

ABSTRACT
Prevotella intermedia readily colonizes healthy dental biofilm and is associated with periodontal diseases. The viscous exopolysaccharide (EPS)-producing capability is known as a major virulence factor of P. intermedia 17 (Pi17). However, the inter-strain difference in P. intermedia regarding virulence-associated phenotype is not well studied. We compared in vivo virulence and whole genome sequences using five wild-type strains: ATCC 49046 (Pi49046), ATCC 15032 (Pi15032), ATCC 15033 (Pi15033), ATCC 25611 (Pi25611), and Pi17. Non-EPS producing Pi25611 was the least virulent in insect and mammalian models. Unexpectedly, Pi49046 did not produce viscous EPS but was the most virulent, followed by Pi17. Genomes of the five strains were quite similar but revealed subtle differences such as copy number variations and single nucleotide polymorphisms. Variations between strains were found in genes encoding glycosyltransferases and genes involved in the acquisition of carbohydrates and iron/haem. Based on these genetic variations, further analyses were performed. Phylogenetic and structural analyses discovered phosphoglycosyltransferases of Pi49046 and Pi17 have evolved to contain additional loops that may confer substrate specificity. Pi17, Pi15032, and Pi15033 displayed increased growth by various carbohydrates. Meanwhile, Pi49046 exhibited the highest activities for haemolysis and haem accumulation, as well as co-aggregation with Porphyromonas gingivalis harbouring fimA type II, which is more tied to periodontitis than other fimA types. Collectively, subtle genetic differences related to glycosylation and acquisition of carbohydrates and iron/haem may contribute to the diversity of virulence and phenotypic traits among P. intermedia strains. These variations may also reflect versatile strategies for within-host adaptation of P. intermedia.
KEYWORDS: P. intermedia, inter-strain difference, virulence, genome, phenotype, within-host adaptation
Introduction
Periodontitis is an infectious-inflammatory disease induced by the interactions between the subgingival polymicrobial biofilm and the host immune system. An earlier DNA-DNA hybridization study of approximately 13,200 plaque samples reported that the ‘red complex’, consisting of Porphyromonas gingivalis, Tannerella forsythia, and Treponema denticola, has a high correlation with clinical measures of periodontitis [1]. Bacteria of the ‘orange complex’, including Fusobacterium nucleatum subspecies, Prevotella intermedia, Prevotella nigrescens, appeared to be more closely related to red complex members than other color-coded (yellow, green, or purple) bacterial complexes [1]. The virulence factors of the red complex have been studied in detail. In particular, the role of P. gingivalis acting as a keystone pathogen has been well established [2]. With advances in microbiome research applying next-generation sequencing technology, numerous additional disease-associated species were identified. This has been the basis for the concept of ‘polymicrobial synergy and dysbiosis’, where synergistic activities of the whole community triggered by keystone pathogens disrupt host immune defenses and cause tissue destruction [3].
Currently, taxonomic characterization of the human microbiome (including oral microbiome) is often limited to the species-level [4]. However, significant genomic and phenotypic variations at the strain-level within a species are common. Inter-strain variations have been well studied especially in terms of pathogenicity. For instances, Escherichia coli and Bacteroides fragilis include both pathogenic and commensal strains [5,6]. Similarly, not all P. gingivalis strains are equally pathogenic and genetic variations exist within the species for many important virulence factors [7]. Many researchers have endeavored to find specific virulence alleles involved in disease and generally have focused on fimbriae, which are filamentous components on the cell surface playing an important role in initiation as well as progression of periodontitis [8]. P. gingivalis has been classified into six types based on nucleotide sequences of fimA encoding fimbrillin (FimA), a subunit protein of fimbriae [9,10]. The most frequently detected genotype in healthy individuals is fimA type I, while Ib, II, and IV are more associated with periodontal disease [9–11]. In particular, the fimA type II allele seems to increase binding affinity for epithelial cell receptors contributing to enhanced virulence [8,12]. Such diversity of the microbial world clearly shows how insufficient it is to define the relationship between human microbiome and host health at species- or higher-level phylogeny.
Our current work focuses on P. intermedia species, a Gram-negative anaerobic rod-shaped bacterium which is a member of the orange complex. It is known that bacterial loads of P. intermedia and P. gingivalis in dental biofilm are strongly associated with each other and their co-colonization is connected to an increase in probing depth [13]. P. intermedia 17 (Pi17) initially isolated from a chronic periodontitis lesion [14] is the most studied strain along with type strain P. intermedia ATCC 25611 (Pi25611). It has been reported that Pi17 produces a viscous exopolysaccharide (EPS), composed mainly of mannose, and induces more severe abscesses in mice than non-EPS-producing Pi25611 [15,16]. Moreover, P17 has been known to be rarely internalized by human polymorphonuclear leukocytes. This may suggest that EPS is a key component contributing to the virulence of Pi17 [15,16]. On the other hand, P. intermedia is also commonly detected in a healthy sulcus [17], an environment very different from a diseased periodontal pocket. The ability of P. intermedia to colonize such distinct niches may reflect inter-strain genetic diversity and elaborate mechanisms for within-host adaptation. However, these are not yet clearly understood and the diversity of virulence-associated phenotypes of P. intermedia strains is not well studied.
The purpose of this study is to evaluate the difference of wild-type P. intermedia strains focusing on virulence-associated phenotypes and genomic sequences. Based on the results, we infer several versatile strategies that P. intermedia uses to continuously adapt to dynamic conditions in the oral cavity. Here, we used Pi17 and publicly available strains ATCC 49046 (Pi49046), ATCC 15032 (Pi15032), ATCC 15033 (Pi15033), and type strain ATCC 25611 (Pi25611). To examine in vivo virulence, we adopted two infection models, a mouse subcutaneous abscess model and a wax moth larvae Galleria mellonella model. Both models have been widely used for the evaluation of bacterial virulence [18,19].
Results and discussion
Inter-strain difference in EPS-forming ability and in vivo virulence
Pi17 is known to produce viscous EPS, a key virulence factor contributing to its survival by interfering with internalization by phagocytes [16]. Pi17 induced more severe abscesses in mice than non-EPS-producing Pi25611 [16], which is consistent with our results (Figures 1 and 2). We observed that viscous materials with fine fibrous structures formed into bundles in the cultures of Pi17, while such viscous materials were not observed in the culture supernatants of the other strains. Unexpectedly, Pi49046 induced the highest larval death (77%) (Figure 2(a)). Strains Pi17, Pi15032, Pi15033, and Pi25611 killed 50%, 43%, 43%, and 10% of the infected larvae, respectively. Inter-strain difference in the virulence in vivo was similarly observed in mice. Pi49046 induced the largest abscesses starting from day 3 (Figure 2(b)). Pi17 induced medium-sized abscesses starting from day 5. However, the other strains did not cause abscesses in mice. These results allude that, beside EPS, other factors influence the in vivo survival and virulence level of P. intermedia.
Figure 1.

