

Abstract
Background:
Cerebrospinal fluid (CSF) microRNA (miRNA) biomarkers of Alzheimer’s disease (AD) have been identified, but have not been evaluated in prodromal AD, including mild cognitive impairment (MCI).
Objective:
To assess whether a set of validated AD miRNA biomarkers in CSF are also sensitive to early-stage pathology as exemplified by MCI diagnosis.
Methods:
We measured the expression of 17 miRNA biomarkers for AD in CSF samples from AD, MCI, and cognitively normal controls (NC). We then examined classification performance of the miRNAs individually and in combination. For each miRNA, we assessed median expression in each diagnostic group and classified markers as trending linearly, nonlinearly, or lacking any trend across the three groups. For trending miRNAs, we assessed multimarker classification performance alone and in combination with apolipoprotein E ε4 allele (APOE ε4) genotype and amyloid-β42 to total tau ratio (Aβ42:T-Tau). We identified predicted targets of trending miRNAs using pathway analysis.
Results:
Five miRNAs showed a linear trend of decreasing median expression across the ordered diagnoses (control to MCI to AD). The trending miRNAs jointly predicted AD with area under the curve (AUC) of 0.770, and MCI with AUC of 0.705. Aβ42:T-Tau alone predicted MCI with AUC of 0.758 and the AUC improved to 0.813 (p = 0.051) after adding the trending miRNAs. Multivariate correlation of the five trending miRNAs with Aβ42:T-Tau was weak.
Conclusion:
Selected miRNAs combined with Aβ42:T-Tau improved classification performance (relative to protein biomarkers alone) for MCI, despite a weak correlation with Aβ42:T-Tau. Together these data suggest that that these miRNAs carry novel information relevant to AD, even at the MCI stage. Preliminary target prediction analysis suggests novel roles for these biomarkers.
Keywords: Alzheimer’s disease, amyloid, APOE ε4, biomarkers, cerebrospinal fluid, microRNA, mild cognitive impairment, tau proteins
INTRODUCTION
As populations age globally and in the United States, the prevalence of people living with Alzheimer’s disease (AD) has increased. Globally AD is the most common cause of dementia, accounting for 60–80% of all cases (http://www.alz.org/alzheimers). In 2019 there were an estimated 5.8 million Americans living with AD, whose care generated a total cost of $290 billion, and it is projected that by 2050 these numbers will grow to approximately 13.8 million individuals costing over $1.1 trillion [1]. Given the enormity of the emotional and economic costs to patients, caregivers, and health systems, there is an urgent need to identify biomarkers that can be used to diagnose AD earlier in disease progression, so that treatments to slow or prevent the disease can be initiated.
Cerebrospinal fluid (CSF) is a well-established biofluid for biomarker studies focused on neuropathological diseases [2]. Among the most extensively studied CSF protein biomarkers for AD are amyloid-β42 (Aβ42), total tau (T-Tau), and phosphorylated tau181 (P-Tau) [3]. These markers are useful for diagnosis, but do not track with the progression of AD in clinical trials, and cannot be used as prognostic tools [4]. The continuum of AD severity is subdivided by cognitive function, and increases from preclinical AD, to mild cognitive impairment (MCI) due to AD, to dementia due to AD [5–7]. However, progression across these stages is highly variable. Outcomes for individuals with MCI are especially difficult to predict: while some patients progress to AD, others remain stable, or even revert to normal cognition. In studies of individuals with MCI, 15% of those over 65 developed dementia [8], while 32% [9] to 38% [10] developed AD dementia within 5 years. Thus, preemptive identification of individuals with MCI that convert to AD is of great importance, as it would allow earlier initiation of treatments to slow brain changes that lead to AD dementia.
Our studies are novel in that we have systematically evaluated the potential of extracellular microRNAs (miRNA) in CSF as biomarkers for AD, and for detection of early brain changes in patients with MCI that progress to AD. We initiated our studies when extracellular RNAs in biofluids were just beginning to be evaluated as biomarkers for human disease [11]. In contrast to prior reports that examined miRNAs in CSF as biomarkers for AD, our initial biomarker discovery studies focused on CSF from living (not postmortem) donors and were comprised of a large sample size from a well-characterized repository of AD patients and cognitively normal controls (NC) [12]. We also utilized a rigorous analytic approach to examine the performance of combinations of miRNAs, and miRNAs in combination with APOE ε4 genotype status, on classification performance [12]. Our discovery studies identified 36 CSF miRNAs that discriminate AD from NC, supporting that measurable differences in miRNAs in CSF from living donors can be exploited for use as clinical biomarkers for AD.
We then moved forward to validate the performance of the AD CSF miRNAs in a new and independent sample cohort, again from a large cohort of living donors [13]. Consistent with the discovery study, we found that miRNA combinations plus APOE ε4 genotype status increased classification performance for AD. We further found that adding the ratio of Aβ42:T-Tau to miRNA combinations increased classification performance and, importantly, that performance of miRNA combinations is better than for Aβ42:T-Tau alone. However, combining all three variables (miRNAs, Aβ42:T-Tau, APOE ε4 status) does not improve performance beyond what is seen for the combination of Aβ42:T-Tau and miRNAs [13]. These findings demonstrated that the miRNAs include significant and novel information not conferred by either APOE ε4 status or Aβ42:T-Tau, and suggest that the information carried by CSF miRNAs could be useful for diagnosing AD.
The purpose of the current study is to assess whether the validated AD miRNA biomarkers are also sensitive to early-stage pathology as exemplified by MCI diagnosis. Thus, here we assessed the performance of 17 AD miRNAs in CSF in patients with MCI in a large sample size of living donors from a well-characterized repository to examine three diagnostic groups: NC, MCI, and AD. Five of the validated miRNAs trended with progression from NC to MCI to AD and showed substantial increases in classification performance in both MCI and AD when added to Aβ42:T-Tau. Notably, the boost in classification performance from the miRNAs was larger for MCI than for AD because Aβ42:T-Tau alone was a weaker predictor of MCI than of AD. Combined with a weak correlation of the trending markers with Aβ42:T-Tau levels, our results suggest that the validated CSF miRNA biomarkers for AD carry independent information that could be relevant for disease characterization even at the MCI stage.
MATERIALS AND METHODS
Participants
All procedures were approved by the Institutional Review Board of the University of California San Diego (UCSD IRB 80012). All participants provided written informed consent and underwent detailed evaluations consisting of medical history, physical and neurological examinations, laboratory tests, and neuropsychological assessments. Cognitively unimpaired NC participants were volunteers in good health with no symptoms of cognitive impairment or neurological disease, and normal performance on a detailed battery of neuropsychological tests. The age distribution and sex ratio of NC samples to that of the MCI and AD diagnostic groups were matched to the best of our ability. Participant samples were banked at the UCSD Shiley-Marcos Alzheimer’s Disease Research Center (UCSD ADRC).
CSF collection
The USCD ADRC uses a standardized CSF collection protocol corresponding to that of other AD centers engaged in biomarker research [14]. Lumbar punctures were done in the morning under fasting conditions, in the lateral decubitus position with a 24-gauge Sprotte spinal needle. The first 2 mL of CSF collected was used for clinical tests, then 10–20 mL of CSF was collected, gently mixed, and transferred to polypropylene tubes in 500 μL aliquots. The tubes included a subject number but no other identifying information. The CSF aliquots were flash frozen on dry ice and stored at −80°C.
APOE ε4 genotyping
APOE ε4 genotypes were determined by polymerase chain reaction (PCR) amplification and restricted fragment length polymorphism [15], as previously described [16]. Venous blood was drawn from participants and genomic DNA was extracted using the QIAamp DNA Blood Mini Kit (Qiagen, Valencia, CA, USA) followed by PCR amplification. The APOE ε4 gene sequences were amplified using forward (5′-ACGCGGGCACGGCTGTCCAAGGA-3′) and reverse (5′-GCGGGCCCCGGCCTGGTACAC-3′) primers. The amplification products were digested with HhaI (restriction enzyme site GCGΛC) and subjected to electrophoresis on polyacrylamide gels. After electrophoresis, the gels were stained with ethidium bromide and the digested fragments were visualized by ultraviolet illumination. A unique combination of HhaI fragment sizes enabled unambiguous typing of all homozygotic and heterozygotic combinations: HhaI cleaves at GCGC encoding 112arg (E4) and 158arg (E3, E4), but does not cut at GTGC encoding 112cys (E2, E3) and 158cys (E2).
CSF Aβ42 and T-Tau measures
Measurements of CSF Aβ42 and T-Tau levels were performed at the UCSD ADRC using enzyme-linked immunosorbent assays (ELISAs) as previously reported [17]. CSF Aβ42 was measured using the Euroimmun ELISA kit EQ 6521-9601-L (ADx Neurosciences, Ghent, Belgium). CSF T-Tau was measured using the ELISA kit EQ 6531-9601-L (ADx Neurosciences).
RNA isolation and qPCR
We instituted safeguards to improve quality control based on findings and outcomes from our initial AD miRNA discovery studies. We included multiple positive controls (CSF miRNAs verified in our previous studies [12, 13] that were unchanged between controls and AD) and negative controls (miRNAs not detected in CSF in our previous studies). We also imposed strict uniformity over manufacturing variables: all kit and reagents were matched by lot, with no exceptions, to minimize variation from sources unrelated to the differentiation of diagnostic groups.
We generated Custom TaqMan® Array MicroRNA Cards which included probes for 17 AD miRNA biomarkers, as well as 4 non-changing CSF miRNAs (positive controls), 2 miRNAs not detected in CSF (negative controls), and U6 snRNA (Supplementary Table 1). Of the 17 biomarker miRNAs in the arrays, 14 appeared as strong biomarker candidates in our validation study [13], and an additional 3 (miR-1291, miR-142-3p, and miR-519b-3p) were strong biomarker candidates in our initial discovery study [12], but did not perform well in the validation study. Each array card was designed to assay 4 individual CSF samples, with 4 technical replicates for all RNA probes on the card. We analyzed miRNA expression in 102 new CSF samples obtained from the UCSD ADRC, and 63 repeated CSF samples from our previous validation studies [13] for reliability testing. As a further reliability check, we obtained duplicate aliquots for 5 CSF samples (4 NC, 1 MCI) and ran each of the duplicates on a different array card. The run order of the samples was arranged so that every single array card contained at least one NC sample and, whenever possible, also included at least one MCI sample, AD sample, or both. This was done to avoid confounding technical card batch differences with sample diagnosis differences, and only 10% of cards failed to meet this arrangement (due to the larger number of NC samples relative to the other two diagnoses). Within diagnosis type, the plating order of samples was fully randomized (under the constraint that we would only accept a randomization where duplicates fell on separate cards). On every card we also included a sample from a homogeneous reservoir of a pooled CSF reference standard obtained from NCs at the Oregon Health & Science University Layton Aging and Alzheimer’s Disease Center in order to monitor the stability of the process over time.
