

Abstract
Background
This study investigated the effect of short-chain fatty acids (SCFAs) on diabetes in a mouse model.
Methods
Autophagy in Akita mice and streptozocin (STZ)-induced diabetic C57BL/6 mice was determined by Western blots and immunohistochemistry (IHC). Western blots, IHC, hematoxylin and eosin staining, Masson staining, periodic acid-Schiff staining, and picrosirius red staining were conducted to detect whether autophagy and renal function improved in Akita mice and STZ-induced diabetic C57BL/6 mice after treatment of SCFAs. Western blots, IHC, and chromatin immunoprecipitation were performed to determine whether SCFAs affected diabetic mice via the histone deacetylase (HDAC2)/unc-51 like autophagy activating kinase 1 (ULK1) axis. Diabetic mice with kidney-specific knockout of HDAC2 were constructed, and IHC, Masson staining, and Western blots were carried out to detect whether the deletion of endogenous HDAC2 contributed to the improvement of autophagy and renal fibrosis in diabetic mice.
Results
Reduced autophagy and severe fibrosis were observed in Akita mice and STZ-induced diabetic C57BL/6 mice. Increased autophagy and reduced renal cell fibrosis were found in SCFA-treated Akita diabetic mice and STZ-induced diabetic C57BL/6 mice. Diabetic mice treated with SCFAs had lower HDAC2 expression and more enriched binding of ULK1 promoter sequences to H3K27Ac. Endogenous knockout of HDAC2 caused enhanced autophagy and decreased renal fibrosis in diabetic mice treated with SCFAs.
Conclusion
SCFAs enhanced autophagy of renal tubular cells and attenuated renal fibrosis in diabetic mice through the HDAC2/ULK1 axis.
Keywords: Diabetes; Diabetic nephropathies; Fatty acids, volatile; Autophagy; Renal fibrosis
GRAPHICAL ABSTRACT

INTRODUCTION
Diabetes mellitus is a common metabolic disease characterized by hyperglycemia, which is related to chronic damage and dysfunction of various tissues, especially the eyes, kidneys, heart, blood vessels, and nerves [1,2]. Diabetes mellitus is a global public health challenge, with a prevalence of about 10% in adults worldwide, and the International Diabetes Federation has predicted that there will be 592 million cases by 2035 [3]. Diabetic nephropathy is a major cause of advanced renal disease, which is a predominant cause of morbidity and death in patients with diabetes mellitus [4,5]. Autophagy is considered a critical regulator of the response to insulin in target tissues, such as the liver, kidney, and adipose tissue, and is related to complications of diabetes mellitus, such as cardiovascular disorders and renal failure [6]. In diabetes, autophagy of renal tubular cells is involved in renal hypertrophy and tissue injury, ultimately leading to the development of diabetic nephropathy [4]. Renal fibrosis, which is characterized by abnormal proliferation of fibroblasts and excessive accumulation of extracellular matrix, is the main pathological feature of diabetic nephropathy [7]. A recent study has reported that the gut microbial environment plays an important role in various metabolic diseases, including intestinal disorders, diabetes mellitus, and kidney diseases [3].
Short-chain fatty acids (SCFAs), including acetic acid, propionic acid, and butyric acid, are important metabolites of gut microbes that are generated predominantly by the gut fermentation of dietary fiber [8,9]. Increasingly many studies have emphasized the significant role of SCFAs as emerging therapeutic targets for inflammatory diseases and metabolic disorders, such as diabetes and diabetic nephropathy [10-12]. It has been reported that SCFA levels dramatically increased when the severity of type 2 diabetes mellitus and diabetes-induced inflammation was attenuated by a flaxseed oil intervention [13]. Moreover, evidence indicated that SCFAs could modulate the inflammatory process, increase tubular proliferating cells, and enhance autophagy [14]. It was found that butyric acid could suppress renal inflammation and reduce the severity of cisplatin-induced renal injuries [15]. Similarly, another study also showed that butyric acid could ameliorate renal fibrosis in mice [16]. A previous report showed that valproic acid was involved in the development of renal fibrosis and kidney injury [17]. Based on the previous findings, this study focused on the explicit role of SCFAs in diabetic kidney injury. Prior evidence has demonstrated the involvement of histone deacetylase (HDAC) in the progression of various metabolic disorders, suggesting that HDAC inhibitors might be an emerging treatment agent for diabetes mellitus [7]. Interestingly, the inhibition of HDACs is a major mechanism underlying SCFA regulation in epithelial cells [18]. It has been reported that SCFAs can inhibit the activity of class-1/2 HDACs [19]. Hence, in this study, we aimed to explore the specific correlations among SCFAs, the autophagy of renal tubular cells, and renal fibrosis in diabetes and to determine the mechanism of SCFAs and HDACs.
METHODS
Animal model and groups
C57BL/6 and Akita mice were purchased from Cyagen Biotechnology Co. Ltd. (Taicang, China). Renal proximal tubule-specific knockout HDAC2 mice and their four genotypes obtained by crossbreeding with Akita mice and a streptozocin (STZ)-induced type I diabetic mouse model were purchased from the Institute of Biomedicine, Nanjing University. Littermates were used as controls in the knockout mouse model experiments. All animal experiments were conducted per the protocols approved by the Institutional Animal Care and Use Committee of Shaanxi Provincial People’s Hospital (No.202201089).
Specific pathogen-free male C57BL/6 mice (n=12, 20–30 g, age 6–8 weeks) were randomly divided into a normal group (control [CT]; n=6) and an STZ group (n=6) after acclimation for 2 weeks. In the STZ group, mice were intraperitoneally injected with 100 mg/kg STZ (dissolved in 0.1 mmol/L, pH 4.8 sodium citrate buffer) once every other day. Instead, in the CT group, mice were only injected with the same amount of sodium citrate buffer. After three STZ injections, the tail was cut for blood sampling and blood glucose measurements. Mice with a blood glucose level over 16.7 mmol/L (300 mg/dL) were considered to have experimentally induced diabetes [20].
