistry 1968, 7, 3734–3739. [DOI] [PubMed] [Google Scholar]; (b) Lee BH; Gerfen GJ; Miller MJ Constituents of Microbial Iron Chelators. Alternate Syntheses of δ-N-Hydroxy-L-ornithine Derivatives and Applications to the Synthesis of Rhodotorulic Acid. J. Org. Chem. 1984, 49, 2418–2423. [Google Scholar]
- (31).Dolence EK; Lin C-E; Miller MJ; Payne SM Synthesis and Siderophore Activity of Albomycin-like Peptides Derived from N5-Acetyl-N5-Hydroxy-L-Ornithine. J. Med. Chem. 1991, 34, 956–968. [DOI] [PubMed] [Google Scholar]
- (32).Dolence EK; Miller MJ Synthesis of Foroxymithine, a Microbial Fermentation Product and Angiotensin I Converting Enzyme Inhibitor. J. Org. Chem. 1991, 56, 492–499. [Google Scholar]
- (33).Fennell KA; Möllmann U; Miller MJ Syntheses and Biological Activity of Amamistatin B and Analogs. J. Org. Chem. 2008, 73, 1018–1024. [DOI] [PubMed] [Google Scholar]
- (34).Maurer PJ; Miller MJ Microbial Iron Chelators: The Total Synthesis of Aerobactin and its Constituent Amino Acid N(6)-acetyl-N(6)-hydroxylysine. J. Am. Chem. Soc. 1982, 104, 3096–3102. [Google Scholar]
- (35).Roosenberg JM II; Miller MJ Total Synthesis of the Siderophore Danoxamine. J. Org. Chem. 2000, 65, 4833–4838. [DOI] [PubMed] [Google Scholar]
- (36).Lee BH; Miller MJ The Natural Ferric Ionophores: The Total Syntheses of Schizokinen, Schizokinen A, and Arthrobactin. J. Org. Chem. 1983, 48, 24–31. [Google Scholar]
- (37).Lee BH; Miller MJ; Prody C; Neilands JB Artificial Siderophores, 2: Synthesis of Trihydroxamate Analogues of Rhodotorulic Acid and their Biological Iron Transport Capabilities in E. coli. J. Med. Chem. 1985, 28, 323–327. [DOI] [PubMed] [Google Scholar]
- (38).Ratledge C A History of Iron Metabolism in the Mycobacteria. Iron Acquisition by the Genus Mycobacterium. History, Mechanisms, Role of Siderocalin, Anti-tuberculosis Drug Development; Byers BR, Ed.; SpringerBriefs in Biometals; Springer, 2013: Chapter 2. [Google Scholar]
- (39).Snow GA Mycobactins: iron-chelating growth factors from mycobacteria. Bacteriol. Rev. 1970, 34, 99–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Maurer PJ; Miller MJ Total Synthesis of a Mycobactin: Mycobactin S2. J. Am. Chem. Soc. 1983, 105, 240–245. [Google Scholar]
- (41).Mattingly PG; Kerwin JF Jr.; Miller MJ A Facile Synthesis of Substituted N-Hydroxy-2-Azetidinones. A Biogenic Type β-Lactam Synthesis. J. Am. Chem. Soc. 1979, 101, 3983–3985. [Google Scholar]
- (42).Miller MJ The Hydroxamate Approach to the Synthesis of β-Lactam Antibiotics. Acc. Chem. Res. 1986, 19, 49–56. [Google Scholar]
- (43).Floyd DM; Fritz AW; Cimarusti CM Monobactams. Stereospecific synthesis of (S)-3-amino-2-oxoazetidine-1-sulfonic acids. J. Org. Chem. 1982, 47, 176–178. [Google Scholar]
- (44).Rajendra G; Miller MJ Intramolecular Electrophilic Additions to Olefins in Organic Synthesis Stereoselective Synthesis of 3,4-Substituted β-Lactams by Bromine-Induced Oxidative Cyclization of O-Acyl β,γ-Unsaturated Hydroxamic Acid Derivatives. J. Org. Chem. 1987, 52, 4471–4477. [Google Scholar]
- (45).Woulfe SR; Miller MJ Synthesis and Biological Activity of [(3(S)-Acylamino-2-Oxo-1-Azetidinyl)oxy] Acetic Acids. A New Class of Heteroatom Activated β-Lactam Antibiotics. J. Med. Chem. 1985, 28, 1447–1453. [DOI] [PubMed] [Google Scholar]
- (46).Decuyper L; Jukic M; Sosic I; Zula A; D’hooghe MD; Gobec S Antibacterial and β-Lactamase Inhibitory Activity of Monocyclic β-Lactams. Med. Res. Rev. 2018, 38, 426–503. [DOI] [PubMed] [Google Scholar]
