

Abstract
Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) is a potent tumor suppressor that regulates several key cellular processes, including proliferation, survival, genomic integrity, migration, and invasion, via PI3K-dependent and independent mechanisms. A subtle decrease in PTEN levels or catalytic activity is implicated not only in cancer but also in a wide spectrum of other diseases, including various respiratory diseases. A systemic overview of the advances in the molecular and cellular mechanisms of PTEN involved in the initiation and progression of respiratory diseases may offer novel targets for the development of effective therapeutics for the treatment of respiratory diseases. In the present review, we highlight the novel findings emerging from current research on the role of PTEN expression and regulation in airway pathological conditions such as asthma/allergic airway inflammation, pulmonary hypertension (PAH), chronic obstructive pulmonary disease (COPD), idiopathic pulmonary fibrosis (IPF), and other acute lung injuries (ALI). Moreover, we discuss the clinical implications of PTEN alteration and recently suggested therapeutic possibilities for restoration of PTEN expression and function in respiratory diseases.
1. Introduction
Chronic lung diseases such as asthma, pulmonary lung injury, chronic obstructive pulmonary disease (COPD), and idiopathic pulmonary fibrosis (IPF) are increasingly becoming an enormous global health concern and economic burden [1]. A growing body of evidence has shown that global exposure to polluted air, particles, toxins, and infectious microorganisms contributes to the development of these pulmonary diseases. Repeated exposure to these agents results in chronic airway inflammation and excessive lung tissue damage [2]. Despite great advances in the treatment of these diseases, there still remains a considerable unmet need for the development of safe and effective therapeutics. COPD is now the third leading cause of death worldwide, and no applicable drugs are available to reduce the mortality of COPD [3]. Although there are two therapeutic agents approved by FDA for IPF therapy, the current clinical therapy is unable to reverse the pathogenesis and only delays the progression of IPF [4].
Identification of novel biomarkers and development of unique gene expression signatures are indispensable to improve early diagnosis and accurate prognosis, as well as to develop effective therapeutics. Accumulating evidence has demonstrated that phosphatase and tensin homolog deleted on chromosome 10 (PTEN) is one of the most important biomarkers that regulate multiple processes associated with the initiation and progression of various chronic lung diseases. It was initially identified as a potent tumor suppressor gene located on chromosome region 10q23, by three different groups in 1997 [5–7]. Canonical PTEN encodes a 403-amino acid peptide composed of five structural-functional domains. A short N-terminal PIP2 binding domain with phosphatase activity is responsible for dephosphorylation of phospholipids, a C2 domain for membrane targeting, a regulatory C-terminal tail containing multiple phosphorylation sites (Ser362, Thr366, Ser370, Ser380, Ser382, Thr382, Thr383, and Ser385), a PEST (Pro, Glu, Ser, and Thr) sequence, and a PDZ domain-binding motif involved in the regulation of PTEN activity/stability (Figure 1(a)). The cellular distribution of PTEN varies among tissues [8]. PTEN is primarily localized in the cytoplasm of most epithelial cells, such as those of the skin, breast, and prostate, whereas it is mostly distributed in the nuclei of neurons, fibroblasts, and thyroid cells [9, 10]. Moreover, differences in the subcellular distribution of PTEN have been observed in normal tissues and in several malignancies [9, 11].
Figure 1.
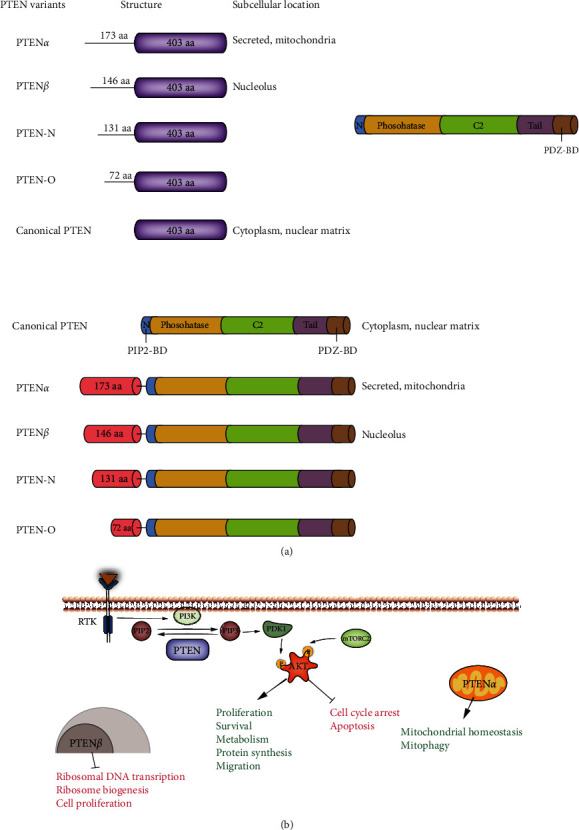
Molecular structure of PTEN (a) and cellular distribution of PTEN variants (b).
A new isoform of PTEN with a 173-amino acid extension at the N-terminus, termed PTEN-long (PTEN-L) or PTENα, was discovered in 2017; it differs from other isoforms in subcellular localization and function. PTENα is secreted from cells and is directly exported into neighboring cells to inhibit cell proliferation [12], or it specifically localizes in the mitochondria to regulate cell metabolism [13]. PTENβ, another PTEN subfamily member, was identified by the same research group. PTENβ has an extension of 146 amino acid residues at the N-terminus and is mainly localized in the nucleolus and regulates preribosomal RNA synthesis [14] (Figure 1(b)). In addition, two other variants (PTEN-N and PTEN-O) have been reported; however, their specific roles remain unclear [15, 16]. Similar to PTEN, the abovementioned variants possess the ability to inhibit PI3K/Akt signaling [16].
PTEN is characterized as a dual-specificity lipid and protein phosphatase that can regulate the signal transduction pathways by both phosphatidylinositol-3,4,5-triphosphate (PIP3)-dependent, and PIP3 independent mechanisms. PIP3 is a potent secondary messenger that binds and activates proteins containing the pleckstrin homology domain, such as members of AKT family (AKT1, AKT2, and AKT3) and phosphoinositide-dependent protein kinase-1 (PDK-1). PTEN dephosphorylates PIP3, thereby inhibiting the activation of PI3K/Akt, NF-κB, and the mammalian target of rapamycin (mTOR) signaling pathways, Akt/GSK-3β pathway, and the Akt/Wnt pathway. In addition, PTEN directly dephosphorylates focal adhesion kinase (FAK) and Src homology 2-containing protein (Shc). PTEN can autodephosphorylate itself on threonine residues within the regulatory C-terminal tail region [17], as well as dephosphorylate Ser, Tyr, and Thr residues of several other protein substrates such as FAK [18], IRS1 [19], CREB1 [20], and DVl2 [21]. In addition to lipid phosphatase activity, numerous studies have reported that PTEN exhibits nonenzymatic functions that are independent of PIP3 and PI3K/Akt pathway in both the cytoplasm and nucleolus. In the nucleolus, PTEN is involved in regulating diverse biological processes, including cell proliferation, transcription, and genomic stability. In the cytoplasm, PTEN was found to promote the activity of IP3R3 by competing with F-box/LRR-repeat protein 2 for IP3R3 binding in the cytosol, leading to Ca2+-mediated apoptosis [22]. Moreover, PTEN can stimulate proteasomal degradation of chromodomain-helicase-DNA-binding protein 1(CHD1) via β-TRCP E3 ubiquitin ligases to suppress CHD1-induced trimethyllysine-4 histone H3 modification, resulting in inhibition of transcription of oncogenic TNF-α/NF-κB pathway [1].
Alterations in PTEN expression and activity are associated with the pathogenesis of cancer as well as a wide spectrum of other diseases. Loss of PTEN function frequently occurs due to a combination of genetic/epigenetic mechanisms, including various mutations, chromosomal deletions, and hypermethylation of gene promoters in various cancers. Moreover, PTEN expression and activity are subject to extremely complex regulation at the transcriptional, posttranscriptional (microRNAs and long noncoding RNAs), translational, and posttranslational levels such as oxidation, S-nitrosylation, acetylation, SUMOylation, ubiquitylation, and phosphorylation [23]. These posttranslational mechanisms impact the conformation, lipid phosphatase activity, subcellular compartmentalization, membrane localization, and stability of PTEN [23, 24]. In addition, PTEN interacts with other cellular proteins, thereby regulating their expression levels, activities, and stability [23, 25]. Alterations of PTEN expression and activity were comprehensively reviewed elsewhere [23, 25]. PTEN plays an important role in the suppression of lung cancer, and the effect of PTEN in lung cancer has been systematically reviewed [26]. Therefore, we only highlight the role of PTEN in diverse lung respiratory diseases in the present review. Additionally, the novel effective therapies by targeting PTEN for lung diseases are summarized.
2. PTEN in Chronic Lung Diseases
2.1. Asthma and Allergic Airway Diseases
Asthma is a chronic inflammatory respiratory disease characterized by airway hyperresponsiveness, airflow obstruction, mucus hypersecretion, and airway remodeling in response to inhaled allergens and nonspecific stimuli [27]. Growing evidence has revealed that significant reduction in the expression and function of PTEN contributes to the pathogenesis of asthma.
Some murine models of asthma induced by ovalbumin have been established to investigate the role of PTEN in the pathogenesis of asthma. Inflammation is the key pathogenesis of asthma. Lee et al. reported that the expression and enzymatic activity of PTEN were significantly reduced, which resulted in an increase in proinflammatory cytokines such as IL-4, IL-5, and eosinophil cationic protein in bronchoalveolar lavage fluids. Restoration of the PTEN expression could significantly reduce bronchial inflammation and airway hyperresponsiveness [28]. They further reported that PPAR-γ activation using rosiglitazone or pioglitazone agonists or adenovirus carrying PPAR-γ cDNA could upregulate the expression levels of PTEN, which inactivated PI3K/Akt pathway to inhibit bronchial inflammation and airway hyperresponsiveness in OVA-induced asthmatic mice [29]. Further, they revealed that restoration of PTEN expression to reduce the symptoms of asthma might be implicated in downregulation of VEGF [30]. In line with the abovementioned findings, Ni et al. reported that glucocorticoid dexamethasone treatment could transcriptionally upregulate the PTEN expression through inhibition of histone acetylation in asthmatic mice [31], suggesting that targeting PPAR-γ and histone deacetylase to restore the expression and function of PTEN is a promising strategy in the treatment of asthma.
Airway wall remodeling is another major pathological feature of asthma, and the increased proliferation and migration of airway smooth cells (ASMCs) play critical roles in these processes [32]. Since PTEN plays an important role in various cell processes, including cell growth, survival, proliferation, and migration, the repressed PTEN expression and activity promote the aggressive proliferation of ASMC that induces the airway remodeling. Numerous findings demonstrated that PTEN silencing promotes ASMC proliferation and induces airway remodeling. A study using a mouse model of OVA-induced asthma showed that loss of PTEN expression accelerates ASMC proliferation and forms a thick smooth muscle layer [33]. Lan et al. showed that the PTEN overexpression inhibits ASMC proliferation and migration by downregulating Akt and FAK signaling [34]. Furthermore, they proved that PTEN overexpression downregulated the levels of p-Akt and cyclin D1, and the enhanced expression of p21 suppresses proliferation and cell cycle arrest of ASMCs [35]. Additionally, Wu et al. demonstrated that the decreased expression of PTEN but increased CD38 expression was observed in TNF-α-induced ASMCs, whereas the overexpression of PTEN could remarkably downregulate CD38 expression, Ca2+ levels and phosphorylation of cyclic AMP response-element binding protein (CREB), and the proliferation and migration of ASMCs, suggesting that regulating PTEN/CD38/Ca2+/CREB signaling could restrict airway remodeling and inflammation in asthma [36]. Moreover, they illustrated that PTEN expression levels are inhibited by the increased expression of Notch1 via Hes1 in TNF-α-induced ASMCs, which facilitates ASMC proliferation, migration, and airway remodeling in asthma [37].
Several studies have demonstrated that the expression levels of PTEN are posttranscriptionally regulated by noncoding RNAs in various smooth muscle cells. Alexandrova et al. investigated the expression signature of small noncoding RNAs (sncRNAs) in asthmatic bronchial smooth muscle (BSM) cells. They found that 32 sncRNAs (26 miRNAs, five piRNAs, and one small nucleolar RNA) were aberrantly expressed in asthmatic patients, and nine miRNAs (miR-27b-3p, miR-92a-3p, miR-30a-5p, miR29a-3p, miR-186-5p, miR-103a-3p, miR-3182, miR-148b-3p, and miR-410-3p) were significantly upregulated in BSM cells from asthmatic patients compared to those in cells from healthy individuals. They identified 38 mRNAs as the major targets of these miRNAs through Ingenuity Pathway Analysis (IPA). Among them, PTEN mRNA was primarily targeted by miR-29a-3p and miR-92a-3p, resulting in abnormal activation of PTEN pathway in cells of asthmatic patients [38]. Hou et al. revealed that stimulation of high-mobility group box protein 1 (HMGB1) increases the expression levels of miR-19, which in turn downregulates PTEN expression, resulting in activation of PI3K/Akt pathway to accelerate ASMC proliferation [39]. Similarly, miR-620 targets PTEN and activates PI3K/Akt signaling to promote cell proliferation in TGF-β1-induced ASMCs [40]. Similarly, Lv et al. reported that TGF-β1 elevates miR-181a expression and suppresses PTEN expression by enhancing Akt/mTOR signaling pathway to promote ASMC proliferation, migration, and extracellular matrix (ECM) secretion [41]. The overexpression of miR-21 also represses the PTEN expression, thereby activating PI3K/Akt pathway and triggering the proliferation and migration of ASMCs [42]. Moreover, PTEN repression by miR-21 activates PI3K/Akt pathway, leading to decreased expression levels of nuclear histone deacetylase (HDAC) 2, thus enhancing inflammation in a severe steroid-insensitive asthma model [43]. Using a murine model of asthma and LPS-induced P815 mast cells, Zhou et al. suggested that PTEN levels are inhibited by the high expression of miR-221, accompanied by activation of p38 via phosphorylation, and NF-κB signaling, which enhances IL-4 secretion [44]. Additionally, miR-21-5p also targets PTEN, resulting in the activation of signal transducer and activator of transcription 3 (STAT3) and PI3K/Akt/mTOR signaling, which aberrantly regulates airway wall remodeling in nonimmune immunoglobulin E- (IgE-) induced ASMCs [45]. It has been reported that long noncoding RNAs (lncRNAs) are involved in the regulation of the PTEN expression. LncRNA-CASC7 and lncRNA H19 upregulate the expression of PTEN by directly targeting miR-21 to inhibit the PI3K/Akt pathway, whereas downregulation of lncRNA-CASC7 and lncRNA H19 was observed in ASMCs from patients with severe asthma, suggesting that both these lncRNAs could be potential targets in the treatment of asthma [46, 47]. As aforementioned, dsyregulation of PTEN plays a key role in the aggressive proliferation of ASMCs, and restoration of PTEN might be an effective strategy to treat asthma. Several studies showed that bronchial epithelial cell injury is regulated by PTEN in asthma. Cui and Yang found that benzo [a] pyrene (Bap) treatment repressed PTEN and FAK expression and activates PI3K/Akt signaling in patients with asthma, which is believed to contribute to the bronchial epithelial injury caused by Bap-mediated ROS generation and cell apoptosis. Additionally, they showed that annexin A1 (ANXA1) protects against bronchial epithelial injury by increasing PTEN and FAK expression and inactivating PI3K/Akt pathway [48], suggesting that restoring PTEN expression might prevent bronchial epithelial apoptosis in asthma. The role of PTEN in regulation of asthma in detail was listed in Table 1.
Table 1.