Comparison of P. intermedia strains grown in liquid medium. In the cultures of Pi17, viscous materials with fine fibrous structures that formed into bundles were observed. On the other hand, such viscous materials were not observed in the culture supernatants of the other strains.The bacterial cells were grown in B-HK medium for 24 h at 37°C anaerobically (80% N2, 10% H2, 10% CO2).
Figure 2.

In vivo virulence of P. intermedia strains. (a) Galleria mellonella infection model. Larvae weighed between 170 and 220 mg at time of inoculation. Bacterial cell suspensions (2 × 107 CFU/larva) of each strain were injected into the larvae. Survival was monitored daily for 72 h and larval death was confirmed by the lack of touch-induced movement. Images are representative of three independent experiments. (b) Murine skin infection model. Male BALB/c mice (4 weeks old; 5 mice per strain) were used. Bacterial cell suspension (5 × 109 CFU/mouse) of each strain was injected into the inguinal area of each mouse to induce abscess.
Overview of comparative genomic analysis
Despite apparent difference between strains in the virulence level, the genomes of the five strains showed approximately 94% average nucleotide identity (in forward direction) (Figure 3(a)). The pangenome of five strains (Figure 3(b)) consisted of 1,615 core gene clusters (60.1%, exist in all strains), 585 dispensable gene clusters (21.8%, shared partially among strains) and 486 strain-specific gene clusters (18.1%). Pi15032 and Pi15033 shared 239 dispensable gene clusters but few strain-specific gene clusters. This indicates their close phylogenetic relationship and also aligns with the results of our in vivo experiments showing the same virulence level between the two strains. Meanwhile, Pi17, Pi25611, and Pi49046 possessed 190, 147, and 140 strain-specific gene clusters, respectively. It is noteworthy that most of the genetic differences between the strains was copy number variations (CNVs) and single nucleotide polymorphisms (SNPs). In total 92,010 SNPs in core genes were identified.
Figure 3.
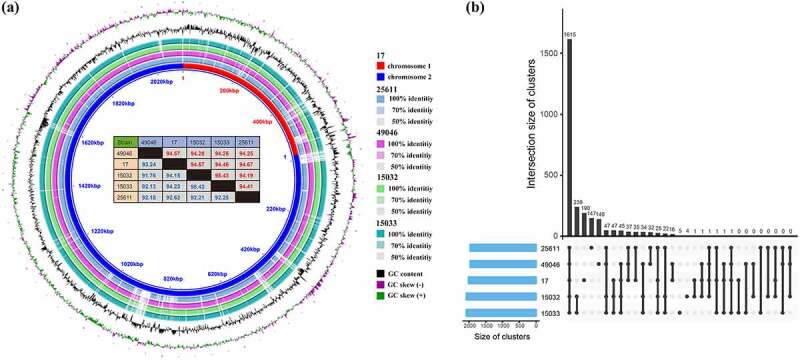
Comparative genomic analysis of five P. intermedia strains. (a) Genome sequence similarity performed by BRIG. Whole genome pairwise alignments were performed using MUMmer. Numbers in red and blue (in the table) represent the similarities between two genomes in forward and reverse direction, respectively. (b) Pangenome analysis using PGAP method. Pangenome gene clusters in intersections are shown as filled (black) circles and the number of clusters is presented in the bar plot above.
Genetic variations associated with glycosyltransferases and nutrient acquisition
Both bacterial pathogens and commensals are surrounded by diverse carbohydrate structures (glycans) which mediate specific interactions with the host and play an important role in all stages of infection [20, 21]. Moreover, protein glycosylation contributes to attachment, infectivity, phenotypic phase-variation, and antigenic diversity, all of which are important for pathogenicity [22–31]. We found inter-strain differences in the genes involved in biosynthesis of glycoconjugates (Table 1). These include several genes encoding glycosyltransferases which catalyze the transfer of sugar moieties. Notably, Pi49046 and Pi17 shared a unique gene encoding phosphoglycosyltransferase (PGT, WP_061869517). We detected strain-specific genes as well as hypervariable genes involved in the acquisition of nutrients, such as carbohydrates (SusC, SusD, polysaccharide export protein) and iron/haem. Among the several hypervariable genes encoding transcriptional regulators, the two-component transcriptional regulators consisting of LytT and LytS was identified in a location immediately upstream of cysteine protease involved in haem acquisition [32]. Some genetic variations were associated with replication, recombination and repair, and defence systems (supplementary data 1).
Table 1.
Inter-strain genetic variations associated with glycosylation, nutrient acquisition and transcriptional regulation.
| Protein ID | Name | Z-score | D | S |
|---|---|---|---|---|
| WP_061869095 | glycosyltransferase involved in cell wall biosynthesis | 2.5716 | - | - |
| WP_061868170 | glycosyltransferase involved in cell wall biosynthesis | - | - | 49046 |
| WP_061869517 | phosphoglycosyl transferase (PGT) family protein | - | 17/ 49046 | - |
| WP_014710060 | polysaccharide pyruvyl transferase | - | - | 17 |
| WP_014710061 | glycosyltransferase involved in cell wall biosynthesis | - | - | 17 |
| WP_014710064 | glycosyltransferase involved in cell wall biosynthesis | - | - | 17 |
| WP_076169367 | glycosyltransferase involved in LPS biosynthesis | - | - | 17 |
| WP_014710262 | glycosyltransferase involved in LPS biosynthesis | 3.1702 | - | |
| WP_028906217 | glycosyltransferase involved in cell wall biosynthesis | - | - | 25611 |
| WP_028906222 | glycosyltransferase involved in cell wall biosynthesis | - | - | 25611 |
| WP_028906219 | glycosyltransferase involved in cell wall biosynthesis | - | - | 25611 |
| WP_028906216 | glycosyltransferase involved in cell wall biosynthesis | - | - | 25611 |
| WP_124140245 | glycosyltransferase involved in cell wall biosynthesis | - | 15032/15033 | |
| WP_061868928 | periplasmic protein involved in polysaccharide export | 2.0949 | - | - |
| WP_061869075 | SusC family | 2.3970 | - | - |
| WP_061869074 | SusD family | 3.8120 | - | - |
| WP_061869484 | haem-binding protein HmuY | 2.3317 | - | - |
| WP_061868464 | outer membrane receptor proteins, mostly Fe transport | 4.4638 | - | - |
| WP_061868395 | outer membrane receptor proteins, mostly Fe transport | 4.1508 | - | - |
| WP_061868524 | outer membrane receptor proteins, mostly Fe transport | 2.7522 | - | - |
| WP_061868193 | outer membrane receptor proteins, mostly Fe transport | S | 49046 | - |
| WP_061869506 | outer membrane receptor proteins, mostly Fe transport | S | 49046 | - |
| WP_061869221 | outer membrane receptor proteins, mostly Fe transport | S | 49046 | - |
| WP_045168284 | outer membrane receptor proteins, mostly Fe transport | 15032/15033 | - | |
| WP_061868482 | sensor histidine kinase LytS | 3.0623 | ||
| WP_061868483 | transcriptional regulator LytT | 5.5683 | ||
| WP_061869463 | hybrid sensor histidine kinase/response regulator | 3.3029 | ||
| WP_061869215 | AraC family transcriptional regulator | 2.9004 | ||
| WP_061869012 | MarR family transcriptional regulator | 2.0301 | ||
| WP_014709474 | AraC family transcriptional regulator | - | 17 | |
| WP_014709590 | AraC family transcriptional regulator | - | 17 | |
| WP_044048113 | TetR/AcrR family transcriptional regulator | - | 17 | |
| WP_061869376 | Predicted transcriptional regulator, contains HTH domain | - | 49046 |
Genes with a z-score ≥1.96 were considered as hypervariable genes between the strains.