Total RNA was extracted from 500 μL of CSF using the mirVana™ PARIS™ Kit (AM1556, Thermo Fisher Scientific (TFS), Waltham, MA) then concentrated using the RNA Clean and Concentrator-5 Kit (R1013, Zymo Research, Irvine CA), as previously described [12, 13]. We previously reported that 500 μL of CSF yields a very low amount of RNA (~1 ng) such that it precludes measuring the concentration [12]. Thus, we instituted a protocol to isolate total RNA from an absolute (500 μL) amount of CSF, then concentrate the RNA to the smallest amount needed for the arrays in order to maximize the amount of RNA going into the PCR. We then controlled for variance in the normalization step. Thus, we used 4.1 μL of total RNA from each sample for reverse transcription (RT) using a custom primer pool generated by TFS to specifically reverse transcribe 23 miRNAs (17 AD miRNA biomarkers, 4 non-changing positive control CSF miRNAs, and 2 negative control miRNAs not detected in CSF), and U6. Based on the recommended protocol by TFS for Custom TaqMan® Array MicroRNA Cards (publication part #4478705), to include a pre-amplification step for RNA inputs <350 ng of total RNA, we then used 7.5 μL of the resulting cDNA for pre-amplification using a custom Pre-Amp primer pool generated by TFS to specifically pre-amplify the 23 miRNAs and U6. The pre-amplification products were diluted 1 : 4 in RNase/DNase-free water, then 9 μL of the diluted samples were mixed with TaqMan® master mix (TFS) and RNase/DNase-free water to a 225 μL final volume, then loaded onto the Custom TaqMan® Array MicroRNA Cards (TFS) containing probes for the 23 miRNAs and U6. We also included parallel reactions that contained only water (no RNA)±RT enzyme, or RNA with no RT enzyme, as controls for spurious amplifications in these experiments (Supplementary Table 1). The quantitative PCR (qPCR) amplifications and acquisition of the resulting data were carried out on a QuantStudio™ 12K Flex Real-Time PCR System (TFS). All NC, MCI, and AD qPCR data files were imported into ExpressionSuite Software v.1.1 (TFS) and processed together to generate automatic baseline and threshold values used to calculate quantification cycle (Cq) values. The Cq value for each well was reported along with the amplification quality score (AmpScore) and Cq confidence (CqConf) score, which serve as metrics for the quality of each amplification.
Preprocessing of Cq values
We applied Cq validity checks to each well individually, and implemented acceptability criteria across the 4 replicate wells per probe per sample that had to be met in order to consider the miRNA measurement valid. We required AmpScore ≥1.0 and CqConf ≥0.8, and based on the rapid decline (with increasing Cq value) that we observed in the fraction of wells meeting these criteria for Cq >34, we set the Cq censoring threshold at 34. If a well failed to meet either the AmpScore or CqConf criteria, we set the Cq value to missing if the assigned Cq was <34; if Cq was ≥34 the well was considered censored instead. After this step, if at least 2 of the 4 replicates for a probe for a sample had a non-missing Cq value, then we considered the measurement valid; otherwise the entire measurement was removed (just 0.16% of measurements failed in this way). The rationale for this approach was that if at least half of the replicates for a sample for a probe indicated some measurable expression, it would be treated as having been measured at a value rather than censored at a lower limit. However, usually either all four replicate wells had valid Cq values, or none of them did. Final Cq values were taken as the median of the miRNA values in the quadruplicate wells; miRNAs with median value ≥34 were labeled censored for the sample, as they were considered to have expression too low for reliable detection in the assay. Censored values were included in the final statistical analysis.
Preprocessing of miRNAs
Our assay included probes for negative controls, positive controls, U6 snRNA, and the AD miRNA biomarkers. We required the negative control miRNAs to show low expression profiles (i.e., uniformly AmpScore <1.0, CqConf <0.8, and Cq ≥34 or missing for every well). Due to their importance for normalization, we applied greater stringency to the positive control miRNAs. Thus, for the positive controls and U6 snRNA we required robust expression in at least 90% of samples, with AmpScore ≥1.0 and CqConf ≥0.8, but with a more stringent average Cq <32 across the cohort than for the biomarker miRNAs. As U6 was used for batch correction of cards and positive controls were used for normalization (see Batch correction and normalization below), we required that every sample have a valid U6 measurement and valid measurements for at least 2 of the 4 non-changing positive control miRNAs. We also verified that the maximum of pairwise mean Cq contrasts between diagnostic groups was no larger than 1 Cq in magnitude for any positive control miRNA (in nearly all cases the maximum contrast magnitude was 0.5 Cq or less). By these rules, all positive controls were retained and no experimental samples were excluded. For biomarker miRNAs we required robust expression in at least 20% of samples with average Cq <34, with AmpScore ≥1.0 and CqConf ≥0.8 for all expressed wells.
Array card batch correction, normalization, and transformation of expression scale
U6 snRNA has shown weaknesses as a normalizer in previous investigations by us and others [18]. However, U6 snRNA showed excellent stability of expression across cards when averaged over all samples on the card and for that reason was suitable as a monitor for variation at the card level. Thus, median Cq values were corrected for array card batch variation by aligning mean values of U6 snRNA across cards using a 3-level random-effects mixed model [19]: sample nested in card nested in RNA isolation batch. We generated best linear unbiased predictions for the batch and card effects from the fitted mixed model and subtracted these from all Cq values in the corresponding batches and cards. Following batch correction, Cq values were normalized relative to a linear combination of our verified “non-changing” positive controls (miR-30e-3p, miR-574-3p, miR-638, and miR-92a) using a crossed random-effects mixed model (miRNAs crossed with samples) for the Cq values of all positive control miRNAs. (Supplementary Table 2 includes the Cq distributional characteristics of the positive control miRNAs, and evidence that the per-card mean of U6, and the distributions of each of the other positive controls, did not differ by diagnosis group.) For normalization we calculated a best linear unbiased prediction for each sample’s offset from the overall mean (across all samples) of positive controls using fitted values from the mixed model, then subtracted this offset from the measured Cq values for the sample.
As in our discovery and validation studies [12, 13], we transformed the normalized Cq values onto an “expression” (i.e., positively oriented) scale so that higher values indicated relatively greater quantities of miRNA expression. (Expression = Cqnormmax – Cqnorm, where Cqnormmax was the largest non-censored normalized Cq value in the data, rounded up to the nearest integer.) Censored Cq values were assigned a value of zero on this expression scale and included in the final analysis. All qPCR data and donor-specific metadata is being prepared for submission into the NCBI Gene Expression Omnibus (GEO) repository.
Exclusion of aberrant samples
We initially examined CSF miRNA expression in 165 samples (49 AD, 38 MCI, and 78 NC). However, after normalization, 32 CSF samples showed normalization offsets that were extremely large in magnitude (~7 Cq), with normalizer values from 3- to 10-fold more extreme than the standard deviation of typical samples. These 32 samples had unusually extreme Cq values across all miRNAs in the array, a finding that was replicated when the assays were rerun using stored RT samples. Investigating further, we identified 14 of the 32 CSF samples as technical failures in the RT that resulted in extremely low miRNA expression. However, no explanation could be found for the remaining 18 CSF samples, most of which showed extremely high miRNA expression. There was also no apparent relationship between an aberrant expression profile and any patient characteristic or design factor, including collection site, RNA isolation batch or date, array card lot or run date, participant diagnosis, sex, age, or any AD-related characteristic such as Aβ42 or T-Tau levels in the CSF. (The minimum p-value across all factors tested for association with the expression profile was >0.2, using a variety of parametric and nonparametric statistical tests.) Thus, we excluded the 14 technical failures and the 18 aberrant samples from further analysis to avoid the risk of bias in the results. Due to the lack of association with any patient factors, these exclusions had no meaningful effect on the distributions of those factors, and had only negligible effect on the Cq values for the remaining 133 samples when these were recalculated in ExpressionSuite; the concordance between calculated Cq values before and after the exclusion of the 32 samples was 0.98 (Lin’s concordance coefficient), showing nearly perfect linear correlation and zero bias. Thus, after exclusion of these samples, 133 unique samples (37 AD, 31 MCI, 65 NC) and 3 duplicates (1 MCI, 2 NC) remained in the analytic cohort.
Statistical analysis
To determine whether miRNA information correlated with diagnostic progression and group ordering (NC to MCI to AD), we first confirmed that the diagnostic groups were different according to established measures of disease severity by visually comparing group distributions of Mini-Mental State Examination (MMSE) scores and Aβ42:T-Tau levels, and tested for natural ordering using ordered logistic regression. Next, we used several visual and analytic methods to assess whether the miRNA expression profiles were naturally ordered across diagnosis groups. We initially plotted the Cq distributions of the miRNAs for each group side by side and looked for trending of the median Cq values. Outcomes were either a linear trend (NC to MCI to AD), a nonlinear trend (2 of 3 groups different from the remaining group), or no trend (an ambiguous pattern). We then calculated approximate Bayesian posterior probabilities (based on approximate Bayes factors from harmonic-mean combining of p-values) that the AD and MCI were both significantly different from the NC, and that the 3 groups were strictly ordered.
We then used receiver operating characteristic (ROC) curve analysis [20] to assess the predictive power of the linear trending miRNAs as a group for discriminating AD and MCI from NC. We report the area under the ROC curve (AUC), a measure of classification performance, for the miRNA classifiers with and without APOE ε4 status, with and without Aβ42:T-Tau levels, and with and without the combination of Aβ42:T-Tau levels and APOE ε4 status to differentiate performance under alternative sets of information. In calculating the ROC curves, we used two different methods to build classifiers: logistic discriminant analysis (similar to logistic regression, but with more than one group) and a smooth k-nearest-neighbor classifier (k = 9) based on Euclidean distance. We then averaged the predicted probabilities using the harmonic mean (the appropriate average for values between zero and one). The ROC curves were smoothed for easier visualization of the differences. (Average spacing between curves and AUC were both well preserved under the smoothing.) We assessed the strength of the canonical (i.e., best linear) correlations between the multivariate set of trending miRNA expression values and diagnosis rank, and with Aβ42:T-Tau levels, to further validate the panel of trending miRNAs, not just for distinguishing AD and MCI from NC, but also for correctly ordering the groups with respect to disease severity.
MiRNA target prediction
We used TargetScan 7.2 [21] and miRDB [22, 23] to predict target mRNAs of the top five trending miRNAs (miR-142-3p, miR-146a-5p, miR-146b-5p, miR-365a-3p, miR-193a-5p). TargetScan and miRDB were chosen as they are both widely used and frequently updated. As subsequent pathway analysis is most effective with a limited gene set, predicted targets were excluded if they had a Cumulative Weighted Context Score above −0.3 in TargetScan or a target score of below 60 in miRDB. The use of a union between the two target prediction algorithm outputs, as opposed to an intersection where only targets predicted by both algorithms are included in the analysis, may increase the sensitivity of the predicted targets and the likelihood of predicting novel targets [24, 25]. Thus to determine whether a union, an intersection, or an individual target list should be used in subsequent pathway analysis for the trending miRNAs, we calculated sensitivity, specificity, and precision values using validated miRNA-target pairs from miRTarBase, an experimentally validated miRNA-target interactions database [26]. The miRTarBase values ranged from 0–1, with high-quality results indicated as closer to 1 [24, 25]. The union of TargetScan and miRDB showed the highest values of sensitivity (0.35), specificity (0.833), and precision (0.98). Thus, pathway analysis was performed on the union set of the unique targets using QIAGEN Ingenuity Pathway Analysis (IPA). For this pathway analysis we excluded cancer related tissue and cell lines to avoid the knowledge bias towards cancer in IPA.