Biological functions of PTEN in the development of asthma.
| Study type | Model/sample | Impact on PTEN | Additional signaling | Biological process | Ref. |
|---|---|---|---|---|---|
| In vivo | Female BALB/c mice/OVA-induced | Decreased PTEN expression and activity | Activated PI3K signaling | Increased bronchial inflammation and airway hyperresponsiveness in asthma | [28] |
| In vivo | Female BALB/c mice/OVA-induced | PTEN expression increased by PPAR-γ | Reduced PI3K activity | Inhibited allergen-induced bronchial inflammation | [29] |
| In vivo | Female C57BL/6 mice | Inhibited PTEN expression | Activated HIF-α and VEGF signaling | Increased inflammation and vascular permeability | [30] |
| In vivo/in vitro | Female BALB/c mice/OVA-induced; A549 lung epithelial cell line | PTEN expression increased by dexamethasone treatment | Histone acetylation inhibition | Dexamethasone treatment upregulated PTEN and exhibited anti-inflammatory effect in asthma | [31] |
| In vivo | Female BALB/c mice/OVA-induced | Decreased PTEN expression | Promoted ASMC proliferation and airway tissue remodeling | [33] | |
| In vitro | Human airway smooth muscle cells (ASMCs) | Overexpression of PTEN | Downregulated Akt and FAK signaling activity | Inhibited ASMC proliferation and migration | [34] |
| In vitro | Human ASMCs | Overexpression of PTEN | Downregulated Akt signaling and cyclin D1 expression, upregulated p21 expression | Inhibited ASMC proliferation and induced cell cycle arrest in the G0/G1 phase | [35] |
| In vivo/in vitro | Female BALB/c mice; mice Airway smooth muscle cells (ASMCs)/ TNF-α | Decreased PTEN expression | Increased CD38-mediated Ca2+/CREB signaling | Promoted ASMC proliferation and airway tissue remodeling | [36] |
| In vitro | Mice airway smooth muscle cells (ASMCs)/TNF-α | Inhibited PTEN expression | Increased Notch1 expression | Facilitated ASMC proliferation and migration | [37] |
| In vivo/in vitro | Lung tissue specimens from asthma patients; bronchial smooth muscle (BSM) cells | Deregulated PTEN signaling | Increased miR-29a-3p and miR-92a-3p expression | Regulated cellular process in asthma | [38] |
| In vitro | Human ASMCs/HMGB1 | Decreased PTEN expression | Activated the PI3K/Akt pathway and upregulated miR-19 | Promoted ASMC proliferation and migration | [39] |
| In vitro | Human ASMCs/ TGF-β1 | Decreased PTEN expression | Activated the PI3K/Akt pathway and upregulated miR-19 | Induced ASMC proliferation and inhibited apoptosis | [40] |
| In vitro | Mice airway smooth muscle cells (ASMCs)/TGF-β1 | Decreased PTEN expression | Upregulated miR-181a and activated the Akt/mTOR pathway | Promoted airway smooth muscle cell proliferation and airway remodeling | [41] |
| In vitro | Human ASMCs/miR-21 lentiviral vector | Decreased PTEN expression | Activated the PI3K/Akt pathway and upregulated miR-21 | Promoted ASMC proliferation and migration | [42] |
| In vivo | Murine model of established allergic airway disease (AAD) | Inhibited PTEN expression | High levels of miR-21 enhanced the PI3K/Akt pathway and suppressed nuclear histone deacetylase (HDAC2)2 levels | Induced airway hyperresponsiveness in severe, steroid-insensitive asthma | [43] |
| In vivo/in vitro | Female BALB/c mice; P815 cells | Suppressed PTEN expression | Increased miR-221 activated p38 and NF-κB signaling | Stimulated IL-4 secretion in mast cells | [44] |
| In vivo/in vitro | Human bronchial biopsies from asthma patients; human ASMCs | Downregulated PTEN expression | Activated STAT3 and miR-21-5p | Induced ASMC remodeling | [45] |
| In vitro | Human ASMCs | Suppressed PTEN expression | LncRNA-CASC7 levels were suppressed, and miR-21 levels were increased; the PI3K/Akt pathway was activated | Enhanced corticosteroid sensitivity in severe asthma | [46] |
| In vivo/in vitro | Serum samples from asthma patients; human ASMCs | Suppressed PTEN expression | LncRNA-H19 levels were suppressed, and miR-21 levels were increased; the PI3K/Akt pathway activated | Promoted ASMC proliferation and migration | [47] |
| In vitro | Human bronchial epithelial cell line (BEAS-2B) | PTEN expression was repressed by Bap treatment | Repressed FAK expression and activated the PI3K/Akt pathway | Induced bronchial epithelial cell apoptosis and cell injury | [48] |
2.2. Acute Lung Injury
Acute lung injury (ALI) is a severe clinical human disorder with high morbidity, caused by various conditions such as pneumonia, systemic inflammation, sepsis, major surgery, mechanical ventilation, or hyperoxia. The most important cell types that contribute to the pathogenesis of ALI are epithelial cells, macrophages, and neutrophils. Loss of PTEN has been observed in alveolar epithelial cells, macrophages, and neutrophils of ALI. Loss of PTEN function resulted in Akt/NF-κB signaling pathway activation triggering lung inflammation-mediated injury. In addition, activation of PI3K/Akt signaling pathway plays an important role in the pathogensis of ALI.
Murine models of ALI induced by various stimuli are commonly used to investigate the role of PTEN in the pathogenesis of ALI. Fu et al. established a lipopolysaccharide- (LPS-) induced ALI mouse model to examine the effect of miRNA expression on PTEN transcription in lung tissues using miRNA microarray analysis. They found that the miR-92a expression was significantly increased in LPS-induced lung tissues; miR-92a suppressed the transcription of PTEN by binding to its 3′-UTR, and low levels of PTEN activated Akt/NF-κB signaling pathway, resulting in lung inflammatory reaction in mice [49]. Another miRNA profile analysis by Lee et al. suggested that miR-21, which is upregulated in oleic acid- (OA-) induced ALI rats, directly repressed PTEN resulting in Akt pathway activation to induce ALI [50] suggesting that silencing miRNA to elevate PTEN expression might prevent ALI induced by various stimuli.
The function of epithelial cells is critical in wound healing in ALI. It was reported that reduction of PTEN levels disturbed the integrity of alveolar epithelial cells (AECs) due to the disassembly of tight junctions of AECs and destruction of the basement membrane via activation of PI3K/Akt signaling in ALI [51]. Conversely, other studies have suggested that PTEN repression has a protective effect on ALI via diverse mechanisms in AECs and lung epithelial cells. In addition, type II alveolar epithelial cell (AECII) apoptosis is an underlying mechanism involved in the pathogenesis of hyperoxia-induced acute lung injury (HALI). Qin et al. reported that elevated miR-21-5p levels in AEC II from lungs of HALI suppress PTEN expression, which in turn activates PI3K/Akt signaling, leading to decreased apoptosis of AEC II, thus ameliorating HALI [52]. Additionally, Wu et al. reported that miR-425 in the bone marrow mesenchymal stem cell- (BMSC-) derived exosomes (BMSC-Exos) inhibits PTEN levels, leading to activation of PI3K/Akt signaling, which attenuates HALI by promoting lung epithelial cell survival [53]. Tiozzo et al. highlighted that PTEN depletion extends tracheal epithelial progenitor cells and inhibits the differentiation of specialized epithelial cell types to confer resistance to lung injury [54]. Consistently, Knoell and colleagues found that PTEN inhibition activates PI3K/Akt pathway to improve wound closure and restore the integrity of lung epithelial monolayer for wound repair [55]. They further confirmed that PTEN repression could prevent ALI by improving epithelial cell tolerance in response to stress by activating PI3K/Akt pathway in a mouse model of OA-induced ALI [56]. They further demonstrated that loss of PTEN led to the activation of ERK and Akt signaling to enhance lung epithelial cell migration, thereby increasing wound healing following lung injury [57].
Inflammation is among the causative factors of lung tissue injury macrophage, alveolar macrophage and neutrophils play demonstrative role during the progression of ALI pathogenesis. A study in a mouse model of ALI by Zhou et al. suggested that HMGB1 induces PTEN expression, which in turn activates Foxo1 and TLR4 expression in alveolar macrophages, thereby triggering inflammation and causing ALI [58]. Furthermore, p38, along with its downstream target protein kinase D1 (PKD1), was reported to conversely regulate PTEN activity in neutrophils, thereby controlling migration of neutrophils in ALI [59].
Accumulation of alveolar edema fluid has been recognized as a common pathology of ALI, and alveolar fluid clearance (AFC) to remove edema fluid from alveolar spaces is critical for ALI recovery [60, 61]. Previous studies have highlighted that PTEN is closely associated with AFC, as it regulates the primary determinant, epithelial sodium channel (ENaC). In LPS-induced inflammatory lung injury, upregulation of miR-21 prevents PTEN expression, thereby resulting in PI3K/Akt activation and suppression of ENaC gamma (ENaC-γ) expression, which is the primary rate-limiting step in AFC. However, lipoxin A4 (LXA4) treatment enhances the ENaCγ expression and promotes AFC via miR-21/PTEN/Akt pathway to protect against LPS-induced ALI [62]. In contrast, Zhang et al. suggested that PTEN repression by miR-130b in BMSCs can promote the expression of ENaC alpha and gamma, which regulates severe pulmonary edema in ALI [63].
Sepsis is the leading cause of lung injury worldwide and occurs mainly due to the dysregulated host response to bacterial infection. In an experimental model of sepsis-induced lung injury, the results revealed that miRNA-23a improved sepsis-induced lung injury by repressing PTEN, leading to PI3K/Akt pathway activation and inhibition of p53 expression [64]. It has been reported that augmentation of PTEN in the leukocytes of both septic patients and mice enhances microbial clearance and inhibits lung damage and cytokine production. Nuclear localization of PTEN and its lipid phosphatase activity contribute to the increased production and maturation of miR-125b and miR-203b by directly regulating the translocation of miRNA-processing enzymes Drosha and DGCR8 in the nucleus, which limits the abundance of MyD88, resulting in the suppression of sepsis-induced lung injury [65]. Zhou et al. found that PTEN is activated by HMGB1, which is induced in endotoxin-stimulated macrophages during sepsis. Activated PTEN inhibits PI3K/PDK1/Akt signaling, which further suppresses β-catenin activity to modulate regulatory T cells, suggesting that HMGB1/PTEN/β-catenin signaling in the induction of regulatory T cells represents a novel therapeutic strategy in the treatment of sepsis-induced lung injury [66]. Intercellular interactions are involved in the regulation of lung injury progression, and miRNA-loaded exosomes play a key role in the transmission of signals between cells. AEC-derived exosomes induce pulmonary inflammation by activating alveolar macrophages via miR-92a-3p. The suppressed NF-κB signaling is activated by miR-92a-3p in macrophages via downregulation of PTEN [67].
Acute respiratory distress syndrome (ARDS) is a severe form of ALI characterized by uncontrolled lung inflammation and lung epithelial and endothelial cell injury with enhanced pulmonary vascular permeability. Wang et al. suggested that PTEN is upregulated in the plasma and peripheral blood mononuclear cells of patients with ARDS, which is probably due to the increased expression of metastasis-associated lung adenocarcinoma transcript 1 (MALAT1), a multifunctional long noncoding RNA that interacts with miR-425 to induce cell apoptosis [68]. Pulmonary sarcoidosis is a systemic inflammatory disease, characterized by parenchymal granulomas with a silent, long-term evolution, and has progressively become the most common cause of sarcoidosis-associated death [69]. Several studies have investigated the role of PTEN in the development and progression of pulmonary sarcoidosis. The protein and mRNA expression levels of PTEN in bronchoalveolar lavage (BAL) cells of sarcoid patients were reported similar to those in the BAL cells of normal patients [70–72]. However, activation of the PI3K/Akt pathway was observed [70]. We presume that PI3K/Akt pathway activation probably occurs due to the reduced enzymatic activity of PTEN after posttranslational modification because PTEN is key suppressor of PI3K/Akt pathway, and the exact mechanism needs to be studied further. The detailed experimental information of PTEN involved in the development of lung injury is summarized in Table 2.
Table 2.
Biological functions of PTEN in the development of lung injury.
| Study type | Model/sample | Impact on PTEN | Additional signaling | Biological process | Ref. |
|---|---|---|---|---|---|
| In vivo/in vitro | Specific pathogen-free (SPF) male BALB/c mice; murine macrophage RAW264.7 cells/LPS | Inhibited PTEN expression | Enhanced the PI3K/Akt and NF-κB pathways and increased miR-92a expression | Induced inflammation in LPS-induced ALI | [49] |
| In vivo | Male Sprague-Dawley rats/OA-induced | Decreased PTEN expression | Activated the PI3K/Akt pathway and increased miR-21 expression | Stimulated airway smooth muscle cell proliferation | [50] |
| In vivo/in vitro | Specific PTEN-deficient mice; lung epithelial cell line (BEAS-2B)/TGF-β | Reduction of PTEN expression | Akt hyperactivation | Controlled alveolar epithelial cell (AEC) integrity | [51] |
| In vivo/in vitro | Sprague-Dawley rats; type II alveolar epithelial cells (AEC II)/ hyperoxia induced | Decreased PTEN expression | Activated the PI3K/Akt pathway and increased miR-21-5p expression | Reduced AEC II apoptosis in hypoxia-induced ALI | [52] |
| In vivo/in vitro | SD rats; bone marrow mesenchymal stem cells/ hyperoxia-induced | Inhibited PTEN expression | Upregulated the PI3K/Akt pathway | Decreased TUNEL-positive cell number, induced cell viability, and reduced apoptosis | [53] |
| In vivo/in vitro | PTENNkx2.1cre mice; PTENNxk2.1-cre epithelial cells | PTEN depletion | Increased p-Akt and β-catenin activation | Expansion of lung epithelial progenitor cells leading to resistance to ALI | [54] |
| In vitro | Primary human upper airway epithelial cells (hUAECs), BEAS-2B, DU145, LNCaP, and PC3 cells | PTEN inhibition | Activated the PI3K/Akt pathway | Restored epithelial monolayer integrity for wound healing | [55] |
| In vivo | Adult C57BL/6 mice/OA-induced | PTEN inhibition | Activated the PI3K/Akt pathway | Enhanced epithelial cell tolerance to stress to mitigate ALI | [56] |
| In vitro | hUAECs and BEAS-2B cells/ mechanical scrap | PTEN inhibition | Activation of Akt and ERK signaling | Enhanced epithelial cell migration to improve wound healing | [57] |
| In vivo/in vitro | Male C57BL/6 mice; alveolar macrophage/ mechanical incision | Increased PTEN expression | Increased Foxo1 expression and NF-κB activation | Activated TLR4-driven inflammatory response in ALI | [58] |
| In vivo/in vitro | p38 knockout mice; HL-60, RAW264.7 cells | Aberrant p38δ–PKD1 signaling | Induced neutrophil migration to inflammatory sites to cause inflammation in lung | [59] | |
| In vivo/in vitro | Specific pathogen-free adult male SD rats; A549 cells/LPS | Inhibition of PTEN expression | Upregulated miR-21, activated PI3K/Akt signaling, reduced the expression of ENaC-γ | Regulated alveolar fluid clearance in ALI | [62] |
| In vivo/in vitro | C57 mice; alveolar type 2 epithelial (AT2) cells | Repressed PTEN expression | Activated PI3K/Akt signaling to increase α/γ-ENaC protein | Regulated alveolar fluid clearance in ALI | [63] |
| In vivo/in vitro | Wistar male rats; BEAS-2B cells/LPS | Repressed PTEN expression | Overexpressed miR-23a, activated PI3K/Akt signaling, and repressed p53 expression | Reduced lung apoptosis and attenuated lung injury | [64] |
| In vivo/in vitro | Female and male C57BL/6 mice; macrophages/cecal ligation and puncture (CLP) | Augmentation of PTEN | miR-125b and miR-203b induction and reduced MyD88 expression | Increased microbial clearance and prevented lung damage | [65] |
| In vivo/in vitro | Floxed β-catenin (β-cateninflox) mice; macrophages/LPS | Activated PTEN | Reduced PI3K/Akt and β-catenin activity, blocked macrophage TGF-β release, and decreased Foxp3+ Treg induction | Enhanced inflammatory response in sepsis-induced ALI | [66] |
| In vivo/in vitro | SD rats/ cecal ligation and puncture NR8383; RLE-6TN cells /LPS | Downregulation of PTEN via posttranscriptional modification | miR-92a-3p overexpression and NF-κB signaling activation | Proinflammatory cytokine release | [67] |
| In vivo/in vitro | ARDS patients; human lung fibroblasts HFL-1 and A549 cells | Protected PTEN expression | Increased MALAT1 expression and decreased miR-425 expression | Promoted cell apoptosis in ARDS | [68] |
2.3. Chronic Obstructive Pulmonary Disease (COPD)
COPD is characterized by the progressive airway limitation, resulting in an abnormal inflammatory response in lung to noxious particles or gases. Cigarette smoke (CS) is considered as the primary cause of COPD, and CS exposure promotes oxidative stress that affects the expression level or function of PTEN by transcriptional and translational modification. In addition, several studies showed that genetic variation in PTEN was closely associated with COPD in substantial coal smoke exposure. Therefore, alterations in PTEN expression and function are closely associated with the development of COPD [73, 74].