S, strain-specific genes.
D, dispensable gene shared between two strains.
Based on these genetic variations, we further conducted phylogenetic and structural analyses of PGTs, and phenotypic evaluations regarding acquisition of nutrients.
Phylogenetic and structural analyses show unique PGTs of Pi49046 and Pi17
PGTs initiate a common step in the biosynthesis of both essential and virulence-associated bacterial glycoconjugates, including glycoproteins, LPS, peptidoglycan [33]. To extensively investigate the inter-strain variation of P. intermedia PGTs, we performed phylogenetic analysis using PGTs of 31 strains, which were downloaded from NCBI genome database. All but one strain (KCOM 1779) harboured two PGTs. Phylogenetic analysis separated the 61 PGTs into three distinct clades A, B, and C (Figure 4(a–c), supplementary data 2 and 3). Clade C consists of well-conserved PGTs across all P. intermedia strains. The other 30 PGTs belong to either clade A or clade B. Besides a well-conserved PGT (clade C), Pi25611, Pi15032, and Pi15033 possess another PGT belonging to clade A. This PGT is phylogenetically close to C. jejuni PglC and N. gonorrhoeae PglB. Both PglC and PglB belong to the second superfamily containing the minimal functional core of WbaP and are involved in N-linked and O-linked protein glycosylation pathways, respectively [33–35]. On the other hand, Pi49046 and Pi17 possessed phylogenetically different PGTs (clade B). The 3D structures of PGTs were modeled by SWISS-MODEL web server [36] (Figure 4(d)). Besides the conserved regions (green color), additional loop regions were found only in Pi49046 PGT and Pi17 PGT (blue and red). We further performed motif analysis of amino acid sequences for the additional loop regions. Both loop region sequences were presumed to have a β-sheet sandwich structure similar to bacterial immunoglobulin-like Ig domain 1 (PS51127, supplementary data 4).
Figure 4.

Phosphoglycosyltransferases (PGTs) of P. intermedia. (a) Phylogenetic analysis using 61 PGTs of 31 strains of P. intermedia. All but one strain (KCOM 1779) harbour two PGTs. The two virulent strains Pi49046 and Pi17 carry PGT belonging to clade B (green lines), while the other three strains (Pi15032, Pi15033 and Pi25611) carry PGT belonging to clade A (red lines). (b) Multiple sequence alignment using PGTs of P. intermedia with well-known PGT subfamilies (PglC and PglB). (c) Phylogenetic tree based on PGT was constructed using multiple sequence alignment. (d) Structural model of PGTs. C. concisus PGT (PDB ID 5W7 L, chain A) was used as the structural template for modelling. Additional loop regions in Pi49046 PGT and Pi17 PGT were coloured as blue and red, respectively. Structurally conserved regions were represented using green colour. Magenta stick configuration residues (K59, D93, E94 and R112) represents the conserved active sites.
Recent structural analyses demonstrated that variable loops inserted on common core structures of glycosyltransferases confer substrate (sugar) specificity and determine the orientation of nucleophilic hydroxyl groups [37]. Although the exact role of the additional loops is not clear at present, it seems, at least, clear that continuous evolution of glycosyltransferases allows these enzymes to assemble diverse glycan structures that are not directly copied from genomic templates [37]. This not only applies to eukaryotic systems but also applies to prokaryotic systems [37]. Meanwhile, regarding the relationship between bacterial glycoconjugates and diseases, a previous study using pooled sequences (not comparison between individual strain) showed that disease-derived and health-derived P. intermedia sequences are different with each other in a manner that was associated with glycoconjugate synthesis [38]. All these results suggest that these subtle but continuous genetic variation in glycosyltransferases (including PGTs) may reflect a strategy by the P. intermedia species to adapt to dynamic oral environments as they transition from healthy to diseased states. It is also plausible that genetic mutations in glycosyltransferases may allow for the synthesis of strain-specific glycan and glycan binding proteins, thus resulting in either increased or decreased virulence. Along with the exact function of the additional loops in PGTs, this hypothesis also needs to be validated in the future studies.
Inter-strain phenotypic difference in harnessing carbohydrates
We found differences amongst strains based on their nutrient acquisition-related genes (Table 1), including SusC and SusD, which are known to play roles in the binding and transport of carbohydrates. Variations in the SusC and SusD orthologs can reflect bacterial adaptations to diet or host-derived glycans [39]. Hence, we further evaluated the phenotypic traits of P. intermedia in carbohydrate utilization. Consistent with a previous report [40], glucose enhanced the growth of Pi25611 (Figure 5). The growth promoting effect of glucose was very high in Pi49046 and Pi25611, but relatively low in Pi17, Pi15032, and Pi15033. Meanwhile, the growth of Pi17, Pi15032 and Pi15033 was also significantly increased by sucrose and raffinose, unlike Pi25611 and Pi49046.
Figure 5.
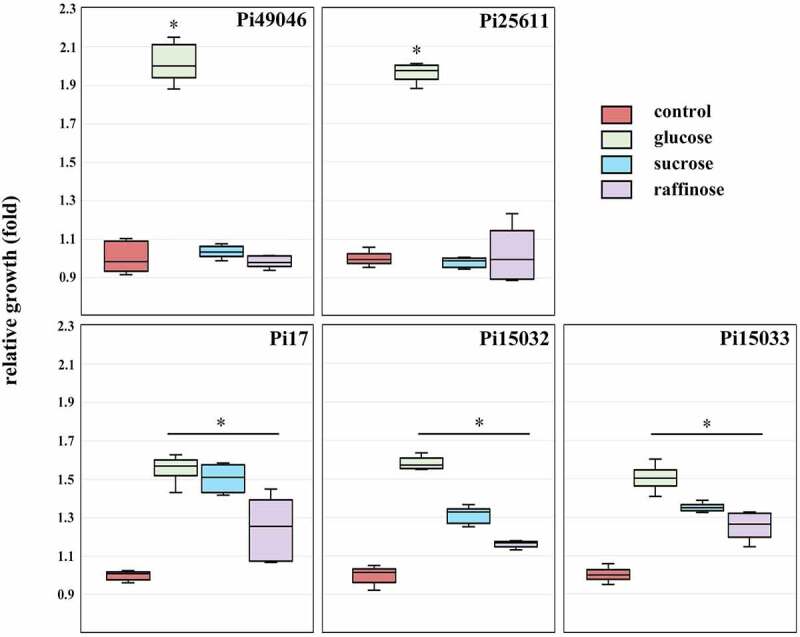
Carbohydrate utilization of P. intermedia strains. Bacterial growth in the presence of carbohydrates is expressed as a ratio relative to growth in the absence of carbohydrates (control). *, P <0.01, versus control. Mann-Whitney U test.