RESULTS
Participant characteristics
Participant characteristics for the 133 total participants in this study (37 AD, 31 MCI, and 65 NC) are shown in Table 1. In the final set of 133 participants the sex ratio was close to a 1 : 1 male to female ratio overall, but varied somewhat from group to group (NC ~2 : 3 but AD and MCI both ~3 : 2). Participants in all 3 groups were well matched for age (mean 73.2 ± 7.1, range 53–89), but the AD group had more variance than the other groups and contained some sex bias (with females tending to be younger than males in that group). NC participants were in good health with a mean MMSE [27] score of 29.1 ± 1.2, Clinical Dementia Rating (CDR) scores of 0, no evidence or personal or family history of cognitive or functional decline, and also performed normally on a neuropsychological test battery. AD patients were diagnosed with probable AD according to ADRDA-NINDS criteria [6, 28], with a mean MMSE score of 21.6 ± 4.5 and CDR scores of 1–2. MCI patients were in between, with mean MMSE score of 26.9 ± 1.5 and CDR scores of 0–0.5. CSF Aβ42 and T-Tau measurements were as expected, with NC having high Aβ42 and low T-Tau, AD having low Aβ42 and high T-Tau, and MCI falling somewhere in the middle (medium values of both Aβ42 and T-Tau). APOE ε4 genotyping was available for 131 of the 133 participants in the study. Within the NC group, 66.2% of participants had no APOE ε4 allele, 27.7% had 1 allele, and 6.2% had 2 alleles. In the AD group 50.0% had no APOE ε4 alleles, 33.3% had 1 allele, and 16.7% had 2 alleles. Thus as expected, APOE ε4 genotype was over-represented in AD [29]. Unsurprisingly, the MCI group was more similar to AD in APOE ε4 allele frequencies: 40.0% with no alleles, 46.7% with 1, and 13.3% with 2. Figure 1 shows the distributions of MMSE scores (A) and Aβ42:T-Tau measures (B) for each of the 3 diagnostic groups.
Table 1. Participant characteristics.
The table includes the sex, age, MMSE, and Aβ42 and T-Tau measures for 133 study participants. APOEε4 genotype was available for 131 participants. Sex ratio and age distribution was matched across groups to the best of our ability in order to mitigate the risk of associations between miRNA markers and diagnosis being confounded by these factors. Examination of each miRNA for association with each factor, both overall and within diagnostic groups, revealed no hints of confounding.
| NC | MCI | AD | All | ||
|---|---|---|---|---|---|
| Sex | Male | 27 (41.5%) | 19 (61.3%) | 23 (62.2%) | 69 (51.9%) |
|
|
|||||
| Female | 38 (58.5%) | 12 (38.7%) | 14 (37.8%) | 64 (48.1%) | |
|
|
|||||
| All | 65 | 31 | 37 | 133 | |
|
| |||||
| Age at LP † | Male | 27 (73.5±5.1) | 19 (75.2±5.8) | 23 (74.2±9.7) | 69 (74.2±7.0) |
|
|
|||||
| Female | 38 (72.7±4.8) | 12 (75.6±7.2) | 14 (67.7±9.8) | 64 (72.1±7.0) | |
|
|
|||||
| All | 65 (73.0±4.9) | 31 (75.4±6.3) | 37 (71.8±10.1) | 133 (73.2±7.1) | |
|
| |||||
| MMSE at LP † | Male | 27 (28.9±1.2) | 19 (26.8±1.5) | 22 (22.7±3.8) | 68 (26.3±3.6) |
|
|
|||||
| Female | 38 (29.2±1.3) | 11 (27.1±1.6) | 14 (19.7±5.1) | 63 (26.7±4.7) | |
|
|
|||||
| All | 65 (29.1±1.2) | 30 (26.9±1.5) | 36 (21.6±4.5) | 131 (26.5±4.1) | |
|
| |||||
| Aβ42 † | Male | 26 (724.8±332.6) | 18 (564.1±351.1) | 21 (337.8±202.4) | 65 (555.2±340.5) |
|
|
|||||
| Female | 33 (708.9±311.8) | 12 (431.0±209.6) | 8 (349.4±177.7) | 53 (591.7±311.6) | |
|
|
|||||
| All | 59 715.9±318.4) | 30 (510.8±305.5) | 29 341.0±192.8) | 118 (571.6±327.0) | |
|
| |||||
| T-Tau † | Male | 26 (291.2±134.2) | 18 (592.8±303.6) | 21 (580.2±278.9) | 65 (468.1±277.6) |
|
|
|||||
| Female | 33 (397.4±237.8) | 12 (538.0±215.5) | 8 (965.3±456.1) | 53 (515.0±335.7) | |
|
|
|||||
| All | 59 (350.6±204.4) | 30 (570.9±269.1) | 29 (686.5±371.8) | 118 (489.2±304.6) | |
|
| |||||
| Aβ42: T-Tau † | Male | 26 (2.8±1.3) | 18 (1.4±1.2) | 21 (0.9±1.4) | 65 (1.8±1.6) |
|
|
|||||
| Female | 33 (2.4±1.7) | 12 (1.0±0.8) | 8 (0.5±0.6) | 53 (1.8±1.6) | |
|
|
|||||
| All | 59 (2.6±1.5) | 30 (1.2±1.1) | 29 (0.8±1.3) | 118 (1.8±1.6) | |
|
| |||||
| Mean (Aβ42): Mean (T-Tau) ‡ | Male | 26 (2.5±0.3) | 18 (1.0±0.2) | 21 (0.6±0.1) | (65) 1.2 ± 0.1 |
|
|
|||||
| Female | 33 (1.8±0.2) | 12 (0.8±0.2) | 8 (0.4±0.1) | (53) 1.1 ± 0.2 | |
|
|
|||||
| All | 59 (2.0±0.2) | 30 (0.9±0.1) | 29 (0.5±0.1) | 118 (1.2±0.1) | |
|
| |||||
| APOEε4 Alleles (0-2) | 0 | 43 (66.2%) | 12 (40.0%) | 18 (50.0%) | 73 (55.7%) |
|
|
|||||
| 1 | 18 (27.7%) | 14 (46.7%) | 12 (33.3%) | 44 (33.6%) | |
|
|
|||||
| 2 | 4 (6.2%) | 4 (13.3%) | 6 (16.7%) | 14 (10.7%) | |
|
|
|||||
| All | 65 | 30 | 36 | 131 | |
reporting N (mean±SD)
reporting N (mean±SE)
Fig. 1.
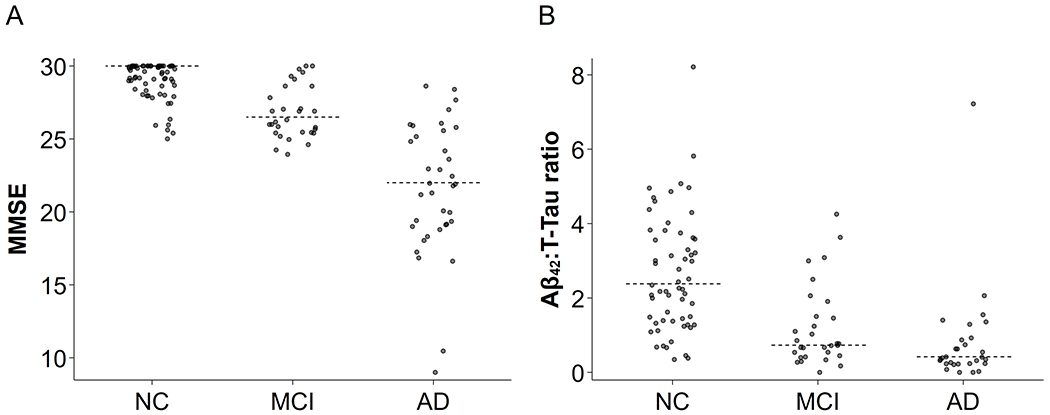
Distributions of MMSE and Aβ42:T-Tau ratio with AD progression. The diagnosis groups trend as expected for measures of disease severity: average MMSE (A) was decreased modestly in MCI and dramatically in AD, and average Aβ42:T-Tau ratio (B) was much lower for MCI and AD than for NC. For both, the MCI profile was intermediate to the NC and AD profiles. Individual participants denoted by points and dotted lines represent group medians. The trend of medians was very apparent for both measures.
Verification of biomarker relevance in the cohort
As a global check on the relevance of the panel of miRNAs for the cohort in the present study (which included both new NC, MCI, and AD samples, as well as repeated NC and AD samples), we verified that the miRNA profiles (taken across the entire panel) for MCI and AD samples consistently differed from those of typical NC samples. Thus, the miRNA profile as a whole is a marker of AD-like disease in our cohort. Details of this analysis can be found in the Supplementary Results.
Quantitative and statistical measures for the individual miRNAs
Next, we report the quantitative and statistical measures of 15 of the 17 miRNAs examined, along with their trend classifications, in Table 2. The 15 miRNAs included all 14 of the validated biomarkers from the previous study, plus 1 of the additional miRNAs (miR-142-3p). The 2 excluded miRNAs were miR-1291, which failed to show expression in most samples (appearing robustly in just 4% of the cohort), and miR-519b-3p, which exhibited universal failure in all samples. Thirteen of the miRNAs were well represented across the samples at close to 80% attestation or better in all 3 diagnostic groups. However, miR-15b-5p and miR-331-3p were poorly attested at <50% each in every group, with the lowest attestation in AD. Inspection of the trend of median fold change across groups showed that, overall, many of the miRNAs had a pattern of decreased expression with increasing disease.
Table 2. Measures for validated AD miRNAs.