Healthy wild-type mice were given distilled water, and diabetic Akita mice (randomly divided into four groups) were given distilled water, sodium propionate (SP), sodium butyrate (SB), and sodium valproate (SV) orally at 300 mg/kg/day for 8 weeks, respectively; these groups were named the wild type (WT) group, diabetes group, D+SP group, D+SB group, and D+SV group (six mice in each group). SP, SB, and SV were dissolved in distilled water for oral administration. The four genotypes of mice, including PT-HDAC2–/– Akita, PT-HDAC2+/+ Akita, PT-HDAC2–/– WT, and PT-HDAC2+/+ WT mice, were obtained by crossbreeding Akita mice with renal proximal tubule-specific knockout HDAC2 mice, and six mice were selected from each group for subsequent experiments.
Renal proximal tubule-specific knockout HDAC2 mice
C57BL/6 mice carrying the floxed HDAC2 allele (HDAC2 flox/flox) were generated by inserting loxp sites between exon 4 and 5 and between exon 6 and 7, respectively [21]. HDAC2flox/flox mice were crossbred with Cre-recombinase expressing transgenic mice, and Cre-recombinase was expressed under the control of a modified PEPCK promoter (PepcK-Cre). PEPCK-Cre is mainly expressed in renal proximal tubule cells and slightly expressed in hepatocytes [22]. After the first round of breeding, heterozygous female offspring (HDAC2flox/+XcreX) were obtained and then crossbred with HDAC2flox/flox male mice, producing HDAC2flox/floxXcreY mice (Pt-HDAC2-KO) with PEPCK-Cre-mediated HDAC2 deficiency in the renal proximal tubules. Since PEPCK-Cre is linked to the X chromosome, we only used male mice for the study to ensure that the genotype was correct.
Immunohistochemistry
Mice were anesthetized by 2 mg/100 g pentobarbital through a tail vein injection. The right atrium was cut immediately after left ventricular puncture. The kidney was infused with phosphate-buffered saline (PBS; pH 7.4) for 2 minutes until the kidney turned white, and then the left kidney was quickly clamped, placed on ice to separate the renal cortex and external and internal medulla, and stored in liquid nitrogen for Western blot analysis. Next, the heart was perfused with 4% paraformaldehyde (pH 7.4) until the tail was upturned. The right kidney was removed and paraffin sections of 4 μm were prepared after the isolated mouse renal medulla tissues were fixed in 4% paraformaldehyde for 48 hours. The sections were baked for 20 minutes and then deparaffinized with conventional xylene and washed once with distilled water. After washing three times with PBS, 3% H2O2 was added in drops. The sections were placed at room temperature for 10 minutes, and then washed again with PBS three times, after which thermal antigen retrieval was performed. After washing again with PBS three times, normal goat serum blocking solution was supplemented, and the sections were placed at room temperature for 20 minutes, with excess fluid shaken off. After the addition of primary antibodies—anti-light chain 3 (LC3) (NB100-2220, 1:300, Novus Biological, Littleton, CO, USA), anti-HDAC2 (51-5100, 1:20, Thermo Fisher, Waltham, MA, USA), and anti-H3acpan-acetyl (PA5-114693, 1:100, Thermo Fisher)—the sections were incubated overnight at 4°C, followed by washing with PBS three times, with supplementation of secondary antibodies and incubation for 1 hour at room temperature. After washing three times with PBS, color development was terminated after 1 to 3 minutes of 3,3´-diaminobenzidine (DAB) development. Five fields were randomly selected for observation, and the sections were scored based on staining intensity combined with the percentage of positive cells. In the tissue sections, positive cells were defined as light yellow or brown ones, and staining intensity was scored based on the staining characteristics exhibited by the majority of cells: 0 points for no staining, 1 point for light yellow cells, 2 points for dull yellowish-brown cells, and 3 points for tan cells. The percentage of positive cells was calculated by counting the number of positive cells in 100 cells in each field: 0 points, 0%–5%; 1 point, 6%–25%; 2 points, 26%–50%; 3 points, 51%–75%; 4 points, >75%. The staining intensity and percentage of positive cells were determined for each field, and the product of staining intensity and percentage of positive cells was calculated. Each experiment was repeated three times.
Picrosirius red staining
The change of type I and type III collagen in renal medulla tissues was observed by picrosirius red staining. Briefly, paraffin sections were routinely dewaxed to water. Picrosirius red solution (0.1% picrosirius red dissolved in saturated picric acid in aqueous solution; G1470) was used to dye the sections for 1 hour. After staining, the sections were rapidly washed twice with distilled water, differentiated with 95% alcohol, dehydrated with anhydrous alcohol, made transparent with xylene, and sealed with gum. Finally, a BX51 polarizing microscope (Olympus Corporation, Tokyo, Japan) was used to observe the sections, and Image Pro Plus version 6.0 (https://image-pro-plus. software.informer.com/download/) was used to calculate the relative area of red or yellow type I collagen and green type III collagen.
Masson staining
On day 7, Masson staining was performed to detect collagen deposition in renal medulla tissues. Specifically, dewaxed sections were first treated with hematoxylin solution (Shanghai Maokang Biotechnology Co. Ltd., Shanghai, China) for 6 minutes. Next, the sections were incubated with carmine solution (Shanghai Maikelin Biochemical Technology Co. Ltd., Shanghai, China) and acid magenta solution (Nanjing reagent) for 1 minute each, and with phosphomolybdate solution (Nanjing SenBeiJia Biotechnology Co. Ltd., Nanjing, China) for 5 minutes. Following incubation for 5 minutes with aniline blue solution (Shanghai Hongshun Biological Technology Co. Ltd., Shanghai, China), a microscope (×200) was used to observe collagen fibers stained blue by the Masson method, and collagen area was visualized under five random fields. The collagen area fraction was defined as the collagen area divided by the total area.
Periodic acid-Schiff staining
Paraffin sections of kidney tissues were routinely dewaxed to water, washed with distilled water for 1 minute, and soaked with periodic acid solution for 30 minutes. After rinsing in distilled water, the sections were soaked in Schiff reagent for 30 minutes in the dark, and then washed in sodium bisulfite solution for three times, 2 minutes each time. After that, the sections were redyed for 1 minute in hematoxylin and washed for 5 minutes, followed by alcohol gradient dehydration, the application of xylene for transparency, and neutral gum sealing. After fixation, the sections were visualized under a light microscope.