In general, the proteolytic ability is believed to be the most important characteristic of periodontal pathogens. This is partly due to the abundance of nitrogen compounds in the diseased pocket and the cytotoxicity of metabolic end products. However, the saccharolytic properties of periodontal pathogens have not attracted much attention and are not well studied. A recent study [41] uncovered that exogenous pyruvate can be a valuable alternative carbon and energy source of P. gingivalis which has long been known to not utilize carbohydrates as carbon and energy sources [40, 42]. Such metabolic plasticity of P. gingivalis appears to be a significant evolutionary advantage supporting the survival and persistence of the bacterium in the oral cavity [41]. Likewise, the metabolic flexibility of P. intermedia that allows it to utilize both nitrogenous compounds and carbohydrates may serve as a beneficial factor for its adaptation to the highly dynamic environment of the oral cavity. Certain strains capable of harvesting various types of carbohydrates may be more favorable for survival in a healthy gingival sulcus, an environment more difficult to acquire nitrogenous compounds than a diseased pocket. It is interesting to speculate that survival in healthy gingival sulcus may favor certain P. intermedia strains that may survive in haem/iron and protein-limited conditions while utilizing diet- or host-derived glycans. Once the strain has successfully colonized the healthy gingival sulcus, genetic variations (such as CNVs or SNPs) can be induced by selective pressures of environmental or host adaptation. The myriad interactions among co-colonizing species and strains may allow for genetic reassortment, and subsequent selection of new variants possessing enhanced suitability for the changing environment [7].
Inter-strain phenotypic difference in haemolysis, haem accumulation, and co-aggregation with P. gingivalis
Haem is an essential growth factor as well as virulence regulator for both P. intermedia and P. gingivalis [43]. They produce black pigments on blood–containing media. The black pigments accumulated on the surface are composed of haem (Fe2+-protoporphyrin IX, also known as
haemin and Fe3+-protoporphyrin IX, depending upon the oxidation state of iron), which are derived via lysis of erythrocytes and proteolytic breakdown of haemoglobin [44–46]. Co-aggregation between P. gingivalis and P. intermedia is known to be attributable to the acquisition of haem. P. gingivalis acquires it through a unique system consisting of the haem-binding lipoprotein HmuY and gingipain proteases [43]. Similarly, a major extracellular cysteine protease (called interpain A) and two HmuY proteins are involved in haemin acquisition of P. intermedia [32]. Importantly, the possibility of a direct collaboration between P. gingivalis HmuY, and P. intermedia cysteine protease has been reported [43]. This suggests that P. gingivalis may benefit from the proteolytic activity of co-aggregated P. intermediate cells during haemin acquisition [43]. In the present study, we found inter–strain genetic variations related to haem/iron acquisition (Table 1). These include two-component transcriptional regulator consisting of LytT and LytS, which are located immediately upstream of gene encoding cysteine protease. Hence, we further evaluated the strains for activities for haemolysis, haem accumulation, and co-aggregation with P. gingivalis.
As shown in Figure 6(a,b), Pi49046 exhibited the largest beta haemolytic area in both types of media (brain heart infusion agar containing horse or sheep blood) and the highest activity for haem accumulation. Pi25611 produced a larger haemolytic area in the horse blood-supplemented agar than sheep blood-supplemented agar, but exhibited lower activities for haemolysis and haem accumulation than Pi49046. The other three strains showed relatively low activity for haemolysis as well as haem accumulation. To evaluate the co-aggregation activity, we used P. gingivalis A7A1–28 (carrying fimA type II) and fimA II-deficient mutant [47]. As described above (introduction), fimA type II is known to be more associated with periodontitis than other fimA types [8,11,12]. Figure 6c shows Pi49046 possesses the highest co-aggregation activity with P. gingivalis A7A1–28, but not with the mutant. By contrast, the other strains showed lower co-aggregation activity with P. gingivalis A7A1–28 than Pi49046. Interestingly, they still possessed a low degree of co-aggregation activity with the mutant. These results demonstrate that co-aggregation between P. gingivalis and P. intermedia is in a strain-specific manner and Pi49046 has a unique surface property that interacts with type II fimA. Further research is needed to elucidate which factors (for example, glycoconjugates) of P. intermedia influence these strain-specific co-aggregation.
Figure 6.
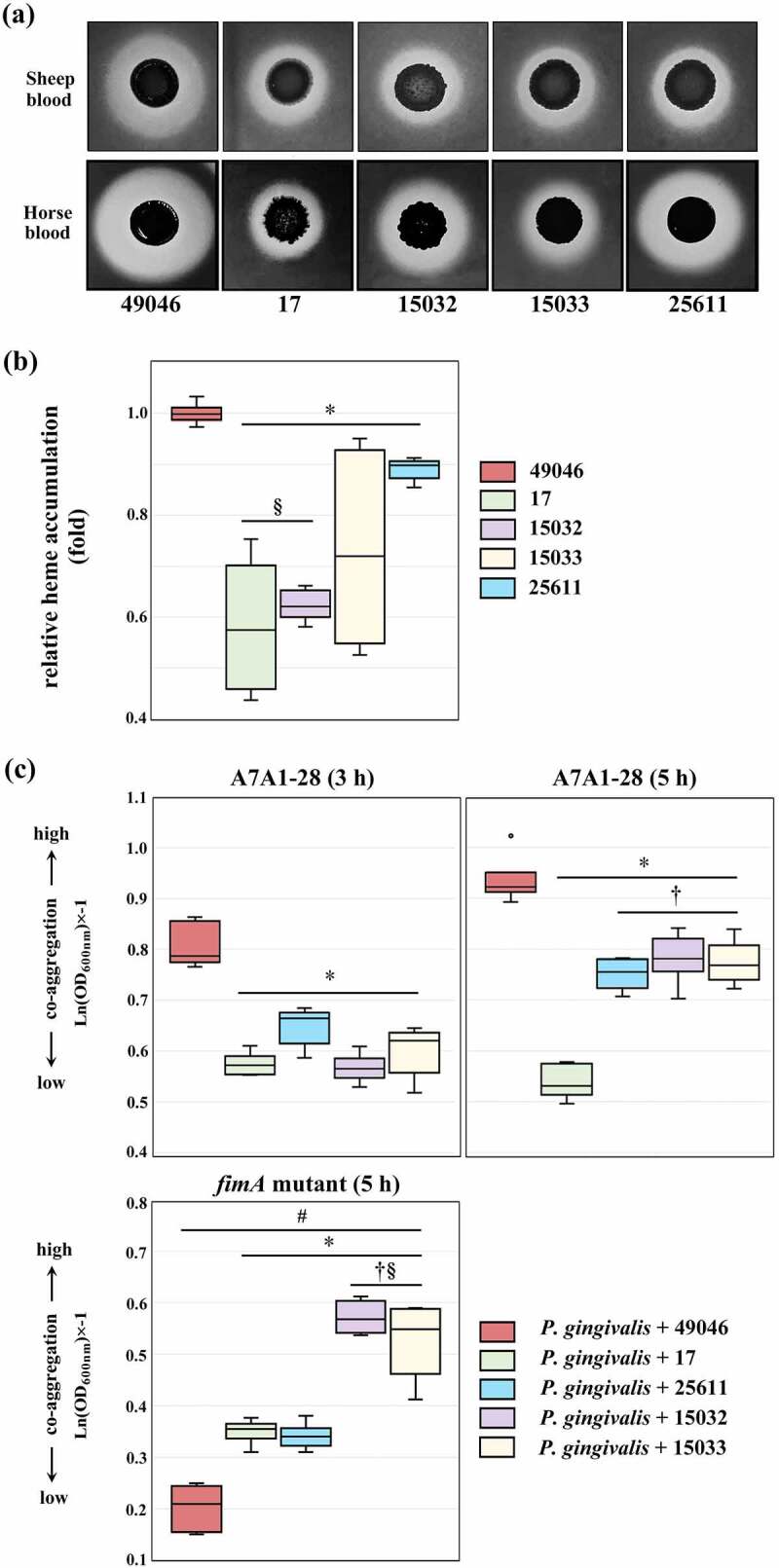
Phenotypic evaluation of nutrient acquisition. (a) Haemolytic activity. Images of beta-haemolysis zones produced after 7 days. (b) Haem accumulating activity. The haem-accumulating activity of each strain is expressed as a ratio relative to the haem-accumulating activity of Pi49046. (c) Co-aggregation activity with P. gingivalis A7A1–28 (carrying fimA type II) and fimA II-deficient mutant. Data are presented as the natural logarithm of OD600 × (‒1). *, P <0.01, versus Pi49046; †, P <0.01, versus Pi17; §, P <0.01, versus Pi25611; #, A7A1–28 (5 h) versus fimA II-deficient mutant. Mann-Whitney U test.