The table reports the quantitative and statistical measures of 15 of the 17 miRNAs examined in each group, along with their Cq trend pattern.
| MiRNA | % Detected | Median ± IQR Expression Level | Median Fold Change vs NC [95% bootstrap CI] | Cq Trend Pattern | |||||
|---|---|---|---|---|---|---|---|---|---|
| NC (n=67) | MCI (n=32) | AD (n=37) | NC | MCI | AD | MCI | AD | ||
| miR-142-3p | 98.5% | 96.9% | 97.3% | 5.16 ± 1.15 | 4.93 ± 1.48 | 4.85 ± 1.26 | 0.83 [0.47,1.44] | 0.74 [0.46,1.18] | TREND |
| miR-146b-5p | 98.5% | 93.8% | 89.2% | 4.64 ± 1.10 | 4.46 ± 1.07 | 4.24 ± 1.28 | 0.81 [0.52,1.24] | 0.67 [0.44,1.02] | TREND |
| miR-146a-5p | 95.5% | 96.9% | 86.5% | 4.14 ± 1.26 | 3.94 ± 1.22 | 3.81 ± 2.24 | 0.80 [0.48,1.32] | 0.72 [0.36,1.43] | TREND |
| miR-365a-3p | 95.5% | 84.4% | 86.5% | 4.18 ± 1.52 | 3.58 ± 2.20 | 3.35 ± 1.23 | 0.48 [0.22,1.04] | 0.43 [0.22,0.85] | TREND |
| miR-193a-5p | 91.0% | 90.6% | 81.1% | 3.82 ± 1.25 | 3.62 ± 1.65 | 3.46 ± 1.24 | 0.80 [0.48,1.33] | 0.70 [0.44,1.12] | TREND |
| miR-19b-3p | 100.0% | 100.0% | 100.0% | 6.46 ± 0.99 | 6.26 ± 1.52 | 6.25 ± 0.70 | 0.90 [0.50,1.63] | 0.81 [0.57,1.14] | AD+MCI vs. NC |
| miR-484* | 100.0% | 96.8% | 100.0% | 5.60 ± 1.34 | 5.89 ± 1.66 | 5.86 ± 0.83 | 1.33 [0.84,2.10] | 1.29 [0.84,1.97] | AD+MCI vs. NC |
| miR-140-5p | 82.1% | 75.0% | 64.9% | 4.03 ± 2.04 | 4.03 ± 2.84 | 2.87 ± 3.74 | 0.94 [0.33,2.68] | 0.31 [0.11,0.89] | AD vs. MCI+NC |
| miR-331-3p | 49.3% | 46.9% | 40.5% | 0.00 ± 3.60 | 0.00 ± 3.69 | 0.00 ± 3.21 | 1.00 [0.02,51.13] | 1.00 [0.04,22.76] | AD vs. MCI+NC** |
| miR-223-3p | 100.0% | 100.0% | 100.0% | 6.43 ± 0.88 | 6.64 ± 1.20 | 6.43 ± 0.77 | 1.24 [0.85,1.82] | 1.00 [0.75,1.34] | NO TREND |
| miR-30a-3p | 97.0% | 100.0% | 91.9% | 4.85 ± 1.19 | 4.69 ± 1.49 | 4.77 ± 1.04 | 0.80 [0.44,1.47] | 0.92 [0.58,1.47] | NO TREND |
| miR-378a-3p | 68.7% | 75.0% | 78.4% | 3.97 ± 4.58 | 4.41 ± 3.82 | 4.17 ± 1.64 | 1.53 [0.42,5.59] | 1.22 [0.44,3.43] | NO TREND |
| miR-584-5p | 100.0% | 100.0% | 97.3% | 3.43 ± 0.56 | 3.54 ± 0.79 | 3.40 ± 0.83 | 1.15 [0.88,1.50] | 0.97 [0.70,1.35] | NO TREND |
| miR-597-5p | 95.5% | 96.9% | 97.3% | 2.38 ± 1.66 | 2.01 ± 1.59 | 2.50 ± 1.27 | 0.74 [0.40,1.34] | 1.14 [0.62,2.08] | NO TREND |
| miR-15b-5p | 40.3% | 46.9% | 29.7% | 0.00 ± 3.44 | 0.00 ± 3.94 | 0.00 ± 2.89 | 1.00 [0.03,39.40] | 1.00 [0.11,8.92] | NO TREND |
5 of 136 samples (2 NC, 1 MCI, 2 AD) failed quality control for this miRNA.
The trend is visible in the normalized Cq values and in the pattern of percent detections, but not on the expression scale where censored values are set to zero.
Expression trends of the individual miRNAs across diagnoses
We show the trend plots for the 15 viable miRNAs in Fig. 2. Five of the AD markers (miR-142-3p, miR-146b-5p, miR-146a-5p, miR-365a-3p, and miR193a-5p) showed a linear trend of median expression across the ordered diagnoses, where miRNA expression decreased as disease severity increased. Four of the remaining markers showed nonlinear trends: miR-19b-3p had higher expression in NC that decreased to similar levels in MCI and AD, whereas miR-484 had lower expression in NC that increased to similar levels in MCI and AD. Both miR-140-5p and miR-331-3p showed similar levels in NC and MCI that decreased in AD. The other 6 markers were classified as having no trend, as their expression in MCI tended to be either higher or lower than in both AD and NC. For each of the trending miRNAs we calculated a Bayesian posterior probability of the hypothesis that both MCI and AD differ from NC and are naturally ordered with respect to average levels of the miRNA. The probabilities were generally close to 2/3 (i.e., approximately 2 : 1 odds in favor).
Fig. 2.
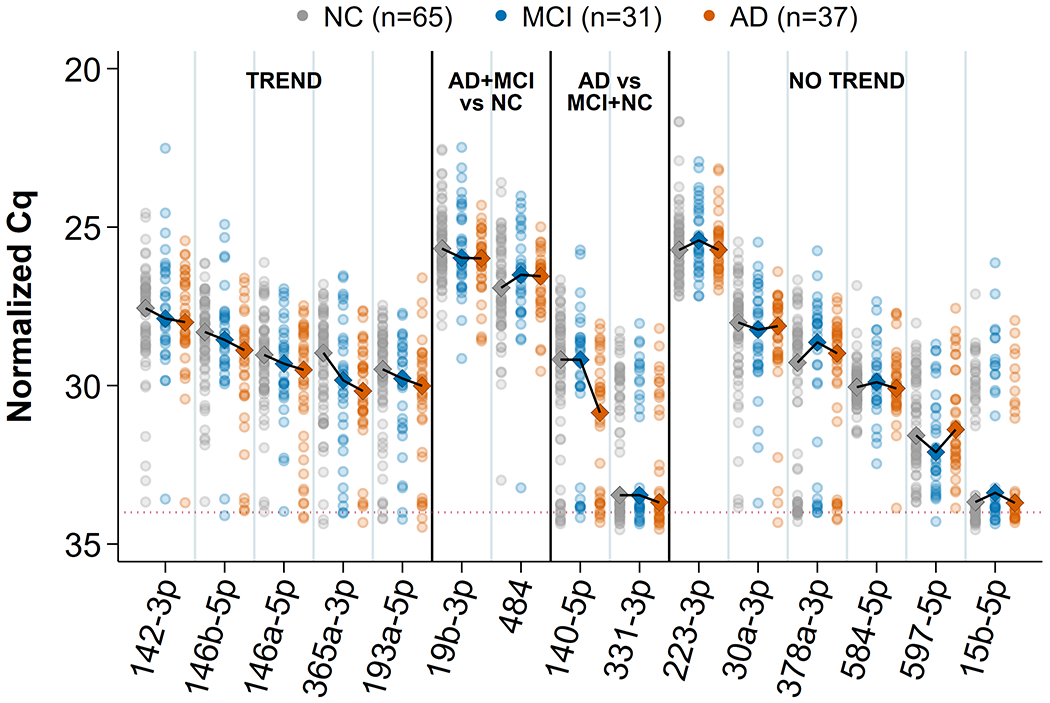
Trend plots of 15 validated miRNAs. All normalized Cq values for samples and median profile across groups are shown for each of the 15 miRNAs that remained viable for this study. The connected diamonds represent medians for 4 kinds of apparent profiles: linear (“TREND”), two kinds of nonlinear (“AD+ MCI versus NC” and “AD versus MCI + NC”), and an ambiguous profile showing a lack of natural diagnosis ordering (“NO TREND”). Multivariate correlation of the trending miRNAs with diagnosis order was −0.36 (p = 0.003).
Classification performance of trending miRNA biomarkers versus APOE ε4 and Aβ42:T-Tau
For detailed evaluation of classification performance (AUC) we used only the 5 trending miRNAs. The nonlinear patterns were interesting, but not directly relevant to our primary goal, which was the identification of CSF miRNAs as markers of disease progression from MCI to AD. We evaluated classification models for MCI versus NC and for AD versus NC. Predictors comprised the 5 trending miRNAs alone and in combination with: 1) APOE ε4, 2) Aβ42:T-Tau, and 3) both Aβ42:T-Tau and APOE ε4. (For comparison, we also assessed AUC for APOE ε4 alone, Aβ42:T-Tau alone, and Aβ42:T-Tau + APOE ε4.) The trending miRNAs achieved an AUC of 0.705 for predicting MCI and 0.770 for predicting AD, compared to NC (Fig. 3 and Table 3). In MCI, adding the trending miRNAs to APOE ε4 showed a large AUC increase (+0.074) over using APOE ε4 alone (Fig. 3A). In AD there was an even more dramatic improvement in AUC (+0.152) from adding the trending miRNAs to APOE ε4 (Fig. 3B). Thus, the miRNA information was not redundant with the APOE ε4 genotype, but provided substantial independent power to differentiate MCI and AD from NC, presumably because the miRNAs reflect the dynamics of the disease state and not mere propensity for disease. Next, adding the trending miRNAs to Aβ42:T-Tau over using Aβ42:T-Tau alone showed substantial AUC increases in both MCI (Fig. 3C) and AD (Fig. 3D). Notably, the boost in classification performance from the miRNAs was larger for MCI (+0.055) than for AD (+0.036) because Aβ42:T-Tau alone was a weaker predictor of MCI (AUC 0.813) than of AD (AUC 0.903). If clinical MCI indeed reflects latent neuropathology, it appears that the information from the CSF protein ratio showed more overlap with the miRNA information when disease was already present, and less overlap when disease was still nascent. Finally, adding the miRNAs to the combination Aβ42:T-Tau + APOE ε4 robustly improved classification performance for MCI (+0.063, Fig. 3E) and modestly for AD (+0.017, Fig. 3F), relative to Aβ42:T-Tau + APOE ε4 alone. However, note that the combination of all 3 markers (trending miRNAs + Aβ42:T-Tau + APOE ε4) provided no meaningful improvement over the combination of trending miRNAs + Aβ42:T-Tau (without APOE ε4): 0.819 versus 0.813 respectively for MCI, and 0.898 versus 0.903 respectively for AD. Our previous discovery and validation studies also showed that adding APOE ε4 or Aβ42:T-Tau to the miRNAs improved classification performance for AD [12, 13]. Together these data suggest that the miRNAs combined with other biomarkers offer a larger gain for predicting MCI, suggesting less overlap and hence greater novelty of relevant information provided by the miRNAs at that stage. Thus, these miRNAs may carry substantial additional information as biomarkers for MCI.
Fig. 3.