- (47).Gordon EM; Ondetti MA; Pluscec J; Cimarusti CM; Bonner DP; Sykes RB O-sulfated β-lactam hydroxamic acids (monosulfactams). Novel monocyclic β-lactam antibiotics of synthetic origin. J. Am. Chem. Soc. 1982, 104, 6053–6060. [Google Scholar]
- (48).Miller MJ; Biswas A; Krook MA Practical synthetic approaches to intermediates for the preparation of the novel O-sulfonated-N-hydroxy-2-azetidinone antibiotics. Tetrahedron 1983, 39, 2571–2575. [Google Scholar]
- (49).Sheehan JC The Enchanted Ring: The Untold Story of Penicillin; MIT Press: Cambridge, MA, 1982. [Google Scholar]
- (50).Hsiao C-N; Liu L; Miller MJ The Use of Cysteine- and Serine-Derived Thiazolidinethiones and Oxazolidine-thiones as Efficient Chiral Auxiliaries in Aldol Condensations. J. Org. Chem. 1987, 52, 2201–2206. [Google Scholar]
- (51).Lotz BT; Gasparski CM; Peterson K; Miller MJ Substrate Specificity Studies of Aldolase Enzymes for use in Organic Synthesis. J. Chem. Soc., Chem. Commun. 1990, 16, 1107–1109. [Google Scholar]
- (52).Bulychev A; O’Brien ME; Massova I; Teng M; Gibson TA; Mobashery S; Miller MJ Potent Mechanism-Based Inhibition of TEM-1 β-Lactamase by Novel N-Sulfonyloxy β-Lactams. J. Am. Chem. Soc. 1995, 117, 5938–5943. [Google Scholar]
- (53).Gasparski CM; Teng M; Miller MJ Functionalization of the β-Lactam Ring: Diastereoselective Azide Transfer and N-O Bond Reduction on C4 Substituted N-Hydroxy-β-lactams in One Step. J. Am. Chem. Soc. 1992, 114, 2741–2743. [Google Scholar]
- (54).(a) Wang DY; Abboud MI; Markoulides MS; Brem J; Schofield CJ The road to avibactam: the first clinically useful non-β-lactam working somewhat like a β-lactam. Future Med. Chem. 2016, 8, 1063–84. [DOI] [PubMed] [Google Scholar]; (b) Miller AA; Shapiro AB; McLeod SM; Carter NM; Moussa SH; Tommasi M; Mueller JP In Vitro Characterization of ETX1317, a Broad-Spectrum β-Lactamase Inhibitor That Restores and Enhances β-Lactam Activity against Multi-Drug-Resistant Enterobacteriales, Including Carbapenem-Resistant Strains. ACS Infect. Dis. 2020, 6, 1389–1397. [DOI] [PubMed] [Google Scholar]
- (55).(a) Polnski T; Chimiak A Oxidation of amino acid esters into N-hydroxy amino acid derivatives. Tetrahedron Lett. 1974, 15, 2453–2456. [Google Scholar]; (b) Naegeli H; Keller-Schierlein W Stoffwechselprodukte von Mikroorganismen. 174. Mitteilung Eine neue Synthese des Ferrichroms; enantio-Ferrichrom. Helv. Chim. Acta 1978, 61, 2088–2095. [Google Scholar]; (c) Bergeron RJ; Phanstiel O IV. The total synthesis of nannochelin: a novel cinnamoyl hydroxamate-containing siderophore. J. Org. Chem. 1992, 57, 7140–7143. [Google Scholar]
- (56).Hu J; Miller MJ A New Method for the Synthesis of Nε-Acetyl- Nε-hydroxy-L-lysine, the Iron-Binding Constituent of Several Important Siderophores. J. Org. Chem. 1994, 59, 4858–4861. [Google Scholar]
- (57).Hu J; Miller MJ An Efficient Synthesis of Cobactin T, A Key Component of the Mycobactin Class of Siderophores. Tetrahedron Lett. 1995, 36, 6379–6382. [Google Scholar]
- (58).Hu J; Miller MJ Total Synthesis of a Mycobactin S, a Siderophore and Growth Promoter of Mycobacterium Smegmatis, and Determination of its Growth Inhibitory Activity against Mycobacterium tuberculosis. J. Am. Chem. Soc. 1997, 119, 3462–3468. [Google Scholar]
- (59).Ji C; Juarez RE; Miller MJ Exploiting Bacterial Iron Acquistion: Siderophore-Conjugates. Future Med. Chem. 2012, 4, 297–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Xu Y; Miller MJ Total Syntheses of Mycobactin Analogs as Potent Antimycobacterial Agents Using a Minimal Protecting Group Strategy. J. Org. Chem. 1998, 63, 4314–4322. [Google Scholar]