At present, strains isolated from deep periodontal pockets are considered more pathogenic than strains that colonize the dental biofilm of a healthy gingival sulcus. Also, it seems very convincing that DNA exchange between strains generates pools of chimeric progeny with diverse phenotypes and subsequent competition between these strains gives rise to the predominance of certain strains suitable for current host conditions [7]. During adaptation to a specific host, some P. intermedia strains can acquire specific allele combinations conferring advantages in both haem acquisition and coaggregation with P. gingivalis possessing type II fimA. Assuming this is correct, it is also possible to presume that, in hosts that have allowed the combination of these strains to dominate, dysbiosis events are more likely to occur leading to serious disease outcomes.
Conclusions
Our current work showed that CNVs and SNPs are considerable sources of genetic diversity of P. intermedia. We recognize the need to link specific genetic mutations more precisely with phenotypic outcomes through further research. Within this limitation, we concluded that the subtle genetic differences related to glycosyltransferase and harnessing of carbohydrates and haem/iron may contribute to difference in the virulence-associated phenotypes of P. intermedia strains. This may also illustrate versatile strategies for within-host adaptation of P. intermedia. Given that within-host adaptation through genomic changes is a distinctive feature of chronic bacterial infection, our findings underline future extensive studies about subspecies level variations within a species of periodontal pathogen. This is essential for establishing the fundamental understanding of periodontitis from the initiation of an infection to host-dependent disease outcomes. In addition, ongoing efforts for strain-level epidemiology of the oral microbiome will provide a practical framework for intervening in host-microbial interactions.
Materials and methods
Bacterial strains and culture condition
Wild type P. intermedia strains (Pi25611, Pi49046, Pi15032, and Pi15033), and P. gingivalis A7A1–28 (ATCC 53977) carrying fimA type II were selected from our culture collections. Pi17 and fimA type II-deficient mutant of P. gingivalis were kindly supplied by Osaka Dental University. The bacterial cells were grown in B-HK medium, comprised of brucella broth (Becton, Dickinson and Company, Sparks, MD, USA), 5 mg/L haemin, and 1 mg/L vitamin K1. To evaluate the effect of carbohydrates on the growth of P. intermedia, the bacterial cells were cultured for 18 h in a 3:1 mixture medium of B-HK and Bacteroides minimal medium (MM) [48], supplemented with 0.5% (w/v) glucose, sucrose, or raffinose. All bacterial cells were cultured at 37°C anaerobically (80% N2, 10% H2, 10% CO2).
Infection of Galleria mellonella larvae
Experiments were performed as previously described with some modifications [49,50]. Briefly, G. mellonella larvae were purchased from S-worm (Cheonan, South Korea), maintained in darkness at room temperature and used within 2 weeks. Only healthy-looking larvae with no melanization were used in the experiments. Larvae weighed between 170 and 220 mg at time of inoculation. Prior to infection, an overnight culture of each strain was inoculated into fresh medium. The initial optical density at 600 nm (OD600) was adjusted to 0.1, which was determined to be equivalent to 2 × 108 colony forming unit (CFU)/mL by plate counting. After 8 h, OD600 of all strains was between 0.6 and 1.0, corresponding to mid-log phase of growth. Each bacterial culture was harvested, washed and adjusted to the required concentration with sterile phosphate-buffered saline (PBS). Randomly assigned sets of 20 larvae were contained in a petri dish. Each larva was each injected with 10 µL of inoculum (2 × 107 CFU/larva) using a hypodermic needle (30 G, 25 mm) and an infusion pump (KDS100, KD Scientific, Holliston, MA, USA), with setup speed to 1 μL/sec. Larvae injected with 10 μL of sterile PBS were included as control group. Post-infection, the larvae were maintained in the darkness at 37°C, monitored for 72 h, and the number of live larvae was recorded every 24 h.
Animal studies
Bacterial virulence was examined using a murine subcutaneous abscess model as previously described [16], with some modifications. Sample size was calculated by applying the acceptable range of degrees of freedom between 10 and 20 [51]. Briefly, 25 male BALB/c mice (4 weeks old) weighing 14–18 g were purchased from Nara biotech (Seoul, South Korea) and randomized into 5 groups (5 mice per bacterial strain). All animals were raised in polycarbonate cages (1 group per cage) housed in well-aerated rooms at approximately 25°C, fed with standard rodent diet and water ad libitum. The inguinal region of each mouse was injected subcutaneously with 500 μL of bacterial suspensions (5 × 109 CFU/mouse). Animals were monitored daily, and changes of abscess lesions were recorded photographically for 8 consecutive days. We performed all animal procedures according to the guidelines of the Institutional Animal Care and Use Committee (IACUC) at Kyung Hee University using approved protocol (KHUASP(SE)-16–155).