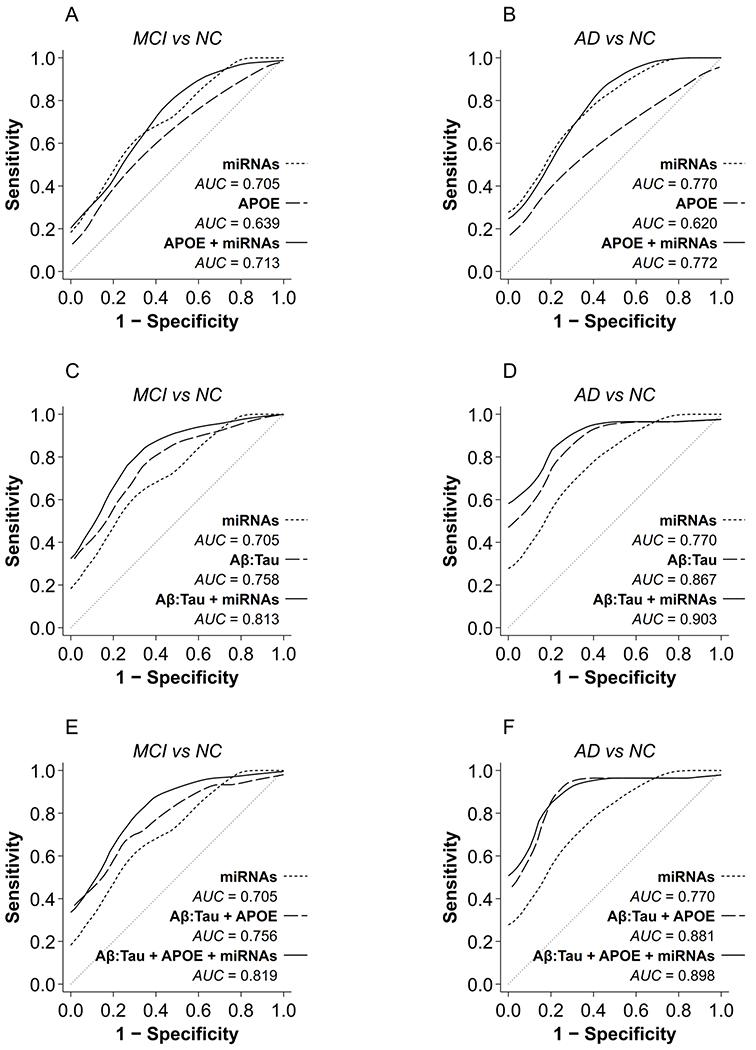
Classification performance of AD biomarkers and trending miRNAs. The effects of combining trending miRNAs with APOE ε4 or Aβ42:T-Tau on classification performance (ROCs) are shown for MCI (A, C, E) and AD (B, D, F). AUC increased from 0.639 for predicting MCI using APOE ε4 alone to 0.713 with the trending miRNAs (A), and from 0.620 for predicting AD using APOE ε4 alone to 0.772 with the trending miRNAs (B).AUC increased from 0.758 using Aβ42:T-Tau alone to predict MCI to 0.813 with trending miRNAs (C), and from 0.867 using Aβ42:T-Tau alone to predict AD to 0.903 with trending miRNAs (D). Combining all 3 markers (Aβ42:T-Tau + APOE ε4 + trending miRNAs) showed no meaningful improvement in classification performance over Aβ42:T-Tau + trending miRNAs: AUC increased from 0.813 to 0.819 for predicting MCI, but decreased from 0.903 to 0.898 for predicting AD (F), in both cases only negligible differences. miRNAs, expression levels of the 5 trending miRNAs; APOE, APOE ε4 status; Aβ42:Tau, Aβ42:T-Tau ratio.
Table 3. Classification performance of combinations of AD-MCI trending miRNA biomarkers, Aβ42:T-Tau, and APOEε4 status.
The boost in the area under the receiver operating characteristic curve (AUC) from adding the trending miRNAs to combinations of the existing predictors is shown as a change in AUC. The miRNAs dramatically outperformed APOEε4 for either diagnosis and added value to an Aβ42:T-Tau prediction of AD. The boost was even more robust for MCI, where Aβ42:T-Tau levels were less predictive of MCI than AD. APOEε4 added little or nothing to models already containing Aβ42:T-Tau information. Omnibus tests of the one-sided hypotheses showed that the trending miRNAs uniformly improved prediction of disease status (i.e. positive change for all 3 models considered, accounting for dependencies among the models) return p-values of 0.024 for MCI and 0.039 for AD.
| Marker Combinations | MCI vs. NC | AD vs. NC | ||||||
|---|---|---|---|---|---|---|---|---|
| AUC | Change | AUC | Change | |||||
| Trending miRNAs | 0.705 | 0.770 | ||||||
|
| ||||||||
| APOEε4 | 0.639 | 0.620 | ||||||
| APOEε4 | + | Trending miRNAs | 0.713 | +0.074 (p=0.176) | 0.772 | +0.152 (p=0.037) | ||
|
| ||||||||
| Aβ42:T-Tau | 0.758 | 0.867 | ||||||
| Aβ42:T-Tau | + | Trending miRNAs | 0.813 | +0.055 (p=0.051) | 0.903 | +0.036 (p=0.113) | ||
|
| ||||||||
| Aβ42:T-Tau | APOEε4 | 0.756 | 0.881 | |||||
| Aβ42:T-Tau | + | APOEε4 | + | Trending miRNAs | 0.819 | +0.063 (p=0.027) | 0.898 | +0.017 (p=0.414) |
Robustness of findings to exclusion of repeated samples
As a sensitivity analysis, we temporarily excluded the repeated 21 NC and 23 AD samples (retaining 44 NC, 31 MCI, and 14 AD), and on the reduced cohort performed the same trend and classification analyses as described above. We found that essentially the same trend profiles emerged for all of the miRNAs, with some attenuation or distortion in a few cases due to the drastically reduced NC and especially AD sample sizes. We further found that the classification performance showed the same rank ordering of models as in the full cohort, as well as strong agreement in AUC values over the scenarios considered. Therefore, we believe these findings are robust whether or not repeated samples are included. Further details of this sensitivity analysis can be found in Supplementary Results.
Weak correlation between Aβ42:T-Tau and trending miRNAs
We employed a multivariate correlation of the trending markers with Aβ42:T-Tau to examine whether there was a correlation between the trending miRNAs and Aβ42:T-Tau. The scatterplot (Fig. 4) showed a modest multivariate correlation of 0.38 (p = 0.004) that exists primarily due to a modest trend in control samples (gray). However, in the “diseased” range of Aβ42:T-Tau levels (between 0 and 2, where most of the AD and MCI samples lie), there was no appreciable correlation between the trending miRNAs and Aβ42:T-Tau levels (0.17, p = 0.922). Empirical trend lines in the plot indicated that a positive relationship only existed for NC samples. This suggests that information provided by the trending miRNAs as markers for MCI was mostly independent of that provided by Aβ42:T-Tau levels, so that the miRNAs carry novel information relevant for prediction of AD-like pathology.
Fig. 4.
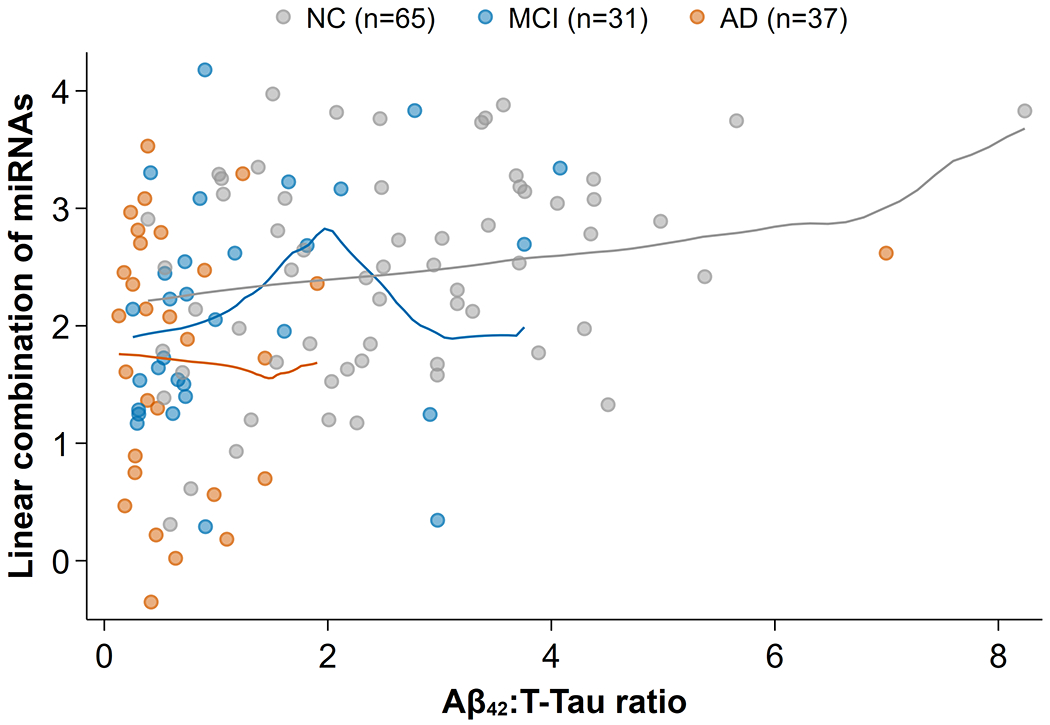
Weak correlation between Aβ42:T-Tau and five trending miRNAs for MCI. The canonical linear combination of miRNAs (i.e., the linear projection of miRNA expression values that best predicts Aβ42:T-Tau) is shown against Aβ42:T-Tau levels. What correlation exists (0.38) was primarily due to a modest trend in control samples (gray). In the “diseased” range of Aβ42:T-Tau between 0 and 2, where most of the AD(orange) and MCI(blue) samples lie, there was no appreciable correlation (0.17) between the trending miRNAs and Aβ42:T-Tau levels. Lines show the empirical trends: for NC there was a weak, but consistent positive relationship, and for AD and MCI the lines show random sampling fluctuations, but no apparent correlation.
Predicted targets of the trending miRNAs
We queried the predicted targets for the top 5 trending miRNAs (miR-142-3p, miR-146a-5p, miR-146b-5p, miR-365a-3p, miR-193a-5p) using TargetScan 7.2 [21] and miRDB [22, 23], as depicted in Fig. 5. Target prediction returned a total of 1,214 predicted mRNA: 367 predicted by TargetScan, 1,096 predicted by miRDB. The two target lists from each algorithm overlapped by 249 targets. We then used IPA to identify pathways that include the 1,214 total predicted targets. IPA reported that 353 of the targets were found in 214 significant pathways. Notably, Ras-related C3 botulinum toxin substrate 1 (RAC1) was predicted to be targeted by 4 of the 5 top miRNAs (miR-142-3p, miR-146a-5p, miR-146b-5p, miR-365a-3p). The significant neural, glial, and immune pathways identified by IPA that include RAC1 are listed in Table 4.
Fig. 5.
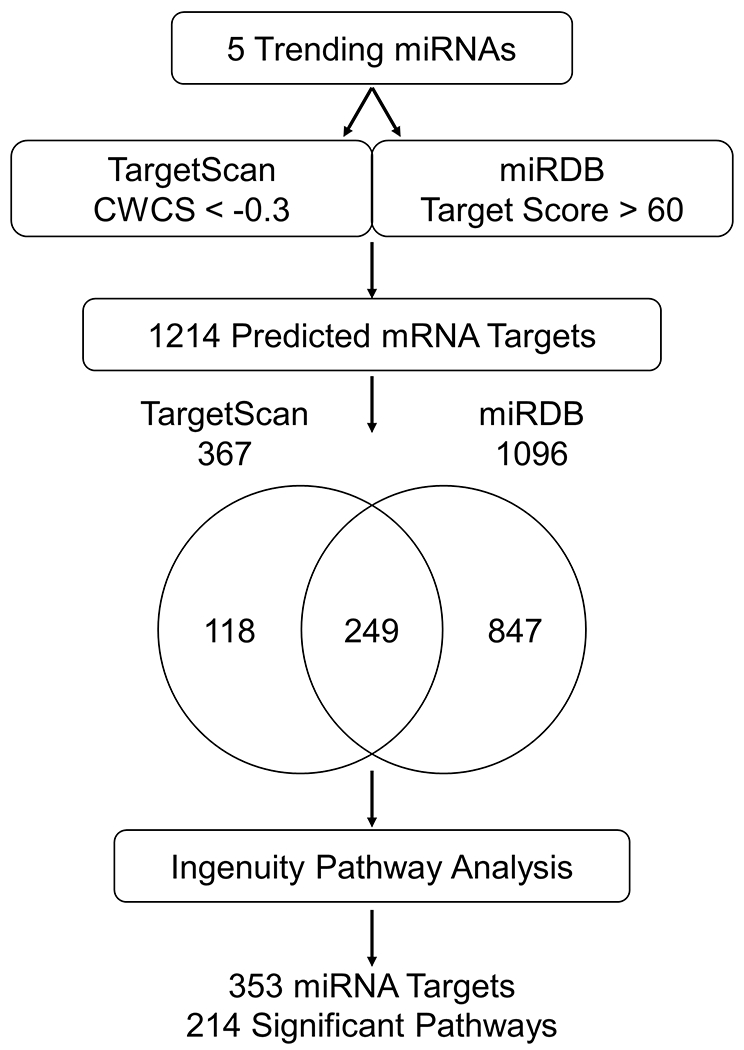
Target prediction pipeline for 5 trending miRNAs. Workflow of the target prediction analysis. The 5 trending miRNAs were queried using TargetScan 7.2 with a CWCS of <−0.3 and miRDB with a target score >60. The Venn diagram shows the overlap in miRNA targets predicted by TargetScan 7.2 and miRDB. The pathways of the 1214 predicted mRNA targets were identified using IPA: 353 of the targets were found in 214 significant pathways.