- (61).Ratledge C; Brown KA Inhibition of mycobactin formation in Mycobacterium smegmatis by p-aminosalicylate. A new proposal for the mode of action of p-aminosalicylate. Am. Rev. Respir. Dis. 1972, 106, 774–776. [DOI] [PubMed] [Google Scholar]
- (62).Juarez-Hernandez RE; Miller PA; Miller MJ Syntheses of Siderophore-Drug Conjugates Using a Convergent Thiol–Maleimide System. ACS Med. Chem. Lett. 2012, 3, 799–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Juárez-Hernández RE; Franzblau SG; Miller MJ Syntheses of Mycobactin Analogs as Potent and Selective Inhibitors of Mycobacterium tuberculosis. Org. Biomol. Chem. 2012, 10, 7584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Juárez-Hernández R; Zhu H; Miller MJ Siderophore-mediated iron acquisition: Target for the development of selective antibiotics towards Mycobacterium tuberculosis. In Iron Acquisition by the Genus Mycobacterium. History, Mechanisms, Role of Siderocalin, Anti-tuberculosis Drug Development; Byers BR, Ed.; SpringerBriefs in Biometals; Springer: 2013; Chapter 5. [Google Scholar]
- (65).Moraski GC; Chang M; Villegas-Estrada A; Franzblau SG; Möllmann U; Miller MJ Syntheses and Biological Evaluation of Novel Heterocyclic Antituberculosis Agents Derived from Oxazoline and Oxazole Benzyl Esters. Eur. J. Med. Chem. 2010, 45, 1703–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Moraski GC; Markley LD; Hipskind PA; Boshoff H; Franzblau SG; Miller MJ Advent of Imidazo[1,2-a]pyridine-3-carboxamides with Potent Multi- and Extended Drug Resistant Antituberculosis Activity. ACS Med. Chem. Lett. 2011, 2, 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Moraski GC; Markley LD; Cramer J; Hipskind PA; Boshoff H; Bailey MA; Alling T; Ollinger J; Parish T; Miller MJ Advancement of Imidazo[1,2-a]pyridine-3-carboxamides with Nanomolar Activity Against Replicating Multi- and Extensively Drug Resistant Mycobacterium tuberculosis. ACS Med. Chem. Lett. 2013, 4, 675–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Cheng Y; Moraski GC; Cramer J; Miller MJ; Schorey JS Bactericidal activity of an imidazo[1,2-a]pyridine using a mouse M. tuberculosis infection model. PLoS One 2014, 9, e87483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Scherr N; Bieri R; Thosmas SS; Chauffour A; Kalia NP; Schneide P; Ruf M-T; Lamelas A; Manimekalai MSS; Gruber G; Ishii N; Suzuki K; Tanner M; Moraski GC; Miller MJ; Witschel M; Jarlier V; Pluschke G; Pethe K Targeting the Mycobacterium ulcerans cytochrome bc1:aa3 for the treatment of Buruli ulcer. Nat. Commun. 2018, 9, 5370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Lin Y-M; Miller MJ Practical Synthesis of Hydroxamate-Derived Siderophore Components by an Indirect Oxidation Method and Syntheses of a DIG-Siderophore Conjugate and a Biotin-Siderophore Conjugate. J. Org. Chem. 1999, 64, 7451–7458. [Google Scholar]
- (71).Minnick AA; McKee JA; Dolence EK; Miller MJ Iron transport-mediated antibacterial activity of and development of resistance to hydroxamate and catechol siderophore-carbacepholosporin conjugates. Antimicrob. Agents Chemother. 1992, 36, 840–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).McKee JA; Sharma SK; Miller MJ Iron Transport Mediated Drug Delivery Systems: Synthesis and Antibacterial Activity of Spermidine- and Lysine-Based Siderophore-β-Lactam Conjugates. Bioconjugate Chem. 1991, 2, 281–291. [DOI] [PubMed] [Google Scholar]
- (73).Ghosh A; Ghosh M; Niu C; Malouin F; Möllmann U; Miller MJ Iron transport mediated drug delivery using mixed-ligand siderophore-β-lactam conjugates. Chem. Biol. 1996, 3, 1011–1019. [DOI] [PubMed] [Google Scholar]