Whole genome sequencing of Pi49046, Pi15032, and Pi15033
A Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA) was used to extract the bacterial genomic DNA. Purified whole genomic DNA was fragmented and the overhangs were cleaved to create blunt ends. A clean up step was performed using the AMPure XP Beads, then the 3’ ends of the DNA fragments were adenylated and ligated to the index adapters. Subsequently, these DNA fragments were separated and extracted purified using a 2% agarose gel. After DNA library construction by PCR using adapter specific primers, paired-end sequencing was performed on the Illumina Hi-Seq 2500 platform. Raw reads with low quality were removed, then the genome assembly was carried out using A5 assembler, as described previously [52]. Gene annotation of the draft genome was performed using the NCBI Prokaryotic Genome Annotation Pipeline [53]. The genome sequences are available at the National Center for Biotechnology Information (NCBI) genome database under the accession NZ_LBGT00000000.1, NZ_QXEM00000000.1, and NZ_QXEN00000000.1. The BioProject IDs are PRJNA281562 and PRJNA488694. Genome information are provided in supplementary data 5.
Comparative genomic analysis
Pairwise genome sequence comparisons were performed using MUMmer (version 3.23) [54]. The overall similarities between any two genomes were calculated by mummerplot. For exploring the genetic contents variation, we applied pan-genomes analysis pipeline (PGAP) [55] and gene family method with default parameters (score: 40, evalue:1 × 10−10, identity: 0.5, and coverage: 0.5). The orthologous clusters (or genes) in each strain from PGAP output results were classified as core, dispensable and strain-specific clusters (or genes). To obtain the comprehensive functional annotation and orthologous gene information, NCBI RefSeq protein coding sequences for the five strains were submitted to the eggNOG-mapper webservice [56]. Functional classification based on Clusters of orthologous groups (COG) was performed using RPSBLAST program on COG database [57] implemented in WebMGA. The results are provided in supplementary data 6.
SNP analysis and Sanger sequencing
SNPs in each core gene were identified using the NUCmer program in the MUMmer (version 3.23). Sequence of Pi49046 was set to the reference sequence and the sequence from the other strains was used as a query sequence. The number of detected SNPs in each core gene was normalized by the total gene length. The normalized SNP rate values of whole core genes (SNP ratios) were tested with R package fitdistrplus. Based on the fitted distribution, z-score of SNP ratio in each core gene was calculated. We identified 92,010 single nucleotide polymorphisms (SNPs) in 1,719 core genes and any gene with a z-score of 1.96 or higher was considered as a hypervariable gene. Five genes were selected among the hypervariable genes, and 25 primer pairs were constructed (supplementary data 7) for confirmation by conventional Sanger sequencing. We used 50 ng of template, 250 nM of primers and PrimeSTAR Max DNA Polymerase (Takara-Bio, Otsu, Japan) for each 20 μL reaction. Cycling conditions comprised of an initial denaturation (94°C for 3 min), and 30 cycles of denaturation (94°C for 30 s), annealing (60°C for 30 s) and extension (72°C for 1 min). PCR amplicons were purified by Promega Wizard SV PCR and Gel Clean Up kits then used for Sanger sequencing performed at Theragen Etex Bio Institute (Suwon, South Korea). The SNPs of the selected genes were confirmed by Sanger sequencing (supplementary data 8).
Phylogenetic analysis of PGT
PGT protein sequences of 31 strains of P. intermedia (Pi17, Pi25611, Pi49046, Pi15032, Pi15033, 17–2, OMA14, KCOM 2033, KCOM 2836, KCOM 1949, KCOM 2837, KCOM 2838, KCOM 1944, KCOM 1741, KCOM 1944, KCOM 2734, DSM 20706, ZT, WW2834, WW855, WW414, KCOM 1653, KCOM 1101, KCOM 2069, KCOM 1779, KCOM 1945, KCOM 2698, KCOM 2833, KCOM 2832, NCTC13070, KCOM 1107) were downloaded from NCBI genome database. The phosphoglycosyl transferase family protein sequences from other bacterial species downloaded from the UniProt database [58] are: Campylobacter concisus PglC (A0A0M4SI81); C. jejuni PglC (O86156); Helicobacter pullorum PglC (E1B268); Neisseria. gonorrhoeae PglB (A0A1D3HQ90); N. meningitidis PglB (Q9RR58); Pseudomonas putida Sugar transferase (A0A0P7CW64). Multiple sequence alignment of the phosphoglycosyl transferase family protein sequences with PGTs from P. intermedia were performed using ClustalW tool. The phylogenetic tree was built with neighbour joining method [59] and the JTT matrix-based method was used to compute the evolutionary distances. The constructed tree was evaluated by bootstrap method with 1,000 bootstrap replications.
Structural modelling and motif analysis
The structures of PGTs from the five strains of P. intermedia were modelled by SWISS-MODEL web server [36]. Briefly, the PGTs were aligned against structural protein sequence databases in Protein Data Bank (PDB). Among the template sequences reported by the server, monotopic PGT from C. concisus (PDB ID 5W7 L) [60] was chosen to serve as a template. The 3D structures were visualized by PyMol software. Motif analysis for the additional loop regions was performed using Motif Scan server (https://myhits.sib.swiss/cgi-bin/motif_scan) and the extracted amino acid sequences.
Evaluation of haemolysis and haem accumulating activity
Haemolytic activity was evaluated as previously described [61]. Briefly, brain heart infusion agar (Becton, Dickinson and Company, Sparks, MD) containing 5% either horse or sheep blood were prepared in the absence of haemin and vitamin K1. The plates were inoculated with each bacterial strain at approximately 108 CFU/spot. The inoculated plates were incubated at 37°C anaerobically for 7 days. Haem-accumulating activity of P. intermedia cells was measured as described previously [46]. The amount of haemin accumulated on the bacterial cell was calculated as the difference between the total amount of haemin added and the amount remaining in the supernatant after the 2-h incubation.
Co-aggregation assays
We performed the experiments as previously described [62] using P. gingivalis A7A1–28, fimA type II-deficient mutant, and the five strains of P. intermedia. Briefly, overnight cultures of each strain were harvested by centrifugation and washed twice with the coaggregation buffer which consisted of 10 mM Tris buffer (pH 8.0), 0.1 mM CaCl2, 0.1 mM MgCl,2, and 150 mM NaCl. Each bacterial suspensionwas adjusted to OD600 of 1.0. Equal volumes (3.0 mL) of each bacterial suspension were added to a glass bottle. All glass bottles were stored aerobically at 37 °C. Every 1 h, over a period of 5 h, the upper phase (0.2 mL) of the bacterial solutions was transferred to measure OD600.