In the cigarette smoke-exposed mouse model, a different alteration of PTEN was found. It has been reported that CS reduced PTEN level in macrophage cells, and the low level of PTEN leads to macrophage M2 polarization by PI3K/Akt activation in emphysematous mice [75], while Li et al. found that CSE exposure enhanced the stability of PTEN and promotes the expression of PI3K regulatory subunit PI3Kp85, which suppresses the phosphorylation of Akt through reducing the expression of protein arginine methyltransferase 6 (PRMT6) leading to CSE-induced epithelial cell apoptosis [76]. The difference of PTEN alteration suggested that PTEN regulated the progression of COPD through multiple mechanisms.
Numerous studies showed that reduced expression or function of PTEN was also found in COPD patients. Besiktepe et al. observed low expression levels of PTEN and HIF-1α in the lung samples of pulmonary emphysema (PE). The decreased PTEN expression may cause protease activation, decrease HIF-1α and lysyl oxidase (LOX), LOX-like protein 1 (LOXL1), LOXL2, and copper metabolism domain containing-1 (COMMD1), thereby contributing to the development of PE [77]. It has been reported that CS-induced oxidative stress inhibited PTEN expression [78], leading to increased PI3K/Akt signaling activity that promoted the production of proinflammatory mediators such as TGF-β, IL-6, CXCL8, CCL2, and CCL5, thereby COPD occurred [79]. Loss of PTEN function might be involved in COPD patients. We found that CSE exposure increased ROS generation that induced PTEN oxidation and impaired Trx-1 activity via dimerization, and the Trx-1 impairment delayed the reduction of PTEN and promoted PI3K/Akt signaling activation in BEAS-2B cells [80]. Additionally, Barnes et al. and Baker et al. illustrated that expression level of PTEN was downregulated by oxidative stress-induced miRNA-34 augmentation in BEAS-2B cells as well as peripheral lung samples from patients. PTEN reduction activated PI3K signaling to accelerate cellular senescence in COPD [81, 82].
Inflammation is recognized as an important driving force in the initiation, regulation, and development of COPD [83], and the repressed PTEN expression or function has been proved to induce inflammation in COPD. Numerous studies have shown that dysregulation of lncRNAs and miRNAs downregulated PTEN expression during the development of COPD. Shen et al. demonstrated that the low expression of lncRNA SHNG5 in COPD tissue was associated with increased inflammation and apoptosis. Downregulation of lncRNA SHNG5 increased the expression of miR-132, resulting in decreased expression of PTEN, which was probably due to miR-132, functions as a competing endogenous RNA for miRNAs [84]. Furthermore, Bozinovski and Anderson concluded that the acquired somatic mutations of PTEN in the epithelium of smokers are the major determinants of COPD. PTEN mutation intensified the inflammatory response by activating NF-κB and AP-1 pathways in COPD [85, 86]. In addition, PTEN gene expression in COPD promoted MMP-9 expression in tumor-associated neutrophils by enhancing the STAT3-AP-1 interaction in bronchial epithelial cells, and loss of epithelial PTEN led to corticosteroid resistance [87].
Maintenance of lung epithelial integrity plays an important role in airway health, and epithelial barrier dysfunction contributes to the pathogenesis of COPD [88, 89]. The apical junctional complex (AJC) formed by tight and adherens junctions is crucial for epithelial barrier function [90]. Transcriptome analysis revealed that the expression levels of physiological AJC genes were globally reduced in the airway epithelia of smokers compared to those of nonsmokers. This was accompanied by a significant decrease in the expression of PTEN and its transcription factors, which was further downregulated in smokers with COPD, indicating that PTEN plays an important role in maintaining the integrity of epithelial junction barrier [78]. Endothelial barrier injury and inflammation are considered to be critical pathophysiological processes in CS-stimulated COPD. Inactivation of PTEN by phosphorylation plays an important role in endothelial dysfunction upon CSE exposure, particularly in the presence of inflammatory cytokines, through β-catenin-dependent gene regulation [91]. Among the various factors implicated in the development of COPD, proteases are specifically relevant to the pathophysiology of this disease. One such protease known to be upregulated in COPD is MMP-9, which regulates disease progression through augmentation of inflammation, extracellular matrix degradation, and neutrophil chemotaxis. A previous study suggested that loss of PTEN in COPD increases MMP-9 expression by enhancing STAT3-AP-1 interaction in bronchial epithelial cells [87]. PTEN plays an important role in preventing COPD, and alterations of PTEN are implicated in the development of COPD. Restoring the PTEN expression and activity is a promising therapeutic approach in COPD treatment. The effect of PTEN in the development and progression of COPD was summarized in Table 3.
Table 3.
Biological functions of PTEN in the development of COPD.
| Study type | Model/sample | Impact on PTEN | Additional signaling | Biological process | Ref. |
|---|---|---|---|---|---|
| In vivo/in vitro | Emphysema mouse model; bone marrow–derived macrophages (BMDMs); CSE-treated RAW264.7 and L929 cell lines | Decreased PTEN expression | Activated the PI3K/Akt pathway | Macrophage polarization toward the M2 phenotype in COPD | [75] |
| In vivo/in vitro | CS-exposed mouse model; BEAS-2B cells | Increased PTEN stability | Inhibited the PI3K/Akt pathway, decreased PRMT6 expression, promoted PI3Kp85 expression, and inhibited PDK1 | Resulted in epithelial cell death in COPD | [76] |
| In vivo | Human lung tissue of COPD | Decreased PTEN expression | Activated HIF-1α signaling and MMP7/9 | Decreased levels of oxidases (LOX, LOXL1, and LOXL2) caused abnormalities in elastic fiber biology | [77–79] |
| In vitro | CSE-exposed BEAS-2B cells | PTEN oxidation | Increased p-Akt level | Impaired Trx-1 activity | [80] |
| In vivo/in vitro | Lung tissues of patients with COPD; cells collected from patients undergoing lung resection surgery; BEAS-2B cells | Loss of PTEN expression | PI3K/mTOR signaling activation and SIRT1/6 inhibition | Caused cell senescence in COPD | [81, 82] |
| In vivo/in vitro | Human peripheral lung tissue; normal human bronchial epithelial cell line (16HEB) | Downregulation of PTEN | Decreased lncRNA SHNG5 sponge miR-132 expression | Regulated effects of CSE on cell proliferation, apoptosis, and inflammation | [84] |
| In vivo | Patients with COPD | Decreased PTEN expression | Enhanced the STAT3-AP-1 interaction | Increased MMP-9 expression to regulate airway remodeling | [87] |
| In vitro | Mouse cardiac endothelial cells (MCECs) | Low expression of PTEN | ROS/Src/EGFR-p38MAPK pathway | PTEN pathway related with AJC transcriptional reprograming to regulate epithelial barrier | [78, 90] |
| In vitro | Human nasal epithelial cell line (RPMI 2650) | Reduced PTEN activity | Increased TLR4/JNK/Bnip3 signaling | Activated mitophagy and induced mitochondrial dysfunction to cause epithelial cell apoptosis, proliferation arrest, and migration inhibition | [91] |
2.4. Pulmonary Fibrosis
Pulmonary fibrosis is a progressive interstitial lung disease. Particularly, idiopathic pulmonary fibrosis (IPF) is characterized by excessive deposition of extracellular matrix molecules, such as collagen and elastin, with overactivation of fibroblasts/myofibroblasts. The pathogenesis of IPF remains largely unknown. Kulkarni et al. comprehensively investigated the target proteins and signaling pathways implicated in the pathogenesis of IPF in bleomycin-induced mice using a label-free LC-MS-based proteomics approach with systembiology. The results showed that mammalian target of rapamycin (mTOR) and extracellular signal-regulated kinase (ERK) are the primary regulators of pro- and antifibrotic responses. PI3K/Akt and Wnt signaling are key profibrotic pathways, whereas PTEN and natural killer cell signaling pathways are the most important antifibrotic pathways [92]. Numerous studies showed that the dysfunction and downregulation of PTEN were observed in IPF indicating that PTEN was involved in regulating the progression of lung fibrosis. The detailed information about the involvement of PTEN in lung fibrosis was listed in Table 4.
Table 4.
Biological functions of PTEN in the development of pulmonary fibrosis.
| Study type | Model/sample | Impact on PTEN | Additional signaling | Biological process | Ref. |
|---|---|---|---|---|---|
| In vivo | Myeloid PTEN-deficient mice/bleomycin | Loss of PTEN expression | Sustained activation of PI3K pathway | Increased TGF-β1 activation, collagen deposition; reduced number of macrophages and T-cells | [93] |
| In vivo/in vitro | Human IPF lung tissue; IPF lung tissue; C57BL/6 and A549 cells/bleomycin | Loss of PTEN | P21WAF1, P16ink4, and SA-β-gal overexpression; NF-κB and Akt activation | Alveolar epithelial cell senescence promotes lung fibrosis | [94, 95] |
| In vivo/in vitro | Human lung tissue; C57BL/6 embryonic mouse fibroblasts and 3T3 murine fibroblasts/TGF-β1; C57BL/6 mice/bleomycin | Diminished PTEN expression and phosphatase activity | Inhibition of PTEN activity in IPF-derived fibroblasts | α-SMA expression, cell proliferation, collagen production, and myofibroblast differentiation | [97] |
| In vivo/in vitro | Primary fibroblast cell lines from IPF and healthy lung/type I collagen–rich matrix; PTEN haploinsufficient and wild-type mice/bleomycin | High phosphatase activity in normal lung fibroblasts, but low activity in IPF-derived fibroblasts | Aberrant activation of the PI3K–Akt–S6K1 signaling pathway in IPF-derived fibroblasts | Enhanced the proliferation of primary lung fibroblasts | [99] |
| In vitro | Fibroblasts and myofibroblasts from patients with IPF; MRC-5 cells/H2O2 | Loss of PTEN expression | Activated the TGF-β1 pathway and increased hyaluronan synthase 2 expression | Increased proliferation, apoptosis resistance, and migration/invasion activities | [100] |
| In vivo/in vitro | Human IPF lung tissue; MRC-5 cells/TGF-β1 | PTEN ubiquitination and degradation | Downregulation of ubiquitin-specific peptidase 13 (USP13) | Enhanced proliferative, migratory, and invasive capacities of lung fibroblasts | [101] |
| In vivo/in vitro | Human IPF lung tissue; HFL-I cells/TGF-β1 | Low expression of PTEN | Enhanced PI3K/Akt and TGF-β/Smad3 signaling | PTEN inhibited the proliferation and myofibroblast differentiation and promoted the apoptosis of fibroblasts | [102] |
| In vitro | Human lung fibroblasts CCL-210/mechanical stretch | Increased PTEN activity | Decreased Akt phosphorylation | Promoted fibroblast apoptosis | [106] |
| In vitro | Primary IPF-derived and normal fibroblasts/polymerized type I collagen | Low phosphatase activity | High Akt activity promoted the inactivation of FoxO3a and downregulation of p27 in IPF-derived fibroblasts | Facilitated fibroblast proliferation | [107] |
| In vitro | Primary control and IPF-derived lung fibroblasts/polymerized type I collagen | Low phosphatase activity | Inactivation of FoxO3a, which downregulated caveolin-1 and Fas expression | Apoptosis-resistant phenotype of IPF-derived fibroblasts | [108] |
| In vitro | Primary IPF-derived lung fibroblasts/polymerized type I collagen | Decreased phosphatase activity | Enhanced p-mTOR expression along with low expression of LC3-2 and FoxO3a | Suppressed autophagic activity | [109, 110] |
| In vivo/in vitro | Primary human alveolar epithelial type II (AEII) cells; small-airway epithelial cells/mechanical stretch | Downregulation of PTEN | miR-19a overexpression | Development of the EMT phenotype and lung fibrosis | [111] |
| In vitro | Murine embryonic fibroblasts/LPS | Low PTEN expression | Upregulation of TLR4 and PI3K/Akt pathway activation | Increased fibroblast proliferation | [112] |
| In vitro | Primary IPF-derived lung fibroblasts; normal human fetal lung fibroblasts (IMR-90) | Low PTEN expression and phosphatase activity | Loss of α4β1 signaling | Migratory/invasive phenotype of fibroblasts | [113] |
| In vitro | IMR-90 cells; murine embryonic fibroblasts/prostaglandin E2 | Increased PTEN phosphatase activity by decreasing the phosphorylation of PTEN | E-prostanoid (EP) 2 receptor | Inhibited fibroblast migration | [115] |
| In vivo/in vitro | Human embryo lung fibroblasts/silica | Loss of PTEN expression due to hypermethylation of its promoter | MAPK and c-Jun methylation | [116] | |
| In vitro | Deletion of PTEN or both PTEN and CCN2 in mouse fibroblasts | Loss of PTEN expression | Overproduction of collagen type I and connective tissue growth factor (CCN2) | Collagen deposition | [117] |
| In vitro | Epithelial H358 cells; normal human adult lung fibroblasts CC2512 and primary mouse lung fibroblasts /unphosphorylated PTEN/TGF-β1 | Loss of PTEN enzymatic activity via phosphorylation of its C-terminus; retention of enzymatic activity in PTEN4A-treated cells | Suppression of β-catenin translocation by PTEN4A treatment | PTEN4A inhibits ECM production | [118] |
In the bleomycin-induced mice model of pulmonary fibrosis, myeloid PTEN-deficient mice exhibited the enhanced TGF-β1 activation and collagen deposition, decreased number of macrophages and T cells, and aberrant macrophage polarization with augmentation of various proinflammatory cytokines such as IL-6 and TNF-α. PTEN deficiency leadsto sustained PI3K activation in myeloid cells, thereby exacerbating IPF progression, suggesting that PTEN inhibits lung fibrosis via immunological mechanism [93]. The impairment of alveolar epithelial cells (AECs) contributes to IPF, and PTEN/PI3K/Akt pathway plays an important role in the maintenance and reconstitution of AEC integrity [94]. Miyoshi et al. reported that both patients with IPF and BLM-induced mice exhibited loss of AEC integrity and destruction of the basement membrane, which was accompanied by the decreased PTEN expression, indicating that PTEN plays a crucial role in the regulation of AEC integrity. PTEN deficiency intensified the disassembly of tight junctions of AECs and increased the abundance of epithelial-derived myofibroblasts and subsequent lung fibrosis [51]. In addition, Qui et al. reported that loss of PTEN promotes the senescence of AECs to induce lung fibrosis. Akt activation is considered to be the major mechanism underlying the reduced expression of PTEN [95]. Furthermore, they found that NF-κB activation was involved in AEC senescence resulted from the reduced PTEN expression, and AEC senescence promoted collagen deposition of fibroblasts through the senescence-associated secretory phenotype [96]. Since cellular senescence is considered as an initial step contributing to lung fibrosis [81]. These findings suggest that therapeutic intervention of PTEN/Akt or PTEN/NF-κB pathways might prevent the occurrence of IPF by suppressing the senescence process of AECs.