Table 4. MiRNA target prediction.
The table lists the significant pathways containing RAC1, a protein known to be involved in AD and predicted to be targeted by 4 of the 5 trending miRNAs.
| Ingenuity Pathways | −log(p-value) | |
|---|---|---|
| Neural | Axonal Guidance Signaling | 6.24 |
| Synaptogenesis Signaling | 4.82 | |
| HGF Signaling | 4.81 | |
| NGF Signaling | 4.65 | |
| Opioid Signaling | 4.06 | |
| Endocannabinoid Developing Neuron | 3.06 | |
| GNRH Signaling | 2.79 | |
| Reelin Signaling in Neurons | 2.55 | |
| Amyotrophic Lateral Sclerosis Signaling | 2.37 | |
| Glucocorticoid Receptor Signaling | 2.29 | |
| Netrin Signaling | 2.22 | |
| Glioma Invasiveness Signaling | 1.86 | |
| Agrin Interactions at Neuromuscular Junction | 1.68 | |
| Semaphorin Signaling in Neurons | 1.46 | |
| Glial | GDNF Family Ligand-Receptor Interactions | 4.51 |
| Immune Related | Systemic Lupus Erythematosus in B Cell Signaling | 6.98 |
| fMLP Signaling in Neutrophils | 4.55 | |
| B Cell Receptor Signaling | 4.49 | |
| STAT3 | 4.16 | |
| CCR3 Signaling in Eosinophils | 4.15 | |
| CXCR4 Signaling | 3.82 | |
| IL-3 Signaling | 3.71 | |
| Fc Epsilon RI Signaling | 3.46 | |
| Natural Killer Cell Signaling | 3.37 | |
| Regulation of IL-2 Expression in Activated and Anergic T Lymphocytes | 3.19 | |
| IL-8 Signaling | 3.15 | |
| PI3K Signaling in B Lymphocytes | 3.10 | |
| Sphingosine-1-phosphate Signaling | 2.98 | |
| Macropinocytosis Signaling | 2.75 | |
| iCOS-iCOSL Signaling in T Helper Cells | 2.31 | |
| Fc Receptor-mediated Phagocytosis in Macrophages and Monocytes | 2.03 | |
| Leukocyte Extravasation Signaling | 1.85 | |
| PKC, Signaling in T Lymphocytes | 1.83 | |
| T Cell Receptor Signaling | 1.70 | |
| HMGB1 Signaling | 1.61 | |
| CD28 Signaling in T Helper Cells | 1.33 |
DISCUSSION
We previously discovered 36 miRNAs in CSF from living donors that differentiate AD from cognitively normal controls [12]. A follow-up study in a new and independent cohort of donors then validated 26 of these miRNAs as potential biomarker candidates for AD in CSF [13]. Our exhaustive classification performance analysis revealed that 14 of the 26 candidates strongly contribute in a mutually complementary and additive manner to AD prediction across a broad range of model scenarios [13]. In this study, we investigated the ability of these 14 validated miRNAs to serve as robust biomarkers for early indications of AD-associated brain changes in patients diagnosed with MCI. One additional miRNA, miR-142-3p, a top-ranking candidate in our discovery study [12], also showed promise as a biomarker for MCI. These 15 biomarkers were then assessed for trending discrimination performance in MCI and AD. In this cohort, 5 miRNAs showed a linear trend of median expression across the ordered diagnoses (miR-142-3p, miR-146b-5p, miR-146a-5p, miR-365a-3p, and miR-193a-5p); this trend reflected decreasing median expression as disease severity increased. Evaluation of the effect of combining the 5 trending miRNAs with current AD biomarkers revealed that adding the trending miRNAs to a Aβ42:T-Tau classifier for MCI offered a 0.055 AUC increase in performance (0.813 versus 0.758). The weak correlation of the trending markers with Aβ42:T-Tau levels suggests that the miRNAs carry substantial additional information relevant for prediction of AD-like pathology, even at the MCI stage. We have summarized findings from our discovery AD study [12], our validation AD study [13], and the present MCI study for each of these 15 biomarkers in Table 5.
Table 5. Performance of CSF miRNAs across biomarker studies.
Rank reflects our initial prioritization of the miRNAs as biomarker candidates in our discovery study (1=highest); the ranks of tied miRNAs were averaged. Viability on retest (Viable) indicates whether or not (Y=yes or N=no) the miRNA proved to be reliably measurable and robustly expressed in a validation cohort to a degree sufficient to warrant further consideration as a biomarker. Validated for AD (Validated) indicates whether or not the miRNA demonstrated continued biomarker potential as a significant contributor to AD classifier models in the validation cohort. The MCI-AD trend (Trend) classification is as in Table 2 and Figure 4. Additional notes reflect our current subjective assessment of the character and value of the miRNA as a biomarker for MCI or AD.
| MiRNA | Rank | Viable | Validated | Trend | Notes |
|---|---|---|---|---|---|
| miR-15b-5p | 25 | Y | Y | NO TREND | Probably decreased in AD, but with large variance and often not expressed |
| miR-16-5p | 12.5 | Y | N | Often decreased in AD, but not consistently across cohorts | |
| miR-19b-3p | 7 | Y | Y | AD+MCI vs. NC | Decreased equally in MCI and AD |
| miR-24-3p | 10 | Y | N | Often decreased in AD, but not consistently across cohorts | |
| miR-26b-5p | 15.5 | N | N | Tended to be decreased in AD, but rarely expressed | |
| miR-27b-3p | 32.5 | N | N | Tended to be decreased in AD, but rarely expressed | |
| miR-28-3p | 23 | Y | N | Unclear directionality of change in AD | |
| miR-29a-3p | 12.5 | Y | N | Often decreased in AD, but not consistently across cohorts | |
| miR-30a-3p | 15.5 | Y | Y | NO TREND | Probably decreased in both MCI and AD, but with unclear ordering |
| miR-30d-5p | 26 | N | N | Tended to be decreased in AD, but rarely expressed | |
| miR-125b-5p | 17 | Y | N | Often decreased in AD, but not consistently across cohorts | |
| miR-140-5p | 12.5 | Y | Y | AD vs. MCI+NC | Decreased in AD, but not MCI |
| miR-142-3p | 1 | Y | N | TREND | Decreasing as NC > MCI > AD; |
| miR-143-3p | 9 | Y | N | Tended to be decreased in AD, but often not expressed | |
| miR-145-5p | 20 | Y | N | Often decreased in AD, but not consistently across cohorts | |
| miR-145-3p | 20 | N | N | Tended to be decreased in AD, but rarely expressed | |
| miR-146a-5p | 18 | Y | Y | TREND | Decreasing as NC > MCI > AD |
| miR-146b-5p | 22 | Y | Y | TREND | Decreasing as NC > MCI > AD |
| miR-193a-5p | 6 | Y | Y | TREND | Decreasing as NC > MCI > AD |
| miR-195-5p | 27 | Y | N | Often decreased in AD, but not consistently across cohorts | |
| miR-202-3p | 32.5 | N | N | Tended to be increased in AD, but rarely expressed | |
| miR-223-3p | 24 | Y | Y | NO TREND | Increased in MCI and AD, but with large variance |
| miR-328-3p | 12.5 | Y | N | Often decreased in AD, but not consistently across cohorts | |
| miR-331-3p | 32.5 | Y | Y | AD vs. MCI+NC |
Decreased in AD but not MCI |
| miR-340-5p | 4 | N | N | Very rarely expressed; unclear directionality of change in AD | |
| miR-365a-3p | 32.5 | Y | Y | TREND | Decreasing as NC > MCI > AD |
| miR-378a-3p | 8 | Y | Y | NO TREND | Increased in MCI and AD, but with large variance |
| miR-484 | 32.5 | Y | Y | AD+MCI vs. NC | Increased equally in MCI and AD |
| miR-519b-3p | 20 | N | N | Technical problems with amplification in all trials; probe seems unusable for CSF | |
| miR-520b | 5 | N | N | Very rarely expressed in any condition, but more likely to appear in AD | |
| miR-532-5p | 28.5 | Y | N | Tended to be decreased in AD, but often not expressed | |
| miR-584-5p | 36 | Y | Y | NO TREND | Probably increased in both MCI and AD, but with unclear ordering |
| miR-590-5p | 32.5 | N | N | Tended to be increased in AD, but rarely expressed | |
| miR-597-5p | 3 | Y | Y | NO TREND | Tended to be increased in AD, but with large variance |
| miR-603 | 28.5 | N | N | Tended to be increased in AD, but rarely expressed | |
| miR-1291 | 2 | N | N | Very rarely expressed in any condition, but more likely to appear in AD |
Our choice to focus on miRNAs that showed a trend across diagnoses was based on the model that relevant disease pathology varies linearly across these groups, with NC < MCI < AD. However, it is also possible that some biological processes will be most robust at the MCI phase of disease initiation, and in fact we also found a class of 6 “no trend” miRNAs where MCI expression was either higher or lower than both AD and NC (Fig. 2). We recognize that these miRNAs may also be informative, and they will thus be the subject of future analyses in longitudinal studies of MCI patients known to either remain stable or progress to AD.
The utility of our analytic approach in the present study is illustrated by the convergence of our findings with others reported in the literature. For example, 3 of our 5 trending miRNAs (miR-142-3p, -146a-5p, -146b-5p) have been identified by others [30–35] as candidate biomarkers for MCI and/or AD. In line with our findings in CSF, the expression level of miR-142-3p was decreased in plasma of MCI and AD relative to control [34]. Also, miR-146b-5p was decreased in the CSF, hippocampus, and medial frontal gyrus of AD compared to normal controls [31]. Furthermore, miR-146b-5p was included in a proposed assay for early diagnosis of AD using fluorescent nanoparticle imaging [35]. While these studies support our findings, alternative miRNAs have been identified as peripheral biomarkers for early detection of AD, then validated in AD cells, postmortem brain, and mouse models. These studies revealed increasing levels of miR-455-3p in the serum of MCI and AD subjects [36]. It is notable that the expression of miR-455-3p was also significantly upregulated in postmortem AD brain, and in fibroblasts from AD patients, relative to controls [37], as well as in amyloid precursor transgenic mice [38]. These data demonstrate the potential to discover biomarkers using peripheral biofluids. In line with this we are currently assessing the performance of not only the trending miRNAs in plasma, but also exploring the potential of additional miRNAs. One can imagine that a robust biomarker assay may consist of combinations of miRNAs, derived from both CSF and the periphery, in order to increase classification performance of specific miRNAs for AD.