Supplementary Material
Funding Statement
This research was supported by the National Research Foundation of Korea (NRF) funded by the Ministry of Science & ICT (2021R1A2C2008180).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Author’s contributions
K.H. Kwack contributed to data acquisition and drafted the manuscript. E.Y. Jang and S.B. Yang contributed to data acquisition. J.H. Lee contributed to conception, design, data analysis, interpretation, and critically revised the manuscript. J.H. Mooncontributed to conception, design, data analysis and interpretation, drafted, and critically revised the manuscript. All authors gave final approval and agreed to be accountable for all aspects of the work.
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/21505594.2022.2095718
References
- [1].Socransky SS, Haffajee AD, Cugini MA, et al. Microbial complexes in subgingival plaque. J Clin Periodontol. 1998;25:134–144. [DOI] [PubMed] [Google Scholar]
- [2].Hajishengallis G, Darveau RP, Curtis MA.. The keystone-pathogen hypothesis. Nat Rev Microbiol. 2012;10:717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lamont RJ, Hajishengallis G. Polymicrobial synergy and dysbiosis in inflammatory disease. Trends Mol Med. 2015;21:172–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Greenblum S, Carr R, Borenstein E. Extensive strain-level copy-number variation across human gut microbiome species. Cell. 2015;160:583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lorenz B, Ali N, Bocklitz T, et al. Discrimination between pathogenic and non-pathogenic E. coli strains by means of Raman microspectroscopy. Anal Bioanal Chem. 2020;412:8241–8247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Pierce JV, and Bernstein HD . Genomic diversity of enterotoxigenic strains of Bacteroides fragilis. PLoS One. 2016;11(6):e0158171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Tribble GD, Kerr JE, Wang BY. Genetic diversity in the oral pathogen Porphyromonas gingivalis: molecular mechanisms and biological consequences. Future Microbiol. 2013;8:607–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Nakagawa I, Amano A, Kuboniwa M, et al. Functional differences among FimA variants of Porphyromonas gingivalis and their effects on adhesion to and invasion of human epithelial cells. Infect Immun. 2002;70:277–285. DOI: 10.1128/Iai.70.1.277-285.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Moon JH, Herr Y, Lee H-W, et al. Genotype analysis of Porphyromonas gingivalis fimA in Korean adults using new primers. J Med Microbiol. 2013;62:1290–1294. DOI: 10.1099/jmm.0.054247-0 [DOI] [PubMed] [Google Scholar]
- [10].Zhao L, Wu Y-F, Meng S, et al. Prevalence of fimA genotypes of Porphyromonas gingivalis and periodontal health status in Chinese adults. J Periodontal Res. 2007;42:511–517. DOI: 10.1111/j.1600-0765.2007.00975.x [DOI] [PubMed] [Google Scholar]
- [11].Amano A, Kuboniwa AM, Nakagawa I, et al. Prevalence of specific genotypes of Porphyromonas gingivalis fimA and periodontal health status. J Dent Res. 2000;79:1664–1668. DOI: 10.1177/00220345000790090501 [DOI] [PubMed] [Google Scholar]
- [12].Zheng C, Wu J, Xie H. Differential expression and adherence of Porphyromonas gingivalis fimA genotypes. Mol Oral Microbiol. 2011;26:388–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Nadkarni MA, Browne GV, Chhour K-L, et al. Pattern of distribution of Prevotella species/phylotypes associated with healthy gingiva and periodontal disease. Eur J Clin Microbiol Infect Dis. 2012;31:2989–2999. DOI: 10.1007/s10096-012-1651-5 [DOI] [PubMed] [Google Scholar]
- [14].Fukushima H, Moroi H, Inoue J, et al. Phenotypic characteristics and DNA relatedness in Prevotella intermedia and similar organisms. Oral Microbiol Immunol. 1992;7:60–64. DOI: 10.1111/j.1399-302x.1992.tb00023.x [DOI] [PubMed] [Google Scholar]
- [15].Yamanaka T, Furukawa T, Matsumoto-Mashimo C, et al. Gene expression profile and pathogenicity of biofilm-forming Prevotella intermedia strain 17. BMC Microbiol. 2009;9:11. DOI: 10.1186/1471-2180-9-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yamanaka T, Yamane K, Furukawa T, et al. Comparison of the virulence of exopolysaccharide-producing Prevotella intermedia to exopolysaccharide non-producing periodontopathic organisms. BMC Infect Dis. 2011;11:228. DOI: 10.1186/1471-2334-11-228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Socransky SS, Haffajee AD. Periodontal microbial ecology. Periodontol 2000. 2005;38:135–187. [DOI] [PubMed] [Google Scholar]
- [18].Wojda I, Staniec B, Sulek M, et al. The greater wax moth Galleria mellonella: biology and use in immune studies. Pathog Dis. 2020;78:ftaa057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lange A, Schäfer A, Bender A, et al. Galleria mellonella: a novel invertebrate model to distinguish intestinal symbionts from pathobionts. Front Immunol. 2018;9:2114. DOI: 10.3389/fimmu.2018.02114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mostowy RJ, Holt KE. Diversity-Generating machines: genetics of bacterial sugar-coating. Trends Microbiol. 2018;26:1008–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Poole J, Day CJ, von Itzstein M, et al. Glycointeractions in bacterial pathogenesis. Nat Rev Microbiol. 2018;16:440–452. [DOI] [PubMed] [Google Scholar]
- [22].Settem RP, Honma K, Stafford GP, et al. Protein-Linked glycans in periodontal bacteria: prevalence and role at the immune interface. Front Microbiol. 2013;4:310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Vik A, Aas FE, Anonsen JH, et al. Broad spectrum O-linked protein glycosylation in the human pathogen Neisseria gonorrhoeae. Proc Natl Acad Sci U S a. 2009;106:4447–4452. DOI: 10.1073/pnas.0809504106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Szymanski CM, Wren BW. Protein glycosylation in bacterial mucosal pathogens. Nat Rev Microbiol. 2005;3:225–237. [DOI] [PubMed] [Google Scholar]
- [25].Fletcher CM, Coyne MJ, Villa OF, et al. A general O-glycosylation system important to the physiology of a major human intestinal symbiont. Cell. 2009;137:321–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kuo C, Takahashi N, Swanson AF, et al. An N-linked high-mannose type oligosaccharide, expressed at the major outer membrane protein of Chlamydia trachomatis, mediates attachment and infectivity of the microorganism to HeLa cells. J Clin Invest. 1996;98:2813–2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Szymanski CM, Burr DH, Guerry P. Campylobacter protein glycosylation affects host cell interactions. Infect Immun. 2002;70:2242–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Karlyshev AV, Everest P, Linton D, et al. The Campylobacter jejuni general glycosylation system is important for attachment to human epithelial cells and in the colonization of chicks. Microbiology. 2004;150:1957–1964. DOI: 10.1099/mic.0.26721-0 [DOI] [PubMed] [Google Scholar]
- [29].Guerry P, Ewing CP, Schirm M, et al. Changes in flagellin glycosylation affect Campylobacter autoagglutination and virulence. Mol Microbiol. 2006;60:299–311. DOI: 10.1111/j.1365-2958.2006.05100.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Young KT, Davis LM, Dirita VJ. Campylobacter jejuni: molecular biology and pathogenesis. Nat Rev Microbiol. 2007;5:665–679. [DOI] [PubMed] [Google Scholar]