Hematoxylin and eosin staining
The renal medulla tissue sections on the 7th day were hydrated by a conventional alcohol gradient, made transparent by xylene, and rinsed with deionized water. The sections were stained by hematoxylin for 3 to 5 minutes, rinsed, and differentiated by 1% hydrochloric acid for 20 seconds, incubated with 1% ammonium hydroxide for 30 seconds, and rinsed. After that, the sections were counterstained with 1% eosin for 5 minutes, washed in running water for 5 minutes and deionized water for 5 minutes, and then dehydrated, made transparent (75% alcohol for 5 minutes, 90% alcohol for 5 minutes, 95% alcohol for 5 minutes, anhydrous alcohol for 5 minutes, xylene for 2×10 minutes), dried, and sealed. Finally, the sections were examined under a microscope and photographed.
Chromatin immunoprecipitation
First, the mouse kidney tissues were isolated and the kidney cells were extracted. A chromatin immunoprecipitation (ChIP) assay was performed to determine the binding of H3K27Ac to the unc-51 like autophagy activating kinase 1 (ULK1) promoter in the kidney cells with a ChIP kit (Millipore, Billerica, MA, USA) according to the manufacturer’s instructions. Cross-linking between DNA and protein was fixed with formaldehyde for 30 minutes and then DNA was isolated and sonicated into 200 to 1,000 bp fragments. After the incubation of fragmented DNA with H3K27Ac or immunoglobulin G antibodies, the pulleddown DNA fragments were analyzed by quantitative polymerase chain reaction.
Western blot
Renal tubule cells obtained from the mouse renal cortex or medulla were lysed by RIPA lysis solution (Beyotime, Shanghai, China) to obtain protein samples. After measuring the protein concentration with a bicinchoninic acid kit (Beyotime), the corresponding volume of protein was taken and added to the sample buffer (Beyotime), followed by mixing well and heating in a boiling water bath for 3 minutes for protein denaturation. Electrophoresis was performed at 80 V for 30 minutes, switching to 120 V after bromophenol blue entered the separation gel, followed by electrophoresis for 1 to 2 hours. The membrane was transferred in an ice bath with a current of 300 mA for 60 minutes. After membrane transfer, the membrane was rinsed in the washing solution for 1 to 2 minutes, and then placed in the blocking solution at room temperature for 60 minutes, or at 4°C overnight. The membrane was incubated with primary antibodies against ULK1 (a7481, 1:1,000), actin (A5441, 1:10,000), cyclophilin B (SAB4200201, 1:5,000, Millipore Sigma, Burlington, MA, USA), and LC3 (NB100-2220, 1:1,000, Novus, Littleton, CO, USA) for 1 hour at room temperature on a shaker, and washed with washing solution (three times, 10 min/time). The membrane was transferred to secondary antibodies, incubated for 1 hour at room temperature and washed three times (10 min/time). After adding the developer dropwise onto the membrane, a chemiluminescence imaging system was used to detect bands on the membrane (GelDocXR, Bio-Rad, Hercules, CA, USA).
Statistical analysis
The statistical analysis was conducted with GraphPad Prism 8, and all data were exhibited as mean±standard deviation. The t test was utilized for inter-group comparisons, and one-way analysis of variance was used for comparisons among multiple groups, with the Tukey multiple-comparison test for post hoc multiple comparisons. P<0.05 was considered indicative of statistical significance.
RESULTS
Decreased autophagy in diabetic mice In this study, we intended to explore the effects of SCFAs on diabetes. Western blots performed with the renal cortical tissue of the diabetic mice revealed that the protein expressions of both LC3-I and LC3-II were clearly down-regulated in the diabetic mice versus the WT mice (P<0.001) (Fig. 1A, C). The immunohistochemistry (IHC) assay results indicated that the expression of LC3 in renal tubular cells was significantly lower in the diabetic mice than in the normal mice (P<0.001) (Fig. 1B, D). Those observations suggest decreased autophagy in diabetic mice.
Fig. 1.

Decreased autophagy in renal tubular cells of diabetic mice. (A) Expression of light chain 3 (LC3)-I and LC3-II in renal cortical tissue cells of 14-week-old Akita mice detected by Western blots. (B) LC3 expression in renal tubular cells of 14-week-old Akita mice measured by immunohistochemistry (IHC). (C) Expression of LC3-I and LC3-II in renal cortical tissue cells of streptozocin (STZ)-treated C57BL/6 diabetic mice measured by Western blots. (D) LC3 expression in renal tubular cells of STZ-treated mice detected by IHC. The t test was utilized for two-group comparisons. One-way analysis of variance was used for comparisons among multiple groups, and the Tukey multiple-comparison test was used for post hoc multiple comparisons. The effect size between different experimental groups and control groups was >0.8. WT, wild-type; CT, control. aP<0.05; bP<0.01; cP<0.001 compared with WT mice (n=6).
SCFAs enhanced autophagy in the renal tubular cells of diabetic mice
The Akita diabetic mice were orally administered a certain amount of SCFAs once a day for 14 weeks, and Western blots demonstrated that the protein expression of LC3-I and LC3-II in the tubular cells of the diabetic mice that were orally administered SP, SB, and SV were markedly higher than in the Akita diabetic mice (P<0.01) (Fig. 2A, B). The C57BL/6 diabetic mice induced with STZ were administered a certain amount of SCFAs for 14 days, and Western blots revealed that the protein expression of LC3-I and LC3-II in renal tubular cells of the diabetic mice that were orally administered SP, SB, and SV were markedly up-regulated versus the C57BL/6 diabetic mice induced by STZ (P<0.01) (Fig. 2C, D). These findings demonstrate that autophagy was clearly enhanced in the renal tubular cells of diabetic mice after a certain period of SCFA administration.
Fig. 2.