Our statistical approach is also consistent with the literature. In one such example, linear combinations of a subset of CSF miRNAs (miR-16-5p, -125-5p, -451a, -605-5p) improved the classification performance between young- or late-onset AD and controls [39]. Also, combining two sets of miRNA pairs (e.g., miR-191/miR-101 plus miR-103/miR-222) was shown to greatly improve classification of MCI from controls [40]. Finally, combining CSF expression levels of let-7b with either Aβ40:Aβ42 or T-Tau:P-Tau also improved the diagnostic potential for AD [41].
A major limitation of this study was the absence of pathologic confirmation of diagnoses or of data on progression to AD among the MCI subjects. We considered reassigning diagnoses according to CSF protein biomarker status, but in the absence of an established cutpoint for the assays available, and in the face of relatively small groups, we elected to confine the analysis to the prespecified clinical diagnoses in order to avoid bias from post-hoc assignments.
One unexpected outcome was the finding of 32 participant samples with aberrant expression profiles in the array data that were uniformly low or high in expression, where “uniformly” means that this pattern was present across all of the miRNA probes but not necessarily by the same amount for each probe. We identified 14 samples that were uniformly very low in expression (Cq values far above those for typical samples, including many or most above the censoring threshold) which were found to be due to technical errors in the RT. We found 18 additional aberrant samples, 14 of which were uniformly very high in expression (Cq values far below those for typical samples), but there was no consideration that could account for these high-expressing aberrant samples. The aberrant pattern was also present in the normalizer values (based on the positive control probes), which is why we chose to present that metric in describing the issue above (see Exclusion of aberrant samples). The scale was on the order of ±7 Cq, which is indicative of hundred-fold or larger differences in total expression between the aberrant samples and the typical samples, for every miRNA. As all of the high-expression (and a few of the low-expression) cases remain unexplained and unrelated to any participant characteristic or design factor, and the criteria for excluding the aberrant data were rigorous and unbiased, we felt compelled to exclude them due to their very extreme response profile, which far exceeds natural variation.
We did examine whether exclusion of the 32 samples impacted the balance of participants in the study. Our intention was to exactly balance all 3 diagnostic groups by sex and age, which our sampling was reasonably successful at prior to the exclusions. We examined each miRNA for association with each factor for the final sample sets, both overall and within diagnostic groups, but found no hints of confounding. Briefly, none of the miRNAs showed any signs of a meaningful age trend, either unconditionally or when adjusting for diagnosis, sex, or both (maximum magnitude of slope much less than 0.5 Cq per decade of age, well within assay noise windows), and only a handful of the miRNAs showed differences between sexes even approaching 0.5 Cq in magnitude, with or without other adjustments. For all the miRNAs we performed sensitivity analyses to rule out confounding of the group effect by age or sex effects, and found only negligible changes to the associations after adjustments. The most affected result under age adjustment was for miR-30a-3p, where the adjusted fold changes were more extreme (i.e., more suggestive of group differences) than the unadjusted estimates by about 20%; however, the direction of changes and the classification of this miRNA as “no trend” remained the same because the MCI change was still larger than the AD change. The most affected result under adjustment for sex was for miR-146a-5p, one of the “trending” miRNAs highlighted in the manuscript; in this case, the trend shallowed under adjustment (also by about 20%) such that all the groups moved closer together, but again, the ordering of the groups as NC > MCI > AD remained stable. None of the miRNAs would be differently classified (as “trend” or otherwise) under these adjustments, and the essential message that the “trending” miRNAs as a set may suggest a pathway for tracking disease progression is also unaffected. Thus, we believe there was no confounding of the findings due to the minor accidental imbalances in age and sex distributions across the diagnostic groups that was unfortunately exacerbated by the need to exclude the aberrant samples from the analytic cohort.
Despite these challenges, our analysis has identified CSF miRNAs that provide additive, novel information which goes beyond the established CSF protein biomarker data to distinguish MCI from control and AD subjects. We consequently attempted to characterize this novel information by applying a target prediction strategy. It is well established that one miRNA can target hundreds of mRNA transcripts, and that several miRNAs can target a single mRNA transcript [42, 43]. In addition, miRNAs can bind to the 3’UTRs, the 5’UTRs, and coding regions, where they can both activate and repress translation [44]. Thus, target prediction has been a major challenge in determining the effect of miRNAs on translation and protein expression. We utilized two well-known target prediction programs with differing algorithms: TargetScan 7.2 [21] and miRDB [22, 23], to identify targets of the 5 trending miRNAs. We then identified the targets that overlapped between the two programs to increase confidence in the predicted targets. We found that RAC1 was among the targets of 4 of the 5 trending miRNAs: miR-142-3p, miR-146a-5p, miR-146b-5p, and miR-365a-3p. RAC1 is a Rho-GTPase involved in many cellular processes that involve cytoskeleton dynamics and stimulate axonal growth, dendritic branching, and spine formation [45]. One study has shown that miR-142-3p acts as a negative regulator of human RAC1 [46], and several others have shown RAC1 regulation by miR-142-3p in rodents.
It is also noteworthy that miR-142-3p has been implicated in human AD processes [47, 48] and that miR-146a-5p is implicated in AD and Tau human [49, 50]. We found it interesting that the most significant neuronal pathway in our analysis is Axonal Guidance Signaling, a process implicated in AD as the loss of dendritic spines is associated with cognitive decline in patients [51]. RAC1 is highly involved in axon guidance and has been shown to be decreased in the frontal cortex of AD patients and increased in their plasma [52]. Additionally, inhibition of RAC1 has also been shown to reduce the activity of gammasecretase, which cleaves amyloid-β protein precursor and has a direct effect on Aβ42 and Tau [52, 53]. These findings show that the predicted target proteins of the trending miRNAs are associated with AD, lending increased validity to our results. This analysis also illustrates that these miRNAs could serve not only as biomarkers, but also as roadmaps to novel targets for further evaluation in AD pathogenesis. Current studies are focused on identifying novel proteins and pathways that may be regulated by the trending miRNAs that contribute to the processes underlying neurodegeneration in AD.
Another important consideration of these findings regards the specificity of the trending miRNAs for AD versus other neurodegenerative diseases or non-AD dementias. While the studies are beyond the current scope of this manuscript, they are important and a focus of our ongoing work to assess the specificity of the trending miRNAs for AD versus Parkinson’s disease, Lewy body dementia, frontotemporal dementia, and vascular impairment. Specificity of the miRNAs for AD would not only strengthen their utility as biomarkers for AD, but may also provide insight into target proteins unique to AD pathology.
In summary, our studies revealed 5 CSF miRNA biomarkers that show a trend of decreased expression in the progression from NC to MCI to AD. Importantly, combining the trending miRNAs with Aβ42:T-Tau levels predicted MCI with increased classification performance, and the weak correlation of the trending markers with Aβ42:T-Tau levels suggests that the miRNAs carry independent information relevant for disease characterization even at the MCI stage. Current studies include assessing the performance of these CSF-validated miRNAs in plasma (where Aβ42:T-Tau has been difficult to measure) and in longitudinal studies of AD and MCI, as well as assessing their specificity for AD versus Parkinson’s disease and other dementias (Lewy body, frontotemporal, and vascular). In addition, the targets of these miRNAs may have potential to serve as novel therapeutic targets for AD.
Supplementary Material
ACKNOWLEDGMENTS
These studies were supported by NIH grants UH3TR000903 (JAS, JFQ), AG08017 (JFQ), and AG005131 (DRG). The UH3 award is supported by the NIH Common Fund, through the Office of Strategic Coordination/Office of the NIH Director. All donor procedures were approved by the Institutional Review Board of the University of California, San Diego (UCSD IRB 80012). We thank the OHSU Gene Profiling Shared Resource for professional advice, technical support, and access to core instrumentation. We also thank Sarah Catherine Baker for critical reading and editing of the manuscript.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/20-0396r1).
Footnotes
SUPPLEMENTARY MATERIAL
The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-200396.