- (74).Wencewicz TA; Miller MJ Biscatecholate-monohydroxamate mixed ligand siderophore-carbacephalosporin conjugates are selective sideromycin antibiotics that target Acinetobacter baumannii. J. Med. Chem. 2013, 56, 4044–4052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Proschak A; Lubuta P; Grun P; Lohr F; Wilharm G; De Berardinis V; Bode HB Structure and biosynthesis of fimsbactins A-F, siderophores from Acinetobacter baumannii and Acinetobacter baylyi. Chem. BioChem. 2013, 14, 633–638. [DOI] [PubMed] [Google Scholar]
- (76).Budzikiewicz H Siderophore-Antibiotic Conjugates Used as Trojan Horses Against Pseudomonas aeruginosa. Curr. Top. Med. Chem. 2001, 1, 73–82. [DOI] [PubMed] [Google Scholar]
- (77).Zheng T; Nolan EM Enterobactin-Mediated Delivery of β-Lactam Antibiotics Enhances Antibacterial Activity against Pathogenic Escherichia coli. J. Am. Chem. Soc. 2014, 136, 9677–9691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Ji C; Miller PA; Miller MJ Iron transportmediated drug delivery: practical syntheses and in vitro antibacterial studies of triscatecholate siderophoreaminopenicillin conjugates reveals selectively potent antipseudomonal activity. J. Am. Chem. Soc. 2012, 134, 9898–9901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Perraud Q; Cantero P; Munier M; Hoegy F; Zill N; Gasser V; Mislin GLA; Ehret-Sabatier L; Schalk IJ Phenotypic Adaptation of Pseudomonas aeruginosa in the Presence of Siderophore-Antibiotic Conjugates during Epithelial Cell Infection. Microorganisms 2020, 8, 1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Mollmann U; Heinisch L; Bauernfeind A; Kohler T; Ankel-Fuchs D Siderophores as drug delivery agents: application of the ‘Trojan Horse’ strategy. BioMetals 2009, 22, 615–624. [DOI] [PubMed] [Google Scholar]
- (81).Ito A; Nishikawa T; Matsumoto S; Yoshizawa H; Sato T; Nakamura R; Tsuji M; Yamano Y Siderophore cephalosporin cefiderocol utilizes ferric iron transporter systems for antibacterial activity against Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2016, 60, 7396–7401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Schalk IJ; Guillon L Fate of ferrisiderophores after import across bacterial outer membranes: different iron release strategies are observed in the cytoplasm or periplasm depending on the siderophore pathways. Amino Acids 2013, 44, 1267–1277. [DOI] [PubMed] [Google Scholar]
- (83).Neumann W; Sassone-Corsi M; Raffatellu M; Nolan EM Esterase-Catalyzed Siderophore Hydrolysis Activates an Enterobactin–Ciprofloxacin Conjugate and Confers Targeted Antibacterial Activity. J. Am. Chem. Soc. 2018, 140, 5193–5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Ji C; Miller MJ Siderophore-fluoroquinolone conjugates containing potential reduction-triggered linkers for drug release: synthesis and antibacterial activity. BioMetals 2015, 28, 541–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Schalk IJ A Trojan-Horse Strategy Including a Bacterial Suicide Action for the Efficient Use of a Specific Gram-Positive Antibiotic on Gram-Negative Bacteria. J. Med. Chem. 2018, 61, 3842–3844. [DOI] [PubMed] [Google Scholar]
- (86).Ghosh M; Miller PA; Miller MJ Antibiotic repurposing: bis-catechol- and mixed ligand (bis-catechol-mono-hydroxamate)-teicoplanin conjugates are active against multidrug resistant Acinetobacter baumannii. J. Antibiot. 2020, 73, 152–157. [DOI] [PubMed] [Google Scholar]
- (87).Boyce JH; Dang B; Ary B; Edmondson Q; Craik CS; DeGrado WF; Seiple IB Platform to Discover Protease-Activated Antibiotics and Application to Siderophore–Antibiotic Conjugates. J. Am. Chem. Soc. 2020, 142, 21310–21321. [DOI] [PMC free article] [PubMed] [Google Scholar]