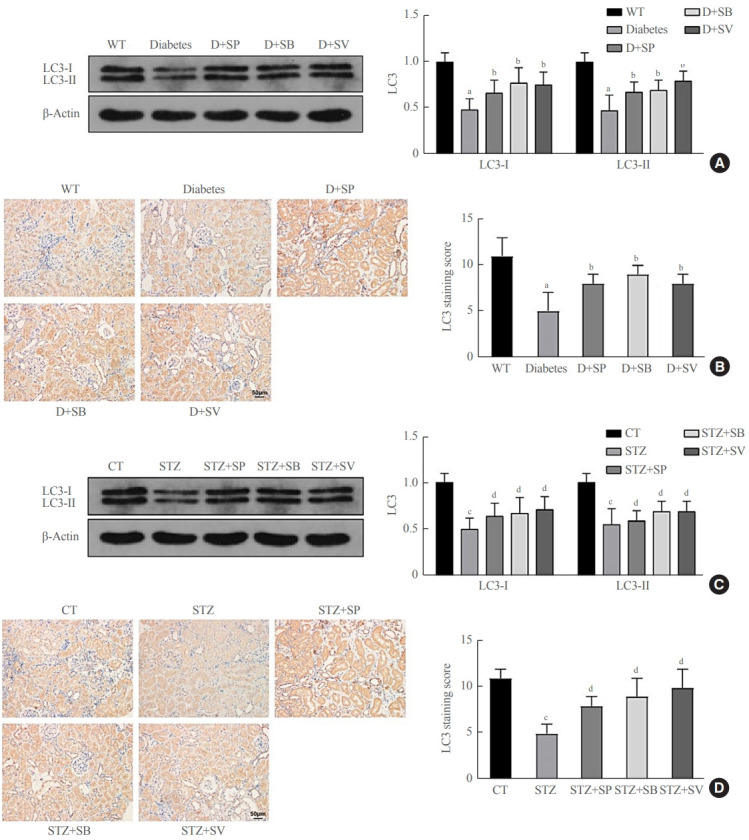
Short-chain fatty acids (SCFAs) enhanced autophagy in renal tubular cells of diabetic mice. Diabetic Akita mice were randomly divided into four groups. Healthy wild-type mice were fed distilled water, and diabetic Akita mice were fed with distilled water, sodium propionate, sodium butyrate, and sodium valproate respectively, per the standard of 300 mg/kg/day for 14 weeks; these were recorded as the wildtype (WT), diabetes, diabetes (D)+sodium propionate (SP), D+sodium butyrate (SB), and D+sodium valproate (SV) groups. (A) Expression of light chain 3 (LC3)-I and LC3-II in renal cortical tissues of WT and Akita mice detected by Western blots. (B) LC3 expression in renal tubular cells of WT and Akita mice measured by immunohistochemistry (IHC). C57BL/6 mice were randomly divided into five groups; after diabetes was induced by streptozocin (STZ) treatment, mice were fed with distilled water, sodium propionate, sodium butyrate, or sodium valproate for 14 weeks. These groups were recorded as control (CT; fed with distilled water without diabetes induction), STZ, STZ+SP, STZ+SB, and STZ+SV. (C) Expressions of LC3-I and LC3-II in renal cortical tissues of C57BL/6 control mice and diabetic mice measured by Western blots. (D) Expression of LC3 in renal tubular cells of C57BL/6 control mice and diabetic mice detected by IHC. One-way analysis of variance was used for comparisons among multiple groups, and the Tukey multiple-comparison test was used for post hoc multiple comparisons. The effect size between different experimental groups and control groups was >0.8. aP<0.01 compared with WT mice fed with distilled water; bP<0.05 compared with diabetic Akita mice fed with distilled water; cP<0.01 compared with WT mice; dP<0.05 compared with STZ-treated mice fed with distilled water (n=6).
SCFAs reduced renal fibrosis in diabetic mice
Previous evidence showed that patients with autophagy had complications of diabetic nephropathy rather than autophagy disorder [23]. We therefore further investigated whether SCFAs had other roles in addition to affecting autophagy in diabetic mice. The histological examination demonstrated that both the Akita mice and the STZ-induced C57BL/6 mice showed more severe mesangial matrix deposition and more tubular vacuolation than the control mice (P<0.001) (Fig. 3), and the kidney injury of the diabetic mice fed SCFAs for 14 weeks exhibited definite improvement compared with diabetic mice fed distilled water (P<0.05) (Fig. 3). Picrosirius red staining demonstrated that the Akita mice and the STZ-induced C57BL/6 mice had more obvious fibrosis than the WT mice, while the symptoms of renal fibrosis were significantly ameliorated after mice were fed SCFAs for 14 weeks (P<0.05) (Fig. 3). The above observations suggest that SCFAs could reduce renal fibrosis in mice with diabetes.
Fig. 3.
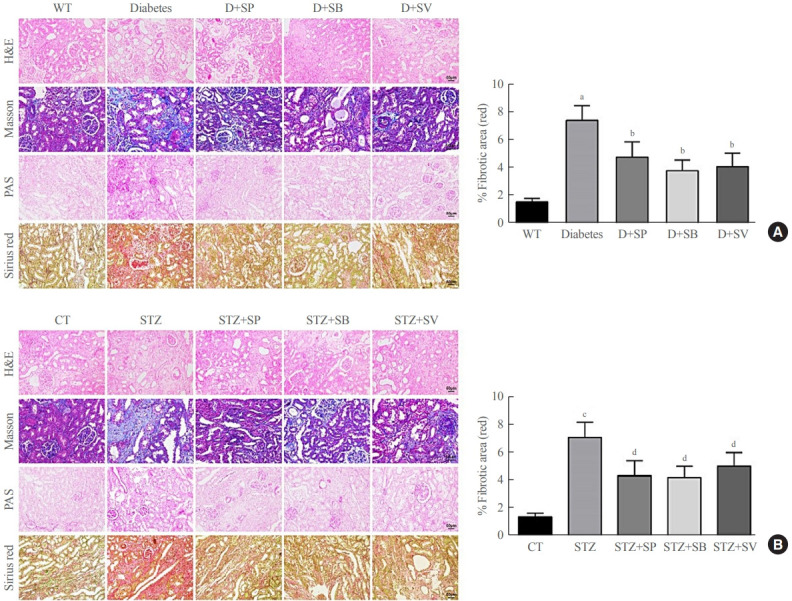
Short-chain fatty acids (SCFAs) reduced renal fibrosis in diabetic mice. hematoxylin-eosin (H&E), Masson, periodic acid-Schiff (PAS), and picrosirius red staining were performed on renal medulla tissue sections in each experimental group and the control group. (A) Staining results of Akita diabetic mice. (B) Staining results of streptozocin (STZ)-induced C57BL/6 mice. One-way analysis of variance was used for comparisons among multiple groups, and the Tukey multiple-comparison test was used for post hoc multiple comparisons. The effect size between different experimental groups and control groups was >0.8. WT, wild type; D, diabetes; SP, sodium propionate; SB, sodium butyrate; SV, sodium valproate; CT, control. aP<0.01 compared with the WT group; bP<0.05 compared with diabetic mice; cP<0.01 compared with the CT group; dP<0.05 compared with STZ mice (n=6).