- [31].Hitchen P, Brzostek J, Panico M, et al. Modification of the Campylobacter jejuni flagellin glycan by the product of the Cj1295 homopolymeric-tract-containing gene. Microbiology. 2010;156:1953–1962. DOI: 10.1099/mic.0.038091-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Bielecki M, Antonyuk S, and Strange RW, et al. Prevotella intermedia produces two proteins homologous to Porphyromonas gingivalis HmuY but with different heme coordination mode. Biochem J. 2020;477:381–405. DOI: 10.1042/BCJ20190607 [DOI] [PubMed] [Google Scholar]
- [33].Lukose V, Walvoort MTC, Imperiali B. Bacterial phosphoglycosyl transferases: initiators of glycan biosynthesis at the membrane interface. Glycobiology. 2017;27:820–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Glover KJ, Weerapana E, Chen MM, et al. Direct biochemical evidence for the utilization of UDP-bacillosamine by PglC, an essential glycosyl-1-phosphate transferase in the Campylobacter jejuni N-linked glycosylation pathway. Biochemistry. 2006;45:5343–5350. [DOI] [PubMed] [Google Scholar]
- [35].Hartley MD, Morrison MJ, Aas FE, et al. Biochemical characterization of the O-Linked glycosylation pathway in Neisseria gonorrhoeae responsible for biosynthesis of protein glycans containing N,N′-Diacetylbacillosamine. Biochemistry. 2011;50:4936–4948. DOI: 10.1021/bi2003372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Waterhouse A, Bertoni M, Bienert S, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:W296–W303. DOI: 10.1093/nar/gky427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Moremen KW, Haltiwanger RS. Emerging structural insights into glycosyltransferase-mediated synthesis of glycans. Nat Chem Biol. 2019;15:853–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zhang Y, Zhen M, Zhan Y, et al. Population-Genomic insights into variation in Prevotella intermedia and Prevotella nigrescens isolates and its association with periodontal disease. Front Cell Infect Microbiol. 2017;7:409. DOI: 10.3389/fcimb.2017.00409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Carrow HC, Batachari LE, Chu HT. Strain diversity in the microbiome: Lessons from Bacteroides fragilis. PLoS Pathog. 2020;16:ARTN e1009056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Takahashi N, Yamada T. Glucose metabolism by Prevotella intermedia and Prevotella nigrescens. Oral Microbiol Immunol. 2000;15:188–195. [DOI] [PubMed] [Google Scholar]
- [41].Moradali MF, Davey ME. Metabolic plasticity enables lifestyle transitions of Porphyromonas gingivalis. NPJ Biofilms Microbiomes. 2021;7:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Shah HN, Williams RAD. Utilization of glucose and amino acids by Bacteroides intermedius and Bacteroides gingivalis. Curr Microbiol. 1987;15:241–246. [Google Scholar]
- [43].Byrne DP, Potempa J, Olczak T, et al. Evidence of mutualism between two periodontal pathogens: co-operative haem acquisition by the HmuY haemophore of Porphyromonas gingivalis and the cysteine protease interpain a (InpA) of Prevotella intermedia. Mol Oral Microbiol. 2013;28:219–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Leung KP, Subramaniam PS, Okamoto M, et al. The binding and utilization of hemoglobin by Prevotella intermedia. FEMS Microbiol Lett. 1998;162:227–233. [DOI] [PubMed] [Google Scholar]
- [45].Byrne DP, Wawrzonek K, Jaworska A, et al. Role of the cysteine protease interpain a of Prevotella intermedia in breakdown and release of haem from haemoglobin. Biochem J. 2009;425:257–264. DOI: 10.1042/BJ20090343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Moon JH, Park JH, Lee JY. Antibacterial action of polyphosphate on Porphyromonas gingivalis. Antimicrob Agents Chemother. 2011;55:806–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Nakano K, Kuboniwa M, Nakagawa I, et al. Comparison of inflammatory changes caused by Porphyromonas gingivalis with distinct fimA genotypes in a mouse abscess model. Oral Microbiol Immunol. 2004;19:205–209. DOI: 10.1111/j.0902-0055.2004.00133.x [DOI] [PubMed] [Google Scholar]
- [48].Joglekar P, Sonnenburg ED, Higginbottom SK, et al. Genetic variation of the SusC/susd homologs from a polysaccharide utilization locus underlies divergent fructan specificities and functional adaptation in Bacteroides thetaiotaomicron strains. mSphere. 2018;3(3):e00185–18. DOI: 10.1128/mSphereDirect.00185-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Barnoy S, Gancz H, and Zhu Y, et al. The Galleria mellonella larvae as an in vivo model for evaluation of Shigella virulence. Gut Microbes. 2017;8(4):335–350. DOI: 10.1080/19490976.2017.1293225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kay S, Edwards J, Brown J, et al. Galleria mellonella infection model identifies both high and low lethality of Clostridium perfringens toxigenic strains and their response to antimicrobials. Front Microbiol. 2019;10:1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Arifin WN, Zahiruddin WM. Sample size calculation in animal studies using resource equation approach. Malays J Med Sci. 2017;24:101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Tritt A, Eisen JA, Facciotti MT, et al. An integrated pipeline for de novo assembly of microbial genomes. PLoS One. 2012;7:e42304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Tatusova T, DiCuccio M, Badretdin A, et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016;44:6614–6624. DOI: 10.1093/nar/gkw569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kurtz S, Phillippy A, Delcher AL, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. DOI: 10.1186/gb-2004-5-2-r12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zhao Y, Wu J, Yang J, et al. PGAP: pan-genomes analysis pipeline. Bioinformatics. 2012;28(3):416–418. DOI: 10.1093/bioinformatics/btr655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Huerta-Cepas J, Forslund K, Coelho LP, et al. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol Biol Evol. 2017;34:2115–2122. DOI: 10.1093/molbev/msx148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Wu S, Zhu Z, Fu L, et al. WebMGA: a customizable web server for fast metagenomic sequence analysis. BMC Genomics. 2011;12:444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].UniProt C. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47:D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Ray LC, Das D, Entova S, et al. Membrane association of monotopic phosphoglycosyl transferase underpins function. Nat Chem Biol. 2018;14:538–541. DOI: 10.1038/s41589-018-0054-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Jang EY, Kim M, Noh MH, et al. In vitro effects of polyphosphate against Prevotella intermedia in planktonic phase and biofilm. Antimicrob Agents Chemother. 2016;60:818–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Barbosa GM, Colombo AV, Rodrigues PH, et al. Intraspecies variability affects heterotypic biofilms of Porphyromonas gingivalis and Prevotella intermedia: evidences of strain-dependence biofilm modulation by physical contact and by released soluble factors. PLoS One. 2015;10:e0138687. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.