Aberrant lung fibroblast/myofibroblast activation and proliferation play important roles in fibrogenesis [97–99]. Decreased PTEN expression and phosphatase activity are frequently observed in fibroblasts or myofibroblasts from lung tissue of IPF patients [100, 101]. Geng et al. reported that the expression of USP13, a deubiquitylase that prevents PTEN ubiquitylation and degradation, is significantly reduced in lung tissues from IPF patients and in primary lung fibroblasts. The decreased expression of USP13 promoted PTEN ubiquitylation, leading to PTEN degradation, thereby contributing to fibroblast activation and pathogenesis of IPF [101]. Xie et al. found that PTEN expression is significantly reduced in Chinese patients with IPF; furthermore, a mechanistic study using TGF-β1-induced human embryonic lung fibroblasts (HFL-1) indicates that loss of PTEN resulted in activation of PI3K/Akt and TFG-β/Smad3 pathways [102]. In addition, PTEN is downregulated in fibroblasts insulted by various initiating factors such as bleomycin, paraquat, radiotherapy, smoking, and PM2.5, thereby causing lung fibrosis [103–105]. Additionally, radiotherapy induces miR-21-mediated PTEN repression, thus promoting EMT and development of lung fibrosis [105].
In an in vitro model of wound repair generated by human lung fibroblasts incorporated into type I collagen matrices, the expression levels of PTEN remain unchanged, whereas its lipid phosphatase activity is amplified in response to collagen matrix contraction. This led to impaired PI3K activity, resulting in reduced phosphorylation of Akt, thus sensitizing fibroblasts to collagen contraction-induced apoptosis [106]. Since IPF-derived fibroblasts were quite different from their normal counterparts, authors further investigated the differences between normal and IPF-derived fibroblasts cultured on polymerized type I collagen. The results showed that β1 integrin interacted with polymerized collagen and suppressed normal fibroblast proliferation through PI3K/Akt/S6K1 pathway owing to the enhanced lipid phosphatase activity of PTEN, whereas IPF-derived fibroblasts had low lipid phosphatase activity that resulted in high activity of PI3K/Akt pathway [99]. The extended study further showed that the expression levels as well as the lipid phosphatase activity of PTEN are necessary to induce myofibroblast differentiation, proliferation, α-smooth muscle actin (α-SMA) expression, and collagen production [97]. Moreover, the decreased phosphatase activity of PTEN was implicated in the low distribution of PTEN on cell membrane of IPF-derived fibroblasts [99]. Further study showed that low membrane-associated PTEN distribution probably resulted from the decreased expression of caveolin-1 that interacted with caveolin-1-binding sequence in PTEN and thus regulated the membrane levels of PTEN [98] suggesting that caveolin-1 is the key therapeutic target to inhibit IPF by regulating PTEN activity. Additionally, the low phosphatase activity of PTEN results in inhibition of downstream transcription factor forkhead box O3a (FoxO3a) via phosphorylation at Ser253, which reduces the expression levels of CDK inhibitor p27 and promoted cell cycle arrest in IPF-derived fibroblasts cultured on polymerized collagen [107]. Furthermore, they showed that the emerging apoptosis-resistant phenotype of IPF-derived fibroblasts is probably due to the aberrant regulation of PTEN/Akt axis, which inactivates FoxO3a, resulting in the downregulation of caveolin-1 suppressing the Fas expression in IPF-derived fibroblasts cultured on polymerized collagen [108]. The aberrantly altered PTEN/Akt axis may desensitize cell apoptosis induced by the polymerized collagen matrix through the inhibition of autophagy and enhancement of mTOR activity [109]. Another study reveals that FoxO3a expression at mRNA and protein levels is significantly suppressed, which caused low autophagic activity through transcriptional suppression of LC3B in IPF-derived fibroblasts cultured on a collagen-rich matrix, thereby promoting IPF progression [110] indicating that upregulation of FoxO3a and Fas to restore phosphatase activity of PTEN might effectively prevent IPF. Except for lung fibroblast activation, lung epithelial-mesenchymal transition (EMT) is one of the primary sources of myofibroblasts. Several studies showed that mechanical ventilation could promote EMT phenotypes of human primary AECIIs, and the augmentation of miR-19b inhibited the PTEN expression by posttranscriptional modification, which results in Akt activation [111].
Excessive proliferation of lung fibroblasts occurs during the early stages of IPF. It has been reported that lipopolysaccharide (LPS) treatment induces aberrant proliferation of mouse lung fibroblasts through activation of TLR4, which downregulates PTEN expression. The reduced PTEN expression results in PI3K/Akt pathway activation, thereby promoting fibroblast proliferation [112]. In addition to proliferation, excessive migration of fibroblasts also contributes to IPF progression, and loss of PTEN function promotes the migration and invasion of fibroblasts. White et al. reported that IPF-derived fibroblasts have low PTEN expression at mRNA and protein levels and decreased lipid phosphatase activity due to loss of alpha4beta1 expression, suggesting that PTEN plays an important role in suppressing fibroblast invasion and migration [113]. Furthermore, they reported that prostaglandin E2 (PGE2) inhibits fibroblast migration by targeting E-prostanoid (EP) 2 receptor, which leads to an increase in the lipid phosphatase activity of PTEN via dephosphorylation [114]. However, fibrotic fibroblasts are easily resistant to PGE2 owing to diminish the EP2 expression, which is attributed to hypermethylation of PGE receptor 2 gene promoter (PTGER2). Furthermore, PTGER2 methylation is probably regulated by the decreased PTEN expression and Akt signaling activation, which suggests that combination treatment using PGE2 and methylation inhibitors may be a potential therapeutic approach for the treatment of IPF [115]. Epigenetic regulation of PTEN gene expression was also found in silica-mediated lung fibrosis. Zhang et al. reported that promoter hypermethylation of PTEN in lung tissues from patients with silicosis might be implicated in the reduction of the PTEN protein expression [116].
The primary hallmark of pulmonary fibrosis is the formation of fibroblastic foci with excessive extracellular matrix (ECM) deposition, including collagen. Parapuram et al. reported that loss of PTEN expression results in excessive collagen deposition, which is closely related to constitutively increased expression of connective tissue growth factor (CCN2), a key regulator of tissue fibrosis [117]. Motohiro et.al demonstrated that TGF-β1 induced phosphorylation of PTEN at the C-terminus decreased its enzymatic activity and failed to inhibit ECM production in human fibroblasts, epithelial cells, and primary mouse lung fibroblasts. However, there was no effect on TGF-β1-induced α-smooth muscle actin expression in fibroblasts [118]. In the BLM-induced model of pulmonary fibrosis, myeloid PTEN-deficient mice exhibited enhanced TGF-β1 activation and collagen deposition, decreased number of macrophages and T cells, and aberrant macrophage polarization with augmentation of various proinflammatory cytokines such as IL-6 and TNF-α. PTEN deficiency led to sustained PI3K activation in myeloid cells, thereby exacerbating IPF, suggesting that PTEN inhibits lung fibrosis via immunological mechanisms [93]. These studies indicate that PTEN is implicated in PF progression by multiple mechanisms, and alterations in PTEN expression or function contribute to lung fibrogenesis.
2.5. Pulmonary Arterial Hypertension (PAH)
Pulmonary arterial hypertension (PAH), a severe and progressive disease characterized by increased pulmonary vascular resistance and pulmonary arterial pressure, is life-threatening. Shortness of breath, fainting episodes, and chest pain are the primary symptoms of PAH, and aberrant vascular remodeling resulting from the increased proliferation and reduced apoptosis of pulmonary arterial smooth muscle cells (PASMCs) is the primary features of PAH. PTEN has been reported to play a crucial role in the regulation of proliferation, differentiation, migration, and apoptosis of various cells. Recent studies have extensively investigated the involvement of PTEN in various murine models of PAH and hypoxia-induced human PASMCs. Dysregulation of PTEN leads to PI3K/Akt signaling activation, which increases the proliferation and migration of PASMCs associated with PAH. The detailed information about the involvement of PTEN in PAH was listed in Table 5.
Table 5.
Biological function of PTEN in the development of PAH.
| Study type | Model/sample | Impact on PTEN | Additional signaling | Biological process | Ref. |
|---|---|---|---|---|---|
| In vivo/in vitro | SD Rat/monocrotaline; human PASMCs/hypoxia | Low levels of PTEN | High expression of miR-132 | Increased proliferation and migration of PASMCs | [119] |
| In vitro | Human PASMCs/hypoxia | Reduction of PTEN expression | High expression of miR-17-5p; down-regulation of p21 | Aberrant proliferation and migration of PASMCs | [120] |
| In vivo/in vitro | SD rats/ monocrotaline; pulmonary arterial endothelial cells/MCT | Increases of PTEN expression | Downregulation of miR-371b-5p | Increased endothelial apoptosis | [121] |
| In vivo/in vitro | SD rats/monocrotaline, hypoxia; human PASMCs/hypoxia | Low expression of PTEN and reduced p-PTEN levels due to ubiquitination | Akt phosphorylation and inactivation of p53, p21, and p27 | Increased proliferation of PASMCs | [122] |
| In vivo | Male SD rats/ monocrotaline, hypoxia | Low expression of PTEN due to its ubiquitination | Aberrant activation of PI3K/Akt signaling | Enhanced vascular remodeling | [123] |
| In vivo | Human PAH lung; PASMCs male SD rats/monocrotaline | Low levels of PTEN due to decreased p-CREB expression | PGE1 induced pCREB expression | Vascular remodeling and improved hemodynamics | [124] |
| In vivo/in vitro | Mice/hypoxia; human PASMCs/hypoxia | Decreased expression of PTEN | miR-19a overexpression enhanced PI3K/Akt signaling | Increased proliferation and migration of PASMCs | [126] |
| In vitro | Human PASMCs/hypoxia | Decreased expression of PTEN | Downregulation of lncRNA MEG3; over-expression of miR-21 | Increased proliferation and migration of PASMCs | [127] |
| In vivo/in vitro | Human HAP; male SD rats/hypoxia | Decreased phosphatase activity or deletion of PTEN | Increased Akt phosphorylation | Increased pressure, extensive, pulmonary vascular remodeling, and increased macrophage accumulation | [128] |
| In vivo /in vitro | Adult female/male Wistar rats/hypoxia; human PASMCs/hypoxia | Low expression of PTEN | Activation of PI3K/Akt signaling | PASMCs resistant to apoptosis | [129] |
| In vivo/in vitro | Male SD rats/permanent ligation of left anterior descending; human PASMCs/hypoxia | Downregulation and decreased phosphatase activity of PTEN | Phosphorylation of Akt | Enhanced proliferative, vascular remodeling | [130] |
Murine models of PAH were usually induced by monocrotaline, hypoxia condition, or monocrotaline combined with hypoxia. Numerous studies have reported that the low expression of PTEN was found in monocrotaline-induced PAH rat, and the decreased expression of PTEN was modulated by multiple mechanisms. Upregulation of miR-132 might repress PTEN expression to promote PASMC proliferation in PAH [119]. Augmentation of miR-17-5p reduced PTEN expression and cyclin-dependent kinase inhibitor 1 (p21) resulting in increased cell proliferation in hypoxia-induced PASMCs [120]. Oppositely, the increased expression of PTEN was also observed in monocrotaline-induced rats, which was associated with downregulation of miR-371b-5p expression. Increased expression of PTEN inactivated PI3K/Akt pathway, promoting apoptosis and reducing the proliferation of pulmonary arterial endothelial cells (PAECs) [121]. Additionally, low level of PTEN and phosphorylated-PTEN was attributed to ubiquitination-mediated degradation in monocrotaline-induced PAH rat, and decreased PTEN expression led to an increase in phosphorylated Akt, inactivation of cell-cycle regulatory proteins p53,p21, and p27, and accumulation of cyclin-D1 [122]. PTEN ubiquitination in monocrotaline-induced PAH rat was also modulated by elevation of NEDD4 [123]. In addition, the low expression of PTEN in human and monocrotaline-induced rat PAH was regulated by cAMP response element binding protein (CREB), a transcription factor that acts as a modulator of the vascular smooth muscle cell phenotype. Phosphorylation of CREB was inhibited with a concomitant decrease of PTEN, whereas a naturally occurring prostanoid prostaglandin E1 (PGE1) treatment could restore PTEN expression by elevating the phosphorylated levels of CREB indicating that PGE1 recruiting CREB/PTEN to inhibit PI3K/Akt signaling pathway can prevent the progression of PAH [124].
In the murine models of PAH induced by hypoxia, dysregulation of miRNAs contributes to the decrease in PTEN expression and function. Liu's group found that hypoxia significantly elevated the expression of miR-214 and miR-19 that targets PTEN to activate PI3K/Akt signaling pathway, thereby promoting cell proliferation of PASMCs [125, 126]. Low expression of PTEN was also implicated in repression of lncRNA MEG3 in hypoxia-induced PASMCs. MEG3 is a proliferation- and migration-inhibiting lncRNA, and it inhibits miR-21 to downregulate PTEN expression, thereby increasing proliferation and migration of PASMCs [127], suggesting that pharmacological intervention of miRNA or lncRNA to regulate PTEN level is an effective strategy for PAH treatment. Except for PTEN reduction in a variety of experimental PAH models, phosphorylation level of PTEN was increased in PAH lesions compared to normal lungs. In the mice model of specific deletion of PTEN in SMCs, they display a much severe PAH phenotype compare to wild type mice under hypoxia exposure for 4 weeks, indicating that PTEN is a key target for therapeutic intervention [128]. Additionally, the overexpression of TGF-β1 results in PTEN downregulation that activates PI3K/Akt signaling pathway to suppress the apoptosis of PASMCs [129]. In the model of PAH secondary to heart failure, high level of peroxynitrite might be responsible for PTEN downregulation, and peroxynitrite induces a significant decrease in the expression and activity of PTEN, which activated Akt pathway to promote proliferation of PASMCs and vascular remodeling, while reducing ROS production could restore PTEN expression and vascular remodeling in PAH secondary to heart failure, indicating that blunting ROS generation is a potential therapeutic approach [130].
2.6. PTEN as a Therapeutic Target
Given the central role of PTEN in a variety of chronic lung diseases, pharmacological modulation of PTEN expression and function at gene and protein levels is considered as a promising strategy in the treatment of chronic lung diseases [131]. Direct and indirect regulation of PTEN expression and activity with numerous modulators has been reported in the treatment of asthma, COPD, pulmonary fibrosis, and lung injury.