Effects of SCFAs on HDAC2 and histone acetylation in diabetic mice
The expression of HDAC and histone acetylation in diabetic mice were detected by IHC and the results manifested that diabetic Akita mice treated with SCFAs had markedly lowered HDAC2 expression than untreated Akita mice, while without obvious change in expressions of HDAC4/5/7 (data not shown). However, diabetic Akita mice after treatment with SCFAs had significantly elevated histone H3 acetylation levels compared with untreated Akita mice (P<0.01) (Fig. 4). Those findings suggested that SCFAs might affect diabetic mice by suppressing HDAC2 expression, enhancing the acetylation of histone H3, and increasing the transcription and protein expressions of related genes.
Fig. 4.
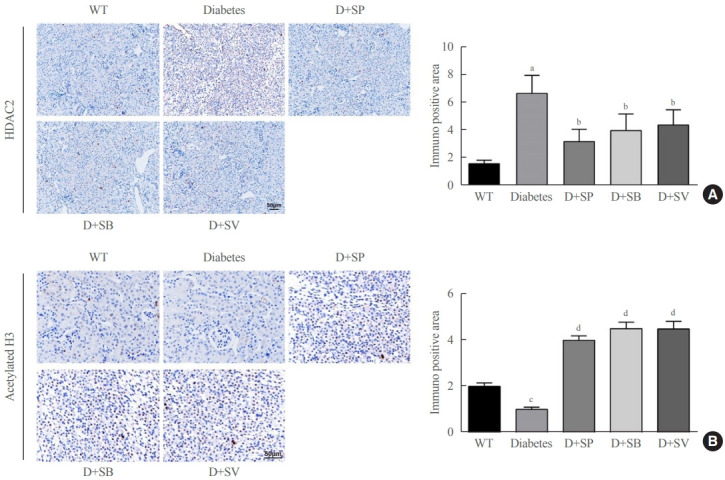
Effects of short-chain fatty acids (SCFAs) on histone deacetylase (HDAC) and histone acetylation in diabetic mice. (A) HDAC2 expression in Akita mice in the five groups detected by immunohistochemistry (IHC). (B) H3 histone acetylation levels in the five groups of Akita mice detected by IHC. One-way analysis of variance was used for comparisons among multiple groups, and the Tukey multiple-comparison test was used for post hoc multiple comparisons. The effect size between different experimental groups and control groups was >0.8. WT, wild type; D, diabetes; SP, sodium propionate; SB, sodium butyrate; SV, sodium valproate. aP<0.01 compared with the WT group; bP<0.05 compared with the diabetes group; cP<0.05 compared with the WT group; dP<0.01 compared with the diabetes group (n=6).
SCFAs affected autophagy and renal fibrosis in diabetic mice through the HDAC2/ULK1 axis
The above experiments found that SCFAs were likely to promote histone H3 acetylation and enhance transcription of related downstream genes by inhibiting the expression of HDAC2. We found that the ULK1 promoter region had a peak for H3K27Ac (Fig. 5A), suggesting that ULK1 might be regulated by histone acetylation. Using Western blots, it was found the expression of ULK1 in Akita diabetic mice was significantly down-regulated compared with that in WT normal mice. In addition, the expression of ULK1 in Akita diabetic mice that were fed SCFAs markedly increased (P<0.05) (Fig. 5B). The ChIP assay also demonstrated that binding of the ULK1 promoter sequences to H3K27Ac was significantly reduced in Akita mice, but significantly up-regulated in Akita mice after treatment with SCFAs (P<0.05) (Fig. 5C). Those results suggested that SCFAs might reduce renal fibrosis and enhance autophagy in diabetic mice via HDAC2/ULK1. For further confirmation of this result, we crossbred Akita mice with renal tubular-specific knockout HDAC2 (PT-HDAC2−/−) mice to obtain PT-HDAC2−/− Akita mice and PT-HDAC2+/+ Akita mice. The kidney-weight ratio was markedly lower in the PT-HDAC2−/− Akita mice than in the PT-HDAC2+/+ Akita mice (P<0.05) (Fig. 5D). Masson staining revealed that kidney fibrosis was markedly alleviated in PT-HDAC2−/− Akita mice versus PT-HDAC2+/+ Akita mice (P<0.05) (Fig. 5E). The IHC results showed significantly higher levels of autophagy in PT-HDAC2−/− Akita mice than in PT-HDAC2+/+ Akita mice (P< 0.05) (Fig. 5F), while the expression of ULK1 was clearly up-regulated in PT-HDAC2−/− Akita mice compared with PT-HDAC2+/+ Akita mice (P<0.05) (Fig. 5G). These observations are consistent with the conclusion that SCFAs could reduce renal fibrosis and enhance autophagy in Akita mice, further illustrating that SCFAs affected autophagy and renal fibrosis in diabetic mice through the HDAC2/ULK1 axis.
Fig. 5.

Short-chain fatty acids (SCFAs) affected autophagy and renal fibrosis in diabetic mice through the histone deacetylase (HDAC2)/unc51 like autophagy activating kinase 1 (ULK1) axis. (A) The ULK1 promoter region in the University of California Santa Cruz (UCSC) database had a peak for H3K27Ac. (B) ULK1 expression in the wild type (WT), diabetes (D)+sodium propionate (SP), D+sodium butyrate (SB), and D+sodium valproate (SV) mice renal medulla tissues and cells obtained from the renal cortex or medulla was measured by Western blots. (C) ULK1 promoter sequences bound to H3K27Ac in mice renal tissues and cells obtained from the renal cortex or medulla from the above five groups detected by chromatin immunoprecipitation (ChIP). (D) The kidneys from 14-week-old mice of four genotypes were weighed, with the ratio of kidney to body weight calculated. (E) The degree of renal medulla fibrosis in mice of four genotypes detected by Masson staining. (F) Light chain 3 (LC3) expression measured by immunochemistry (IHC) in mice of the four genotypes. (G) ULK1 expression in mice of the four genotypes detected by Western blots. One-way analysis of variance was used for comparisons among multiple groups, and the Tukey multiple-comparison test was used for post hoc multiple comparisons. The effect size between different experimental groups and control groups was >0.8. aP<0.05 compared with the WT group; bP<0.05 compared with the diabetes group; cP<0.05; dP<0.01; eP<0.01 (n=6).