REFERENCES
- [1].Alzheimer’s Association (2019) Alzheimer’s disease facts and figures. Alzheimers Dement 15, 321–387. [Google Scholar]
- [2].Ghidoni R, Benussi L, Paterlini A, Albertini V, Binetti G, Emanuele E (2011) Cerebrospinal fluid biomarkers for Alzheimer’s disease: The present and the future. Neurodegener Dis 8, 413–420. [DOI] [PubMed] [Google Scholar]
- [3].Cummings J (2019) The National Institute on Aging-Alzheimer’s Association Framework on Alzheimer’s disease: Application to clinical trials. Alzheimers Dement 15, 172–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Quinn JF (2013) Biomarkers for Alzheimer’s disease: Showing the way or leading us astray? J Alzheimers Dis 33 Suppl 1, S371–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, Gamst A, Holtzman DM, Jagust WJ, Petersen RC, Snyder PJ, Carrillo MC, Thies B, Phelps CH (2011)The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7, 270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr., Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, Mohs RC, Morris JC, Rossor MN, Scheltens P, Carrillo MC, Thies B, Weintraub S, Phelps CH (2011) The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR Jr., Kaye J, Montine TJ, Park DC, Reiman EM, Rowe CC, Siemers E, Stern Y, Yaffe K, Carrillo MC, Thies B, Morrison-Bogorad M, Wagster MV, Phelps CH (2011) Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7, 280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Petersen RC, Lopez O, Armstrong MJ, Getchius TSD, Ganguli M, Gloss D, Gronseth GS, Marson D, Pringsheim T, Day GS, Sager M, Stevens J, Rae-Grant A (2018) Practice guideline update summary: Mild cognitive impairment: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology. Neurology 90, 126–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ward A, Tardiff S, Dye C, Arrighi HM (2013) Rate of conversion from prodromal Alzheimer’s disease to Alzheimer’s dementia: A systematic review of the literature. Dement Geriatr Cogn Dis Extra 3, 320–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mitchell AJ, Shiri-Feshki M (2009) Rate of progression of mild cognitive impairment to dementia–meta-analysis of 41 robust inception cohort studies. Acta Psychiatr Scand 119, 252–265. [DOI] [PubMed] [Google Scholar]
- [11].Quinn JF, Patel T, Wong D, Das S, Freedman JE, Laurent LC, Carter BS, Hochberg F, Van Keuren-Jensen K, Huentelman M, Spetzler R, Kalani MY, Arango J, Adelson PD, Weiner HL, Gandhi R, Goilav B, Putterman C, Saugstad JA (2015) Extracellular RNAs: Development as biomarkers of human disease. J Extracell Vesicles 4, 27495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lusardi TA, Phillips JI, Wiedrick JT, Harrington CA, Lind B, Lapidus JA, Quinn JF, Saugstad JA (2017) MicroRNAs in human cerebrospinal fluid as biomarkers for Alzheimer’s disease. J Alzheimers Dis 55, 1223–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wiedrick JT, Phillips JI, Lusardi TA, McFarland TJ, Lind B, Sandau US, Harrington CA, Lapidus JA, Galasko DR, Quinn JF, Saugstad JA (2019) Validation of microRNA biomarkers for Alzheimer’s disease in human cerebrospinal fluid. J Alzheimers Dis 67, 875–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Shi M, Bradner J, Hancock AM, Chung KA, Quinn JF, Peskind ER, Galasko D, Jankovic J, Zabetian CP, Kim HM, Leverenz JB, Montine TJ, Ginghina C, Kang UJ, Cain KC, Wang Y, Aasly J, Goldstein D, Zhang J (2011) Cerebrospinal fluid biomarkers for Parkinson disease diagnosis and progression. Ann Neurol 69, 570–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hixson JE, Vernier DT (1990) Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res 31, 545–548. [PubMed] [Google Scholar]
- [16].Wierenga CE, Dev SI, Shin DD, Clark LR, Bangen KJ, Jak AJ, Rissman RA, Liu TT, Salmon DP, Bondi MW (2012) Effect of mild cognitive impairment and APOE genotype on resting cerebral blood flow and its association with cognition. J Cereb Blood Flow Metab 32, 1589–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lehmann S, Delaby C, Boursier G, Catteau C, Ginestet N, Tiers L, Maceski A, Navucet S, Paquet C, Dumurgier J, Vanmechelen E, Vanderstichele H, Gabelle A (2018) Relevance of Abeta42/40 ratio for detection of Alzheimer disease pathology in clinical routine: The PLMR Scale. Front Aging Neurosci 10, 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gevaert AB, Witvrouwen I, Vrints CJ, Heidbuchel H, Van Craenenbroeck EM, Van Laere SJ, Van Craenenbroeck AH (2018) MicroRNA profiling in plasma samples using qPCR arrays: Recommendations for correct analysis and interpretation. PLoS One 13, e0193173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Raudenbush SW, Bryk AS (2002) Hierarchical Linear Models: Applications and Data Analysis Methods, Sage Publishing. [Google Scholar]
- [20].Pepe MS (2003) The Statistical Evaluation of Medical Tests for Classification and Prediction, Oxford University Press, Oxford, New York. [Google Scholar]
- [21].Agarwal V, Bell GW, Nam JW, Bartel DP (2015) Predicting effective microRNA target sites in mammalian mRNAs. Elife 4, e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chen Y, Wang X (2020) miRDB: An online database for prediction of functional microRNA targets. Nucleic Acids Res 48, D127–D131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Liu W, Wang X (2019) Prediction of functional microRNA targets by integrative modeling of microRNA binding and target expression data. Genome Biol 20, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Fan X, Kurgan L (2015) Comprehensive overview and assessment of computational prediction of microRNA targets in animals. Brief Bioinform 16, 780–794. [DOI] [PubMed] [Google Scholar]
- [25].Oliveira AC, Bovolenta LA, Nachtigall PG, Herkenhoff ME, Lemke N, Pinhal D (2017) Combining results from distinct microRNA target prediction tools enhances the performance of analyses. Front Genet 8, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chou CH, Shrestha S, Yang CD, Chang NW, Lin YL, Liao KW, Huang WC, Sun TH, Tu SJ, Lee WH, Chiew MY, Tai CS, Wei TY, Tsai TR, Huang HT, Wang CY, Wu HY, Ho SY, Chen PR, Chuang CH, Hsieh PJ, Wu YS, Chen WL, Li MJ, Wu YC, Huang XY, Ng FL, Buddhakosai W, Huang PC, Lan KC, Huang CY, Weng SL, Cheng YN, Liang C, Hsu WL, Huang HD (2018) miRTarBase update 2018: A resource for experimentally validated microRNA-target interactions. Nucleic Acids Res 46, D296–D302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Folstein MF, Robins LN, Helzer JE (1983) The Mini-Mental State Examination. Arch Gen Psychiatry 40, 812. [DOI] [PubMed] [Google Scholar]
- [28].McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM (1984) Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34,939–944. [DOI] [PubMed] [Google Scholar]
- [29].Seshadri S, Drachman DA, Lippa CF (1995) Apolipoprotein E epsilon 4 allele and the lifetime risk of Alzheimer’s disease. What physicians know, and what they should know. Arch Neurol 52, 1074–1079. [DOI] [PubMed] [Google Scholar]
- [30].Alexandrov PN, Dua P, Hill JM, Bhattacharjee S, Zhao Y, Lukiw WJ (2012) microRNA (miRNA) speciation in Alzheimer’s disease (AD) cerebrospinal fluid (CSF) and extracellular fluid (ECF). Int J Biochem Mol Biol 3,365–373. [PMC free article] [PubMed] [Google Scholar]
- [31].Cogswell JP, Ward J, Taylor IA, Waters M, Shi Y, Cannon B, Kelnar K, Kemppainen J, Brown D, Chen C, Prinjha RK, Richardson JC, Saunders AM, Roses AD, Richards CA (2008) Identification of miRNA changes in Alzheimer’s disease brain and CSF yields putative biomarkers and insights into disease pathways. J Alzheimers Dis 14, 27–41. [DOI] [PubMed] [Google Scholar]
- [32].Denk J, Boelmans K, Siegismund C, Lassner D, Arlt S, Jahn H (2015) MicroRNA profiling of CSF reveals potential biomarkers to detect Alzheimer’s disease. PLoS One 10, e0126423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kiko T, Nakagawa K, Tsuduki T, Furukawa K, Arai H, Miyazawa T (2014) MicroRNAs in plasma and cerebrospinal fluid as potential markers for Alzheimer’s disease. J Alzheimers Dis 39, 253–259. [DOI] [PubMed] [Google Scholar]
- [34].Nagaraj S, Laskowska-Kaszub K, Debski KJ, Wojsiat J, Dabrowski M, Gabryelewicz T, Kuznicki J, Wojda U (2017) Profile of 6 microRNA in blood plasma distinguish early stage Alzheimer’s disease patients from non-demented subjects. Oncotarget 8, 16122–16143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Park JS, Kim ST, Kim SY, Jo MG, Choi MJ, Kim MO (2019) A novel kit for early diagnosis of Alzheimer’s disease using a fluorescent nanoparticle imaging. Sci Rep 9, 13184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kumar S, Vijayan M, Reddy PH (2017) MicroRNA-455-3p as a potential peripheral biomarker for Alzheimer’s disease. Hum Mol Genet 26, 3808–3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kumar S, Reddy PH (2018) MicroRNA-455-3p as a potential biomarker for Alzheimer’s disease: An update. Front Aging Neurosci 10, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kumar S, Vijayan M, Bhatti JS, Reddy PH (2017) MicroRNAs as peripheral biomarkers in aging and age-related diseases. Prog Mol Biol Transl Sci 146, 47–94. [DOI] [PubMed] [Google Scholar]
- [39].McKeever PM, Schneider R, Taghdiri F, Weichert A, Multani N, Brown RA, Boxer AL, Karydas A, Miller B, Robertson J, Tartaglia MC (2018) MicroRNA expression levels are altered in the cerebrospinal fluid of patients with young-onset Alzheimer’s disease. Mol Neurobiol 55, 8826–8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kayano M, Higaki S, Satoh JI, Matsumoto K, Matsubara E, Takikawa O, Niida S (2016) Plasma microRNA biomarker detection for mild cognitive impairment using differential correlation analysis. Biomark Res 4, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Liu Y, He X, Li Y, Wang T (2018) Cerebrospinal fluid CD4+T lymphocyte-derived miRNA-let-7b can enhances the diagnostic performance of Alzheimer’s disease biomarkers. Biochem Biophys Res Commun 495, 1144–1150. [DOI] [PubMed] [Google Scholar]
- [42].Bartel DP (2009) MicroRNAs: Target recognition and regulatory functions. Cell 136, 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lewis BP, Burge CB, Bartel DP (2005) Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15–20. [DOI] [PubMed] [Google Scholar]
- [44].Ni WJ, Leng XM (2015) Dynamic miRNA-mRNA paradigms: New faces of miRNAs. Biochem Biophys Rep 4, 337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Aguilar BJ, Zhu Y, Lu Q (2017) Rho GTPases as therapeutic targets in Alzheimer’s disease. Alzheimers Res Ther 9, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wu L, Cai C, Wang X, Liu M, Li X, Tang H (2011) MicroRNA-142-3p, a new regulator of RAC1, suppresses the migration and invasion of hepatocellular carcinoma cells. FEBS Lett 585, 1322–1330. [DOI] [PubMed] [Google Scholar]
- [47].Cosin-Tomas M, Antonell A, Llado A, Alcolea D, Fortea J, Ezquerra M, Lleo A, Marti MJ, Pallas M, Sanchez-Valle R, Molinuevo JL, Sanfeliu C, Kaliman P (2017) Plasma miR-34a-5p and miR-545-3p as early biomarkers of Alzheimer’s disease: Potential and limitations. Mol Neurobiol 54, 5550–5562. [DOI] [PubMed] [Google Scholar]
- [48].Ghanbari M, Munshi ST, Ma B, Lendemeijer B, Bansal S, Adams HH, Wang W, Goth K, Slump DE, van den Hout M, van IWFJ, Bellusci S, Pan Q, Erkeland SJ, de Vrij FMS, Kushner SA, Ikram MA (2019) A functional variant in the miR-142 promoter modulating its expression and conferring risk of Alzheimer disease. Hum Mutat 40, 2131–2145. [DOI] [PubMed] [Google Scholar]
- [49].Sierksma A, Lu A, Salta E, Vanden Eynden E, Callaerts-Vegh Z, D’Hooge R, Blum D, Buee L, Fiers M, De Strooper B (2018) Deregulation of neuronal miRNAs induced by amyloid-beta or TAU pathology. Mol Neurodegener 13, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wu Y, Xu J, Xu J, Cheng J, Jiao D, Zhou C, Dai Y, Chen Q (2017) Lower serum levels of miR-29c-3p and miR-19b-3p as biomarkers for Alzheimer’s disease. Tohoku J Exp Med 242, 129–136. [DOI] [PubMed] [Google Scholar]
- [51].Falke E, Nissanov J, Mitchell TW, Bennett DA, Trojanowski JQ, Arnold SE (2003) Subicular dendritic arborization in Alzheimer’s disease correlates with neurofibrillary tangle density. Am J Pathol 163, 1615–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Borin M, Saraceno C, Catania M, Lorenzetto E, Pontelli V, Paterlini A, Fostinelli S, Avesani A, Di Fede G, Zanusso G, Benussi L, Binetti G, Zorzan S, Ghidoni R, Buffelli M, Bolognin S (2018) Rac1 activation links tau hyperphosphorylation and Abeta dysmetabolism in Alzheimer’s disease. Acta Neuropathol Commun 6, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Boo JH, Sohn JH, Kim JE, Song H, Mook-Jung I (2008) Rac1 changes the substrate specificity of gamma-secretase between amyloid precursor protein and Notch1. Biochem Biophys Res Commun 372, 913–917. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.