2.6.1. Asthma
As mentioned above, chronic inflammatory respiratory and airway remodeling are the major pathological feature of asthma, and the mainstay therapy of asthma in clinic is anti-inflammatory and bronchodilator therapy. Natural products exhibit promising anti-inflammatory effect in diverse human diseases including asthma. Epigallo-catechin-3-gallate (EGCG) is a common polyphenol found abundantly in green tea and has diverse biological activities. Recently, it was reported that EGCG supplementation has a protective effect in ovalbumin- (OVA-) induced asthmatic mice. In term of mechanism, it inhibits EMT and migration via augmentation of PTEN expression in TGF-β1-induced 16HBE cells, thereby reducing inflammation and airway remodeling [132]. Resveratrol, another natural polyphenolic compound rich in red grapes, attenuates airway inflammation and remodeling in OVA-induced murine model of asthma through augmentation of PTEN mRNA and protein expression levels via SIRT1 activation [133]. A monoterpenoid, borneol, has been reported to delay the OVA-induced asthma progression in mice through downregulation of miR-26a and miR-142-3p that blocks PTEN transcription [134]. In addition, α- and γ-mangostin, major xanthones present in mangosteen fruit (Garcinia mangostana Linn), mitigate airway inflammation in OVA-induced allergic asthmatic mice by restoration of PTEN expression, which inactivates PI3K/Akt pathway and its downstream molecule NF-κB, consequently reducing inflammatory cell recruitment into the airway and elevating the Th2 cytokine expression [135]. Lee's group reported that the Korean red ginseng extract and its component nepetin could attenuate Th2-derived cytokines, eosinophil infiltration, and airway remodeling in the OVA-induced allergic asthma model, and the mechanism is implicated in PPARγ upregulation and PTEN-PI3K/Akt pathway inhibition by reducing the phosphorylation level of PTEN [136]. Although Korean ginseng extract and its component nepetin showed a promising antiasthmatic activity, no evidence for the active compounds and how to regulate PTEN phoshorylation was provided in this study. Pharmacological modulation of PTEN expression to inhibit PI3K/Akt pathway with natural products is an effective strategy in the treatment of asthma; however, whether those compounds directly regulate PTEN remains unknown. Dexamethasone is one of the most potent corticosteroids for asthmatic therapy in clinic, and it exerts the anti-inflammatory effect upon binding to the glucocorticoid receptor. It has been reported that dexamethasone inhibits inflammation partly by restoring PTEN expression and suppressing histone acetylase activity, which is usually decreased in the lung tissues of asthmatic mice [31] suggesting that dexamethasone regulates the PTEN expression level.
2.6.2. Lung Fibrosis
Numerous studies exhibited that naturally occurring products have potential in the treatment of lung fibrosis by regulation of PTEN. Resveratrol inhibits the activation of TGF-β1-induced normal fibroblasts and IPF-derived fibroblasts, and the mechanism is probably related to upregulation of PTEN, which inhibits the phosphorylation of Akt and extracellular signal-regulated kinases (ERK1/2) [137]. Dasatinib, a platelet-derived growth factor receptor (PDGFR) and Src-kinase inhibitor, showed antifibrotic effect in bleomycin- (BLM-) induced mice and upregulation of PTEN expression after dasatinib treatment, which was decreased in BLM-treated mice [103]. It has been reported that paraquat (PQ) induces reduction of PPAR-γ, PTEN, TGF-β1, and α-SMA expression at both protein and mRNA levels, resulting in lung fibrosis in rats. The PPAR-γ agonist rosiglitazone attenuates PQ-induced pulmonary fibrosis by upregulating PTEN and decreasing the expression of TGF-β1 in a PPAR-γ-dependent manner [104]. Although restoring PTEN expression level by various natural products is accompanied by suppression of lung fibrosis, the exact mechanism remains unclear. The endogenous lipid mediator prostaglandin E2 (PGE2) showed increased apoptosis in normal and fibrotic lung fibroblasts against pulmonary fibrosis. This was associated with increased activity of PTEN, which inhibited the Akt signaling pathway. Furthermore, PGE2 reduced survivin expression and increased Fas expression in lung fibroblasts [138]. In addition, it inhibited fibroblast migration by restoring the lipid phosphatase activity of PTEN in an E-prostanoid (EP) 2 receptor-dependent manner [114]. The endogenous compound is nontoxic and further revealing the mechanism is necessary to find a potent lead compound. Berberine, an isoquinoline alkaloid, exhibited a marked protective effect against BLM-induced mouse pulmonary fibrosis in a gut-dependent manner; however, only oral administration was effective but not intravenous injection. The underlying molecular mechanism involves PPAR-γ activation that promotes the expression of HGF in colonic fibroblasts and increases HGF levels, which reaching the lung tissues through blood circulation to attenuate IPF. Moreover, berberine augments PTEN mRNA and protein expression levels in BML-induced mice [139]. Another study showed that, mechanistically, berberine attenuated lung fibrosis by suppressing the phosphorylation of Smad 2/3, augmenting Smad 7, and blocking FAK-dependent PI3K/Akt-mTOR signaling pathway [140]. Berberine is a multitarget drug and shows great potential in the treatment of lung fibrosis.
Silica exposure triggers lung inflammation and pulmonary fibrosis. Alveolar macrophages play a key role in the inflammatory response during silicosis. Coelonin, a dihydrophenanthrene compound isolated from Bletilla striata, inhibits inflammation in lipopolysaccharide- (LPS-) induced alveolar macrophages. The anti-inflammatory reaction of coelonin is probably associated with augmentation of PTEN expression and inhibition of its phosphorylation at Ser380 site, thereby inactivating PI3K/Akt signaling pathway, as well as inhibition of G1 phase cell cycle arrest by preventing p27kip1 degradation and proinflammatory cytokine gene expression through suppression of NF-κB activation [141]. In addition, Kimura et al. found that exogenous administration of mutant PTEN at the C-terminus (S380A, T382A, T383A, and S385A) could restore the enzymatic activation of PTEN insulted by TGF-β1 resulting in inhibition of ECM overproduction in epithelial cells and fibroblasts in lung fibrosis [118].
2.6.3. COPD
Mucus hypersecretion is a major pathology of COPD, cystic fibrosis (CF), and chronic bronchitis. FOXA2 is a key transcriptional regulator that maintains airway mucus homeostasis and is inhibited in airway diseases. Recently, it was reported that exendin-4, an analog of glucagon peptide-1 (GLP-1), could attenuate the production of excessive mucus by restoring the FOXA2 expression in COPD, CF-diseased cells, and mouse lungs infected by P. aeruginosa. It was found that exendin-4 triggered GLP-1R-dependent PKA and PPAR-γ activation, which in turn increased the expression of PTEN and PTP1B phosphatases. Both PTEN and PTP1B inactivated the key kinases STAT6 and EGFR via dephosphorylation, thereby restoring FOXA2 function and mucus homeostasis [142]. Curcumin, a natural polyphenol compound abundantly found in the rhizome of Curcuma longa, could suppress acute lung injury and inflammatory cytokines in rats with acute pulmonary embolism (APE). It probably inhibited the expression of Sp1, resulting in the downregulation of miR-21, which enhanced the posttranscriptional regulation of PTEN, subsequently inhibiting NF-κB signaling pathway-mediated lung inflammation in rats with APE [143]. Cefminox, a dual agonist of the prostacyclin receptor and PPAR-γ identified by virtual screening, could attenuate hypoxia-induced pulmonary hypertension in rats. The mechanism is associated with PTEN upregulation, which suppresses activation of Akt/mTOR signaling pathway. Moreover, it could enhance the production of cyclic adenosine monophosphate (cAMP), suggesting that cefminox has a promising potential in the treatment of pulmonary hypertension, as it targets prostacyclin receptor and PPAR-γ [144]. Ivacaftor, approved by FDA for cystic fibrosis therapy, is a potentiator of cystic fibrosis transmembrane conductance regulator (CFTR). Riquelme and colleagues revealed that ivacaftor could potentiate CFTR activity by promoting the membrane distribution of PTEN and increasing its function via direct interaction with CFTR, thereby suppressing PI3K/Akt signaling to inhibit hyperinflammation in response to P. aeruginosa infection [145]. DNA methyltransferase inhibitor 5-Aza-2′-deoxycytidine (5-Aza-dC) has been reported to attenuate hypoxic pulmonary hypertension through demethylation of PTEN promoter [146]. Although numerous potential therapeutic agents were identified to be effective against COPD by modulating the expression and function of PTEN, there is still no drug reaching to clinical trial.
2.6.4. Others
MicroRNAs (miRNAs) have shown great therapeutic potential in diverse lung diseases. For example, miR-486 protects against PM2.5-induced cytotoxicity in human lung alveolar epithelial A549 cells by inhibiting the expression of PTEN and FOXO1 [147]. It has also been reported that intratracheal administration of exosomes secreted by mesenchymal stromal cells (MSCs) ameliorates lung edema and dysfunction as well as the production of various proinflammatory cytokines in a mouse lung ischemia/reperfusion (I/R) model by transporting miR-21-5p. The in vitro study showed that MSC-secreted exosomes improved the endothelial cell apoptosis by suppressing both intrinsic and extrinsic apoptotic pathways through inhibition of PTEN and PDCD4 via miR-21-5p in hypoxia/reoxygenation condition [148]. In addition, the exosome miR-371b-5p derived from the human alveolar progenitor type II cell (ATIIC) line (A549) alleviates bleomycin-induced mouse lung injury. This event is mediated by negative regulation of PTEN expression, resulting in the phosphorylation of Akt and its downstream signaling molecules, such as GSK3β and FOXOs, to promote ATIIC cell proliferation and survival. This indicates that ATIIC-derived exosome miR-371b-5p could be a promising therapeutic candidate for augmenting ATIIC proliferation/survival and promoting the reepithelialization of injured alveolar cells in various incurable lung diseases [149].
Moreover, resveratrol can also protect against methamphetamine-induced high permeability and apoptosis of alveolar epithelial cells by reducing ROS levels and activating SIRT1, which leads to PTEN upregulation, thereby repressing Akt activation [150]. Resveratrol inhalation can slow down age-related degeneration of lung structure and function by maintaining the integrity of alveolar epithelial type 2 cells. Since resveratrol has been identified as an agonist of deacetylase, it may mainly augment the SIRT1 expression, which leads to the increased phosphorylation of Akt and Mdm2, thereby promoting p53 destabilization, decreased Bax expression, and inactivation of PTEN by phosphorylation [151]. The cannabinoid Δ9-tetrahydrocannabinol (THC), which is the active component of cannabis, was found to suppress pulmonary inflammation in mice induced by staphylococcal enterotoxin B (SEB) through downregulation of miRNA 17-92 cluster, particularly miRNA-18a that represses posttranscriptional regulation of PTEN gene. This indicates that the increased expression of PTEN inactivates PI3K/Akt signaling pathway, thereby reversing SEB-induced toxicity and death [152]. Modulation of PTEN with numerous modulators has been reported in the treatment of asthma, COPD, pulmonary fibrosis, and lung injury. Undoubtedly, restoring PTEN expression/function is a prime interest in the treatment of chronic lung diseases; however, it is extremely challenging to find a potent and direct PTEN modulators. The reported therapeutic agents as presented in Table 6 might indirectly regulate PTEN, and more studies are needed to fully clarify on the molecular mechanism.
Table 6.
List of therapeutic drugs that modulate PTEN expression and activity in various lung diseases.
| Diseases | Drug candidates | Models/samples | Effect on PTEN and target | Pharmacological effects | Ref. |
|---|---|---|---|---|---|
| Asthma | Resveratrol | Mouse/ovalbumin;16HBE cells | Restoration of PTEN expression and activation of SIRT1 | Airway inflammation and airway remodeling | [132] |
| Asthma | Epigallo-catechin-3-gallate | Mouse/ovalbumin;16HBE cells/TGF-β1 | Upregulation of PTEN and inhibition of PI3K/Akt | Suppresses inflammation and inflammatory cell infiltration; reduces airway remodeling by inhibiting EMT | [133] |
| Asthma | Borneol | Mouse/ovalbumin | Downregulation of miR-26a and miR-142-3p to upregulate PTEN expression | CD4+ T cell infiltration and proliferation | [134] |
| Asthma | α- and γ-mangostin | Mouse/ovalbumin | Upregulation of PTEN to suppress PI3K/Akt and NF-κB signaling | Reduces inflammatory cell recruitment into the airway, airway hyperresponsiveness (AHR), and increased levels of Th2 cytokines | [135] |
| Asthma | Korean red ginseng and Salvia plebeia R.Br. | Mouse/ovalbumin | Downregulation of phosphorylated PTEN and Akt and upregulation of PPAR-γ | Reduces the levels of Th2 cytokines IL-4, IL-5, and IL-13 in BALF and splenocytes and downregulates the IL-4, IL-13, IL-17, T-NF-α, and MUC5AC genes | [136] |
| Fibrosis | Dasatinib | Mice/bleomycin | Upregulation of PTEN and inhibition of PDGFR-alpha; Src and c-Abl activation | Myofibroblast activation and collagen-1 accumulation | [105] |
| Fibrosis | Unphosphorylated PTEN | H358 cells, fibroblast CC2512 cells and mouse primary lung fibroblasts/TGF-β1 | Restores the loss of PTEN activity | Reduces fibronectin expression and ECM production | [118] |
| Fibrosis | Resveratrol | Normal and IPF-derived lung fibroblasts/TGF-β1 | Upregulation of PTEN and downregulation of p-ERK and Akt | Inhibits cell proliferation of both normal and IPF-derived fibroblasts, α-SMA expression, and intracellular collagen deposition | [137] |
| Fibrosis | Prostaglandin E2 | Primary normal fetal lung fibroblasts IMR-90 | Increases PTEN activity and decreases p-Akt; downregulates survivin expression; increases Fas expression | Fibroblast apoptosis | [138] |
| Fibrosis | Berberine | Mice/bleomycin | Upregulation of PTEN in the colon; activation of PPAR-γ | Promotes HGF expression in colonic fibroblasts, which arrive in the lungs to palliate IPF | [139] |
| Fibrosis | Berberine | Wistar rats/bleomycin | Amplifies PTEN expression to inhibit FAK and PI3K/Akt/mTOR signaling; inhibits p-Smad 2/3 and enhances Smad 7 expression | Inhibits fibrotic markers, α-SMA, fibronectin, and collagens I and III and reverses bleomycin-induced ultrastructural alterations in the lungs | [140] |
| Inflammation | Coelonin | Raw264.7 cells/LPS | Upregulation of PTEN and inhibition of PTEN phosphorylation, resulting in suppressed NF-κB activation and p27kip1 degradation | Cell-cycle arrest in the G1 phase | [141] |
| Acute pulmonary embolism | Curcumin | Sprague–Dawley rats | Downregulation of miR-21 expression via inhibition of Sp1 to upregulate PTEN and impair the NF-κB signaling pathway | Reduces mPAP and RVSP levels, W/D ratio, thrombus volume, and inflammatory factors | [143] |
| Pulmonary arterial hypertension | Cefminox | Primary rat pulmonary artery smooth muscle cells (PASMCs)/hypoxia | Upregulation of PTEN by inhibiting Akt/mTOR signaling and enhanced cAMP production | Inhibits growth of PASMCs as a dual agonist of prostacyclin receptor (IP) and PPAR-γ | [144] |
| Hypoxic pulmonary hypertension (HPH) | 5-Aza-2′-deoxycytidine | Sprague–Dawley rats/hypoxia; PASMCs/hypoxia | Rescues the decreased PTEN expression by inhibiting hypermethylation | Proliferation, migration, and induction of apoptosis in PASMCs; pulmonary artery pressure and right ventricular hypertrophy index in HPH | [146] |
| Lung injury | miR-486 mimic | A549/PM2.5 | Negative regulation of PTEN and FOXO1 | Reduces cell apoptosis and ROS generation | [147] |
| Lung injury by ischemia/reperfusion | miR-21-5p | Mice/(I/R); primary murine pulmonary endothelial cells/H/R | miR-21-5p targeting PTEN and PDCD4 | Reduces lung edema and dysfunction, M1 polarization of alveolar macrophages, and secretion of proinflammatory cytokines | [148] |
| Lung injury | miR-371b-5p | A549 cells | Targets PTEN to inhibit phosphorylation of Akt and its downstream substrates, GSK3β and FOXOs | Augments ATIIC survival/proliferation, thereby promoting reepithelialization of injured alveoli | [149] |
| Chronic lung injury | Resveratrol | Mouse/methamphetamine | Activation of Sirt1 to downregulate PTEN and upregulation of p-Akt | Reduces oxidative stress and reverses MA-induced higher permeability and apoptosis of alveolar epithelium | [150] |
| Lung injury | Inhaled resveratrol | terc−/−F2 C57Bl/6J mice | Inactivates p-PTEN and activates p-Akt and p-Mdm2 via activation of SIRT1 | Maintaining AECII integrity and prevent deterioration of lung function | [151] |
| Lung injury | Δ9 Tetrahydrocannabinol | C3H/HeJ mice/Staphylococcal enterotoxin B | Posttranscriptional upregulation of PTEN via inhibition of miR-18a | Prevents SEB-induced mortality and alleviates symptoms of toxic shock | [152] |
3. Conclusion and Further Direction
In the present review, we highlighted the efforts made in the past few decades to investigate the role of PTEN in various chronic lung diseases such as asthma, COPD, IPF, PAH, and acute lung injury. PTEN is implicated in the regulation of various biological functions in lungs, ranging from inflammatory reactions, cell apoptosis, proliferation, differentiation, and so on. PTEN is abundantly expressed in various cell types of lung tissues such as macrophages, epithelial cells, endothelial cells, and smooth muscle cells. Both clinical and preclinical studies have shown that PTEN exerts a protective effect during the development and progression of diverse chronic respiratory diseases. Downregulation or inactivation of PTEN has been identified as the leading cause of almost all these diseases. The expression and activity of PTEN are subject to an extremely complex regulation at the transcriptional, posttranscriptional, translational, and posttranslational levels in various lung diseases. The specific mechanisms may vary in different types of diseases or different lung cell types. A variety of therapeutic agents such as noncoding RNAs, natural products, and clinical therapeutics aiming to restore PTEN expression/activity have showed great potential in the tested lung diseases in vitro and in vivo, but their exact mechanisms of action have not been fully elucidated. Further clinical trials are needed for these therapeutic agents. We conclude that PTEN plays a multifaceted role in the pathogenesis of lung disorders and is a promising therapeutic target for chronic lung diseases.