DISCUSSION
Autophagy and renal fibrosis are critical processes in the disease progression of diabetes mellitus [6,7]. Patients with diabetes mellitus are also prone to intestinal flora disorders, such as the reduction of beneficial bacteria and/or the increase of harmful bacteria [24]. As gut microbiota have been reported to be involved in the development of metabolic disorders, in this study, we investigated the effects of SCFAs on diabetes in a mouse model.
First, our study found that autophagy of renal tubular cells decreased in diabetic mice. Consistent with our result, a previous study indicated that tubule epithelial cell autophagy was defective in both types of diabetes at early and advanced stages and that autophagy may play a protective role in diabetes [25]. Autophagy impairment in diabetes might lead to renal hypertrophy, pathological changes, and the development of diabetic nephropathy [4]. A subsequent experiment suggested that SCFAs enhanced autophagy in renal tubular cells of diabetic mice. SCFAs, which are products of dietary fiber fermentation, are crucial mediators of host-microbial interactions, such as the regulation of inflammation by lowering stress in epithelium and gut immunity with secondary bile acids and tryptophan [26]. In a mouse model of chronic kidney disease (CKD) transplanted with microbiota from CKD patients, butyrate supplementation repressed CKD development and reduced the production of trimethylamine N-oxide, a product of gut microbiota-mediated metabolism that is correlated with renal function impairment [27]. In diabetic mice, dietary fiber protects against nephropathy by modulating the gut microbiota and enriching SCFA-producing bacteria, and SCFAs are key regulators of the renoprotective effects of dietary fiber [10]. Andrade-Oliveira et al. [14] reported that the presence of SCFAs, especially acetate, lowered apoptosis, increased the number of tubular proliferating cells, and activated the autophagy pathway in mice with acute kidney injury. Hence, treatment with SCFAs may activate the autophagy pathway and thus provide benefits in the progression of diabetes. Another important observation in our study was that SCFAs reduced renal fibrosis in mice with diabetes. Renal fibrosis is a common pathway in CKD that eventually leads to terminal renal failure [7,28]. Published evidence showed that SCFAs were beneficial for cystic fibrosis-specific improvement and attenuated inflammation [29,30]. Another study also demonstrated that SCFAs and fecal microbiota were correlated with fibrosis in primary biliary cholangitis [31]. Those reports all provide evidence that supports our findings. Briefly, the presence of SCFAs might ameliorate the progression of diabetes by enhancing autophagy and reducing renal fibrosis.
Once absorbed into the host bloodstream, SCFAs exert cellular effects through two major categories: G-protein coupled receptor (GPCR)-regulated and non-GPCR-mediated effects, and the latter includes the transportation of SCFAs across membranes by transporters and SCFA-mediated inhibition of HDACs [32]. Notably, HDACs have been found to be involved in the progression of various metabolic disorders, including diabetes mellitus, and SCFAs were related to the activity of class-1/2 HDACs [33,34]. Our study found that SCFAs promoted histone H3 acetylation in the ULK1 promoter and enhanced transcript levels of related downstream genes by inhibiting the expression of HDAC2. Similar to our findings, a previous study indicated that SCFAs (SP and SB) decreased inflammation by suppressing HDAC activity and elevating histone H3 acetylation [19]. Previous evidence also showed that SCFAs inhibited the activity of class-1/2 HDACs and might act as inhibitors of HDAC, suggesting their potential as a promising treatment for various disorders [35]. For example, butyrate was metabolized less in tumors, where it accumulated and functioned as an HDAC inhibitor that stimulated histone acetylation, affected apoptosis and cell proliferation, and reduced colorectal tumorigenesis [36]. As regulatory proteins involved in glucose metabolism, HDACs have been confirmed to regulate insulin gene transcription [37]. SB treatment significantly elevated insulin receptor substrate-1 phosphorylation at a tyrosine residue, a key mediator of insulin signaling, and improved beta-cell survival and glucose metabolism through the regulation of p38/extracellular signal-regulated kinase (ERK)/mitogen-activated protein kinase (MAPK) pathway [38]. We found that the ULK1 promoter region had an H3K27Ac peak, suggesting that ULK1 might be regulated by histone acetylation. The results of our experiment revealed that ULK1 was down-regulated in Akita diabetic mice and treatment with SCFAs enhanced the expression of ULK1. Knockout of endogenous HDAC2 in diabetic mice could also rescue ULK1 expression. RNA-sequencing analysis has shown that HDAC2 is a negative target regulatory molecule of ULK1, and HDAC2 antagonizes the effect of ULK1 during the process of pyroptosis [39]. A further experiment found that SCFAs might affect autophagy and renal fibrosis in diabetic mice through the HDAC2/ULK1 axis. Evidence demonstrated that induction of autophagy was regulated predominantly via ULK1 in diabetes [6]. The impairment of autophagy in diabetic nephropathy was linked with low ULK1 expression [4]. However, few studies have investigated the association between SCFAs and ULK1. In addition to HDAC2 inhibition, SCFAs could regulate autophagy by stabilizing intestinal epithelial hypoxia-inducible factor-1α (HIF-1α) and thereby alleviating experimental-induced colitis [40]; SCFAs induced autophagy in hepatic cells by stimulating peroxisome proliferator activated receptor gamma (PPARγ)-dependent uncoupling protein 2 expression [41].
In summary, our study found that SCFAs might enhance autophagy and reduce renal fibrosis in diabetic mice by inhibiting HDAC2 and up-regulating ULK1. Nevertheless, this study has some limitations. First, the occurrence and progression of diabetes is a complex process and many factors may affect the disease course other than autophagy and renal fibrosis. It remains unclear whether SCFAs have effects on other risk factors of diabetes. Although it has been confirmed that SCFAs could be transported into the apical membrane in the colon by a sodiumcoupled mono-carboxylate transporter (SLA5A8) and that SCFAs could also be transported by SCFA-permeant ion channels, organic anion transporters, or monocarboxylate transporter 1 [32], there is a paucity of research on the transporters on kidney cells for transportation of SCFAs. Additionally, the presence of SCFAs may regulate the expression of other molecules and pathways; however, our study only focused on HDAC2 and ULK1. In brief, the findings of our study may provide insights onto promising therapeutic strategies for diabetes.
Acknowledgments
We would like to thank Dr. Le Ji from the Department of Orthopedics of Shaanxi Provincial People’s Hospital for the valuable comments on the experimental design and to thank the Clinical Experimental Center for providing the experimental site free of charge. We would like to thank our family and colleagues for their support.
Footnotes
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
AUTHOR CONTRIBUTIONS
Conception or design: X.M., Q.W. Acquisition, analysis, or interpretation of data: X.M. Drafting the work or revising: X.M. Final approval of the manuscript: X.M., Q.W.
REFERENCES
- 1.ValdezGuerrero AS, Quintana-Perez JC, Arellano-Mendoza MG, Castaneda-Ibarra FJ, Tamay-Cach F, Aleman-Gonzalez-Duhart D. Diabetic retinopathy: important biochemical alterations and the main treatment strategies. Can J Diabetes. 2021;45:504–11. doi: 10.1016/j.jcjd.2020.10.009. [DOI] [PubMed] [Google Scholar]
- 2.Muriach M, Flores-Bellver M, Romero FJ, Barcia JM. Diabetes and the brain: oxidative stress, inflammation, and autophagy. Oxid Med Cell Longev. 2014;2014:102158. doi: 10.1155/2014/102158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aw W, Fukuda S. Understanding the role of the gut ecosystem in diabetes mellitus. J Diabetes Investig. 2018;9:5–12. doi: 10.1111/jdi.12673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma Z, Li L, Livingston MJ, Zhang D, Mi Q, Zhang M, et al. p53/microRNA-214/ULK1 axis impairs renal tubular autophagy in diabetic kidney disease. J Clin Invest. 2020;130:5011–26. doi: 10.1172/JCI135536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin YC, Chang YH, Yang SY, Wu KD, Chu TS. Update of pathophysiology and management of diabetic kidney disease. J Formos Med Assoc. 2018;117:662–75. doi: 10.1016/j.jfma.2018.02.007. [DOI] [PubMed] [Google Scholar]
- 6.Barlow AD, Thomas DC. Autophagy in diabetes: β-cell dysfunction, insulin resistance, and complications. DNA Cell Biol. 2015;34:252–60. doi: 10.1089/dna.2014.2755. [DOI] [PubMed] [Google Scholar]
- 7.Xu BH, Sheng J, You YK, Huang XR, Ma R, Wang Q, et al. Deletion of Smad3 prevents renal fibrosis and inflammation in type 2 diabetic nephropathy. Metabolism. 2020;103:154013. doi: 10.1016/j.metabol.2019.154013. [DOI] [PubMed] [Google Scholar]
- 8.Kobayashi M, Mikami D, Kimura H, Kamiyama K, Morikawa Y, Yokoi S, et al. Short-chain fatty acids, GPR41 and GPR43 ligands, inhibit TNF-α-induced MCP-1 expression by modulating p38 and JNK signaling pathways in human renal cortical epithelial cells. Biochem Biophys Res Commun. 2017;486:499–505. doi: 10.1016/j.bbrc.2017.03.071. [DOI] [PubMed] [Google Scholar]
- 9.Mikami D, Kobayashi M, Uwada J, Yazawa T, Kamiyama K, Nishimori K, et al. Short-chain fatty acid mitigates adenine-induced chronic kidney disease via FFA2 and FFA3 pathways. Biochim Biophys Acta Mol Cell Biol Lipids. 2020;1865:158666. doi: 10.1016/j.bbalip.2020.158666. [DOI] [PubMed] [Google Scholar]
- 10.Li YJ, Chen X, Kwan TK, Loh YW, Singer J, Liu Y, et al. Dietary fiber protects against diabetic nephropathy through short-chain fatty acid-mediated activation of G protein-coupled receptors GPR43 and GPR109A. J Am Soc Nephrol. 2020;31:1267–81. doi: 10.1681/ASN.2019101029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kasubuchi M, Hasegawa S, Hiramatsu T, Ichimura A, Kimura I. Dietary gut microbial metabolites, short-chain fatty acids, and host metabolic regulation. Nutrients. 2015;7:2839–49. doi: 10.3390/nu7042839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wen L, Wong FS. Dietary short-chain fatty acids protect against type 1 diabetes. Nat Immunol. 2017;18:484–6. doi: 10.1038/ni.3730. [DOI] [PubMed] [Google Scholar]
- 13.Zhu L, Sha L, Li K, Wang Z, Wang T, Li Y, et al. Dietary flaxseed oil rich in omega-3 suppresses severity of type 2 diabetes mellitus via anti-inflammation and modulating gut microbiota in rats. Lipids Health Dis. 2020;19:20. doi: 10.1186/s12944-019-1167-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andrade-Oliveira V, Amano MT, Correa-Costa M, Castoldi A, Felizardo RJ, de Almeida DC, et al. Gut bacteria products prevent AKI induced by ischemia-reperfusion. J Am Soc Nephrol. 2015;26:1877–88. doi: 10.1681/ASN.2014030288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsiao YP, Chen HL, Tsai JN, Lin MY, Liao JW, Wei MS, et al. Administration of Lactobacillus reuteri combined with Clostridium butyricum attenuates cisplatin-induced renal damage by gut microbiota reconstitution, increasing butyric acid production, and suppressing renal inflammation. Nutrients. 2021;13:2792. doi: 10.3390/nu13082792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang H, Qian J, Zhao X, Xing C, Sun B. β-Aminoisobutyric acid ameliorates the renal fibrosis in mouse obstructed kidneys via inhibition of renal fibroblast activation and fibrosis. J Pharmacol Sci. 2017;133:203–13. doi: 10.1016/j.jphs.2016.12.005. [DOI] [PubMed] [Google Scholar]
- 17.Khan S, Jena G, Tikoo K. Sodium valproate ameliorates diabetes-induced fibrosis and renal damage by the inhibition of histone deacetylases in diabetic rat. Exp Mol Pathol. 2015;98:230–9. doi: 10.1016/j.yexmp.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 18.Tan J, McKenzie C, Potamitis M, Thorburn AN, Mackay CR, Macia L. The role of short-chain fatty acids in health and disease. Adv Immunol. 2014;121:91–119. doi: 10.1016/B978-0-12-800100-4.00003-9. [DOI] [PubMed] [Google Scholar]
- 19.Silva LG, Ferguson BS, Avila AS, Faciola AP. Sodium propionate and sodium butyrate effects on histone deacetylase (HDAC) activity, histone acetylation, and inflammatory gene expression in bovine mammary epithelial cells. J Anim Sci. 2018;96:5244–52. doi: 10.1093/jas/sky373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Orinska Z, Maurer M, Mirghomizadeh F, Bulanova E, Metz M, Nashkevich N, et al. IL-15 constrains mast cell-dependent antibacterial defenses by suppressing chymase activities. Nat Med. 2007;13:927–34. doi: 10.1038/nm1615. [DOI] [PubMed] [Google Scholar]