Acknowledgments
This work was financially supported by grants (No. 2020M682311, No. 00104311-2020-1, No. 82104561) from the China Postdoctoral Science Foundation, Doctoral Starting Fund of Henan University of Chinese Medicine, and National Natural Science Fund, respectively.
Data Availability
The relevant data used to support the findings of this study are included within the article.
Conflicts of Interest
The authors declare no conflicts of interest regarding this work.
Authors' Contributions
Bangrong Cai contributed to the methodology, investigation, editing, and writing-original draft. Liu Yang contributed to the investigation and partial original draft writing. Young Do Jung and Ying Zhang contributed to the editing and software. Xinguang Liu and Peng Zhao contributed to the literature retrieval and visualization. Jiansheng Li contributed to the supervision, writing-review and editing, and project administration. Bangrong Cai and Liu Yang contributed equally to this work. All authors read and approved the final manuscript.
References
- 1.Zhao D., Lu X., Wang G., et al. Synthetic essentiality of chromatin remodelling factor CHD1 in PTEN- deficient cancer. Nature . 2017;542(7642):484–488. doi: 10.1038/nature21357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arias-Pérez R. D., Taborda N. A., Gómez D. M., Narvaez J. F., Porras J., Hernandez J. C. Inflammatory effects of particulate matter air pollution. Environmental Science and Pollution Research International . 2020;27(34):42390–42404. doi: 10.1007/s11356-020-10574-w. [DOI] [PubMed] [Google Scholar]
- 3.Vlahos R., Bozinovski S. Recent advances in pre-clinical mouse models of COPD. Clinical Science . 2014;126(4):253–265. doi: 10.1042/CS20130182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Phan T. H. G., Paliogiannis P., Nasrallah G. K., et al. Emerging cellular and molecular determinants of idiopathic pulmonary fibrosis. Cellular and Molecular Life Sciences . 2021;78(5):2031–2057. doi: 10.1007/s00018-020-03693-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li J., Yen C., Liaw D., et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science . 1997;275(5308):1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 6.Li D. M., Sun H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Research . 1997;57(11):2124–2129. [PubMed] [Google Scholar]
- 7.Steck P. A., Pershouse M. A., Jasser S. A., et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nature Genetics . 1997;15(4):356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 8.Sano T., Lin H., Chen X., et al. Differential expression of MMAC/PTEN in glioblastoma multiforme: relationship to localization and prognosis. Cancer Research . 1999;59(8):1820–1824. [PubMed] [Google Scholar]
- 9.Gimm O., Perren A., Weng L. P., et al. Differential nuclear and cytoplasmic expression of PTEN in normal thyroid tissue, and benign and malignant epithelial thyroid tumors. The American Journal of Pathology . 2000;156(5):1693–1700. doi: 10.1016/S0002-9440(10)65040-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lachyankar M. B., Sultana N., Schonhoff C. M., et al. A role for nuclear PTEN in neuronal differentiation. The Journal of neuroscience . 2000;20(4):1404–1413. doi: 10.1523/JNEUROSCI.20-04-01404.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perren A., Komminoth P., Saremaslani P., et al. Mutation and expression analyses reveal differential subcellular compartmentalization of PTEN in endocrine pancreatic tumors compared to normal islet cells. The American Journal of Pathology . 2000;157(4):1097–1103. doi: 10.1016/S0002-9440(10)64624-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hopkins B. D., Fine B., Steinbach N., et al. A secreted PTEN phosphatase that enters cells to alter signaling and survival. Science . 2013;341(6144):399–402. doi: 10.1126/science.1234907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liang H., He S., Yang J., et al. PTENα, a PTEN isoform translated through alternative initiation, regulates mitochondrial function and energy metabolism. Cell Metabolism . 2014;19(5):836–848. doi: 10.1016/j.cmet.2014.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liang H., Chen X., Yin Q., et al. PTENβ is an alternatively translated isoform of PTEN that regulates rDNA transcription. Nature Communications . 2017;8(1):p. 14771. doi: 10.1038/ncomms14771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malaney P., Uversky V. N., Davé V. PTEN proteoforms in biology and disease. Cellular and Molecular Life Sciences . 2017;74(15):2783–2794. doi: 10.1007/s00018-017-2500-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tzani I., Ivanov I. P., Andreev D. E., et al. Systematic analysis of the PTEN 5' leader identifies a major AUU initiated proteoform. Open Biology . 2016;6(5) doi: 10.1098/rsob.150203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tibarewal P., Zilidis G., Spinelli L., et al. PTEN protein phosphatase activity correlates with control of gene expression and invasion, a tumor-suppressing phenotype, but not with AKT activity. Science Signaling . 2012;5(213):p. ra18. doi: 10.1126/scisignal.2002138. [DOI] [PubMed] [Google Scholar]
- 18.Tamura M., Gu J., Matsumoto K., Aota S., Parsons R., Yamada K. M. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science . 1998;280(5369):1614–1617. doi: 10.1126/science.280.5369.1614. [DOI] [PubMed] [Google Scholar]
- 19.Shi Y., Wang J., Chandarlapaty S., et al. PTEN is a protein tyrosine phosphatase for IRS1. Nature Structural & Molecular Biology . 2014;21(6):522–527. doi: 10.1038/nsmb.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gu T., Zhang Z., Wang J., Guo J., Shen W. H., Yin Y. CREB is a novel nuclear target of PTEN phosphatase. Cancer Research . 2011;71(8):2821–2825. doi: 10.1158/0008-5472.CAN-10-3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shnitsar I., Bashkurov M., Masson G. R., et al. PTEN regulates cilia through Dishevelled. Nature Communications . 2015;6(1):p. 8388. doi: 10.1038/ncomms9388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuchay S., Giorgi C., Simoneschi D., et al. PTEN counteracts FBXL2 to promote IP3R3- and Ca2+-mediated apoptosis limiting tumour growth. Nature . 2017;546(7659):554–558. doi: 10.1038/nature22965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee Y. R., Chen M., Pandolfi P. P. The functions and regulation of the PTEN tumour suppressor: new modes and prospects. Nature Reviews Molecular Cell Biology . 2018;19(9):547–562. doi: 10.1038/s41580-018-0015-0. [DOI] [PubMed] [Google Scholar]
- 24.Kotelevets L., Trifault B., Chastre E., Scott M. G. H. Posttranslational Regulation and Conformational Plasticity of PTEN. Cold Spring Harbor Perspectives in Medicine . 2020;10(7) doi: 10.1101/cshperspect.a036095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hopkins B. D., Hodakoski C., Barrows D., Mense S. M., Parsons R. E. PTEN function: the long and the short of it. Trends in Biochemical Sciences . 2014;39(4):183–190. doi: 10.1016/j.tibs.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gkountakos A., Sartori G., Falcone I., et al. PTEN in lung cancer: dealing with the problem, building on new knowledge and turning the game around. Cancers . 2019;11(8):p. 1141. doi: 10.3390/cancers11081141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kay A. B. Asthma and inflammation. The Journal of Allergy and Clinical Immunology . 1991;87(5):893–910. doi: 10.1016/0091-6749(91)90408-G. [DOI] [PubMed] [Google Scholar]
- 28.Kwak Y. G., Song C. H., Yi H. K., et al. Involvement of PTEN in airway hyperresponsiveness and inflammation in bronchial asthma. The Journal of Clinical Investigation . 2003;111(7):1083–1092. doi: 10.1172/JCI16440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee K. S., Park S. J., Hwang P. H., et al. PPAR-gamma modulates allergic inflammation through up-regulation of PTEN. FASEB Journal . 2005;19(8):1033–1035. doi: 10.1096/fj.04-3309fje. [DOI] [PubMed] [Google Scholar]
- 30.Lee K. S., Kim S. R., Park S. J., et al. Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) reduces vascular endothelial growth factor expression in allergen-induced airway inflammation. Molecular Pharmacology . 2006;69(6):1829–1839. doi: 10.1124/mol.106.022228. [DOI] [PubMed] [Google Scholar]
- 31.Ni Z., Tang J., Cai Z., et al. A new pathway of glucocorticoid action for asthma treatment through the regulation of PTEN expression. Respiratory Research . 2011;12(1):p. 47. doi: 10.1186/1465-9921-12-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang W. X., Li C. C. Airway remodeling: a potential therapeutic target in asthma. World Journal of Pediatrics . 2011;7(2):124–128. doi: 10.1007/s12519-011-0264-x. [DOI] [PubMed] [Google Scholar]
- 33.Wen X., Yan J., Han X. R., et al. PTEN gene silencing contributes to airway remodeling and induces airway smooth muscle cell proliferation in mice with allergic asthma. Journal of Thoracic Disease . 2018;10(1):202–211. doi: 10.21037/jtd.2017.12.104. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Lan H., Zhong H., Gao Y., et al. The PTEN tumor suppressor inhibits human airway smooth muscle cell migration. International Journal of Molecular Medicine . 2010;26(6):893–899. doi: 10.3892/ijmm_00000539. [DOI] [PubMed] [Google Scholar]
- 35.Luo L., Gong Y. Q., Qi X., Lai W., Lan H., Luo Y. Effect of tumor suppressor PTEN gene on apoptosis and cell cycle of human airway smooth muscle cells. Molecular and Cellular Biochemistry . 2013;375(1-2):1–9. doi: 10.1007/s11010-012-1484-7. [DOI] [PubMed] [Google Scholar]
- 36.Wu Y., Lu Y., Zou F., et al. PTEN participates in airway remodeling of asthma by regulating CD38/Ca2+/CREB signaling. Aging . 2020;12(16):16326–16340. doi: 10.18632/aging.103664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li X., Zou F., Lu Y., et al. Notch1 contributes to TNF-α-induced proliferation and migration of airway smooth muscle cells through regulation of the Hes1/PTEN axis. International Immunopharmacology . 2020;88, article 106911 doi: 10.1016/j.intimp.2020.106911. [DOI] [PubMed] [Google Scholar]
- 38.Alexandrova E., Miglino N., Hashim A., et al. Small RNA profiling reveals deregulated phosphatase and tensin homolog (PTEN)/phosphoinositide 3-kinase (PI3K)/Akt pathway in bronchial smooth muscle cells from asthmatic patients. The Journal of Allergy and Clinical Immunology . 2016;137(1):58–67. doi: 10.1016/j.jaci.2015.05.031. [DOI] [PubMed] [Google Scholar]
- 39.Hou C., Chen Y., Huang X., Huang Q., Li M., Tan X. miR-19 targets PTEN and mediates high mobility group protein B1(HMGB1)-induced proliferation and migration of human airway smooth muscle cells. PLoS One . 2019;14(6, article e0219081) doi: 10.1371/journal.pone.0219081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen H., Guo S. X., Zhang S., Li X. D., Wang H., Li X. W. MiRNA-620 promotesTGF-β1-induced proliferation of airway smooth muscle cell through controllingPTEN/AKTsignaling pathway. The Kaohsiung Journal of Medical Sciences . 2020;36(11):869–877. doi: 10.1002/kjm2.12260. [DOI] [PubMed] [Google Scholar]
- 41.Lv X., Li Y., Gong Q., Jiang Z. TGF-β1 induces airway smooth muscle cell proliferation and remodeling in asthmatic mice by up-regulating miR-181a and suppressing PTEN. International Journal of Clinical and Experimental Pathology . 2019;12(1):173–181. [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Y., Yang K., Shi H., et al. MiR-21 modulates human airway smooth muscle cell proliferation and migration in asthma through regulation of PTEN expression. Experimental Lung Research . 2015;41(10):535–545. doi: 10.3109/01902148.2015.1090501. [DOI] [PubMed] [Google Scholar]
- 43.Kim R. Y., Horvat J. C., Pinkerton J. W., et al. MicroRNA-21 drives severe, steroid-insensitive experimental asthma by amplifying phosphoinositide 3-kinase-mediated suppression of histone deacetylase 2. The Journal of Allergy and Clinical Immunology . 2017;139(2):519–532. doi: 10.1016/j.jaci.2016.04.038. [DOI] [PubMed] [Google Scholar]
- 44.Zhou Y., Yang Q., Xu H., et al. miRNA-221-3p Enhances the Secretion of Interleukin-4 in Mast Cells through the Phosphatase and Tensin Homolog/p38/Nuclear Factor-kappaB Pathway. PLoS One . 2016;11(2, article e0148821) doi: 10.1371/journal.pone.0148821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fang L., Wang X., Sun Q., et al. IgE Downregulates PTEN through MicroRNA-21-5p and Stimulates Airway Smooth Muscle Cell Remodeling. International Journal of Molecular Sciences . 2019;20(4):p. 875. doi: 10.3390/ijms20040875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu J. H., Li C., Zhang C. H., Zhang Z. H. LncRNA-CASC7 enhances corticosteroid sensitivity via inhibiting the PI3K/AKT signaling pathway by targeting miR-21 in severe asthma. Pulmonology . 2020;26(1):18–26. doi: 10.1016/j.pulmoe.2019.07.001. [DOI] [PubMed] [Google Scholar]
- 47.Yu H., Qi N., Zhou Q. LncRNA H19 inhibits proliferation and migration of airway smooth muscle cells induced by PDGF-BB through miR-21/PTEN/Akt axis. Journal of Asthma and Allergy . 2021;14:71–80. doi: 10.2147/JAA.S291333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cui Y., Yang S. Overexpression of Annexin A1 protects against benzo[a]pyrene-induced bronchial epithelium injury. Molecular Medicine Reports . 2018;18(1):349–357. doi: 10.3892/mmr.2018.8998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fu L., Zhu P., Qi S., Li C., Zhao K. MicroRNA-92a antagonism attenuates lipopolysaccharide (LPS)-induced pulmonary inflammation and injury in mice through suppressing the PTEN/AKT/NF-κB signaling pathway. Biomedicine & Pharmacotherapy . 2018;107:703–711. doi: 10.1016/j.biopha.2018.08.040. [DOI] [PubMed] [Google Scholar]
- 50.Lee S. M., Choi H., Yang G., Park K. C., Jeong S., Hong S. microRNAs mediate oleic acid-induced acute lung injury in rats using an alternative injury mechanism. Molecular Medicine Reports . 2014;10(1):292–300. doi: 10.3892/mmr.2014.2155. [DOI] [PubMed] [Google Scholar]
- 51.Miyoshi K., Yanagi S., Kawahara K., et al. Epithelial Pten controls acute lung injury and fibrosis by regulating alveolar epithelial cell integrity. American Journal of Respiratory and Critical Care Medicine . 2013;187(3):262–275. doi: 10.1164/rccm.201205-0851OC. [DOI] [PubMed] [Google Scholar]
- 52.Qin S., Wang H., Liu G., Mei H., Chen M. miR-21-5p ameliorates hyperoxic acute lung injury and decreases apoptosis of AEC II cells via PTEN/AKT signaling in rats. Molecular Medicine Reports . 2019;20(6):4953–4962. doi: 10.3892/mmr.2019.10779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu Y., Li J., Yuan R., Deng Z., Wu X. Bone marrow mesenchymal stem cell-derived exosomes alleviate hyperoxia-induced lung injury via the manipulation of microRNA-425. Archives of Biochemistry and Biophysics . 2021;697, article 108712 doi: 10.1016/j.abb.2020.108712. [DOI] [PubMed] [Google Scholar]
- 54.Tiozzo C., De Langhe S., Yu M., et al. Deletion of Pten expands lung epithelial progenitor pools and confers resistance to airway injury. American Journal of Respiratory and Critical Care Medicine . 2009;180(8):701–712. doi: 10.1164/rccm.200901-0100OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lai J. P., Dalton J. T., Knoell D. L. Phosphatase and tensin homologue deleted on chromosome ten (PTEN) as a molecular target in lung epithelial wound repair. British Journal of Pharmacology . 2007;152(8):1172–1184. doi: 10.1038/sj.bjp.0707501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lai J. P., Bao S., Davis I. C., Knoell D. L. Inhibition of the phosphatase PTEN protects mice against oleic acid-induced acute lung injury. British Journal of Pharmacology . 2009;156(1):189–200. doi: 10.1111/j.1476-5381.2008.00020.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mihai C., Bao S., Lai J. P., Ghadiali S. N., Knoell D. L. PTEN inhibition improves wound healing in lung epithelia through changes in cellular mechanics that enhance migration. American Journal of Physiology Lung Cellular and Molecular Physiology . 2012;302(3):L287–L299. doi: 10.1152/ajplung.00037.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou M., Zhang Y., Chen X., et al. PTEN-Foxo1 signaling triggers HMGB1-mediated innate immune responses in acute lung injury. Immunologic Research . 2015;62(1):95–105. doi: 10.1007/s12026-015-8639-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ittner A., Block H., Reichel C. A., et al. Regulation of PTEN activity by p38δ-PKD1 signaling in neutrophils confers inflammatory responses in the lung. The Journal of Experimental Medicine . 2012;209(12):2229–2246. doi: 10.1084/jem.20120677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eaton D. C., Helms M. N., Koval M., Bao H. F., Jain L. The contribution of epithelial sodium channels to alveolar function in health and disease. Annual Review of Physiology . 2009;71(1):403–423. doi: 10.1146/annurev.physiol.010908.163250. [DOI] [PubMed] [Google Scholar]
- 61.Buck T. M., Brodsky J. L. Epithelial sodium channel biogenesis and quality control in the early secretory pathway. Current Opinion in Nephrology and Hypertension . 2018;27(5):364–372. doi: 10.1097/MNH.0000000000000438. [DOI] [PubMed] [Google Scholar]
- 62.Qi W., Li H., Cai X. H., et al. Lipoxin A4 activates alveolar epithelial sodium channel gamma via the microRNA-21/PTEN/AKT pathway in lipopolysaccharide- induced inflammatory lung injury. Laboratory Investigation . 2015;95(11):1258–1268. doi: 10.1038/labinvest.2015.109. [DOI] [PubMed] [Google Scholar]
- 63.Zhang H., Ding Y., Hou Y., Liu Y., Zhou Z., Nie H. Bone marrow mesenchymal stem cells derived miRNA-130b enhances epithelial sodium channel by targeting PTEN. Respiratory Research . 2020;21:p. 329. doi: 10.1186/s12931-020-01595-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang J., Mao M., Zhen Y. Y. miRNA-23a has effects to improve lung injury induced by sepsis in vitro and vivo study. Biomedicine & Pharmacotherapy . 2018;107:81–89. doi: 10.1016/j.biopha.2018.07.097. [DOI] [PubMed] [Google Scholar]
- 65.Sisti F., Wang S., Brandt S. L., et al. Nuclear PTEN enhances the maturation of a microRNA regulon to limit MyD88-dependent susceptibility to sepsis. Science Signaling . 2018;11(528) doi: 10.1126/scisignal.aai9085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhou M., Fang H., Du M., et al. The modulation of regulatory T cells via HMGB1/PTEN/β-catenin axis in LPS induced acute lung injury. Frontiers in Immunology . 2019;10:p. 1612. doi: 10.3389/fimmu.2019.01612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu F., Peng W., Chen J., et al. Exosomes derived from alveolar epithelial cells promote alveolar macrophage activation mediated by miR-92a-3p in sepsis-induced acute lung injury. Frontiers in Cellular and Infection Microbiology . 2021;11, article 646546 doi: 10.3389/fcimb.2021.646546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang L., Liu J., Xie W., et al. Overexpression of MALAT1 relates to lung injury through sponging miR-425 and promoting cell apoptosis during ARDS. Canadian Respiratory Journal . 2019;2019:9. doi: 10.1155/2019/1871394.1871394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ramos-Casals M., Retamozo S., Sisó-Almirall A., Pérez-Alvarez R., Pallarés L., Brito-Zerón P. Clinically-useful serum biomarkers for diagnosis and prognosis of sarcoidosis. Expert Review of Clinical Immunology . 2019;15(4):391–405. doi: 10.1080/1744666X.2019.1568240. [DOI] [PubMed] [Google Scholar]
- 70.Ding J., Dai J., Cai H., Gao Q., Wen Y. Extensively disturbance of regulatory T cells - Th17 cells balance in stage II pulmonary sarcoidosis. International Journal of Medical Sciences . 2017;14(11):1136–1142. doi: 10.7150/ijms.18838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bhargava M., Viken K. J., Barkes B., et al. Novel protein pathways in development and progression of pulmonary sarcoidosis. Scientific Reports . 2020;10(1, article 13282) doi: 10.1038/s41598-020-69281-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Navratilova Z., Novosadova E., Hagemann-Jensen M., et al. Expression profile of Six RNA-binding proteins in pulmonary sarcoidosis. PLoS One . 2016;11(8, article e0161669) doi: 10.1371/journal.pone.0161669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kirkham P. A., Barnes P. J. Oxidative stress in COPD. Chest . 2013;144(1):266–273. doi: 10.1378/chest.12-2664. [DOI] [PubMed] [Google Scholar]
- 74.Barnes P. J. Oxidative stress-based therapeutics in COPD. Redox Biology . 2020;33, article 101544 doi: 10.1016/j.redox.2020.101544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lu J., Xie L., Liu C., Zhang Q., Sun S. PTEN/PI3k/AKT Regulates Macrophage Polarization in Emphysematous mice. Scandinavian Journal of Immunology . 2017;85:395–405. doi: 10.1111/sji.12545. [DOI] [PubMed] [Google Scholar]
- 76.Li T., Fanning K. V., Nyunoya T., Chen Y., Zou C. Cigarette smoke extract induces airway epithelial cell death via repressing PRMT6/AKT signaling. Aging . 2020;12(23):24301–24317. doi: 10.18632/aging.202210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Besiktepe N., Kayalar O., Ersen E., Oztay F. The copper dependent-lysyl oxidases contribute to the pathogenesis of pulmonary emphysema in chronic obstructive pulmonary disease patients. Journal of Trace Elements in Medicine and Biology . 2017;44:247–255. doi: 10.1016/j.jtemb.2017.08.011. [DOI] [PubMed] [Google Scholar]
- 78.Shaykhiev R., Otaki F., Bonsu P., et al. Cigarette smoking reprograms apical junctional complex molecular architecture in the human airway epithelium in vivo. Cellular and Molecular Life Sciences . 2011;68(5):877–892. doi: 10.1007/s00018-010-0500-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yanagisawa S., Baker J. R., Vuppusetty C., et al. Decreased phosphatase PTEN amplifies PI3K signaling and enhances proinflammatory cytokine release in COPD. American Journal of Physiology Lung Cellular and Molecular Physiology . 2017;313(2):L230–l239. doi: 10.1152/ajplung.00382.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cai B., Liu M., Li J., Xu D., Li J. Cigarette smoke extract amplifies NADPH oxidase-dependent ROS production to inactivate PTEN by oxidation in BEAS-2B cells. Food and Chemical Toxicology . 2021;150, article 112050 doi: 10.1016/j.fct.2021.112050. [DOI] [PubMed] [Google Scholar]
- 81.Barnes P. J., Baker J., Donnelly L. E. Cellular senescence as a mechanism and target in chronic lung diseases. American Journal of Respiratory and Critical Care Medicine . 2019;200(5):556–564. doi: 10.1164/rccm.201810-1975TR. [DOI] [PubMed] [Google Scholar]
- 82.Baker J. R., Vuppusetty C., Colley T., et al. Oxidative stress dependent microRNA-34a activation via PI3Kα reduces the expression of sirtuin-1 and sirtuin-6 in epithelial cells. Scientific Reports . 2016;6(1):p. 35871. doi: 10.1038/srep35871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Barnes P. J. Cellular and molecular mechanisms of chronic obstructive pulmonary disease. Clinics in Chest Medicine . 2014;35(1):71–86. doi: 10.1016/j.ccm.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 84.Shen Q., Zheng J., Wang X., Hu W., Jiang Y., Jiang Y. LncRNA SNHG5 regulates cell apoptosis and inflammation by miR-132/PTEN axis in COPD. Biomedicine & Pharmacotherapy . 2020;126, article 110016 doi: 10.1016/j.biopha.2020.110016. [DOI] [PubMed] [Google Scholar]
- 85.Anderson G. P., Bozinovski S. Acquired somatic mutations in the molecular pathogenesis of COPD. Trends in Pharmacological Sciences . 2003;24(2):71–76. doi: 10.1016/S0165-6147(02)00052-4. [DOI] [PubMed] [Google Scholar]
- 86.Hosgood H. D., 3rd, Menashe I., He X., Chanock S., Lan Q. PTEN identified as important risk factor of chronic obstructive pulmonary disease. Respiratory Medicine . 2009;103(12):1866–1870. doi: 10.1016/j.rmed.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vannitamby A., Seow H. J., Anderson G., et al. Tumour-associated neutrophils and loss of epithelial PTEN can promote corticosteroid-insensitive MMP-9 expression in the chronically inflamed lung microenvironment. Thorax . 2017;72(12):1140–1143. doi: 10.1136/thoraxjnl-2016-209389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jones J. G., Minty B. D., Lawler P., Hulands G., Crawley J. C., Veall N. Increased alveolar epithelial permeability in cigarette smokers. The Lancet . 1980;315(8159):66–68. doi: 10.1016/S0140-6736(80)90493-6. [DOI] [PubMed] [Google Scholar]
- 89.Knowles M. R., Buntin W. H., Bromberg P. A., Gatzy J. T., Boucher R. C. Measurements of transepithelial electric potential differences in the trachea and bronchi of human subjects in vivo. The American Review of Respiratory Disease . 1982;126(1):108–112. doi: 10.1164/arrd.1982.126.1.108. [DOI] [PubMed] [Google Scholar]
- 90.Ivanov A. I., Nusrat A., Parkos C. A. Endocytosis of the apical junctional complex: mechanisms and possible roles in regulation of epithelial barriers. BioEssays . 2005;27(4):356–365. doi: 10.1002/bies.20203. [DOI] [PubMed] [Google Scholar]
- 91.Barbieri S. S., Ruggiero L., Tremoli E., Weksler B. B. Suppressing PTEN activity by tobacco smoke plus interleukin-1beta modulates dissociation of VE-cadherin/beta-catenin complexes in endothelium. Arteriosclerosis, Thrombosis, and Vascular Biology . 2008;28(4):732–738. doi: 10.1161/ATVBAHA.107.159434. [DOI] [PubMed] [Google Scholar]
- 92.Kulkarni Y. M., Dutta S., Iyer A. K., et al. A proteomics approach to identifying key protein targets involved in VEGF inhibitor mediated attenuation of bleomycin-induced pulmonary fibrosis. Proteomics . 2016;16(1):33–46. doi: 10.1002/pmic.201500171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kral J. B., Kuttke M., Schrottmaier W. C., et al. Erratum: Sustained PI3K Activation exacerbates BLM-induced Lung Fibrosis via activation of pro-inflammatory and pro-fibrotic pathways. Scientific Reports . 2016;6(1):p. 23034. doi: 10.1038/srep26048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yanagi S., Tsubouchi H., Miura A., Matsumoto N., Nakazato M. Breakdown of epithelial barrier integrity and overdrive activation of alveolar epithelial cells in the pathogenesis of acute respiratory distress syndrome and lung fibrosis. BioMed Research International . 2015;2015:12. doi: 10.1155/2015/573210.573210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Qiu T., Tian Y., Gao Y., et al. PTEN loss regulates alveolar epithelial cell senescence in pulmonary fibrosis depending on Akt activation. Aging . 2019;11(18):7492–7509. doi: 10.18632/aging.102262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tian Y., Li H., Qiu T., et al. Loss of PTEN induces lung fibrosis via alveolar epithelial cell senescence depending on NF-κB activation. Aging Cell . 2019;18(1, article e12858) doi: 10.1111/acel.12858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.White E. S., Atrasz R. G., Hu B., et al. Negative regulation of myofibroblast differentiation by PTEN (phosphatase and tensin homolog deleted on chromosome 10) American Journal of Respiratory and Critical Care Medicine . 2006;173(1):112–121. doi: 10.1164/rccm.200507-1058OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Xia H., Khalil W., Kahm J., Jessurun J., Kleidon J., Henke C. A. Pathologic caveolin-1 regulation of PTEN in idiopathic pulmonary fibrosis. The American Journal of Pathology . 2010;176(6):2626–2637. doi: 10.2353/ajpath.2010.091117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Xia H., Diebold D., Nho R., et al. Pathological integrin signaling enhances proliferation of primary lung fibroblasts from patients with idiopathic pulmonary fibrosis. The Journal of Experimental Medicine . 2008;205(7):1659–1672. doi: 10.1084/jem.20080001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Geng J., Huang X., Li Y., et al. Phosphatase and tensin homolog deleted on chromosome 10 contributes to phenotype transformation of fibroblasts in idiopathic pulmonary fibrosis via multiple pathways. Experimental Biology and Medicine . 2016;241(2):157–165. doi: 10.1177/1535370215600100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Geng J., Huang X., Li Y., et al. Down-regulation of USP13 mediates phenotype transformation of fibroblasts in idiopathic pulmonary fibrosis. Respiratory Research . 2015;16(1):p. 124. doi: 10.1186/s12931-015-0286-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Xie B., Zheng G., Li H., et al. Effects of the tumor suppressor PTEN on the pathogenesis of idiopathic pulmonary fibrosis in Chinese patients. Molecular Medicine Reports . 2016;13(3):2715–2723. doi: 10.3892/mmr.2016.4852. [DOI] [PubMed] [Google Scholar]
- 103.Yilmaz O., Oztay F., Kayalar O. Dasatinib attenuated bleomycin-induced pulmonary fibrosis in mice. Growth Factors . 2015;33(5-6):366–375. doi: 10.3109/08977194.2015.1109511. [DOI] [PubMed] [Google Scholar]
- 104.Zhang H., You L., Zhao M. Rosiglitazone attenuates paraquat-induced lung fibrosis in rats in a PPAR gamma-dependent manner. European Journal of Pharmacology . 2019;851:133–143. doi: 10.1016/j.ejphar.2019.02.037. [DOI] [PubMed] [Google Scholar]