- 21.Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rankin EB, Tomaszewski JE, Haase VH. Renal cyst development in mice with conditional inactivation of the von Hippel-Lindau tumor suppressor. Cancer Res. 2006;66:2576–83. doi: 10.1158/0008-5472.CAN-05-3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Riser BL, Najmabadi F, Perbal B, Rambow JA, Riser ML, Sukowski E, et al. CCN3/CCN2 regulation and the fibrosis of diabetic renal disease. J Cell Commun Signal. 2010;4:39–50. doi: 10.1007/s12079-010-0085-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma Q, Li Y, Li P, Wang M, Wang J, Tang Z, et al. Research progress in the relationship between type 2 diabetes mellitus and intestinal flora. Biomed Pharmacother. 2019;117:109138. doi: 10.1016/j.biopha.2019.109138. [DOI] [PubMed] [Google Scholar]
- 25.Sakai S, Yamamoto T, Takabatake Y, Takahashi A, Namba-Hamano T, Minami S, et al. Proximal tubule autophagy differs in type 1 and 2 diabetes. J Am Soc Nephrol. 2019;30:929–45. doi: 10.1681/ASN.2018100983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gasaly N, de Vos P, Hermoso MA. Impact of bacterial metabolites on gut barrier function and host immunity: a focus on bacterial metabolism and its relevance for intestinal inflammation. Front Immunol. 2021;12:658354. doi: 10.3389/fimmu.2021.658354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang S, Lv D, Jiang S, Jiang J, Liang M, Hou F, et al. Quantitative reduction in short-chain fatty acids, especially butyrate, contributes to the progression of chronic kidney disease. Clin Sci (Lond) 2019;133:1857–70. doi: 10.1042/CS20190171. [DOI] [PubMed] [Google Scholar]
- 28.Zheng C, Huang L, Luo W, Yu W, Hu X, Guan X, et al. Inhibition of STAT3 in tubular epithelial cells prevents kidney fibrosis and nephropathy in STZ-induced diabetic mice. Cell Death Dis. 2019;10:848. doi: 10.1038/s41419-019-2085-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghorbani P, Santhakumar P, Hu Q, Djiadeu P, Wolever TM, Palaniyar N, et al. Short-chain fatty acids affect cystic fibrosis airway inflammation and bacterial growth. Eur Respir J. 2015;46:1033–45. doi: 10.1183/09031936.00143614. [DOI] [PubMed] [Google Scholar]
- 30.Ohira H, Tsutsui W, Fujioka Y. Are short chain fatty acids in gut microbiota defensive players for inflammation and atherosclerosis? J Atheroscler Thromb. 2017;24:660–72. doi: 10.5551/jat.RV17006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lammert C, Shin A, Xu H, Hemmerich C, O’Connell TM, Chalasani N. Short-chain fatty acid and fecal microbiota profiles are linked to fibrosis in primary biliary cholangitis. FEMS Microbiol Lett. 2021;368:fnab038. doi: 10.1093/femsle/fnab038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pluznick JL. Gut microbiota in renal physiology: focus on short-chain fatty acids and their receptors. Kidney Int. 2016;90:1191–8. doi: 10.1016/j.kint.2016.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Makkar R, Behl T, Arora S. Role of HDAC inhibitors in diabetes mellitus. Curr Res Transl Med. 2020;68:45–50. doi: 10.1016/j.retram.2019.08.001. [DOI] [PubMed] [Google Scholar]
- 34.Ho RH, Chan J, Fan H, Kioh D, Lee BW, Chan E. In silico and in vitro interactions between short chain fatty acids and human histone deacetylases. Biochemistry. 2017;56:4871–8. doi: 10.1021/acs.biochem.7b00508. [DOI] [PubMed] [Google Scholar]
- 35.Yu X, Shahir AM, Sha J, Feng Z, Eapen B, Nithianantham S, et al. Short-chain fatty acids from periodontal pathogens suppress histone deacetylases, EZH2, and SUV39H1 to promote Kaposi’s sarcoma-associated herpesvirus replication. J Virol. 2014;88:4466–79. doi: 10.1128/JVI.03326-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Donohoe DR, Holley D, Collins LB, Montgomery SA, Whitmore AC, Hillhouse A, et al. A gnotobiotic mouse model demonstrates that dietary fiber protects against colorectal tumorigenesis in a microbiota- and butyrate-dependent manner. Cancer Discov. 2014;4:1387–97. doi: 10.1158/2159-8290.CD-14-0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Christensen DP, Dahllof M, Lundh M, Rasmussen DN, Nielsen MD, Billestrup N, et al. Histone deacetylase (HDAC) inhibition as a novel treatment for diabetes mellitus. Mol Med. 2011;17:378–90. doi: 10.2119/molmed.2011.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khan S, Jena GB. Protective role of sodium butyrate, a HDAC inhibitor on beta-cell proliferation, function and glucose homeostasis through modulation of p38/ERK MAPK and apoptotic pathways: study in juvenile diabetic rat. Chem Biol Interact. 2014;213:1–12. doi: 10.1016/j.cbi.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 39.Wang Y, Chen Q, Jiao F, Shi C, Pei M, Wang L, et al. Histone deacetylase 2 regulates ULK1 mediated pyroptosis during acute liver failure by the K68 acetylation site. Cell Death Dis. 2021;12:55. doi: 10.1038/s41419-020-03317-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou C, Li L, Li T, Sun L, Yin J, Guan H, et al. SCFAs induce autophagy in intestinal epithelial cells and relieve colitis by stabilizing HIF-1α. J Mol Med (Berl) 2020;98:1189–202. doi: 10.1007/s00109-020-01947-2. [DOI] [PubMed] [Google Scholar]
- 41.Iannucci LF, Sun J, Singh BK, Zhou J, Kaddai VA, Lanni A, et al. Short chain fatty acids induce UCP2-mediated autophagy in hepatic cells. Biochem Biophys Res Commun. 2016;480:461–7. doi: 10.1016/j.bbrc.2016.10.072. [DOI] [PubMed] [Google Scholar]