- 105.Liu Z., Liang X., Li X., et al. MiRNA-21 functions in ionizing radiation-induced epithelium-to-mesenchymal transition (EMT) by downregulating PTEN. Toxicology Research . 2019;8(3):328–340. doi: 10.1039/C9TX00019D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nho R. S., Xia H., Diebold D., et al. PTEN Regulates Fibroblast Elimination during Collagen Matrix Contraction. Journal of Biological Chemistry . 2006;281(44):33291–33301. doi: 10.1074/jbc.M606450200. [DOI] [PubMed] [Google Scholar]
- 107.Nho R. S., Hergert P., Kahm J., Jessurun J., Henke C. Pathological alteration of FoxO3a activity promotes idiopathic pulmonary fibrosis fibroblast proliferation on type i collagen matrix. The American Journal of Pathology . 2011;179(5):2420–2430. doi: 10.1016/j.ajpath.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nho R. S., Peterson M., Hergert P., Henke C. A. FoxO3a (forkhead box O3a) deficiency protects idiopathic pulmonary fibrosis (IPF) fibroblasts from type I polymerized collagen matrix-induced apoptosis via caveolin-1 (cav-1) and Fas. PLoS One . 2013;8(4, article e61017) doi: 10.1371/journal.pone.0061017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nho R. S., Hergert P. IPF fibroblasts are desensitized to type I collagen matrix-induced cell death by suppressing low autophagy via aberrant Akt/mTOR kinases. PLoS One . 2014;9(4, article e94616) doi: 10.1371/journal.pone.0094616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Im J., Hergert P., Nho R. S. Reduced FoxO3a expression causes low autophagy in idiopathic pulmonary fibrosis fibroblasts on collagen matrices. American Journal of Physiology-Lung Cellular and Molecular Physiology . 2015;309(6):L552–L561. doi: 10.1152/ajplung.00079.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mao P., Li J., Huang Y., et al. MicroRNA-19b mediates lung epithelial-mesenchymal transition via phosphatidylinositol-3,4,5-trisphosphate 3-phosphatase in response to mechanical stretch. American Journal of Respiratory Cell and Molecular Biology . 2017;56(1):11–19. doi: 10.1165/rcmb.2015-0377OC. [DOI] [PubMed] [Google Scholar]
- 112.He Z., Gao Y., Deng Y., et al. Lipopolysaccharide induces lung fibroblast proliferation through Toll-like receptor 4 signaling and the phosphoinositide3-kinase-Akt pathway. PLoS One . 2012;7(4, article e35926) doi: 10.1371/journal.pone.0035926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.White E. S., Thannickal V. J., Carskadon S. L., et al. Integrin α4β11 regulates migration across basement membranes by lung fibroblasts: a role for phosphatase and tensin homologue deleted on chromosome 10. American Journal of Respiratory and Critical Care Medicine . 2003;168(4):436–442. doi: 10.1164/rccm.200301-041OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.White E. S., Atrasz R. G., Dickie E. G., et al. Prostaglandin E2 inhibits fibroblast migration by E-prostanoid 2 receptor-mediated increase in PTEN activity. American Journal of Respiratory Cell and Molecular Biology . 2005;32:135–141. doi: 10.1165/rcmb.2004-0126oc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Huang S. K., Fisher A. S., Scruggs A. M., et al. Hypermethylation of PTGER2 confers prostaglandin E2 resistance in fibrotic fibroblasts from humans and mice. The American Journal of Pathology . 2010;177:2245–2255. doi: 10.2353/ajpath.2010.100446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhang X., Jia X., Mei L., Zheng M., Yu C., Ye M. Global DNA methylation and PTEN hypermethylation alterations in lung tissues from human silicosis. Journal of Thoracic Disease . 2016;8(8):2185–2195. doi: 10.21037/jtd.2016.07.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Parapuram S. K., Thompson K., Tsang M., et al. Loss of PTEN expression by mouse fibroblasts results in lung fibrosis through a CCN2-dependent mechanism. Matrix Biology . 2015;43:35–41. doi: 10.1016/j.matbio.2015.01.017. [DOI] [PubMed] [Google Scholar]
- 118.Kimura M., Hashimoto N., Kusunose M., et al. Exogenous induction of unphosphorylated PTEN reduces TGFβ-induced extracellular matrix expressions in lung fibroblasts. Wound Repair and Regeneration . 2017;25(1):86–97. doi: 10.1111/wrr.12506. [DOI] [PubMed] [Google Scholar]
- 119.Zeng Z. H., Wu W. H., Peng Q., Sun Y. H., Liu J. X. MicroRNA-132 mediates proliferation and migration of pulmonary smooth muscle cells via targeting PTEN. Molecular Medicine Reports . 2019;19(5):3823–3830. doi: 10.3892/mmr.2019.10053. [DOI] [PubMed] [Google Scholar]
- 120.Liu G., Hao P., Xu J., et al. Upregulation of microRNA-17-5p contributes to hypoxia-induced proliferation in human pulmonary artery smooth muscle cells through modulation of p21 and PTEN. Respiratory Research . 2018;19:p. 200. doi: 10.1186/s12931-018-0902-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhu G., Zhang W., Liu Y., Wang S. miR-371b-5p inhibits endothelial cell apoptosis in monocrotaline-induced pulmonary arterial hypertension via PTEN/PI3K/Akt signaling pathways. Molecular Medicine Reports . 2018;18(6):5489–5501. doi: 10.3892/mmr.2018.9614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ravi Y., Selvendiran K., Meduru S., et al. Dysregulation of PTEN in cardiopulmonary vascular remodeling induced by pulmonary hypertension. Cell Biochemistry and Biophysics . 2013;67(2):363–372. doi: 10.1007/s12013-011-9332-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhu Y., Wu Y., Shi W., et al. Inhibition of ubiquitin proteasome function prevents monocrotaline-induced pulmonary arterial remodeling. Life Sciences . 2017;173:36–42. doi: 10.1016/j.lfs.2017.02.007. [DOI] [PubMed] [Google Scholar]
- 124.Lai Y. J., Hsu H. H., Chang G. J. Prostaglandin E1 Attenuates Pulmonary Artery Remodeling by Activating Phosphorylation of CREB and the PTEN Signaling Pathway. Scientific Reports . 2017;7:p. 9974. doi: 10.1038/s41598-017-09707-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Liu H., Yin T., Yan W., et al. Dysregulation of microRNA-214 and PTEN contributes to the pathogenesis of hypoxic pulmonary hypertension. International Journal of Chronic Obstructive Pulmonary Disease . 2017;12:1781–1791. doi: 10.2147/COPD.S104627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhao M., Chen N., Li X., Lin L., Chen X. MiR-19a modulates hypoxia-mediated cell proliferation and migration via repressing PTEN in human pulmonary arterial smooth muscle. Life Sciences . 2019;239, article 116928 doi: 10.1016/j.lfs.2019.116928. [DOI] [PubMed] [Google Scholar]
- 127.Zhu B., Gong Y., Yan G., et al. Down-regulation of lncRNA MEG3 promotes hypoxia-induced human pulmonary artery smooth muscle cell proliferation and migration via repressing PTEN by sponging miR-21. Biochemical and Biophysical Research Communications . 2018;495(3):2125–2132. doi: 10.1016/j.bbrc.2017.11.185. [DOI] [PubMed] [Google Scholar]
- 128.Horita H., Furgeson S. B., Ostriker A., et al. Selective inactivation of PTEN in smooth muscle cells synergizes with hypoxia to induce severe pulmonary hypertension. Journal of the American Heart Association . 2013;2(3, article e000188) doi: 10.1161/JAHA.113.000188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Liu Y., Cao Y., Sun S., et al. Transforming growth factor-beta1 upregulation triggers pulmonary artery smooth muscle cell proliferation and apoptosis imbalance in rats with hypoxic pulmonary hypertension via the PTEN/AKT pathways. The International Journal of Biochemistry & Cell Biology . 2016;77(Part A):141–154. doi: 10.1016/j.biocel.2016.06.006. [DOI] [PubMed] [Google Scholar]
- 130.Ravi Y., Selvendiran K., Naidu S. K., et al. Pulmonary hypertension secondary to left-heart failure involves peroxynitrite-induced downregulation of PTEN in the lung. Hypertension . 2013;61(3):593–601. doi: 10.1161/HYPERTENSIONAHA.111.00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.McLoughlin N. M., Mueller C., Grossmann T. N. The therapeutic potential of PTEN modulation: targeting strategies from gene to protein. Cell Chemical Biology . 2018;25(1):19–29. doi: 10.1016/j.chembiol.2017.10.009. [DOI] [PubMed] [Google Scholar]
- 132.Yang N., Zhang H., Cai X., Shang Y. Epigallocatechin-3-gallate inhibits inflammation and epithelial-mesenchymal transition through the PI3K/AKT pathway via upregulation of PTEN in asthma. International Journal of Molecular Medicine . 2018;41(2):818–828. doi: 10.3892/ijmm.2017.3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chen G., Tang J., Ni Z., et al. Antiasthmatic effects of resveratrol in ovalbumin-induced asthma model mice involved in the upregulation of PTEN. Biological & Pharmaceutical Bulletin . 2015;38(4):507–513. doi: 10.1248/bpb.b14-00610. [DOI] [PubMed] [Google Scholar]
- 134.Wang J. Y., Dong X., Yu Z., et al. Borneol inhibits CD4 + T cells proliferation by down-regulating miR-26a and miR-142-3p to attenuate asthma. International Immunopharmacology . 2021;90, article 107223 doi: 10.1016/j.intimp.2020.107223. [DOI] [PubMed] [Google Scholar]
- 135.Jang H. Y., Kwon O. K., Oh S. R., Lee H. K., Ahn K. S., Chin Y. W. Mangosteen xanthones mitigate ovalbumin-induced airway inflammation in a mouse model of asthma. Food and Chemical Toxicology . 2012;50(11):4042–4050. doi: 10.1016/j.fct.2012.08.037. [DOI] [PubMed] [Google Scholar]
- 136.Lee Y. S., Yang W. K., Yee S. M., et al. KGC3P attenuates ovalbumin-induced airway inflammation through downregulation of p-PTEN in asthmatic mice. Phytomedicine . 2019;62, article 152942 doi: 10.1016/j.phymed.2019.152942. [DOI] [PubMed] [Google Scholar]
- 137.Fagone E., Conte E., Gili E., et al. Resveratrol inhibits transforming growth factor-β-induced proliferation and differentiation of ex vivo human lung fibroblasts into myofibroblasts through ERK/Akt inhibition and PTEN restoration. Experimental Lung Research . 2011;37(3):162–174. doi: 10.3109/01902148.2010.524722. [DOI] [PubMed] [Google Scholar]
- 138.Huang S. K., White E. S., Wettlaufer S. H., et al. Prostaglandin E2 induces fibroblast apoptosis by modulating multiple survival pathways. FASEB Journal . 2009;23(12):4317–4326. doi: 10.1096/fj.08-128801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Guan C., Qiao S., Lv Q., et al. Orally administered berberine ameliorates bleomycin-induced pulmonary fibrosis in mice through promoting activation of PPAR-γ and subsequent expression of HGF in colons. Toxicology and Applied Pharmacology . 2018;343:1–15. doi: 10.1016/j.taap.2018.02.001. [DOI] [PubMed] [Google Scholar]
- 140.Chitra P., Saiprasad G., Manikandan R., Sudhandiran G. Berberine inhibits Smad and non-Smad signaling cascades and enhances autophagy against pulmonary fibrosis. Journal of Molecular Medicine . 2015;93(9):1015–1031. doi: 10.1007/s00109-015-1283-1. [DOI] [PubMed] [Google Scholar]
- 141.Jiang F., Li M., Wang H., et al. Coelonin, an anti-inflammation active component of Bletilla striata and its potential mechanism. International journal of molecular sciences . 2019;20(18, article 4422) doi: 10.3390/ijms20184422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Choi W., Choe S., Lin J., et al. Exendin-4 restores airway mucus homeostasis through the GLP1R-PKA-PPARγ- FOXA2-phosphatase signaling. Mucosal Immunology . 2020;13(4):637–651. doi: 10.1038/s41385-020-0262-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Liang D., Wen Z., Han W., Li W., Pan L., Zhang R. Curcumin protects against inflammation and lung injury in rats with acute pulmonary embolism with the involvement of microRNA-21/PTEN/NF-κB axis. Molecular and Cellular Biochemistry . 2021;476(7):2823–2835. doi: 10.1007/s11010-021-04127-z. [DOI] [PubMed] [Google Scholar]
- 144.Xia J., Yang L., Dong L., et al. Cefminox, a dual agonist of prostacyclin receptor and peroxisome proliferator-activated receptor-gamma identified by virtual screening, has therapeutic efficacy against hypoxia-induced pulmonary hypertension in rats. Frontiers in Pharmacology . 2018;9:p. 134. doi: 10.3389/fphar.2018.00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Riquelme S. A., Hopkins B. D., Wolfe A. L., et al. Cystic Fibrosis Transmembrane Conductance Regulator Attaches Tumor Suppressor PTEN to the Membrane and Promotes Anti Pseudomonas aeruginosa Immunity. Immunity . 2017;47(6):1169–1181.e7. doi: 10.1016/j.immuni.2017.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Xing X. Q., Li B., Xu S. L., et al. 5-Aza-2′-deoxycytidine, a DNA methylation inhibitor, attenuates hypoxic pulmonary hypertension via demethylation of the PTEN promoter. European Journal of Pharmacology . 2019;855:227–234. doi: 10.1016/j.ejphar.2019.05.021. [DOI] [PubMed] [Google Scholar]
- 147.Li J., Zhou Q., Liang Y., et al. miR-486 inhibits PM2.5-induced apoptosis and oxidative stress in human lung alveolar epithelial A549 cells. Annals of Translational Medicine . 2018;6(11):p. 209. doi: 10.21037/atm.2018.06.09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Li J. W., Wei L., Han Z., Chen Z. Mesenchymal stromal cells-derived exosomes alleviate ischemia/reperfusion injury in mouse lung by transporting anti-apoptotic miR-21-5p. European Journal of Pharmacology . 2019;852:68–76. doi: 10.1016/j.ejphar.2019.01.022. [DOI] [PubMed] [Google Scholar]
- 149.Quan Y., Wang Z., Gong L., et al. Exosome miR-371b-5p promotes proliferation of lung alveolar progenitor type II cells by using PTEN to orchestrate the PI3K/Akt signaling. Stem Cell Research & Therapy . 2017;8(1):p. 138. doi: 10.1186/s13287-017-0586-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wang X., Liu M., Zhu M. J., et al. Resveratrol protects the integrity of alveolar epithelial barrier via SIRT1/PTEN/p-Akt pathway in methamphetamine-induced chronic lung injury. Cell Proliferation . 2020;53(3, article e12773) doi: 10.1111/cpr.12773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Navarro S., Reddy R., Lee J., Warburton D., Driscoll B. Inhaled resveratrol treatments slow ageing-related degenerative changes in mouse lung. Thorax . 2017;72(5):451–459. doi: 10.1136/thoraxjnl-2016-208964. [DOI] [PubMed] [Google Scholar]
- 152.Rao R., Nagarkatti P. S., Nagarkatti M. Δ(9) Tetrahydrocannabinol attenuates Staphylococcal enterotoxin B-induced inflammatory lung injury and prevents mortality in mice by modulation of miR-17-92 cluster and induction of T-regulatory cells. British Journal of Pharmacology . 2015;172(7):1792–1806. doi: 10.1111/bph.13026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The relevant data used to support the findings of this study are included within the article.